

Abstract
Gulf War Illness (GWI) refers to a multi-system disorder that afflicts approximately 30% of First Gulf War (GW) veterans. Amongst the symptoms exhibited, mood and memory impairment are commonly reported by GW veterans. Exposure to organophosphate (OP) compounds which target the cholinergic system is considered a leading cause for GWI symptoms. It is hypothesized that chronic OP-based war-time stimulation of cholinergic signaling led to recruitment of excitatory glutamatergic signaling and other downstream signaling cascades leading to neuronal injury, neuroinflammation, generation of reactive oxygen species, oxidative stress, and mitochondrial damage within the central nervous system. These findings have been observed in both experimental models and GWI veterans. In this context the role of calcium (Ca2+) signaling in GWI has come to the forefront. Here we present our Ca2+ hypothesis of GWI that suggests sustained neuronal Ca2+ elevations serve as a molecular trigger for pathological synaptic plasticity that has allowed for the persistence of GWI symptoms. Subsequently we discuss that therapeutic targeting of Ca2+ homeostatic mechanisms provides novel targets for effective treatment of GWI-related neurological signs in our rodent model.
Keywords: Organophosphates, DFP, depression, calcium dynamics, levetiracetam, ketamine
Gulf War Illness
In early 1991, the United States led an international coalition of forces against Iraqi occupation of Kuwait. Code-named Operation Desert Storm, the U.S. and Allied forces claimed victory with the liberation of Kuwait after only 4 days of ground and air combat. However, the troop buildup and subsequent withdrawal from the War theatre lasted over a 4-month period. Of the 700 000 troops deployed, there were less than 250 causalities. For all intents and purposes, the First Gulf War was considered a success. Yet, upon their return stateside, Gulf War (GW) veterans reported of a range of complaints. The constellation of symptoms included fatigue, headache, memory and attention problems, muscle and joint pain, and gastrointestinal distress. These chronic and debilitating symptoms could not be explained by other medical or psychiatric diagnoses. In 2014, the Institute of Medicine declared this chronic multi-symptom illness to be called Gulf War Illness (GWI).1 Today, approximately one-third of GW veterans are estimated to suffer from GWI.2
The Institute of Medicine recommends 2 case definitions for diagnosing GWI.1-3 The CDC definition is more commonly used clinically and for epidemiological research. To meet the criteria for this definition, a GW veteran must experience at least 1 symptom for more than 6 months in 2 or more of the following 3 categories: (1) Fatigue, (2) Mood and cognitive dysfunction, and (3) Musculoskeletal problems including pain. The Kansas definition, a more restrictive definition recommended for use in research, requires that a GW veteran experience symptom in 3 of the following 6 domains: (1) Pain, (2) Cognitive and mood symptoms, (3) Fatigue and sleep problems, (4) Gastrointestinal distress, (5) Respiratory symptoms, and (6) Skin symptoms. While GWI affects multiple physiological systems, the nervous system is predominantly impacted. Indeed, the Veterans Affairs (VA) Research Advisory Committee on Gulf War Veterans’ Illnesses (RAC) issued a report in 20084 that states that GWI “most prominently affects the brain and nervous system.”
Causes for GWI: Role of Organophosphate Exposures
The etiology of GWI has been a matter of debate for some time. Service members in the GW theatre were exposed to an environment of toxic exposures unlike any other war-time theatre. Environmental exposures included widespread use of pesticides, low-level exposure to nerve agents, prophylactic use of pyridostigmine bromide (PB), oil-well fire smoke, sand, dust, and particulate matter, and depleted uranium. In addition, a large percentage of troops from US and UK also received anthrax vaccination before deployment. Combat stress experienced during deployment is also a possible factor. It is believed that a complex interplay between environmental, biological, and psychological factors could underlie the development of a constellation of symptoms experienced by GWI suffering veterans.2,5
Among these, exposure to cholinergic agents has been strongly implicated as a chief cause of GWI.2,5,6 Acetylcholinesterase (AChE) inhibitors comprise a class of chemicals that can be classified as therapeutics, pesticides, and nerve agents. Troops serving in the GW had the potential to be exposed to all 3 categories. Pyridostigmine Bromide (PB), which has been used to treat myasthenia gravis since 1955, was administered to an estimated 250 000 personnel during the GW.7 PB is a reversible inhibitor of AChE used as a pretreatment against potential exposure to nerve agents. When administered prophylactically, it prevents nerve agents such as sarin and soman from irreversibly binding to and inhibiting AChE. More than 35 types of pesticides were used during the GW.8 Permethrin, DEET, and organophosphate (OP) pesticides chlorpyrifos and malathion were reported to be widely used by all GW personnel to combat mosquitos, biting flies, and ticks.9 In addition, troops were also likely exposed to low levels of the nerve agent sarin and cyclosarin, resulting from destruction of Iraqi chemical weapons storage facilities.10,11
Interestingly, there are several studies and case reports in civilian population for the development of symptoms that share features of GWI following accidental, occupational, or terrorism-related OP exposures. For example, survivors of the 1995 Tokyo Subway Sarin gas attack continue to show decline of psychomotor function and memory function.12 Further, agriculture workers and pesticide applicators exhibit neuropsychiatric symptoms, mood destabilization, and suicidal ideations that could be correlated to their history of exposure to OPs13,14 thus providing a strong evidence in favor of a causal relationship between OP exposure and significant neuropsychiatric disorder.
Rodent Model for GWI
The Vol. 10 Committee on Gulf War and Health2 recognizes that given the complexities surrounding First Gulf War deployment, no one perfect animal model that accounts for all GWI related exposures could exist. Yet, the Committee also concludes that an animal model is advantageous for identifying and evaluating mechanisms and treatment strategies for GWI. Investigators have developed elegant GWI animal models by mimicking exposure to various GWI causative factor and study its pathology. Common models use varying combinations of PB, permethrin, the sarin surrogate DFP, and stress.15-20 In 2016, our lab reported a rat model of GWI-like neurological signs that employed repeated, low-dose exposure to an OP compound- diisopropyl fluorophosphate (DFP), a surrogate of sarin used in civilian laboratories.21
In this model, adult, male rats were repeatedly exposed to a low DFP dose (0.5 mg/kg, s.c.) over a 5-day period to mimic GW-related nerve gas OP exposures.21 These doses were 1/5th the LD50 value in rodents and did not produce any symptoms of overt cholinergic stimulation. When tested 3-months following OP exposure, these rats exhibited significant psychiatric impairments including signs of chronic depression-like condition, anxiety, and cognitive deficits.21 Interestingly, these signs were observed in the absence of any other GWI-implicated stressor. Further, neurological impairments persisted 6-months following DFP exposures.22 At this point, rats were at a human equivalent age of 45 to 50 years, which aligns with the current age of GW veterans. This indicates that low-dose, sub-chronic exposure to OP agents can reproduce long-lasting neurological deficits that are associated with GWI. This OP-based DFP rat model has been helpful in identifying molecular mechanisms underlying the development of GWI22,23 and has been used to screen drugs for effective treatment of GWI-related neurological signs.24,25
Mechanism Underlying Neurological Dysfunction in DFP-Based GWI Rat Model
Research advances made over the past 2 decades has greatly informed our understanding of the pathophysiology of GWI.5 Yet, this is still a work in progress and, new knowledge gained will aid in identifying biomarkers or therapeutic targets for GWI treatments. Inhibition of AChE, the mechanism by which nerve agents and OP compounds work, leads to a buildup of ACh in the synaptic cleft. ACh is a key neurotransmitter in the central and peripheral nervous system and has a wide range of physiological effects, which accounts for the variety of symptoms that result from OP intoxication.26 Unsurprisingly, GWI research has focused on the consequences of disrupting normal cholinergic synaptic transmission. Yet, little is understood about the pathophysiology that results from low-dose OP exposure that does not produce visible symptoms such as observed in our animal model using DFP. This presented the question of what molecular changes occur upon transient interruptions of normal cholinergic signaling that may lead to the chronic neurological deficits that characterize GWI. AChE inhibition leads to tissue hyperexcitability as well as a reduction of oxidative phosphorylation. The concomitant high rate of ATP consumption and compromised ability to maintain energy levels results in mitochondrial dysfunction and oxidative stress. AChE inhibition has also been proposed to trigger an inflammatory response.27 Several mechanistic hypotheses have been proposed that include mitochondrial dysfunction28-30 and chronic inflammation15,19,29,31 as potential contributors to the GWI pathophysiology.
Neuronal Calcium Levels in DFP-Based GWI Rat Model
Considering this evidence, we hypothesized that a factor upstream of mitochondrial dysfunction played a role in the pathophysiology of GWI neurological symptoms. Under normal circumstances, cholinergic activity recruits glutamatergic activation in brain areas including the amygdala and hippocampus. Glutamatergic activity involving N-methyl-D-aspartate (NMDA) receptor activation results in intracellular calcium (Ca2+) influx, which thereby activates a host of downstream effectors. Calcium serves as a charge carrier and unlike other bivalent cations is a ubiquitous signaling molecule. Transient elevations in intracellular Ca2+ are essential for normal neurological functions including memory and synaptic plasticity.32 However, protracted elevations in neuronal Ca2+ can trigger neuro-degradative process and produce pathological synaptic plasticity that has been observed in many neurological disorders including Alzheimer’s,33 Parkinson’s,34 Epilepsy,35 and Traumatic Brain Injury (TBI)36 that exhibit co-morbid conditions similar to GWI neurological dysfunction.
To estimate intracellular Ca2+ levels ([Ca2+]i) in our repeated, low-dose DFP model, we isolated hippocampal CA1 neurons from rats expressing GWI-like neurological morbidities at 3- and 6 months following OP exposures. Ratiometric Fura-2 imaging exhibited significantly elevated [Ca2+]i levels in these hippocampal neurons at both 3- and 6-months post DFP exposures. Not only were these [Ca2+]i levels higher than age-matched controls, we also observed that a significantly greater proportion of hippocampal neurons manifested these [Ca2+]i elevations22, 24. These experiments indicated presence of protracted elevated hippocampal [Ca2+]i levels in a GWI rodent model.
Molecular Mechanism for Neuronal Ca2+ Dysfunction in DFP-Based GWI Rat Model
The molecular mechanism that underpins the protracted intracellular [Ca2+]i elevations may be an important therapeutic target in GWI as well as other neurological disorders that display this so-called “calcium plateau” along with behavioral comorbidities (Figure 1). Neuronal Ca2+ homeostasis is a highly regulated process involving a dynamic interplay between influx, efflux, release, and buffering mechanisms within the cytoplasm. Calcium entry from the extracellular milieu is mediated by ionotropic voltage-gated Ca2+ channels and ligand-gated AMPA and NMDA channels. In our low-dose DFP model of GWI, acutely isolated CA1 hippocampal neurons exhibited long-lasting elevations in intracellular [Ca2+]i levels.22,24 Various pharmacological inhibitors of Ca2+ entry was applied to isolated hippocampal neurons to determine the source of Ca2+ entry. In the presence of effective concentrations of Nifedipine, a voltage-gated Ca2+ channel blocker, 6,7-dinitroquinoxaline-2,3-dione (DNQX), an AMPA/kainite channel inhibitor, and the nonspecific cation channel blocker gadolinium chloride (GdCl3), neuronal Ca2+ levels did not significantly decrease indicating that extracellular Ca2+ entry via these channels were not responsible for the chronic Ca2+ elevations observed in DFP rats. Application of MK-801, an NMDA receptor antagonist, produced a small but significant drop in [Ca2+]i.24 This is in line with reports that provide evidence for increased glutamatergic neurotransmission following repeated low-dose OP exposures37 leading to excitotoxic activation of NMDA receptor (NMDAR) signaling in DFP rats. Thus, NMDAR blockade could be a therapeutic target in GWI. Unfortunately, NMDAR antagonists have not been able to be safely translated for therapy.38 Although the decrease in Ca2+ levels were significant, pharmacological inhibition of NMDAR did not return Ca2+ to baseline levels, suggesting that there could be mechanisms beyond Ca2+ influx contributing to chronic Ca2+ dys-homeostasis in DFP rats.24
Figure 1.
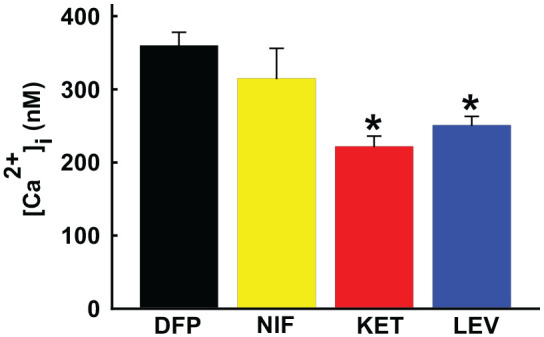
Effect of calcium channel antagonists on calcium levels in GWI. DFP exposure was associated with significant elevations in Ca2+ levels. Treatment with Ca2+ channel blocker nifedipine (NIF) did not significantly affect DFP-induced Ca2+ elevations. Treatment with NMDAR antagonist ketamine (KET) or CICR antagonist levetiracetam (LEV) produced significant reductions in DFP-induced Ca2+ elevations. Data adapted from References.22-24
Another important component of neuronal Ca2+ homeostasis revolves around regulation of Ca2+ release via intracellular stores. Calcium is stored within the endoplasmic reticulum (ER), and it can be released intracellularly upon activation of receptors embedded in the ER membrane. Ca2+-induced Ca2+-release (CICR) is one such process that is mediated by ryanodine receptors (RyRs) and inositol tris-phosphate receptors (IP3Rs) that amplify the Ca2+ signal by releasing additional Ca2+ from the ER in response to Ca2+ influx via plasmalemmal channels.32 Like elevated neuronal Ca2+ levels, aberrant CICR mechanisms have also been implicated in many neurological conditions.33-36 CICR dysfunction may also be implicated in GWI. In the presence of either the RyR antagonist dantrolene or the RyR- and IP3R-activated CICR inhibitor levetiracetam (LEV), intracellular Ca2+ levels in hippocampal neurons from DFP rats were reduced by almost 60%. This was further confirmed using in vivo administration of LEV which produced significant reductions in intracellular Ca2+ compared to DFP alone24. While dantrolene and LEV did not restore [Ca2+]i to baseline control levels, the substantial decrease in Ca2+ levels in the presence of dantrolene or LEV suggests that IP3Rs and RyRs are significantly contributing to the maintenance of DFP-induced sustained Ca2+ elevations, and CICR is likely a dominant source of neuronal Ca2+ dysregulation in DFP rats24.
Identifying the cause of aberrant CICR function could lead to new therapeutic targets. Of the 3 RyR isoforms in the brain, RyR2 is reported to be intricately involved in modulating behavior39 and has been implicated in stress-induced cognitive dysfunction.40 RyR2 is stabilized in a closed state by the regulatory subunit Calstabin2 (also known as FKBP12.6).41 It has been reported that under stress-induced conditions, posttranslational modification of RyR2 results in dissociation of Calstabin2 rendering RyR2 “leaky” and releasing Ca2+ from ER40,41. Indeed, we observed significant reduction in Calstabin2 levels within the hippocampus of DFP rats.24 Thus, “leaky” neuronal RyR could also underlie GWI neuropathology and could possibly prove to be effective targets for therapeutic interventions.
As discussed above, neuronal Ca2+ homeostasis is regulated by a complex interplay between various entry and release mechanisms, efflux pathways, and buffering proteins. Our work thus far has identified dysfunction related to Ca2+ influx and release mechanisms, namely aberrant activity of NMDAR and RyR. However, alterations in efflux mechanisms and Ca2+ buffering proteins may also play a critical role in the pathophysiology of GWI and needs further investigation. This is particularly relevant considering many neurological diseases that shares co-morbid symptoms with GWI are also known to exhibit Ca2+ handling defects. For example, dysregulations of Calbindin D-28K, a major Ca2+ buffering protein, are reported in stress-related psychiatric conditions42 and in patients with depression.43 Decreased calbindin expression has also been reported in patients with temporal lobe epilepsy with comorbid depression.44 Another important Ca2+ handling protein is the sarco/endoplasmic reticulum Ca2+ ATPase (SERCA), an ATP-dependent pump that facilitates transport of cytosolic Ca2+ into the ER. Sequestration of free cytosolic Ca2+ into the ER is critical for the maintenance of cytosolic Ca2+ homeostasis. Alterations in SERCA activity have been reported in the pathogenesis of schizophrenia and bipolar disorder as well as neurodegenerative diseases including Alzheimer disease and Parkinson disease.45 Future studies that investigate the role of Ca2+ binding proteins will therefore yield important insight regarding Ca2+ dyshomeostasis in GWI.
Ca2+ Signaling Based Therapeutics for GWI Neurological Symptoms
It has been approximately 30 years since the U.S. declared victory in the First Gulf War. Unfortunately, GW veterans continue to suffer debilitating symptoms of GWI in the absence of effective therapeutic options. Mood alterations including depression along with memory impairment are among commonly reported neurological symptoms in GWI suffering veterans.2,46 Studies and meta-analyses have shown that that GW veterans were more than twice as likely to experience depression compared with military personnel who were not deployed to the First GW.46 The recommended treatment for depression among GWI patients does not differ from the general population. The Institute of Medicine recommends antidepressant medications and psychotherapy.2,4 However, GWI patients suffering from depression experience the same challenges as much of the general population with depression, namely, a delay in therapeutic onset and inability to achieve sustained remission of depressive symptoms.47 Therefore, identifying novel antidepressant treatments that are fasting-acting and mechanistically different from standard antidepressants is of paramount importance. Considering the evidence supporting the role of elevated intracellular Ca2+ in GWI pathology in our DFP model, two promising pharmacological treatments have been under investigation in our laboratory.24,25
Levetiracetam in a DFP-Based Rat Model for GWI
Based on the results that demonstrate impaired CICR in our GWI rat model, targeting mechanisms of CICR may improve neurological symptoms of GWI. LEV is an FDA-approved anti-epileptic drug (Keppra®) that has been shown to inhibit both RyR- and IP3R-activated CICR in hippocampal neurons.48 It has been reported to improve neurological outcomes in Alzheimer’s49 and following TBI,50 2 neurological conditions that are also associated Ca2+ dysregulation.34,36 When DFP rats exhibiting symptoms of depression, anxiety, and memory impairment were administered a therapeutic dose of LEV (50 mg/kg, i.p.), signs of cognitive and mood deficits improved significantly without altering baseline behavior in naïve rats24 (Figure 2). Based on the human to rat dose translation equation,51 our dose of LEV in rats (50 mg/kg) is significantly lower than the 1000-3000 mg/day dose recommended in adult humans. Unfortunately, there are reports of increased irritability and aggression in some epileptic patients medicated with LEV.52 There is a wide range of clinical factors that may predispose a patient to developing these behavioral effects including the epileptic disorder itself.53 The beneficial effects of LEV observed in our GWI model occur at low doses. However, there remains a possibility that continual use of LEV may produce some of the adverse behavioral effects observed in some epileptic patients. While only a randomized clinical trial could confirm our pre-clinical observations regarding LEV and GWI, these studies indicate that targeting CICR via LEV-like compounds could be a viable therapeutic option for GWI-related depression and cognitive deficits.
Figure 2.
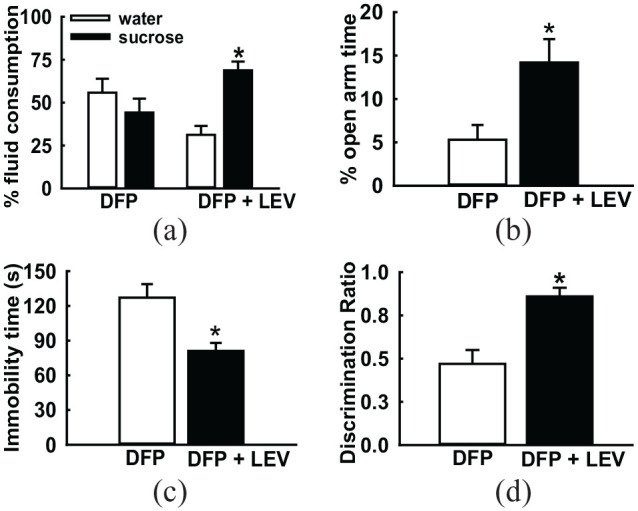
Effect of levetiracetam treatment on GWI neurological morbidities. DFP-exposed rats were treated with therapeutic doses of levetiracetam (LEV, 50 mg/kg, twice daily for 4 days). On the fifth day, these rats were subjected to a battery of behavioral tests for assessment of mood and memory function. On the sucrose preference test (a), LEV-treated rats displayed significantly greater consumption of sucrose over water. On the elevated plus maze (b), LEV-treated rats spend more time in the open-arm of the maze. On the forced swim test (c), LEV-treated rats displayed significant reductions in immobility time and on the novel-object recognition task (d), LEV-treated rats showed higher discrimination ratios compared to saline-treated age-matched DFP rats. These observations indicated that LEV treatment significantly improved neurological signs of memory deficits and depression in DFP-treated GWI rats. Data adapted from Phillips et al.24
Ketamine in a DFP-Based Rat Model for GWI
Another promising therapeutic option that has emerged from our Ca2+ homeostatic studies in GWI rat model is ketamine, an NMDA receptor antagonist with widely reported fast-acting and sustained antidepressant effects in both human and animal studies.54 Ketamine is a schedule III drug that has been FDA-approved as an anesthetic since 1970. While anesthetic doses of ketamine produce dissociative effects, Berman and colleagues reported rapid and long-lasting antidepressant effects in patients with treatment-resistant depression using an IV infusion of a sub-anesthetic dose of ketamine.54 The recent FDA approval of esketamine (Spravato®) saw the first new antidepressant mechanism on the market in half a century. Ketamine has predominantly been assessed in patients with diagnosed treatment-resistant depression. Based on reports of high prevalence of depression in GWI,46 we sought to determine whether ketamine would extend a similar robust antidepressant profile in a depression phenotype that has been proven difficult to manage with current therapies. In rats displaying symptoms of despair and anhedonia 3 months post-DFP exposure, ketamine administration at sub-anesthetic doses (5 and 10 mg/kg) produced a significant and rapid antidepressant response without causing locomotor retardation23,25 (Figure 3). In line with previous reports, when individual enantiomers were assessed, (R)-ketamine produced a more robust antidepressant effect compared to (S)-ketamine. The antidepressants effects produced by (R)-ketamine were sustained for 24 h unlike (S)-ketamine, further supporting evidence that (R)-ketamine produces long-lasting antidepressant effects.25 In humans, for the treatment of depression, ketamine is administered between 0.5 and 0.75 mg/kg as an intravenous infusion across a 40 min session.55 This human dose agrees with the rat dose (5-10 mg/kg) administered as an intraperitoneal injection. Both the human and rat doses are well below the anesthetic doses for ketamine and are well tolerated. Given the potential abuse liability associated with ketamine, it is recommended that long-term ketamine treatment, if warranted, be closely monitored.56
Figure 3.
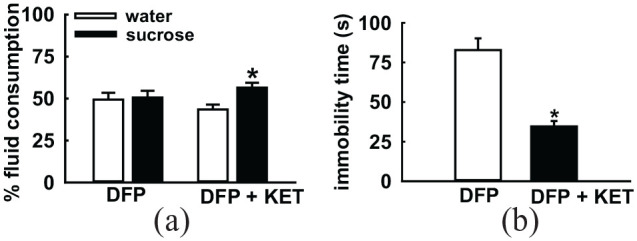
Effect of ketamine on depressive symptoms in GWI. DFP-exposed rats were treated with ketamine (KET, 10 mg/kg). Rats were then subjected to behavioral assays for assessment of signs of depression. On the sucrose preference test (a), KET-treated rats displayed significantly greater consumption of sucrose over water. On the forced swim test (b), KET-treated rats displayed significant reductions in immobility time. These observations indicated that KET significantly improved signs of depression in DFP-treated GWI rats. Data adapted from Zhu et al.25
Though ketamine is traditionally considered an NMDA receptor antagonist, various non-NMDAR mechanisms have also been proposed to underlie ketamine’s therapeutic action. For example, recent studies have proposed the role of ketamine metabolites in sustained activation of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPARs) for the long-term antidepressant effect. In addition, the role of transcription factor eukaryotic elongation factor-2 (eEF2) in upregulating brain-derived neurotrophic factor (BDNF) expression has also been suggested for ketamine’s antidepressant effect (reviewed in57). Interestingly we observed that BDNF expression was significantly lower in the hippocampus of DFP-rats compared to age-matched control rats.23 Given the pleiotropic actions of BDNF signaling on neuronal survival and synaptic plasticity pathways, BDNF dysregulation could be an important downstream mechanism for GWI neuropathology (Also see Figure 4). In the context of GWI, we recently investigated molecular mechanisms for ketamine’s antidepressant action using our DFP-based rat model of GWI-like depressive signs. Our results indicated that ketamine exhibited dual modes of action. While the inhibition of the NMDAR-Ca2+ pathway contributed to the rapid onset antidepressant effects of ketamine while, the long-lasting antidepressant actions involved upregulation of BDNF signaling.23 Like the LEV studies, these pre-clinical studies provide impetus for further assessment of ketamine in clinical trials with GWI veterans for treatment of GWI-related depression.
Figure 4.

Calcium Hypothesis of GWI neurological morbidities. GW-related OP exposures cause chronic elevations in hippocampal Ca2+ levels from enhanced ER Ca2+ release (CICR). Sustained Ca2+ elevations trigger second-messenger pathways that affect gene expression of key proteins (eg, BDNF) by repressing transcription factor (eg, CREB) leading to pathological neuronal plasticity that exhibits itself as GWI neurological signs.
Our data also raises an interesting possibility that a combination therapy of LEV and ketamine, each targeting different aspects of Ca2+ handling within the neuron, may more effectively lower elevated Ca2+ levels in GWI neurons. Ketamine blocks Ca2+ entry via NMDAR antagonism, while LEV blocks intracellular Ca2+ release via CICR antagonism. Individually, these agents significantly lowered Ca2+ levels but could not restore them to baseline (see Figure 1). Thus, combining LEV and ketamine could offer greater suppression of Ca2+ levels in GWI. However, it is also possible that the combination may exacerbate the risk profile of individual drugs and produce greater adverse effects and unexpected interactions. These possibilities need to be tested in future studies.
Calcium Hypothesis of GWI
This evidence led us to propose the “Ca2+ hypothesis of GWI” that suggests a role for Ca2+ ions in the expression of GWI neurological morbidities (Figure 4). Our hypothesis states that low-dose, OP-induced sustained Ca2+ elevations could activate multiple signaling pathways that produce neuronal injury and trigger nuclear signaling that may result in long-term pathological plasticity changes which manifest as behavioral morbidities observed in this GWI model.21,22,24 Evidence thus far supports our hypothesis that a factor upstream of mitochondrial dysfunction28 is involved in the underlying pathology of GWI, and intracellular Ca2+ homeostatic mechanisms serves as a novel target. Based on this hypothesis, we targeted the molecular pathways underlying the chronic Ca2+ elevations and observed abnormal CICR mechanisms in GWI neurons. Together with LEV and ketamine data, our studies provide the first proof-of-concept data that targeting OP-induced neuronal Ca2+ alterations could potentially offer an effective approach to identify novel drugs for the treatment of GWI neurological symptoms.23-25 It will be important to investigate whether Ca2+ dysfunction is present in other animal models of GWI and whether targeting intracellular Ca2+ in these models continues to be an effective approach.
Future Directions
It has become clear that GWI is a multi-system disorder with many different signaling pathways that are disrupted simultaneously which could act individually or in concert to produce the multi-symptom pathology. Recent papers have reviewed mechanisms underlying brain dysfunction and therapeutic strategies in animal models of GWI and GWI veterans.58,59 The focus of this paper was to discuss findings related to alterations in neuronal Ca2+ levels and subsequently review how restoration of protracted Ca2+ levels using FDA approved therapeutics alleviates neurological signs of GWI in our rat DFP model which, then led us to propose the Ca2+ hypothesis of GWI. Whether the protracted Ca2+ elevations reported in our repeated, low-dose DFP model are also observed in other GWI models would help validate and expand this Ca2+ hypothesis as a major mechanism for GWI neurological dysfunctions.
Our work has begun to explore an important aspect of cellular communication that could have major implications for the persistence of GWI. How OP-induced alterations in Ca2+ signaling interacts with other signaling mechanisms in the development, expression, and maintenance of GWI condition will be the focus of future studies.
Footnotes
Funding:The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Office of the Assistant Secretary of Defense for Health Affairs, through the Gulf War Illness Research Program under Award No. W81XWH-14-1-0478, W81XWH-17-1-0573, and W81XWH-20-1-0278. Opinions, interpretations, conclusions, and recommendations are those of the authors and are not necessarily endorsed by the Department of Defense.
Declaration of conflicting interests:The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions: K.F.P.: Writing-Review and Editing; L.S.D.: Conceptualization, Writing-original draft preparation, Project administration, Funding acquisition. All authors have read and agreed to the published version of the manuscript.
ORCID iD: Laxmikant S Deshpande  https://orcid.org/0000-0003-1491-1561
https://orcid.org/0000-0003-1491-1561
References
- 1. Institute of Medicine. Chronic Multisymptom Illness in Gulf War Veterans: Case Definitions Reexamined. The National Academies Press; 2014. [PubMed] [Google Scholar]
- 2. Institute of Medicine. Gulf War and Health: Volume 10: Update of Health Effects of Serving in the Gulf War. The National Academies Press; 2016. [PubMed] [Google Scholar]
- 3. Steele L. Prevalence and patterns of Gulf War illness in Kansas veterans: association of symptoms with characteristics of person, place, and time of military service. Am J Epidemiol. 2000;152:992-1002. [DOI] [PubMed] [Google Scholar]
- 4. U.S. Department of Veterans Affairs, Research Advisory Committee on Gulf War Veterans’ Illnesses. Gulf War Illness and the health of Gulf War veterans, scientific findings and recommendations. U.S. Government Printing Office; 2008. [Google Scholar]
- 5. White RF, Steele L, O’Callaghan JP, et al. Recent research on Gulf War illness and other health problems in veterans of the 1991 Gulf War: effects of toxicant exposures during deployment. Cortex. 2016;74:449-475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Golomb BA. Acetylcholinesterase inhibitors and Gulf War illnesses. Proc Natl Acad Sci. 2008;105:4295-4300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Institute of Medicine. Gulf War and Health: Volume 1. Depleted Uranium, Sarin, Pyridostigmine Bromide, Vaccines. The National Academies Press; 2000. [PubMed] [Google Scholar]
- 8. Fricker RD, Reardon E, Spektor DM, et al. Pesticide use during the Gulf War: a survey of Gulf War Veterans, Santa Monica, Calif.: RAND corporation, MR-1018/12-OSD, 2000. Accessed June 17, 2020 https://www.rand.org/pubs/monograph_reports/MR1018z12.html
- 9. Institute of Medicine. Gulf War and Health: Volume 2: Insecticides and Solvents. The National Academies Press; 2003. [Google Scholar]
- 10. Haley RW, Tuite JJ. Epidemiologic evidence of health effects from long-distance transit of chemical weapons fallout from bombing early in the 1991 Persian Gulf War. Neuroepidemiology. 2013;40:178-189. [DOI] [PubMed] [Google Scholar]
- 11. Directorate for Deployment Health Support of the Special Assistant to the Under Secretary of Defense (Personnel and Readiness) for Gulf War Illness Medical Readiness and Military Deployments US demolition operations at the Khamisiyah ammunition point (case narrative), 2002. https://gulflink.health.mil/library/kham_infojsp.shtml
- 12. Nishiwaki Y, Maekawa K, Ogawa Y, et al. 2001. Effects of sarin on the nervous system in rescue team staff members and police officers 3 years after the Tokyo subway sarin attack. Environ Health Perspect. 2001;109:1169-1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wesseling C, Keifer M, Ahlbom A, et al. Long-term neurobehavioral effects of mild poisonings with organophosphate and n-methyl carbamate pesticides among banana workers. Int J Occup Environ Health. 2002;8:27-34. [DOI] [PubMed] [Google Scholar]
- 14. Cherry N, Mackness M, Durrington P, et al. Paraoxonase (PON1) polymorphisms in farmers attributing ill health to sheep dip. Lancet (London, England). 2002;359:763-764. [DOI] [PubMed] [Google Scholar]
- 15. Parihar VK, Hattiangady B, Shuai B, et al. Mood and memory deficits in a model of Gulf War illness are linked with reduced neurogenesis, partial neuron loss, and mild inflammation in the hippocampus. Neuropsychopharmacology. 2013;38:2348-2362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Abdel-Rahman A, Abou-Donia S, El-Masry E, et al. Stress and combined exposure to low doses of pyridostigmine bromide, DEET, and permethrin produce neurochemical and neuropathological alterations in cerebral cortex, hippocampus, and cerebellum. J Toxicol Environ Health A. 2004;67:163-192. [DOI] [PubMed] [Google Scholar]
- 17. Abdel-Rahman A, Shetty AK, Abou-Donia MB. Disruption of the blood-brain barrier and neuronal cell death in cingulate cortex, dentate gyrus, thalamus, and hypothalamus in a rat model of Gulf-War syndrome. Neurobiol Dis. 2002;10:306-326. [DOI] [PubMed] [Google Scholar]
- 18. Zakirova Z, Tweed M, Crynen G, et al. Gulf War agent exposure causes impairment of long-term memory formation and neuropathological changes in a mouse model of Gulf War illness. PLoS One. 2015;10:e0119579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Koo BB, Michalovicz LT, Calderazzo S, et al. Corticosterone potentiates DFP-induced neuroinflammation and affects high-order diffusion imaging in a rat model of Gulf War illness. Brain Behav Immun. 2017;67:42-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. O’Callaghan JP, Kelly KA, Locker AR, et al. Corticosterone primes the neuroinflammatory response to DFP in mice: potential animal model of Gulf War illness. J Neurochem. 2015;133:708-721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Phillips KF, Deshpande LS. Repeated low-dose organophosphate DFP exposure leads to the development of depression and cognitive impairment in a rat model of Gulf War illness. Neurotoxicology. 2016;52:127-133. [DOI] [PubMed] [Google Scholar]
- 22. Phillips KF, Deshpande LS. Chronic neurological morbidities and elevated hippocampal calcium levels in a DFP-based rat model of Gulf War illness. Mil Med. 2018;183:552-555. [DOI] [PubMed] [Google Scholar]
- 23. Ribeiro ACR, Zhu J, Kronfol MM, et al. Molecular mechanisms for the antidepressant-like effects of a low-dose ketamine treatment in a DFP-based rat model for Gulf War illness. Neurotoxicology. 2020;80:52-59. [DOI] [PubMed] [Google Scholar]
- 24. Phillips KF, Santos E, Blair RE, et al. Targeting intracellular calcium stores alleviates neurological morbidities in a DFP-based rat model of Gulf War illness. Toxicol Sci. 2019;169:567-578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhu J, Hawkins E, Phillips K, et al. Assessment of ketamine and its enantiomers in an organophosphate-based rat model for features of Gulf War illness. Int J Environ Res Public Health. 2020;17:4710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bajgar J. Organophosphates/nerve agent poisoning: mechanism of action, diagnosis, prophylaxis, and treatment. Adv Clin Chem. 2004;38:151-216. [DOI] [PubMed] [Google Scholar]
- 27. Michalovicz LT, Kelly KA, Sullivan K, et al. Acetylcholinesterase inhibitor exposures as an initiating factor in the development of Gulf War illness, a chronic neuroimmune disorder in deployed veterans. Neuropharmacology. 2020;171:108073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Koslik HJ, Hamilton G, Golomb BA. Mitochondrial dysfunction in Gulf War illness revealed by 31Phosphorus magnetic resonance spectroscopy: a case-control study. PLoS One. 2014;9:e92887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Shetty GA, Hattiangady B, Upadhya D, et al. Chronic oxidative stress, mitochondrial dysfunction, Nrf2 activation and inflammation in the hippocampus accompany heightened systemic inflammation and oxidative stress in an animal model of Gulf War illness. Front Mol Neurosci. 2017;10:182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chen Y, Meyer JN, Hill HZ, et al. Role of mitochondrial DNA damage and dysfunction in veterans with Gulf War illness. PLoS One. 2017;12:e0184832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Locker AR, Michalovicz LT, Kelly KA, et al. Corticosterone primes the neuroinflammatory response to Gulf War illness-relevant organophosphates independently of acetylcholinesterase inhibition. J Neurochem. 2017;142:444-455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bengtson CP, Bading H. Nuclear calcium signaling. Adv Exp Med Biol. 2012;970:377-405. [DOI] [PubMed] [Google Scholar]
- 33. Popugaeva E, Pchitskaya E, Bezprozvanny I. Dysregulation of neuronal calcium homeostasis in Alzheimer’s disease - A therapeutic opportunity? Biochem Biophys Res Commun. 2017;483:998-1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Surmeier DJ, Schumacker PT. Calcium, bioenergetics, and neuronal vulnerability in Parkinson’s disease. J Biol Chem. 2013;288:10736-10741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Nagarkatti N, Deshpande LS, DeLorenzo RJ. Development of the calcium plateau following status epilepticus: role of calcium in epileptogenesis. Expert Rev Neurother. 2009;9:813-824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tehse J, Taghibiglou C. The overlooked aspect of excitotoxicity: glutamate-independent excitotoxicity in traumatic brain injuries. Eur J Neurosci. 2018;49:1157-1170. [DOI] [PubMed] [Google Scholar]
- 37. Torres-Altoro MI, Mathur BN, Drerup JM, et al. Organophosphates dysregulate dopamine signaling, glutamatergic neurotransmission, and induce neuronal injury markers in striatum. J Neurochem. 2011;119:303-313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Williams NR, Schatzberg AF. NMDA antagonist treatment of depression. Curr Opinion Neurobiol. 2016;36:112-117. [DOI] [PubMed] [Google Scholar]
- 39. Kushnir A, Wajsberg B, Marks AR. Ryanodine receptor dysfunction in human disorders. Biochim Biophys Acta Mol Cell Res. 2018;1865:1687-1697. [DOI] [PubMed] [Google Scholar]
- 40. Liu X, Betzenhauser MJ, Reiken S, et al. Role of leaky neuronal ryanodine receptors in stress-induced cognitive dysfunction. Cell. 2012;150:1055-1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yuan Q, Deng KY, Sun L, et al. Calstabin 2: an important regulator for learning and memory in mice. Sci Rep. 2016;6:21087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Li JT, Xie XM, Yu JY, et al. Suppressed calbindin levels in hippocampal excitatory neurons mediate stress-induced memory loss. Cell Rep. 2017;21:891-900. [DOI] [PubMed] [Google Scholar]
- 43. Maciag D, Hughes J, O’Dwyer G, et al. Reduced density of calbindin immunoreactive GABAergic neurons in the occipital cortex in major depression: relevance to neuroimaging studies. Biol Psychiatry. 2010;67:465-470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. D’Alessio L, Konopka H, Solís P, et al. Depression and temporal lobe epilepsy: expression pattern of calbindin immunoreactivity in hippocampal dentate gyrus of patients who underwent epilepsy surgery with and without comorbid depression. Behav Neurol. 2019;2019:7396793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Britzolaki A, Saurine J, Klocke B, Pitychoutis PM. A role for SERCA pumps in the neurobiology of neuropsychiatric and neurodegenerative disorders. Adv Exp Med Biol. 2020;1131:131-161. [DOI] [PubMed] [Google Scholar]
- 46. Blore JD, Sim MR, Forbes AB, et al. Depression in Gulf War veterans: a systematic review and meta-analysis. Psychol Med. 2015;45:1565-1580. [DOI] [PubMed] [Google Scholar]
- 47. Ferrari F, Villa RF. The neurobiology of depression: an integrated overview from biological theories to clinical evidence. Mol Neurobiol. 2016;54:4847-4865 [DOI] [PubMed] [Google Scholar]
- 48. Nagarkatti N, Deshpande LS, DeLorenzo RJ. Levetiracetam inhibits both ryanodine and IP3 receptor activated calcium induced calcium release in hippocampal neurons in culture. Neurosci Lett. 2008;436:289-293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Musaeus CS, Shafi MM, Santarnecchi E, Herman ST, Press DZ. Levetiracetam alters oscillatory connectivity in Alzheimer’s disease. J Alzheimers Dis. 2017;58:1065-1076. [DOI] [PubMed] [Google Scholar]
- 50. Zou H, Brayer SW, Hurwitz M, et al. Neuroprotective, neuroplastic, and neurobehavioral effects of daily treatment with levetiracetam in experimental traumatic brain injury. Neurorehabil Neural Repair. 2013;27:878-888. [DOI] [PubMed] [Google Scholar]
- 51. Nair AB, Jacob S. A simple practice guide for dose conversion between animals and human. J Basic Clin Pharm. 2016;7:27-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Mula M, Agrawal N, Mustafa Z, et al. Self-reported aggressiveness during treatment with levetiracetam correlates with depression. Epilepsy Behav. 2015;45:64-67. [DOI] [PubMed] [Google Scholar]
- 53. Mbizvo GK, Dixon P, Hutton JL, Marson AG. The adverse effects profile of levetiracetam in epilepsy: a more detailed look. Int J Neurosci. 2014;124:627-634. [DOI] [PubMed] [Google Scholar]
- 54. Berman RM, Cappiello A, Anand A, et al. Antidepressant effects of ketamine in depressed patients. Biol Psychiatry. 2000;47:351-354. [DOI] [PubMed] [Google Scholar]
- 55. Andrade C. Ketamine for depression, 4: in what dose, at what rate, by what route, for how long, and at what frequency? J Clin Psychiatry. 2017;78:e852-e857. [DOI] [PubMed] [Google Scholar]
- 56. Schak KM, Vande Voort JL, Johnson EK, et al. Potential risks of poorly monitored ketamine use in depression treatment. Am J Psychiatry. 2016;173:215-218. [DOI] [PubMed] [Google Scholar]
- 57. Yang C, Yang J, Luo A, et al. Molecular and cellular mechanisms underlying the antidepressant effects of ketamine enantiomers and its metabolites. Transl Psychiatry. 2019;9:280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Dickey B, Madhu LN, Shetty AK. Gulf War illness: mechanisms underlying brain dysfunction and promising therapeutic strategies. Pharmacol Ther. 2020:107716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Nugent SM, et al. A systematic review of therapeutic interventions and management strategies for Gulf War illness. Mil Med. Published online October 30, 2020. doi: 10.1093/milmed/usaa260. [DOI] [PubMed] [Google Scholar]