

Abstract
Cancer‐derived myocardial damage is an important cause of death in cancer patients. However, the development of dietary interventions for treating such damage has not been advanced. Here, we investigated the effect of dietary intervention with lauric acid (LAA) and glucose, which was effective against skeletal muscle sarcopenia in a mouse cachexia model, on myocardial damage. Treatment of H9c2 rat cardiomyoblasts with lauric acid promoted mitochondrial respiration and increased ATP production by Seahorse flux analysis, but did not increase oxidative stress. Glycolysis was also promoted by LAA. In contrast, mitochondrial respiration and ATP production were suppressed, and oxidative stress was increased in an in vitro cachexia model in which cardiomyoblasts were treated with mouse cachexia ascites. Ascites‐treated H9c2 cells with concurrent treatment with LAA and high glucose showed that mitochondrial respiration and glycolysis were promoted more than that of the control, and ATP was restored to the level of the control. Oxidative stress was also reduced by the combined treatment. In the mouse cachexia model, myocardiac atrophy and decreased levels of a marker of muscle maturity, SDS‐soluble MYL1, were observed. When LAA in CE‐2 diet was orally administered alone, no significant rescue was observed in the cancer‐derived myocardial disorder. In contrast, combined oral administration of LAA and glucose recovered myocardial atrophy and MYL1 to levels observed in the control without increase in the cancer weight. Therefore, it is suggested that dietary intervention using a combination of LAA and glucose for cancer cachexia might improve cancer‐derived myocardial damage.
Keywords: atrophy, cachexia, mitochondria, myocardium, oxidative stress
We elucidated that cancer‐derived myocardial damage results mainly from oxidative stress. The combined administration of LAA and glucose can effectively provide protective effects against myocardial damage and promote recovery.
Abbreviations
- ECAR
extracellular acidification rate
- FCCP
carbonyl cyanide‐p‐trifluoromethoxy phenylhydrazone
- HMGB
high mobility group box
- HNE
hydroxynonenal
- LAA
lauric acid
- LCFA
long‐chain fatty acid
- MCFA
medium‐chain fatty acid
- MYL
myosin light chain
- OCR
oxygen consumption rates
- QFM
quadriceps femoris muscle
- ROS
reactive oxygen species
- TNF
tumor necrosis factor
- UCP
uncoupling protein
1. INTRODUCTION
Cachexia is reported to be present in 40%‐80% of all patients with advanced cancer, 1 , 2 and it accounts for 20%‐30% of all cancer‐related deaths. 3 Weight loss in patients with cancer‐related cachexia is associated with myocardial atrophy, 4 , 5 which is a major cause of death in cancer patients. 6 Hence, investigating preventive or therapeutic methods for cancerous myocardial damage that can greatly affect the prognosis of cancer patients, would be useful for medical treatment.
The potential causes of cancer‐derived myocardial damage include direct effects of the cancer, any underlying heart disease in the patient, and effects of cancer treatment. 7 One of the causes of cancer‐derived myocardial injury derived from the cancer itself is considered to be energy metabolism disorder due to mitochondrial dysfunction 7 ; particularly, reduced mitochondrial uncoupling and ATP production have been reported. 8 , 9
Medium‐chain fatty acids (MCFAs) show a faster intestinal absorption and uptake into tissues than long‐chain fatty acids (LCFAs). 10 , 11 , 12 Furthermore, unlike LCFAs, MCFAs migrate into mitochondria independently of the carnitine shuttle and rapidly undergo β‐oxidation to promote oxidative phosphorylation. 12 , 13 Therefore, in cancer cells with an impaired electron transport system complex, abnormal oxidative stress is enhanced by the loading of MCFAs, resulting in cell death. 14 In contrast, MCFAs and short‐chain fatty acids have been reported to increase skeletal muscle in animals, 15 and MCFAs showed an inhibitory effect on cancer‐related sarcopenia in a mouse model. 16
Various dietary interventions have been attempted for cancer‐related sarcopenia. Previously, we succeeded in suppressing sarcopenia in a mouse cachexia model using glucose, which has a skeletal muscle protective effect but also a tumor‐promoting effect, in combination with the MCFA, lauric acid (LAA), which has a skeletal muscle promoting effect. 16 Furthermore, in nutritional intervention for myocardial disorders, the exacerbation of the disorder due to calorie restriction has been reported. 17 In a mouse heart failure model, fatty acid intake promotes mitochondrial fragmentation and improves cardiac dysfunction. 18 Furthermore, ketogenic diet with MCFAs improves succinic dehydrogenase activity, subsequently improving the myocardial atrophy and dysfunction. 19 In contrast, excessive LAA intake induces oxidative stress in the myocardium and leads to myocardial atrophy. 20 However, there are only a few reports on nutritional interventions for cancer‐derived myocardial disorders. Therefore, we investigated the in vitro and in vivo effects of a combination of LAA and glucose on cancer‐derived myocardial damage and evaluated its efficacy against cancer‐related sarcopenia.
2. MATERIALS AND METHODS
2.1. Cell culture
The CT26 mouse colon cancer cell line was a kind gift from Professor IJ Fidler (MD Anderson Cancer Center). CT26 cells were cultured in Dulbecco's modified Eagle's medium (DMEM; Wako Pure Chemical Industries, Ltd.) supplemented with 10% fetal bovine serum (Sigma‐Aldrich Chemical Co.). The embryonic rat heart‐derived H9c2 cardiomyoblast cells were purchased from American Type Culture Collection, and cultured in DMEM with 10% fetal bovine serum (Sigma‐Aldrich). Cell proliferation was assessed by counting cell number with a hemocytometer (Sysmex). H9c2 cardiomyoblast cells were treated with LAA (40 μg/mL) 16 and/or high glucose (450 mg/dL). 14 , 16
2.2. Animals
Five‐wk‐old male BALB/c mice were purchased from SLC Japan. The animals were maintained in a pathogen‐free animal facility under a 12 h : 12 h, light : dark cycle in a temperature‐controlled (22°C) and humidity‐controlled environment, in accordance with the institutional guidelines approved by the Committee for Animal Experimentation of Nara Medical University, Kashihara, Japan, following current regulations and standards of the Japanese Ministry of Health, Labor and Welfare (approval nos. 11812, 11857, 11916, 12043, and 12262). Animals were acclimated to their housing for 7 d before the start of the experiment. Mice were fed with CE‐2 diet (containing 5% crude fat, mainly derived from soy bean oil; CLEA Japan, Inc).
To measure tumor weight, mice were euthanized by aortic blood removal under the anesthesia sevoflurane (Maruishi Pharmaceutical Co. Ltd.) and the peritoneal tumors were dissected from the intestine, mesenterium, diaphragm, and abdominal wall, grossly removing non‐tumoral tissues.
For preparation of the heart, after euthanasia, the heart was excised, its weight was measured and then divided into 2 at 2/5 from the apex of the heart. The upper part was used for histological analysis, and the lower part was used for analyzing protein expression. The image of the cut surface of the heart was captured on a computer, and the areas of the myocardium and the lumen of the heart were traced using ImageJ (GitHub).
For preparation of skeletal muscles, the QFM was cut at the muscle end on the upper edge of the patella, peeled off from the femur, and separated at the muscle origin on the frontal surface of the anterior lower iliac spine. The excised QFM was weighed immediately, avoiding drying, and then stored at −80°C.
2.3. Diet and drink
Glucose solution (50% glucose for injection, Otsuka Pharmaceutical Co. Ltd.) was used directly or diluted to 10% with distilled water for drinking. 16 CE‐2 diet (CLEA Japan, Inc) was used as control diet. LAA diet was prepared by mixing 0% or 2% (w/w) LAA, Tokyo Chemical Industry Co., Ltd.) with control diet (CE‐2). 16 Glucose drink and LAA diet were administered by free intake. 16
2.4. Histological analysis
Myocardial tissues were fixed in 4% paraformaldehyde, dehydrated, and embedded in paraffin. After slicing the created block to 3 μm, hematoxylin and eosin staining was performed to observe the morphology.
2.5. Protein extraction
The lower part of the excised heart stored at −80°C was crushed with a hammer to remove tendons and fascia. Only the muscle tissue was washed with cold phosphate‐buffered saline and pelleted with a sonicator (QSONICA, WakenBtech Co. Ltd.). Whole‐cell lysates were prepared as previously described using 0.1% SDS‐added RIPA‐buffer (Thermo Fisher Scientific). Protein assay was performed using a Protein Assay Rapid Kit (Wako Pure Chemical Corporation).
2.6. Enzyme‐linked immunosorbent assay (ELISA) and colorimetric assay
ELISA kits were used to measure the concentration of myosin light chain (MYL)‐1 (Cusabio Biotech Co., Ltd.), high mobility group box (HMGB)‐1 (Shino‐Test Co.), mouse tumor necrosis factor (TNF)‐α (R&D Systems, Inc), 4‐hydroxynonenal (HNE) and ATP (Abcam). Ascites lactate was measured using a colorimetric assay kit (BioVision, Inc). The assays were performed in accordance with the manufacturers' instructions, and whole‐cell lysates were used for the measurements.
2.7. Mitochondrial stress test (Seahorse assay)
To simulate the cachectic condition, the ascites of CT26‐induced cachexia mice were collected. The ascites were filtered with the Sterile Millex Filter (pore size 0.22 µm, Sigma). H9c2 cells were cultured in a regular medium for 48 h and this culture medium was also filtered. The H9c2 cells were cultured in a growth medium in 6‐well plates before the Seahorse assay with the ascites (20% v/v) or the collected cultured medium (20% v/v). OCR of 1 × 104 viable H9c2 cells per well were measured using the Seahorse XFe24 Extracellular Flux Analyzer with Seahorse XF24 FluxPaks (Agilent Technologies). Seahorse assays were carried out as follows: OCR in pmol/min were measured before (basal OCR) and after successive injection of 1‐µmol/L oligomycin (ATP synthase inhibitor), 2‐µmol/L FCCP (carbonyl cyanide‐p‐trifluoromethoxy phenylhydrazone, an uncoupling protonophore), 1‐µmol/L rotenone (Complex I inhibitor), and 5‐µmol/L antimycin A (Complex III inhibitor). From the resulting data, we determined the OCR associated with respiratory ATP synthesis (oligomycin‐sensitive), the maximum OCR in FCCP‐uncoupled mitochondria, the rotenone‐sensitive OCR attributable to uncoupled Complex I activity, the antimycin‐sensitive Complex II/III activity, and the OCR by mitochondrial functions other than ATP synthesis, including OCR that is mitochondrial membrane potential‐driven (proton leak), non‐respiratory oxygen consumption, and the respiratory “spare capacity” (excess capacity of the respiratory electron transport chain that is not being used in basal respiration).
2.8. Glycolytic stress test
The extracellular acidification rate (ECAR) of H9c2 cells was measured using a modified glycolytic stress test in the Seahorse XFe24 Extracellular Flux Analyzer with Seahorse XF24 FluxPaks (Agilent Technologies). H9c2 cells were cultured in a growth medium in 6‐well plates with the ascites or the cultured medium before Seahorse experiments. H9c2 cells (1 × 104 cells/well) were later plated in the XF base medium (Agilent Technologies) containing 200 mmol/L l‐glutamine and 5 mmol/L HEPES, as recommended by the manufacturer for glycolytic assays. The sensor cartridge apparatus was rehydrated 1 d in advance by adding 1‐mL XF Calibrant to each well and incubating at 37°C until needed. The injection ports of the sensor cartridge apparatus were loaded with the following drugs, in chronological order of 4 injections, to meet the indicated final concentrations in the wells: 10 mmol/L glucose, 1 µmol/L oligomycin, 1 µmol/L rotenone, and 5 µmol/L antimycin A (combined injection), and 50 mmol/L 2‐deoxyglucose. Treatment with the rotenone/antimycin combination allowed assessment of the impact of electron transport on ECAR by respiratory acidification coupled to passage of some glycolytic pyruvate through the tricarboxylic acid (TCA) cycle to supply respiration.
2.9. Statistical analysis
Statistical significance was calculated using unpaired Student t tests using InStat software (version 3.0; GraphPad Software, Inc). Data were expressed as the mean ± standard deviation of 3 independent experiments. P < .05 (two‐sided) was considered to indicate statistical significance.
3. RESULTS
3.1. Effect of LAA on H9c2 cardiomyoblasts
The effect of LAA on H9c2 cardiomyoblasts was examined for various treatment concentrations (Figure 1A). An LAA concentration of 40 μg/mL promoted cardiomyoblast proliferation, whereas 120 μg/mL LAA inhibited cell growth. Therefore, the concentration of LAA treatment was set to 40 μg/mL in subsequent experiments.
Figure 1.
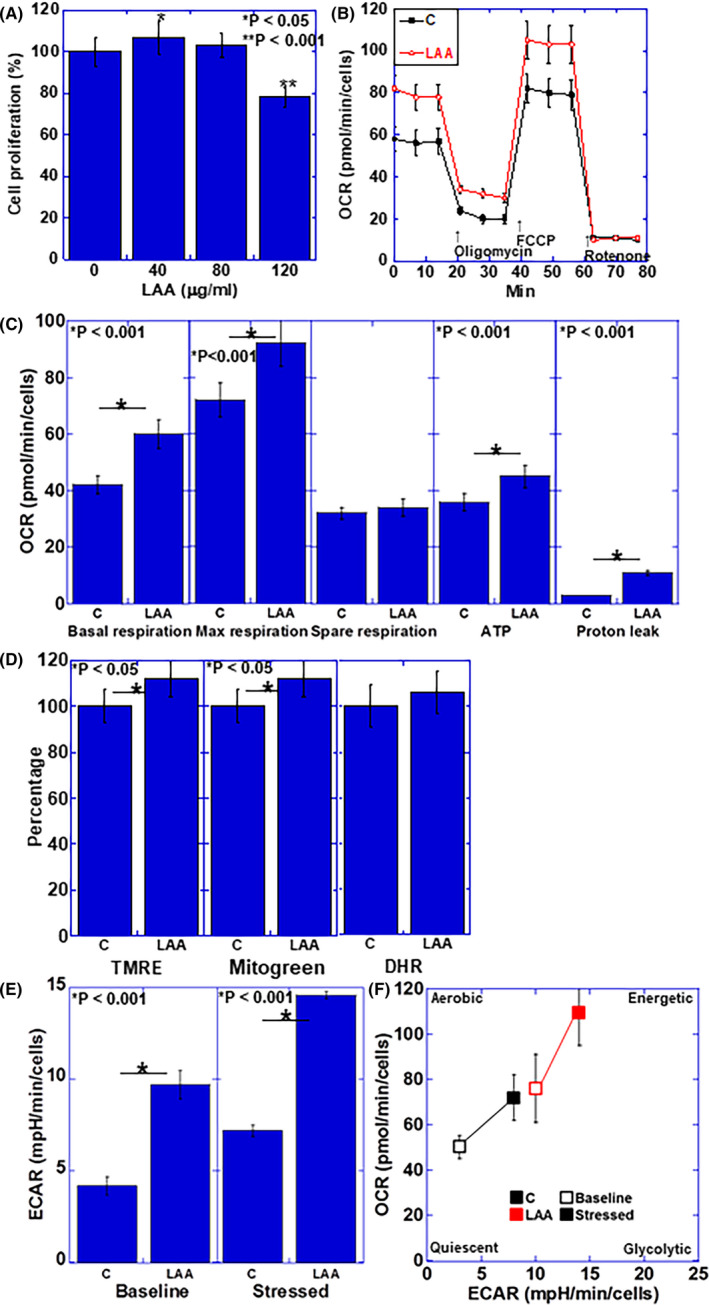
Effect of LAA in H9c2 cardiomyoblasts. A, Cell proliferation in LAA‐treated H9c2 cells. B, Flux analysis of LAA‐treated H9c2 cells. C, Effect of LAA on mitochondrial respiration, ATP production, and proton leak. D, Effect of LAA on mitochondrial membrane voltage (evaluated using TMRE), mitochondrial volume (evaluated using Mitogreen) and oxidative stress (evaluated using DHR). E, Effect of LAA on glycolytic activity (determined by ECAR measurement). F, Effect of LAA on cell energy phenotype profile. Error bars, standard error from 3 trials. The statistical significance was calculated by Student t test. C, control; DHR, dihydrorhodamine 123; ECAR, extracellular acidification rate; LAA, lauric acid; OCR, oxygen consumption rate; TMRE, tetramethyl rhodamine
The effect of LAA on the energy production of H9c2 cardiomyoblasts was examined by flux analysis (Figure 1B,C). Spare respiration was not altered compared to the control; however, basal respiration, maximum respiration, and ATP production were all promoted by LAA. The proton leak increased upon LAA treatment.
Next, the effect of LAA treatment on mitochondrial potential, mitochondrial volume, and oxidative stress of H9c2 cardiomyoblasts was examined (Figure 1D). LAA did not affect the oxidative stress, whereas LAA increased the mitochondrial membrane potential and the mitochondrial volume. In contrast, ECAR was increased by LAA in both baseline and stressed phases (Figure 1E). When the cell energy phenotype profile was obtained (Figure 1F), LAA was found to promote both oxidative phosphorylation and glycolysis in baseline and stressed phases.
3.2. Effects of LAA and glucose on energy metabolism in H9c2 cardiomyoblasts in in vitro cachexia model
We used an in vitro cachexia model wherein 20% of ascites of a mouse cachexia model 16 was added to H9c2 cardiomyoblasts, and the effect of LAA (40 μg/mL) and glucose (450 mg/dL) was examined (Figure 2A). As a control, a culture medium containing 20% of the culture supernatant of H9c2 cardiomyoblasts was used. Table 1 shows the composition of the culture solution used for the treatment, however no significant difference was observed among the treatment media with regards to sugar, pyruvic acid, and l‐glutamine. TNFα and HMGB1 were detected in the ascites‐supplemented culture medium. Ascites treatment markedly suppressed the proliferation of H9c2 cardiomyoblasts, whereas LAA + glucose completely rescued the proliferation.
Figure 2.
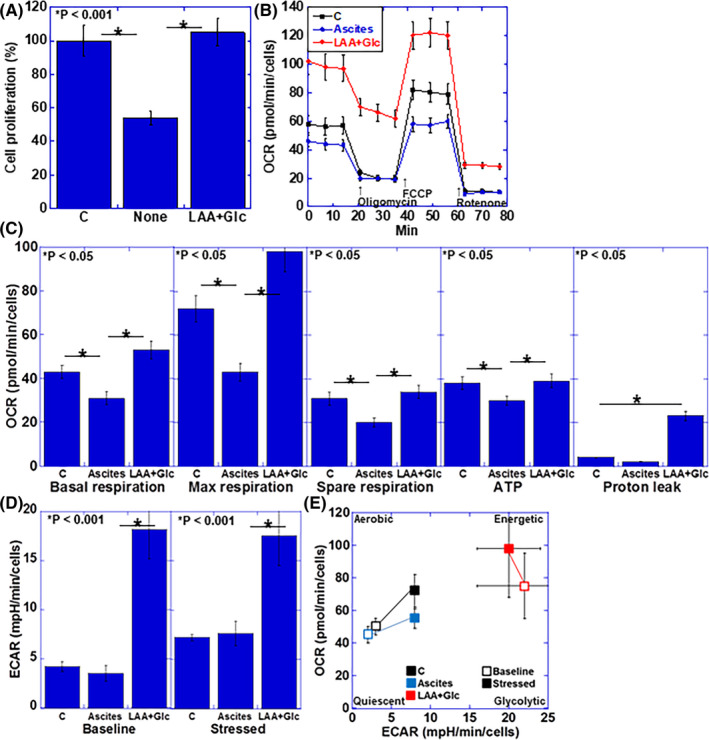
Effect of LAA and glucose on energy metabolism in cachectic ascites‐treated H9c2 cardiomyoblasts. A, Cell proliferation in ascites‐treated H9c2 cells. B, Flux analysis of ascites‐treated H9c2 cells. C, Effect of LAA + Glc on mitochondrial respiration, ATP production, and proton leak. D, Effect of LAA + Glc on glycolytic activity (determined by ECAR measurement). E, Effect of LAA + Glc on cell energy phenotype profile. Error bars, standard error from 3 trials. The statistical significance was calculated by Student t test. C, control; ECAR, extracellular acidification rate; Glc, glucose; LAA, lauric acid; OCR, oxygen consumption rate
Table 1.
Components of medium for treatment
| Component | Medium a | ||
|---|---|---|---|
| D‐MEM a | Ascites added b | CM added c | |
| Glucose (mg/dL) | 450 ± 2 | 361 ± 8 | 378 ± 6 |
| Pyruvate (mg/dL) | 11 ± 0.1 | 9 ± 1 | 9 ± 1 |
| Glutamine (mg/dL) | 58 ± 0.2 | 49 ± 4 | 50 ± 3 |
| Lactate (pmol) | 0 | 6.1 ± 1.2 | 1.2 ± 0.2 |
| HMGB1 (μg/mL) | ND d | 12 ± 0.8 | ND |
| TNFα (pg/mL) | ND | 9 ± 0.1 | ND |
Regular medium: D‐MEM (WAKO, Osaka, Japan) supplemented with 10% fetal bovine serum.
Regular medium with added 30% (v/v) mouse ascites of the cachexia model.
Regular medium with added 30% (v/v) culture medium obtained from culturing H9c2 for 48 h.
ND, not detected.
The effects of LAA + glucose in the in vitro cachexia model of H9c2 cardiomyoblasts were examined from the viewpoint of mitochondrial energy metabolism by flux analysis (Figure 2B,C). Compared with the control, ascites treatment led to decreased basal respiration, maximum respiration, spare respiration, and ATP production. In contrast, treatment with LAA + glucose could recover the basal respiration and maximum respiration to levels above those of the control, while the spare respiration and ATP production levels were recovered to similar levels as in the control. However, the proton leak increased significantly upon LAA + glucose treatment.
The ECAR upon ascites treatment showed no significant change from the control, however a marked increase in both baseline and stressed phases was observed upon treatment with LAA + glucose (Figure 2D). When the cell energy phenotype profile was obtained (Figure 2E), no significant change was observed in the ascites treatment; however, upon treatment with LAA + glucose, both oxidative phosphorylation and glycolysis were promoted, resulting in increased energetic phenotype.
3.3. Effects of LAA and glucose on mitochondria in H9c2 cardiomyoblasts in in vitro cachexia model
Next, the effects of LAA + glucose on mitochondrial membrane potential, mitochondrial volume, and oxidative stress in H9c2 cardiomyoblasts treated with cachectic ascites were examined (Figure 3A,B). Ascites treatment decreased the membrane potential and mitochondrial volume of H9c2 cardiomyoblasts and increased the oxidative stress. In contrast, treatment with LAA + glucose improved the membrane potential, mitochondrial volume, and oxidative stress, along with restoration of the membrane potential and mitochondrial volume to levels found in the control.
Figure 3.
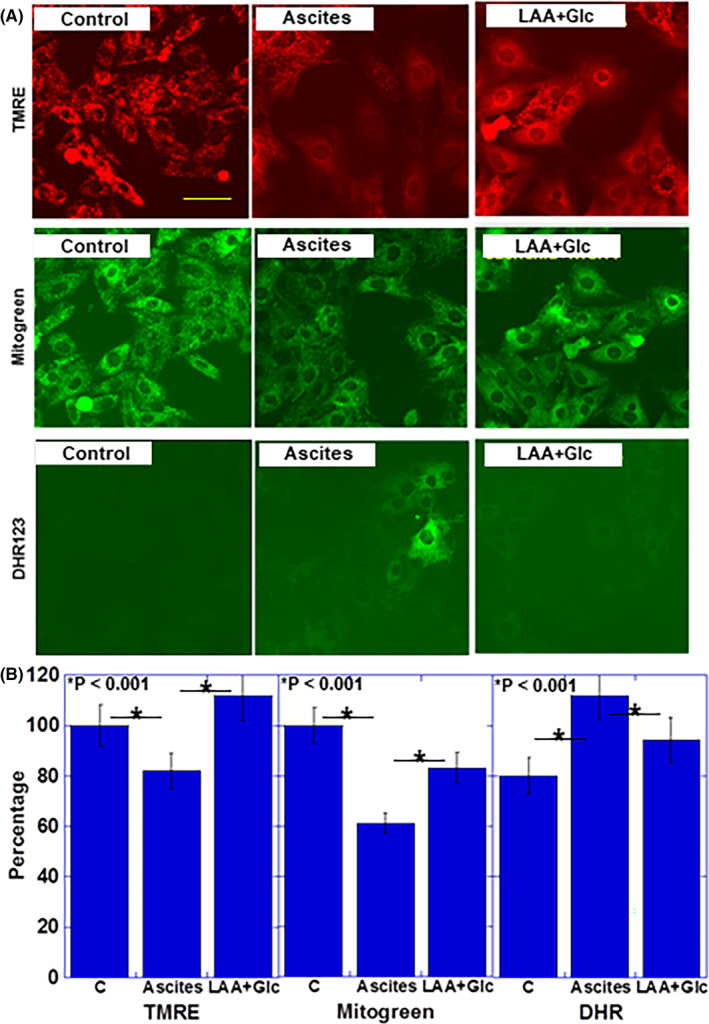
Effect of LAA and glucose on mitochondria in cachectic ascites‐treated H9c2 cardiomyoblasts. A, B, Effect of LAA + Glc on mitochondrial membrane voltage (evaluated using TMRE), mitochondrial volume (evaluated using Mitogreen) and oxidative stress (evaluated using DHR). Scale bar, 50 μm. Error bars, standard error from 3 trials. C, control; cachexia, ascites‐treated; DHR, dihydrorhodamine 123; Glc, glucose; LAA, lauric acid; MCFA, TMRE, tetramethyl rhodamine
3.4. Effects of LAA and glucose administration on myocardial atrophy in murine cachexia model
Next, we examined the effect of combined use of LAA and glucose on myocardial atrophy using the cachexia model, 16 prepared by inoculating mouse CT26 colon cancer cells into the peritoneal cavity of syngeneic BALB/c mice (Figures 4 and 5). In the cross‐section of the heart, dilation of the left ventricular lumen was observed in the cachexia group, and the dilation was reduced in the LAA + glucose combination group (Figure 4A). In the histology of the left ventricle, the density of the nuclei in the cachexia group was increased and the cardiomyocytes were atrophied. In contrast, the nuclear density decreased in the LAA + glucose group (Figure 4B).
Figure 4.
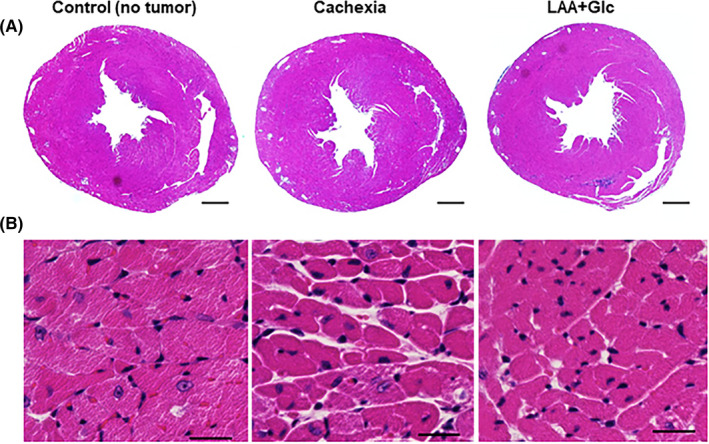
Morphological alteration of myocardium and effect of LAA and glucose in mouse cachectic model. A, Image of cut surface of the heart stained with hematoxylin and eosin. Scale bars, 0.3 mm. B, Photomicrogram of myocardium of the left ventricle stained with hematoxylin and eosin. Scale bars, 50 μm. Glc, glucose; LAA, lauric acid
Figure 5.
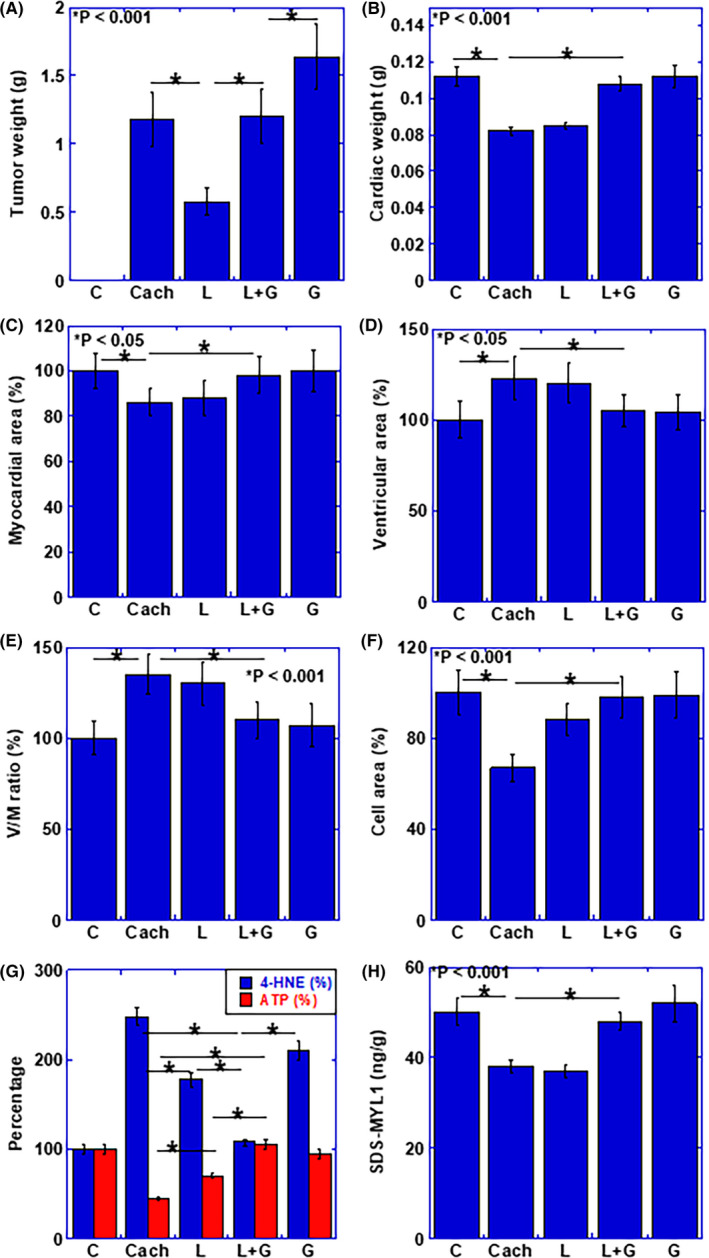
Alteration of myocardium and effect of LAA and glucose in mouse cachectic model. A, Weight of peritoneal tumor. B–H, Effect of LAA and glucose in cachectic mice on B, cardiac weight, C, myocardial area, D, ventricular area, E, ventricular area to myocardial ratio (V/M ratio), F, mean cardiomyocyte area (cell area), G, concentrations of ATP and 4‐HNE in myocardium, and H, protein level of SDS‐soluble myosin light chain (SDS‐MYL1). Error bars, standard error from 3 mice. Statistical significance was calculated by Student t test. C, no tumor control; Cach, cachexia mice; HNE, hydroxynonenal; G, glucose; SDS,‐MYL1, L, lauric acid; sodium dodecyl sulfate‐soluble myosin light chain‐1
The tumor weight was markedly reduced by the administration of LAA, but was similar to the cachexia group in the LAA + glucose combination group. In contrast, glucose alone enhanced tumor weight (Figure 5A). Cardiac weight was decreased in the cachexia group and not improved in the LAA alone group. In contrast, in the combination group, the cardiac weight was improved to the level of the control group (Figure 5B). Furthermore, as shown in Figure 5C‐E, in the cachexia group, the myocardial area was decreased and the intraventricular luminal area was increased. As a result, the lumen‐ventricular area ratio (V/M ratio) increased. No significant improvement was observed in these changes in the LAA alone group. In contrast, the cardiac alterations in cachexia in the LAA + glucose group were improved to the same levels as those in the control group. Furthermore, the area of cardiomyocytes was decreased in cachexia, and incomplete improvement was observed in the LAA alone group. In contrast, the LAA + glucose group showed similar levels as those in the control group (Figure 5F). As glucose alone, morphological alterations due to cachexia were improved at the similar levels as those in the combination group (Figure 5B‐F). To assess the energy metabolism and oxidative stress in myocardium, ATP and 4‐HNE levels were examined (Figure 5G). In cachexia, the ATP level was decreased and that of 4‐HNE was increased. However, these alterations were recovered in the LAA + glucose group; LAA alone group showed partial recovery of the ATP and 4‐HNE levels. In glucose group, ATP level was rescued in the similar level as that in the LAA + glucose group. However, 4‐HNE was still higher than that in the LAA + glucose group. Finally, the SDS‐soluble MYL1 protein level, which is considered to be an index of muscle cell maturity, 20 , 21 was examined (Figure 5H). Levels of SDS‐MYL1 were decreased in the cachexia group, and no improvement was observed in the LAA alone group. In contrast, the LAA + glucose group showed similar levels as in the control group. In glucose alone group, SDS‐MYL1 was increased in comparison with that in the cachexia group, however it was still lower than that in the LAA + glucose group.
4. DISCUSSION
In the present study, we clarified the alterations in energy metabolism in cancer‐derived myocardial disorder by treating cardiomyoblasts with ascites from cachexic mice in which weight loss, skeletal muscle atrophy, ascites, and movement disorder were observed. 16 As a result, it was elucidated that oxidative phosphorylation was reduced with mitochondrial damage, and oxidative stress was generated. These in vitro findings were also confirmed in the mouse cachexia model, evident from a decrease in ATP and an increase in 4‐HNE levels in the myocardium.
In cancer‐derived myocardial disorder, cardiac atrophy, myocardial volume reduction, myocardial remodeling, and dysfunction are observed in human patients. 7 , 22 In the present study, decreased cardiac weight and myocardial mass, and dilated ventricular lumen were observed, which are considered to be alterations corresponding to cancer‐derived myocardial disorder in humans. Myocardial fibrosis, which is observed in myocardial remodeling, 23 was not clear in our model. It is considered that this is because the cachexia model used in this study is a relatively acute alteration that reaches the moribund stage in a few weeks, such that fibrosis could not occur.
In heart failure in individuals without cancer, metabolic shift of mitochondrial oxidative metabolism to glycolysis and uncoupling between glycolysis and glucose oxidation are considered to play important roles in the development of heart failure and dysfunction. 24 In the rat heart failure model, cardiac glycolysis rates increase with decrease in diastolic function. 25 However, no increase in acetyl coenzyme A (CoA) production is observed, and β‐oxidation of lipids and ATP production also decrease. 25 Thus, in heart failure myocardium, energy in the remaining mitochondria is produced predominantly by fatty acid oxidation and not glucose oxidation. 26 Therefore, inhibition of fatty acid oxidation and improvement of mitochondrial glucose oxidation are important for the correction of energy metabolism in heart failure. 24
In contrast, in our cachexia model, in vitro analysis showed a marked decrease in oxidative phosphorylation, whereas decrease in glycolysis was not significant. Decrease in mitochondrial membrane potential and mitochondrial volume suggested that cancer‐derived myocardial damage is mainly due to mitochondrial dysfunction, which is a different pathological condition from heart failure in non‐tumor bearing bodies. The marked increase in oxidative stress suggested that the production of ROS due to mitochondrial dysfunction was the main cause of cancer‐derived myocardial damage. Therefore, the promotion of fatty acid oxidation by administration of LAA and glucose seemed to be contrary to the correction of energy metabolism in non‐cancer heart failure, however both oxidative phosphorylation and glycolytic energy production pathways were activated and the energy profile was energetic in LAA and glucose‐treated cardiomyoblasts. Treatment of LAA and glucose also reduced oxidative stress and reduced mitochondrial damage. In contrast, glucose alone increased ATP production to provide morphological improvement, however glucose alone still retained high levels of oxidative stress not to succeed myocardial maturation.
In the mouse model, administration of LAA alone slightly increased the myocardial area, however recovery of heart weight, myocardial atrophy, ventricular dilation, and myocardial SDS‐MYL1 were not observed. Furthermore, in vitro experiments showed an improvement in the energy metabolism of cardiomyoblasts, but in vivo experiments did not show sufficient recovery. In skeletal muscle disorders in the same cachexia model, LAA alone showed amelioration of atrophy compared with myocardium. 16 The possible cause is the glycolysis‐promoting effect of LAA. In flux analysis, treatment with LAA alone promoted mitochondrial respiration and increased glycolysis. It is also known that LAA promotes hexokinase activity. 27 In contrast, under LAA and high glucose conditions, the ECAR at baseline was higher than that after treatment with LAA alone. From this, it is considered that in a high glucose environment that leads to an increase in glucose consumption and activation of glycolysis, 28 the glycolytic activity of LAA is more markedly expressed.
Our data showed an increase in proton leak when cardiomyoblasts were treated with LAA alone, and a more prominent increase when treated with LAA and high glucose in the in vitro cachexia model. LAA has been reported to cause mitochondrial uncoupling in C2C12 myocytes. 29 Inducible proton leak is caused by uncoupling proteins (UCPs) existing in the inner mitochondrial membrane. 30 LAA induces UCP3 and pyruvate dehydrogenase kinase‐4 expression in C2C12 myocytes 31 and increases inducible proton leak. Conversely, mitochondrial uncoupling due to fatty acids was observed even in brown fat of UCP1 knockout mice, 32 indicating the existence of UCP‐independent proton leak. The UCP‐independent proton leak is largely dependent on the fatty acyl composition of phospholipids in the inner mitochondrial membrane. 33 As proton permeability is enhanced by LCFAs due to lipid composition change in the inner mitochondrial membrane, it is possible that similar changes occurred after treatment with LAA. LAA‐induced proton leak is considered to be the cause of weight loss due to high‐dose LAA intake. 20 In the myocardium, proton leak reduces the mitochondrial electrochemical proton gradient and reduces ROS production. 34 In fact, our data also showed that LAA treatment promoted mitochondrial respiration without increasing oxidative stress. These findings suggested that LAA might promote energy production with a protective action on myocardium.
In our in vitro cachexia model, cachexic mouse ascites was used to treat rat cardiomyoblasts. TNFα and HMGB1, which are strongly associated with sarcopenia in patients with colorectal cancer, 35 were confirmed in the ascites. Based on the difference in species, HMGB1 was considered to be directly acting on rat cells and activation of NFκB, which is a signal pathway of HMGB1, was also observed (data not shown). As HMGB1 induces the expression of inflammatory cytokines such as TNFα, 36 , 37 it is possible that inflammatory cytokines were induced in cardiomyoblasts. Furthermore, HMGB1 is a DNA‐binding nuclear protein and a typical damage‐associated molecular pattern (DAMP) molecule 38 that is passively released during cell death. 39 HMGB1 decreases myocardial contractility, induces cardiomyocyte apoptosis, and stimulates cardiac fibroblast activity. 40 Furthermore, HMGB1 causes oxidative damage to the mitochondria of vascular endothelial cells. 41 HMGB1 also inhibits cardiomyocyte calcium signaling and suppresses oxidative phosphorylation. 42 Inhibition of extracellular HMGB1 exhibits myocardial protection against various myocardial damages such as ischemia and myocarditis. 40 CT26 cells secrete high levels of HMGB1 and promote secretion of other inflammatory cytokines such as TNFα. In addition, HMGB1 impairs mitochondrial energy metabolism as DAMP. It is considered that lauric acid + glucose normalizes the energy metabolism of myocardial cells by promoting oxidative phosphorylation from β‐oxidation of fatty acids and accompanying promotion of mitochondrial glucose oxidation. Furthermore, it is considered that HMGB1 secretion from cancer cells is suppressed by the antitumor effect of lauric acid.
In this study, we treated rat cardiomyoblasts with ascitic fluid of cachexic mice, however a combination of originally syngeneic cardiomyocytes and ascites was preferable. From these results we expected to clarify the findings such as the action of cytokines, which can be studied only in syngeneic systems. In the future, we would like to produce a rat cachexia model and study it using ascites. Abnormalities in energy metabolism of cardiomyoblasts in cachexia were also revealed, but the underlying molecular mechanism remains unknown. Therefore, further comprehensive analysis including expression and activity of key enzymes of energy metabolism will be required. In this study, morphological static analysis in a mouse model was the main focus of the in vivo studies. It is expected that future cardiac functional analysis will clarify the functional effect of nutritional intervention on cancer‐derived myocardial damage.
Cardiotoxicity due to anticancer drugs is relevant to myocardial damage in cancer patients. 43 We examined myocardial damage due to cancer itself, but it will be also necessary to examine myocardial damage due to chemotherapy, which could further clarify, from a more clinical viewpoint, the significance of nutritional intervention using glucose and MCFAs on myocardial damage in cancer patients.
DISCLOSURE
The authors have no conflict of interest to declare.
ACKNOWLEDGMENTS
This work was supported by MEXT KAKENHI Grant Number 20K11260 (OH), 19K16564 (RFT), 19K19332 (CN), 19K19915 (KG), 20K18007 (KS), 20K19349 (IK), 18K10788 (KF), and the Natural Science Foundation of Jiangsu Education Department Project (17KJB320010) and the National Natural Science Foundation of China (81702723). The authors thank Ms Tomomi Masutani for expert assistance with the preparation of this manuscript.
Nukaga S, Mori T, Miyagawa Y, et al. Combined administration of lauric acid and glucose improved cancer‐derived cardiac atrophy in a mouse cachexia model. Cancer Sci. 2020;111:4605–4615. 10.1111/cas.14656
Contributor Information
Yi Luo, Email: lynantong@hotmail.com, Email: cooninh@zb4.so-net.ne.jp.
Hiroki Kuniyasu, Email: cooninh@zb4.so-net.ne.jp.
REFERENCES
- 1. Blum D, Stene GB, Solheim TS, et al. Validation of the Consensus‐Definition for Cancer Cachexia and evaluation of a classification model–a study based on data from an international multicentre project (EPCRC‐CSA). Ann Oncol. 2014;25:1635‐1642. [DOI] [PubMed] [Google Scholar]
- 2. Fearon KC, Voss AC, Hustead DS. Definition of cancer cachexia: effect of weight loss, reduced food intake, and systemic inflammation on functional status and prognosis. Am J Clin Nutr. 2006;83:1345‐1350. [DOI] [PubMed] [Google Scholar]
- 3. Tisdale MJ. Cachexia in cancer patients. Nat Rev Cancer. 2002;2:862‐871. [DOI] [PubMed] [Google Scholar]
- 4. Argilés JM, Stemmler B, López‐Soriano FJ, Busquets S. Nonmuscle tissues contribution to cancer cachexia. Mediators Inflamm. 2015;2015:182872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kazemi‐Bajestani SM, Becher H, Fassbender K, Chu Q, Baracos VE. Concurrent evolution of cancer cachexia and heart failure: bilateral effects exist. J Cachexia Sarcopenia Muscle. 2014;5:95‐104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mamidanna R, Nachiappan S, Bottle A, Aylin P, Faiz O. Defining the timing and causes of death amongst patients undergoing colorectal resection in England. Colorectal Dis. 2016;18:586‐593. [DOI] [PubMed] [Google Scholar]
- 7. Murphy KT. The pathogenesis and treatment of cardiac atrophy in cancer cachexia. Am J Physiol Heart Circ Physiol. 2016;310:H466‐H477. [DOI] [PubMed] [Google Scholar]
- 8. Tzika AA, Fontes‐Oliveira CC, Shestov AA, et al. Skeletal muscle mitochondrial uncoupling in a murine cancer cachexia model. Int J Oncol. 2013;43:886‐894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fermoselle C, García‐Arumí E, Puig‐Vilanova E, et al. Mitochondrial dysfunction and therapeutic approaches in respiratory and limb muscles of cancer cachectic mice. Exp Physiol. 2013;98:1349‐1365. [DOI] [PubMed] [Google Scholar]
- 10. Scheig R. Absoption of dietary fat: use of medium‐chain triglycerides in malabsorption. Am J Clin Nutr. 1968;21:300‐304. [DOI] [PubMed] [Google Scholar]
- 11. Hagenfeldt L, Wahren J, Pernow B, Räf L. Uptake of individual free fatty acids by skeletal muscle and liver in man. J Clin Invest. 1972;51:2324‐2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Papamandjaris AA, MacDougall DE, Jones PJ. Medium chain fatty acid metabolism and energy expenditure: obesity treatment implications. Life Sci. 1998;62:1203‐1215. [DOI] [PubMed] [Google Scholar]
- 13. Metges CC, Wolfram G. Medium‐ and long‐chain triglycerides labeled with 13C: a comparison of oxidation after oral or parenteral administration in humans. J Nutr. 1991;121:31‐36. [DOI] [PubMed] [Google Scholar]
- 14. Kadochi Y, Mori S, Fujiwara‐Tani R, et al. Remodeling of energy metabolism by a ketone body and medium‐chain fatty acid suppressed the proliferation of CT26 mouse colon cancer cells. Oncol Lett. 2017;14:673‐680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bedford A, Yu H, Hernandez M, Squires EJ, Leeson S, Gong J. Effects of fatty acid glyceride product SILOhealth 104 on the growth performance and carcass composition of broiler chickens. Poult Sci. 2018;97:1315‐1323. [DOI] [PubMed] [Google Scholar]
- 16. Mori T, Ohmori H, Luo Y, et al. Giving combined medium‐chain fatty acids and glucose protects against cancer‐associated skeletal muscle atrophy. Cancer Sci. 2019;110:3391‐3399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gruber C, Nink N, Nikam S, et al. Myocardial remodelling in left ventricular atrophy induced by caloric restriction. J Anat. 2012;220:179‐185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Guo Y, Wang Z, Qin X, et al. Enhancing fatty acid utilization ameliorates mitochondrial fragmentation and cardiac dysfunction via rebalancing optic atrophy 1 processing in the failing heart. Cardiovasc Res. 2018;114:979‐991. [DOI] [PubMed] [Google Scholar]
- 19. Balietti M, Fattoretti P, Giorgetti B, et al. A ketogenic diet increases succinic dehydrogenase activity in aging cardiomyocytes. Ann N Y Acad Sci. 2009;1171:377‐384. [DOI] [PubMed] [Google Scholar]
- 20. Miyagawa Y, Mori T, Goto K, et al. Intake of medium‐chain fatty acids induces myocardial oxidative stress and atrophy. Lipids Health Dis. 2018;17:258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yazaki Y, Mochinaga S, Raben MS. Fractionation of the light chains from rat and rabbit cardiac myosin. Biochim Biophys Acta. 1973;328:464‐469. [DOI] [PubMed] [Google Scholar]
- 22. Springer J, Tschirner A, Haghikia A, et al. Prevention of liver cancer cachexia‐induced cardiac wasting and heart failure. Eur Heart J. 2014;35:932‐941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Li L, Zhao Q, Kong W. Extracellular matrix remodeling and cardiac fibrosis. Matrix Biol. 2018;69:490‐506. [DOI] [PubMed] [Google Scholar]
- 24. Fukushima A, Milner K, Gupta A, Lopaschuk GD. Myocardial energy substrate metabolism in heart failure : from pathways to therapeutic targets. Curr Pharm Des. 2015;21:3654‐3664. [DOI] [PubMed] [Google Scholar]
- 25. Fillmore N, Levasseur JL, Fukushima A, et al. Uncoupling of glycolysis from glucose oxidation accompanies the development of heart failure with preserved ejection fraction. Mol Med. 2018;24:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fillmore N, Mori J, Lopaschuk GD. Mitochondrial fatty acid oxidation alterations in heart failure, ischaemic heart disease and diabetic cardiomyopathy. Br J Pharm. 2014;171:2080‐2090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Stewart JM, Blakely JA. Long chain fatty acids inhibit and medium chain fatty acids activate mammalian cardiac hexokinase. Biochim Biophys Acta. 2000;1484:278‐286. [DOI] [PubMed] [Google Scholar]
- 28. Depré C, Rider MH, Hue L. Mechanisms of control of heart glycolysis. Eur J Biochem. 1998;258:277‐290. [DOI] [PubMed] [Google Scholar]
- 29. Samartsev VN, Simonyan RA, Markova OV, Mokhova EN, Skulachev VP. Comparative study on uncoupling effects of laurate and lauryl sulfate on rat liver and skeletal muscle mitochondria. Biochim Biophys Acta. 2000;1459:179‐190. [DOI] [PubMed] [Google Scholar]
- 30. Parker N, Vidal‐Puig A, Brand MD. Stimulation of mitochondrial proton conductance by hydroxynonenal requires a high membrane potential. Biosci Rep. 2008;28:83‐88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Abe T, Hirasaka K, Kohno S, et al. Capric acid up‐regulates UCP3 expression without PDK4 induction in mouse C2C12 myotubes. J Nutr Sci Vitaminol. 2016;62:32‐39. [DOI] [PubMed] [Google Scholar]
- 32. Shabalina IG, Kramarova TV, Nedergaard J, Cannon B. Carboxyatractyloside effects on brown‐fat mitochondria imply that the adenine nucleotide translocator isoforms ANT1 and ANT2 may be responsible for basal and fatty‐acid‐induced uncoupling respectively. Biochem J. 2006;399:405‐414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Brookes PS, Buckingham JA, Tenreiro AM, Hulbert AJ, Brand MD. The proton permeability of the inner membrane of liver mitochondria from ectothermic and endothermic vertebrates and from obese rats: correlations with standard metabolic rate and phospholipid fatty acid composition. Comp Biochem Physiol B Biochem Mol Biol. 1998;119:325‐334. [DOI] [PubMed] [Google Scholar]
- 34. Cadenas S. Mitochondrial uncoupling, ROS generation and cardioprotection. Biochim Biophys Acta Bioenerg. 2018;1859:940‐950. [DOI] [PubMed] [Google Scholar]
- 35. Ohmori H, Kawahara I, Mori T, et al. Evaluation of parameters for cancer‐induced sarcopenia in patients autopsied after death from colorectal cancer. Pathobiology. 2019;86:306‐314. [DOI] [PubMed] [Google Scholar]
- 36. Tripathi A, Shrinet K, Kumar A. HMGB1 protein as a novel target for cancer. Toxicol Rep. 2019;6:253‐261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lu H, Zhang Z, Barnie PA, Su Z. Dual faced HMGB1 plays multiple roles in cardiomyocyte senescence and cardiac inflammatory injury. Cytokine Growth Factor Rev. 2019;47:74‐82. [DOI] [PubMed] [Google Scholar]
- 38. Sims GP, Rowe DC, Rietdijk ST, Herbst R, Coyle AJ. HMGB1 and RAGE in inflammation and cancer. Ann Rev Immunol. 2010;28:367‐388. [DOI] [PubMed] [Google Scholar]
- 39. Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature. 2002;418:191‐195. [DOI] [PubMed] [Google Scholar]
- 40. Raucci A, Di Maggio S, Scavello F, D'Ambrosio A, Bianchi ME, Capogrossi MC. The Janus face of HMGB1 in heart disease: a necessary update. Cell Mol Life Sci. 2019;76:211‐229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Feng Z, Wang JW, Wang Y, Dong WW, Xu ZF. Propofol protects lung endothelial barrier function by suppression of High‐Mobility Group Box 1 (HMGB1) release and mitochondrial oxidative damage catalyzed by HMGB1. Med Sci Monit. 2019;25:3199‐3211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zhang C, Mo M, Ding W, et al. High‐mobility group box 1 (HMGB1) impaired cardiac excitation‐contraction coupling by enhancing the sarcoplasmic reticulum (SR) Ca(2+) leak through TLR4‐ROS signaling in cardiomyocytes. J Mol Cell Cardiol. 2014;74:260‐273. [DOI] [PubMed] [Google Scholar]
- 43. Babiker HM, McBride A, Newton M, et al. Cardiotoxic effects of chemotherapy: A review of both cytotoxic and molecular targeted oncology therapies and their effect on the cardiovascular system. Crit Rev Oncol Hematol. 2018;126:186‐200. [DOI] [PubMed] [Google Scholar]