

Abstract
Isatuximab, an anti‐CD38 monoclonal antibody, targets cells that strongly express CD38 including malignant plasma cells. This open‐label, single‐arm, multicenter, phase 1/2 trial investigated the tolerability/safety and efficacy of isatuximab monotherapy in Japanese patients with heavily pretreated, relapsed/refractory multiple myeloma (RRMM). In Phase 1, patients were sequentially assigned to receive isatuximab once weekly (QW) in cycle 1 (4 weeks) and every 2 weeks (Q2W) in subsequent cycles. Cohort 1 (n = 3) received 10 mg/kg QW/Q2W; cohort 2 (n = 5) received 20 mg/kg QW/Q2W. No dose‐limiting toxicities occurred; the recommended dose for the single‐arm phase 2 study (n = 28) was 20 mg/kg QW/Q2W. The overall safety profile was consistent with the current knowledge of isatuximab. The most common adverse events were infusion reactions (42.9%; 12/28); all were grade 1/2 and generally occurred during the first infusion. The overall response rate with 20 mg/kg QW/Q2W isatuximab was 36.4% (12/33); patients with high‐risk cytogenetic abnormalities had comparable results. In phase 2, the median progression‐free survival was 4.7 (95% confidence interval, 3.75 to not reached) months. Median overall survival was not reached. Isatuximab monotherapy was well tolerated and effective in patients with heavily pretreated RRMM including high‐risk cytogenetic patients. This trial is registered at ClinicalTrials.gov as NCT02812706.
Keywords: clinical trial, isatuximab, multiple myeloma, safety, survival
This open‐label, single‐arm, multicenter phase 1/2 trial investigated the tolerability/safety and efficacy of isatuximab monotherapy in Japanese patients with heavily pretreated, relapsed/refractory multiple myeloma (RRMM). The results demonstrated that isatuximab monotherapy was well tolerated and effective in patients with heavily pretreated RRMM, including high‐risk cytogenetic patients.

Abbreviations
- ADI
actual dose intensity
- AE
adverse event
- AUC1week
area under the plasma concentration versus time in the 1‐week dosing interval
- CBR
clinical benefit rate
- Ceoi
concentration at the end of infusion
- CI
confidence interval
- Cmax
maximum concentration
- CR
complete response
- DLT
dose‐limiting toxicity
- Ig
immunoglobulin
- IMiD
immunomodulatory drug
- ISS
International Staging System
- MM
multiple myeloma
- MR
minimal response
- MRD
minimal residual disease
- NE
not evaluable
- ORR
overall response rate
- OS
overall survival
- PD
progressive disease
- PFS
progression‐free survival
- PI
proteasome inhibitor
- PK
pharmacokinetic
- PR
partial response
- PS
performance status
- Q2W
every 2 weeks
- QW
every week
- RD
receptor density
- RDI
relative dose intensity
- RRMM
relapsed and refractory multiple myeloma
- sCR
stringent complete response
- SD
stable disease
- TEAE
treatment‐emergent adverse event
- tmax
time to reach Cmax
- VGPR
very good partial response
- β2‐MG
β2‐microglobulin
1. INTRODUCTION
Multiple myeloma (MM) is difficult to treat, and many patients experience disease relapse or become refractory to conventional therapy, including immunomodulatory drugs (IMiDs; eg, lenalidomide and pomalidomide) and proteasome inhibitors (PIs; eg, bortezomib and carfilzomib). 1 Owing to the high relapse rate, patients typically require multiple lines of therapy, often with combinations of drugs. Three monoclonal antibodies, daratumumab (anti‐CD38 antibody), elotuzumab (anti‐SLAMF7 antibody), and isatuximab (anti‐CD38 antibody), were recently approved owing to their efficacy and safety in patients with relapsed/refractory multiple myeloma (RRMM). 2 , 3 , 4 , 5 , 6 , 7
Isatuximab targets a specific epitope and shows potent antitumor activity in CD38+ hematologic malignancies, including MM. 8 The epitope recognized by isatuximab differs from that recognized by daratumumab. 8 Isatuximab induces cell death via IgG Fc–dependent mechanisms including antibody‐dependent cellular cytotoxicity, complement‐dependent cytotoxicity, and antibody‐dependent cellular phagocytosis. 8 , 9 , 10 , 11 Isatuximab also inhibits ectoenzymatic function and directly induces apoptosis of MM cells without crosslinking. 12 Thus, isatuximab may have a different mechanism of action from that of daratumumab.
Several phase 1‐3 studies in the US and EU have examined the efficacy of isatuximab as monotherapy 13 or in combination with pomalidomide or lenalidomide and dexamethasone. 7 , 14 , 15 Isatuximab was recently approved in the United States, the European Union, Canada, Australia, and Switzerland for use in combination with pomalidomide and dexamethasone in patients with RRMM who have received at least two prior therapies including lenalidomide and a PI (https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/761113s000lbl.pdf; https://www.ema.europa.eu/en/documents/product‐information/sarclisa‐epar‐product‐information_en.pdf). In June 2020, isatuximab was also approved in Japan (in combination with pomalidomide and dexamethasone) for the treatment of RRMM (https://www.genome.jp/kegg/drug/br08318.html) and is now available to Japanese clinicians. We performed a combined phase 1/2 trial of isatuximab in Japan to evaluate its safety and efficacy as monotherapy in patients with RRMM.
2. MATERIALS AND METHODS
Additional methods, including statistical analyses, can be found in the Supporting Information (Document S1).
2.1. Patients
Eligible patients were aged ≥20 years, with a diagnosis of symptomatic MM, 16 , 17 measurable disease, and had received ≥3 prior lines of treatment with minimal response (MR) or better to ≥1 line. Prior therapies must have included either an IMiD or a PI, or IMiD plus PI, for ≥2 cycles or ≥2 months of treatment. Patients who had received more than one type of IMiD and/or PI were required to be refractory to the most recent regimen used (defined as progression during or within 60 days of completion of treatment).
2.2. Study design
This open‐label, nonrandomized, single‐arm, local multicenter trial, conducted in Japan, comprised a dose‐escalation phase (phase 1) to determine the maximum tolerated dose based on dose‐limiting toxicities (DLTs), followed by a confirmatory phase (phase 2).
Phase 1 included two cohorts. Patients in cohort 1 received half of the highest administered dose tested in the US phase 1 study 13 , 18 (10 mg/kg every week [QW] for 4 weeks in cycle 1, followed by 10 mg/kg every 2 weeks [Q2W] in subsequent 4‐week cycles; 10 mg/kg QW/Q2W). Patients in cohort 2 received the dose recommended in the US study (20 mg/kg QW in cycle 1 followed by 20 mg/kg Q2W in subsequent 4‐week cycles; 20 mg/kg QW/Q2W). All patients in phase 2 received the dose established in cohort 2 in phase 1.
This trial was performed in accordance with the Declaration of Helsinki, Good Clinical Practice, and relevant local/international guidelines, and was approved by independent ethics committees/institutional review boards at all participating sites. All patients provided informed consent prior to initiation of any study procedures.
2.3. Assessments
The primary objectives were to evaluate the safety and tolerability of isatuximab in phase 1 and to evaluate the efficacy of isatuximab at the recommended dose in phase 2. A secondary objective was to evaluate the safety including the pharmacokinetic profile of isatuximab. Other secondary objectives included response, overall survival (OS), progression‐free survival (PFS), and the relationship between response and baseline CD38 receptor density (RD) on MM cells. As an exploratory objective, we assessed minimal residual disease (MRD) in patients achieving complete response (CR) and its correlation with clinical outcomes.
DLTs were assessed in cycle 1 in phase 1 to make decisions regarding dose escalation and phase transition. Possible DLTs included hematological adverse events (AEs) attributed to isatuximab (Table S1).
Pharmacokinetic parameters were assessed by noncompartmental analysis in cycle 1, phase 1.
Disease assessments were performed every 4 weeks in all enrolled patients. Responses were evaluated using modified International Myeloma Working Group uniform response criteria 17 as stringent CR (sCR), CR, very good partial response (VGPR), partial response (PR), MR, stable disease (SD), or progressive disease (PD) (Tables S2 and S3). 16 The primary efficacy endpoint was overall response rate (ORR), calculated as the proportion of patients whose best response was sCR, CR, VGPR, or PR. Secondary efficacy variables were the clinical benefit rate (CBR; best response of sCR, CR, VGPR, PR, or MR), duration of follow‐up, duration of response, time to response, OS, and PFS. Responses were assessed by an Independent Adjudication Committee based on central laboratory M protein assessments. The best percent change in paraprotein was determined for all patients with measurable paraprotein at baseline. The CD38 RD was determined using bone marrow samples at screening, and its correlation with clinical responses was determined.
3. RESULTS
3.1. Patients and treatments
Eight patients were enrolled in phase 1 (three received 10 mg/kg QW/Q2W and five received 20 mg/kg QW/Q2W) and 28 in phase 2 (Figure 1).
Figure 1.
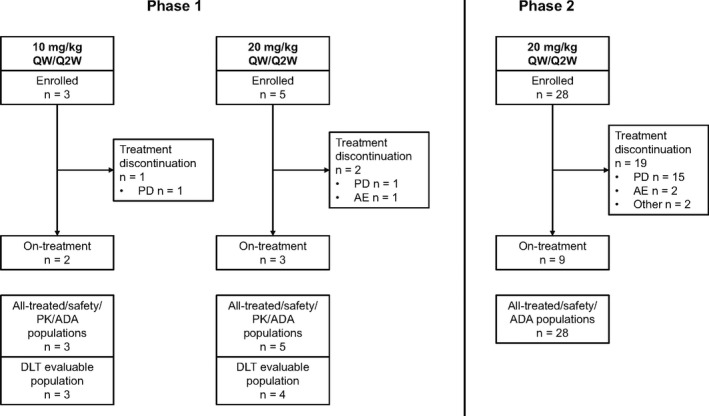
Patient disposition. Five patients were enrolled at 20 mg/kg Q2W in phase 1 because preregistration was continued while the third patient was in the screening period in case the patient withdrew during screening. ADA, anti‐drug antibody; AE, adverse event; DLT, dose‐limiting toxicity; PD, progressive disease; PK, pharmacokinetic; Q2W, every 2 weeks; QW, every week
Table 1 presents the baseline characteristics of patients enrolled in both phases of the study. Most patients had MM of heavy chain IgG, light chain kappa subtype, with measurable M‐protein at baseline. Two patients in phase 1 and five in phase 2 had plasmacytomas, while seven and 15 had bone lesions. Eleven patients (31%) had at least one high‐risk cytogenetic abnormality determined by fluorescence in situ hybridization. In phase 1, two patients had two high‐risk cytogenetic abnormalities, which were 17p deletion and t(4;14) translocation. In phase 2, three patients had two high‐risk cytogenetic abnormalities, which were 17p deletion and t(14;16) translocation in one patient and 17p deletion and t(4;14) translocation in the other two patients.
Table 1.
Patient characteristics at baseline
| Phase 1 | Phase 2 | All‐treated | ||
|---|---|---|---|---|
| 10 mg/kg QW/Q2W (n = 3) | 20 mg/kg QW/Q2W (n = 5) | 20 mg/kg QW/Q2W (n = 28) | 20 mg/kg QW/Q2W (n = 33) | |
| Sex, n (%) | ||||
| Male | 1 (33) | 1 (20) | 18 (64) | 19 (58) |
| Female | 2 (67) | 4 (80) | 10 (36) | 14 (42) |
| Median age (range), years | 69.0 (59‐74) | 76.0 (69‐80) | 71.5 (48‐82) | 72.0 (48‐82) |
| Median weight (range), kg | 44.40 (43.6‐73.4) | 48.70 (37.6‐66.0) | 56.30 (38.8‐75.0) | 55.3 (37.6‐75.0) |
| ECOG PS, n (%) | ||||
| 0 | 2 (67) | 2 (40) | 15 (54) | 17 (52) |
| 1 | 1 (33) | 2 (40) | 9 (32) | 11 (33) |
| 2 | 0 | 1 (20) | 4 (14) | 5 (15) |
| Presence of anemia, n (%) | 3 (100) | 5 (100) | 28 (100) | 33 (100) |
| Median time from diagnosis to first dose of isatuximab (range), years | 6.69 (4.8‐18.0) | 4.25 (1.6‐6.6) | 6.24 (1.4‐18.6) | 5.46 (1.4‐18.6) |
| ISS at initial diagnosis, n (%) | ||||
| I | 0 | 1 (20) | 10 (36) | 11 (33) |
| II | 2 (67) | 2 (40) | 11 (39) | 13 (39) |
| III | 0 | 2 (40) | 4 (14) | 6 (18) |
| Unknown | 1 (33) | 0 | 3 (11) | 3 (9) |
| Multiple myeloma subtype, n (%) | ||||
| Heavy chain | ||||
| IgA | 0 | 0 | 6 (21) | 6 (18) |
| IgD | 0 | 0 | 1 (4) | 1 (3) |
| IgG | 3 (100) | 4 (80) | 19 (68) | 23 (70) |
| Not applicable | 0 | 0 | 1 (4) | 1 (3) |
| Undetected | 0 | 1 (20) | 1 (4) | 2 (6) |
| Light chain | ||||
| Kappa | 2 (67) | 3 (60) | 17 (61) | 20 (61) |
| Lambda | 1 (33) | 2 (40) | 11 (39) | 13 (39) |
| Biclonal, no | 3 (100) | 5 (100) | 28 (100) | 33 (100) |
| Measurable paraprotein, n (%) | ||||
| Serum M‐protein | 3 (100) | 3 (60) | 21 (75) | 24 (73) |
| Urine M‐protein | 0 | 1 (20) | 3 (11) | 4 (12) |
| Both | 0 | 1 (20) | 4 (14) | 5 (15) |
| Median plasma cells in marrow (range), % | 6.20 (0.0‐45.8) | 15.80 (6.6‐81.8) | 14.50 (0.4‐84.6) | 15.60 (0.4‐84.6) |
| Patients with plasmacytomas, n (%) | 1 (33) | 1 (20) | 5 (18) | 6 (18) |
| Patients with bone lesions, n (%) | 2 (67) | 5 (100) | 15 (54) | 20 (61) |
| Derived ISS at study entry, n (%) | ||||
| I | 1 (33) | 1 (20) | 14 (50) | 15 (45) |
| II | 1 (33) | 2 (40) | 9 (32) | 11 (33) |
| III | 1 (33) | 2 (40) | 5 (18) | 7 (21) |
| Median serum β2‐MG (range), mg/L | 5.10 (2.8‐5.8) | 4.50 (2.5‐10.4) | 3.30 (1.9‐12.7) | 3.40 (1.9‐12.7) |
| Median albumin (range), g/L | 35.00 (34.0‐37.0) | 38.00 (23.0‐40.0) | 36.50 (18.0‐42.0) | 37.00 (18.0‐42.0) |
| High‐risk cytogenetic abnormalities at study entry, n (%) | ||||
| At least one cytogenetic abnormality | 2 (67) | 1 (20) | 8 (29) | 9 (27) |
| At least two cytogenetic abnormalities | 1 (33) | 1 (20) | 3 (11) | 4 (12) |
| 17p deletion (TP53) | 2 (67) | 1 (20) | 5 (18) | 6 (18) |
| t(4;14) translocation (FGFR3/IGH) | 1 (33) | 1 (20) | 5 (18) | 6 (18) |
| t(14;16) translocation (IGH/MAF) | 0 | 0 | 1 (4) | 1 (3) |
Abbreviations: ECOG, Eastern Cooperative Oncology Group; Ig, immunoglobulin; ISS, International Staging System; PS, performance status; Q2W, every 2 weeks; QW, every week; β2‐MG, β2‐microglobulin.
Patients had received a median of five prior treatment lines, including an IMiD and a PI, and the majority were refractory to an IMiD and/or a PI (Table 2). Thirty patients (91%) were refractory to IMiD (including 23 patients [70%] refractory to pomalidomide) and 29 patients (88%) were refractory to PI at baseline. Six patients (75%) in phase 1 and 22 patients (79%) in phase 2 were refractory to both IMiD and PI.
Table 2.
Prior treatments recorded at baseline
| Phase 1 | Phase 2 | All‐treated | ||
|---|---|---|---|---|
| 10 mg/kg QW/Q2W (n = 3) | 20 mg/kg QW/Q2W (n = 5) | 20 mg/kg QW/Q2W (n = 28) | 20 mg/kg QW/Q2W (n = 33) | |
| Median prior treatment lines (range) | 5.0 (4‐12) | 4.0 (3‐6) | 5.0 (2‐11) | 5.0 (2‐11) |
| Prior therapies, n (%) | ||||
| IMiD | 3 (100) | 5 (100) | 28 (100) | 33 (100) |
| Lenalidomide | 3 (100) | 5 (100) | 27 (96) | 32 (97) |
| Pomalidomide | 3 (100) | 3 (60) | 22 (79) | 25 (76) |
| Thalidomide | 1 (33) | 0 | 8 (29) | 8 (24) |
| PI | 3 (100) | 5 (100) | 28 (100) | 33 (100) |
| Bortezomib | 3 (100) | 5 (100) | 27 (96) | 32 (97) |
| Carfilzomib | 1 (33) | 2 (40) | 9 (32) | 11 (33) |
| Ixazomib | 0 | 1 (20) | 3 (11) | 4 (12) |
| Other | 1 (33) | 2 (40) | 10 (36) | 12 (36) |
| Panobinostat | 1 (33) | 1 (20) | 7 (25) | 8 (24) |
| Elotuzumab | 0 | 1 (20) | 6 (21) | 7 (21) |
| IMiD plus PI | 3 (100) | 5 (100) | 28 (100) | 33 (100) |
| Lenalidomide and bortezomib | 3 (100) | 5 (100) | 26 (93) | 31 (94) |
| Lenalidomide, bortezomib, pomalidomide and carfilzomib | 1 (33) | 1 (20) | 6 (21) | 7 (21) |
| Prior transplant | 2 (67) | 1 (20) | 9 (32) | 10 (30) |
| Patients refractory to prior therapies, n (%) | ||||
| Refractory to IMiD | 3 (100) | 5 (100) | 25 (89) | 30 (91) |
| Lenalidomide | 3 (100) | 5 (100) | 24 (86) | 29 (88) |
| Pomalidomide | 3 (100) | 3 (60) | 20 (71) | 23 (70) |
| Refractory to PI | 2 (67) | 4 (80) | 25 (89) | 29 (88) |
| Bortezomib | 2 (67) | 4 (80) | 20 (71) | 24 (73) |
| Carfilzomib | 1 (33) | 2 (40) | 8 (29) | 10 (30) |
| Ixazomib | 0 | 1 (20) | 3 (11) | 4 (12) |
| Refractory to IMiD and PI | 2 (67) | 4 (80) | 22 (79) | 26 (79) |
| Lenalidomide and bortezomib | 2 (67) | 4 (80) | 17 (61) | 21 (64) |
| Lenalidomide, bortezomib, pomalidomide and carfilzomib | 1 (33) | 1 (20) | 4 (14) | 5 (15) |
Abbreviations: IMiD, immunomodulatory drug; PI, proteasome inhibitor; Q2W, every 2 weeks; QW, every week.
Granulocyte colony–stimulating factor was administered in five of 36 patients. Two of these received it as prophylactic treatment.
The median (range) number of treatment cycles was 22.0 (2‐24) in the 10 mg/kg QW/Q2W group in phase 1, 15.0 (1‐21) in the 20 mg/kg QW/Q2W group in phase 1, and 6.0 (1‐13) in the 20 mg/kg QW/Q2W group in phase 2. The median (range) duration of exposure in the three groups was 90.4 (6‐96), 57.9 (2‐82), and 22.0 (4‐50) weeks, respectively, and the median (range) cumulative dose was 449.10 (50.0‐490.0), 619.30 (40.0‐859.3), and 259.60 (77.8‐520.3) mg/kg, respectively. Five patients in phase 1 and nine patients in phase 2 were still on treatment at the cut‐off date (31 July 2018). The median duration of the first infusion and subsequent infusions was 2.6 h and 2.1 h at the 10 mg/kg dose level, and 3.8 h and 3.9 h at the 20 mg/kg dose level in phase 1. In phase 2 at 20 mg/kg, the median duration of the first infusion and subsequent infusions was 4.3 and 3.9 hours, respectively.
In the 10 mg/kg QW/Q2W group in phase 1, the median (range) actual dose intensity (ADI) was 10.00 (7.4‐10.0) mg/kg/week in cycle 1 and 4.89 (4.8‐5.0) mg/kg/week in subsequent cycles; median (range) relative dose intensity (RDI) was 100.00 (73.7‐100.0)% in cycle 1 and 97.83 (96.3‐100.0)% in subsequent cycles. In the 20 mg/kg QW/Q2W group in phase 1, respective ADI and RDI values were 19.95 (15.3‐20.7) mg/kg/week, 9.93 (9.8‐10.0) mg/kg/week, 99.75 (76.3‐103.6)%, and 99.27 (98.0‐99.9)%; in the 20 mg/kg QW/Q2W group in phase 2, respective ADI and RDI values were 20.00 (13.4‐21.3) mg/kg/week, 10.00 (6.8‐10.9) mg/kg/week, 100.00 (66.8‐106.4)%, and 100.00 (68.0‐108.5)%.
3.2. Safety
3.2.1. DLTs
One patient was excluded from the DLT evaluable population owing to AEs that led to treatment discontinuation after two doses of isatuximab (diplegia and neurogenic bladder; both unrelated to isatuximab; caused by progression of primary disease). There were no DLTs at either dose in phase 1. Therefore, the dose selected for phase 2 was 20 mg/kg QW/Q2W, consistent with the dose recommended in the US study 11 and the exposure‐response analysis performed to select the optimal dosing regimen. 19
3.2.2. AEs
Rates of treatment‐emergent adverse events (TEAEs), drug‐related TEAEs, serious TEAEs, and infusion‐related reactions are summarized in Table 3, along with rates of individual TEAEs (any grade and grade ≥3). Serious TEAEs occurred in one patient treated with 10 mg/kg QW/Q2W (pneumonia and deep vein thrombosis) and one patient treated with 20 mg/kg QW/Q2W (diplegia, neurogenic bladder, and disease progression) in phase 1. TEAEs of grade ≥3 included pneumonia in two patients, and intervertebral discitis, lung infection, disseminated intravascular coagulation, seizure, thrombotic cerebral infarction, ileus, and synovial cyst in one patient each. The only serious TEAE classified as related to isatuximab was grade ≥3 pneumonia, which occurred in one patient treated with 10 mg/kg QW/Q2W in phase 1 and in two patients in phase 2.
Table 3.
Overview of treatment‐emergent adverse events (any grade and grade 3/4) by study phase and dose
| n (%) | Phase 1 | Phase 2 | ||||
|---|---|---|---|---|---|---|
| 10 mg/kg QW/Q2W (n = 3) | 20 mg/kg QW/Q2W (n = 5) | 20 mg/kg QW/Q2W (n = 28) | ||||
| Any grade | Grade ≥3 | Any grade | Grade ≥3 | Any grade | Grade ≥3 | |
| Any TEAE | 3 (100) | 1 (33) | 4 (80) | 2 (40) | 25 (89) | 12 (43) |
| Drug‐related TEAE | 2 (67) | 1 (33) | 1 (20) | 0 | 18 (64) | 3 (11) |
| Serious TEAE | 1 (33) | 1 (20) | 7 (25) | |||
| Serious drug‐related TEAE | 1 (33) | 0 | 2 (7) | |||
| TEAE leading to death | 0 | 1 (20) | 0 | |||
| TEAE leading to discontinuation | 0 | 1 (20) | 2 (7) | |||
| At least one DLT | 0 | 0 | – | |||
| At least one infusion‐related reaction | 2 (67) | 0 | 1 (20) | 0 | 12 (43) | 0 |
| TEAEs in ≥1 patient in phase 1 or ≥5% of patients in phase 2 | ||||||
| Infusion‐related reactions | 2 (67) | 0 | 1 (20) | 0 | 12 (43) | 0 |
| Pyrexia | 0 | 0 | 0 | 0 | 6 (21) | 1 (4) |
| Nasopharyngitis | 2 (67) | 0 | 1 (20) | 0 | 6 (21) | 0 |
| Vomiting | 1 (33) | 0 | 2 (40) | 0 | 1 (4) | 0 |
| Pneumonia | 1 (33) | 1 (33) | 1 (20) | 1 (20) | 3 (11) | 2 (7) |
| Rhinorrhea | 1 (33) | 0 | 1 (20) | 0 | 3 (11) | 0 |
| Cataract | 1 (33) | 0 | 0 | 0 | 3 (11) | 1 (4) |
| Diarrhea | 1 (33) | 0 | 0 | 0 | 3 (11) | 0 |
| Influenza | 0 | 0 | 0 | 0 | 2 (7) | 0 |
| Pharyngitis | 1 (33) | 0 | 0 | 0 | 2 (7) | 0 |
| Sinusitis | 1 (33) | 0 | 0 | 0 | 2 (7) | 0 |
| Leukopenia | 1 (33) | 0 | 0 | 0 | 2 (7) | 1 (4) |
| Back pain | 1 (33) | 0 | 0 | 0 | 4 (14) | 0 |
| Deep vein thrombosis | 1 (33) | 1 (33) | 0 | 0 | 1 (4) | 0 |
| Lymphopenia | 1 (33) | 1 (33) | 0 | 0 | 1 (4) | 0 |
| Hypertension | 1 (33) | 0 | 0 | 0 | 1 (4) | 1 (4) |
| Upper respiratory tract infection | 1 (33) | 0 | 0 | 0 | 1 (4) | 0 |
| Cough | 1 (33) | 0 | 0 | 0 | 1 (4) | 0 |
| Conjunctivitis | 1 (33) | 0 | 0 | 0 | 0 | 0 |
| Bone pain | 1 (33) | 0 | 0 | 0 | 0 | 0 |
| Dermatitis contact | 1 (33) | 0 | 0 | 0 | 0 | 0 |
| Nausea | 0 | 0 | 1 (20) | 0 | 2 (7) | 0 |
| Upper respiratory tract inflammation | 0 | 0 | 1 (20) | 0 | 1 (4) | 0 |
| Platelet count decreased | 0 | 0 | 1 (20) | 0 | 1 (4) | 0 |
| Diplegia | 0 | 0 | 1 (20) | 1 (20) | 0 | 0 |
| Neurogenic bladder | 0 | 0 | 1 (20) | 1 (20) | 0 | 0 |
| Disease progression | 0 | 0 | 1 (20) | 1 (20) | 0 | 0 |
| Vertigo | 0 | 0 | 1 (20) | 0 | 0 | 0 |
| Hot flush | 0 | 0 | 1 (20) | 0 | 0 | 0 |
| Pathological fracture | 0 | 0 | 1 (20) | 0 | 0 | 0 |
| Fatigue | 0 | 0 | 1 (20) | 0 | 0 | 0 |
| Edema peripheral | 0 | 0 | 0 | 0 | 3 (11) | 1 (4) |
| Bronchitis | 0 | 0 | 0 | 0 | 2 (7) | 0 |
| Chest discomfort | 0 | 0 | 0 | 0 | 2 (7) | 0 |
| Chills | 0 | 0 | 0 | 0 | 2 (7) | 0 |
| Decreased appetite | 0 | 0 | 0 | 0 | 2 (7) | 0 |
| Hypoxia | 0 | 0 | 0 | 0 | 2 (7) | 0 |
| Nasal congestion | 0 | 0 | 0 | 0 | 2 (7) | 0 |
| Pruritus | 0 | 0 | 0 | 0 | 2 (7) | 0 |
| Rhinitis allergic | 0 | 0 | 0 | 0 | 2 (7) | 0 |
| Stomatitis | 0 | 0 | 0 | 0 | 2 (7) | 0 |
Abbreviations: DLT, dose‐limiting toxicity; Q2W, every 2 weeks; QW, every week; TEAE, treatment‐emergent adverse event.
Infusion‐related reactions were assessed as AEs of special interest, and occurred in three patients (two events in two patients at 10 mg/kg and two events in one patient at 20 mg/kg) in phase 1 and 12 patients (13 events) in phase 2 (Table 3). None of the reactions were of grade ≥3. When a reaction occurred, it was at the first infusion in all patients in phase 1 and in 11 patients in phase 2. One patient in phase 2 experienced reactions at the first and third infusion. All infusion‐related reactions resolved within 1 day, except two patients with reactions that lasted 2 days. No patients discontinued treatment due to infusion reactions.
Respiratory infection and laboratory neutropenia were evaluated as significant AEs. Among all 36 patients enrolled in both phases, respiratory infections occurred in 19, with lower respiratory TEAEs in eight patients, including five patients who experienced pneumonia. Fifteen patients experienced neutropenia as a laboratory abnormality during the on‐treatment period in phase 2, with most patients (n = 9, 32.1%) experiencing a grade 2 episode and four patients having a grade 3 episode. No neutropenic complications (neutropenic infection or febrile neutropenia) were reported during the on‐treatment period. AEs leading to treatment discontinuation were observed in three patients. In phase 1, one patient (20%) experienced diplegia and neurogenic bladder of grade ≥3 that led to treatment discontinuation. In phase 2, two patients (7.1%) experienced the following AEs of all grades and grade ≥3 leading to treatment discontinuation: intervertebral discitis, pneumonia, disseminated intravascular coagulation, and thrombotic cerebral infarction (one patient each, 3.6%). An AE leading to death was reported in one patient in phase 1 (20 mg/kg QW/Q2W) with disease progression. There were no treatment‐related deaths.
3.3. Efficacy
Among 33 patients who received isatuximab at 20 mg/kg QW/Q2W in phases 1 and 2, the ORR (≥PR) was 36.4% (95% CI: 20.4 to 54.9; 12/33 patients), which exceeded the null hypothesis rate of < 10% (P < 0.0001). The CBR (≥MR) was 54.5% (95% CI: 36.4‐71.9; 18/33 patients) (Table 4). The ORR (≥PR) in phase 2 was 32%. Figure 2 shows the best response as a function of time on treatment in phase 2, including in patients with high‐risk cytogenetic abnormalities. There appeared to be no differences in the response rate according to the number of prior lines or cytogenetic risk. Among eight patients with high‐risk cytogenetic abnormalities, the response was ≥PR in three patients including VGPR in two patients. All three patients with ≥PR had the t(4;14) cytogenetic abnormality. Regarding prior treatment, PR was achieved in two of six patients previously treated with elotuzumab. All six patients discontinued elotuzumab due to PD, including three who received combination therapy between discontinuing elotuzumab and starting isatuximab (Table S4). One of three patients achieved PR.
Table 4.
Best overall responses by study phase and dose
| n (%) | Phase 1 | Phase 2 | All‐treated | |
|---|---|---|---|---|
| 10 mg/kg QW/Q2W (n = 3) | 20 mg/kg QW/Q2W (n = 5) | 20 mg/kg QW/Q2W (n = 28) | 20 mg/kg QW/Q2W (n = 33) | |
| ORR (≥PR) | 2 (67) | 3 (60) | 9 (32) | 12 (36.4) (95% CI: 20.4‐54.9, P < 0.0001) |
| CBR (≥MR) | 2 (67) | 3 (60) | 15 (54) | 18 (54.5) (95% CI: 36.4‐71.9) |
| sCR | 0 | 0 | 0 | 0 |
| CR | 0 | 1 (20) | 1 (4) | 2 (6) |
| VGPR | 1 (33) | 1 (20) | 3 (11) | 4 (12) |
| PR | 1 (33) | 1 (20) | 5 (18) | 6 (18) |
| MR | 0 | 0 | 6 (21) | 6 (18) |
| SD | 0 | 0 | 7 (25) | 7 (21) |
| PD | 0 | 1 (20) | 3 (11) | 4 (12) |
| Unconfirmed PD | 1 (33) | 0 | 2 (7) | 2 (6) |
| NE | 0 | 1 (20) | 1 (4) | 2 (6) |
Abbreviations: CBR, clinical benefit rate; CI, confidence interval; CR, complete response; MR, minimal response; NE, not evaluable; ORR, overall response rate; PD, progressive disease; PR, partial response; Q2W, every 2 weeks; QW, every week; sCR, stringent complete response; SD, stable disease; VGPR, very good partial response.
Figure 2.
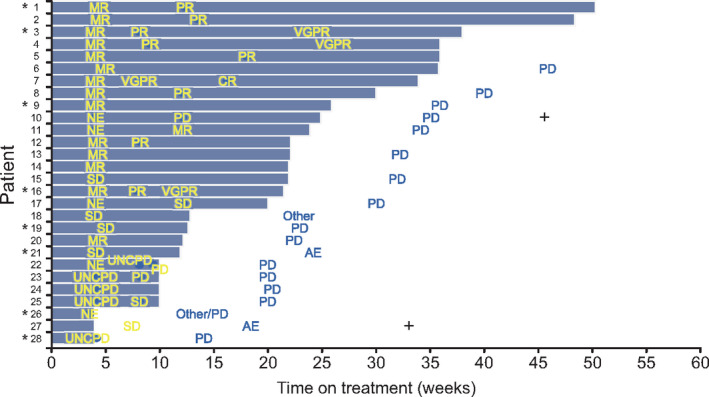
Best response and time on treatment in phase 2 (20 mg/kg QW/Q2W). The median follow‐up was 19.21 weeks (range 3.6‐44.6 weeks). Patient 18 withdrew consent and refused to return to the hospital according to the study schedule. Patient 21 withdrew due to pneumonia. Patient 26 decided to withdraw due to no treatment effect. Patient 27 withdrew due to intervertebral discitis, disseminated intravascular coagulation, and thrombotic cerebral infarction. CR, complete response; MR, minimal response; NE, not evaluable; ORR, overall response rate; PD, progressive disease; PR, partial response; sCR, stringent complete response; SD, stable disease; UNCPD, unconfirmed PD; VGPR, very good partial response. *Indicates patients with cytogenetic abnormalities. Patient 1: t(4;14) translocation (FGFR3/IGH); Patient 3: t(4;14) translocation (FGFR3/IGH); Patient 9: 17p deletion (TP53) and t(4;14) translocation (FGFR3/IGH); Patient 16: t(4;14) translocation (FGFR3/IGH); Patient 19: 17p deletion (TP53) and t(14;16) translocation (IGH/MAF); Patient 21: 17p deletion (TP53); Patient 26: 17p deletion (TP53) and t(4;14) translocation (FGFR3/IGH); Patient 28: 17p deletion (TP53). +Indicates deaths
In other subgroups of patients, response rates tended to be greater in patients with low ECOG performance status, low ISS grade, baseline creatinine clearance ≥60 mL/min/1.73 m2, and absence of plasmacytoma at screening (Table S5).
The median (range) follow‐up from the start of isatuximab therapy was 84.57 (4.1‐90.1) weeks in the 10 mg/kg QW/Q2W group in phase 1, 52.00 (5.0‐76.1) weeks in the 20 mg/kg QW/Q2W group in phase 1, and 19.21 (3.6‐44.6) weeks in the 20 mg/kg QW/Q2W group in phase 2. The median (range) duration of response in the three groups was 82.64 (79.1‐86.1), 48.14 (48.1‐50.3), and 24.14 (11.6‐36.3) weeks, respectively. The follow‐up period differed between the phases and the data are immature for the phase 2 cohort. The median (range) time to first response was comparable in all three groups at 4.86 (4.1‐5.6), 5.43 (4.0‐28.1), and 4.29 (4.1‐12.1) weeks, respectively.
PFS and OS were assessed in 28 patients in phase 2 (Figure 3A, B). The median PFS was 4.7 months (95% CI: 3.75 to not reached) while median OS was not reached. The OS probabilities at 6 months and 1 year were 1.000 and 0.781, respectively. There were two deaths in phase 2. Both patients died during the posttreatment period and the causes of death were not related to AEs with the study treatment. One of these patients did not receive subsequent therapy and the other was treated with carfilzomib and dexamethasone after isatuximab discontinuation.
Figure 3.
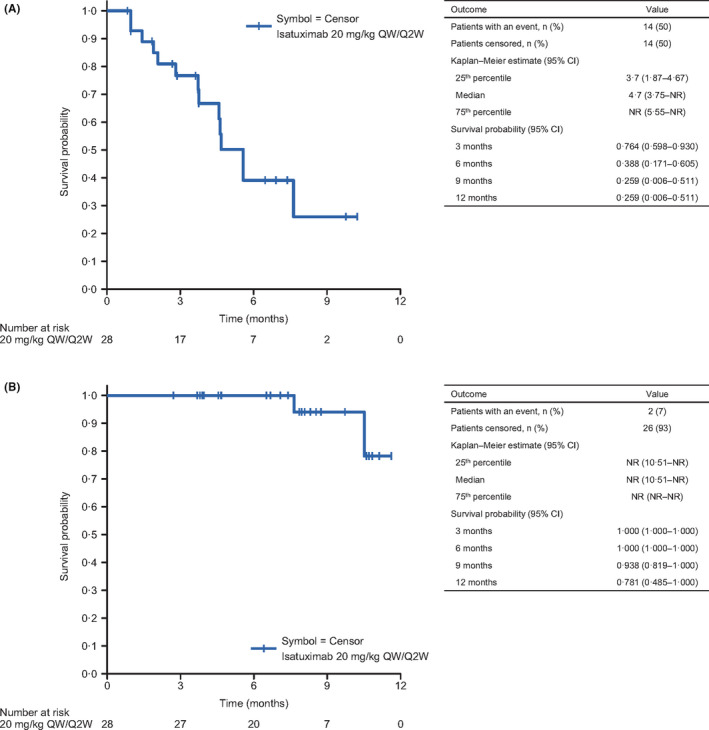
Kaplan‐Meier plots of (A) progression‐free survival and (B) overall survival in patients treated with isatuximab at 20 mg/kg QW/Q2W in phase 2. CI, confidence interval; NR, not reached; Q2W, every 2 weeks; QW, every week
About half of all patients had a ≥50% reduction in paraprotein, with a reduction of ≥90% in four patients in phase 1 (one at 10 mg/kg QW/Q2W and three at 20 mg/kg QW/Q2W) and six patients in phase 2. The best percent change in paraprotein and overall response in individual patients is shown in Figure S1.
MRD status was assessed in three patients. Of two patients achieving CR, one patient in the 20 mg/kg group in phase 1 was MRD negative and one patient in phase 2 was MRD positive at the 10−5 threshold. The patient with VGPR in the 10 mg/kg group in phase 1 was MRD positive at 10−5.
3.4. Biomarkers
CD38 RD data were available for 32 patients. As shown in Figure 4, CD38 RD was slightly higher in responders than in nonresponders, with median (range) values of 122 313.5 (71 808 to 232 958) among 14 responders and 72 731.0 (26 921 to 394 910) among 18 nonresponders. When patients were divided according to CD38 RD thresholds (Figure S2), the ORR tended to be greater in patients whose RD was above the threshold value. However, some patients with lower RD values showed responses to isatuximab.
Figure 4.
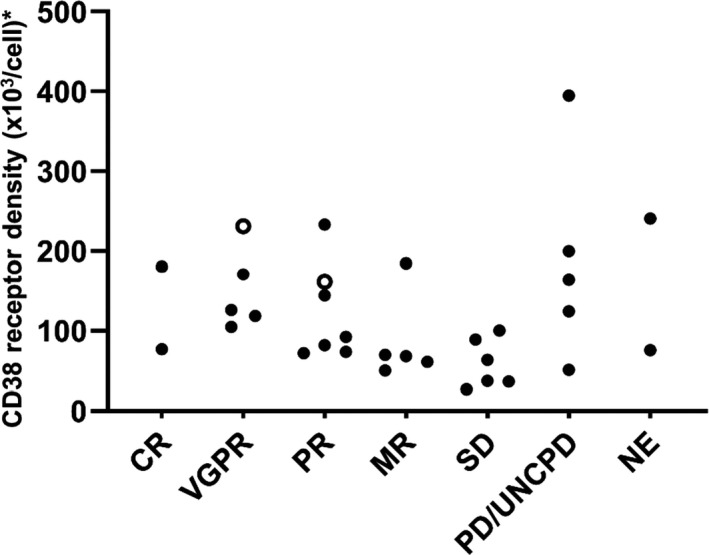
Relationship between CD38 receptor density and clinical response in the all‐treated population. White symbols, 10 mg/kg QW/Q2W; black symbols, 20 mg/kg QW/Q2W. CD38 receptor density was unavailable for four patients (UNCPD in 10 mg/kg QW/Q2W; MR, SD, and PD in 20 mg/kg QW/Q2W). CR, complete response; MR, minimal response; NE, not evaluable; PD, progressive disease; PR, partial response; Q2W, every 2 weeks; QW, every week; SD, stable disease; UNCPD, unconfirmed PD; VGPR, very good partial response. *CD38 receptor density (×103/cell) was reported as the specific molecule equivalent per cell (sMEC), using the conversion formula: sMEC = MEC (selected antibody) − MEC (negative isotypic control), where MEC (molecule equivalent per cell) = 10(log[MFI] × a + b ), in which a and b are the slope and the y‐intercept of the calibration curve equation, respectively
3.5. Pharmacokinetics of isatuximab
Pharmacokinetic properties of isatuximab on cycle 1 in phase 1 are shown in Table 5. The total variability of exposure parameters was low to moderate, with coefficients of variability of 18%‐32%. For a twofold dose increase (from 10 to 20 mg/kg), isatuximab exposure increased 2.3‐fold (based on the geometric mean ratio).
Table 5.
Isatuximab plasma pharmacokinetic parameters at cycle 1 of phase 1
| Phase 1 | ||
|---|---|---|
| 10 mg/kg QW/Q2W (n = 3) | 20 mg/kg QW/Q2W (n = 5) | |
| Patients with evaluable PK, n | 3 | 4 |
| Infusion duration, h | ||
| Median (range) | 2.63 (2.32‐3.23) | 3.82 (3.28‐6.05) a |
| Ceoi, µg/mL | ||
| Mean (SD) | 122 (21.6) | 246 (51.8) |
| Geometric mean (CV%) | 121 (18) | 242 (21) |
| Cmax, µg/mL | ||
| Mean (SD) | 124 (22.9) | 280 (64.4) |
| Geometric mean (CV%) | 123 (18) | 274 (23) |
| AUC1week, (μg•hr/mL) | ||
| Mean (SD) | 9300 (3010) | 21 300 (5520) |
| Geometric mean (CV%) | 8970 (32) | 20 800 (26) |
| tmax, h | ||
| Median (range) | 2.68 (2.32‐7.25) | 5.56 (3.28‐8.48) |
Abbreviations: AUC1week, area under the plasma concentration versus time in the 1‐week dosing interval; Ceoi, concentration at the end of infusion; Cmax, maximum concentration; CV, coefficient of variation; PK, pharmacokinetic; Q2W, every 2 weeks; QW, every week; tmax, time to reach Cmax.
n = 5.
4. DISCUSSION
This study evaluated the maximum tolerated dose, safety, and efficacy of isatuximab monotherapy in heavily pretreated Japanese patients with RRMM. No DLTs were observed at either dose in phase 1, and 20 mg/kg QW/Q2W was the recommended dose in patients, consistent with recommendations in other non‐Japanese studies, 13 , 19 in which the dose of 20 mg/kg QW/Q2W was recommended in monotherapy considering the receptor occupancy and the PK/PD modeling and simulations. Pharmacokinetic data showed that, considering the total variability, isatuximab exposure was consistent in Japanese and non‐Japanese patients. 13 Isatuximab monotherapy at a dose of 20 mg/kg QW/Q2W showed promising efficacy with an ORR of 36.4%, a response rate of ≥VGPR of 18%, and one MRD negative CR. The observed responses in phase 2 were durable considering the median duration of response (24.14 weeks) and occurred quickly considering the median time to first response (4.29 weeks).
Of note, as the majority of patients entered the study with IgGκ myeloma, the same isotype as isatuximab, it is likely that isatuximab interference with immunofixation may have resulted in an underestimation of the CR rate. An in vitro diagnostic test that mitigates the potential interference caused by isatuximab in immunofixation electrophoresis is currently in development (https://www.pharmiweb.com/press‐release/2020‐01‐07/sebia‐enters‐into‐agreement‐with‐sanofi‐to‐develop‐multiple‐myeloma‐diagnostic‐test), and it is anticipated that this will facilitate accurate measurement of CR in future patients with myeloma receiving isatuximab.
It is well acknowledged that physicians should pay attention to the risk of infection due to leukopenia in patients with MM. In the present study, decreases in the neutrophil count were observed in all patients in phase 1 and in approximately half of the patients (15 of 28 patients) in phase 2. However, only one patient postponed treatment due to a reduction in neutrophil count. Infection occurred in 19 patients, including lower respiratory tract infection in eight and pneumonia in five patients. Antimicrobial agents were administered to 31 patients, which included prophylactic administration in 20 patients. These findings suggest that neutropenia and infection, while common, are generally manageable in patients. Anemia, which has previously been reported in isatuximab‐treated patients with RRMM, 13 was not reported in our study as a TEAE, likely because all patients already had anemia at baseline. Beyond these, other AEs of interest were infusion‐related reactions, and there were few serious TEAEs. Overall, isatuximab was generally well tolerated, with low rates of treatment discontinuation due to AEs and none due to infusion‐related reactions. Indeed, in this analysis, no infusion‐related reactions of grade ≥3 were observed; this is in contrast to a prior phase 1 study of isatuximab, in which two patients discontinued treatment due to grade 4 infusion‐related reactions. 13 However, clinical trials of daratumumab have reported rates of infusion‐related reactions comparable with those in our study: In a phase 1 study in nine Japanese patients with RRMM, 44% of patients reported an infusion‐related reaction, and none were grade ≥3 or resulted in discontinuation. 20 Similarly, in a pooled analysis of two global phase 2 studies, infusion‐related reactions were reported in 48% of daratumumab‐treated patients, with most occurring during the first infusion; all were considered to be manageable with pre‐ and postinfusion medications (antihistamines, corticosteroids, and paracetamol/acetaminophen). 21 , 22 These data suggest that although physicians should remain vigilant for infusion‐related reactions and infections, the majority of events appear to be manageable, and that routine use of these drugs will further help by familiarizing health care professionals with warning signs and mitigation procedures.
The results of this study demonstrate the efficacy of isatuximab monotherapy in terms of a high response rate and a durable response, as well as its safety, among heavily pretreated patients with RRMM. It is notable that the responses were observed in patients with high‐risk cytogenetics and in patients with more than six prior lines, including patients refractory to both a PI and IMiD. Heavily pretreated patients frequently show deteriorations in renal function and bone marrow function due to the primary disease and it is often difficult to continue treatment in such patients for reasons of safety. The current findings are clinically relevant and suggest the possibility of using isatuximab in these patients, for whom there may be few alternatives. Moreover, although a detailed population PK analysis is not yet available, the available PK data accruing from this study, and from previously published isatuximab monotherapy studies, 13 , 23 indicate that exposure parameters between Japanese and non‐Japanese patients with RRMM are broadly consistent. Although area under the curve and maximum concentration values were slightly lower in our study, this may have been a result of interstudy variability in the duration of the isatuximab infusions administered, and we consider that the differences were minor and within the acceptable range.
CD38 is expressed in several hematological malignancies, including MM, and represents a key therapeutic target. 24 , 25 The relationship between CD38 RD and the response was investigated in this study. The CD38 RD levels substantially overlapped between responders and nonresponders, suggesting it is unhelpful to use CD38 RD as a predictive biomarker of isatuximab response, despite slight numerical difference in the median CD38 RD between the two populations. Some patients with high CD38 RD did not respond. Considering these findings, we think it is necessary to elucidate the mechanisms that regulate sensitivity to antibody‐mediated cytotoxicity through complement, 26 NK cells, 27 phagocytes, and apoptosis as well as adaptive immunity. 28 , 29
Other recent global trials have examined isatuximab as monotherapy in RRMM, 13 , 23 in combination with pomalidomide plus dexamethasone in RRMM (ICARIA‐MM), 7 , 15 and in combination with carfilzomib and dexamethasone in relapsed MM (IKEMA). 30 In the phase 1 study of isatuximab monotherapy in patients with RRMM, the ORR was 23.8% (including one CR) in patients receiving doses of ≥10 mg/kg with a median duration of response of 36 weeks, and in high‐risk patients, the ORR was 16.7% with a median duration of response of 25 weeks. 13 Data from the pivotal phase 3 ICARIA‐MM trial revealed that isatuximab in combination with standard of care (pomalidomide and low‐dose dexamethasone) prolonged PFS (11.5 months) and ORR (60.4%) compared with standard of care (6.5 months and 35.3%, respectively). 7 The results indicate that isatuximab is a promising treatment option for RRMM. The ORR was greater when isatuximab was administered in combination with lenalidomide plus dexamethasone and with pomalidomide plus dexamethasone, which might be due to the different targets of these drugs providing possible additive effects. IMiDs may increase CD38 expression and hence prime cells for anti‐CD38 antibody–mediated cytotoxicity. 7 , 11 The safety profile of this study was generally similar to that observed in prior studies. 13 , 14 Ongoing phase 3 trials are investigating isatuximab in newly diagnosed myeloma, including in combination with lenalidomide, bortezomib, and dexamethasone.
Several studies of daratumumab and elotuzumab in MM have also been published. Elotuzumab monotherapy was shown to be well tolerated, but efficacy data showed no objective responses. 31 For daratumumab, in a phase 2 trial of monotherapy, the median PFS and the 12‐month OS were 3.7 months and 64.8%, respectively. 32 Daratumumab was also well tolerated in these patients. In a recently published combined analysis of two daratumumab monotherapy studies of heavily pretreated patients, the ORR was 30.4% with 20 (13.5%) patients achieving ≥VGPR and 7 (4.7%) achieving ≥CR; the rate of serious drug‐related TEAEs was 9%. 22 Similar results were seen in our study, although the mechanisms of action are thought to differ between isatuximab and daratumumab. 33 One study has compared the mechanisms of action of isatuximab and daratumumab, but further studies are required to investigate how differences in their mechanisms of action, including ectoenzyme modulation activity and programmed cell death activity, which are characteristics of isatuximab, may influence its clinical effects and resistance. 34
Limitations of this study are its small sample size, inclusion of selected patients, absence of a control group, and the small number of patients with high‐risk cytogenetic abnormalities. Despite these limitations, this study provided evidence on the effectiveness and safety of isatuximab with central review of outcomes. Therefore, the results are clinically informative.
In conclusion, in this study of patients with heavily pretreated MM, isatuximab monotherapy was well tolerated, and there were no DLTs at either dose in phase 1. Approximately one‐third of patients experienced a partial or better response and half of patients experienced a MR or better. These data demonstrate the efficacy of isatuximab in this setting, including in patients with more than six prior lines of treatment and in patients with high‐risk cytogenetic abnormalities.
CONFLICT OF INTEREST
Kazutaka Sunami has received research grants and personal fees from Takeda, Ono, BMS, and Celgene; and research grants from Sanofi, Novartis, GSK, Janssen, AbbVie, MSD, Alexion Pharma, and Daiichi‐Sankyo. Kenshi Suzuki has received personal fees from Janssen, Novartis, Celgene, Ono, Fujimoto, Takeda, Dainippon Sumitomo, BMS, and Sanofi; and is an employee of SRL. Masaki Ri has received research grants and personal fees from Ono, Janssen, Celgene, BMS, Sanofi, and Takeda; and research grants from Chugai, Kyowa Hakko Kirin, MSD, Gilead, Novartis, Daiichi‐Sankyo, and AbbVie. Morio Matsumoto has received personal fees from Celgene, Janssen, BMS, and Ono. Chihiro Shimazaki has received personal fees from Ono, Celgene, and Fujimoto. Hideki Asaoku has nothing to disclose. Hirohiko Shibayama has received research grants from AbbVie, AstraZeneca, Celgene, Chugai, Ono, Janssen, and Sanofi; honoraria from Celgene, Chugai, Kyowa Kirin, Janssen, Novartis, and Takeda; and scholarship endowment from Astellas, Eisai, Nippon Shinyaku, Sanofi, Shionogi, Taiho, and Teijin. Kenichi Ishizawa has received research grants and personal fees from Chugai, Kyowa Hakko Kirin, Eisai, and Otsuka; research grants from Sanofi, AbbVie, and SymBio; and personal fees from Janssen, Ono, Celgene, and MSD. Hiroyuki Takamatsu has acted as a consultant for Sanofi and has received grants and personal fees from Celgene, BMS and Ono; grants from CSL Behring and SRL; and personal fees from Janssen, Takeda, Fujimoto, and Becton, Dickinson and Company. Takashi Ikeda received research grants from Pfizer, Sanofi, and Takeda and lecture fees from Takeda. Dai Maruyama has received research grants and personal fees from Takeda, Janssen, Eisai, Celgene, Kyowa Hakko Kirin, Ono, Mundipharma, Chugai, MSD, Zenyaku, BMS, Daiichi‐Sankyo, and AstraZeneca; research grants from Sanofi, AbbVie, Astellas, Amgen Astellas Biopharma, Otsuka, Novartis, Pfizer, Solasia, Bayer, SymBio, CMIC, Quintiles, and IQVIA; and personal fees from Sumitomo Dainippon, Asahi Kasei, Linical, Pharma International, and Fujimoto. Hitomi Kaneko has nothing to disclose. Michihiro Uchiyama has nothing to disclose. Toru Kiguchi has received research grants and personal fees from BMS, Otsuka, Kyowa Hakko Kirin, MSD, Takeda, Nippon Shinyaku, Novartis, Sumitomo Dainippon, Janssen, and Celgene; research grants from Daiichi‐Sankyo, Astellas, SymBio, Taiho, Teijin, Sanofi, and Celltrion; and personal fees from Chugai, Pfizer, Eisai, Mochida, Ono, and Asahi Kasei. Satoshi Iyama has received research grants from Otsuka Pharmaceutical, Otsuka Pharmaceutical Factory, Astellas Pharma, Daiichi Sankyo, Alexion Pharma, CSL Behring, and Dainippon Sumitomo. Hirokazu Murakami has received research grants from Fujimoto and personal fees from Celgene and Sanofi. Keishiro Takahashi, Keisuke Tada, Sandrine Macé, and Helene Guillemin‐Paveau are employees of Sanofi. Shinsuke Iida has received research grants from Abbvie, Bristol‐Myers Squibb, Janssen, MSD, and Takeda; personal fees from Bristol‐Myers Squibb, Celgene, Daiichi Sankyo, Janssen, Ono, Sanofi, and Takeda; and scholarship endowments from Chugai, Kyowa Kirin, Ono, Sanofi, and Takeda.
Supporting information
Fig S1
Fig S2
Table S1
Table S2
Table S3
Table S4
Table S5
Doc S1
ACKNOWLEDGEMENTS
This study was funded by Sanofi. The authors would like to thank the participating patients and their families, the study centers and investigators for their contributions to the study, and the following individuals from Sanofi: T. Sasaki, R. Onishi, K. Aoki, and S. Sugihara for the study operation support from research and development and S. Tsukube for the editorial support. The authors thank Nicholas D. Smith (EMC KK) and Sally‐Anne Mitchell, PhD (McCANN HEALTH CMC, Japan) for medical writing support, which was funded by Sanofi.
Sunami K, Suzuki K, Ri M, et al. Isatuximab monotherapy in relapsed/refractory multiple myeloma: A Japanese, multicenter, phase 1/2, safety and efficacy study. Cancer Sci 2020;111:4526–4539. 10.1111/cas.14657
DATA AVAILABILITY STATEMENT
Qualified researchers may request access to patient‐level data and related study documents including the clinical study report, study protocol with any amendments, blank case report form, statistical analysis plan, and dataset specifications. Patient‐level data will be anonymized, and study documents will be redacted to protect the privacy of trial participants. Further details on Sanofi's data sharing criteria, eligible studies, and process for requesting access can be found at https://www.clinicalstudydatarequest.com.
REFERENCES
- 1. Chim CS, Kumar SK, Orlowski RZ, et al. Management of relapsed and refractory multiple myeloma: novel agents, antibodies, immunotherapies and beyond. Leukemia. 2018;32:252‐262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Chari A, Suvannasankha A, Fay JW, et al. Daratumumab plus pomalidomide and dexamethasone in relapsed and/or refractory multiple myeloma. Blood. 2017;130:974‐981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Dimopoulos MA, San‐Miguel J, Belch A, et al. Daratumumab plus lenalidomide and dexamethasone versus lenalidomide and dexamethasone in relapsed or refractory multiple myeloma: updated analysis of POLLUX. Haematologica. 2018;103:2088‐2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Facon T, Kumar S, Plesner T, et al. MAIA Trial Investigators. Daratumumab plus lenalidomide and dexamethasone for untreated myeloma. N Engl J Med. 2019;380:2104‐2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lonial S, Dimopoulos M, Palumbo A, et al. Elotuzumab therapy for relapsed or refractory multiple myeloma. N Engl J Med. 2015;373:621‐631. [DOI] [PubMed] [Google Scholar]
- 6. Mateos MV, Dimopoulos MA, Cavo M, et al. ALCYONE Trial Investigators. Daratumumab plus bortezomib, melphalan, and prednisone for untreated myeloma. N Engl J Med. 2018;378:518‐528. [DOI] [PubMed] [Google Scholar]
- 7. Attal M, Richardson PG, Rajkumar SV, et al. Isatuximab plus pomalidomide and low‐dose dexamethasone versus pomalidomide and low‐dose dexamethasone in patients with relapsed and refractory multiple myeloma (ICARIA‐MM): a randomised, multicentre, open‐label, phase 3 study. Lancet. 2019;394:2096‐2107. [DOI] [PubMed] [Google Scholar]
- 8. Deckert J, Wetzel MC, Bartle LM, et al. SAR650984, a novel humanized CD38‐targeting antibody, demonstrates potent antitumor activity in models of multiple myeloma and other CD38+ hematologic malignancies. Clin Cancer Res. 2014;20:4574‐4583. [DOI] [PubMed] [Google Scholar]
- 9. Feng X, Zhang L, Acharya C, et al. Targeting CD38 suppresses induction and function of T regulatory cells to mitigate immunosuppression in multiple myeloma. Clin Cancer Res. 2017;23:4290‐4300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jiang H, Acharya C, An G, et al. SAR650984 directly induces multiple myeloma cell death via lysosomal‐associated and apoptotic pathways, which is further enhanced by pomalidomide. Leukemia. 2016;30:399‐408. [DOI] [PubMed] [Google Scholar]
- 11. Moreno L, Perez C, Zabaleta A, et al. The mechanism of action of the anti‐CD38 monoclonal antibody isatuximab in multiple myeloma. Clin Cancer Res. 2019;25:3176‐3187. [DOI] [PubMed] [Google Scholar]
- 12. van de Donk NWCJ, Usmani SZ. CD38 antibodies in multiple myeloma: mechanisms of action and modes of resistance. Front Immunol. 2018;9:2134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Martin T, Strickland S, Glenn M, et al. Phase I trial of isatuximab monotherapy in the treatment of refractory multiple myeloma. Blood Cancer J. 2019;9:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Martin T, Baz R, Benson DM, et al. A phase 1b study of isatuximab plus lenalidomide and dexamethasone for relapsed/refractory multiple myeloma. Blood. 2017;129:3294‐3303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mikhael J, Richardson P, Usmani SZ, et al. A phase Ib study of isatuximab plus pomalidomide/dexamethasone in relapsed/refractory multiple myeloma. Blood. 2019;134:123‐133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Palumbo A, Rajkumar SV, San Miguel JF, et al. International Myeloma Working Group consensus statement for the management, treatment, and supportive care of patients with myeloma not eligible for standard autologous stem‐cell transplantation. J Clin Oncol. 2014;32:587‐600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Greipp PR, San Miguel J, Durie BG, et al. International staging system for multiple myeloma. J Clin Oncol. 2005;23:3412‐3420. [DOI] [PubMed] [Google Scholar]
- 18. Martin TG, Hsu K, Strickland SA, Glenn MJ, Mikhael J, Charpentier E. A phase I trial of SAR650984, a CD38 monoclonal antibody, in relapsed or refractory multiple myeloma. J Clin Oncol. 2014;32(suppl):8532. [Google Scholar]
- 19. Thai HT, Liu L, Koiwai K, et al. Exposure‐response analysis & disease modeling for selection of optimal dosing regimen of isatuximab as single agent in patients with multiple myeloma. Abstract PF645. 24th EHA Annual Congress. Amsterdam; 2019.
- 20. Iida S, Suzuki K, Kusumoto S, et al. Safety and efficacy of daratumumab in Japanese patients with relapsed or refractory multiple myeloma: a multicenter, phase 1, dose‐escalation study. Int J Hematol. 2017;106:541‐551. [DOI] [PubMed] [Google Scholar]
- 21. Usmani SZ, Weiss BM, Plesner T, et al. Clinical efficacy of daratumumab monotherapy in patients with heavily pretreated relapsed or refractory multiple myeloma. Blood. 2016;128:37‐44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Usmani SZ, Nahi H, Plesner T, et al. Daratumumab monotherapy in patients with heavily pretreated relapsed or refractory multiple myeloma: Final results from the phase 2 GEN501 and SIRIUS trials. Lancet Haematol. 2020;7:e447‐e455. [DOI] [PubMed] [Google Scholar]
- 23. Mikhael J, Richter J, Vij R, et al. A dose‐finding Phase 2 study of single agent isatuximab (anti‐CD38 mAb) in relapsed/refractory multiple myeloma. Leukemia. 2020; 10.1038/s41375-020-0857-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Frerichs KA, Nagy NA, Lindenbergh PL, et al. CD38‐targeting antibodies in multiple myeloma: mechanisms of action and clinical experience. Expert Rev Clin Immunol. 2018;14:197‐206. [DOI] [PubMed] [Google Scholar]
- 25. Morandi F, Horenstein AL, Costa F, Giuliani N, Pistoia V, Malavasi F. CD38: a target for immunotherapeutic approaches in multiple myeloma. Front Immunol. 2018;9:2722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nijhof IS, Casneuf T, van Velzen J, et al. CD38 expression and complement inhibitors affect response and resistance to daratumumab therapy in myeloma. Blood. 2016;128:959‐970. [DOI] [PubMed] [Google Scholar]
- 27. Iyoda T, Yamasaki S, Hidaka M, et al. Amelioration of NK cell function driven by Vα24+ invariant NKT cell activation in multiple myeloma. Clin Immunol. 2018;187:76‐84. [DOI] [PubMed] [Google Scholar]
- 28. Krejcik J, Casneuf T, Nijhof IS, et al. Daratumumab depletes CD38+ immune regulatory cells, promotes T‐cell expansion, and skews T‐cell repertoire in multiple myeloma. Blood. 2016;128:384‐394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Fedele PL, Willis SN, Liao Y, et al. IMiDs prime myeloma cells for daratumumab‐mediated cytotoxicity through loss of Ikaros and Aiolos. Blood. 2018;132:2166‐2178. [DOI] [PubMed] [Google Scholar]
- 30. Moreau P, Dimopoulos M‐A, Mikhael J, et al. Isatuximab plus carfilzomib and dexamethasone vs carfilzomib and dexamethasone in relapsed/refractory multiple myeloma (IKEMA): Interim analysis of a phase 3, randomized, open‐label study. Abstract LB2603. 25th EHA Annual Congress. Virtual Edition; 2020.
- 31. Zonder JA, Mohrbacher AF, Singhal S, et al. A phase 1, multicenter, open‐label, dose escalation study of elotuzumab in patients with advanced multiple myeloma. Blood. 2012;120:552‐559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lonial S, Weiss BM, Usmani SZ, et al. Daratumumab monotherapy in patients with treatment‐refractory multiple myeloma (SIRIUS): an open‐label, randomised, phase 2 trial. Lancet. 2016;387:1551‐1560. [DOI] [PubMed] [Google Scholar]
- 33. Touzeau C, Moreau P, Dumontet C. Monoclonal antibody therapy in multiple myeloma. Leukemia. 2017;31:1039‐1047. [DOI] [PubMed] [Google Scholar]
- 34. Malavasi F, Faini AC. Mechanism of action of a new anti‐CD38 antibody: enhancing myeloma immunotherapy. Clin Cancer Res. 2019;25:2946‐2948. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1
Fig S2
Table S1
Table S2
Table S3
Table S4
Table S5
Doc S1
Data Availability Statement
Qualified researchers may request access to patient‐level data and related study documents including the clinical study report, study protocol with any amendments, blank case report form, statistical analysis plan, and dataset specifications. Patient‐level data will be anonymized, and study documents will be redacted to protect the privacy of trial participants. Further details on Sanofi's data sharing criteria, eligible studies, and process for requesting access can be found at https://www.clinicalstudydatarequest.com.