

Abstract
Small molecule potent IRAK4 inhibitors from a novel bicyclic heterocycle class were designed and synthesized based on hits identified from Aurigene’s compound library. The advanced lead compound, CA-4948, demonstrated good cellular activity in ABC DLBCL and AML cell lines. Inhibition of TLR signaling leading to decreased IL-6 levels was also observed in whole blood assays. CA-4948 demonstrated moderate to high selectivity in a panel of 329 kinases as well as exhibited desirable ADME and PK profiles including good oral bioavailability in mice, rat, and dog and showed >90% tumor growth inhibition in relevant tumor models with excellent correlation with in vivo PD modulation. CA-4948 was well tolerated in toxicity studies in both mouse and dog at efficacious exposure. The overall profile of CA-4948 prompted us to select it as a clinical candidate for evaluation in patients with relapsed or refractory hematologic malignancies including non-Hodgkin lymphoma and acute myeloid leukemia.
Keywords: IRAK4, bicyclic heterocycles, SAR, CA-4948, AML, DLBCL
Interleukin-1 receptor associated kinases (IRAKs) are serine/threonine protein kinases belonging to the tyrosine-like kinase (TLK) family. Among the members of the IRAK family, only IRAK1 and IRAK4 exhibit kinase activity.1,2 IRAKs are downstream effectors of Toll-like receptor (TLR) and interleukin-1 receptor (IL-1R) pathways and play an important role in innate immune signaling. TLR stimulation leads to recruitment of MYD88, an adaptor molecule, to the activated receptor complex, which then complexes with IRAK4 and activates IRAK1. TRAF6 is then activated by IRAK1 leading to NFkB activation.2 Dysregulated activation of the IRAK pathway in cancer cells further contributes to disease progression through inflammation of the tumor microenvironment. Waldenstrom’s Macroglobulinemia (WM) and a subset of activated B cell such as diffuse large B cell lymphomas (ABC DLBCLs) are characterized by oncogenic mutations in MYD88 that result in constitutive activation of the NFkB pathway.3,4 TLRs and their associated signal transducers are frequently overexpressed and/or constitutively activated in myelodysplastic syndromes (MDS). IRAK1 overexpression and activation are also observed in acute myeloid leukemia (AML).5 Finally, the overexpression of the oncogenic long form of IRAK4 (IRAK4-L) has been found in over half of cases of AML and MDS and portends worse prognosis.6,7 Thus, IRAKs are attractive therapeutic targets for the treatment of MDS, AML, and other tumors with altered innate immune signaling.
Since the validation of IRAK4 as a drug target,8,9 there have been a number of public disclosures on potent, selective, and structurally diverse inhibitors that can be potentially utilized for treatment of different disease conditions such as chronic inflammatory diseases, myeloproliferative disorders, and cancer.10−15 Among these, Pfizer’s PF-0665083313 (Figure 1) was the first inhibitor to enter the clinic16 and is currently under investigation in Phase 2 trials for the treatment of rheumatoid arthritis and hidradenitis suppurativa.17,18 Apart from PF-06650833, four other compounds have also entered clinical development during the past few years and are currently in phase 1 trials, viz., BAY-1830839,19 BAY-183484520 (Bayer), R-83521 (Rigel), and our chemical matter, CA-494822 (Figure 1). The Bayer and Rigel compounds are currently under evaluation for treatment of autoimmunity, inflammation, psoriasis, and ABC-DLBCL21,23,24 whereas CA-4948 is being tested in patients with relapsed or refractory hematologic malignancies including non-Hodgkin lymphoma, MDS, and AML.22 Herein, we disclose the design strategies and SAR optimization that led to identification of CA-4948.
Figure 1.

Clinical stage IRAK4 inhibitors with published chemical structures.
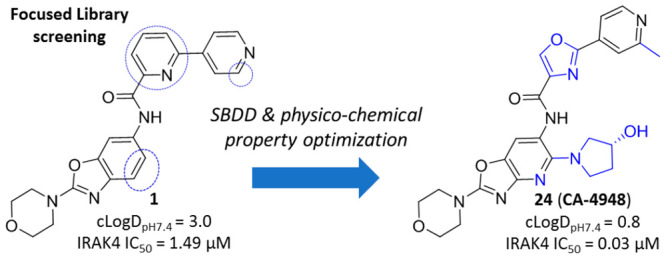
The in-house kinase focused library screening campaign at Aurigene resulted in identification of a few moderately potent IRAK-4 inhibitors. Among these, the cocrystal structure of an early benzoxazole amide hit (1) in complex with IRAK4 revealed the binding mode and critical active-site interactions of this class of compounds (Figure 2a). The amide carbonyl was observed to be the kinase hinge interacting moiety, and the benzoxazole oxygen was noticed to be within modest hydrogen bonding distance (3.4 Å) from Tyr264 in the same region. Apart from these, the bipyridyl appendage connected to the amide was found to be occupying a region close to the gatekeeper residue Tyr262 with the distal 4-pyridyl making a hydrogen bonding interaction with catalytic Lys213. Comparison with the binding mode of AMP-PNP in IRAK4 led to the identification of several potential design opportunities in order to improve the binding affinity of 1, e.g., by optimizing the interactions around Lys213, occupying the ribose pocket, etc. (Figure 2a). Therefore, a few analogs (2–5) were synthesized based on this strategy and interestingly all these compounds showed significant improvement in target inhibition with respect to 1, thereby validating these hypotheses. Out of these, 4 and 5 with ribose pocket extensions showed significantly improved target inhibition by over 150-fold. Among these, 5 showed IRAK4 IC50 < 5 nM and cellular IC50 < 100 nM (Table 1) but demonstrated poor aqueous solubility, pH 7.4 (<2 μM) and Caco-2 permeability (0.4 × 10–6 cm/s).
Figure 2.

Comparison of crystallographic binding modes in the IRAK4 kinase domain showing different active-site interactions in dotted lines: (a) 1 (magenta, RCSB PDB ID: 7C2W) and AMP-PNP (yellow, PDB ID: 2OID). Blue highlighted regions signify possible regions for modification of 1. (b) 1 (magenta, RCSB PDB ID: 7C2W) and CA-4948 (cyan, RCSB PDB ID: 7C2V). (c) Key interaction of CA-4948 in the IRAK4 kinase domain (cyan, RCSB PDB ID: 7C2V).
Table 1. Optimization of Benzoxazole Amide Hit and Evolution of Aza-benzoxazole Series.

Calculated using MarvinView6.1.7.
Inhibition of LTA induced TNFα release in THP-1 cells.
Not determined.
IC50’s not determined since the maximum IRAK4 inhibitions observed were less than 90%.
Shown as IC50 ± standard deviation (N = 2).
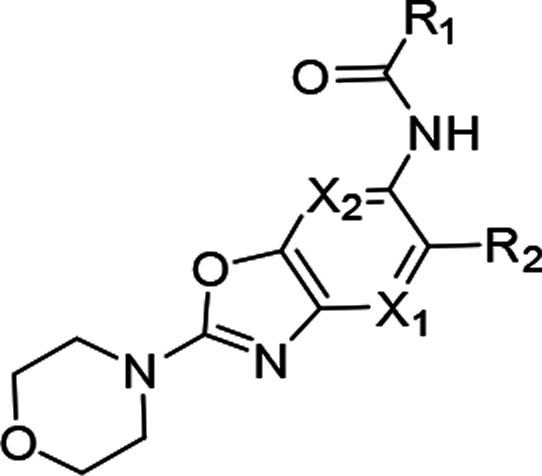
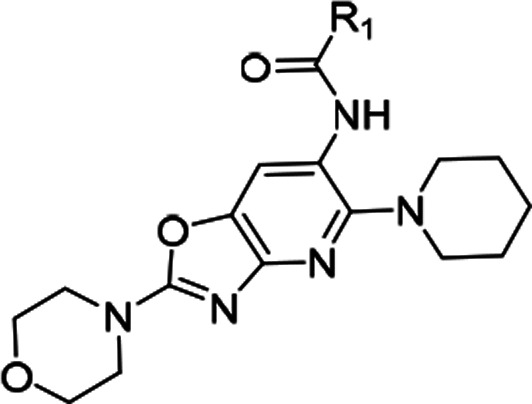
Compound 3 having marginally reduced lipophilicity (cLogDpH7.4 < 3) (Table 1) showed improvement in aqueous solubility, pH 7.4 (2.4 μM), and Caco-2 permeability (21.4 × 10–6 cm/s) relative to 5. This observation led us to explore the introduction of polarity in different parts of the molecule. We chose the core region first to start this exploration. Careful analysis of the binding mode of 1 in IRAK4 (Figure 2a) revealed that introduction of a polar atom in position 4 of the benzoxazole core is unlikely to affect the binding affinity because this region appeared to be far from any protein residues whereas the 7 position may not be amenable to such changes since it is closer to the hinge and may result in an unfavorable steric interference with the Met265 backbone carbonyl. We synthesized compounds 6 and 7 based on this hypothesis, and the results showed that introduction of the N atom at position 4 is most likely tolerated but not at position 7, since 6 showed significantly higher IRAK4 inhibition than 7 at 10 μM (Table 1). This modification also led to significant reduction of lipophilicity of the molecule (cLogDpH7.4 1.8) and consequent improvement in aqueous solubility, pH 7.4 (9.7 μM). We then considered 4-aza benzoxazole to be our core of choice, and compounds 8 and 9 (aza-analogs of compounds 5 and 4) were then synthesized with this core. Both retained potent IRAK4 inhibitory activity but 8 demonstrated better aqueous solubility, pH 7.4 (14.6 μM), in comparison to 9 (<2 μM). As a proof of this excellent IRAK4 inhibition, compound 8 also showed excellent inhibition of LTA induced TNFα in THP-1 cells. Given such good translation of enzymatic to cellular potency, some additional modifications were explored on 8 to lower cLogDpH7.4, and in the process, different modifications around the gatekeeper, Lys213-Asp329, and ribose pockets were designed. Among the different gatekeeper modifications explored (Table 2), 10 and 13–15 showed IRAK4 inhibition as good as 8 but 11 and 12 with a bulkier pyridyl in between the hydroxyl or amino pyrrolidine and hinge interacting moieties showed somewhat inferior inhibition likely due to steric interference of the terminal hydroxyl and primary amine respectively with the positively charged Lys213. Out of these, 13 and 15 turned out to be the best taking into account both cLogDpH7.4 and target inhibition. Although 14 also showed IRAK4 inhibition as good as 13 and 15 but cLogDpH7.4 was observed to be somewhat inferior (Table 2).
Table 2. Optimization of Compound 8 in the Gate-Keeper-Lys213-Asp329 Pocket.

Calculated using MarvinView6.1.7.
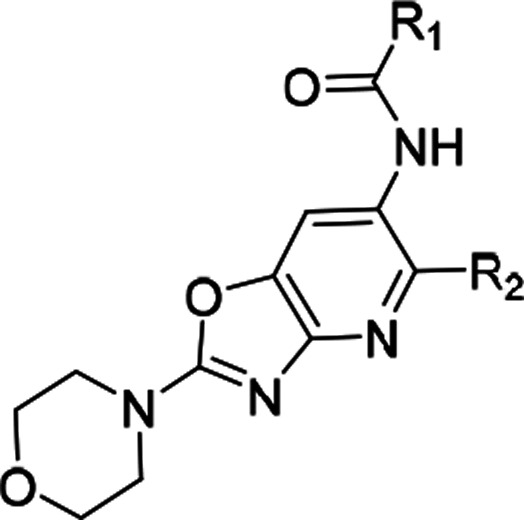
At this stage we chose these two gate-keeper region moieties and proceeded to explore SAR in the ribose pocket. A small library of analogs was synthesized taking these two chosen gate-keeper region moieties, and it was observed that polar and nonpolar groups were tolerated in this region, which was helpful for optimization of overall lipophilicity of these molecules. Initially 16 and 17 with a small ribose pocket group (R2) but different gate-keeper/back-pocket moieties (R1) were synthesized (Table 3). Since 16 showed comparatively better IRAK4 inhibition, this gate-keeper/back-pocket moiety (2-methylpyridine) was prioritized for further SAR generation. Compounds 16–25 (Table 3) were considered for in vitro ADME evaluation in order to identify leads for in vivo studies. Based on the ADME profile, we shortlisted compounds 24 and 25 as the most active analogs (IRAK4 IC50 < 30 nM) having reasonable solubility, good permeability, and desirable microsomal stability across tested species. Both of these enantiomeric analogs 24 and 25 showed good LTA induced TNFα inhibition in THP-1 cells (IC50 514 and 233 nM, respectively). However, 24 was found to be superior to 25 both in terms of selectivity in a small in-house kinase panel (Table 4) as well as in mouse PK (Table 5). 24 showed better clearance as well as AUC than 25, and moreover the excellent oral PK profile of 24 across preclinical species (Table 5) encouraged us to consider 24 for further evaluation.
Table 3. Ribose Pocket Modification of Compounds 13 and 15.

Calculated using MarvinView6.1.7.
Shown as IC50 ± standard deviation (N = 4).
Table 4. Percentage Inhibitions Measured for Key Compounds in Table 3 against Different Kinases in-House.
| FLT3 |
CDK2 |
AURA |
GSK3B |
MUSK |
||||||
|---|---|---|---|---|---|---|---|---|---|---|
| ID | 0.1 μMa | 1.0 μMa | 0.1 μMa | 1.0 μMa | 0.1 μMa | 1.0 μMa | 0.1 μMa | 1.0 μMa | 0.1 μMa | 1.0 μMa |
| 19 | 87 | 94 | 44 | 81 | 3 | 19 | 46 | 86 | 14 | 28 |
| 22 | 82 | 92 | 20 | 69 | 0 | 13 | 47 | 84 | 13 | 18 |
| 24 | 42 | 70 | 0 | 10 | 0 | 0 | 0 | 6 | 0 | 4 |
| 25 | 34 | 64 | 9 | 15 | 6 | 5 | 12 | 16 | 7 | 10 |
Indicates compound concentration.
Table 5. Pharmacokinetic Properties of 24 and 25a.
| Compound | Species | Dose (mg/kg) | Route | C0 (ng/mL) | AUC0-t (h × ng/mL) | VD (mL/kg) | CL (mL/h/kg) | T1/2 (h) | Cmax (ng/mL) | AUC0-t (h × ng/mL) | Tmax (h) | %F |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 24 | Mice | 3 | iv | 7482 | 5274 | 661 | 556 | 2.6 | ||||
| 25 | 788 | 928 | 5573 | 3134 | 0.8 | |||||||
| 24 | 10 | po | 6618 | 12464 | 0.5 | 71 | ||||||
| 25 | 2531 | 4489 | 0.3 | 100 | ||||||||
| 24 | Rat | 3 | iv | 27659 | 111567 | 2000 | 24 | 5.3 | ||||
| 24 | 10 | po | 22147 | 171258 | 1.2 | 46 | ||||||
| 24 | Dog | 1 | iv | 1963 | 6064 | 500 | 186 | 2.6 | ||||
| 24 | 10 | po | 12823 | 86465 | 1.17 | 100 |
Values represented as mean with sparse sampling technique. iv formulation (solution) includes 10% v/v ethanol, 30% v/v PEG, and 60% v/v water. po formulation (suspension) contains 50 parts of 1% v/v Tween 20 in water and 50 parts of 0.5% w/v hydroxy ethyl cellulose.
A cocrystal structure of 24 in complex with IRAK4 was resolved where the binding mode was observed to be similar to that of 1. Nevertheless, the Lys213 and Tyr264 polar contacts observed in the case of 1 were found to be absent here, and instead this compound was observed to be making a water mediated contact from the hydroxy group with Glu194 backbone and a direct Asp329 backbone interaction with the pyridyl nitrogen (Figure 2b). 24 was further evaluated in a broad kinome panel (Eurofins, 329 kinase). Apart from IRAK4, this compound was also observed to be exhibiting significant activity (≥50% inhibition at 1 μM) in a handful of other kinases such as CLK1, CLK2, CLK4, FLT3, DYRK1A, DYRK1B, TrkA, TrkB, Haspin, and NEK11. The compound also showed good modulation of IL6 in human whole blood assay (Table 6) and pIRAK1 modulation (IC50 of 270 nM) in MV4-11 cell-line, as cellular proof of mechanism. 24 when screened in cellular growth inhibition assay in a short panel of lymphoma cell lines demonstrated moderate potency in OCI-Ly10 (IC50 = 1.5 μM) harboring MYD88 mutation.
Table 6. IL-6 Release Inhibition Observed with 24 under Different Stimulation Conditions in Whole Blood Assays.
| Stimulation Condition for IL-6 release | IC50a (nM) |
|---|---|
| TLR2 (LTA) induced | 989 ± 686 (N = 3) |
| IL-1R (IL-1β) induced | 1375 ± 272 (N = 2) |
| TLR5 (FLA-ST) induced | 696 (N = 1) |
Blood from different donors was used in each experiment/each replicate. IC50 values are represented as mean ± SD wherever applicable.
In vivo efficacy of 24 was evaluated in an OCI-Ly3 xenograft model through oral route of administration in a once daily dosing regimen (Figure 3). A dose of 200 mg/kg q.d. showed partial tumor regression and a 100 mg/kg q.d. dose showed >90% tumor growth inhibition. The compound was well-tolerated, and there were no overt toxicities at these efficacious doses. Drug concentrations from plasma and tumors were determined at 2 h post final dose administration on day 15. Compound 24 treatment resulted in dose proportional increase in plasma and tumor exposures (Table 7). Activated TLR/IL1R results in the assembly of the Myddosome, a multiprotein complex composed of MyD88, IRAK4, and IRAK2 proteins. Myddosome protein complex activates the serine/threonine kinase, IRAK1, through IRAK4-dependent phosphorylation; therefore, we monitored phosphorylation of IRAK1 as a PD marker of IRAK4 inhibition.2,3,25 In a PK-PD study in OCI-Ly3 tumor bearing animals (Figure 4), 24 (200 mg/kg) showed 78% and 56% inhibition of pIRAK1, respectively, in tumor tissues at 12 and 24 h, respectively (Figure 4a), which correlated well with observed plasma and tumor exposures at these time-points (Figure 4b). Low pIRAK1 exposures observed at 4 and 8 h time-points (Figure 4b) likely due to IRAK1 being downstream in MYD88-IRAK signaling hence taking more time for demonstrating modulation of pIRAK1. Similar observations were noted in in vitro experiments wherein pIRAK1 modulation was observed only at later time points (data not shown). 24 also exhibited good CYP inhibition profile (IC50 > 50 μM in CYP2C19 and <5% inhibition at 1 μM across CYP3A4, 2D6, 2C9, 2C8, 2B6, and 1A2) and microsomal stability in mouse, rat, and human. 24 was not genotoxic in the in vitro bacterial mutagenesis assay and did not produce any biologically relevant clastogenicity changes in the in vivo micronucleus test in mice. Based on its overall profile including efficacy and desirable tolerability in toxicology studies in both mouse and dog, 24 was advanced into clinical development26 and later code-named CA-4948.
Figure 3.
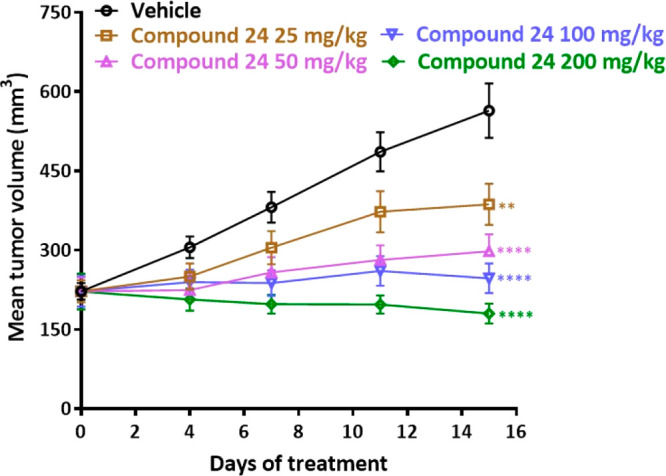
In vivo efficacy of 24 in OCI-Ly3 xenograft model. One-way ANOVA, Dunnett’s multiple comparison’s test: **p < 0.01, ****p < 0.0001.
Table 7. Mean Plasma and Tumor Concentrations of Compound 24 after Repeated Oral Administration in OCI-Ly3 Tumor Bearing Mice.
| Treatment groups | Mean plasma conc (ng/mL) | Mean tumor conc (ng/g) |
|---|---|---|
| 25 mg/kg | 5077.8 ± 587.5 | 2667.2 ± 399.7 |
| 50 mg/kg | 11565.9 ± 1074.7 | 8291.2 ± 1013.3 |
| 100 mg/kg | 23268.2 ± 2280.2 | 17943.3 ± 2122.6 |
| 200 mg/kg | 39123.0 ± 3101.2 | 36973.7 ± 2622.5 |
Figure 4.

In vivo PK/PD of 24 showing (a) pIRAK1 modulation and (b) plasma–tumor exposures. Mean mPPB of 24 = 92.8% (Average of 2 determinations). Unbound plasma exposures at 12 and 24 h estimated to be 868.0 and 49.3 ng/mL, respectively.
24 (CA-4948) was synthesized starting with 2-chloro-5-hydroxy pyridine (I) which was nitrated with nitrating mixture followed by reduction with Raney Ni to get compound III (Scheme 1). Compound III was cyclized with potassium ethyl xanthate followed by methylation of thiol to yield compound V which was refluxed with morpholine to obtain compound VI. Compound VI was nitrated with nitrating mixture to obtain compound VII which was coupled with (R)-3-hydroxy pyrrolidine to obtain compound VIII. Compound VIII was reduced with Raney Ni followed by HATU coupling with intermediate E resulting in CA-4948. Other compounds described here were synthesized using similar procedures described in the Supporting Information.
Scheme 1. Synthetic Route of 24 (CA-4948).

In conclusion, we have optimized a series of aza-benzoxazoles and identified a compound demonstrating potent IRAK4 inhibition with favorable selectivity over other kinases, cellular activity, pharmacokinetics, efficacy, and safety. The lead compound, CA-4948 (24), showed tumor regression in a rodent DLBCL xenograft model (OCI-Ly3) without any overt toxicities. In a PK-PD study it also showed significant inhibition of pIRAK1 which correlated well with plasma and tumor exposures. While 24 showed significant inhibition of a few other kinases (Table S1), there is no explicit literature evidence to support contribution of such effects toward the observed efficacy and pIRAK1 modulation. CA-4948 is currently being evaluated in phase-1 clinical trials, in patients with Relapsed or Refractory Hematologic Malignancies including non-Hodgkin lymphoma, MDS, and AML.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.0c00255.
Experimental details for synthesis, protein expression, assay methods, modeling, and crystallography (PDF)
The authors declare the following competing financial interest(s): All authors are current or former employees of Aurigene Discovery Technologies Limited or Curis Inc.
Supplementary Material
References
- Kawasaki T.; Kawai T. Toll-Like Receptor Signaling Pathways. Front. Immunol. 2014, 5, 1–8. 10.3389/fimmu.2014.00461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhyasen G. W.; Starczynowski D. T. IRAK signalling in cancer. Br. J. Cancer 2015, 112, 232–237. 10.1038/bjc.2014.513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngo V. N.; Young R. M.; Schmitz R.; Jhavar S.; Xiao W.; Lim K. H.; Kohlhammer H.; Xu W.; Yang Y.; Zhao H.; Shaffer A. L.; Romesser P.; Wright G.; Powell J.; Rosenwald A.; Muller-Hermelink H. K.; Ott G.; Gascoyne R. D.; Connors J. M.; Rimsza L. M.; Campo E.; Jaffe E. S.; Delabie J.; Smeland E. B.; Fisher R. I.; Braziel R. M.; Tubbs R. R.; Cook J. R.; Weisenburger D. D.; Chan W. C.; Staudt L. M. Oncogenically active MYD88 mutations in human lymphoma. Nature 2011, 470, 115–119. 10.1038/nature09671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang G.; Zhou Y.; Liu X.; Xu L.; Cao Y.; Manning R. J.; Patterson C. J.; Buhrlage S. J.; Gray N.; Tai Y. T.; Anderson K. C.; Hunter Z. R.; Treon S. P. A mutation in MYD88 (L265P) supports the survival of lymphoplasmacytic cells by activation of Bruton tyrosine kinase in Waldenström macroglobulinemia. Blood 2013, 122, 1222–1232. 10.1182/blood-2012-12-475111. [DOI] [PubMed] [Google Scholar]
- Rhyasen G. W.; Bolanos L.; Fang J.; Jerez A.; Wunderlich M.; Rigolino C.; Mathews L.; Ferrer M.; Southall N.; Guha R.; Keller J.; Thomas C.; Beverly L. J.; Cortelezzi A.; Oliva E. N.; Cuzzola M.; Maciejewski J. P.; Mulloy J. C.; Starczynowski D. T. Targeting IRAK1 as a therapeutic approach for myelodysplastic syndrome. Cancer Cell 2013, 24, 90–104. 10.1016/j.ccr.2013.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith M. A.; Choudhary G. S.; Pellagatti A.; Choi K.; Bolanos L. C.; Bhagat T. D.; Gordon-Mitchell S.; Ahrens D. V.; Pradhan K.; Steeples V.; Kim S.; Steidl U.; Walter M.; Fraser I. D. C.; Kulkarni A.; Salomonis N.; Komurov K.; Boultwood J.; Verma A.; Starczynowski D. T. U2AF1 mutations induce oncogenic IRAK4 isoforms and activate innate immune pathways in myeloid malignancies. Nat. Cell Biol. 2019, 21, 640–650. 10.1038/s41556-019-0314-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhary G. S.; Smith M. A.; Pellagatti A.; Bhagat T. D.; Gordon S.; Pandey S.; Shah N.; Aluri S.; Booher R. N.; Ramachandra M.; Samson M. E.; Pradhan K.; Bowman T. V.; Pillai M. M.; Guha C.; Wickrema A.; Will B.; Shastri A.; Steidl U. G.; Boultwood J.; Starczynowski D. T.; Verma A. SF3B1Mutations Induce Oncogenic IRAK4 Isoforms and Activate Targetable Innate Immune Pathways in MDS and AML. Blood 2019, 134 (Supplement_1), 4224. 10.1182/blood-2019-124458. [DOI] [Google Scholar]
- Wietek C.; O’Neill L. A. IRAK-4: a new drug target in inflammation, sepsis, and autoimmunity. Mol. Interventions 2002, 2, 212–215. 10.1124/mi.2.4.212. [DOI] [PubMed] [Google Scholar]
- Li S.; Strelow A.; Fontana E. J.; Wesche H. IRAK-4: a novel member of the IRAK family with the properties of an IRAK-kinase. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 5567–5572. 10.1073/pnas.082100399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powers J. P.; Li S.; Jaen J. C.; Liu J.; Walker N. P.; Wang Z.; Wesche H. Discovery and initial SAR of inhibitors of interleukin-1 receptor-associated kinase-4. Bioorg. Med. Chem. Lett. 2006, 16, 2842–2845. 10.1016/j.bmcl.2006.03.020. [DOI] [PubMed] [Google Scholar]
- Buckley G. M.; Gowers L.; Higueruelo A. P.; Jenkins K.; Mack S. R.; Morgan T.; Parry D. M.; Pitt W. R.; Rausch O.; Richard M. D.; Sabin V.; Fraser J. L. IRAK-4 inhibitors. Part 1: a series of amides. Bioorg. Med. Chem. Lett. 2008, 18, 3211–3214. 10.1016/j.bmcl.2008.04.058. [DOI] [PubMed] [Google Scholar]
- Scott J. S.; Degorce S. L.; Anjum R.; Culshaw J.; Davies R. D. M.; Davies N. L.; Dillman K. S.; Dowling J. E.; Drew L.; Ferguson A. D.; Groombridge S. D.; Halsall C. T.; Hudson J. A.; Lamont S.; Lindsay N. A.; Marden S. K.; Mayo M. F.; Pease J. E.; Perkins D. R.; Pink J. H.; Robb G. R.; Rosen A.; Shen M.; McWhirter C.; Wu D. Discovery and Optimization of Pyrrolopyrimidine Inhibitors of Interleukin-1 Receptor Associated Kinase 4 (IRAK4) for the Treatment of Mutant MYD88L265P Diffuse Large B-Cell Lymphoma. J. Med. Chem. 2017, 60, 10071–10091. 10.1021/acs.jmedchem.7b01290. [DOI] [PubMed] [Google Scholar]
- Lee K. L.; Ambler C. M.; Anderson D. R.; Boscoe B. P.; Bree A. G.; Brodfuehrer J. I.; Chang J. S.; Choi C.; Chung S.; Curran K. J.; Day J. E.; Dehnhardt C. M.; Dower K.; Drozda S. E.; Frisbie R. K.; Gavrin L. K.; Goldberg J. A.; Han S.; Hegen M.; Hepworth D.; Hope H. R.; Kamtekar S.; Kilty I. C.; Lee A.; Lin L. L.; Lovering F. E.; Lowe M. D.; Mathias J. P.; Morgan H. M.; Murphy E. A.; Papaioannou N.; Patny A.; Pierce B. S.; Rao V. R.; Saiah E.; Samardjiev I. J.; Samas B. M.; Shen M. W. H.; Shin J. H.; Soutter H. H.; Strohbach J. W.; Symanowicz P. T.; Thomason J. R.; Trzupek J. D.; Vargas R.; Vincent F.; Yan J.; Zapf C. W.; Wright S. W. Discovery of Clinical Candidate 1-{[(2S,3S,4S)-3-Ethyl-4-fluoro-5-oxopyrrolidin-2-yl]methoxy}-7-methoxyisoquinoline-6-carboxamide (PF-06650833), a Potent, Selective Inhibitor of Interleukin-1 Receptor Associated Kinase 4 (IRAK4), by Fragment-Based Drug Design. J. Med. Chem. 2017, 60, 5521–5542. 10.1021/acs.jmedchem.7b00231. [DOI] [PubMed] [Google Scholar]
- Degorce S. L.; Anjum R.; Dillman K. S.; Drew L.; Groombridge S. D.; Halsall C. T.; Lenz E. M.; Lindsay N. A.; Mayo M. F.; Pink J. H.; Robb G. R.; Scott J. S.; Stokes S.; Xue Y. Optimization of permeability in a series of pyrrolotriazine inhibitors of IRAK4. Bioorg. Med. Chem. 2018, 26, 913–924. 10.1016/j.bmc.2018.01.008. [DOI] [PubMed] [Google Scholar]
- Bryan M. C.; Drobnick J.; Gobbi A.; Kolesnikov A.; Chen Y.; Rajapaksa N.; Ndubaku C.; Feng J.; Chang W.; Francis R.; Yu C.; Choo E. F.; DeMent K.; Ran Y.; An L.; Emson C.; Huang Z.; Sujatha-Bhaskar S.; Brightbill H.; DiPasquale A.; Maher J.; Wai J.; McKenzie B. S.; Lupardus P. J.; Zarrin A. A.; Kiefer J. R. Development of Potent and Selective Pyrazolopyrimidine IRAK4 Inhibitors. J. Med. Chem. 2019, 62, 6223–6240. 10.1021/acs.jmedchem.9b00439. [DOI] [PubMed] [Google Scholar]
- ClinicalTrials.gov. National Library of Medicine (U.S.) . (2016, August) Study for MR Formulation of PF-06650833 in Healthy Adult Japanese Subjects. Identifier: NCT02936154. Retrieved from https://clinicaltrials.gov/ct2/show/NCT02936154.
- ClinicalTrials.gov. National Library of Medicine (U.S.) . (2016, December) Safety and Efficacy of Pf-06650833 In Subjects With Rheumatoid Arthritis, With An Inadequate Response To Methotrexate. Identifier: NCT02996500. Retrieved from https://clinicaltrials.gov/ct2/show/NCT02996500.
- ClinicalTrials.gov. National Library of Medicine (U.S.) . (2019, September) A Study to Evaluate the Safety and Efficacy of PF-06650833, PF-06700841, and PF 06826647 in Adults With Hidradenitis Suppurativa. Identifier: NCT04092452. Retrieved from https://clinicaltrials.gov/ct2/show/NCT04092452.
- ClinicalTrials.gov. National Library of Medicine (U.S.) . (2018, May)BAY1830839: First in Man, Single Dose Escalation, Safety & Tolerability and Pharmacokinetics. Identifier: NCT03540615. Retrieved from https://clinicaltrials.gov/ct2/show/NCT03540615.
- ClinicalTrials.gov. National Library of Medicine (U.S.) . (2017, February) First in Human Study to Investigate the Safety, Tolerability, Pharmacokinetics and to Explore Pharmacodynamics of BAY1834845. Identifier: NCT03054402. Retrieved from https://clinicaltrials.gov/ct2/show/NCT03054402.
- Rigel Pharmaceuticals Inc. Pipeline Page https://www.rigel.com/index.php/pipeline/irak/ (accessed 2019-11-28).
- Curis, Inc. Pipeline Page http://www.curis.com/pipeline/ca-4948 (accessed 2019-11-28).
- Lange M.; Wengner A. M.; Bothe U.; Boemer U.; Nubbemeyer R.; Siebeneicher H.; Steuber H.; Guenther J.; Potze L.; Schmidt N.; Politz O.; Doecke W.-D.; Lagkadinou E.; Zollner T. M.; von Nussbaum F.; Mumberg D.; Steinmeyer A.; Brands M.; Ziegelbauer K.. Preclinical evaluation of a novel interleukin-1 receptor-associated kinase 4 (IRAK4) inhibitor in combination with PI3K inhibitor copanlisib or BTK inhibitors in ABC-DLBCL. Cancer Res.. 2018, 78 ( (13 Suppl), ), Abstract nr 1887. [Google Scholar]
- ClinicalTrials.gov. National Library of Medicine (U.S.) . (2018, April) A Multiple Dose Study of BAY1834845 in Healthy Male Subjects and in Patients With Psoriasis. Identifier: NCT03493269. Retrieved from https://clinicaltrials.gov/ct2/show/NCT03493269.
- Yu X.; Li W.; Deng Q.; Li L.; Hsi E. D.; Young K. H.; Zhang M.; Li Y. MYD88 L265P Mutation in Lymphoid Malignancies. Cancer Res. 2018, 78, 2457–2462. 10.1158/0008-5472.CAN-18-0215. [DOI] [PubMed] [Google Scholar]
- ClinicalTrials.gov. National Library of Medicine (U.S.) . (2017, November) A Study of CA-4948 in Patients With Relapsed or Refractory Hematologic Malignancies. Identifier: NCT03328078. Retrieved from https://clinicaltrials.gov/ct2/show/NCT03328078.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.