

Abstract
A series of (−)-β-d-(2R,4R)-dioxolane-thymine-5′-O-aliphatic acid esters as well as amino acid esters were synthesized as prodrugs of (−)-β-d-(2R,4R)-dioxolane-thymine (DOT). The compounds were evaluated for anti-HIV activity against HIV-1LAI in human peripheral blood mononuclear (PBM) cells as well as for their cytotoxicity in PBM, CEM and Vero cells. Improved anti-HIV potency in vitro was observed for the compound 2-4 (5′-O-aliphatic acid esters) without increase in cytotoxicity in comparison to the parent drug. Chemical and enzymatic hydrolysis of the prodrugs was also studied, in which the prodrugs exhibited good chemical stability with the half-lives from 3 h to 54 h at pH 2.0 and 7.4 phosphate buffer. However, the prodrugs were relatively labile to porcine esterase with the half-lives from 12.3 to 48.0 min.
Keywords: Anti-HIV activity, Nucleoside prodrugs, Dioxolane thymine (DOT), 5′-O-aliphatic acid esters, 5′-O-amino acid esters
1. Introduction
Nucleoside analogues continue to play important role as antiviral agents,1 however, the therapeutic potential is often hampered by their poor biopharmaceutical properties.2 These properties may be due to their high polarity as well as low bio-availability. Thus, various prodrugs of nucleosides have been synthesized by chemical modification to improve their physicochemical and/or pharmacokinetic properties.3,4 Among the nucleoside prodrugs, 5′-O-esters5,6 and amino acid esters7,8 are common to improve their physicochemical properties.3,9 The rationale for this approach is to alter the lipophilicity, permeability, stability and tissue specificity. Therefore, prodrugs could potentially enhance the overall pharmaceutical properties over the parent drugs which produce the active drugs to exert their pharmacological action in vivo. Various esters, carbamates and amide prodrugs can be hydrolyzed by esterases and amidases, which are ubiquitous in vivo with a broad substrate specificity.10
As a part of our continuing efforts to discover novel and effective antiviral agents, we have reported the synthesis and antiviral activity of various 1,3-oxathioalane and 1,3-dioxolane nucleosides.11–16 From these studies, we discovered several clinical candidates, which include (−)-β-d-(2R,4R)-2,6-diaminopurine-dioxolane (amdoxovir or DAPD)17 and (−)-β-d-(2R,4R)-dioxolane-thymine (DOT).18,19 DOT is a pyrimidine 1,3-dioxolane nucleoside, exhibiting interesting activity against drug resistant mutants of HIV-1.18,19 Another dioxolane nucleoside, amdoxovir is undergoing Phase II clinical studies as an anti-HIV agent.20 The significant antiviral activity of the parent nucleoside of DAPD, (−)-β-d-(2R,4R)–dioxolane-guanine (DXG)21,22 along with its prodrug, amdoxovir (DAPD)17 and 2-aminopurine dioxolane (APD)23 (another DXG prodrug) prompted us to further enhance the antiviral potency of DOT24,18 by a prodrug strategy, as it is a moderately potent anti-HIV agent by itself, but with unique resistant profiles.18,19 Previously, we have reported the synthesis and anti-HIV activity of dendrimer, phosphoramidate and phosphate prodrugs of DOT.25,26
Aliphatic and amino acids have been extensively used for prodrugs due to their well-established chemistry, structural diversities as well as lack of toxicity concerns.27,28 Lipophilic aliphatic esters are known to preferentially be taken up by mononuclear phagocyte system (MPS) after intravenous injection. Apart from CD4+ T-lymphocytes, cells of MPS play a decisive role in the pathogenesis of AIDS and are considered as a major reservoir for HIV.29 The use of amino acid for a prodrug strategy also provides advantages in the cellular transport system.30 In view of these information, DOT-prodrugs with 5′-O-aliphatic and amino acid esters were synthesized and evaluated for their anti-HIV activity in vitro in efforts to improve the pharmaceutical and pharmacological properties of DOT.
2. Results and discussion
2.1. Chemistry
The chemical structure of synthesized aliphatic acid ester (1-10) and amino acid ester prodrugs (11-22) of DOT are shown in Figure 1. Eight aliphatic acids (acetic, valeric, octanoic, lauric, palmitic, pivalic, cyclopropanecarboxylic and cyclohexanecarboxylic acid) were chosen and were treated with DOT at room temperature in the presence of DMAP and 1,3-diisopropylcarbodiimide (DIC) to yield the prodrugs 1-8 (Scheme 1). Compound 9 was synthesized by the reaction of chloromethyl pivalate with DOT in the presence of NaH. The pivaloyl moiety is linked to DOT by an ether bond. The DOT was reacted with Bis-dPEG8 acids in the presence of DMAP and DIC to provide compound 10 as a dinucleoside. For the synthesis of amino acid ester prodrugs of DOT (Scheme 1, compounds 11-22), aromatic amino acids (l-Phe, d-Phe and l-Trp), aliphatic amino acids (l-Val, d-Val, l-Ala, l-Leu, l-Ile, Gly and 2-aminoisobutyric acid) and the polar amino acids (l-Pro and l-Gln) were selected. In each case, Boc-protected amino acids were used to react with DOT in the presence of DMAP and DIC to provide the Boc-protected intermediates. The intermediates were purified by silica gel flash chromatography. Further, deprotection of the Boc group was achieved by treating with 50% TFA in DCM for 1 hour at 0 °C. After removal of the excess TFA, the resulting residue was dissolved in anhydrous methanol and treated with anhydrous HCl/ether to give the HCl salt of amino acid esters. Subsequently, the pure product was obtained by crystallization from methanol. The synthesized DOT-prodrugs (1-22) were characterized by mass spectrometry (ESI-MS), 1H NMR and elemental analysis.
Figure 1.
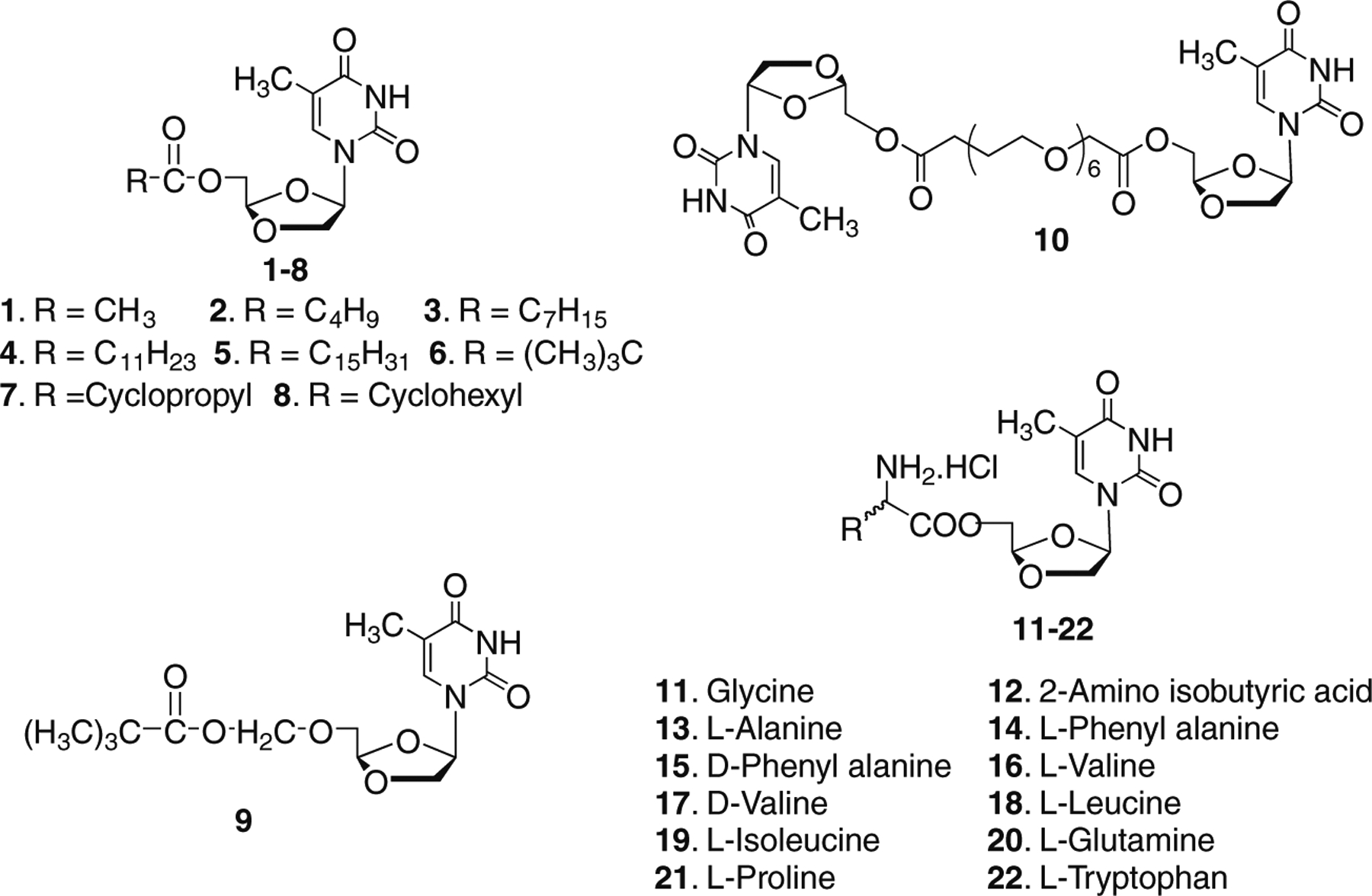
Structures of 5′-O-aliphatic and amino acid esters of (−)-β-d-(2R,4R)-dioxolane-thymine (DOT).
Scheme 1.
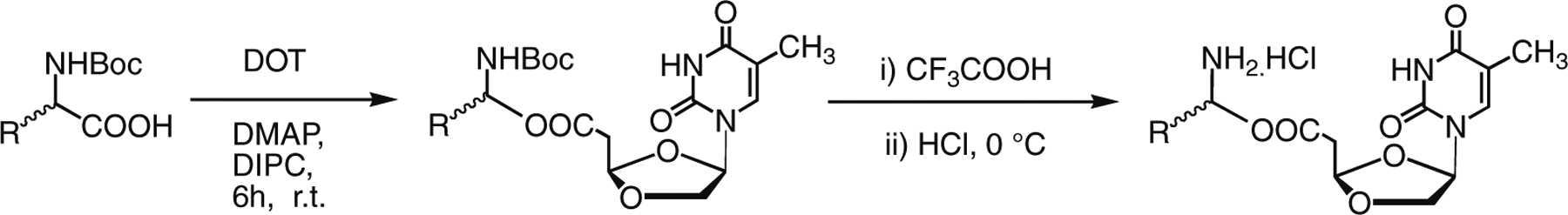
Synthesis of 5′-O-amino acid esters of DOT.
2.2. Anti-HIV activity and cytotoxicity
The anti-HIV activity of the prodrugs were evaluated against HIV-1LAI in PBM cells and the cytotoxicity was evaluated in PBM, CEM and Vero cells as shown in Table 1. Most of the synthesized prodrugs exhibited potent anti-HIV activity against HIV-1LAI without increased cytotoxicity in comparison to the parent drug, DOT. Among the synthesized prodrugs, valeric, octanoic and lauric acid ester prodrugs (2-4) exhibited improved antiviral activity with an EC50 < 0.6 μM (EC90 < 4.3 μM) in comparison to DOT (EC50 = 0.68 μM, EC90 = 4.0 μM). The prodrug with a t-butyl acid group (6) showed enhanced cytotoxicity to Vero cells (IC50 = 18.1 μM). Compound 9 and compound 10 have shown slightly reduced antiviral activity. Among the synthesized amino acid esters, compounds 13-17 exhibited similar anti-HIV potency as the parent drug without any cytotoxicity to PBM, CEM and Vero cells.
Table 1.
Anti-HIV activity and cytotoxicity of 5′-O-aliphatic and amino acid esters of (−)-β-d-(2R,4R)-dioxolane-thymine (DOT).
| Compounds | HIV-1LAI (μM)a | Cytotoxicity (IC50, μM)b | |||
|---|---|---|---|---|---|
| EC50 | EC90 | PBM | CEM | Vero | |
| 1 | 2.9 | 12.3 | >100 | 99.4 | >100 |
| 2 | 0.4 | 2.5 | >100 | >100 | 49.0 |
| 3 | 0.6 | 4.3 | >100 | >100 | >100 |
| 4 | 0.4 | 3.2 | >100 | >100 | >100 |
| 5 | 23.5 | >100 | >100 | >100 | 92.2 |
| 6 | 1.3 | 14.5 | >100 | >100 | 18.1 |
| 7 | 4.7 | 15.1 | >100 | >100 | >100 |
| 8 | 1.3 | 3.9 | >100 | >100 | >100 |
| 9 | 1.4 | 9.7 | >100 | >100 | >100 |
| 10 | 1.5 | 10.2 | >100 | >100 | >100 |
| 11 | 2.5 | 8.4 | >100 | >100 | >100 |
| 12 | 2.2 | 7.6 | >100 | >100 | >100 |
| 13 | 0.98 | 8.3 | >100 | >100 | >100 |
| 14 | 0.88 | 4.4 | >100 | >100 | >100 |
| 15 | 1.0 | 5.2 | >100 | >100 | >100 |
| 16 | 0.56 | 4.9 | >100 | >100 | >100 |
| 17 | 0.98 | 6.5 | >100 | >100 | >100 |
| 18 | 3.3 | 12.4 | >100 | >100 | >100 |
| 19 | 3.0 | 9.9 | >100 | >100 | >100 |
| 20 | 0.89 | 6.3 | >100 | >100 | >100 |
| 21 | 1.4 | 7.3 | >100 | >100 | >100 |
| 22 | 1.7 | 8.0 | >100 | >100 | >100 |
| DOT | 0.68 | 4.0 | >100 | >100 | >100 |
| AZT | 0.007 | 0.04 | >100 | 14.3 | 50.6 |
Anti-HIV activity against HIV-1LAI in PBM cells.
Assay in PBM, CEM and Vero cells.
2.3. Hydrolysis of prodrugs
An ideal prodrug should exhibit a good chemical and enzymatic stability before entering the cell, while requires enzymatic conversion to the parent drug after being transported across the biological membrane.31,32 In order to access the chemical and enzymatic stability, we have conducted chemical and enzymatic stability studies of the synthesized prodrugs and the results are shown in Table 2. The rate of chemical hydrolysis of the synthesized prodrug in phosphate buffer at pH 2.0 and pH 7.4 (Table 2) were obtained from the linear regression of pseudo first order plot using concentration vs. time. The most of the prodrugs showed a good chemical stability, and it reveals that the structure of the prodrugs influences the rate of hydrolysis (Table 2). In general, prodrugs with aliphatic esters were hydrolyzed slower than the prodrugs with amino acid esters. For aliphatic esters, the rates of chemical hydrolysis were reduced with an increased length of aliphatic chains. Bis-PEG8 ester of DOT (10) showed an increased rate of chemical hydrolysis in comparison to the prodrugs with aliphatic acids. Among amino acid prodrugs, the rate of chemical hydrolysis varied according to the structural features. The stereochemistry of amino acids appeared to affect the chemical stability. The l-amino acid prodrugs showed higher stability than the d-amino acid prodrugs (14 vs 15 and 16 vs 17). In general, l-Gln, l-Pro and l-Trp prodrugs (20-22) were hydrolyzed faster than the other amino acid prodrugs. The prodrug with l-Gln (20) showed the shortest half-life (190 min at pH 2.0 and 240 min at pH 7.4). The l-Val prodrug (16) showed the longest half-life (960 min at pH 2.0 and 2,200 min at pH 7.4).
Table 2.
Half-lives (t1/2, min) of the chemical and enzymatic hydrolysis of 5′-O-aliphatic and amino acid esters of (−)-β-d-(2R,4R)-dioxolane-thymine (DOT).
| Compound | Chemical hydrolysisa | Enzymatic hydrolysisb | |
|---|---|---|---|
| pH 2.0 | pH 7.4 | ||
| 1 | 1580 ± 320c | 2650 ± 560 | 16.3±1.1 |
| 2 | 1600 ±320 | 3180 ±620 | 18.5 ± 0.4 |
| 3 | 1800 ±380 | 3250 ± 670 | 29.0 ± 0.3 |
| 4 | NDd | ND | 41.0 ±0.4 |
| 5 | ND | ND | 48.0 ±1.6 |
| 6 | 1300 ±280 | 2780±470 | 17.0 ± 0.7 |
| 7 | 1200 ±270 | 2800 ± 480 | 17.3 ± 0.3 |
| 8 | 1850 ±320 | 3200 ± 580 | 19.3 ± 0.3 |
| 9 | ND | ND | 17.6 ± 0.5 |
| 10 | 380 ± 40 | 520 ± 65 | 18.3 ± 0.5 |
| 11 | 520 ± 90 | 1320 ±180 | 12.4 ± 0.4 |
| 12 | 920 ± 90 | 1870 ±280 | 13.4 ± 0.5 |
| 13 | 870±58 | 1600 ±234 | 13.9 ± 0.4 |
| 14 | 550 ± 65 | 1070 ±28 | 14.6 ± 0.6 |
| 15 | 430 ± 50 | 940 ± 25 | 15.6 ± 0.6 |
| 16 | 960 ± 80 | 2200 ± 450 | 18.9 ± 0.7 |
| 17 | 730±48 | 1800 ±270 | 19.4 ± 0.8 |
| 18 | 870±55 | 2150 ±310 | 17.9 ± 0.9 |
| 19 | 800 ± 60 | 2040 ± 290 | 17.5±1.2 |
| 20 | 190 ±40 | 240 ± 60 | 13.4 ± 0.5 |
| 21 | 230 ± 40 | 380 ± 50 | 13.6 ± 0.4 |
| 22 | 320 ± 35 | 420 ± 40 | 17.8 ± 0.3 |
At pH 2.0 and pH at 7.4 phosphate buffer at 37 ± 0.5 °C.
At pH 8.0 buffer solution of porcine esterase at 37 ± 0.5 °C.
Half-lives expressed as mean ± SEM.
Not determined due to solubility.
The rates of enzymatic hydrolysis were also determined and are also listed in the Table 2. As expected, the data show that the prodrugs were less stable (t1/2: from 13 to 48 min) in the presence of porcine esterase than in phosphate buffer. For aliphatic acid prodrugs, the rates of hydrolysis were decreased with the increase of the length of the aliphatic chain, in which the palmitic acid prodrug hydrolyzed with a t1/2 of 48 min while the prodrug with an acetic acid has a t1/2 of 16.3 min. The prodrugs with amino acids showed shorter half-lives (t1/2 12.4 to 19.4 min) than the aliphatic acid prodrugs (t1/2 from 16.3 to 48 min). The Gly prodrug 11 showed the shortest half-lives (12.4 min) while the d-Val prodrug 17 showed the longest half-lives (19.4 min). The results suggest that the synthesized prodrugs are good substrates for the esterase and can efficiently generate the parent drug. Although the structural features influenced the chemical and enzymatic stability of two types of DOT-prodrugs, no clear correlation between the anti-HIV activity and stability was observed in these series.
In conclusion, some of the synthesized aliphatic and amino acid prodrugs of DOT have shown enhanced anti-HIV activity in vitro without increase in cytotoxicity. In comparison to the previously reported phosphoramidate, phosphate and dendrimer prodrugs, the newly synthesized prodrugs have similar enhanced anti-HIV activity, however, the overall cytotoxicity profile was more favorable. The synthesized prodrugs possess good chemical stability in the buffer systems, and they are efficient substrates for porcine esterase. The present study suggests that a certain class of DOT-prodrugs may improve the overall biological profile. The detailed pharmacokinetic and further biological evaluations may be warranted to assess the full potential of these prodrugs.
3. Materials and methods
3.1. Chemistry
DOT was synthesized in our laboratory as described previously.11 Bis-dPEG8 acid was purchased from Quanta BioDesign, Ltd. (Powell, Ohio). Porcine esterase and other chemicals were obtained from Sigma Chemical Company (St. Louis, MO) and Advanced Chem. Tech. (Louisville, KY). Melting points were determined on a MELl-TEMPII apparatus and were uncorrected. TLC was performed on Uniplate (silica gel) purchased from Anal-tech Co. Column chromatography was performed using either silica gel-60 (220–240 mesh) for flash chromatography or silica gel G (TLC grade >440 mesh) for vacuum flash column chromatography. NMR spectra were recorded on Varian Inova 500 spectrometer at 500 MHz with software VNMRC6–1 with Me4Si as the internal standard. Chemical shifts (d) are reported as s (single), d (doublet), t (triplet), q (quartet), m (multiplet, or b (broad). UV spectra were recorded on a Beckman DU-650 spectrophotometer. Optical rotations were measured on a Jasco DIP-370 digital polarimeter. Mass spectra were measured on a Micromass Autospec mass spectrometer. Elemental analyses were performed by Atlantic Microlab, Inc. (Norcross, GA). HPLC were performed using Waters 2996 instrument manufactured by Waters Corporation with an analytical symmetry C18 column (4.6 × 75 mm) and acetonitrile/water mixture as eluent.
3.2. General procedure for the synthesis of 5′-O-aliphatic acid ester of DOT (1–8)
A solution of DOT (1 mmol), aliphatic acid (2 mmol), DMAP (2 mmol) and 1,3-diisopropylcarbodiimide (DIC) (2 mmol) in dichloromethane (10 mL) was stirred at room temperature for 6 to 15 h. Dichloromethane (10 mL) was added and the reaction mixture was washed with aqueous sodium bicarbonate solution (2 × 10 mL) followed by water (2 × 10 mL). The organic layer was dried over anhydrous Na2SO4, and concentrated under reduced pressure. The residue was purified using flash silica gel chromatography to provide the desired product. The pure product was obtained by crystallizing from the mixture of ethyl acetate and hexane (1:1) solution.
3.2.1. Dioxolane thymine-5′-O-acetic ester (1)
Yield: 98% Mp: 190.5 °C. 1H NMR (CDCl3): δ 1.91 (s, 3H, 5-Me), 2.05 (s, 3H, CH3COO-), 4.16–4.22 (m, 2H, H5′), 4.34–4.43 (m, 2H, H2′), 5.17 (s, 1H, H4′), 6.21 (m, 1H, H1′), 7.31 (s, 1H, H6), 8.44 (s, 1H, H3). MS: m/z 271 (M+1)+ Anal. calcd for C11H14N2O6: C 48.89; H 5.22; N 10.37; Found: C 48.80; H 5.26; N 10.17.
3.2.2. Dioxolane thymine-5′-O-valeric ester (2)
Yield: 95% Mp: 162.9 °C. 1H NMR (CDCl3): δ 0.92 (t, 3H, CH3-), 1.25–1.52 (m, 4H, −CH2-), 1.90 (s, 3H, 5-Me), 2.35 (m, 2H, CH2-COO), 4.18–4.20 (m, 2H, H5′), 4.30–4.40 (m, 2H, H2′), 5.15 (s, 1H, H4′), 6.20 (m, 1H, H1′), 7.28 (s, 1H, H6), 8.40 (s, 1H, H3). MS: m/z 313 (M+1)+ Anal. calcd for C14H20N2O6: C 53.84; H 6.45; N 8.97; Found: C 53.86; H 6.52; N 8.89.
3.2.3. Dioxolane thymine-5′-O-octanotic ester (3)
Yield: 94% Mp: 158.0 °C. 1H NMR (CDCl3): δ 0.92 (t, 3H, CH3), 1.30–1.52 (m, 10H, CH2), 1.91 (s, 3H, 5-Me), 2.33 (m, 2H, CH2COO), 4.16–4.24 (m, 2H, H5′), 4.34–4.44 (m, 2H, H2′), 5.16 (s, 1H, H4′), 6.24 (m, 1H, H1′), 7.34 (s, 1H, H6), 8.46 (s, 1H, H3). MS: m/z 355 (M+1)+ Anal. calcd for C17H26N2O6: C 57.61; H 7.39; N 7.90; Found: C 57.63; H 7.47; N 7.84.
3.2.4. Dioxolane thymine-5′-O-dodecanoic ester (4)
Yield: 94% Mp: 146.5 °C. 1H NMR (CDCl3): δ 0.92 (t, 3H, CH3), 1.26 (m, 16H, CH2), 1.53 (m, 2H, CH2-CH2-COO), 1.91 (s, 3H, 5-Me), 2.33 (m, 2H, CH2COO-), 4.14–4.21(m, 2H, H5′), 4.37–4.45 (m, 2H, H2′), 5.21 (s, 1H, H4′), 6.23 (m, 1H, H1′), 7.36 (s, 1H, H6), 8.78(s, 1H, H3). MS: m/z 412 (M+1)+ Anal. calcd for C20H34N2O6: C 61.44; H 8.35; N 6.82; Found: C 61.17; H 8.36; N 6.58.
3.2.5. Dioxolane thymine-5′-O-palmitic ester (5)
Yield: 93% Mp: 142.2 °C. 1H NMR (CDCl3): δ 0.90 (t, 3H, CH3), 1.28–1.34 (m, 24H, CH2), 1.54 (m, 2H, CH2-CH2-COO), 1.91 (s, 3H, 5-Me), 2.29 (m, 2H, CH2COO), 4.18–4.25 (m, 2H, H5′), 4.33–4.46 (m, 2H, H2′), 5.19 (s, 1H, H4′), 6.20 (m, 1H, H1′), 7.35 (s, 1H, H6), 8.48 (s, 1H, H3). MS: m/z 468 (M+1)+ Anal. calcd for C25H42N2O6: C 64.35; H 9.07; N 6.00; Found: C 64.44; H 9.20; N 6.05.
3.2.6. Dioxolane thymine-5′-O-pivalic ester (6)
Yield: 91% Mp: 172.3 °C. 1H NMR (CDCl3): δ 0.98 (s, 9H, (CH3)3-), 1.92 (s, 3H, 5-Me), 4.16–4.21 (m, 2H, H5′), 4.33–4.41(m, 2H, H2′), 5.17 (s, 1H, H4′), 6.23 (m, 1H, H1′), 7.32 (s, 1H, H6), 8.79 (s, 1H, H3). MS: m/z 313 (M+1)+ Anal. calcd for C14H20N2O6: C 53.84; H 6.45; N 8.97; Found: C 53.94; H 6.47; N 8.87.
3.2.7. Dioxolane thymine-5′-O-cyclopropanecarboxylic ester (7)
Yield: 87% Mp: 171.5 °C. 1H NMR (CDCl3): δ 0.94 (m, 2H, CH2), 1.07 (m, 2H, CH2), 1.66 (m, 1H, −CHCOO), 1.91 (s, 3H, 5-Me), 4.17–4.24 (m, 2H, H5′), 4.31–4.41 (m, 2H, H2′), 5.19 (s, 1H, H4′), 6.24 (m, 1H, H1′), 7.33 (s, 1H, H6), 8.93(s, 1H, H3). MS: m/z 297 (M+1)+ Anal. calcd for C13H16N2O6 0.2%H2O: C 52.82; H 5.65; N 8.92; Found: C 53.21; H 5.54; N 9.28.
3.2.8. Dioxolane thymine-5′-O-cyclohexanecarboxylic ester (8)
Yield: 90%. Mp: 176.5 °C. 1H NMR (CDCl3): δ 1.20–1.88 (m, 10H, CH2), 2.34 (m, 1H, CHCOO), 1.89 (s, 3H, 5-Me), 4.15–4.20 (m, 2H, H5′), 4.34–4.45 (m, 2H, H2′), 5.18 (s, 1H, H4′), 6.22 (m, 1H, H1′), 7.35 (s, 1H, H6), 8.95 (s, 1H, H3). MS: m/z 339 (M+1)+ Anal. calcd for C16H22N2O6: C 56.80; H 6.55; N 8.28; Found: C 57.01; H 6.58; N 8.26.
3.2.9. Dioxolane thymine-5′-O-pivaloyloxymethyl ether (9)
A solution of DOT (1 mmol), chloromethyl pivalate (1.5 mmol), NaH (1.5 mmol) in DMF (10 mL) was stirred at room temperature for 5 h. Reaction mixture was cooled to 0 °C and methanol (2 mL) was added. After usual work-up, the residue was purified using flash silica gel chromatography to afford the desired product. The compound was obtained by crystallizing with the mixture of ethyl acetate and hexane (1:1) solution. Yield: 86%. Mp: 115.3 °C. 1H NMR (CDCl3): δ 0.98 (s, 9H, CH3), 1.92 (s, 3H, 5-Me), 4.12–4.22 (m, 2H, H5′), 4.41 (s, 2H, OCH2O), 4.36–4.45 (m, 2H, H2′), 5.17 (s, 1H, H4′), 6.20 (m, 1H, H1′), 7.35 (s, 1H, H6), 8.44 (s, 1H, H3). MS: m/z 343 (M+1)+ Anal. calcd for C15H22N2O7: C 52.63; H 6.48; N 8.18; Found: C 52.44; H 6.41; N 8.10.
3.2.10. Dioxolane thymine-5′-O-bis-PEG8 ester (10)
The same procedure given for the compounds 1-8 was followed with bis-dPEG8 acid/DOT (1/2, mol/mol). Yield: 95% (for bis-dPEG8 acid). Mp: 85.2 °C. 1H NMR (CDCl3): δ 1.92 (s, 3H, 5-Me), 2.24 (m, 4H, CH2), 3.89 (m, 28H, CH2O), 4.12–4.20 (m, 4H, H5′), 4.31–4.40 (m, 4H, H2′), 5.15 (s, 2H, H4′), 6.31 (m, 2H, H1′), 7.28 (s, 2H, H6), 8.38 (s, 2H, H3). MS: m/z 848 (M+1)+Anal. calcd for C36H54N4O19 0.3%H2O: C 50.74; H 6.46; N 6.57; Found: C 50.38; H 6.46; N 6.56.
3.2.11. General procedure for the syntheses of 5′-O-amino acid ester of DOT (11–22)
A solution of DOT (1 mmol), Boc-protected amino acid (2.5 mmol), DMAP (2.5 mmol) and 1,3-diisopropylcarbodiimide (DIC) (2.5 mmol) in dichloromethane (10 mL) was stirred at room temperature from 6 to 16 h. Dichloromethane (10 mL) was added and the mixture was washed with aqueous sodium bicarbonate solution (2 × 10 mL) and water (2 × 10 mL). The organic layer was dried over anhydrous Na2SO4, and concentrated under reduced pressure. The residue was purified by flash silica gel chromatography to provide dioxolane thymine 5′-Boc-protected amino acid esters. The 5′-Boc-protected amino acid prodrug (100 mg) was dissolved in dichloromethane (10 mL) and the solution was cooled to 0 °C. Trifluoroacetic acid (5 mL) was added to the solution with vigorous stirring. The mixture was warmed to room temperature and then stirred for 1 h. The excess trifluoroacetic acid was removed under reduced pressure. The solid was dissolved in methanol (10 mL) and the solution was cooled to 0 °C. A mixture of 1.0 M HCl/ether (5 mL) was added with a vigorous stirring. After 20 min, the excess HCl/ether was removed and the solution was concentrated under reduced pressure. The pure DOT-5′-amino acid ester hydrochloride was obtained as an amorphous powder from methanol.
3.2.12. Dioxolane thymine-5′-O-glycyl ester hydrochloride (11)
Yield: 91% 1H NMR (CDCl3): δ 1.95 (s, 3H, 5-Me), 3.82–3.96 (m, 2H, CH2N), 4.21–4.42 (m, 2H, H5′ and H′), 5.17 (s, 1H, H4′), 6.38 (m, 1H, H1′), 7.55 (s, 1H, H6). MS: m/z 286 (M+1)+ Anal. calcd for C11H16ClN3O6 0.1% H2O: C 41.43; H 5.12; N 12.71; Found: C 41.30; H 5.11; N 12.52.
3.2.13. Dioxolane thymine-5′-O-isobutyric ester hydrochloride (12)
Yield: 94% 1H NMR (MeOH-d4): δ 1.61 (s, 3H, CH3), 1.64 (s, 3H, CH3), 1.91 (s, 3H, 5-Me), 4.21–4.57 (m, 4H, H2′ and H5′), 5.26 (s, 1H, H4′), 6.39 (m, 1H, H1′), 7.48 (s, 1H, H6). MS: m/z 314 (M+1)+ Anal. calcd for C13H20ClN3O6 0.2 H2O%: C 44.19; H 5.82; N 11.89; Found: C 43.90; H 5.91; N 11.75.
3.2.14. Dioxolane thymine-5′-O-l-alanyl ester hydrochloride (13)
Yield: 92% 1H NMR (MeOH-d4): δ 1.58 (d, 3H, CH3), 1.92 (s, 3H, 5-Me), 4.12 (m, 1H, CHN), 4.21–4.61 (m, 4H, H2′ and H5′), 5.23 (s, 1H, H4′), 6.39 (m, 1H, H1′), 7.55 (s, 1H, H6). MS: m/z 300 (M+1)+Anal. calcd for C12H18ClN3O6 0.3H2O%: C 42.25; H 5.50; N 12.32; Found: C 42.50; H 5.26; N 11.97.
3.2.15. Dioxolane thymine-5′-O-l-phenylalanyl ester hydrochloride (14)
Yield: 98% 1H NMR (CDCl3): δ 1.92 (s, 3H, 5-Me), 3.01 (m, 2H, PhCH2), 3.74 (m, 1H, CHN), 4.17–4.45 (m, 4H, H5′ and H2′), 5.17 (s, 1H, H4′), 6.37 (m, 1H, H1′), 7.18–7.32 (m, 5H, Ph), 7.51 (s, 1H, H6). MS: m/z 376 (M+1)+ Anal. calcd for C18H22ClN3O6 1.1 H2O% C 50.02; H 5.65; N 9.73; Found: C 49.75; H 5.23; N 9.58.
3.2.16. Dioxolane thymine-5′-O-d-phenylalanyl ester hydrochloride (15)
Yield: 97% 1H NMR (CDCl3): δ 1.91 (s, 3H, 5-Me), 3.08 (m, 2H, PhCH2), 3.72 (m, 1H, CHN), 4.15–4.47 (m, 4H, H5′ and H2′), 5.16 (s, 1H, H4′), 6.38 (m, 1H, H1′), 7.17–7.34 (m, 5H, Ph), 7.52 (s, 1H, H6). MS: m/z 376 (M+1)+ Anal. calcd for C18H22ClN3O6 1.0 H2O%: C 50.09; H 5.65; N 9.73; Found: C 49.97; H 5.54; N 9.42.
3.2.17. Dioxolane thymine-5′-O-l-valyl ester hydrochloride (16)
Yield: 96% 1H NMR (CDCl3): δ 1.10–1.12 (d, 6H, CH3), 1.91 (s, 3H, 5-Me), 2.61 (m, 1H, CH), 3.96 (m, 1H, CHN), 4.31–4.61 (m, 4H, H5′ and H2′), 5.32 (s, 1H, H4′), 6.36(m, 1H, H1′), 7.52 (s, 1H, H6). MS: m/z 328 (M+1)+ Anal. calcd for C14H22ClN3O6: C 46.22; H 6.10; N 11.55; Found: C 46.37; H 6.18; N 11.35.
3.2.18. Dioxolane thymine-5′-O-d-valyl ester hydrochloride (17)
Yield: 93% 1H NMR (CDCl3): δ 1.19 (d, 3H, CH3), 1.21 (d, 3H, CH3), 1.91 (s, 3H, 5-Me), 2.33 (m, 1H, CH), 3.98 (m, 1H, CHN), 4.2–4.55 (m, 4H, H5′ and H2′), 5.17 (s, 1H, H4′), 6.40 (m, 1H, H1′), 7.51 (s, 1H, H6). MS: m/z 328 (M+1)+ Anal. calcd for C14H22ClN3O6 0.7H2O%: C 44.46; H 6.29; N 11.11; Found: C 44.37; H 6.30; N 11.09.
3.2.19. Dioxolane thymine-5′-O-l-leucinyl ester hydrochloride (18)
Yield: 96% 1H NMR (CDCl3): δ 1.01–1.04 (dd, 6H, CH3), 1.73 (m, 1H, CH), 1.82 (m, 2H, CH2), 1.93 (s, 3H, 5-Me), 4.08 (m, 1H, CHN), 4.22–4.57 (m, 4H, H5′ and H2′), 5.27 (s, 1H, H4′), 6.37 (m, 1H, H1′), 7.48 (s, 1H, H6). MS: m/z 342 (M+1)+ Anal. calcd for C15H24ClN3O6 0.5H2O%: C 46.57; H 6.51; N 10.86; Found: C 46.49; H 6.19; N 10.52.
3.2.20. Dioxolane thymine-5′-O-l-isoleucinyl ester hydrochloride (19)
Yield: 97% 1H NMR (CDCl3): δ 1.01 (t, 3H, CH3), 1.32 (d, 3H, CH3), 1.70 (m, 2H, CH2), 1.94 (s, 3H, 5-Me), 2.05 (m, 1H, CH), 4.08 (m, 1H, CHN), 4.22–4.61 (m, 4H, H5′ and H2′), 5.27 (s, 1H, H4′), 6.37 (m, 1H, H1′), 7.52 (s, 1H, H6). MS: m/z 342 (M+1)+ Anal. calcd for C14H21ClN4O7: C 47.68; H 6.40; N 11.12; Found: C 47.69; H 6.46; N 11.02.
3.2.21. Dioxolane thymine-5′-O-l-Glutminyl ester hydrochloride (20)
Yield: 91% 1H NMR (MeOH-d4): δ 1.93 (s, 3H, 5-Me), 2.17 (m 2H, CH2), 2.34 (m, 2H, CH2), 4.18 (m, 1H, CHN), 4.22–4.55 (m, 4H, H5′ and H2′), 5.19 (s, 1H, H4′), 6.38 (m, 1H, H1′), 7.48 (s, 1H, H6), 9.80 (s, 1H, 3-NH). MS: m/z 357 (M+1)+ Anal. calcd for C14H21ClN4O7 0.2H2O%: C 42.42; H 5.44; N 14.13; Found: C 42.15; H 5.34; N 13.94.
3.2.22. Dioxolane thymine-5′-O-l-prolyl ester hydrochloride (21)
Yield: 90% 1H NMR (MeOH-d4): δ 1.97 (s, 3H, 5-Me), 2.24 and 2.45 (m, 4H, CH2-CH2), 3.42 (m, 2H, CH2-N), 4.22 (m, 1H, CHN), 4.37–4.61 (m, 4H, H5′ and H2′), 5.27 (s, 1H, H4′), 6.37 (m, 1H, H1′), 7.52 (s, 1H, H6). MS: m/z 355 (M+1)+ Anal. calcd for C15H22ClN4O6 0.4H2O%: C 45.57; H 5.68; N 11.39; Found: C 45.28; H 5.95; N 10.99.
3.2.23. Dioxolane thymine-5′-O-l-tryptophanyl ester hydrochloride (22)
Yield: 87% 1H NMR (CD3OD): δ 1.98 (s, 3H, 5-Me), 3.42–3.50 (m, 2H, CH2), 4.17 (m, 1H, CHN), 4.27–4.54 (m, 4H, H5′ and H2′), 5.17 (s, 1H, H4′), 6.37 (m, 1H, H1′), 7.02–7.60 (m, 6H, Try H). MS: m/z 415 (M+1)+ Anal. calcd for C20H24Cl2N4O6: C 49.29; H 4.96; N 11.50; Found: C 49.67; H 5.33; N 11.42.
3.3. Virology
3.3.1. Antiviral assays
The procedures for the antiviral assays in human PBM cells have been published previously.33,34 Briefly, PBM cells were isolated by Ficoll-Hypaque discontinuous gradient centrifugation of whole blood samples (obtained from Atlanta Red Cross) from healthy seronegative donors. Cells were stimulated with phytohemagglutinin A (3 μg/mL; Sigma-Aldrich, St. Louis, MO) for 2–3 days prior to use. HIV-1LAI obtained from the Centers for Disease Control and Prevention (Atlanta, GA) was used as the standard reference virus for the antiviral assays. Infections were done in bulk for 1 h, either with 100 TCID50/1 × 107 cells for a flask (T25) assay or with 200 TCID50/2 × 106 cells/well for 24 well plate assay. Previous studies indicated that this was the optimum virus concentration in order to obtain good replication on day 5 when the virus is harvested from the supernatant. Cells were added to a flask or plate containing a 10-fold serial dilution of the test compound. Assay medium was RPMI-1640 supplemented with heat inactivated 16% fetal bovine serum, 1.6 mM l-glutamine, 80 IU/mL penicillin, 80 μg/mL streptomycin, 0.0008% DEAE-Dextran, 0.045% sodium bicarbonate, and 26 IU/mL recombinant interleukin-2 (Chiron Corp., Emeryville, CA). AZT was used as the positive control for all the virologic assays. Uninfected PBM cells were grown in parallel at equivalent cell concentrations as a control. The cell cultures were maintained in a humidified 5% CO2-air at 37 °C for 5 day, and supernatants were collected for reverse transcriptase activity. One mL of each supernatant was centrifuged at 9740g for 2 h to pellet the virus. The pellet was solubilized with vortexing in 100 μL of virus solubilization buffer (0.5% Triton X-100, 0.8 M NaCl, 0.5 mM phenylmethylsulfonyl chloride, 20% glycerol, and 0.05 M Tris, pH 7.8). Each sample (10 μL) was added to 75 μL of RT reaction mixture (0.06 M Tris, pH 7.8, 0.012 M MgCl2, 0.006 M dithiothreitol, 0.006 mg/mL poly (rA)n oligo (dT)12–18, 96 μg/mL dATP, and 1 μM of 0.08 mCi/mL 3H-thymidine-05′-triphosphate) (Perkin Elmer, Boston, MA) and incubated at 37 °C for 2 h. The reaction was stopped by the addition of 100 μL of 10% trichloroacetic acid containing 0.05% sodium pyrophosphate. The acid-insoluble product was harvested onto filter paper using a Packard Harvester (Meriden, CT), and RT activity was read on a Packard Direct Beta Counter (Meriden, CT). RT results were expressed in counts per minute (CPM) per milliliter. The antiviral 50% effective concentration (EC50) and 90% effective concentration (EC90) was determined from the concentration-response curve using the median effect method.35
3.3.2. Cytotoxicity assays
The compounds were evaluated for their potential toxic effects on uninfected PHA-stimulated human PBM cell, CEM (T-lymphoblastoid cell line obtained from American Type Culture Collection, Rockville, MD) and Vero (African green monkey kidney) cells. The log phase Vero, CEM and PHA-stimulated human PBM cells were seeded at a density of 5 × 103, 2.5 × 103, and 5 × 104 cells/well, respectively. All of the cells were plated in 96-well cell culture plates containing 10-fold serial dilutions of the test drug. The cultures were incubated for 2, 3, and 4 days for Vero, CEM, and PBM cells, respectively, in a humidified 5% CO2-air at 37 °C. At the end of incubation, MTT tetrazolium dye solution (cell titer 96, Promega, Madison, WI) was added to each well and incubated overnight. The reaction was stopped with stop solubilization solution (Promega, Madison, WI). The plates were incubated for 5 h to ensure that the formazan crystals were dissolved. The plates were read at 570 nm using an ELISA plate reader (Bio-Tek Instruments, Inc., Winooski, VT, Model EL 312e). The 50% inhibition concentration (IC50) was determined from the concentration-response curve using the median effect method.35,36
3.4. Hydrolysis of prodrugs
3.4.1. Kinetics in aqueous buffer
Hydrolysis of prodrugs was studied at pH 2.0 and pH 7.4. An aliquot (1 mL) of 1 mM solution of prodrugs in DMSO was diluted to 10 mL using phosphate buffer (10 mM). A constant ionic strength (μ) of 0.1 was maintained by the addition of an appropriate quantity of NaCl to the solutions. The double dilution with the buffer solutions yielded to ester prodrugs solution, which contain 5% DMSO (v/v) and were maintained at 37 ± 0.5 °C in screw-capped vials in a water bath. The samples were withdrawn at appropriate time intervals, filtered and the filtrate was analyzed by HPLC.
3.4.2. Enzyme study
The prodrug stock solution (2.8 mM) was prepared by dissolving into 1:0.1 mixture of Tris-HCl buffer (pH = 8.0) and DMSO. Procine esterase stock solution (40 μg/mL) was made using Tris-HCl buffer (pH = 8.0). The mixing of the equal amount of nucleoside prodrug solution and procine esterase solution yielded to nucleoside prodrugs solution (1.4 mM) in Tris-HCl buffer (pH = 8.0) containing 5% DMSO (v/v) along with 20 μg/mL porcine esterase. The final nucleoside solution was further incubated at 37 ± 0.5 °C. Aliquots were removed at intervals, quenched with ice-cold methanol/acetonitrile 1/1 (v/v), filtered and analyzed by HPLC. The enzyme activity was determined using ethyl butyrate as substrate with HPLC to monitor substrate disappearance.
Acknowledgments
This research was supported by the US Public Health Service Grant 4R37-AI25899 and by the Department of Veterans Affairs.
References and notes
- 1.De Clercq E J. Clin. Virol 2001, 22, 73. [DOI] [PubMed] [Google Scholar]
- 2.Rautio J; Kumpulainen H; Heimbach T; Oliyai R; Oh D; Jarvinen T; Savolainen J Nat. Rev. Drug Discov 2008, 7, 255. [DOI] [PubMed] [Google Scholar]
- 3.Li F; Maag H; Alfredson T J. Pharm. Sci 2008, 97, 1109. [DOI] [PubMed] [Google Scholar]
- 4.Heimbach T; Fleisher D; Kaddoumi A In Prodrugs, 2007; pp. 157–215. [Google Scholar]
- 5.Kryczka T; Kazimierczuk Z; Kozlowska M; Chrapusta SJ; Vilpo L; Vilpo J; Stachnik K; Janisz M; Grieb P Anticancer Drugs 2007, 18, 301. [DOI] [PubMed] [Google Scholar]
- 6.Beaumont K; Webster R; Gardner I; Dack K Curr. Drug Metab 2003, 4, 461. [DOI] [PubMed] [Google Scholar]
- 7.Song X; Vig BS; Lorenzi PL; Drach JC; Townsend LB; Amidon GL J. Med. Chem 2005, 48, 1274. [DOI] [PubMed] [Google Scholar]
- 8.Lorenzi PL; Landowski CP; Song X; Borysko KZ; Breitenbach JM; Kim JS; Hilfinger JM; Townsend LB; Drach JC; Amidon GL J. Pharmacol. Exp. Ther 2005, 314, 883. [DOI] [PubMed] [Google Scholar]
- 9.Ettmayer P; Amidon GL; Clement B; Testa B J. Med. Chem 2004, 47, 2393. [DOI] [PubMed] [Google Scholar]
- 10.Satoh T; Taylor P; Bosron WF; Sanghani SP; Hosokawa M; La Du BN Drug Metab. Dispos 2002, 30, 488. [DOI] [PubMed] [Google Scholar]
- 11.Chu CK; Ahn SK; Kim HO; Beach JW; Alves AJ; Jeong LS; Islam Q; Roey PV; Schinazi RF Tetrahedron Lett. 1991, 32, 3791. [Google Scholar]
- 12.Jeong LS; Schinazi RF; Beach JW; Kim HO; Nampalli S; Shanmuganathan K; Alves AJ; McMillan A; Chu CK; Mathis R J. Med. Chem 1993, 36, 181. [DOI] [PubMed] [Google Scholar]
- 13.Jeong LS; Schinazi RF; Beach JW; Kim HO; Shanmuganathan K; Nampalli S; Chun MW; Chung WK; Choi BG; Chu CK J. Med. Chem 1993, 36, 2627. [DOI] [PubMed] [Google Scholar]
- 14.Kim HO; Schinazi RF; Nampalli S; Shanmuganathan K; Cannon DL; Alves AJ; Jeong LS; Beach JW; Chu CK J. Med. Chem 1993, 36, 30. [DOI] [PubMed] [Google Scholar]
- 15.Kim HO; Schinazi RF; Shanmuganathan K; Jeong LS; Beach JW; Nampalli S; Cannon DL; Chu CK J. Med. Chem 1993, 36, 519. [DOI] [PubMed] [Google Scholar]
- 16.Kim HO; Ahn SK; Alves AJ; Beach JW; Jeong LS; Choi BG; Van Roey P; Schinazi RF; Chu CK J. Med. Chem 1992, 35, 1987. [DOI] [PubMed] [Google Scholar]
- 17.Furman PA; Jeffrey J; Kiefer LL; Feng JY; Anderson KS; Borroto-Esoda K; Hill E; Copeland WC; Chu CK; Sommadossi JP; Liberman I; Schinazi RF; Painter GR Antimicrob. Agents Chemother 2001, 45, 158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lennerstrand J; Chu CK; Schinazi RF Antimicrob. Agents Chemother 2007, 51, 2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chu CK; Yadav V; Chong YH; Schinazi RF J. Med. Chem 2005, 48, 3949. [DOI] [PubMed] [Google Scholar]
- 20.Murphy R; Zala C; Ochoa C; Tharnish P; Mathew J; Fromentin E; Asif G; Hurwitz S; Kivel N; Schinazi R In 15th Conference on Retroviruses and Opportunistic Infections: Boston, MA, USA, 2008. [Google Scholar]
- 21.Chen H; Boudinot FD; Chu CK; McClure HM; Schinazi RF Antimicrob. Agents Chemother 1996, 40, 2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen H; Schinazi RF; Rajagopalan P; Gao Z; Chu CK; McClure HM; Boudinot FD AIDS Res. Hum. Retroviruses 1999, 15, 1625. [DOI] [PubMed] [Google Scholar]
- 23.Menne S; Asif G; Narayanasamy J; Butler SD; George AL; Hurwitz SJ; Schinazi RF; Chu CK; Cote PJ; Gerin JL; Tennant BC Antimicrob. Agents Chemother 2007, 51, 3177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Asif G; Hurwitz SJ; Obikhod A; Delinsky D; Narayanasamy J; Chu CK; McClure HM; Schinazi RF Antimicrob. Agents Chemother 2007, 51, 2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liang Y; Narayanasamy J; Schinazi RF; Chu CK Bioorg. Med. Chem 2006, 14, 2178. [DOI] [PubMed] [Google Scholar]
- 26.Liang Y; Narayanasamy J; Rapp KL; Schinazi RF; Chu CK Antiviral Chem. Chemother 2006, 17, 321. [DOI] [PubMed] [Google Scholar]
- 27.Ibrahim SS; Boudinot FD; Schinazi RF; Chu CK Antiviral Chem. Chemother 1996, 7, 167. [Google Scholar]
- 28.Menger FM; Guo Y; Lee AS Bioconjug. Chem 1994, 5, 162. [DOI] [PubMed] [Google Scholar]
- 29.von Briesen H; Ramge P; Kreuter J AIDS Rev. 2000, 2, 31. [Google Scholar]
- 30.Lai L; Xu Z; Zhou J; Lee K-D; Amidon GL J. Biol. Chem 2008, 283, 9318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Han HK; Amidon GL AAPS PharmSci 2000, 2, E6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Denny WA Eur. J. Med. Chem 2001, 36, 577. [DOI] [PubMed] [Google Scholar]
- 33.Schinazi RF; Sommadossi JP; Saalmann V; Cannon DL; Xie MY; Hart GC; Smith GA; Hahn EF Antimicrob. Agents Chemother 1990, 34, 1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schinazi RF; Cannon DL; Arnold BH; Martino-Saltzman D Antimicrob. Agents Chemother 1988, 32, 1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Belen’kii MS; Schinazi RF Antiviral Res. 1994, 25, 1. [DOI] [PubMed] [Google Scholar]
- 36.Stuyver LJ; Lostia S; Adams M; Mathew JS; Pai BS; Grier J; Tharnish PM; Choi Y; Chong Y; Choo H; Chu CK; Otto MJ; Schinazi RF Antimicrob. Agents Chemother 2002, 46, 3854. [DOI] [PMC free article] [PubMed] [Google Scholar]