

Abstract
The present investigation was envisaged to elucidate the neuroprotective effect of Higenamine (HGN) against aluminum chloride (AlCl3) triggered experimental Alzheimer’s disease (AD) rat model. Thirty-six male albino Wister rats were randomized and divided in 6 groups and subjected to experimentation for 6 weeks. Control group, AlCl3 (100 mg/kg orally), HGN (50 mg/kg orally), HGN25, HGN50, HGN75 (HGN 25, 50 and 75 mg/kg respectively and AlCl3 100 mg/kg orally). After completion of 42 days protocol, the animals were subjected to passive avoidance test. The animals were then anesthetized by intramuscularly injecting ketamine hydrochloride (24 mg/kg body weight) and euthanized by cervical amputation. Cortical and hippocampal tissues were carefully removed and were employed for quantification of aluminum and acetylcholinesterase. The tissues were quantified using Western blotting and detection kits for APP, Aβ1-42, β and γ secretases, Bax, Bad, caspases-9, cyto-c, pAkt and pGSK-3β, and oxidative markers. HGN significantly protected AlCl3 induced memory and learning impairments, Al overload, AChE hyperactivity, amyloid β (Aβ) burden and apoptosis in brain tissues via activating Akt/GSK3β pathway. HGN attenuated oxidative damage induced by Al by modulation of oxidative markers. Our findings advocate the neuroprotective effect of HGN in AlCl3 induced AD rat model.
Keywords: Alzheimer’s disease, higenamine, neuroprotective, cognitive impairment
Introduction
Alzheimer’s disease (AD) is regarded as the most generalized form of dementia that eventually lowers cognitive functionality, encompassing memory, speech, language, learning capacity, alignment, and decision-making capability.1 Global prevalence of dementia is around 36 million individuals, of which 75% suffer from AD.2 AD is marked by 3 critical structural changes revealed in the brain, loss of neurons, generation and deposition of hyperphosphorylated τ-proteins referred as neurofibrillary tangles (NFT), β-amyloid (Aβ) peptide accretion referred as senile or amyloid plaques.3 Brain areas associated with loss of memory and cognitive impairment, including cortical and hippocampal regions, which are cholinergic systems are predominantly affected in the course of AD.4
In-depth AD investigations revealed a series of elevated Aβ peptides responsible for triggering inflammation of neurons, coupled with neuroapoptosis, that progresses to cognitive impairment and advances to AD.5,6 The Aβ peptide is generated by splitting amyloid precursor protein (APP), a transmembrane glycoprotein, by β and γ secretases enzyme.7 This initiates a cascade of serious responses extending to synaptic deficits, spinal functionality loss, and collapse of Ca2+ balance, and finally, neuroapoptosis in individuals with AD.8 It has thus been evidenced from several investigations that Aβ1-42 is crucially involved in the pathogenesis of AD, and induces serious effects by various cascades, essentially via reducing spinal density, spinal damage, neuroapoptosis, attenuation of synaptic function induced memory, and lowering of excitatory signals at the synapse.9 These updates made on Aβ peptides have exposed a practical molecular foundation for the diagnosis and treatment of AD.10
Though precise molecular mechanisms involved in AD are still uncovered, but it is thought to be due to intricate involvement of several players such as genetics, inflammation, oxidation and environmental influencers.2 Several investigations however have claimed participation by an environmental contaminant, aluminum (Al), as a contributory factor to AD.11,12 As a contaminant, Al have highest probability to enter humans through food, dust, air, water and medicines. Al is known to be consumed in food cooked in Al containers and foils, used in the paper industry, water purification, fire extinguishers, food, and pharmaceutical additives.11 Food items through which Al enters humans include shellfish, cheese, milk and bakery products, sausages, tea, and various cosmetics. Also, AI derivates are used in phosphate binders, antacids, vaccines, and biologicals injections.12 Several animal studies have advocated Al-induced damage of neurons resulting in pathological, chemical, and behavioral alterations, closely similar to AD.13,14
Elevated consumption of Al leads to Aβ accumulation in the hippocampal and cortical regions of the brain, thereby progressing to learning and memorizing deficits in rats.14 After accumulation, Aβ activates the apoptotic pathway, through deposition into endoplasmic reticulum, or by blocking cellular receptors, that may induce stress in endoplasmic reticulum or mitochondria.15 During AD, the APP levels are increased due to disturbed processing of RNA with dismantled RNA species, such as myc box dependent-interacting protein-1, clusterin and presenilin-1.16 Neuroapoptosis triggered by Aβ induction may involve various signaling cascades such as MAPK, Wnt, Nf-kB, PI3K/Akt.17-19 Al is also known to induce neuroapoptosis by enhancing caspase-3 activation and modulating Akt and p-GSK-3β expression.20
Higenamine (HGN), chemically called as dl-demethylcoclaurine, is a monobenzylisoquinoline alkaloid member of structural class of protoberberines, and had been isolated as a potent cardiotonic component from Aconitum in 1976.21,22 Aconitum japonicum, a source of HGN has been employed as folk medicine in China and Japan for the treatment of rheumatic fever, syncope, joint pain, collapse, gastroenteritis, bronchial asthma, diarrhea, tumors and menoxenia.23 Several other investigations have revealed to be useful in conditions such as heart failure, breathing difficulties, arthritis, shortness of breath, sepsis, arrhythmia, heart failure and erectile dysfunction.24 HGN modulates β1 and β2 adrenergic receptors and induces a positive ionotropic effect. HGN exhibits an anti-thrombotic effect and inhibits aggregation of rat platelets.25 Investigations also advocate anti-oxidative potential of HGN by inhibition of lipid peroxidation and potential superoxide anion radical scavenging activity.26 The antioxidant activity of HGN is attributed to the reduction of reactive oxygen species and increasing the heme oxygenase-1 activity.27 Anti-apoptotic activity of HGN may also be due to its antioxidant potential. HGN pre-treatment in rats potentially reduced caspase-3 and elevated the expression of Bcl-2/Bax ratio. Anti-apoptotic efficacy of HGN has been attributed to modulation of PI3K/Akt signaling cascade, wherein PI3 K inhibition could downregulate Akt. HGN could significantly reduce NO production by lowering iNOS mRNA gene expression, thereby inhibiting the NF-κB activation and contributing to its anti-inflammatory effect.28
Our investigation into the literature associated with HGN revealed that, despite such broad-spectrum therapeutic efficacy, no exploration has been done to study the effect of HGN regarding its potential to treat AD and associated cognitive impairment. In accordance of the above discussed therapeutic capacities of HGN, the present research was conducted to study the possible effect of HGN in AD induced in Wister rats by exposure to Al, and investigate its underlying mechanism.
Material and Methods
Chemicals
Higenamine hydrochloride (purity ≥ 95%), Aβ, γ and β-secretase (anti-rabbit), and APP was procured from Merck Life Science Co. Ltd., Shanghai, China. Other compounds used for the investigation where, Bax, Bad, Bcl-2, Bcl-xL, cyto-c, pro and cleaved caspase-3 and -9 (anti-rabbit), GSK-3β and p-GSK-3β (ser 9), p-Akt (ser 473), voltage-dependent anion channel (VDAC), mouse anti-b-actin, and horseradish peroxidase-conjugated goat anti-rabbit IgG were purchased from Cell Signaling, Jiangxi, China.
Animals
Eighty-four male albino Wister rats (weighing between 200-225 g) procured from the Central Animal Laboratory and breeding center of Capital Medical University, Beijing, China, were divided into 2 major groups based on the phase 1 and 2 experimentation. For conducting animal experiments, guidelines provided by Guide for the Care and Use of Laboratory Animals from the National Institutes of Health were followed. During both the phases, the animals were housed in individual cages with unrestricted access to food and water and maintained at standard conditions—22 ± 2 oC temperature, 50 ± 10% relative humidity, and 12 hours light/dark cycle. For both the phases, the animals were allowed to acclimatize in laboratory conditions for 1 week.
For the phase 1 of the investigation, 36 male albino Wister rats were randomized and divided into 6 groups, each having 6 animals. The animals were then subjected to the following protocol for 6 weeks.
Group I: Control group (10 ml distilled water orally).
Group II: AlCl3 (100 mg/kg orally).
Group III: HGN (50 mg/kg orally, dissolved in 10 ml distilled water).
Group IV: AlCl3+HGN25 (25 mg/kg orally, dissolved in 10 ml distilled water + AlCl3 100 mg/kg orally).
Group V: AlCl3+HGN50 (50 mg/kg orally, dissolved in 10 ml distilled water + AlCl3 100 mg/kg orally).
Group VI: AlCl3+HGN75 (75 mg/kg orally, dissolved in 10 ml distilled water + AlCl3 100 mg/kg orally).
The dose of Al used in rats to induce AD was comparatively higher than in humans; it is linked to overdose of Al in humans that is experienced under certain situations.2 However, the dose is based on previous investigations, duration, and age of animals. Hence it was selected to confirm Al triggered molecular, behavioral, biochemical and neuronal discrepancies similar to AD in humans.29,30 Changes in the pattern of food and water intake coupled with bodyweight variations were recorded during 42 days of investigation. After completion of 42 days protocol, the animals were subjected to a passive avoidance test. The animals were then anesthetized by intramuscularly injecting ketamine hydrochloride (24 mg/kg body weight) and euthanized by cervical amputation.31 Cortical and hippocampal tissues were carefully removed and employed for quantification of Al and AChE. There was no remarkable change in the levels of Al and AChE in the brain tissues of rats administered with 50 mg/kg and 75 mg/kg of HGN, hence for further investigation 50 mg/kg HGN dose was used.
The phase 2 of the investigation was initiated by equally randomizing 48 rats into 4 groups and conducting the experimental protocol for 6 weeks: control group, AlCl3 group (AlCl3 100 mg/kg orally), HGN50 (HGN 50 mg/kg), and HGN50+AlCl3 (HGN 50 mg/kg + AlCl3 100 mg/kg orally). To elucidate whether HGN can induce a neuroprotective effect in AlCl3 induced AD model, Morris test was used together with the estimation of Aβ related protein expression and apoptotic protein expressions. During phase 2 of the investigation, body weight, fluctuations in normal behavior, and physical appearance observation were recorded. Also, there were no uncharacteristic sign and symptoms and mortality during the entire duration of the investigation.
Passive Avoidance Test
Passive avoidance is a fear-driven analysis to investigate the association of short- and long-term memories. The test essentially analyses the preferential behavior to dark. The assembly essentially consists of 2 chambers- light and dark, separated by a wall having a retractable door and with floor embedded with a metal grid through which current could be supplied. Initially for the acquisition trail, the rats were individually the rats were positioned in the light chamber. As the animals entered the darkroom, the animals experienced an electric shock of 0.5 mA, 40 V intensity for 1 second, through their feet. The animals are instantly removed and were placed in their respective cages. After 24 hours, the rats were placed in the chamber with light, and the time duration the animals utilize to get into the dark section is recorded, and considered as step-through latency. The animal was visualized for maximum 5 minutes, and if it did not step into the darkroom for the entire 5 minutes duration, the step-through latency was registered as 300 seconds, and further observation is stopped.32
Morris Water Maze (MWM)
The assembly consists of a round swimming pool (diameter 150 cm and height 45 cm), filled with water at 28 ± 1 oC and 30 cm depth, with well-defined 4 equal quadrants and a rigid platform. During the training session, a small platform was placed slightly (1 cm) above the water’s surface. Each rat was supposed to complete 4 training sessions, at an interval of 5 minutes. For each trial, an individual animal was slowly left in a variable quadrant and allowed to trace the platform. The animal was then allowed to stay over the platform for the next 20 seconds. In case any animal was unable to trace the platform in 120 seconds, it was helped to reach the platform and allowed to stay there for 20 seconds. On the 19th and 20th day of the study protocol, the animals were subjected to 2 more acquisitions, and an average time to reach the platform was recorded as acquisition latency. After AlCl3 administration, on 21st and 42nd day of the study, the average time to trace the platform was recorded as the retention.29
Determination of Aluminum Content
The brain tissues collected cortical and hippocampal regions were weighed and transferred to polytetrafluoroethane, to this was added 0.05 ml nitric acid and 0.2 ml hydrogen peroxide for each 30 mg tissue homogenate, and preserved at 120 oC for further 2 hours. The aluminum content was quantified by atomic absorption spectrophotometer.13
Determination of Acetylcholinesterase Activity
The hippocampal and cortical tissues were subjected to the estimation of AChE activity using AchE activity assay kit (Bio Vision, USA), following the instruction manual provided with the kit.
Western Blot Analysis
The brain collected from the hippocampus and cortical regions were subjected to homogenization in a buffer for extracting proteins using Pro-Prep solution according to the manufacturer’s manual (iNtRON, Biotechnology, Inc.). The mixture was then centrifuged (750 × g) at 4 oC for 10 minutes, to collect the nuclear portion, and further at 10,000 × g for 20 minutes at 4 oC to collect the mitochondrial part. The supernatant collected comprises the nuclear fraction, and the pellet at the bottom contains the mitochondrial remains. The pellet was further subjected for 60 minutes to cold centrifugation at 100,000 × g at 4 oC, and the resulting sediment collected was the cytosolic portion.33 The protein fraction collected was quantified by employing the Bio-Rad protein assay kit (Rad Laboratories, USA).
The protein fraction collected after lysis of the cells was loaded onto 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and then transferred over to polyvinylidene fluoride (PVF) membrane after separation. The PVF membranes were initially preserved for 2 hours with 5% skimmed milk, followed by overnight incubation with rabbit monoclonal (1:250) APP, pro and cleaved caspase-3, caspase-9, β-amyloid, γ− and β- secretases, p-Akt, p-GSK-3β, tGSK-3β, Bad, Bax, Bcl-xL, Bcl-2, VDAC and β-actin, in a blend of bovine serum albumin (5% in tris-buffered saline) and 0.05% Tween-20 at 4 oC. The membranes were then incubated at room temperature for 2 hours with secondary antibodies (IgG linked to horseradish peroxidase). The membranes were further rinsed 3 times with a mixture of bovine serum albumin (5% in tris-buffered saline) and 0.05% Tween-20. The specimens were then visualized by chemiluminescent method (GeScript ECL, Piscataway, USA). For densitometric measurements, gel image analysis software package was employed. The data collected was standardized by β-actin for cytosolic and the anti-VDAC antibodies for mitochondrial portions.34
Determination of Oxidative Stress Markers
The tissues collected from hippocampal and cortical region were quantified for oxidative stress markers namely, reactive oxygen species (ROS), malonaldehyde (MDA), superoxide dismutase (SOD) and glutathione peroxidase (GPx). For measurement of ROS, the hippocampal and cortical neurons were preserved for 30 minutes at room temperature with 2′,7′-dichlorofluorescein diacetate (10 μM). The neuronal cells were visualized through a fluorescence microscope, and the intensity of fluorescence was quantified using ImageJ software program (National Institute of Health, MD, USA). For detection of MDA, SOD and GPx, the neurons were subjected to lysis using radioimmunoprecipitation assay (RIPA) buffer. The lipid peroxidation assay was employed for quantifying MDA. The content was MDA was detected by employing thiobarbituric acid procedure using lipid peroxidation MDA detection kit (Beyotime, USA). SOD content was detected using the colorimetric cell viability kit-1 (WST-8) (PromoCell GmbH, Germany) method using the SOD estimation kit (Beyotime, USA). Glutathione peroxidase detection kit (Beyotime, USA) was employed for the determination of the cellular GPx in the brain cell lysates.
Statistical Data Analysis
The data collected was presented as mean ± standard error of mean obtained from all the experiments within respective group. The statistically significant values were calculated using 1-way analysis of variance (ANOVA), using SPSS 15.0 software package (Windows operating system), and independent assessments were performed using Duncan’s multiple range test. Statistically significant values are considered where p < 0.05.
Results
HGN Administration Lowers Weight Reduction Caused by AlCl3
Rats exposed to AlCl3 revealed a statistically marked (p < 0.05) reduction in the body weight in comparison to the animals from the control group (Figure 1). Animals administered with HGN coupled with AlCl3, revealed amelioration in the body weight, proportionately with the HGN dose administered. Compared to the control group, there was no remarkable change in the bodyweight of rats administered with HGN alone. Also, there was insignificant variation among the control and the test group animals related to water and food consumption.
Figure 1.

AlCl3-treated rats indicated significant (P < 0.05) reduction in body weight when compared with rats from control group. Oral treatment with HGN to AlCl3 induced rats significantly (P < 0.05) increased the body weight dose dependently. There are no significant changes in weight gain of HGN alone treated rats when compared with control rats. Data are expressed as mean ± SEM (1-way ANOVA followed by Duncan’s multiple range test) for 6 rats in each group. a—p < 0.05—comparison between initial and final body weight, b—p < 0.05 comparison between HGN50 treated and AlCl3 treated group, c—p < 0.05- comparison between AlCl3 treated and AlCl3 + HGN treated group.
HGN Administration Reduces Learning and Memory Impairment Induced by AlCl3
Memory is referred as persistent behavioral alterations resulted due to similar environmental stimuli exposure, leading to maintenance of constant behavior.35 There is no direct method for scaling the magnitude if memory however, relative measurement may be done by determining spatial memory, associative learning tests, alteration tests, recognition memory tests, attentional tests, set-shifting tests and reverse learning tests. Such measurements signifying behavioral changes are considered as crucial parameters for assessment of neurotoxicity induced by Al exposure.36
In the investigation presented herewith, for the determination of the associative learning, a passive avoidance test was performed. The study involved animals’ learning capacity to reveal quick responses to unpleasant stimuli, such as exposure and dark environment coupled with an electric shock. Rodents, such as rats, are night loving animals and hence prefer dark environment. In the passive avoidance test, the animal has to reduce the habit by carefully memorizing the negative stimuli, which is an electric shock. Results indicated that in comparison to the control group animals, those exposed to AlCl3 had a reduced step though latency, signifying deterioration of memory. Contrary to this, animals receiving HGN and AlCl3 markedly everted the memory and learning inadequacy induced by AlCl3 alone. There was a statistically insignificant difference in memory enhancement between HGN 50 mg/kg and 75 mg/kg dose but statistically significant compared to 25 mg/kg. (Figure 2).
Figure 2.

AlCl3 treated rats revealed enhanced stepthrough latency in passive avoidance test. AlCl3 induced step-through latency was reduced dose dependently by HGN co-treatment. Data are expressed as mean ± SEM (1-way ANOVA followed by Duncan’s multiple range test) for 6 rats in each group. a—p < 0.05—comparison against Control group, b—p < 0.05 comparison against AlCl3 group.
Hippocampal functionality is specifically affected during AD or with an increase in the age, which can be experimentally determined by performing the Morris water maze test, which measures spatial learning and memory.37 In this test, the rats were permitted to swim in a water-filled tank and encouraged to depart water by and reach a hidden platform. The rats were guided to trace the platform using structural clues. The spatial memory of rats could be measured by recording the time required to spot the hidden platform. In the present investigation, the rats exposed to AlCl3 alone took more time to reach the platform than the control group animals on 20th day of the training, revealing memory impairment. Consecutively, HGN exposure in AlCl3 treated animals revealed significantly enhanced memory performance on 20th day compared to AlCl3 treated group alone. Results of retention latency, measured on 21st and 40th, indicated significant lowering in animals exposed to AlCl3 alone. Long-term HGN (50 mg/kg) treatment in AlCl3 treated rats exhibited considerable enhancement in the retention latency, compared to rats exposed to AlCl3 only (Figure 3).
Figure 3.
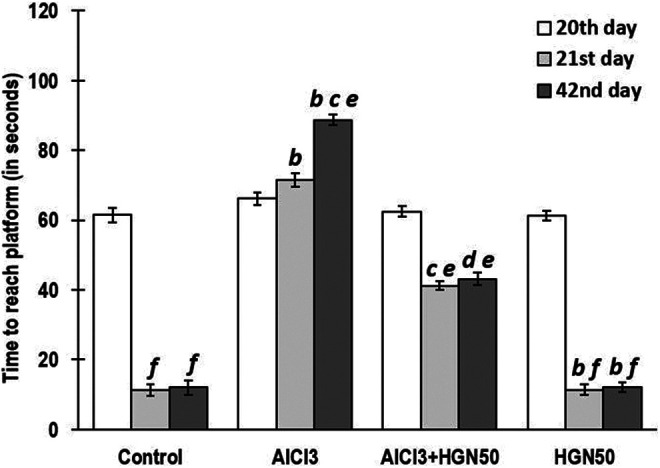
AlCl3 rats took more time to reach both the visible (on day 20) and hidden (on day 21 and 42) indicating memory deficits. Co-treatment of HGN50 (AlCl3+50 mg/kg HGN) significantly enhanced memory performance on day 20, 21 and 42 in both training and retention phase. Data are expressed as mean ± SEM (a repeated-measured ANOVA followed by Duncan’s multiple range test) for 6 rats in each group. a—p < 0.05 compared to the control, b—p < 0.01 compared to the control, c—p < 0.05 compared to AlCl3 treated rats, d—p < 0.01 compared to AlCl3 treated rats, e—p < 0.05 compared to 20th day of treatment, f—p < 0.01 compared to 20th day of treatment.
HGN Attenuates AlCl3 Triggered Al Loading Quantum and AChE Levels
Previous investigation strongly advocates possible involvement of Al in AD, which can be observed as higher Al quantum in the hippocampal and cortical region of brains in humans as well as experimental animals.38 Even though Al accumulation sites predominantly are the forebrain, cerebellum, and brainstem, but the most affected and susceptible Al affected areas include cortical and hippocampal regions, which are important for learning and memory responses. Acetylcholine and the corresponding cholinergic neurons are strongly associated with learning, memory, displacement, and control of blood flow to the brain.39 Al is responsible for the deterioration of memory by weakening cholinergic function. The acetylcholinesterase (AChE) enzyme is involved in the degradation of acetylcholine, whereas choline acetyltransferase is accountable for generation of acetylcholine.40 Measurement of activity of both these enzymes could be employed for investigation of memory impairment caused due to cholinergic functionality. Hippocampal and cortical AChE activity and Al concentration were significantly increased in rats treated with AlCl3, compared to control group animals. Co-treatment of HGN (25, 50, and 75 mg/kg) and AlCl3 revealed marked reduction in the AChE activity and Al quantum in the hippocampus and cortex compared to those treated with AlCl3 alone. However, there was no significant variability in attenuation of AChE activity and Al concentration on treatment with 50 mg/kg and 75 mg/kg HGN, but highly influential compared to 25 mg/kg HGN administration (Figure 4 and Figure 5). Considering the least significant difference between effects of 50 mg/kg and 75 mg/kg dose of HGN, for further investigation, we had considered HGN 50 mg/kg as optimal dose.
Figure 4.
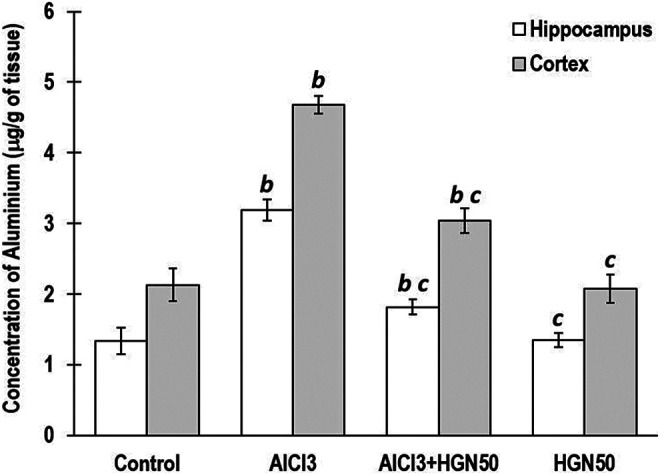
AlCl3 animals exhibited enhanced levels of Al in hippocampus and cortex. Cotreatment of HGN (AlCl3+50 mg/kg HGN) attenuated the AlCl3 mediated Al burden. Data are expressed as mean ± SEM (1-way ANOVA followed by Duncan’s multiple range test) for 6 rats in each group. a—p < 0.05 compared to the control, b—p < 0.01 compared to the control, c—p < 0.05 compared to the AlCl3 treated rats, d—p < 0.01 compared to the AlCl3 treated rats.
Figure 5.
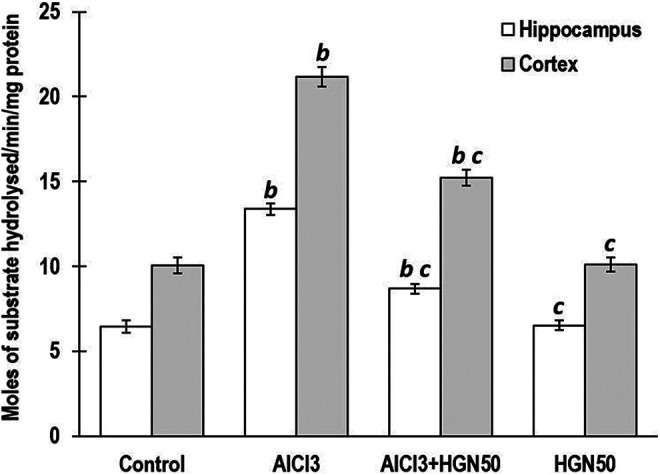
AlCl3 group showed significantly enhanced AchE activity in hippocampus and cortex. HGN50 (AlCl3+50 mg/kg HGN)co-treatment significantly attenuated the AchE hyperactivity in both regions of the brain. HGN50 (AlCl3+50 mg/kg HGN) treatment showed a reduction in Al levels and AChE activity, but more significant than HGN50. Data are expressed as mean ± SEM (1-way ANOVA followed by Duncan’s multiple range test) for 6 rats in each group. a—p < 0.05 compared to the control, b—p < 0.01 compared to the control, c—p < 0.05 compared to the AlCl3 treated rats, d—p < 0.01 compared to the AlCl3 treated rats.
HGN Abolishes AlCl3 Triggered Apoptosis and Aβ Biosynthesis
Observations collected from the Western blotting to analyze the expression of proteins related to Aβ biosynthesis and apoptosis in the hippocampal and cortical regions are indicated in Figure 6. The Aβ levels are enhanced in the brain of experimental animals exposed to Al, either directly by increasing its biosynthesis or indirectly by reducing its breakdown.41 In comparison to the control group rats, long-term AlCl3 administration revealed higher expression of APP, Aβ1-42, β- and γ-secretases, that may aid in developing of Aβ plaque. However, HGN (50 mg/kg) treatment to AlCl3 exposed rats indicated a significant reduction in APP, Aβ1-42, β- and γ-secretases expression (Figure 6).
Figure 6.

AlCl3 treatment significantly enhanced the protein expressions of APP, Aβ1-42, β and γ-secretases and favors amyloid biosynthesis. Coadministration of HGN50 (AlCl3+50 mg/kg HGN) attenuated the AlCl3 mediated amyloid biosynthesis. (a) Representative Western blots, (b) Corresponding graph of relative Western blot intensity in the hippocampus and (c) Corresponding graph of relative Western blot intensity in the cortex. Data are expressed as mean ± SEM (1-way ANOVA followed by Duncan’s multiple range test) for 3 rats in each group. a—p < 0.05 compared to the control, b—p < 0.01 compared to the control, c—p < 0.05 compared to the AlCl3 treated rats, d—p < 0.01 compared to the AlCl3 treated rats.
The most prominent cell extinction mechanism in neurodegenerative disorders such as Parkinson’s disease and AD is apoptosis.42 A report revealed that Al exposure essentially triggers apoptosis of brain cells by upregulating the Bax and caspase expression and downregulating the Bcl-2 expression.32 Treatment of rats with AlCl3 for 42 days markedly enhanced the expressions of Bax, Bad, cyto-c and caspase-9 in the mitochondrial portion, and downregulated the expressions of Bcl-2, Bcl-xL, and cyto-c in the cytosolic part, collected from hippocampal and cortical regions. Treatment with HGN (mg/kg) attenuated the alteration of the protein expression triggered by AlCl3 in experimental rats. Negligible intergroup variability was observed in the pro-caspase-3 expression in the hippocampus and cortex brain areas. However, caspase-3, which is an activated derivative of caspase-3, is least expressed in control group animals but is significantly enhanced in Al-treated animals. HGN treatment exhibits a marked reduction in the pro-caspase-3 breakdown to active caspase-3 (Figure 7).
Figure 7.

Chronic treatment of AlCl3 significantly increased the protein expressions of Bax, Bad, cyto c, caspases -9 and cyto c (mitochondrial fraction) and decreased the expressions of Bcl-2, Bcl-xL and cyto c (cytosolic fraction) in the hippocampus and cortex and favors apoptosis. HGN50 (AlCl3+50 mg/kg HGN) supplementation attenuated the AlCl3 induced apoptosis. (a) and (b) Representative Western blots, (c) and (e), Corresponding graph of relative Western blot intensity in the hippocampus and (d) and (f) Corresponding graph of relative Western blot intensity in the cortex. No significant changes in the expressions of pro-caspase-3 were found in control and experimental groups. The activated caspase-3 expression is enhanced following aluminium treatment and inhibited by the HGN50 co-treatment, which further proves the antiapoptotic property of HGN. Data are expressed as mean ± SEM (1-way ANOVA followed by Duncan’s multiple range test) for 3 rats in each group. a—p < 0.05 compared to the control, b—p < 0.01 compared to the control, c—p < 0.05 compared to the AlCl3 treated rats, d—p < 0.01 compared to the AlCl3 treated rats.
HGN Normalizes Disturbed Akt/pGSK-3β Signaling Markers Caused by AlCl3
Al potentially results in apoptosis by modulation of a series of signalling cascades. Exposure to Al leads to dephosphorylation, Akt inactivation, and triggering of pro-apoptotic mediators such as Bad. Also, Al results in dephosphorylation and stimulation of GSK-3β, known to regulate apoptosis. Activation of GSK-3β and lowering of Akt activity is correlated with depolarization and permeabilization of mitochondria, coupled with activation of caspase-3 and release of cytochrome-c. Al exposure results in apoptosis by downregulation of Akt signalling cascade. In the present investigations, rats exposed to AlCl3, revealed significant lowering of p-Akt and p-GSK-3β expression in the hippocampal and cortical tissues, whereas HGN (50 mg/kg) co-treatment markedly enhanced their expressions. Compared to the control group, the expression of p-Akt, GSK-3β, and p-GSK-3β did not indicate any significant alteration when HGM was administered alone (Figure 8). These results strongly advocate the neuroprotective efficacy of HGM (50 mg/kg) via normalizing the AlCl3 induced altered expression of p-Akt and p-GSK-3β.
Figure 8.

AlCl3 rats exhibited significantly lowered the expressions of pAkt and pGSK-3β in hippocampus and cortex. Western blot studies indicated that their expressions were significantly attenuated by co-treatment with HGN50 (AlCl3+50 mg/kg HGN). (a) Representative Western blots, (b) Corresponding graph of relative Western blot intensity in the hippocampus and (c) Corresponding graph of relative Western blot intensity in the cortex. Data are expressed as mean ± SEM (1-way ANOVA followed by Duncan’s multiple range test) for 3 rats in each group. a—p < 0.05 compared to the control, b—p < 0.01 compared to the control, c—p < 0.05 compared to the AlCl3 treated rats, d—p < 0.01 compared to the AlCl3 treated rats.
HGN Confers Protection Against Oxidative Stress
Oxidative stress worsens cognitive impairment in AD. The study presented herewith reveals a marked reduction of oxidative stress via modulation of the investigated markers- ROS, MDA, SOD and GPx. Compared to the control group animals, MDA and ROS production in AlCl3 induced neurotoxicity was significantly increased. HGN administration lowered the MDA and ROS levels significantly compared to the animals exposed to AlCl3 exposure. Furthermore, AlCl3 exposure revealed a significant reduction of SOD and GPx activity, which was normalized by chronic HGN treatment (Figure 9).
Figure 9.

AlCl3 rats exhibited significantly elevated levels of ROS and MDA, and reduced the levels of SOD and GPx in hippocampus and cortex. Administration of HGN50 (AlCl3+50 mg/kg HGN) normalized the levels of oxidative markers. ROS, MDA, SOD and GPx levels were quantified in (a) hippocampus and (b) cortex using commercially available detection kits. Data are expressed as mean ± SEM (1-way ANOVA followed by Duncan’s multiple range test) for 3 rats in each group. a—p < 0.05 compared to the control, b—p < 0.01 compared to the control, c—p < 0.05 compared to the AlCl3 treated rats, d—p < 0.01 compared to the AlCl3 treated rats.
Discussion
Previous investigation results indicating increased levels of Al in hippocampal and cortical regions caused by ingestion of AlCl3 dissolved in distilled water, are in confirmation with that obtained in the present study.13 HGN markedly lowered Al level in the tissue homogenates collected from hippocampal and cortical brain areas. Reduction in the Al quantum after HGN administration in rats may be attributed to the binding capacity of HGN to Al, which needs more in-depth investigation.
Chronic AlCl3 administration revealed a significant reduction in the contextual memory indicated through spatial memory test and passive avoidance test. The spatial memory test was conducted using Morris maze, wherein animals treated with AlCl3 alone revealed less tendency to recall and retain the hidden platform’s position, even after being trained for several days. HGN co-administration reverted the AlCl3 induced memory deficiency, indicating enhancement of memorizing capacity of HGN. The passive avoidance test results suggest that AlCl3 treated rats were unable to recall the unpleasant stimuli and hence entered the dark compartment quickly wherein they experienced electric shock earlier, compared to the control group animals. Animals co-treated with HGN and AlCl3 indicated delay to promptly enter the dark area wherein they already had experienced electric shock. These findings demonstrated that HGN enhanced the recalling capacity of animals even though they were exposed to AlCl3.
The primary cholinesterase enzyme, responsible for the breakdown of acetylcholine choline, is acetylcholinesterase (AChE). The enzyme’s functionality ensures that acetylcholine’s effect subsides after activation and comes to the resting stage. Around 25000 acetylcholine molecules/second are degraded by one AChE molecule in neuronal and non-neuronal tissues.43 Also, the activity of ACheE is vulnerable to external considerations such as diet and the presence of Al.44 In accordance with the previous investigations, the present study also indicated that on the administration of AlCl3 in rats, there is a marked elevation in the activity of AChE.13,45 Modification of AChE structure has been reported by binding with Al ions, thereby increasing its activity.46 Administration of HGN in AlCl3 induced AD rat model revealed a neuroprotective effect by downregulation of the AChE effect. Reduction in the AChE potential elevates the level of acetylcholine and confers favorable effects on cognitive functionality.47
AD represents complex pathophysiology encompassing τ-pathology, Aβ accumulation, oxidative burden, loss of mitochondrial and proteasome functionality, inflammation, and damage to the cholinergic neurons due to interaction of Aβ and metal.48 Drugs that could provoke cholinergic functionality, such as rivastigmine and galantamine, could be reasonably useful at the initial AD stage. Parallel strategies apart from cholinergic targets could also be adopted for the treatment of AD, such as metal chelation, growth factors, hormones, non-steroidal anti-inflammatory drugs, antioxidants, anti-hyperlipidaemic drugs, phosphodiesterase inhibiting drugs, Vitamin B6, B12, E, and folic acid, anti-Aβ therapies, and drugs targeting neurotransmitters or selective neuropeptide. Thus, improvement of memory may be attributed to AChE inhibitory effect of HGN.
Initial stages of AD are marked with abundant deposition of Ab that further progresses to formation of neuritic amyloid plaques. Subsequent breakdown of APP occurs by several protease enzymes, including β and γ-secretases. Enzyme β-secretase trims APP and generates APP C-terminal fraction, which is further broken down in the transmembrane region by γ-secretase into Aβ1-40 and Aβ1-42 C-terminal fractions.49,50 Amyloid plaques predominantly exist as Aβ1-42 moiety, and the vascular amyloid consists of Aβ1-40 fraction. AlCl3 treated rats revealed increased amyloid generation indicated by enhanced activity of markers related to the biosynthesis of APP, Aβ1-42, β- and γ- secretases.49 Previous studies have claimed markedly higher expression of Aβ1-42 in the cortical and hippocampal regions, whereas unaltered Aβ1-40 expression.51 Hence, focusing Aβ generation and its production cascade could be regarded as a crucial therapeutic approach in AD therapy.52 HGN remarkably reduced the amyloid generation by attenuating the APP, Aβ1-42, β- and γ-secretases, that could progressively reduce the Aβ biosynthesis.
Apoptosis triggered by Al exposure in the hippocampal and cortical areas results from inhibition of anti-apoptotic proteins and stimulation of pro-apoptotic proteins.53 Anti-apoptotic markers include Bcl-2 and Bcl-xL, whereas the pro-apoptotic markers include Bax, Bak, and Bad, which regulate the innate apoptotic cascade involving mitochondria. A critical harmony between the pre-apoptotic and the anti-apoptotic proteins ensures the continuity or termination of cells. Cytochrome-c release is inhibited by Bcl-2 that leads to inhibition of apoptosis, and Bax leads to apoptosis by attenuating anti-apoptotic protein Bcl-2, which subsequently enhances the release of cytochrome-c. Cytochrome-c further stimulates caspases, that ultimately induces cell death. Caspases belong to a class of proteases that provoke apoptosis, inflammation, and resulting necrosis. In the apoptotic process, caspase-9 functions as an initiator, and caspase-3 acts as an effector, and together they induce apoptosis. Our findings revealed that AlCl3 exposure dramatically upregulated the activity of pro-apoptotic markers (Bad, Bax, caspase-3, and caspase-9), and downregulated the activity of anti-apoptotic markers (Bcl-2 and Bcl-xL). Co-administration of HGN and AlCl3 significantly prevented the apoptosis induced by AlCl3 alone by attenuation of the activity of Bax, caspase-3, caspase-9, and cytosolic cyto-c, and inhibited the mitochondrial cyto-c release coupled with enhancing the Bcl-2 expression.
A crucial kinase signaling cascade regulates cell regulatory processes and apoptosis. Neuronal apoptosis is coordinated by following signaling cascades, c-Jun N-terminal kinase (JNK), protein kinase B (Akt), and glycogen synthase kinase-3 (GSK-3).54-56 Stimulation of Akt signaling cascade influences cell proliferation in several types of neurons, whereas its suppression induces the death of neurons.57-59 In the present report, AlCl3 alone attenuated the pAkt expression, but when co-administered with HGN, the pAkt expression is stimulated. The active version of Akt is pAkt, which has been known to cease the apoptosis process by modulation of Bcl-2 representatives involving Bad, caspase-9 and GSK-3β.60- 62 Stimulation of Akt results in attenuation of GSK-3β via Ser9 phosphorylation.63 One of the predominant τ-kinase, GSK-3β, is known to participate in the τ-protein phosphorylation that further leads to the formation of neurofibrillary tangles and amyloid plaques in the course of AD.64 The performance of GSK-3β is influenced by Ser9 and Tyr216 phosphorylation.65,66 The phosphorylation of Tyr216 triggers the activity, whereas phosphorylation of Ser9 attenuates its activity.20 In the present investigation, the results indicate that AlCl3 administration reduced the Ser9 pGSK-3β expression, eventually augmenting the GSK-3β kinase expression and increasing the τ-phosphorylation, which is similar to earlier recommendations. HGN (50 mg/kg) administration in AlCl3 rats indicated significantly higher Ser9 pGSK-3β expression, which results in attenuation of GSK-3β kinase activity and high t-phosphorylation.
During neurodegenerative events such as AD, a large number of free oxygen radicals are produced. These free oxygen radicals or oxidants lead to oxidative stress and thereby result in neuronal death.67 Hence oxidative stress has been regarded as a critical factor in the progression of AD. During stressful situations and the aging process that leads to AD, the ROS generation is elevated in mitochondria. Loss of mitochondrial functionality and oxidative stress both contribute to neurodegenerative diseases such as AD.68,69 Enzyme SOD triggers the dismutation of oxygen and generate hydrogen peroxide. Glutathione peroxidase (GPx) can decompose free oxygen and hydroxyl radical, thereby reducing oxidative stress.70 Polyunsaturated fatty acids under lipid peroxidation and produce an organic compound MDA.71 Generation of MDA and ROS, and SOD and GPx levels are prominent markers used to as oxidative stress markers.72,73 AlCl3 alone significantly enhanced the MDA and ROS production compared to the animals from the control group. Co-administration of HGN and AlCl3 reduced the MDA and SOD levels significantly in rats. SOD and GPx levels were markedly reduced after exposure to AlCl3, whereas HGN-AlCl3 co-administration normalized the SOD and GPx levels, similar to those in the control group. The findings advocated convincing activity of HGN against AlCl3 induced oxidative stress in rats. Altogether, HGN (50 mg/kg) assisted in attenuating the devastative effects of Al-induced AD-like symptoms, related to cognitive impairment and loss of memory, and profoundly influenced molecular mechanisms involved in the severity of AD.
Conclusion
Our findings exhibited that HGN could normalize memory and learning affected by Al exposure in rats. HGN also modulates the AChE activity and Al burden, and Aβ load and apoptosis through triggering of Akt/GSK-3β signaling cascade, and attenuation of oxidative stress in AlCl3 administered rats.
Footnotes
Declaration of Conflicting Interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The author(s) received no financial support for the research, authorship, and/or publication of this article.
ORCID iD: Xiaona Yang  https://orcid.org/0000-0002-0462-0253
https://orcid.org/0000-0002-0462-0253
References
- 1. Mani V, Parle M. Memory enhancing activity of Coriandrum sativum in rats. Pharmacologyonline. 2009;2:827–839. [Google Scholar]
- 2. Walton J. Cognitive deterioration and associated pathology induced by chronic low-level aluminum ingestion in a translational rat model provides an explanation of Alzheimer’s disease, tests for susceptibility and avenues for treatment. Int J Alzheimers Dis. 2012;2012:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Humpel C. Chronic mild cerebrovascular dysfunction as a cause for Alzheimer’s disease. Exp Gerontol. 2011;46(4):225–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Justin Thenmozhi A, William Raja TR, Manivasagam T, Janakiraman U, Essa MM. Hesperidin ameliorates cognitive dysfunction, oxidative stress and apoptosis against aluminium chloride induced rat model of Alzheimer’s disease. Nutr Neurosci. 2017;20(6):360–368. [DOI] [PubMed] [Google Scholar]
- 5. Li X, Zhao X, Xu X, et al. Schisantherin A recovers Aβ-induced neurodegeneration with cognitive decline in mice. Physiol Behav. 2014;132:10–16. [DOI] [PubMed] [Google Scholar]
- 6. Yang H, Wang S, Yu L, Zhu X, Xu Y. Esculentoside A suppresses Aβ (1-42)-induced neuroinflammation by down-regulating MAPKs pathways in vivo. Neurol Res. 2015;37(10):859–866. [DOI] [PubMed] [Google Scholar]
- 7. Lazzari C, Kipanyula MJ, Agostini M, Pozzan T, Fasolato C. Aβ42 oligomers selectively disrupt neuronal calcium release. Neurobiol Aging. 2015;36(2):877–885. [DOI] [PubMed] [Google Scholar]
- 8. Pozueta J, Lefort R, Shelanski ML. Synaptic changes in Alzheimer’s disease and its models. Neuroscience. 2013;251:51–65. [DOI] [PubMed] [Google Scholar]
- 9. Varga E, Juhász G, Bozsó Z, Penke B, Fülöp L, Szegedi V. Amyloid-β1-42 disrupts synaptic plasticity by altering glutamate recycling at the synapse. J Alzheimers Dis. 2015;45(2):449–456. [DOI] [PubMed] [Google Scholar]
- 10. Viola KL, Klein WL. Amyloid β oligomers in Alzheimer’s disease pathogenesis, treatment, and diagnosis. Acta Neuropathol. 2015;129(2):183–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sharma P, Mishra KP. Aluminum-induced maternal and developmental toxicity and oxidative stress in rat brain: response to combined administration of tiron and glutathione. Reprod Toxicol. 2006;21(3):313–321. [DOI] [PubMed] [Google Scholar]
- 12. Al-Hashem F. Camel’s milk protects against aluminum chloride-induced toxicity in the liver and kidney of white albino rats. Am J Biochem Biotechnol. 2009;5(3):98–108. [Google Scholar]
- 13. Thenmozhi A, Raja T, Janakiraman U, Manivasagam T. Neuroprotective effect of hesperidin on aluminium chloride induced Alzheimer’s disease in Wistar rats. Neurochem Res. 2015;40(4):767–776. [DOI] [PubMed] [Google Scholar]
- 14. Justin Thenmozhi A, Dhivyabharathi M, William Raja T, Manivasagam T, Essa MM. Tannoid principles of Emblica officinalis renovate cognitive deficits and attenuate amyloid pathologies against aluminum chloride induced rat model of Alzheimer’s disease. Nutr Neurosci. 2016:19(6):269–278. [DOI] [PubMed] [Google Scholar]
- 15. Muirhead KE, Borger E, Aitken L, Conway SJ, Gunn-Moore FJ. The consequences of mitochondrial amyloid β-peptide in Alzheimer’s disease. Biochem J. 2010;426(3):255–270. [DOI] [PubMed] [Google Scholar]
- 16. Bai B, Hales CM, Chen PC, et al. U1 small nuclear ribonucleoprotein complex and RNA splicing alterations in Alzheimer’s disease. Proc Natl Acad Sci USA. 2013;110(41):16562–16567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Modi PK, Komaravelli N, Singh N, Sharma P. Interplay between MEK-ERK signalling, cyclin D1 and cyclin-dependent kinase 5 regulates cell cycle reentry and apoptosis of neurons. Mol Biol Cell. 2012;23(18):3722–3730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Liang J, Liu L, Xing D. Photobiomodulation by low-power laser irradiation attenuates Aβ induced cell apoptosis through the Akt/GSK3β/β-catenin pathway. Free Radic Biol Med. 2012;53(7):1459–1467. [DOI] [PubMed] [Google Scholar]
- 19. Garrido JL, Godoy JA, Alvarez A, Bronfman M, Inestrosa NC. Protein kinase C inhibits amyloid beta peptide neurotoxicity by acting on members of the Wnt pathway. FASEB J. 2002;16(14):1982–1984. [DOI] [PubMed] [Google Scholar]
- 20. Zhang H, Yang X, Qin X, Niu Q. Caspase-3 is involved in aluminum-induced impairment of long-term potentiation in rats through the Akt/GSK-3β pathway. Neurotox Res. 2016;29(4):484–494. [DOI] [PubMed] [Google Scholar]
- 21. Kosuge T, Yokota M. Letter: studies on cardiac principle of aconite root. Chem Pharm Bull (Tokyo). 1976;24(1):176–178. [DOI] [PubMed] [Google Scholar]
- 22. Kosuge TYM. Cardioactive principle of Aconitum japonicum Thunb. Chem Pharm Bull. 1976;24(8):1768–1772. [Google Scholar]
- 23. Singhuber J, Zhu M, Prinz S, Kopp B. Aconitum in traditional Chinese medicine: a valuable drug or an unpredictable risk. J Ethnopharmacol. 2009;126(1):18–30. [DOI] [PubMed] [Google Scholar]
- 24. Zhanga N, Liana Z, Penga X, Li Z, Zhu H. Applications of Higenamine in pharmacology and medicine. J Ethnopharmacol. 2017;196(1):242–252. [DOI] [PubMed] [Google Scholar]
- 25. Wu M, Zhang Y, Zhou Q, Xiong J, Dong YR, Yan C. Higenamine protects ischemia/reperfusion induced cardiac injury and myocyte apoptosis through activation of 2-AR/PI3K/AKT signaling pathway. Pharmacol Res. 2016;104:115–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhang Y, Zhang J, Wu C, et al. Higenamine protects neuronal cells from oxygen-glucose deprivation/reoxygenation-induced injury. J Cell Biochem. 2019;120(3):3757–3764. [DOI] [PubMed] [Google Scholar]
- 27. Lee Y, Kang Y, Kim H, et al. Higenamine reduces apoptotic cell death by induction of heme oxygenase-1 in rat myocardial ischemia-reperfusion injury. Apoptosis. 2006;11(7):1091–1100. [DOI] [PubMed] [Google Scholar]
- 28. Ha Y, Kim M, Park M, et al. Higenamine reduces HMGB1 during hypoxia-induced brain injury by induction of heme oxygenase-1 through PI3K/Akt/Nrf-2 signal pathways. Apoptosis. 2012;17(5):463–474. [DOI] [PubMed] [Google Scholar]
- 29. Prakash A, Kumar A. Mitoprotective effect of Centella asiatica against aluminum-induced neurotoxicity in rats: possible relevance to its anti-oxidant and anti-apoptosis mechanism. Neurol Sci. 2013;34(8):1403–1409. [DOI] [PubMed] [Google Scholar]
- 30. Kumar A, Dogra S, Prakash A. Protective effect of curcumin (Curcuma longa), against aluminum toxicity: possible behavioral and biochemical alterations in rats. Behav Brain Res. 2009;205(2):384–390. [DOI] [PubMed] [Google Scholar]
- 31. Prema A, Thenmozhi A, Manivasagam T, Essa M, Akbar M, Akbar M. Fenugreek seed powder nullified aluminium chloride induced memory loss, biochemical changes, Aβ burden and apoptosis via regulating Akt/GSK3β signaling pathway. PLoS One. 2016;11(11):e0165955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mathiyazahan D, Justin Thenmozhi A, Manivasagam T. Protective effect of black tea extract against aluminium chloride-induced Alzheimer’s disease in rats: a behavioural, biochemical and molecular approach. J Funct Foods. 2015;16:423–435. [Google Scholar]
- 33. Janakiraman U, Manivasagam T, Justin Thenmozhi A, et al. Influences of chronic mild stress exposure on motor, non-motor impairments and neurochemical variables in specific brain areas of MPTP/probenecid induced neurotoxicity in mice. PLoS One. 2016;11:e0146671. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 34. Dhanalakshmi C, Janakiraman U, Manivasagam T, et al. Vanillin attenuated behavioural impairments, neurochemical deficits, oxidative stress and apoptosis against rotenone induced rat model of Parkinson’s disease. Neurochem Res. 2016;41(8):1899–1910. [DOI] [PubMed] [Google Scholar]
- 35. Heise GA. Behavioral methods for measuring effects of drugs on learning and memory in animals. Med Res Rev. 1984;4(4):535–558. [DOI] [PubMed] [Google Scholar]
- 36. Baydar T, Papp A, Aydin A, et al. Accumulation of aluminum in rat brain: does it lead to behavioral and electrophysiological changes? Biol Trace Elem Res. 2003;92(3):231–244. [DOI] [PubMed] [Google Scholar]
- 37. West MJ. Regionally specific loss of neurons in the aging human hippocampus. Neurobiol Aging. 1993;14(4):287–293. [DOI] [PubMed] [Google Scholar]
- 38. Jelenkovic A, Jovanovic MD, Stevanovic I, et al. Influence of the green tea leaf extract on neurotoxicity of aluminium chloride in rats. Phytother Res. 2014;28(1):82–87. [DOI] [PubMed] [Google Scholar]
- 39. Mesulam MM, Guillozet A, Shaw P, Levey A, Duysen EG, Lockridge O. Acetylcholinesterase knockouts establish central cholinergic pathways and can use butyrylcholinesterase to hydrolyse acetylcholine. Neuroscience. 2002;110(4):627–639. [DOI] [PubMed] [Google Scholar]
- 40. Yellamma K, Saraswathamma S, Kumar BN. Cholinergic system under aluminium toxicity in rat brain. Toxicol Int. 2010;17(2):106–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Clauberg M, Joshi JG. . Regulation of serine protease activity by Al: implications for Alzheimer disease. Proc Natl Acad Sci USA. 1993;90(3):1009–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Marx J. Neuroscience. New leads on the “how’ of Alzheimer’s”. Science. 2001;293(5538):2192–2194. [DOI] [PubMed] [Google Scholar]
- 43. Taylor P, Radic Z. The cholinesterases: from genes to proteins. Annu Rev Pharmacol Toxicol. 1994;34(1):281–320. [DOI] [PubMed] [Google Scholar]
- 44. Kaizer RR, da Silva AC, Morsch VM, Corrêa MC, Schetinger MR. Diet-induced changes in AChE activity after long-term exposure. Neurochem Res. 2004;29(12):2251–2255. [DOI] [PubMed] [Google Scholar]
- 45. Kumar A, Dogra S, Prakash A. Effect of carvedilol on behavioral, mitochondrial dysfunction and oxidative damage against D-galactose induced senescence in mice. Naunyn Schmiedebergs Arch Pharmacol. 2009;380(5):431–441. [DOI] [PubMed] [Google Scholar]
- 46. Zatta P, Zambenedetti P, Bruna V, Filippi B. Activation of acetylcholinesterase by aluminium (III): the relevance of the metal species. Neuroreport. 1994;5(14):1777–1780. [DOI] [PubMed] [Google Scholar]
- 47. Giacobini E, Spiegel R, Enz A, Veroff AE, Cutler NR. Inhibition of acetyl- and butyryl-cholinesterase in the cerebrospinal fluid of patients with Alzheimer’s disease by rivastigmine: correlation with cognitive benefit. J Neural Transm. 2002;109(7-8):1053–1065. [DOI] [PubMed] [Google Scholar]
- 48. Carreiras MC, Mendes E, Perry MJ, Francisco AP, Marco-Contelles J. The multifactorial nature of Alzheimer’s disease for developing potential therapeutics. Curr Top Med Chem. 2013;13(15):1745–1770. [DOI] [PubMed] [Google Scholar]
- 49. Jarrett JT, Berger EP, Lansbury PT. The C-terminus of the β protein is critical in amyloidogenesis. Ann N Y Acad Sci. 1993;695(1):144–148. [DOI] [PubMed] [Google Scholar]
- 50. Tamagno E, Parola M, Bardini P, et al. β-site APP cleaving enzyme up-regulation induced by 4-hydroxynonenal is mediated by stress-activated protein kinases pathways. J Neurochem. 2005;92(3):628–636. [DOI] [PubMed] [Google Scholar]
- 51. Wang L, Hu J, Zhao Y, Lu X, Zhang Q, Niu Q. Effects of aluminium on β-Amyloid1-42 and secretases (APP-Cleaving Enzymes) in rat brain. Neurochem Res. 2014;39(7):1338–1345. [DOI] [PubMed] [Google Scholar]
- 52. Roberson ED, Mucke L. 100 years and counting: prospects for defeating Alzheimer’s disease. Science. 2006;314(5800):781–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Chaudhary M, Joshi DK, Tripathi S, Kulshrestha S, Mahdi AA. Docosahexaenoic acid ameliorates aluminum induced biochemical and morphological alteration in rat cerebellum. Ann Neurosci. 2014;21(1):5–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Brunet A, Datta SR, Greenberg ME. Transcription-dependent and -independent control of neuronal survival by the PI3K-Akt signaling pathway. Curr Opin Neurobiol. 2001;11(3):297–305. [DOI] [PubMed] [Google Scholar]
- 55. Beurel E, Jope RS. The paradoxical pro- and anti-apoptotic actions of GSK3 in the intrinsic and extrinsic apoptosis signaling pathways. Prog Neurobiol. 2006;79(4):173–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Borsello T, Forloni G. JNK signalling. A possible target to prevent neurodegeneration. Curr Pharm Des. 2007;13(18):1875–1886. [DOI] [PubMed] [Google Scholar]
- 57. Namikawa K, Honma M, Abe K, et al. Akt/protein kinase B prevents injury-induced motor neuron death and accelerates axonal regeneration. J Neurosci. 2000;20(8):2875–2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Zhao H, Sapolsky RM, Steinberg GK. Phosphoinositide-3-kinase/Akt survival signal pathways are implicated in neuronal survival after stroke. Mol Neurobiol. 2006;34(3):249–270. [DOI] [PubMed] [Google Scholar]
- 59. Ji L, Chauhan A, Wegiel J, Essa MM, Chauhan V. Gelsolin is proteolytically cleaved in the brains of individuals with Alzheimer’s disease. J Alzheimers Dis. 2009;18(1):105–111. [DOI] [PubMed] [Google Scholar]
- 60. Datta SR, Dudek H, Tao X, et al. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91(2):231–241. [DOI] [PubMed] [Google Scholar]
- 61. Cardone MH, Roy N, Stennicke HR, et al. Regulation of cell death protease caspase-9 by phosphorylation. Science. 1998;282(5392):1318–1321. [DOI] [PubMed] [Google Scholar]
- 62. Pap M, Cooper GM. Role of glycogen synthase kinase-3 in the phosphatidylinositol 3-Kinase/Akt cell survival pathway. J Biol Chem. 1998;273(32):19929–19932. [DOI] [PubMed] [Google Scholar]
- 63. Hu YS, Long N, Pigino G, Brady ST, Lazarov O. Molecular mechanisms of environmental enrichment: Impairments in AKT/GSK3β, neurotrophin-3 and CREB signaling. PLoS One. 2013;8(5):e64460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Takashima A. GSK-3 is essential in the pathogenesis of Alzheimer’s disease. J Alzheimers Dis. 2006;9(s3):309–317. [DOI] [PubMed] [Google Scholar]
- 65. Sutherland C, Leighton IA, Cohen P. Inactivation of glycogen synthase kinase-3 by phosphorylation: new kinase connections in insulin and growth-factor signaling. Biochem J. 1993;296(1):15–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Wang QM, Fiol CJ, DePaoli-Roach AA, Roach PJ. Glycogen synthase kinase-3 is a dual specificity kinase differentially regulated by tyrosine and serine/threonine phosphorylation. J Biol Chem. 1994;269:14566–14574. [PubMed] [Google Scholar]
- 67. Coyle JT, Puttfarcken P. Oxidative stress, glutamate, and neurodegenerative disorders. Science. 1993;262(5134):689–695. [DOI] [PubMed] [Google Scholar]
- 68. Chauhan V, Chauhan A. Oxidative stress in Alzheimer’s Disease. Pathophysiology. 2006;13(3):195–208. [DOI] [PubMed] [Google Scholar]
- 69. Di CM, Giacomazza D, Picone P, Nuzzo D, San Biagio PL. Are oxidative stress and mitochondrial dysfunction the key players in the neurodegerative diseases? Free Radic Res. 2012;46(11):1327–1338. [DOI] [PubMed] [Google Scholar]
- 70. Akbas A, Inanir A, Benli I, Onder Y, Aydogan L. Evaluation of some antioxidant enzyme activities (SOD and GPX) and their polymorphisms (MnSOD2 Ala9Val, GPX1 Pro198Leu) in fibromyalgia. Eur Rev Med Pharmacol Sci. 2014;18(8):1199–1203. [PubMed] [Google Scholar]
- 71. Kowalczuk K, Stryjecka-Zimmer M. The influence of oxidative stress on the level of malondialdehyde (MDA) in different areas of the rabbit brain. Ann Univ Mariae Curie Sklodowska Med. 2002;57(2):160–164. [PubMed] [Google Scholar]
- 72. Engin KN, Yemişci B, Yiğit U, Ağaçhan A, Coşkun C. Variability of serum oxidative stress biomarkers relative to biochemical data and clinical parameters of glaucoma patients. Mol Vis. 2010;16(7):1260–1271. [PMC free article] [PubMed] [Google Scholar]
- 73. Khoubnasab Jafari M, Ansarin K, Jouyban A. Comments on “Use of malondialdehyde as a biomarker for assesing oxidative stress in different disease pathologies: a review.” Iran J Public Health. 2015;44(5):714–715. [PMC free article] [PubMed] [Google Scholar]