

Abstract
A complete understanding of the plant microbiome has not yet been achieved due to its complexity and temporal shifts in the community structure. To overcome these issues, we created a synthetic bacterial community of the aquatic plant, duckweed. The synthetic community established with six bacterial strains showed a stable composition for 50 days, which may have been because duckweed maintains a similar physiological status through its clonal reproduction. Additionally, the synthetic community reflected the taxonomic structure of the natural duckweed microbiome at the family level. These results suggest the potential of a duckweed-based synthetic community as a useful model system for examining the community assembly mechanisms of the plant microbiome.
Keywords: synthetic ecology, duckweed, plant-microbe interaction, SynCom, bacterial community assembly
Plants form taxonomically-structured microbiomes; however, the mechanisms underlying community assembly have not yet been elucidated by microbiome research. The use of synthetic communities, i.e. a mixture of isolated strains mimicking the natural microbiome, recently emerged as a novel experimental approach. The merits of using synthetic communities include their simplicity, reproducibility, and ease of detecting causality (Castrillo et al., 2017; Vorholt et al., 2017; Durán et al., 2018; Herrera Paredes et al., 2018; Liu et al., 2019). Previous studies that utilized plant-associated synthetic communities (often referred to as SynCom) provided high-confidence data on the mechanisms by which specific genes and metabolites influence microbial community assembly (Bodenhausen et al., 2014; Lebeis et al., 2015; Bai et al., 2015; Carlström et al., 2019; Voges et al., 2019). Niu et al. (2017) created a seven-membered synthetic bacterial community of maize roots, and investigated the mechanisms by which bacterial interspecies interactions affect community assembly.
We herein report the establishment of a synthetic bacterial community associated with the aquatic plant, duckweed. Duckweed is an attractive model plant for examining plant-microbe interactions due to its easy gnotobiotic cultivation and manipulation (Appenroth et al., 2016; Yamakawa et al., 2018; Ishizawa et al., 2019). More importantly, duckweed populations maintain a certain age structure and physiological status over long periods of time through their budding-based growth (Datko et al., 1980), whereas most plants markedly change their morphology, physiology, and associated microbiome with development and senescence (Zhalnina et al., 2018; Hara et al., 2019). Therefore, we hypothesized that the first “stable” synthetic community, which maintains similar properties irrespective of the culture time, may be established with duckweed. Since current analytical technologies for genes and metabolites provide only a “snapshot” information at the time of measurement, temporal shifts in plant and microbial physiologies have been a major obstacle to detailed analyses. Therefore, a stable synthetic system may facilitate clearer investigations of plant-microbe interactions, leading to unique insights in this field.
In the present study, we initially grew duckweed in a natural pond environment and isolated duckweed-associated bacteria as candidate members of the synthetic community. A laboratory stock of sterilized duckweed (Lemna minor RDSC5512), which was previously sterilized (Suzuki et al., 2014) and routinely cultured with modified Hoagland (MH) medium (Toyama et al., 2006) in a growth chamber (28°C, photon flux of 80 μmol m–2 s–1, 16-h light cycle), was floated on the Inukai pond (Suita, Osaka, Japan) within a stainless steel mesh box (20×20×20 cm, opening of 1×1 mm) from October 8th to 18th, 2018. Approximately 1,000 fronds were transplanted, and 50 were collected after 1, 3, 5, 7, and 10 days and then preserved at –80°C until DNA extraction. Another 50 fronds were collected after 7 days as the isolation source of co-existing bacteria.
A culture-independent analysis of the duckweed microbiome was performed as described previously (Ishizawa et al., 2020). Briefly, the DNA of epiphytic bacteria was extracted using the Cica Genius DNA extraction kit (Kanto Chemical) and the V4 region of the bacterial 16S rRNA gene was amplified with two-step PCR using 5′-tailed primers (515F–805R; Caporaso et al., 2010) and Illumina index PCR primers (Illumina). Multiplex sequencing was performed on an Illumina Miseq platform (300-bp paired-end).
Raw sequence reads were demultiplexed in Qiime v1.9.1. Quality filtering, the merging of paired-end reads, chimera removal, singleton removal, and taxonomy assignments were performed with dada2 (Callahan et al., 2016) in R v3.5.2. Forward and reverse sequences were trimmed from 20 to 280 bp and from 20 to 160 bp, respectively. The SILVA database (v138; Quast et al., 2013) was used for the taxonomic assignment of amplicon sequence variants (ASVs). The relative abundance of bacterial taxa was estimated with 35,386–40,233 reads per sample after removing non-prokaryotic ASVs. The Shannon index was calculated using the “vegan” package in R.
Regarding bacterial isolation, fresh plant samples (50 fronds) were gently washed with ca. 40 mL of sterile MH medium and homogenized with a disposable homogenizer (Biomasher II; Nippi) until no large plant tissues (>ca. 0.5 mm) remained. The homogenate was diluted and spread on R2A (Daigo, Nihon Pharmaceutical), tryptic soy broth (TSB; Difco), 1/100 R2A, or 1/100 TSB medium with 1.5% agar or 1.0% gellan gum (with 0.3 g L–1 MgSO4 as the cation source) and 100 mg L–1 cycloheximide. Morphologically distinct colonies were exhaustively selected, purified, and preserved at –80°C with 15% glycerol for further use. Regarding taxonomic identification, partial 16S rRNA gene sequences (primers 27F–1392R) were amplified and sequenced as described previously (Ishizawa et al., 2017). The sequences obtained were compared to those in the GenBank/EMBL/DDBJ databases using BLAST. The neighbor-joining phylogenetic trees of the isolates were constructed using MEGA7 software v7.0.14 with dominant ASVs in the pond-grown duckweed microbiome.
The synthetic bacterial community was constructed by co-culturing duckweed with 16, 6, or 5 bacterial isolates. Each isolate was cultivated overnight (28°C, 120 rpm) in 10 mL of R2A medium (supplemented with 2% methanol for Methylophilaceae strains) in a vial, pelleted (10,000×g, 4°C, 5 min), re-suspended three times in 5 mL of sterile MH medium, and then mixed at approximately the same cell densities based on the optical density at 600 nm (OD600). The mixture with a known OD600 was added to 60 mL MH medium in the flask to adjust the final OD600 to ca. 0.0005. Ten fronds of sterile L. minor were transplanted into the flask to initiate the co-cultivation under controlled conditions (28°C, 80 μmol m–2 s–1, 16-h light cycle). Under our growth conditions, duckweed grew exponentially to ca. 50–60 fronds in 5 days. Hence, the synthetic system was maintained by transplanting ten duckweed fronds (with bacteria attached) into new medium every 5 days. In the experiments using 16- and 5-membered communities, plant samples for the DNA analysis were collected at the end of the second batch cultivations. In the six-membered community, samples were collected from each of the triplicate flasks after the 1st, 2nd, 5th, and 10th batch cultivations. We also estimated the total bacterial colonization density (CFU per frond) at each sampling point by plating diluted homogenates of plant samples (from each flask) on R2A agar supplemented with 2% methanol. The number of colonies was counted after a 3-day incubation at 28°C.
The relative abundance of bacterial strains in the synthetic communities was evaluated by Illumina Miseq sequencing as described above, and the V5-V6 region of the bacterial 16S rRNA gene (primers 799F–1185mR) (Chelius and Triplett, 2001; Hodkinson and Lutzoni, 2009) was analyzed to distinguish all bacterial strains. Reads of ASVs with sequences that perfectly matched that of or had a single base mismatch from the inoculated strains (43,484–56,546 reads per sample) were used to calculate relative abundance. The family level composition of the synthetic communities was compared to that of the natural duckweed microbiome and other plant-associated microbiomes sequenced in previous studies (Schlaeppi et al., 2014; Xie et al., 2015; Agler et al., 2016; Niu et al., 2017; Toju et al., 2019; Acosta et al., 2020; Huang et al., 2020; Ishizawa et al., 2020). Renkonnen dissimilarity (1 – the Renkonnen similarity index) was used for comparisons because this metric is robust against differences in sample sizes and diversity (Wolda, 1981). Non-metric multidimensional scaling (NMDS) was performed for visualization using the “vegan” package in R.
Figure 1 shows the bacterial community composition associated with the pond-grown duckweed. Duckweed assembled a unique microbial community within 1 day, and the dominant bacterial families (>2% relative abundance on average, shown in Fig. 1) remained unchanged throughout the culture period. These dominant families are consistent with previous findings on the duckweed microbiome (Xie et al., 2015; Acosta et al., 2020; Huang et al., 2020; Ishizawa et al., 2020; Iwano et al., 2020; Iwashita et al., 2020), indicating that duckweed attracts specific bacterial taxa irrespective of the environmental context. On the other hand, the dominant genera or ASVs within each family were repeatedly replaced over 10 days (Fig. S1). Hence, the composition at finer phylogenetic levels may be determined by rather stochastic processes.
Fig. 1.
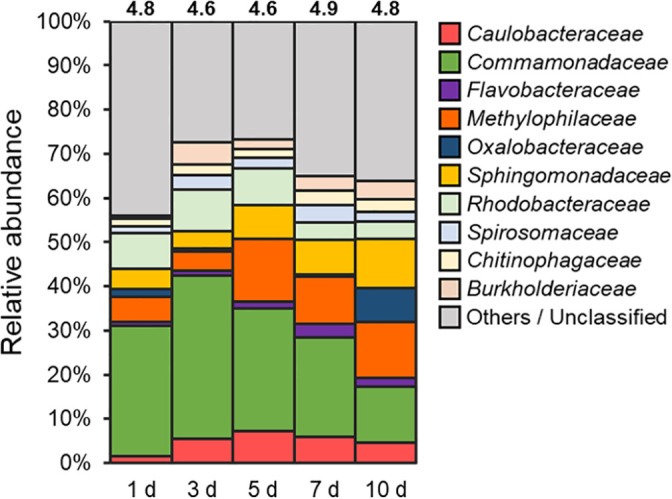
Phylogenetic distribution of duckweed-associated bacterial communities in the natural pond environment. Lemna minor plants were grown in the Inukai pond and the epiphytic bacterial community was analyzed at various times. Values above the bars indicate the alpha diversity (Shannon index). Families with average relative abundance <2.0% were assembled as “Others”.
Given the consistent family-level composition of the duckweed microbiome, we attempted to establish a synthetic community with a similar family-level structure to natural communities. To achieve this, we selected 16 bacterial isolates belonging to the six dominant families (Caulobacteraceae, Comamonadaceae, Flavobacteriaceae, Methylophilaceae, Oxalobacteraceae, and Sphingomonadaceae) for further experiments (Table S1, Fig. S2, S3, S4, S5, S6, and S7). These families were selected based on their common presence at relatively high abundance in the L. minor microbiome. Collectively, they accounted for 44.1–58.5% of the pond-grown duckweed microbiome analyzed in the present study (Fig. 1), and on average 78.3% of that previously observed for the same L. minor clone (Ishizawa et al., 2020).
When we inoculated sterile duckweed with the 16 bacterial strains (2–3 strains per family), all six families co-existed at a certain abundance (2.0–23.5%), whereas the four strains (DW043, DW096, DW159, and DW160) failed to survive in the community (<0.1%) (Fig. 2A). Moreover, only a single strain appeared to thrive from each of the six families. These results suggest that bacterial strains in the same family are more likely to compete for similar niches, while strains in different families co-exist due to the different niche preference (Martiny et al., 2015; Goldford et al., 2018).
Fig. 2.

Relative abundance of bacterial strains in synthetic communities of Lemna minor. (A) The 16-membered synthetic community analyzed after the 2nd batch (10 days). (B) The six-membered synthetic community monitored up to the 10th batch (50 days). (C) Drop-out communities of the six-membered synthetic community analyzed after the 2nd batch (10 days). Values above the bars indicate the alpha diversity (Shannon index).
We then constructed the synthetic community with six bacterial strains selected from the 16-membered community (DW039, DW067, DW100, DW102, DW145, and DW155) for further simplification, and found similar communities among triplicates throughout the 50-day cultivation period (Fig. 2B). The bacterial colonization density was also maintained at between 106 to 107 CFU per frond for 50 days (Table S2). This high stability over 50 days was outstanding because the plant microbiome inevitably exhibits time-dependent community shifts with host development and senescence (Sugiyama et al., 2014; Xu et al., 2018; Zhalnina et al., 2018; Hara et al., 2019). The synthetic community of maize roots established by Niu et al. (2017) also showed a dynamic community shift during their 15-day monitoring period. Therefore, as our initial expectation, a static synthetic community was established, which appeared to be aided by the unchanging physiological status of duckweed. In addition, the synthetic community reflected the taxonomic profile of the natural duckweed microbiome, in that Caulobacteraceae, Comamonadaceae, and Methylophilaceae were the most dominant and the other three families co-existed at a specific relative abundance (>0.3%). The results of the comparative analysis indicated that our synthetic community structure at the bacterial family level reflected the unique structure of the natural duckweed microbiome (Fig. 3A), and was the most similar to those associated with the same L. minor clone grown under the same light, thermal, and nutrient conditions (Ishizawa et al., 2020; Fig. 3B).
Fig. 3.

Comparison of taxonomic profiles of synthetic communities with several plant-associated microbiomes (A) and natural duckweed-associated microbiomes (B). The first two dimensions of a non-metric multidimensional scaling analysis were plotted based on Renkonnen dissimilarity at the bacterial family level.
We also performed drop-out experiments on the six-membered community. When strong interactions (e.g. competition or metabolite exchange) exist among the six strains, the deletion of a single strain is expected to impact the community structure of the remaining members. Similar experiments on maize, alfalfa, and Arabidopsis-based synthetic communities identified some keystone species that conspicuously distorted the remaining community upon deletion (Niu et al., 2017; Carlström et al., 2019; Moccia et al., 2020). However, in the present study, all six drop-out communities showed similar structures to those expected from the original community (Fig. 2C). Nevertheless, one of the three dominant strains (DW039, DW102, and DW145) specifically increased upon the deletion of another dominant strain, suggesting that multiple ecological processes, including niche segregation and interspecies interactions, are involved in the formation of our synthetic community.
In conclusion, we herein established a synthetic bacterial community of duckweed that maintained its community structure irrespective of the culture time. This high stability is a unique feature of our synthetic system that will enable a more detailed understanding of plant-microbe interactions with existing technologies. The community assembly of the plant microbiome is currently described as a two-step selection model, in which plant-derived substrates induce the initial taxonomic selection, and undefined genotypic factors fine-tune the co-existing members (Bulgarelli et al., 2013). Goldford et al. (2018) recently postulated that substrate-driven selection mainly occurs for functions conserved at the bacterial family level. Based on these concepts, the assembly mechanisms of the duckweed microbiome warrant further study from two aspects: (i) the functions of the dominant families that make them competent in the duckweed microbiome and (ii) the factors influencing the winners of within-family competition. Future analyses on bacterial functions and interactions in the duckweed-based synthetic community will contribute to a more detailed understanding of the plant microbiome.
All raw sequence data related to the present study are available in the DDBJ Sequence Read Archive under accession numbers DRA010591 and LC573430–LC573445.
Citation
Ishizawa, H., Tada, M., Kuroda, M., Inoue, D., Futamata, H., and Ike, M. (2020) Synthetic Bacterial Community of Duckweed: A Simple and Stable System to Study Plant-microbe Interactions. Microbes Environ 35: ME20112.
https://doi.org/10.1264/jsme2.ME20112
Supplementary Material
Acknowledgements
This work was supported by the Advanced Low Carbon Technology Research and Development Program (ALCA) of the Japan Science and Technology Agency (JST) grant number JPMJAL1108 and the Japan Society for the Promotion of Science (JSPS) KAKENHI grant numbers JP18J10181, JP19K22930, and JP20J00210.
References
- Acosta K., Xu J., Gilbert S., Denison E., Brinkman T., Lebeis S., and Lam E. (2020) Duckweed hosts a taxonomically similar bacterial assemblage as the terrestrial leaf microbiome. PLoS One 15: e0228560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agler M.T., Ruhe J., Kroll S., Morhenn C., Kim S.T., Weigel D., et al. (2016) Microbial hub taxa link host and abiotic factors to plant microbiome variation. PLoS Biol 14: e1002352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appenroth K.J., Ziegler P., and Sree K.S. (2016) Duckweed as a model organism for investigating plant-microbe interactions in an aquatic environment and its applications. Endocytobiosis Cell Res 27: 94–106. [Google Scholar]
- Bai Y., Müller D.B., Srinivas G., Garrido-Oter R., Potthoff E., Rott M., et al. (2015) Functional overlap of the Arabidopsis leaf and root microbiota. Nature 528: 364–369. [DOI] [PubMed] [Google Scholar]
- Bodenhausen N., Bortfeld-Miller M., Ackermann M., and Vorholt J.A. (2014) A synthetic community approach reveals plant genotypes affecting the phyllosphere microbiota. PLoS Genet 10: e1004283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulgarelli D., Schlaeppi K., Spaepen S., van Themaat E.V.L., and Schulze-Lefert P. (2013) Structure and functions of the bacterial microbiota of plants. Annu Rev Plant Biol 64: 807–838. [DOI] [PubMed] [Google Scholar]
- Callahan B.J., McMurdie P.J., Rosen M.J., Han A.W., Johnson A.J.A., and Holmes S.P. (2016) DADA2: high-resolution simple inference from Illumina amplicon data. Nat Methods 13: 581–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso J.G., Kuczynski J., Stombaugh J., Bittinger K., Bushman F.D., Costello E.K., et al. (2010) QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7: 335–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlström C.I., Field C.M., Bortfeld-Miller M., Müller B., Sunagawa S., and Vorholt J.A. (2019) Synthetic microbiota reveal priority effects and keystone strains in the Arabidopsis phyllosphere. Nat Ecol Evol 3: 1445–1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castrillo G., Teixeira P.J.P.L., Paredes S.H., Law T.F., de Lorenzo L., Feltcher M.E., et al. (2017) Root microbiota drive direct integration of phosphate stress and immunity. Nature 543: 513–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chelius M.K., and Triplett E.W. (2001) The diversity of archaea and bacteria in association with the roots of Zea mays L. Microb Ecol 41: 252–263. [DOI] [PubMed] [Google Scholar]
- Datko A.H., Mudd S.H., and Giovanelli J. (1980) Lemna paucicostata Hegelm. 6746: life cycle and characterization of the colony types in a population. Plant Physiol 65: 913–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durán P., Thiergart T., Garrido-Oter R., Agler M., Kemen E., Schulze-Lefert P., and Hacquard S. (2018) Microbial interkingdom interactions in roots promote Arabidopsis survival. Cell 175: 973–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldford J.E., Lu N., Bajić D., Estrela S., Tikhonov M., Sanchez-Gorostiaga A., et al. (2018) Emergent simplicity in microbial community assembly. Science 6401: 469–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara S., Matsuda M., and Minamisawa K. (2019) Growth stage-dependent bacterial communities in soybean plant tissues: Methylobrum transiently dominated in the flowering stage on the soybean shoot. Microbes Environ 34: 446–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrera Paredes S., Gao T., Law T.F., Finkel O.M., Mucyn T., Teixeira P.J.P.L., et al. (2018) Design of synthetic bacterial communities for predictable plant phenotypes. PLoS Biol 16: e2003962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodkinson B.P., and Lutzoni F. (2009) A microbiotic survey of lichen-associated bacteria reveals a new linage from the Rhizobiales. Symbiosis 49: 163–180. [Google Scholar]
- Huang W., Gilbert S., Poulev A., Acosta K., Lebeis S., Long C., and Lam E. (2020) Host-specific and tissue-dependent orchestration of microbiome community structure in traditional rice paddy ecosystems. Plant Soil 452: 379–395. [Google Scholar]
- Ishizawa H., Kuroda M., Morikawa M., and Ike M. (2017) Evaluation of environmental bacterial communities as a factor affecting the growth of duckweed Lemna minor. Biotechnol Biofuels 10: 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishizawa H., Kuroda M., Inoue K., Inoue D., Morikawa M., and Ike M. (2019) Colonization and competition dynamics of plant growth-promoting/inhibiting bacteria in the phytosphere of duckweed Lemna minor. Microb Ecol 77: 440–450. [DOI] [PubMed] [Google Scholar]
- Ishizawa H., Kuroda M., Inoue D., Morikawa M., and Ike M. (2020) Community dynamics of duckweed-associated bacteria upon inoculation of plant growth-promoting bacteria. FEMS Microbiol Ecol 96: fiaa101. [DOI] [PubMed] [Google Scholar]
- Iwano H., Hatohara S., Tagawa T., Tamaki H., Li Y., and Kubota K. (2020) Effect of treated sewage characteristics on duckweed biomass production and microbial communities. Water Sci Technol 82: 292–302. [DOI] [PubMed] [Google Scholar]
- Iwashita T., Tanaka Y., Tamaki H., Yoneda Y., Makino A., Tateno Y., et al. (2020) Comparative analysis of microbial communities in fronds and roots of three duckweed species: Spirodela polyrhiza, Lemna minor, Lemna aequinoctialis. Microbes Environ 35: ME20081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebeis S.L., Paredes S.H., Lundberg D.S., Breakfield N., Gehring J., Mcdonald M., et al. (2015) Salicylic acid modulates colonization of the root microbiome by specific bacterial taxa. Science 349: 860–864. [DOI] [PubMed] [Google Scholar]
- Liu Y.X., Qin Y., and Bai Y. (2019) Reductionist synthetic community approaches in root microbiome research. Curr Opin Microbiol 49: 97–102. [DOI] [PubMed] [Google Scholar]
- Martiny J.B.H., Jones S.E., Lennon J.T., and Martiny A.C. (2015) Microbiomes in light of traits: A phylogenetic perspective. Science 350: aac9323. [DOI] [PubMed] [Google Scholar]
- Moccia K., Willems A., Papoulis S., Flores A., Forister M.L., Fordyce J.A., and Lebeis S.L. (2020) Distinguishing nutrient-dependent plant driven bacterial colonization patterns in alfalfa. Environ Microbiol Rep 12: 70–77. [DOI] [PubMed] [Google Scholar]
- Niu B., Paulson J.N., Zheng X., and Kolter R. (2017) Simplified and representative bacterial community of maize roots. Proc Natl Acad Sci U S A 114: E2450–E2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quast C., Pruesse E., Yilmaz P., Gerken J., Schweer T., Yarza P., et al. (2013) The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res 41: D590–D596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlaeppi K., Dombrowski N., Garrido-Oter R., van Themaat E.V.L., and Schulze-Lefert P. (2014) Quantitative divergence of the bacterial root microbiota in Arabidopsis thaliana relatives. Proc Natl Acad Sci U S A 111: 585–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiyama A., Ueda Y., Zushi T., Takase H., and Yazaki K. (2014) Changes in the bacterial community of soybean rhizospheres during growth in the field. PLoS One 9: e100709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki W., Sugawara M., Miwa K., and Morikawa M. (2014) Plant growth-promoting bacterium Acinetobacter calcoaceticus P23 increases the chlorophyll content of the monocot Lemna minor (duckweed) and the dicot Lactuca sativa (lettus). J Biosci Bioeng 118: 41–44. [DOI] [PubMed] [Google Scholar]
- Toju H., Okayasu K., and Notaguchi M. (2019) Leaf-associated microbiomes of grafted tomato plants. Sci Rep 9: 1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyama T., Yu N., Kumada H., Sei K., Ike M., and Fujita M. (2006) Accelerated aromatic compounds degradation in aquatic environment by use of interaction between Sphrodela polyrrhiza and bacteria in its rhizosphere. J Biosci Bioeng 101: 346–353. [DOI] [PubMed] [Google Scholar]
- Voges M.J.E.EE., Bai Y., Schulze-Lefert P., and Sattely E.S. (2019) Plant-derived coumarins shape the composition of an Arabidopsis synthetic root microbiome. Proc Natl Acad Sci U S A 116: 12558–12565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vorholt J.A., Vogel C., Carlström C.I., and Müller C.B. (2017) Establishing causality: Opportunities of synthetic communities for plant microbiome research. Cell Host Microbe 22: 142–155. [DOI] [PubMed] [Google Scholar]
- Wolda H. (1981) Similarity indices, sample size and diversity. Oecologia 50: 296–302. [DOI] [PubMed] [Google Scholar]
- Xie W., Su J., and Zhu Y. (2015) Phyllosphere bacterial community of floating macrophytes in paddy soil environments as revealed by Illumina high-throughput sequencing. Appl Environ Microbiol 81: 522–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L., Naylor D., Dong Z., Simmons T., Pierroz G., Hixon K.K., et al. (2018) Drought delays development of the sorghum root microbiome and enriches for monoderm bacteria. Proc Natl Acad Sci U S A 115: E4284–E4293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamakawa Y., Jog R., and Morikawa M. (2018) Effects of co-inoculation of two different plant growth-promoting bacteria on duckweed. Plant Growth Regul 86: 287–296. [Google Scholar]
- Zhalnina K., Louie K.B., Hao Z., Mansoori N., de Rocha U.N., Shi S., et al. (2018) Dynamic root exudate chemistry and microbial substrate preferences drive patterns in rhizosphere microbial community assembly. Nat Microbiol 3: 470–480. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.