

Abstract
Diagnostic markers for both colorectal cancer (CRC) and its precursor lesions are lacking. Although aberrant methylation of the secretin receptor (SCTR) gene was observed in CRC, the diagnostic performance has not been evaluated. Therefore, this study aimed to assess and verify the diagnostic value of SCTR methylation of CRC and its precursor lesions through integrating the largest methylation data. The diagnostic performance of SCTR methylation was analyzed in the discovery set from The Cancer Genome Atlas (TCGA) CRC methylation data (N = 440), and verified in a large‐scale test set (N = 938) from the Gene Expression Omnibus (GEO). Targeted bisulfite sequencing analysis was developed and applied to detect the methylation status of SCTR in our independent validation set (N = 374). Our findings revealed that the SCTR gene was frequently hypermethylated at its CpG islands in CRC. In the TCGA discovery set, the diagnostic score was constructed using 4 CpG sites (cg01013590, cg20505223, cg07176264, and cg26009192) and achieved high diagnostic performance (area under the ROC curve [AUC] = 0.964). In the GEO test set, the diagnostic score had robust diagnostic ability to distinguish CRC (AUC = 0.948) and its precursor lesions (AUC = 0.954) from normal samples. Moreover, hypermethylation of the SCTR gene was also found in cell‐free DNA samples collected from CRC patients, but not in those from healthy controls. In the validation set, consistent results were observed using the targeted bisulfite sequencing array. Our study highlights that hypermethylation at CpG islands of the SCTR gene is a potential diagnostic biomarker in CRCs and its precursor lesions.
Keywords: biomarker, colorectal cancer, diagnosis, DNA methylation, SCTR
Hypermethylation at CpG island of the SCTR gene holds great promise as a diagnostic biomarker of CRC and its precursor lesions.

Abbreviations
- AJCC
American Joint Commission on Cancer
- AUC
area under the ROC curve
- cfDNA
cell‐free DNA
- CIMP
CpG island methylator phenotype
- CRC
colorectal cancer
- FIT
fecal immunochemical test
- FOBT
fecal occult blood testing
- GEO
Gene Expression Omnibus
- LRES
long‐range epigenetic silencing
- ROC
receiver operating characteristic
- TCGA
The Cancer Genome Atlas
1. INTRODUCTION
Colorectal cancer (CRC) is currently the third most commonly diagnosed cancer and the second leading cause of mortality in the world based on GLOBOCAN 2018, with an estimate of 1.85 million diagnosed novel cases and 0.88 million deaths. 1 Both hereditary and non‐hereditary factors have been linked to the development of CRC. 2 , 3 Hereditary and genetic factors, including a family history of CRC and inherited mutations, account for a small fraction of CRC cases, however more than half of CRC cases are attributable to non‐hereditary factors. Despite improved medications, CRC is still the main cause of cancer death because of advanced‐stage diagnosis and the few number of effective drugs. Survival rate of CRC sharply declines at an advanced tumor stage, whereas patients diagnosed at stage I or II disease have a 5‐y survival rate of 91% and 82%, and those at stage IV disease have 5‐y survival rate of 12%. 4 Early detection of CRC is crucial to reduction in CRC‐related mortality. Currently, a various CRC screening tests are available, including colonoscopy, FOBT, and FIT, however the various limitations of these tests have led to poor patient compliance and low participation rates. 5 , 6 For instance, colonoscopy is invasive and expensive despite being the gold standard for CRC screening. By contrast, blood CRC screening tests may improve participation rates, given the advantage of less invasive procedure and complications.
DNA methylation alterations are involved in initiation and progression of cancers by reprogramming of the epigenetic landscape. DNA methylation has been proposed as a potential biomarker for clinical diagnosis, prognosis, and prediction of treatment responses in various types of cancers, including CRC. 7 , 8 With respect to CRC diagnosis, some aberrantly methylated genes, such as SEPT9, SFRP2, NDRG4, BMP3, and IKZF1, have been suggested as potential markers. 9 , 10 , 11 , 12 , 13 , 14 One of the most widely studied methylation‐based markers for the diagnosis of CRC is hypermethylation of the SEPT9 gene, which was commercialized (Epi proColon) and approved by the United States Food and Drug Administration (US FDA) in 2016. 15 The diagnostic performance of this methylation‐based test varied in multiple studies, with sensitivity ranging from 48% to 90% and specificity from 73% to 97%, respectively. 7 One of the major limitations is its poor sensitivity for the detection of colorectal adenomas, as detection and removal of these precursor lesions are essential for reducing the incidence of CRC. 9 , 16 , 17 , 18 Therefore, methylation‐based markers for the diagnosis of CRC and its precursor lesions are still required.
The secretin receptor (SCTR), which was the first to be discovered in duodenal mucosa, is a G protein‐coupled receptor and a member of the glucagon‐VIP‐secretin receptor. 19 The SCTR gene is located on chromosome 2q14.2, which contains 3 hypermethylation genes (EN1, SCTR, and INHBB) in correlation with LRES in CRC. 20 Previous studies have shown that hypermethylated EN1 in stool DNA may have application as a non‐invasive biomarker of CRC. 21 SCTR has been found to be hypermethylated in CRC, and was associated with downregulation of gene expression. 20 , 21 , 22 However, the potential of SCTR methylation in the diagnosis of CRC has not been previously evaluated. We therefore investigated whether hypermethylation of SCTR is a promising early detection molecular marker in CRC and its precursor lesions.
2. MATERIALS AND METHODS
2.1. Public data collection
To identify the methylation profile of the SCTR gene in CRCs, 10 public methylation datasets were obtained from TCGA and the GEO, https://www.ncbi.nlm.nih.gov/geo/), totaling 1378 samples (Table 1). TCGA CRC methylation dataset (N = 440) of the Illumina 450k array was download from the UCSC Xena Browser (https://xena.ucsc.edu/), and was used as the discovery set. The methylation level of CpG site was expressed as a β value, and calculated as M/(M + U), where M and U represent methylated intensity and unmethylated intensity, respectively. Ten CpG sites throughout the SCTR gene were available after removing 12 CpG sites with missing data larger than 20% (Table S1). Furthermore, TCGA CRC gene expression profile was also downloaded from the UCSC Xena Browser. By a systematic search in the GEO database, 8 methylation datasets of the Illumina 450k array were obtained as test set (N = 930): GSE42752, GSE48684, GSE68060, GSE77718, GSE77954, GSE101764, GSE107352, and GSE129364. 23 , 24 , 25 , 26 , 27 , 28 , 29 An additional methylation dataset for the Illumina EPIC array with 4 cell‐free DNA (cfDNA) samples from CRC and 4 samples from healthy controls were obtained from GSE122126. 30
TABLE 1.
Overview of the datasets used in this study
| Dataset | Source | Assay | Sample type | Number of samples |
|---|---|---|---|---|
| Discovery set (N = 440) | ||||
| TCGA CRC | Infinium 450K | Tissue | Normal = 45, CRC = 395 | |
| Test set (N = 938) | ||||
| Test set A | GSE42752 | Infinium 450K | Tissue | Normal = 41, CRC = 22 |
| GSE48684 a | Infinium 450K | Tissue | Normal = 41, CRC = 64 | |
| GSE68060 a | Infinium 450K | Tissue | Normal = 36, CRC = 82 | |
| GSE77718 | Infinium 450K | Tissue | Normal = 96, CRC = 96 | |
| GSE77954 | Infinium 450K | Tissue | Normal = 11, CRC = 13 | |
| GSE101764 | Infinium 450K | Tissue | Normal = 149, CRC = 112 | |
| GSE107352 | Infinium 450K | Tissue | Normal = 21, CRC = 30 | |
| Test set B | GSE48684 a | Infinium 450K | Tissue | Normal = 41, Adenoma = 42 |
| GSE77954 a | Infinium 450K | Tissue | Normal = 11, Adenoma = 12 | |
| GSE129364 | Infinium 450K | Tissue | Normal = 3, Adenoma = 59 | |
| Test set C | GSE122126 | Infinium EPIC | cfDNA | Normal = 4, CRC = 4 |
| Validation set (N = 374) | ||||
| Inhouse study | Targeted bisulfite sequencing | Tissue | Normal = 23, Polyp = 10, Adenoma = 8, CRC = 275 | |
| Inhouse study | Targeted bisulfite sequencing | WBC | Normal = 29, CRC = 29 | |
2.2. Construction and evaluation of methylation‐based diagnostic score
The workflow is described in Figure 1. Differential methylation analyses between CRC tissues and normal tissues were performed for 10 CpG sites at SCTR in the TCGA discovery set. The diagnostic performance of individual CpG site for distinguishing CRC tissues and normal tissues was assessed by ROC curves. Candidate CpG sites were selected based on an AUC of >0.90. Using the logistic regression model, a 4‐CpG diagnostic score was constructed in the discovery set. As the test set, 8 GEO methylation datasets were used to evaluate the robustness and generalizability of the 4‐CpG diagnostic score. The GEO test set was divided into 2 parts: test set A (CRC tissues and normal tissues) and test set B (adenoma tissues and normal tissues).
FIGURE 1.
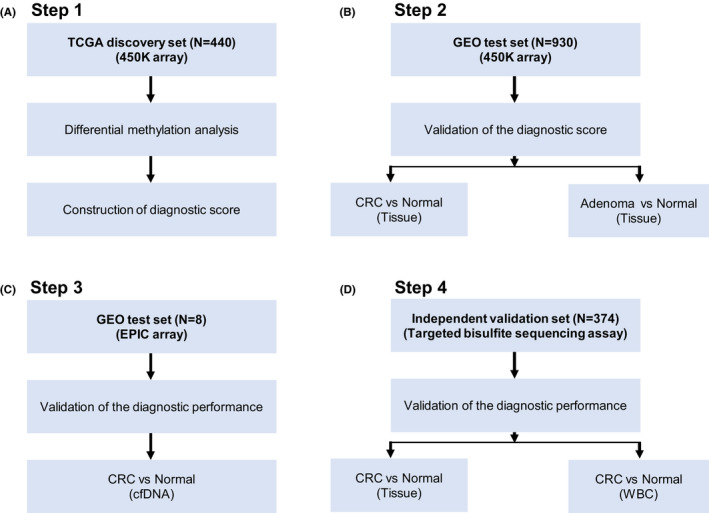
Overall workflow of the various analyses in different methylation datasets
2.3. Inhouse validation study
To further assess the SCTR methylation, we performed a quantitative methylation analysis in an independent validation set (N = 374). Briefly, our study collected 10 tissues of hyperplastic polyps, 8 tissues of adenomas, 275 tissues of CRC, 23 adjacent normal tissues, 29 white blood cell samples from CRC, and 29 white blood cell samples from healthy controls. These samples were collected at the Third Affiliated Hospital of Harbin Medical University. All patients with CRC were newly diagnosed and underwent surgery without neoadjuvant chemotherapy, and were confirmed by postoperative pathology. Clinicopathological information including age, gender, tumor location, and tumor stage, were obtained from clinical records and pathological report. 31 Tumor stage was determined using the AJCC staging system for patients with CRC. Written informed consent was obtained from all study participants and the study protocol was approved by the Medical Ethics Committee of Harbin Medical University.
2.4. SCTR quantitative methylation analysis
Quantitative methylation analysis of SCTR was evaluated using MethylTarget sequencing (Genesky Biotechnologies Inc). As previously described, this targeted bisulfite sequencing assay is a next‐generation sequencing technology for methylation profiling of targeted genomic regions. 32 Genomic DNA from tissues and white blood cells were extracted using the classic phenol‐chloroform procedure and a QIAamp DNA Blood Mini Kit (Qiagen). DNA samples were bisulfite‐converted with a EpiTect Fast DNA Bisulfite Kit (QIAGEN GmbH) using the manufacturer's protocol. Corresponding primers for SCTR were designed using Primer3 software (Figure S1). 33 A two‐step PCR approach was performed for each bisulfite‐converted DNA sample, with the first PCR for amplifying the targeted DNA sequence and the second PCR for adding barcodes. Sequencing was performed on an Illumina HiSeq 2000 system using a 150‐bp paired‐end mode.
2.5. Statistical analysis
All statistical analyses were performed in R version 3.5.1 software, and a P‐value < .05 was considered significant. All genomic coordinates referred to the human genome version GRCh37/hg19. Student t test was used to evaluate the significant difference of methylation levels and expression levels of the SCTR gene between 2 groups. Spearman correlation method was applied to assess coordinated methylation (co‐methylation) of adjacent CpG sites. The association between SCTR methylation and expression was confirmed by Pearson correlation coefficients in 391 samples from TCGA CRC dataset. ROC and AUC were used to verify the discriminative performance of SCTR methylation. An optimized cut‐off value was selected at the maximal Youden index in ROC. The binary logistic regression model was fitted to construct the diagnostic score using multiple CpG sites. The trend test for SCTR methylation at different disease stages during neoplastic progression was determined using a linear regression model.
3. RESULTS
3.1. SCTR methylation and expression in colorectal cancer tissues from the TCGA discovery set
In the TCGA discovery set, differential methylation analyses were performed between 395 CRC and 45 normal tissues in 10 CpG sites of the SCTR gene. Based on location relative to CpG islands, 4 CpG sites were located in the Open Sea (cg19706534, cg13880193, cg10944063, and cg19576736), one in North Shelf (cg24549161), one in North Shore (cg16943697) and 4 in CpG islands (cg01013590, cg20505223, cg07176264, and cg26009192). Using Student t test, methylation levels for 4 CpG sites at CpG islands were significantly higher in CRC than those in normal samples (Figure 2). Four (cg13880193, cg10944063, cg24549161, and cg16943697) out of 6 residual CpG sites were significantly lower in CRC than those in normal samples. Spearman correlation coefficients were calculated to investigate the methylation patterns of 10 CpG sites on SCTR in 395 CRC samples. Strong correlations in 4 CpG sites at CpG islands indicated that these CpG sites shared a similar methylation status, which was termed co‐methylation (Figure S2). In TCGA dataset, SCTR expression was downregulated in 380 CRC samples compared with 51 normal samples (Figure S3). Moreover, methylation levels of 4 CpG sites at CpG islands were negatively correlated with its gene expression in 391 paired methylation and expression data (Figure S4). Collectively, CpG islands of SCTR were significantly hypermethylated in CRC and were associated with downregulation of gene expression.
FIGURE 2.
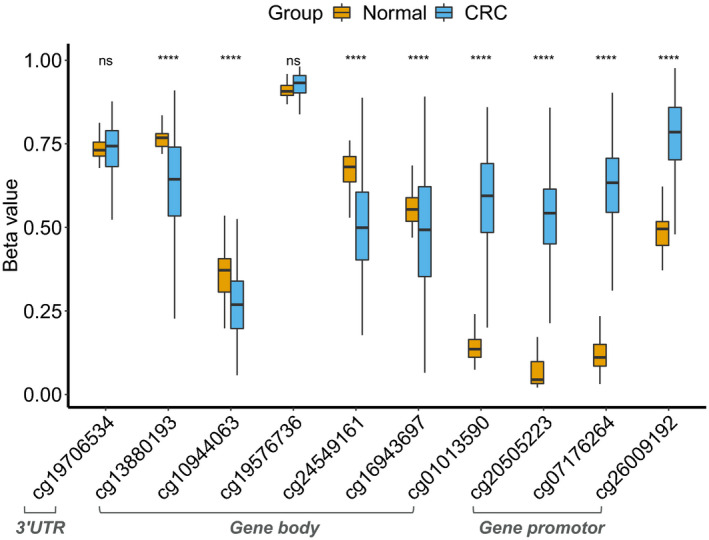
Differential methylation analysis for 10 CpG sites of the SCTR gene between colorectal cancer tissues and normal tissues in TCGA discovery set. Gene promoter was defined as the region containing TSS200, TSS1500, 5′UTR, and the first exon. Symbols indicated statistical significance for the t test: ns, P > .05; **** P ≤ .0001
3.2. SCTR methylation as detection biomarker for colorectal cancer
We investigated the diagnostic performance of 10 CpG sites as detection markers for CRC. Therefore, we calculated the AUC values of individual CpG sites to discriminate CRC (N = 395) from normal samples (N = 45). Sensitivities and specificities under the different cut‐off value for methylation levels are shown in Figure S5. Four CpG sites at CpG islands showed high discriminative ability, with AUC values in the range 0.919‐0.953.
These 4 CpG sites with high AUC values (>0.90) were then selected to construct the diagnostic score using a logistic regression method. The 4‐CpG diagnostic score was a sum of the methylation levels of 4 CpG sites with weights given by the corresponding coefficient from the logistic regression model (Figure S6): (7.501 × cg01013590) + (1.522 × cg20505223) + (3.454 × cg07176264) + (3.682 × cg26009192). ROC analyses showed that the diagnostic score yielded an AUC of 0.964 in TCGA discovery set (Figure 3A). Using a fixed cut‐off value of 6.154, the diagnostic score yielded an accuracy of 94.5% (416 of 440) in the discovery set; the sensitivity and specificity were 93.9% and 100% (Table 2), respectively.
FIGURE 3.
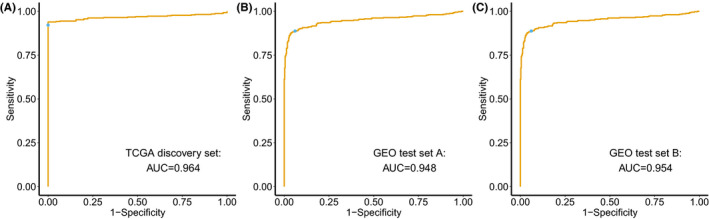
Diagnostic performance of the 4‐CpG diagnostic score for colorectal cancer and its precursors. ROC curves of the diagnostic score constructed by the logistic regression model in (A) the TCGA discovery set, (B) the GEO test set A, and (C) the GEO test set B. The blue point indicates sensitivity and specificity at the fixed cut‐off value of 6.154
TABLE 2.
Performance metrics of 4‐CpG diagnostic score in TCGA discovery set and GEO test sets
|
TCGA discovery set CRC vs Normal |
GEO test set A CRC vs Normal |
GEO test set B Adenoma vs Normal |
|
|---|---|---|---|
| Accuracy (95% CI) | 0.945 (0.920, 0.965) | 0.914 (0.893, 0.932) | 0.917 (0.864, 0.954) |
| Sensitivity | 0.939 | 0.888 | 0.903 |
| Specificity | 1.000 | 0.942 | 0.946 |
| Kappa | 0.760 | 0.828 | 0.818 |
Abbreviations: CRC, colorectal cancer; GEO, Gene Expression Omnibus; TCGA, The Cancer Genome Atlas.
We first applied the diagnostic score of TCGA discovery set to the GEO test set A. The diagnostic score had robust performance (AUC = 0.948) to distinguish 419 CRC tissues from 395 normal tissues in the GEO test set A (Figure 3B). Using the same cut‐off value of 6.154 in the GEO test set A, the accuracy was 91.4% (744 of 814); the sensitivity and specificity were 88.8% and 94.2%, respectively.
3.3. SCTR methylation as detection biomarker for precursor lesions of colorectal cancer
Next, we assessed the performance of the diagnostic score in the GEO test set B for differentiating between 113 adenomas and 55 normal samples. The diagnostic score could effectively differentiate adenoma from normal samples (AUC = 0.954, Figure 3C). Applying the cut‐off value of 6.154, the accuracy was 91.7% (154 of 168); the sensitivity and specificity were 90.3% and 94.6%, respectively. Unsupervised hierarchical clustering of 4 CpG site could separate adenoma from normal samples (Figure S7).
3.4. SCTR methylation in cell‐free DNA samples from colorectal cancer
Moreover, we investigated whether CpG islands of the SCTR gene were hypermethylated in cfDNA from CRC patients. Three CpG sites were available in a dataset with 8 cfDNA samples (Figure S8). Hypermethylation of 3 CpG sites was found in cfDNA samples collected from CRC (3 out of 4), but not in those from healthy individuals (0 of 4).
3.5. SCTR‐specific sequencing array in colorectal tissue samples of the validation set
For clinical application, a cost‐effective detection technology is needed to detect the methylation level of the SCTR gene. Thus, the diagnostic performance of SCTR methylation was further assessed in our independent validation set using target bisulfite sequencing, which is a reliable technology and is less costly than Infinium 450K array. Based on the location of cg20505223 from the Infinium 450K array, we designed a SCTR‐specific sequencing array to detect methylation status at CpG islands. The target region covered 27 CpG sites. Genomic locations of the 27 CpG sites are described in Table S2. Three CRC tissue samples with a bisulfite conversion rate below 98% were filtered out after quality control. Using t test, differential methylation analyses were performed for cg20505223, as well as 26 adjacent CpG sites. Methylation levels of cg20505223 (mean β value = 0.331) were significantly higher in 272 CRC tissues compared with those (mean β value = 0.043) in 23 normal tissues (Figure 4A). Similar results from differential methylation analyses were also observed in 26 adjacent CpG sites (Figure S9 and Table S2).
FIGURE 4.
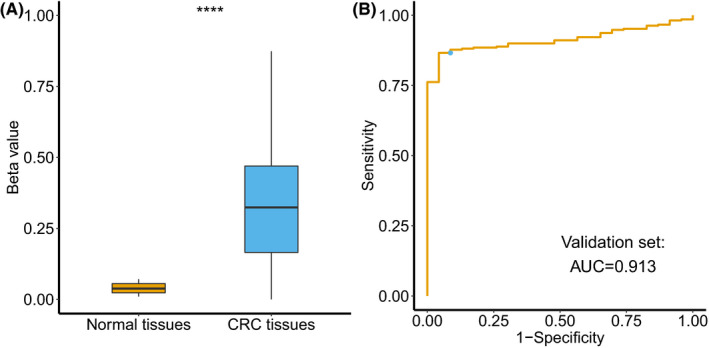
Methylation status of cg20505223 in the validation set. A, Differential methylation analysis of cg20505223 between 272 colorectal cancer tissues and 23 normal tissues, **** P ≤ .0001. B, Receiver operating characteristic curve analysis and Youden index analysis of cg20505223 for discriminating colorectal cancer tissues and normal tissues. The blue point indicates sensitivity and specificity at the cut‐off value of 0.071
With regards to diagnostic performance, the AUC value was 0.913 using cg20505223 to discriminate between CRC and normal tissues (Figure 4B), which was slightly lower compared with that (AUC = 0.919) in TCGA discovery set. Under a cut‐off value of 0.071 at the maximal Youden index, the sensitivity and specificity were 86.6% and 95.7% (Table S3). When subgroup analyses were performed by clinicopathological variables, methylation levels (Table S4) and AUC values (Figure S10) for cg20505223 were consistent in different subgroups. In particular, no significant differences (P = .880) of the diagnostic performance were found between the early‐stage group (AJCC stage I/II) and the advanced‐stage group (AJCC stage III/IV). Similar diagnostic performance was also observed in 26 adjacent CpG sites, with AUC values ranging from 0.907 to 0.948 (Table S3). Therefore, results from the validation set confirmed that SCTR was hypermethylated at its CpG islands region, which could distinguish CRC tissues from normal tissues using a cost‐effective method.
3.6. SCTR‐specific sequencing array in white blood cell samples of the validation set
As white blood cells contribute the largest proportion of background DNA in cfDNA samples, methylation difference in white blood cells samples between CRC patients and healthy controls may confound the methylation profile in cfDNA. We applied SCTR‐specific sequencing array in white blood cell samples from 29 CRC patients and 29 healthy controls. White blood cell samples had low methylation levels for cg20505223 for both CRC patients (mean β value = 0.024) and healthy controls (mean β value = 0.038). Although a significant difference was found in 2 groups (P = .011), the direction of differences was such that methylation levels in CRC patients were lower than those in healthy controls (Figure 5). Twenty‐six adjacent CpG sites showed low methylation levels in white blood cell samples from CRC patients and healthy controls (Table S5). Therefore, SCTR hypermethylation in cfDNA samples from CRC patients was not derived from white blood cells.
FIGURE 5.
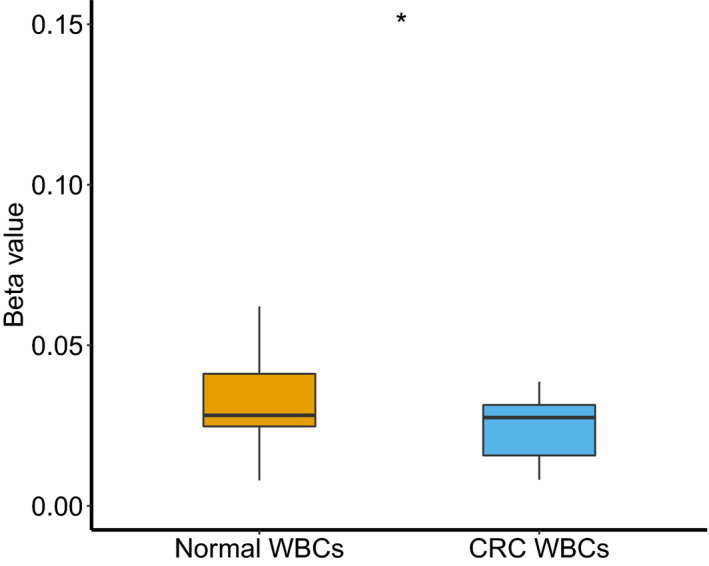
Methylation levels of cg20505223 in white blood cell samples from 29 colorectal cancer patients and 29 healthy controls. * P ≤ .05
3.7. SCTR methylation in neoplastic progression in the validation set
To determine the methylation status of the SCTR gene at different disease stages during neoplastic progression, the methylation status of 313 colorectal tissues, including 23 normal tissues, 10 hyperplastic polyps, 8 adenomas, and 272 CRCs, were evaluated in the validation set. As shown in Figure 6, the medians of average methylation level of targeted regions in different disease stages were as follows: normal tissues, 0.063; hyperplastic polyps, 0.080; adenomas, 0.141; and CRCs, 0.370. The trend test showed that methylation levels in CpG islands of SCTR increased sequentially from normal to hyperplastic polyp, to adenoma, and to CRC (trend test P < .001).
FIGURE 6.
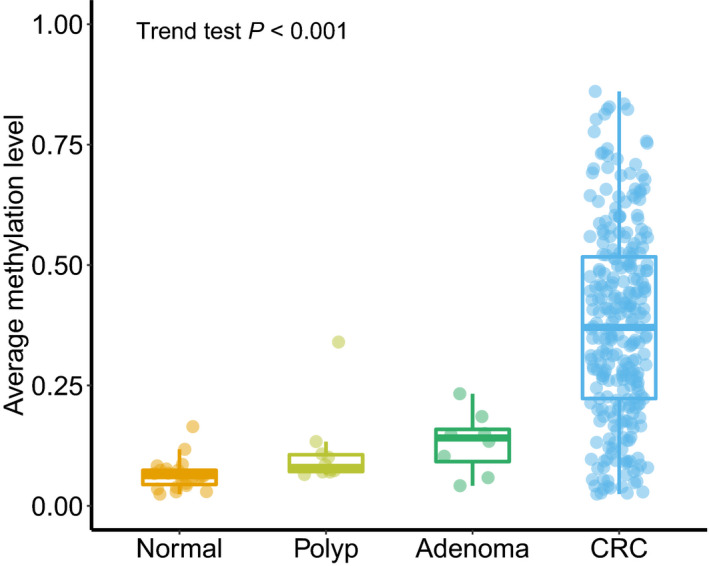
Average methylation levels of the SCTR gene at different disease stages during neoplastic progression. * P ≤ .05
4. DISCUSSION
In this present study, we investigated the potential use of SCTR methylation as a diagnostic marker for CRC and its precursor lesions by integrating the largest methylation data (N = 1752). Our study indicated that methylation levels of 4 CpG sites at CpG islands of the SCTR gene were significantly higher in CRC samples than those in normal samples. Overall, the 4‐CpG diagnostic score had an excellent performance to distinguish CRC and adenomas from normal samples, with AUC values over 0.948. Hypermethylation of the SCTR gene was also found in cfDNA samples from CRC patients and could be detected by a cheaper PCR‐based technology. These results highlighted that SCTR methylation had a strong potential as a novel marker for the detection of CRC and its precursor lesions in clinical applications.
Our study validated findings of previous mechanism studies that found that the SCTR gene was hypermethylated at CpG sites in its CpG island in CRC, and therefore was negatively associated with gene expression. A study showed that SCTR was hypermethylated in 88% (23 of 26) of CRC, and was associated with gene silencing. 20 Another study reported similar results of SCTR methylation in CRC, but a lower frequency (48 of 90, 53%) of SCTR methylation in CRC. 21 Currently, greater numbers of genome‐wide methylation data could be accessible from publicly available datasets such as TCGA and GEO; this provides the opportunity to quantitatively evaluate methylation change of the SCTR gene in CRC. Integrating the largest genome‐wide methylation data, our study demonstrated that 4 CpG sites at CpG islands of the SCTR gene were highly hypermethylated in both CRC and adenomas.
The main finding of this study was the identification of SCTR methylation as a promising marker for detection of CRC; this achieved sufficient discrimination accuracy to distinguish CRC from normal samples. The diagnostic score with 4 CpG sites achieved an AUC of 0.964 in TCGA discovery set. Moreover, the diagnostic score was externally verified in large‐scale data from the GEO database. The AUC for discriminating CRC and normal tissues was 0.948 in the GEO test set, and proved generalizability of the 4‐CpG diagnostic markers across different settings. Our study of SCTR methylation was extended to biological fluids of CRC patients by analyzing a GEO dataset. Hypermethylation of the SCTR gene was also found in cfDNA samples collected from CRC patients. Due to complex technology and cost‐effectiveness, candidate gene method is more suitable for clinical application than a genome‐wide method. Therefore, targeted bisulfite sequencing was used to verify SCTR methylation in our inhouse study, which had the advantages of being highly specific for targeted CpG sites with small genomic DNA amounts, and accurate methylation profiling of single CpG sites in targeted genomic regions. Our study found that the diagnostic performance of SCTR methylation in the validation set was similar to that of the discovery set, suggesting that it had potential for clinical transformation. Our inhouse study also ruled out the possible noise effects from white blood cells on the observed hypermethylation of cfDNA samples from CRC. Altogether, our study revealed that SCTR methylation had a strong potential as a diagnostic biomarker for CRC.
In screening settings, the detection of precursor lesions, which would allow earlier intervention by removal of these diseases, had significant importance in the prevention of CRC. Previous studies have indicated that various methylated genes were potential biomarkers for detection of CRC, however methylation‐based markers that could diagnose both CRC and its precursor lesions are lacking. 7 As an example, the plasma‐based SEPT9 methylation marker has been commercialized for clinical use. Although the SEPT9 methylation marker in plasma displayed high specificities in large studies, it showed poor sensitivities (7.9%‐38.7%) for the detection of precursor lesions (adenomas). 9 , 16 , 17 , 18 Our study found that SCTR methylation achieved high accuracy in the detection of both CRC and adenomas.
As with most epithelial cancers, CRC results from an accumulative change of genetics and epigenetics that transforms precursor lesions in the colon and rectum into carcinomas. However, the role of DNA methylation in CRC formation is still not well understood. Previous studies have identified that methylated genes (ITGA4, MGMT, SLC5A8, SFRP2, and MINT1) are involved in this progression sequence. 34 In this study, methylation levels at CpG islands of the SCTR gene increased sequentially in the polyp to a carcinoma progression sequence. In addition, hypermethylation of the SCTR gene was observed in even the earliest lesions of CRC, suggesting that it plays a role in the initiation of tumorigenesis.
SCTR methylation has been described in the development and progression of CRC. SCTR gene locations at the boundaries of chromosome 2q14.2, where gene suppression commonly occurs in CRC through a mechanism of LRES. Downregulation of genes at the chromosome 2q14.2 region has been associated with coordinated hypermethylation of 3 CpG islands of EN1, SCTR, and INHBB. 20 , 22 Of these, the methylated EN1 CpG island in stool DNA has been shown to have application as a non‐invasive biomarker of CRC. 21 Although there is a consistent finding for the association between cg20505223 and overall survival of CRC, this was not observed in TCGA discovery set and our inhouse validation set (Figure S11); the significant methylation difference of the SCTR gene may contribute to the specific characteristics of the CIMP and its clinicopathologic features in CRC patients. A previous study reported that SCTR hypermethylation had potential influence on specific characteristics of CIMP‐positive CRC. 35 Further mechanism studies are warranted to provide a better understanding of the biological role of SCTR methylation on the development and progression of CRC.
Our study has some advantages. SCTR methylation was assessed in the largest study with different genome‐wide methylation datasets that provided promising preliminary data on its diagnostic utility. SCTR methylation was further validated by targeted bisulfite sequencing, which would be a clinically applicable method given the advantages of competitive pricing over genome‐wide approaches and accurate methylation profiling. Our study also has several limitations. First, although hypermethylation of the SCTR gene was observed in cfDNA samples from CRC patients, the number of samples of both CRC and heathy control was limited. However, we believe that SCTR methylation is worthy of further evaluation in prospective studies due to the excellent performance for discriminating CRC and its precursor lesions from normal tissues. Second, all available samples for the validation set were collected from a single center.
In conclusion, our study discovered and validated that hypermethylation at CpG islands of the SCTR gene holds great promise as a diagnostic biomarker for CRC and its precursor lesions based on integration analysis of large‐scale methylation data from TCGA, GEO, and an inhouse database. Although SCTR hypermethylation was observed in the cfDNA samples from CRC patients, whether SCTR methylation can be applied as a non‐invasive biomarker should be further determined in larger studies.
DISCLOSURE
The authors have no conflict of interest.
Supporting information
Supplementary Material
Figures S1‐S11
Tables S1‐S5
ACKNOWLEDGMENTS
We would like to thank all the patients who participated the studies from TCGA and GEO datasets and our study.
Li D, Zhang L, Fu J, et al. SCTR hypermethylation is a diagnostic biomarker in colorectal cancer. Cancer Sci 2020;111:4558–4566. 10.1111/cas.14661
DaPeng Li and Lei Zhang are contributed equally to the article.
Contributor Information
YaShuang Zhao, Email: zhao_yashuang@263.net.
BinBin Cui, Email: cuibinbin@hrbmu.edu.cn.
REFERENCES
- 1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394‐424. [DOI] [PubMed] [Google Scholar]
- 2. Dekker E, Tanis PJ, Vleugels JLA, Kasi PM, Wallace MB. Colorectal cancer. Lancet. 2019;394:1467‐1480. [DOI] [PubMed] [Google Scholar]
- 3. Ding W, Ma Y, Zhu W, et al. MICA ( *)012:01 Allele Facilitates The Metastasis of KRAS‐mutant colorectal cancer. Front Genet. 2020;11:511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Miller KD, Nogueira L, Mariotto AB, et al. Cancer treatment and survivorship statistics, 2019. CA Cancer J Clin. 2019;69:363‐385. [DOI] [PubMed] [Google Scholar]
- 5. Lin JS, Piper MA, Perdue LA, et al. Screening for colorectal cancer: updated evidence report and systematic review for the US preventive services task force. JAMA. 2016;315:2576‐2594. [DOI] [PubMed] [Google Scholar]
- 6. Schreuders EH, Ruco A, Rabeneck L, et al. Colorectal cancer screening: a global overview of existing programmes. Gut. 2015;64:1637‐1649. [DOI] [PubMed] [Google Scholar]
- 7. Jung G, Hernandez‐Illan E, Moreira L, Balaguer F, Goel A. Epigenetics of colorectal cancer: biomarker and therapeutic potential. Nat Rev Gastroenterol Hepatol. 2020;17:111‐130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wang C, Pu W, Zhao D, et al. Identification of hyper‐methylated tumor suppressor genes‐based diagnostic panel for esophageal squamous cell carcinoma (ESCC) in a Chinese Han population. Front Genet. 2018;9:356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. He N, Song L, Kang Q, et al. The pathological features of colorectal cancer determine the detection performance on blood ctDNA. Technol Cancer Res Treat. 2018;17:1533033818791794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tang D, Liu J, Wang DR, Yu HF, Li YK, Zhang JQ. Diagnostic and prognostic value of the methylation status of secreted frizzled‐related protein 2 in colorectal cancer. Clin Investig. 2011;34:E88‐E95. [DOI] [PubMed] [Google Scholar]
- 11. Imperiale TF, Ransohoff DF, Itzkowitz SH, et al. Multitarget stool DNA testing for colorectal‐cancer screening. N Engl J Med. 2014;370:1287‐1297. [DOI] [PubMed] [Google Scholar]
- 12. Symonds EL, Pedersen SK, Baker RT, et al. A blood test for methylated BCAT1 and IKZF1 vs. a fecal immunochemical test for detection of colorectal neoplasia. Clin Transl Gastroenterol. 2016;7:e137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Guo S, Diep D, Plongthongkum N, Fung HL, Zhang K, Zhang K. Identification of methylation haplotype blocks aids in deconvolution of heterogeneous tissue samples and tumor tissue‐of‐origin mapping from plasma DNA. Nat Genet. 2017;49:635‐642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fan J, Li J, Guo S, et al. Genome‐wide DNA methylation profiles of low‐ and high‐grade adenoma reveals potential biomarkers for early detection of colorectal carcinoma. Clin Epigenet. 2020;12:56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lamb YN, Dhillon S. Epi proColon 2.0 CE: a blood‐based screening test for colorectal cancer. Mol Diagn Ther. 2017;21:225‐232. [DOI] [PubMed] [Google Scholar]
- 16. Wu D, Zhou G, Jin P, et al. Detection of colorectal cancer using a simplified SEPT9 gene methylation assay is a reliable method for opportunistic screening. J Mol Diagn. 2016;18:535‐545. [DOI] [PubMed] [Google Scholar]
- 17. Fu B, Yan P, Zhang S, et al. Cell‐free circulating methylated SEPT9 for noninvasive diagnosis and monitoring of colorectal cancer. Dis Markers. 2018;2018:6437104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Song L, Peng X, Li Y, et al. The SEPT9 gene methylation assay is capable of detecting colorectal adenoma in opportunistic screening. Epigenomics. 2017;9:599‐610. [DOI] [PubMed] [Google Scholar]
- 19. Bayliss WM, Starling EH. The mechanism of pancreatic secretion. J Physiol. 1902;28:325‐353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Frigola J, Song J, Stirzaker C, Hinshelwood RA, Peinado MA, Clark SJ. Epigenetic remodeling in colorectal cancer results in coordinate gene suppression across an entire chromosome band. Nat Genet. 2006;38:540‐549. [DOI] [PubMed] [Google Scholar]
- 21. Mayor R, Casadome L, Azuara D, et al. Long‐range epigenetic silencing at 2q14.2 affects most human colorectal cancers and may have application as a non‐invasive biomarker of disease. Br J Cancer. 2009;100:1534‐1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Forn M, Munoz M, Tauriello DV, et al. Long range epigenetic silencing is a trans‐species mechanism that results in cancer specific deregulation by overriding the chromatin domains of normal cells. Mol Oncol. 2013;7:1129‐1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Naumov VA, Generozov EV, Zaharjevskaya NB, et al. Genome‐scale analysis of DNA methylation in colorectal cancer using Infinium HumanMethylation450 BeadChips. Epigenetics. 2013;8:921‐934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Luo Y, Wong CJ, Kaz AM, et al. Differences in DNA methylation signatures reveal multiple pathways of progression from adenoma to colorectal cancer. Gastroenterology. 2014; 147(2):418‐429.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. McInnes T, Zou D, Rao DS, et al. Genome‐wide methylation analysis identifies a core set of hypermethylated genes in CIMP‐H colorectal cancer. BMC Cancer. 2017;17:228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Qu X, Sandmann T, Frierson H Jr, et al. Integrated genomic analysis of colorectal cancer progression reveals activation of EGFR through demethylation of the EREG promoter. Oncogene. 2016;35:6403‐6415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Barrow TM, Klett H, Toth R, et al. Smoking is associated with hypermethylation of the APC 1A promoter in colorectal cancer: the ColoCare Study. J Pathol. 2017;243:366‐375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Damaso E, Castillejo A, Arias MDM, et al. Primary constitutional MLH1 epimutations: a focal epigenetic event. Br J Cancer. 2018;119:978‐987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Fiedler D, Hirsch D, El Hajj N, et al. Genome‐wide DNA methylation analysis of colorectal adenomas with and without recurrence reveals an association between cytosine‐phosphate‐guanine methylation and histological subtypes. Gene Chromosomes Canc. 2019;58:783‐797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Moss J, Magenheim J, Neiman D, et al. Comprehensive human cell‐type methylation atlas reveals origins of circulating cell‐free DNA in health and disease. Nat Commun. 2018;9:5068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Liu X, Fu J, Bi H, et al. DNA methylation of SFRP1, SFRP2, and WIF1 and prognosis of postoperative colorectal cancer patients. BMC Cancer. 2019;19:1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Pu W, Wang C, Chen S, et al. Targeted bisulfite sequencing identified a panel of DNA methylation‐based biomarkers for esophageal squamous cell carcinoma (ESCC). Clin Epigenet. 2017;9:129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Untergasser A, Cutcutache I, Koressaar T, et al. Primer3–new capabilities and interfaces. Nucleic Acids Res. 2012;40:e115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lao VV, Grady WM. Epigenetics and colorectal cancer. Nat Rev Gastroenterol Hepatol. 2011;8:686‐700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Karpinski P, Ramsey D, Grzebieniak Z, Sasiadek MM, Blin N. The CpG island methylator phenotype correlates with long‐range epigenetic silencing in colorectal cancer. Mol Cancer Res. 2008;6:585‐591. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Figures S1‐S11
Tables S1‐S5