

Abstract
Trastuzumab deruxtecan (T‐DXd: DS‐8201a) is an anti‐human epidermal growth factor receptor 2 (HER2) Ab–drug conjugated with deruxtecan (DXd), a derivative of exatecan. The objective of this study was to characterize T‐DXd‐induced lung toxicity in cynomolgus monkeys. Trastuzumab deruxtecan was injected i.v. into monkeys once every 3 weeks for 6 weeks (10, 30, and 78.8 mg/kg) or for 3 months (3, 10, and 30 mg/kg). To evaluate the involvement of DXd alone in T‐DXd‐induced toxicity, DXd monohydrate was given i.v. to monkeys once a week for 4 weeks (1, 3, and 12 mg/kg). Interstitial pneumonitis was observed in monkeys given T‐DXd at 30 mg/kg or more. The histopathological features of diffuse lymphocytic infiltrates and slight fibrosis were similar to interstitial lung diseases (ILD)/pneumonitis related to anticancer drugs in patients, with an incidence that was dose‐dependent and dose‐frequency‐dependent. Monkeys receiving DXd monohydrate did not suffer lung toxicity, although the DXd exposure level was higher than that of DXd in the monkeys given T‐DXd. The HER2 expression in monkey lungs was limited to the bronchial level, although the lesions were found at the alveolar level. Immunohistochemical analysis confirmed that T‐DXd localization was mainly in alveolar macrophages, but not pulmonary epithelial cells. These findings indicate that monkeys are an appropriate model for investigating T‐DXd‐related ILD/pneumonitis. The results are also valuable for hypothesis generation regarding the possible mechanism of T‐DXd‐induced ILD/pneumonitis in which target‐independent uptake of T‐DXd into alveolar macrophages could be involved. Further evaluation is necessary to clarify the mechanism of ILD/pneumonitis in patients with T‐DXd therapy.
Keywords: cynomolgus monkey, DS‐8201a, HER2, interstitial lung disease, trastuzumab deruxtecan
Trastuzumab deruxtecan (T‐DXd; DS‐8201a), an anti‐human epidermal growth factor receptor 2 Ab–drug conjugate with a derivative of exatecan (DXd), has been associated with interstitial lung diseases (ILD)/pneumonitis in clinical trials. This work indicates that the histopathological features of T‐DXd‐induced lung toxicity in monkeys are similar to ILD/pneumonitis associated with anticancer drugs in patients.

1. INTRODUCTION
Alterations of Erb‐b2 receptor tyrosine kinase 2 (ERBB2, or human epidermal growth factor receptor 2 [HER2]), including overexpression, amplification, and other mutations, are found in some solid tumors including breast, gastric, colorectal, and lung cancers. 1 Trastuzumab deruxtecan (T‐DXd: DS‐8201a) is composed of: HER2‐targeting human IgG1, which has the same amino acid sequence as trastuzumab; deruxtecan (DXd), a derivative of exatecan; a topoisomerase I inhibitor as the payload; and a cleavable peptide‐based linker. 2 , 3 Trastuzumab deruxtecan is considered to have several anticancer mechanisms of action. These include HER2‐dependent cytotoxicity, effector functions (ie Ab‐dependent cell‐mediated cytotoxicity) through FcγRs, and “bystander killing” effects in a heterogeneous tumor environment due to the moderate membrane‐permeability of DXd and a high drug‐to‐Ab ratio (DAR, ca. 8). 4 , 5 Trastuzumab deruxtecan has shown anticancer activity in patients with breast, gastric, lung (non–small cell lung cancer), and colorectal, and other cancers. 6 , 7 , 8
A toxicity study program to support the clinical development of T‐DXd included 6‐week toxicity studies in rats as a non‐cross‐reactive species and cynomolgus monkeys as a cross‐reactive species, as well as a 3‐month toxicity study in monkeys. In the 6‐week toxicity studies, T‐DXd induced lung toxicity in monkeys at 30 mg/kg or more, but not in rats up to the highest dose of 198 mg/kg. 3 , 4 In a global phase II study of T‐DXd in patients with HER2‐positive metastatic breast cancer who had been previously treated with trastuzumab emtansine (T‐DM1), interstitial lung diseases (ILD)/pneumonitis assessed as related to T‐DXd were reported in 25 of 184 patients (13.6%). 7
Irinotecan, a different topoisomerase I inhibitor, 9 and other HER2‐directed therapies including trastuzumab and T‐DM1 have also been associated with ILD/pneumonitis in patients: at rates of less than 0.5% in trastuzumab 10 and almost 1% in T‐DM1. 11 In the toxicity studies, trastuzumab did not cause lung toxicity in cynomolgus monkeys when treated once a week for 6 months up to 25 mg/kg. 12 However, mononuclear infiltration of the lung interstitium was found in monkeys treated with T‐DM1 once every 3 weeks for 3 months at dose levels of 3 mg/kg or more, which predicted adverse pulmonary events in patients. 13
Drug‐induced ILD/pneumonitis in humans is caused by numerous drugs, with anticancer drugs being the most common causative agents. 14 They are classified into several types, such as noncardiogenic pulmonary edema, diffuse alveolar damage (DAD), nonspecific interstitial pneumonia (NSIP), and organizing pneumonia according to the clinicopathologic features. 15 Little is known about the initial trigger for drug‐induced ILD/pneumonitis, but it has generally been divided into a direct (dose‐dependent) cytotoxic action or an immune‐mediated mechanism. 16
The purpose of this study was to characterize the pulmonary toxicity observed in toxicity studies of T‐DXd in monkeys from a pathological perspective. The relationship between the plasma concentration of T‐DXd or DXd and the pathogenesis of lung injury in monkeys was also assessed. Furthermore, the uptake and distribution of T‐DXd into the lungs were also evaluated by immunohistochemistry.
2. MATERIALS AND METHODS
2.1. Test article
Two T‐DXd batches were manufactured to be used in the preclinical assessments. One was formulated in 10 mmol/L histidine buffer (pH 5.8), 10% trehalose, and 0.02% polysorbate 20 (concentration of 19.7 mg/mL), and its DAR was 7.78; the other was formulated in 25 mmol/L histidine buffer (pH 5.5) and 9% sucrose (concentration of 20.1 mg/mL), and its DAR was 7.82. Deruxtecan monohydrate was also synthesized and was dissolved in physiological saline at 3 mg/mL as a free form of DXd. The vehicle control consisted of the corresponding formulation buffer or physiological saline.
2.2. Animals
Male and female cynomolgus monkeys (Macaca fascicularis) were obtained from commercial suppliers. The experiments were approved by the Institutional Animal Care and Use Committee in Daiichi Sankyo Co., Ltd. and were undertaken in accordance with the animal welfare by‐laws at the study site.
2.3. Study design
2.3.1. Six‐week toxicity study of T‐DXd
Monkeys in this 6‐week study were 3‐7 years of age and 2.98‐6.85 kg in weight at the initiation of acclimation. The animals (three animals/dose/sex) received three i.v. injections of T‐DXd every 3 weeks for 6 weeks (days 1, 22, and 43) at dose levels of 0 (vehicle control), 10, 30, and 78.8 mg/kg. Two animals/dose/sex given T‐DXd at dose levels of 30 and 78.8 mg/kg for 6 weeks were added to assess the reversibility of toxic changes following a 6‐week recovery period. The vehicle control and T‐DXd were given at a dose volume of 4 mL/kg and a dose rate of 3 mL/min.
2.3.2. Three‐month toxicity study of T‐DXd
Monkeys in this 3‐month study were 3‐10 years of age and 2.62‐8.85 kg in weight at the initiation of acclimation. The animals (four animals/dose/sex) received five i.v. injections of T‐DXd every 3 weeks for 3 months (days 1, 22, 43, 64, and 85) at dose levels of 0 (vehicle control), 3, 10, and 30 mg/kg. Two animals/sex given T‐DXd at a dose level of 30 mg/kg for 3 months were added to assess the reversibility of toxic changes following a 3‐month recovery period. The dosing conditions were the same as indicated in the section “Six‐week toxicity study of T‐DXd.”
2.3.3. Four‐week toxicity study of DXd monohydrate
Monkeys in this 4‐week study were 3‐8 years of age and 3.03‐6.98 kg in weight at the initiation of acclimation. The animals (three animals/dose/sex) received five i.v. injections of DXd once a week for 4 weeks (days 1, 8, 15, 22, and 29) at dose levels of 0 (vehicle control), 1, 3, and 12 mg/kg. Two animals/sex given DXd at a dose level of 12 mg/kg for 4 weeks were added to assess the reversibility of toxic changes following a 4‐week recovery period. The dosing conditions were the same as indicated in section “Six‐week toxicity study of T‐DXd.”
2.4. Examinations
2.4.1. Pathological examination
The animals were euthanized by exsanguination under anesthesia by sodium pentobarbital (Tokyo Chemical Industry Co., Ltd.) the day after the final treatment (for T‐DXd, day 44 in the 6‐week study, and day 86 in the 3‐month study; and for DXd, day 30 in the 4‐week study) or after the respective recovery periods. The lungs including the bronchi were fixed in 10% neutral buffered formalin. Both lower lobes and the additional lobes with a gross lesion were sectioned, embedded in paraffin, and stained with H&E using a routine procedure. Furthermore, Masson‐trichrome staining was carried out in the selected formalin‐fixed paraffin‐embedded (FFPE) sections of the lungs.
2.4.2. Immunohistochemistry
Immunohistochemistry (IHC) was carried out in the lung FFPE sections from all animals in the vehicle control and high‐dose groups of the 6‐week and 3‐month studies of T‐DXd using a rabbit polyclonal anti‐human HER2 Ab (A0485; Agilent Technologies), a mouse monoclonal anti‐cathepsin B Ab (clone CA10; Abcam), and a mouse monoclonal anti‐DXd Ab (clone 1A3; Immuno‐Biological Laboratories). Immunohistochemistry was also carried out in the liver FFPE sections in two animals/sex of the vehicle control and high‐dose groups of the 6‐week and 3‐month studies of T‐DXd using the mouse monoclonal anti‐DXd Ab.
The sections were heated in target retrieval solution pH 6 (Agilent Technologies) for 10 minutes at 95°C for DXd or autoclaved in target retrieval solution pH 9 for 10 minutes at 121°C for cathepsin B. They were immersed in 3% hydrogen peroxide for 10 minutes at room temperature. After protein blocking with ready‐to‐use solution (Agilent Technologies), they were incubated with 3 μg/mL anti‐HER2 Ab, 1 μg/mL anti‐DXd Ab, or 0.3 μg/mL anti‐cathepsin B Ab for 60 minutes at room temperature, followed by incubation with Dako Envision+ polymer (Agilent Technologies) for 30 minutes at room temperature. They were incubated with 3,3′‐diaminobenzidine and then counterstained with hematoxylin.
The specimens from two males in the vehicle control and high‐dose groups of the 6‐week and 3‐month studies of T‐DXd were used for double IHC. They were autoclaved in target retrieval solution pH 6 for 10 minutes at 121°C. They were immersed in 3% hydrogen peroxide for 20 minutes at room temperature. After protein blocking with ready‐to‐use solution, they were incubated with 1 μg/mL anti‐DXd Ab for 60 minutes at room temperature, followed by incubation with Dako Envision+ polymer for 30 minutes at room temperature. They were next immersed in Opal 520 fluorophore in an Opal 4 color manual IHC kit (PerkinElmer) for 10 minutes at room temperature. The specimens were autoclaved in AR6 buffer working solution in the Opal 4 color manual IHC kit for 10 minutes at 121°C. After protein blocking with ready‐to‐use solution, they were incubated with mouse monoclonal cytokeratin (clone AE1/AE3; Agilent Technologies) or 1 μg/mL rabbit polyclonal Iba‐1 Ab (Fujifilm Wako Pure Chemical) for 60 minutes at room temperature, followed by incubation with Opal 570 Fluorophore for 10 minutes at room temperature. The specimens were then mounted in Vectashield mounting medium with DAPI (Vector Laboratories).
The sections stained with anti‐HER2, cathepsin B, and DXd Abs were evaluated semiquantitatively under a light microscope. The double IHC specimens were evaluated under a fluorescence microscope (BZ‐X700; Keyence).
2.4.3. Western blot analysis for cathepsin B
Tissue lysates of the lung and liver from an untreated female monkey were prepared with T‐PER Tissue Protein Extraction Reagent (Thermo Fisher Scientific). The equivalent amounts of protein were separated by SDS‐PAGE and transferred to membranes. The membranes were incubated with the cathepsin B Ab (0.25 μg/mL) or a mouse monoclonal anti‐proliferating cell nuclear antigen Ab (0.3 μg/mL, clone PC10; Agilent Technologies) for 2 hours at room temperature, then with biotin‐labelled secondary Abs for 1 hour at room temperature. The signals were detected with Amersham Imager 680 RGB (Cytiva).
2.4.4. Quantification of T‐DXd and DXd in plasma
Blood samples of approximately 0.4 mL were drawn from the femoral vein of all animals 5 minutes and 1, 7, 24, 72, 168, and 504 hours after the first and second dosing (6‐week study) or after the first and fourth dosing (3‐month study) in the T‐DXd studies, and 5 and 30 minutes, and 2, 7, and 24 hours after the first and fourth dosing in the DXd study. Plasma T‐DXd concentrations were quantified by a ligand binding assay with Gyrolab xP Workstation (Gyros Protein Technology). Trastuzumab deruxtecan was detected using biotinylated recombinant human HER2 protein and an Alexa Fluor 647‐labeled anti‐DXd mouse mAb. Plasma DXd concentrations were quantified using a high‐performance‐liquid chromatography tandem mass spectrometer (Nexera X2 system, Shimadzu/QTRAP 5500; Sciex). Toxicokinetic (TK) parameters, such as initial plasma concentration (C0: T‐DXd), maximum plasma concentration (Cmax: DXd), area under the plasma concentration‐time curve (AUC21d or AUC1d), and terminal elimination half‐life (t 1/2) were calculated individually with Phoenix WinNonlin version 6.1 (Certara) or Microsoft Excel 2013.
2.5. Statistical analysis
The TK parameters are expressed as mean ± SD. The AUC21d of T‐DXd and DXd in monkeys with or without lung toxicity at the same dose level, 30 or 78.8 mg/kg, was analyzed by F test to evaluate the homogeneity of variance (5% significance level), followed by Student’s t test when the variance was homogeneous or by Welch’s t test when it was not. The results obtained by t test were classified as significant for P values of less than 5%.
3. RESULTS
3.1. Interstitial pneumonitis in toxicity studies of T‐DXd in monkeys
In the 6‐week toxicity study of T‐DXd in monkeys, lung macroscopic findings related with T‐DXd treatment were observed in one female monkey at 30 mg/kg after the recovery period, one male monkey after the dosing period, and one male and two female monkeys after the recovery period (Figure S1). Histopathological findings in the lungs related with T‐DXd treatment were found in one male monkey at 78.8 mg/kg after the dosing period, and in one female monkey at 30 mg/kg and in one male and two female monkeys at 78.8 mg/kg after the recovery period, as reported in the previous study (Table 1). 4 Lung histopathology results of monkeys given T‐DXd at 78.8 mg/kg indicated relatively subacute lung toxicity as represented by alveolar edema and aggregates of alveolar macrophages in some lung lobes. Slight interstitial inflammation in the alveolar septa and intraalveolar fibrosis were also shown (Figure 1). Some of the alveolar epithelial cells were enlarged and showed a regenerative response. One female monkey given 30 mg/kg and necropsied after the recovery period had less severe interstitial inflammation than those at 78.8 mg/kg. No apparent change in the lungs was noted at 10 mg/kg.
Table 1.
Lung histopathology in a 6‐week study of trastuzumab deruxtecan in cynomolgus monkeys
| Necropsy | 6‐wk dosing period | 6‐wk recovery period | ||||||
|---|---|---|---|---|---|---|---|---|
| Dose (mg/kg) | 30 | 78.8 | 30 | 78.8 | ||||
| Sex: no. of animals | M: 3 | F: 3 | M: 3 | F: 3 | M: 2 | F: 2 | M: 2 | F: 2 |
| Lung histopathological findings | ||||||||
| Alveolar edema | − | − | 1/3 | − | − | − | 1/2 | − |
| Interstitial inflammation, focal | − | − | 1/3 | − | − | 1/2 | 1/2 | 2/2 |
| Aggregation, alveolar macrophages | − | − | 1/3 | − | − | 1/2 | 1/2 | 2/2 |
−, no change; F, female; M, male.
Figure 1.
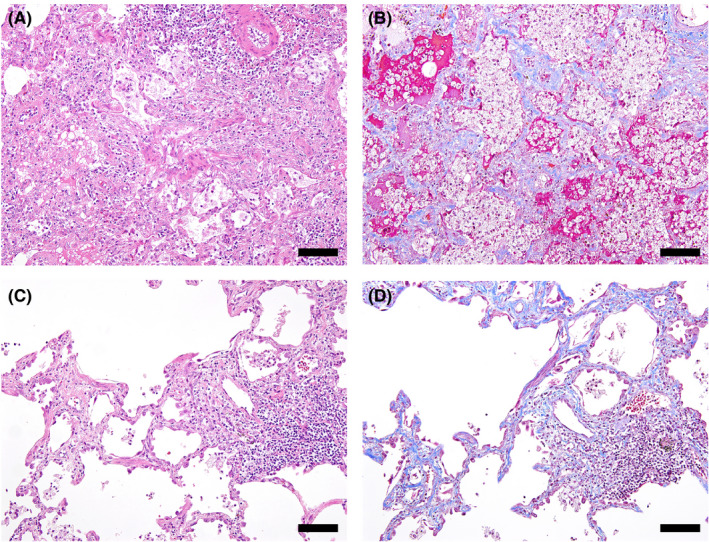
Lung interstitial inflammation associated with trastuzumab deruxtecan (T‐DXd) treatment in cynomolgus monkeys. A, B, Inflammatory cell infiltrates such as neutrophils and lymphocytes in the alveolar wall, intraalveolar fibrosis, alveolar edema, and aggregates of foamy alveolar macrophages were observed at 78.8 mg/kg T‐DXd in the 6‐week toxicity study (A, H&E; B, Masson‐trichrome). C, D, Thickening of the alveolar wall with lymphocytic inflammation and fibrosis was observed at 30 mg/kg T‐DXd in the 3‐month toxicity study (C, H&E; D, Masson trichrome). Bar, 100 μm
In the 3‐month study of T‐DXd in monkeys, lung macroscopic findings related with T‐DXd treatment were observed in one male monkey at 30 mg/kg after the dosing period, and one male and one female monkey after the 3‐month recovery period (Figure S2). In the lung histopathology, 30 mg/kg T‐DXd caused interstitial pneumonitis in three male and one female monkey after the dosing period and one male monkey after the recovery period (Table 2). The pulmonary lesions were less severe than those at 78.8 mg/kg after the 6‐week dosing period; however, lymphocytic interstitial inflammation was more apparent than in the 6‐week study (Figure 1). The incidence of lung toxicity in the 3‐month study (ca. 40%) was higher than that in the 6‐week study (10%) when comparing the same dose level of 30 mg/kg. There were no clear toxicity findings at 3 or 10 mg/kg.
Table 2.
Lung histopathology in a 3‐month study of trastuzumab deruxtecan in cynomolgus monkeys
| Necropsy | 3‐mo dosing period | 3‐mo recovery period | ||
|---|---|---|---|---|
| Dose (mg/kg) | 30 | |||
| Sex: no. of animals | M: 4 | F: 4 | M: 2 | F: 2 |
| Lung histopathological findings | ||||
| Interstitial inflammation, focal | 3/4 | 1/4 | 1/2 | − |
| Alveolar inflammation, focal | 1/4 | − | − | − |
| Aggregation, alveolar macrophages | 3/4 | 1/4 | 1/2 | − |
−, no change; F, female; M, male.
3.2. Relationship between lung toxicity and systemic exposure
The systemic exposure to T‐DXd in monkeys was proportional to the dose levels ranging from 3 to 78.8 mg/kg, although t1/2 at 3 mg/kg tended to be shorter than those at the higher doses (Tables S1 and S2). 17 No changes in TK parameters were noted after repeated dosing. Systemic DXd exposure in the monkeys given T‐DXd i.v. was markedly lower (<10−6) than T‐DXd exposures at all doses because of the stable linker in plasma. 17
The relationship of T‐DXd AUC21d or DXd AUC1d with lung toxicity was evaluated. No apparent differences in AUC values were noted within any dose level of T‐DXd, regardless of the occurrence of lung toxicity (Table 3).
Table 3.
Relationship between drug exposure and lung toxicity in cynomolgus monkeys treated with i.v. trastuzumab deruxtecan (T‐DXd)
| Dose (mg/kg) | 30 | 78.8 | ||
|---|---|---|---|---|
| Lung toxicity | − (n = 16) | + (n = 6) | − (n = 6) | + (n = 4) |
| T‐DXd AUC21d (µg∙d/mL) | ||||
| 1st dose | 3640 ± 403 | 4032 ± 794 | 8316 ± 1427 | 8921 ± 1775 |
| t test (P value) | P = .292 | P = .566 | ||
| 2nd or 4th dose | 4614 ± 731 | 4852 ± 1228 | 9895 ± 791 a | 10 306 ± 1064 |
| t test (P value) | P = .579 | P = .526 | ||
| DXd AUC1d (ng∙d/mL) | ||||
| 1st dose | 9.78 ± 2.38 | 11.41 ± 4.03 | 66.2 ± 46.6 | 16.8 ± 0.123 |
| t test (P value) | P = .387 | P = .514 | ||
| 2nd or 4th dose | 10.55 ± 2.57 | 12.43 ± 5.13 | 34.2 ± 9.94 a | 32.9 ± 9.05 |
| t test (P value) | P = .424 | P = .844 | ||
Values are expressed as the mean ± SD in the 6‐week and 3‐month studies.
−, negative; +, positive; AUC, area under the plasma concentration‐time curve.
n = 5.
3.3. No lung toxicity in a toxicity study of DXd in monkeys
Monkeys given bolus injection of DXd once weekly for 4 weeks or following the 4‐week recovery period did not show notable lung findings at doses up to 12 mg/kg. When DXd was given to monkeys, plasma DXd was rapidly excreted into the feces as its unmetabolized form. 17 In the 4‐week study of DXd in monkeys, t 1/2 values at dose levels ranging from 1 to 12 mg/kg were approximately 2‐4 hours (Table S3). 17 The C0 and AUC1d values after the bolus injection of DXd were much higher at 1 mg/kg than the Cmax and AUC21d of DXd in T‐DXd‐treated monkeys at 30 or 78.8 mg/kg, doses that are toxic to the lungs in some monkeys.
3.4. Immunohistochemistry for HER2
In the lung specimens obtained from the 6‐week and 3‐month studies in monkeys, positive staining of HER2 was observed on the cellular membrane of the bronchial, bronchiolar, and bronchial gland epithelium (Figure 2). No clear difference of distribution or intensity of the positive signals between the vehicle control and T‐DXd‐treated groups was observed, although positive staining on the cellular membrane was observed in a limited number of the alveolar epithelial cells showing regenerative changes in the pulmonary lesions of a few animals given a dose of 30 or 78.8 mg/kg.
Figure 2.
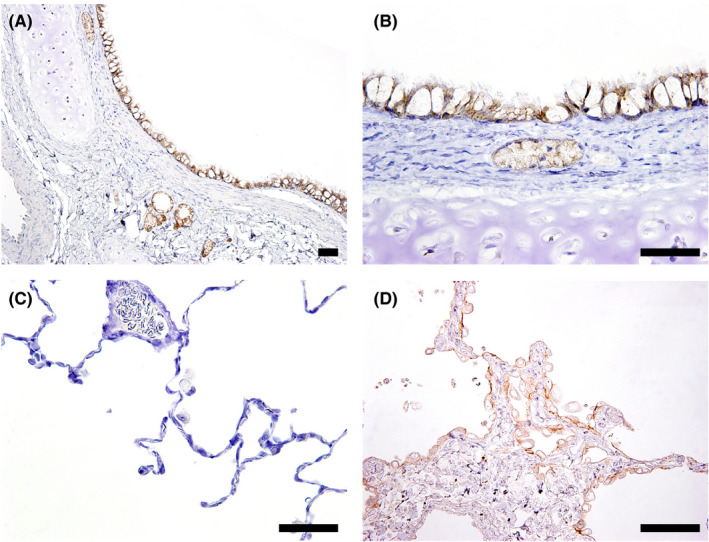
Representative photomicrographs of immunohistochemistry for human epidermal growth factor receptor 2 (HER2) in monkey lungs. A‐C, Bronchial and bronchial gland epithelium showed positive for HER2 in an animal of the vehicle control group (6‐week toxicity study). D, Regenerative alveolar epithelium in the affected areas showed positive for HER2 in an animal given 30 mg/kg trastuzumab deruxtecan (3‐month toxicity study). Bar, 50 μm
3.5. Immunohistochemistry and western blot analysis for cathepsin B
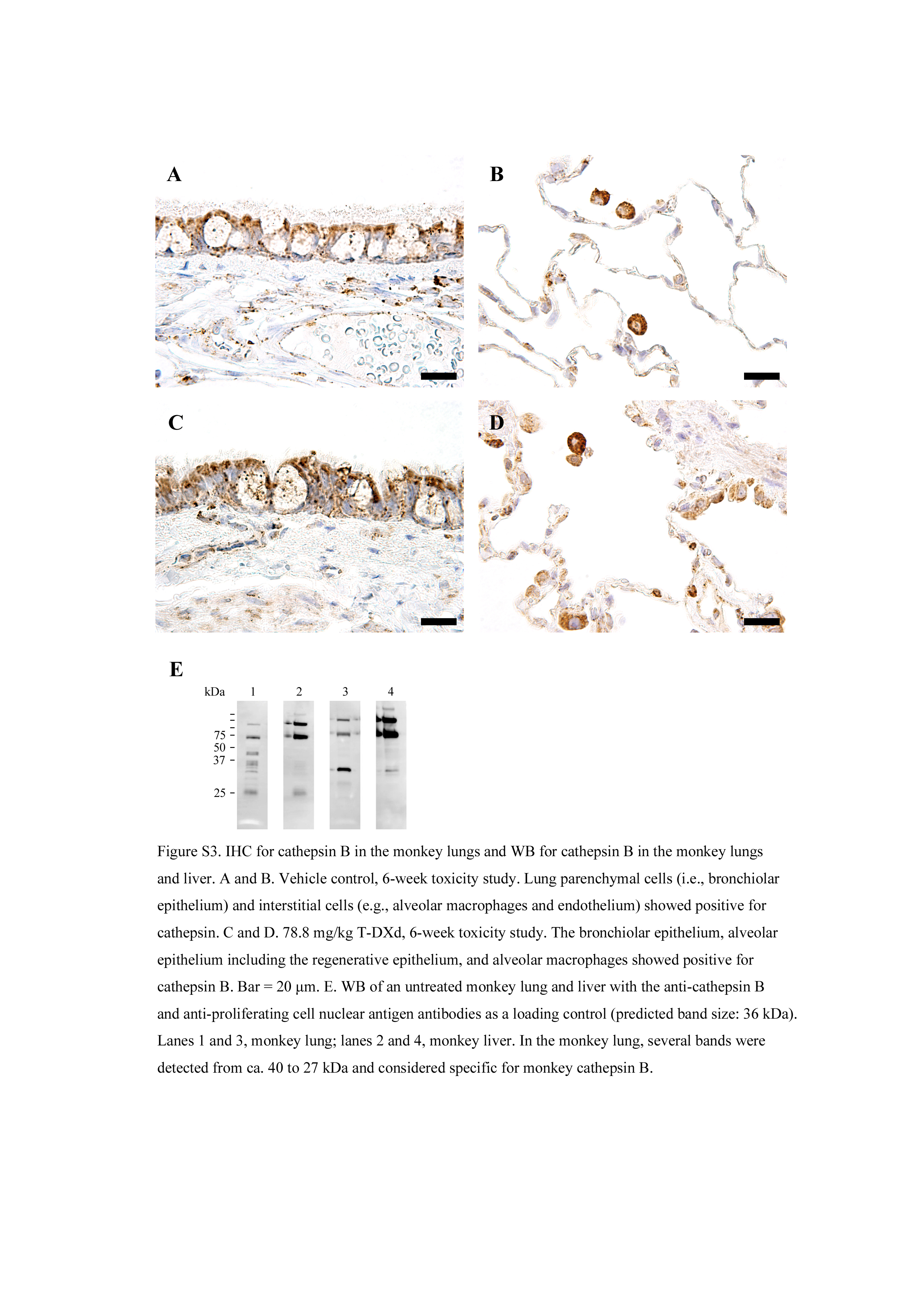
Positive staining of cathepsin B was found in the cytoplasm of epithelial components (ie alveolar and bronchiolar epithelium) as well as nonparenchymal cells such as alveolar macrophages, lymphocytes, and endothelium in the lungs of monkeys (Figure S3). Alveolar macrophages had the strongest intensity of positive staining among the cells. There was no clear difference in the distribution or intensity of the positive signals in the vehicle control and T‐DXd‐treated groups, including the monkeys experiencing or not experiencing interstitial pneumonitis related to T‐DXd.
Western blot analysis of the lung and liver from an untreated female monkey was applied to confirm the cross‐reactivity of the anticathepsin B Ab in cynomolgus monkeys. Several bands were detected from approximately 40 to 27 kDa in the monkey lung (Figure S3). It was considered these bands corresponded with procathepsin B, mature cathepsin B, and a heavy chain of cathepsin B, 18 , 19 and therefore specific for the monkey cathepsin B.
3.6. Immunohistochemistry for DXd
The distribution of DXd in the lungs and liver was evaluated using the specific Ab against DXd and FFPE sections from the 6‐week and 3‐month toxicity studies of T‐DXd in monkeys. In the lungs, alveolar macrophages infiltrating into the alveolar space were positive for DXd in monkeys given 30 mg/kg or more T‐DXd for 6 weeks and 3 months (Figure 3). No obvious localization in pulmonary epithelial cells was detected, although the intercellular space of bronchial epithelium was occasionally positive. In addition, DXd‐positive responses were observed in some of the regenerative alveolar epithelium. In the liver, Kupffer cells were also positive for DXd in monkeys given 30 mg/kg or more T‐DXd for 6 weeks and 3 months (Figure S4). Some hepatocytes also showed positive staining for DXd. In the lungs and liver, there was no positive signal in any animal of the vehicle control group or after the recovery period.
Figure 3.
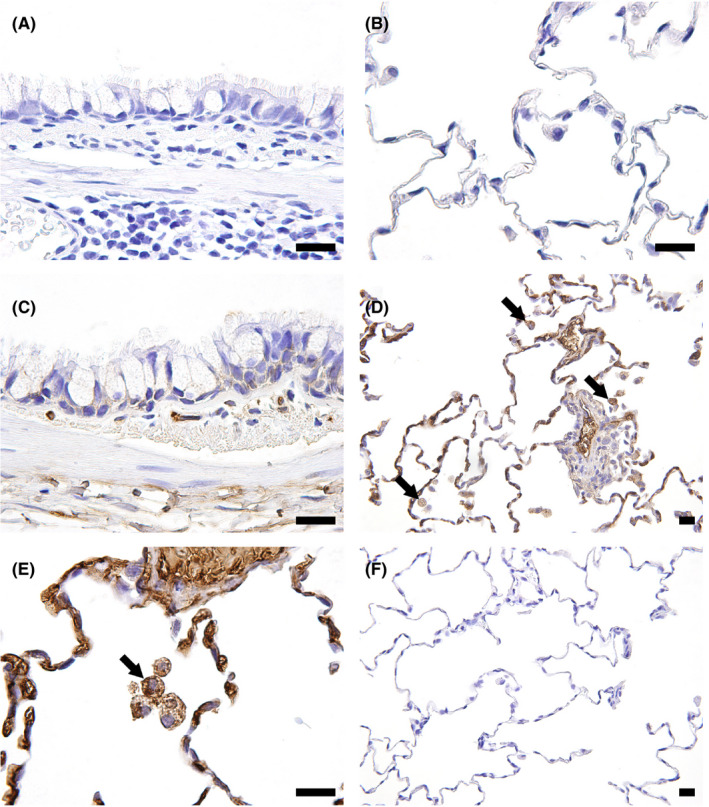
Representative photomicrographs of immunohistochemistry for deruxtecan (DXd) in monkey lungs. A, B, Vehicle control, 6‐week toxicity study. No positive staining of DXd was observed. C, 78.8 mg/kg trastuzumab deruxtecan (T‐DXd), 6‐week toxicity study. No obvious DXd positive signal in the bronchiolar epithelium was detected. D, E, 30 mg/kg T‐DXd, 3‐month toxicity study. Alveolar macrophages in the alveolus as well as blood components were positive for DXd (arrows). F, 30 mg/kg T‐DXd, 3‐month toxicity study. No positive staining of DXd was detected after the 3‐month recovery period. Bar, 20 μm
3.7. Double IHC for evaluating pulmonary drug distribution
To further evaluate the distribution of T‐DXd in the lungs, double IHC for DXd and cytokeratin or macrophage marker Iba‐1 was further carried out in the FFPE sections from the 6‐week and 3‐month studies. No clear colocalization of positive staining for DXd or cytokeratin was observed in cytoplasm of the bronchiolar epithelium and alveolar epithelium (Figure 4). However, Iba‐1‐positive macrophages also showed positive for DXd, indicating that T‐DXd was distributed in alveolar macrophages.
Figure 4.
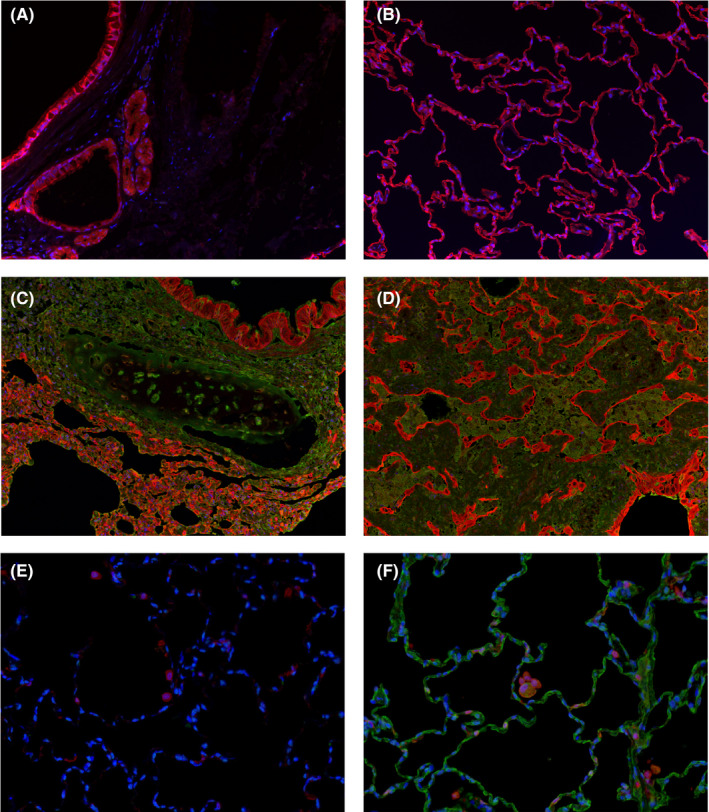
Representative photomicrographs of double immunohistochemistry (IHC) in monkey lungs. Double IHC for deruxtecan (DXd) and cytokeratin (A‐D) and for DXd and Iba‐1 (E, F) was undertaken in monkey lungs from the 6‐week toxicity study of trastuzumab deruxtecan (T‐DXd). Blue, DAPI; green, deruxtecan (DXd); red, cytokeratin or Iba‐1. A, B, E, No DXd staining was observed in a vehicle control animal. C, D, No clear colocalization of DXd and cytokeratin was found in the bronchial epithelium or alveolar epithelium of the animal given 78.8 mg/kg T‐DXd, although edematous fluid in the alveolus and a few regenerative alveolar epithelial cells showed positive for DXd. F, Iba‐1‐positive macrophages were also positive for DXd
4. DISCUSSION
Lung toxicity in monkeys dosed with T‐DXd at 30 mg/kg or more was characterized by diffuse lymphocytic infiltrates and slight fibrosis. These changes were considered to be similar to what is seen, generally, in human NSIP based on the histopathological features. The typical finding of human NSIP is uniform and diffuse thickening of the alveolar walls due to inflammatory cell infiltration and fibrosis, accompanied by the hyperplasia of type 2 pneumocytes. 20 , 21 Inferentially, this suggests that the T‐DXd‐induced lung toxicity in monkeys could be comparable to that seen, generally, in some patients with T‐DXd‐related ILD. Therefore, monkeys appear to be an appropriate model to elucidate the mechanisms behind T‐DXd‐related ILD/pneumonitis in patients.
Possible mechanisms of toxicity caused by Ab–drug conjugates (ADC), in general, are considered to be the following: (a) target‐dependent uptake and catabolism of ADC; (b) permeability of free payload resulting from deconjugation during circulation of ADC; (c) target‐independent uptake and catabolism of ADC in normal cells; and (d) “bystander killing” by free payload released from cells following catabolism of ADC. 22 , 23 The ADCs targeting HER2, T‐DM1, and T‐DXd caused ILD/pneumonitis in patients. 7 , 9 , 10 Although the underlying mechanism of T‐DXd‐related injury is not clearly understood, a cytotoxic effect by the payload is likely the final step in the pathway.
It has been reported that HER2 expression in human lungs is limited to the bronchial epithelium, and is not found in the alveolar epithelium. 24 The results of this study showed that the expression pattern of HER2 in monkey lungs was comparable to that in humans. However, IHC for DXd did not indicate apparent colocalization of positive signals in the bronchial epithelium. As free DXd would be distributed in parenchymal cells at a very low level or be dissolved during fixation and tissue processing, considering the physicochemical and absorption, distribution, metabolism, and excretion properties, DXd IHC is likely to detect T‐DXd, but not free DXd. 17 In addition, T‐DXd‐related lung toxicity in monkeys was diffusely distributed and mainly localized in the alveolar region. These results suggest that the uptake through HER2 in the pulmonary epithelial cells might not be involved in T‐DXd‐induced lung toxicity in monkeys. An additional assessment in monkeys with a nonbinding ADC containing the same linker, DXd, and DAR as T‐DXd would clarify the validity of this hypothesis.
Target‐independent uptake of ADC to normal cells is supposed to occur through receptor‐mediated (eg FcγR, mannose receptors) and pinocytic processes. 23 , 25 Although the target organs of toxicity by ADC depend on the payload and linker technology, it is believed that most adverse events would be caused by off‐target effects originating from target‐independent uptake. 26 The present data suggested that T‐DXd was mainly distributed to macrophages in the lungs and liver of monkeys. Macrophages in monkeys also expressed cathepsin B as well as other pulmonary components. Cathepsin B is a lysosomal cysteine protease that is constitutively expressed in macrophages in humans. 27 , 28 It is considered to be one of the enzymes responsible for linker cleavage of T‐DXd. 29 , 30 Therefore, target‐independent uptake of T‐DXd in macrophages followed by the release of free DXd might be associated with the lung toxicity caused by T‐DXd in monkeys.
In the lungs, there are at least two macrophage populations, alveolar macrophages and interstitial macrophages. Each macrophage type is distinguished by a unique combination of surface markers and also is characterized as having different immune functions, such as phagocytic capacity, cytokine production, and regulation of wound healing and fibrosis. 31 Further analysis regarding macrophage subtypes in the lungs of monkeys would provide information to help understand T‐DXd‐induced lung toxicity. In addition, the mechanism of uptake of T‐DXd in monkey pulmonary macrophages might be revealed by additional assessments with Fc‐engineered IgG, which can abolish binding to FcγR. 32 , 33 , 34
The T‐DXd‐induced lung toxicity in monkeys showed apparent dose‐dependent and dose‐frequency‐dependent patterns in terms of the incidence and severity. This suggests that T‐DXd induces lung injury through direct cytotoxicity rather than an immune‐mediated action. In ILD caused by chemotherapeutic agents, the dose level is thought to be one of the risk factors. 20 Studies of the biodistribution of T‐DXd in monkeys revealed that intact T‐DXd was present mostly in blood, with no tissue‐specific retention, including in the lungs 17 ; therefore, the blood exposure might be a factor contributing to ILD in monkeys. The relevance of this in patients could be important to explore. Cumulative doses of some anticancer drugs, such as bleomycin, are an important risk factor for drug‐induced ILD. 14 In the nonclinical safety studies of T‐DXd in monkeys, the dosing durations were 6 weeks (three doses) and 3 months (five doses); therefore, it is unclear whether longer term exposures of T‐DXd in monkeys increase the incidence of interstitial pneumonitis. The accumulation of clinical data with a longer term of treatment might provide valuable information to discuss the dependency of T‐DXd‐related ILD on cumulative doses in patients.
When the relationship between individual drug exposure and lung toxicity in monkeys was evaluated, no statistically significant difference in blood exposure of T‐DXd was detected in monkeys with or without lung toxicity. Susceptibility to drug‐induced ILD, then, might be related to genetic variability as some individuals could have higher sensitivity to certain anticancer drugs due to their genetic background. 15 There have been few detailed investigations of drug‐induced ILD and associated genetic variants, although some candidate genetic factors involved in drug‐induced DAD are now being considered and investigated. 15 , 35 It is challenging to take into account genetic polymorphism in cynomolgus monkeys, including in Mafa MHC, in safety assessments 36 ; however, it could provide new insight to discuss individual susceptibility to ILD caused by anticancer drugs, including T‐DXd.
In the phase II study at a dose level of 5.4 mg/kg T‐DXd, 25 of 184 patients (13.6%) had ILD related to the receipt of T‐DXd. 7 Doi et al 6 reported that the mean AUClast of 5.4 mg/kg T‐DXd in patients was approximately 500 μg·day/mL. The interstitial pneumonitis related with T‐DXd treatment occurred at dose levels of 30 mg/kg and higher in monkeys. The human equivalent dose of 30 mg/kg in monkeys was calculated as ca. 9.7 mg/kg based on body surface area (divided by a conversion factor of 3.1). The dose and exposure levels of monkey lung toxicity are twice and seven times as high as those in humans, respectively. One of the apparent differences between monkeys and patients is physical conditions. In nonclinical safety studies, healthy and young adult animals are generally used. Patients are considered to have medical history (eg chemotherapy, radiation) or aging, and pulmonary illness such as infection, asthma, and tumor metastasis. Further information including potential risk factors of T‐DXd‐related ILD is needed to better understand the species difference and the mechanism of action.
In summary, T‐DXd induced interstitial pneumonitis in monkeys in a manner dependent on both the dose level and the dosing frequency. The histopathological features in the monkeys were similar to ILD associated with anticancer drugs in patients, in particular NSIP, indicating that monkeys could be an appropriate model for investigating drug‐induced ILD in humans. In addition, target‐independent uptake of T‐DXd in pulmonary macrophages could be involved in T‐DXd‐induced ILD. Assessing the mechanisms behind lung injury in cynomolgus monkeys by further studies could inform our understanding of the mechanism of T‐DXd‐related ILD/pneumonitis in patients.
DISCLOSURE
The authors are employees of Daiichi Sankyo Co., Ltd.
Supporting information
Fig S1
Fig S2
Fig S3
{kind=link}
Fig S4
{kind=link}
Table S1
Table S2
Table S3
ACKNOWLEDGMENTS
The authors would like to thank the global team of AstraZeneca and Daiichi Sankyo for critical review of this manuscript, Shinobu Hakamata and Ruriko Ishida for supporting IHC, and Junya Matsushita for supporting WB.
Kumagai K, Aida T, Tsuchiya Y, Kishino Y, Kai K, Mori K. Interstitial pneumonitis related to trastuzumab deruxtecan, a human epidermal growth factor receptor 2‐targeting Ab–drug conjugate, in monkeys. Cancer Sci. 2020;111:4636–4645. 10.1111/cas.14686
REFERENCES
- 1. Oh DY, Bang YJ. HER2‐targeted therapies ‐ a role beyond breast cancer. Nat Rev Clin Oncol. 2020;17(1):33‐48. [DOI] [PubMed] [Google Scholar]
- 2. Nakada T, Masuda T, Naito H, et al. Novel antibody drug conjugates containing exatecan derivative‐based cytotoxic payloads. Bioorg Med Chem Lett. 2016;26(6):1542‐1545. [DOI] [PubMed] [Google Scholar]
- 3. Nakada T, Sugihara K, Jikoh T, et al. The latest research and development into the antibody‐drug conjugate, [fam‐] trastuzumab deruxtecan (DS‐8201a), for HER2 cancer therapy. Chem Pharm Bull (Tokyo). 2019;67(3):173‐185. [DOI] [PubMed] [Google Scholar]
- 4. Ogitani Y, Aida T, Hagihara K, et al. DS‐8201a, a novel HER2‐targeting ADC with a novel DNA topoisomerase I inhibitor, demonstrates a promising antitumor efficacy with differentiation from T‐DM1. Clin Cancer Res. 2016;22(20):5097‐5108. [DOI] [PubMed] [Google Scholar]
- 5. Ogitani Y, Hagihara K, Oitate M, et al. Bystander killing effect of DS‐8201a, a novel anti‐human epidermal growth factor receptor 2 antibody‐drug conjugate, in tumors with human epidermal growth factor receptor 2 heterogeneity. Cancer Sci. 2016;107(7):1039‐1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Doi T, Shitara K, Naito Y, et al. Safety, pharmacokinetics, and antitumour activity of trastuzumab deruxtecan (DS‐8201), a HER2‐targeting antibody‐drug conjugate, in patients with advanced breast and gastric or gastro‐oesophageal tumours: a phase 1 dose‐escalation study. Lancet Oncol. 2017;18(11):1512‐1522. [DOI] [PubMed] [Google Scholar]
- 7. Modi S, Saura C, Yamashita T, et al. Trastuzumab deruxtecan in previously treated HER2‐positive breast cancer. N Engl J Med. 2020;382(7):610‐621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tsurutani J, Iwata H, Krop I, et al. Targeting HER2 with trastuzumab deruxtecan: a dose‐expansion, phase i study in multiple advanced solid tumors. Cancer Discov. 2020;10(5):688‐701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dimopoulou I, Bamias A, Lyberopoulos P, et al. Pulmonary Toxicity From Novel Antineoplastic Agents. Ann Oncol. 2006;17(3):372‐379. [DOI] [PubMed] [Google Scholar]
- 10. Costa R, Costa‐Filho RB, Talamantes SM, et al. Interstitial pneumonitis secondary to trastuzumab: A case report and literature review. Case Rep Oncol. 2017;10(2):524‐530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. US Food and Drug Administration . Kadcyra USPI. https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/125427s105lbl.pdf. Accessed September 24, 2020. [Google Scholar]
- 12. European Medicines Agency . Herceptin (trastuzumab) Scientific Discussion. https://www.ema.europa.eu/en/documents/scientific‐discussion/herceptin‐epar‐scientific‐discussion_en.pdf. Accessed September 24, 2020. [Google Scholar]
- 13. US Food and Drug Administration . Kadcyra Pharmacology Review. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2013/125427Orig1s000PharmR.pdf. Accessed September 24, 2020. [Google Scholar]
- 14. Skeoch S, Weatherley N, Swift AJ, et al. Drug‐induced interstitial lung disease: a systematic review. J Clin Med. 2018;7(10):356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ogura T, Takigawa N, Tomii K, et al. Summary of the Japanese Respiratory Society statement for the treatment of lung cancer with comorbid interstitial pneumonia. Respir Investig. 2019;57(6):512‐533. [DOI] [PubMed] [Google Scholar]
- 16. Matsuno O. Drug‐induced interstitial lung disease: mechanisms and best diagnostic approaches. Respir Res. 2012;13:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nagai Y, Oitate M, Shiozawa H, et al. Comprehensive preclinical pharmacokinetic evaluations of trastuzumab deruxtecan (DS‐8201a), a HER2‐targeting antibody‐drug conjugate, in cynomolgus monkeys. Xenobiotica. 2019;49(9):1086‐1096. [DOI] [PubMed] [Google Scholar]
- 18. Hanewinkel H, Glössl J, Kresse H. Biosynthesis of cathepsin B in cultured normal and I‐cell fibroblasts. J Biol Chem. 1987;262(25):12351‐12355. [PubMed] [Google Scholar]
- 19. Colletti GA, Miedel MT, Quinn J, et al. Loss of lysosomal ion channel transient receptor potential channel mucolipin‐1 (TRPML1) leads to cathepsin B‐dependent apoptosis. J Biol Chem. 2012;287(11):8082‐8091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Schwaiblmair M, Behr W, Haeckel T, et al. Drug induced interstitial lung disease. Open Respir Med J. 2012;6:63‐74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zaizen Y, Fukuoka J. Pathology of idiopathic interstitial pneumonias. Surg Pathol Clin. 2020;13(1):91‐118. [DOI] [PubMed] [Google Scholar]
- 22. Polakis P. Antibody drug conjugates for cancer therapy. Pharmacol Rev. 2016;68(1):3‐19. [DOI] [PubMed] [Google Scholar]
- 23. de Goeij BE, Lambert JM. New developments for antibody‐drug conjugate‐based therapeutic approaches. Curr Opin Immunol. 2016;40:14‐23. [DOI] [PubMed] [Google Scholar]
- 24. Press MF, Cordon‐Cardo C, Slamon DJ. Expression of the HER‐2/neu proto‐oncogene in normal human adult and fetal tissues. Oncogene. 1990;5(7):953‐962. [PubMed] [Google Scholar]
- 25. Gorovits B, Krinos‐Fiorotti C. Proposed mechanism of off‐target toxicity for antibody‐drug conjugates driven by mannose receptor uptake. Cancer Immunol Immunother. 2013;62(2):217‐223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mahalingaiah PK, Ciurlionis R, Durbin KR, et al. Potential mechanisms of target‐independent uptake and toxicity of antibody‐drug conjugates. Pharmacol Ther. 2019;200:110‐125. [DOI] [PubMed] [Google Scholar]
- 27. Aggarwal N, Sloane BF. Cathepsin B: multiple roles in cancer. Proteomics Clin Appl. 2014;8(5–6):427‐437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gondi CS, Rao JS. Cathepsin B as a cancer target. Expert Opin Ther Targets. 2013;17(3):281‐291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Inoue K, Kumazawa E, Kuga H, et al. CM‐dextran‐polyalcohol‐camptothecin conjugate: DE‐310 with a novel carrier system and its preclinical data. Adv Exp Med Biol. 2003;519:145‐153. [DOI] [PubMed] [Google Scholar]
- 30. Shiose Y, Ochi Y, Kuga H, et al. Relationship between drug release of DE‐310, macromolecular prodrug of DX‐8951f, and cathepsins activity in several tumors. Biol Pharm Bull. 2007;30(12):2365‐2370. [DOI] [PubMed] [Google Scholar]
- 31. Byrne AJ, Maher TM, Lloyd CM. Pulmonary macrophages: a new therapeutic pathway in fibrosing lung disease? Trends Mol Med. 2016;22(4):303‐316. [DOI] [PubMed] [Google Scholar]
- 32. Saunders KO. Conceptual approaches to modulating antibody effector functions and circulation half‐life. Front Immunol. 2019;10:1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Schlothauer T, Herter S, Koller CF, et al. Novel human IgG1 and IgG4 Fc‐engineered antibodies with completely abolished immune effector functions. Protein Eng Des Sel. 2016;29(10):457‐466. [DOI] [PubMed] [Google Scholar]
- 34. Lo M, Kim HS, Tong RK, et al. Effector‐attenuating substitutions that maintain antibody stability and reduce toxicity in mice. J Biol Chem. 2017;292(9):3900‐3908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Furukawa H, Oka S, Shimada K, et al. Genetics of interstitial lung disease: Vol de nuit (night flight). Clin Med Insights Circ Respir Pulm Med. 2015;9(suppl 1):1‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Shiina T, Blancher A. The cynomolgus macaque MHC polymorphism in experimental medicine. Cells. 2019;8(9):978. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1
Fig S2
Fig S3
Fig S4
Table S1
Table S2
Table S3