

Abstract
Balancing cell death is essential to maintain healthy tissue homeostasis and prevent disease. Tumor necrosis factor (TNF) not only activates nuclear factor κB (NFκB), which coordinates the cellular response to inflammation, but may also trigger necroptosis, a pro‐inflammatory form of cell death. Whether TNF‐induced NFκB affects the fate decision to undergo TNF‐induced necroptosis is unclear. Live‐cell microscopy and model‐aided analysis of death kinetics identified a molecular circuit that interprets TNF‐induced NFκB/RelA dynamics to control necroptosis decisions. Inducible expression of TNFAIP3/A20 forms an incoherent feedforward loop to interfere with the RIPK3‐containing necrosome complex and protect a fraction of cells from transient, but not long‐term TNF exposure. Furthermore, dysregulated NFκB dynamics often associated with disease diminish TNF‐induced necroptosis. Our results suggest that TNF's dual roles in either coordinating cellular responses to inflammation, or further amplifying inflammation are determined by a dynamic NFκB‐A20‐RIPK3 circuit, that could be targeted to treat inflammation and cancer.
Keywords: A20, computational modeling, necroptosis fate decisions, NFκB dynamics, TNF
Subject Categories: Autophagy & Cell Death, Signal Transduction
Tumor necrosis factor (TNF) activates NFκB to counteract necrotic cell death. This study reveals that inducible A20 expression provides an incoherent feedforward loop that protects cells from transient, but not long‐lasting TNF exposure.
Introduction
The cytokine tumor necrosis factor (TNF) mediates diverse cell fate decisions in response to inflammation (Fig EV1A) (Beutler et al, 1985a; Newton & Dixit, 2012). TNF‐induced activation of nuclear factor κB (NFκB) regulates the expression of hundreds of inflammatory response genes involved in eliminating pathogens, resolving inflammation and healing (Wallach et al, 1999; Cheng et al, 2017). However, TNF is also a cell‐killing agent (Carswell et al, 1975; Beutler et al, 1985a) and may trigger apoptotic or necroptotic cell death programs with distinct pathophysiological consequences (Vandenabeele et al, 2010). While apoptotic cells fragment into membrane bound vesicles, which allows their removal and resolution of inflammation (Galluzzi et al, 2018), necroptotic cells spill damage‐associated molecular patterns (DAMPs) into the microenvironment, which promotes inflammation (Pasparakis & Vandenabeele, 2015; Wallach et al, 2016). Indeed, necroptosis has been linked to acute and chronic inflammatory diseases (Ito et al, 2016; Newton et al, 2016; Shan et al, 2018), and inhibition may be a promising therapeutic strategy (Degterev et al, 2019). Conversely, necroptosis may be beneficial in apoptosis‐resistant cancer (Hanahan & Weinberg, 2011; Fulda, 2015; Brumatti et al, 2016; Oliver Metzig et al, 2016) and to evoke an anti‐tumor immune response (Aaes et al, 2016; Krysko et al, 2017; Najafov et al, 2017). However, too little is known about the regulatory network controlling necroptosis to allow for predictable manipulation as a therapeutic strategy (Annibaldi & Meier, 2018).
Figure EV1. NECtrack measures pro‐inflammatory necroptotic cell death.
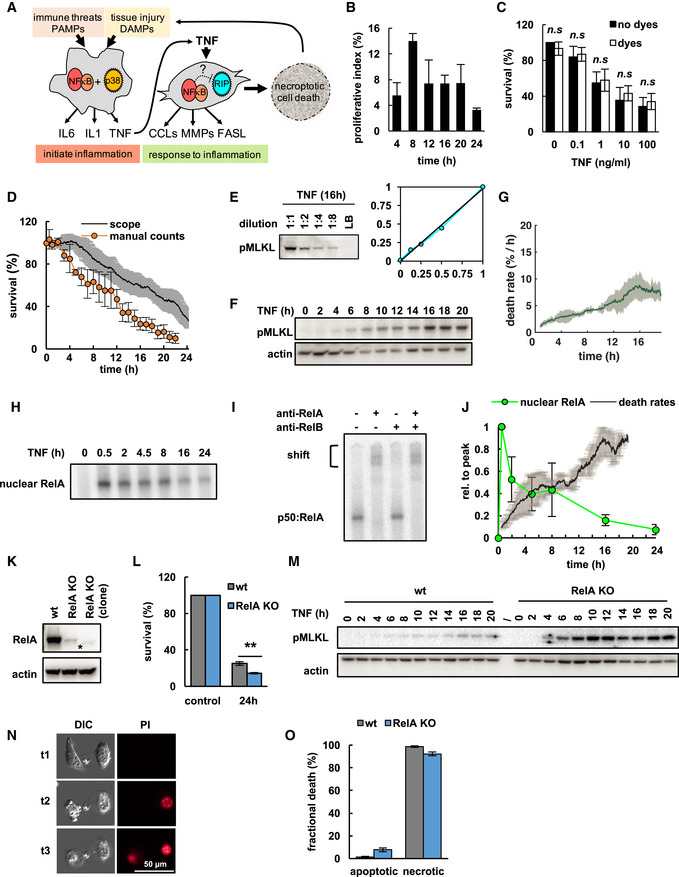
- Immune threats or tissue injury generate pathogen‐associated molecular patterns (PAMPs) or damage‐associated molecular patterns (DAMPs) to initiate inflammatory response. Tissue‐resident macrophages respond with combined activation of NFκB and MAPK p38 pathways, and produce key inflammatory cytokines including IL6, IL1, and TNF. Stromal cells including fibroblasts perceive those cytokines and participate in response to inflammation. TNF‐induced NFκB signaling drives release of chemokines (CCLs), matrix metalloproteinases (MMPs), and death ligands (FASL) to coordinate and resolve inflammatory cell infiltration. Alternatively, TNF may induce necroptotic cell death via RIP signaling, which generates more DAMPs to amplify inflammatory response. Whether TNF‐induced NFκB cross‐regulates necroptosis is unclear.
- Proliferative index of L929 wild‐type (wt) cells treated with TNF obtained from live‐cell microscopy (mean of three independent experiments ± standard deviation).
- Fractional cellular survival after 24 h TNF in the presence or absence of fluorescent DNA staining (Hoechst and PI) quantified by crystal violet assay (mean of three independent experiments ± standard deviation; two‐tailed Student's t‐test revealed no statistically significant differences, n.s., P > 0.05).
- Fractional survival of TNF‐treated L929 wt cells obtained via two independent methods (mean of three independent experiments ± standard deviation).
- L929 wt cells were treated with 10 ng/ml TNF for 16 h. Cell lysates were diluted in lysis buffer (LB) and analyzed for pMLKL via immunoblot using the primary antibody ab196436 (Abcam). Relative quantification of the signal demonstrates an approximate linear relationship between pMLKL concentrations and detected signal.
- Immunoblot for pMLKL in L929 wt cells (representative data of three independent experiments).
- Two‐phased death rates in a clonal L929 wt cell population (mean of three independent experiments ± standard deviation).
- TNF‐induced NFκB activity in L929 wt cells measured via EMSA (representative data of three independent experiments).
- Antibody against RelA, but not RelB induces shift of bands, revealing they largely consist of p50:RelA (representative data of three independent experiments).
- Normalized RelA activity measured via EMSA and death rates in time course of TNF treatment (means of three independent experiments ± standard deviation).
- Immunoblot for RelA in L929 wt and CRISP/Cas9 RelA‐knockout (RelA KO) cell lines including clonal RelA KO population (asterisk indicates unspecific band present in all lanes).
- Fractional survival after 24 h of TNF treatment (mean of three independent experiments ± standard deviation; two‐tailed Student's t‐test **P < 0.01).
- Immunoblot for pMLKL (dash indicates empty lane; representative data of three independent experiments).
- Representative images illustrating morphological characteristics of apoptosis vs. necrosis in RelA KO cells. Apoptotic cell (t1, left) shows cytoplasmic blebbing (t2) and delayed PI positivity of nucleus (t3) indicative of secondary necrosis. Necrotic cell (right, t1) shows cytoplasmic swelling and rapid PI positivity (t2). Differential interference contrast, DIC.
- Quantification of apoptotic vs. necrotic death in L929 wt and RelA KO cells manually curated from live‐cell microscopy experiments using morphological criteria shown in (N) (200 cells analyzed per experiment; mean of three independent experiments ± standard deviation).
Source data are available online for this figure.
When a monoclonal cell population is challenged with a cytotoxic stimulus, not all cells make the decision to die at the same time, and some cells may even survive altogether (Albeck et al, 2008; Spencer et al, 2009; Paek et al, 2016; Mitchell et al, 2018; Green, 2019). In principle, stimulus‐induced cell fate decisions may merely be a function of the cell's pre‐existing propensity to die or to survive. For instance, TNF‐related apoptosis‐inducing ligand (TRAIL) sorts cells into survivors or non‐survivors based on the state of the molecular signaling network (Spencer et al, 2009), and thus, the fate decision of an individual cell is predictable prior to administering the stimulus (Loriaux & Hoffmann, 2009). Alternatively, the pro‐death stimulus may also trigger the de novo expression of an inhibitor of the cell fate decision. In this case, the fate decision of an individual cell is affected by molecular stochasticity that governs gene induction and the interactions of pro‐ and anti‐death regulators, and by the dynamics of those activities. The regulatory motif, known as an Incoherent Feedforward Loop, is thus known to have the capacity for differentiating the duration of the incoming stimulus (Alon, 2007).
Tumor necrosis factor's cytotoxic activity was initially described in the L929 fibroblast cell line (Carswell et al, 1975) leading to the characterization of necroptotic cell death (Degterev et al, 2005). TNF is now known to first trigger the formation of signaling complex I by recruiting receptor interacting serine/threonine kinase 1 (RIPK1) to TNF receptor 1 (TNFR1), leading to the activation of the inhibitor κB kinase (IKK) and transcription factor NFκB (Hoffmann & Baltimore, 2006). Dissociation of RIPK1 from the plasma membrane‐bound complex I then allows for the recruitment of RIPK3 (complex IIb or the necrosome), which leads to RIPK3 oligomerization and phosphorylation of mixed lineage kinase like (pMLKL), causing plasma membrane rupture and necroptotic cell death (He et al, 2009; Ofengeim & Yuan, 2013; Gong et al, 2017; Tang et al, 2019). Unlike experimental cell systems that rely on co‐treatment with sensitizing agents, L929 have a natural propensity for necroptosis (Vanhaesebroeck et al, 1992; Vanlangenakker et al, 2011a), possibly due to high RIPK3 expression, and are thus an appropriate model system for studying the regulatory network that controls TNF‐mediated cell fate decisions.
Prior studies investigated NFκB as a potential necroptosis inhibitor (Thapa et al, 2011; Vanlangenakker et al, 2011a; Shindo et al, 2013; Xu et al, 2018), but in certain circumstances, NFκB may even promote necroptosis, e.g., by contributing to TNF production (Oliver Metzig et al, 2016). Furthermore, it remains unclear whether prior NFκB activity determines the propensity for cells to die, or whether TNF‐induced NFκB activation may determine the decision‐making, which would require de novo protein synthesis to be induced rapidly enough to affect the activity of signaling complexes I and II. Only the latter would constitute an incoherent feed forward loop capable of distinguishing stimulus dynamics.
Here, we sought to determine whether and how TNF‐induced NFκB activation regulates TNF‐induced necroptosis decisions. Given the potential of perturbation studies to skew the true regulatory network (Kreuz et al, 2001; Poukkula et al, 2005), we developed a live‐cell microscopy workflow to study unperturbed L929 cells and obtain time‐resolved quantitative necroptosis rates following TNF exposure. A conceptual mathematical model informed us how these death rate dynamics can be interpreted, leading us to identify TNF‐induced and NFκB‐responsive TNFAIP3/A20 as a key regulator of necroptotic fate decisions. The A20 circuit forms an incoherent feedforward loop to protect a fraction of cells from transient TNF doses, but renders them sensitive to long‐term TNF exposure. As predicted by a more detailed mathematical model, dysregulated NFκB dynamics diminish the cell's ability to make necroptosis decisions based on the duration of TNF exposure.
Results
Necroptosis kinetics are reflective of an incoherent feedforward loop
To distinguish whether TNF‐induced necroptosis decisions are merely a function of a pre‐existing propensity or the dynamics of stimulus‐induced regulators, we constructed two simple conceptual models. In the first, TNF induces activation of RIPK1/3 and the necroptosis effector pMLKL, but this signaling is counteracted by an unknown, constitutively present survival factor (Fig 1A). In the second model, TNF also induces inhibitor of κB (IκB)‐controlled NFκB (O'Dea & Hoffmann, 2010), which in turn induces expression of the survival factor, thus forming an incoherent feedforward loop (Fig 1B) (Kaern et al, 2003; Alon, 2007; Tyson & Novak, 2010). To account for cell‐to‐cell heterogeneity, we assumed stochastic gene expression (Friedman et al, 2006; Shalek et al, 2013) of the survival factor, either constitutive or induced, and applied a threshold for pMLKL (Gong et al, 2017; Samson et al, 2020) corresponding to irreversible cell death (Fig 1C and D; Appendix; Appendix Tables S1–S4).
Figure 1. Necroptosis kinetics are reflective of an incoherent feedforward loop.
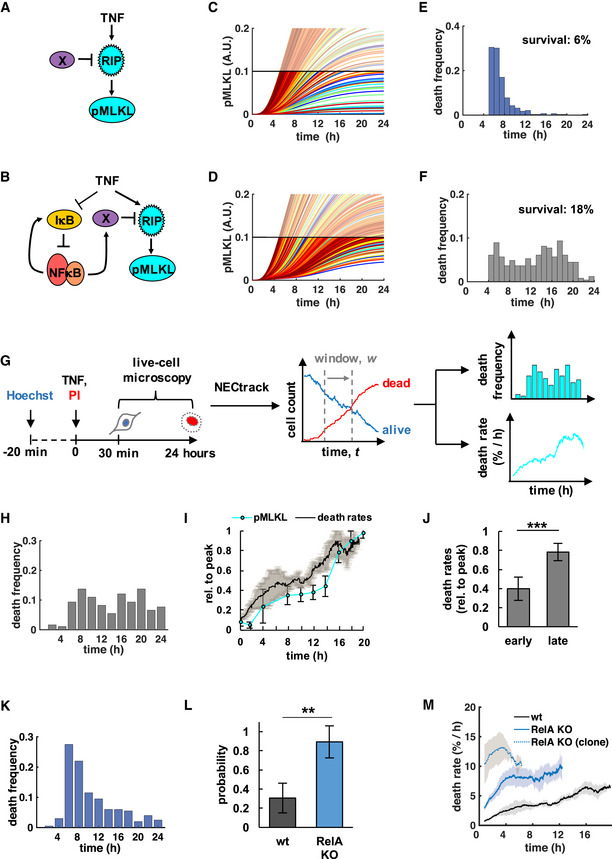
-
A, BConceptual mathematical modeling schematics depict TNF‐induced necroptosis signaling via RIPK1/3 (RIP) and phosphorylation of MLKL (pMLKL). RIP is counteracted by a putative (A) constitutive, or (B) stimulus‐induced, NFκB‐dependent survival factor X.
-
C, DTime course simulations of pMLKL levels in 300 single cells where each trajectory crossing a threshold represents a cell death event. Simulations for (C) constitutive, or (D) stimulus‐induced survival mechanism.
-
E, FDistributions of death times that result from simulations in (C, D), respectively. Fractional survival indicated after 24‐h time course simulation.
-
GLive‐cell microscopy workflow and automated image analysis via NECtrack to quantify TNF‐induced necroptosis kinetics in L929 cells. Distributions of death times and death rates are computed from raw counts of live and dead cells based on nuclear propidium iodide (PI) staining.
-
HDistribution of death times in TNF‐treated L929 wild‐type (wt) cells (representative data of three independent experiments).
-
INormalized death rates in L929 wt cells plotted with pMLKL protein levels measured via immunoblot (mean of three independent experiments ± standard deviation; corresponding images of representative Western blot experiment in Fig EV1F).
-
JAverage death rates of the early (< 12 h) and late phase of the TNF time course data in (I) (mean of three independent experiments ± standard deviation; two‐tailed Student’s t‐test ***P < 0.001).
-
KDistribution of death times in L929 RelA‐knockout (RelA KO) cells treated with TNF (representative data of three independent experiments).
-
LProbability of unimodal distributions of death times calculated by Hartigan’s dip significance (mean of three independent experiments ± standard deviation; two‐sample t‐test **P < 0.01).
-
MDeath rates in TNF‐treated cell lines including clonal RelA‐knockout population (mean of three independent experiments ± standard deviation).
Source data are available online for this figure.
Plotting the cell death time course by hourly binning the number of simulations in which pMLKL exceeds the threshold, we found that when the survival factor is pre‐existing and the mechanism constitutive, death times followed a unimodal distribution (Fig 1E). In contrast, TNF‐induced, NFκB‐dependent expression of a pro‐survival factor produced a non‐unimodal, apparently bimodal death time distribution (Fig 1F). While exact death times are a function of the particular parameters chosen in this analysis, the distinction between unimodal vs. non‐unimodal death time distributions was a robust feature of pre‐existing vs. inducible survival mechanisms (Appendix Fig S1).
Next, we established the live‐cell microscopy workflow and automated image analysis tool NECtrack to measure TNF‐induced necroptosis dynamics (Fig 1G, Movie EV1). TNF‐treated L929 cells were imaged and tracked for 24 h by nuclear Hoechst staining, and new necroptotic death events were identified by nuclear uptake of propidium iodide (PI) added to the culture medium. This workflow quantified necroptosis without being confounded by concurrent cell proliferation (Fig EV1B), a common bias of bulk readout assays based on fractional survival (Harris et al, 2016). Average necroptosis rates per hour were obtained by normalizing new death events to the number of present live cells. The use of nuclear dyes at low concentrations had no significant effect on cell numbers (Fig EV1C). To address the concern of phototoxicity, we compared different counting protocols and found that the microscopy workflow actually preserved cell viability better than a parallel, but independent counting protocol that required trypsinization (Fig EV1D).
Our measurements indicated a bimodal distribution of death times in L929 cell populations undergoing TNF‐induced necroptosis (Fig 1H), reflected by two‐phased death rate dynamics (Fig 1I). Indeed, death rates were about twofold higher in the late vs. early (< 12 h) phase (Fig 1J) and correlated with levels of pMLKL, the molecular marker for necroptosis (Fig 1I and EV1E and F). Similar biphasic necroptosis kinetics were observed in a newly cloned L929 cell population treated with TNF (Fig EV1G). These experimental results suggested that TNF does not only trigger pro‐death signaling leading to necroptosis, but also activates a mechanism that provides for transient protection of a fraction of cells within the population.
We asked whether TNF‐induced activation of NFκB RelA (Fig EV1H and I) may be responsible. Interestingly, over a time course of 24 h, RelA activity and death dynamics were inversely correlated (Fig EV1J). Depriving L929 cells of RelA via CRISPR/Cas9 (Fig EV1K) led to similar fractional survival after 24 h (Fig EV1L), but shifted necroptosis to the early phase resulting in a largely unimodal distribution of death times (P = 0.9, Fig 1K and L) and single‐phased death rates (Fig 1M). Increased necroptotic cell death during the early phase was correlated with detection of pMLKL (Fig EV1M) with most cells displaying morphological characteristics of necrotic cell death as expected (Fig EV1N and O, Movie EV2). This sensitizing effect was even more pronounced in a clonal knockout population (Fig 1M) that was confirmed to be fully RelA‐deficient (Fig EV1K). Together, these results support that TNF‐induced RelA is a potent inhibitor of necroptosis transiently protecting a fraction of L929 cells from necroptosis.
Rapid induction of A20 transiently inhibits the RIPK1‐RIPK3 complex and necroptosis
Next, we sought to identify the inducible mechanism by which RelA transiently protects a fraction of L929 cells from necroptosis. NFκB‐induced gene products may limit necroptosis by inhibiting reactive oxygen species (ROS) production or pro‐death c‐Jun N‐terminal kinase (JNK) signaling (Sakon et al, 2003; Kamata et al, 2005; Shindo et al, 2013; Zhang et al, 2017; Yang et al, 2018). While TNF‐induced generation of ROS may amplify necroptosis signaling (Vanlangenakker et al, 2011b), addition of the anti‐oxidant butylated hydroxyanisole (BHA; Fig EV2A), or the specific JNK inhibitor SP600125 (Fig EV2B) had limited effect in RelA‐knockout cells. This suggested that RelA‐mediated inhibition of necroptosis was not critically mediated by ROS or JNK.
Figure EV2. RelA mediates protection from necroptosis through TNF‐inducible A20.
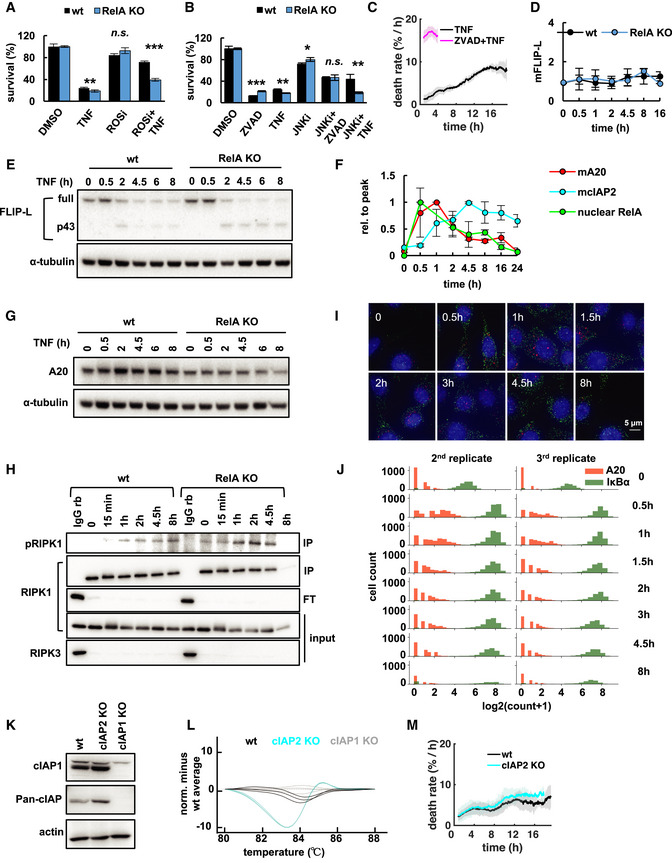
-
A, BFractional survival quantified by crystal violet assay after treatment with TNF and (A) ROS inhibitor (ROSi), (B) JNK inhibitor (JNKi), and/or ZVAD (mean of three independent experiments ± standard deviation; two‐tailed Student's t‐test *P < 0.05, **P < 0.01, ***P < 0.001, or no statistically significant difference, n.s., P > 0.05).
-
CTNF‐induced death rates in L929 wild‐type cells with and without ZVAD pre‐treatment obtained via live‐cell microscopy (mean of three independent experiments ± standard deviation).
-
DTNF‐induced mRNA expression measured via qRT–PCR (mean of three independent experiments ± standard deviation; two‐tailed Student's t‐test revealed no statistically significant results, P > 0.05).
-
EImmunoblot for FLIP‐L (full) and its cleavage product (p43; representative data of three independent experiments).
-
FRelative mRNA expression of A20 or cIAP2 measured via qRT–PCR, and normalized RelA activity measured via EMSA in wt cells treated with TNF (mean of three independent experiments ± standard deviation).
-
GImmunoblot for A20 protein (representative data of three independent experiments).
-
HWhole cell lysates depleted of RIPK3‐containing protein complexes using anti‐RIPK3 served as input for RIPK1 co‐immunoprecipitation (IP) and subsequent immunoblot analysis. Flow through, FT. Isotype control, IgG rb.
-
IRepresentative microscopy images of smFISH experiments for the NFκB‐responsive target genes A20 (red spots) and IκBα (green spots) in wt cells treated with TNF (contrast set to same limits for every time point).
-
JHistogram of volume normalized mRNA copy numbers measured by smFISH (two independent experiments; first independent experiment shown in Fig 2D).
-
K, L(K) Immunoblot for cIAP2 using commercially available Pan‐cIAP antibody and cIAP1‐knockout cell line as control. As cIAP2 protein is below detection limit, CRISPR/Cas9‐mediated knockout was validated (L) via high‐resolution melt (HRM) analysis.
-
MTNF‐induced death rates obtained via live‐cell microscopy (mean of three independent experiments ± standard deviation).
Source data are available online for this figure.
Several potential NFκB‐responsive target genes have been described to modulate TNFR‐induced complex I, complex II, and/or the necrosome (Vanlangenakker et al, 2011a; Dondelinger et al, 2016). In complex IIa, FLIP‐L has been implicated in restricting the proteolytic activity of pro‐caspase‐8 and directing its substrate specificity toward RIPK1 to disassemble the RIPK1‐RIPK3‐complex and inhibit necroptosis (Oberst et al, 2011; Tsuchiya et al, 2015; Hughes et al, 2016; Newton et al, 2019). In line with this, pre‐treatment with the pan‐caspase inhibitor ZVAD accelerated TNF‐induced necroptosis rates (Fig EV2C). However, FLIP‐L mRNA was not induced by TNF in L929 cells (Fig EV2D), and similar dynamics of FLIP‐L cleavage were observed in wild‐type and RelA‐knockout cells (Fig EV2E), indicative of comparable proteolytic activity of pro‐caspase 8 in complex II (Yu et al, 2009; Tsuchiya et al, 2015). Similarly, mRNA levels of CYLD or cIAP1, which are involved in the regulation of complex I activity (Annibaldi & Meier, 2018), were not significantly induced by TNF or reduced in RelA‐knockout cells (Fig 2A). In contrast, TNFAIP3/A20 and cIAP2 were significantly induced within 0.5 or 1 h of TNF treatment in a RelA‐dependent manner (Fig 2A), whereas only A20 expression was transient and correlated with dynamics of RelA activity (Fig EV2F). Inducible mRNA expression was accompanied by elevated A20 protein detected after 2 h of TNF treatment in wild‐type, but not RelA‐knockout cells, while constitutive expression levels were not significantly affected (Fig EV2G). Thus, inducible A20 appeared to be a promising candidate to mediate RelA‐dependent transient protection from necroptosis.
Figure 2. Rapid induction of A20 transiently inhibits the RIPK1‐RIPK3 complex and necroptosis.
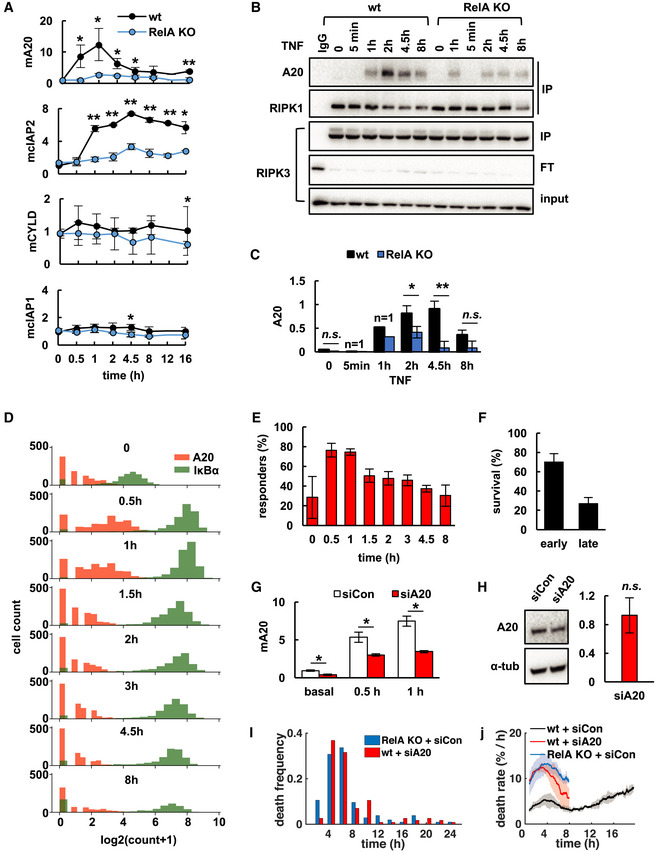
-
ATNF‐induced mRNA expression in L929 wild‐type (wt), or RelA‐knockout (RelA KO) cells measured via qRT–PCR (mean of three independent experiments ± standard deviation, two‐tailed Student’s t‐test *P < 0.05, **P < 0.01).
-
BImmunoblot after co‐immunoprecipitation (IP) of RIPK3 in indicated cell lines. FT, flow through.
-
CRelative quantification of A20 in RIPK3‐IP fraction in (B) (means and statistical significance established for 0, 2, 4.5, and 8 h time points from three independent experiments ± standard deviation; two‐tailed Student’s t‐test *P < 0.05, **P < 0.01, n.s., P > 0.05).
-
DHistogram of volume normalized mRNA copy numbers measured by smFISH of NFκB target genes A20 and IκBα in L929 wt cells treated with TNF (representative data of three independent experiments; two additional independent experiments in Fig EV4H).
-
EFractions of “responder” cells in (D) and Fig EV4H (A20 count > 1 per cell; mean of all three independent experiments ± standard deviation).
-
FFractional survival of wt cells after the early (< 12 h) and late phase of TNF time course obtained by microscopy (mean of three independent experiments ± standard deviation).
-
GA20 mRNA expression via qRT–PCR in TNF‐treated wt cells transfected with targeting (siA20) or non‐targeting (siCon) siRNA (mean of three independent experiments ± standard deviation; two‐tailed Student's t‐test *P < 0.05).
-
HImmunoblot and relative quantification of basal A20 protein after siRNA‐knockdown normalized to non‐targeting siRNA control (mean of three independent experiments ± standard deviation; two‐tailed Student's t‐test revealed no statistically significant difference between targeting and non‐targeting siRNA treatment, n.s., P > 0.05).
-
I, J(I) TNF‐induced distributions of death times (representative data of three independent experiments) and (J) death rates obtained by live‐cell microscopy (mean of three independent experiments ± standard deviation).
Source data are available online for this figure.
Several A20‐dependent mechanisms to limit TNF‐induced cell death have previously been reported (Draber et al, 2015; Onizawa et al, 2015; Polykratis et al, 2019; Martens et al, 2020; Razani et al, 2020). In complex I, A20 binds and stabilizes M1‐ubiquitin chains, which may limit formation of death‐inducing complex II (Draber et al, 2015; Polykratis et al, 2019; Martens et al, 2020; Razani et al, 2020). In addition, A20 integrates into the downstream necrosome and restricts RIPK3 activation to limit necroptosis (Onizawa et al, 2015). Whereas complex I forms rapidly (Micheau & Tschopp, 2003), the necrosome is activated slower (Vanlangenakker et al, 2011a), potentially allowing de novo expressed A20 to interfere with its activity. Indeed, we found that inducible expression of A20 coincided with its increased dynamic integration into RIPK3 immuno‐precipitates at 2 and 4.5 h in wild‐type compared with RelA‐knockout cells (Fig 2B and C). This was accompanied by decreased binding of RIPK1 in wild‐type cells (Fig 2B), indicating destabilization of the necrosome (Onizawa et al, 2015). Similar levels of phosphorylated RIPK1 (Ser166; Fig EV2H) and cleaved FLIP‐L (Fig EV2E) were detected in wild‐type and RelA‐knockout cells, indicating that complex I and II activities (Yu et al, 2009; Dondelinger et al, 2015; Tsuchiya et al, 2015; Dondelinger et al, 2017; Laurien et al, 2020) were less affected by inducible A20.
As biphasic necroptosis kinetics had indicated that the RelA‐dependent survival mechanism only protected a fraction of cells from premature necroptosis, we asked whether cell‐to‐cell heterogeneity in TNF‐induced A20 expression may be responsible. Analyzing TNF‐induced gene expression in single cells via smFISH revealed that A20 mRNA was only upregulated in 76% and 75% of “responder” cells at 0.5 and 1 h, respectively, whereas the NFκB‐responsive target IκBα was induced in nearly all cells (Figs 2D and E, and EV2I and J). In fact, the fraction of “responder” cells coarsely correlated with fractional survival after the early phase of TNF treatment (Fig 2F).
To further confirm the functional requirement of inducible A20, we performed siRNA‐mediated knockdown of A20. Knockdown conditions were optimized to significantly decrease TNF‐induced expression of the A20 mRNA (Fig 2G), but—due to protein half‐life—without having a significant effect on basal A20 protein levels present at the start of the TNF stimulation (Fig 2H). These conditions strongly sensitized L929 wild‐type cells to TNF leading to the majority of cells dying during the early phase, which resulted in unimodal death time distributions (Fig 2I) and single‐phased death rate dynamics comparable to RelA‐knockout cells (Fig 2J). In contrast, loss of cIAP2 (Fig EV2K and L) had no significant effect on necroptosis rates (Fig EV2M), which was in line with previous findings (Vanlangenakker et al, 2011b).
Together, our results indicated that following TNF‐induced, RelA‐responsive A20 expression in a subset of cells, A20 binds to and subsequently inhibits RIPK1‐RIPK3 complexes, thereby transiently protecting these cells from necroptosis. However, as RelA activity and A20 expression subside, these cells may become sensitive again and undergo necroptosis in the later phase of the time course.
The NFκB‐A20‐RIPK3 incoherent feedforward loop discriminates TNF stimulus dynamics
To further investigate the crosstalk between TNF‐induced NFκB and necroptosis fate decisions, we constructed a mathematical model that integrates the diverse roles of A20 in NFκB activation and necroptosis control (Fig 3A) (Lee et al, 2000; Werner et al, 2008; Harhaj & Dixit, 2012; Draber et al, 2015; Onizawa et al, 2015; Polykratis et al, 2019; Martens et al, 2020; Razani et al, 2020; Sun, 2020). The model consists of 41 species and 98 reactions (Fig EV3A, Appendix, Appendix Tables S5–S9) and combines the previously published modules for TNFR‐IKK and IκB‐NFκB signaling (Werner et al, 2008; Shih et al, 2009) with a newly constructed necroptosis module depicting activation of RIPK1, RIPK3, and the effector pMLKL. We parameterized the model based on literature and to fit our experimental measurements in L929 cells (Appendix). Simulation of 24 h of TNF treatment accurately recapitulated measurements of A20 expression at the mRNA and protein level (Fig EV3B and C), NFκB activation (Fig EV3D), and necrosome activity (Fig EV3E–G) (Li et al, 2012). Accounting for cell‐to‐cell heterogeneity in A20 expression as measured in smFISH experiments, as well as in RIPK1 activation (Appendix), we tested whether the model also recapitulated necroptosis kinetics in TNF‐treated L929 cell populations, and the NFκB‐dependent regulation. Indeed, simulations of wild‐type (Fig 3B) or RelA‐knockout cell populations (Fig 3C) showed the distinctive bimodal vs. unimodal distributions of death times as well as the two‐ vs. single‐phased death rates, respectively, that we had observed in experiments. These results provided quantitative support to the notion that TNF‐induced, RelA‐mediated expression of A20 provides transient protection from necroptosis.
Figure 3. The NFκB‐A20‐RIPK3 incoherent feedforward loop discriminates TNF stimulus dynamics.
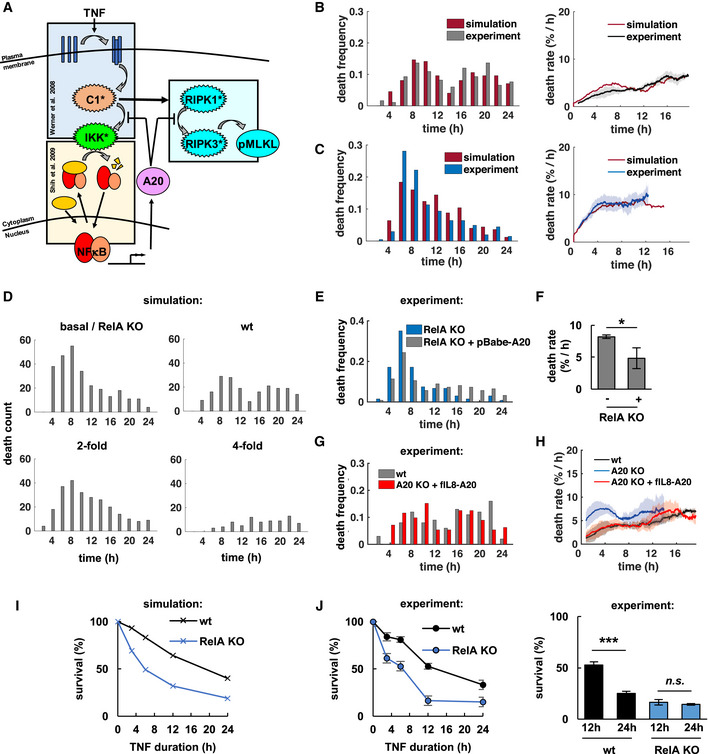
-
AModeling schematics depict TNF‐induced activation (*) of complex I (C1) and IKK to induce transcriptional activity of NFκB. C1 can also initiate activation of RIPK1 and RIPK3 to induce phosphorylation of necroptosis executor MLKL (pMLKL). TNF‐induced expression of IκB attenuates NFκB, whereas A20 inhibits IKK and RIPK3.
-
B, CComputational simulations and microscopy analysis of death time distributions (left, representative data of three independent experiments) and death rates (right, mean of three independent experiments ± standard deviation) in TNF‐treated L929 (B) wild‐type (wt), or (C) RelA‐knockout (KO) cells.
-
DSimulated death time distributions with twofold or fourfold increased constitutive A20 expression in the absence of inducible transcription (300 simulated cells per condition).
-
EDeath time distributions in TNF‐treated parental RelA KO cells (−) or RelA KO cells expressing A20 from a constitutive transgene (pBabe‐A20, +, representative data of three independent experiments).
-
FAverage death rates (< 12 h) in TNF‐treated cells (mean of three independent experiments ± standard deviation; two‐tailed Student's t‐test *P < 0.05, or no statistically significant difference, n.s., P > 0.05).
-
G, H(G) Distribution of death times (representative data of three independent experiments), or (H) death rates in TNF‐treated wt, parental A20 KO cells, or A20 KO cells reconstituted with an NFκB‐inducible transgene (fIL8‐A20; mean of three independent experiments ± standard deviation).
-
I, J(I) Simulations and (J) experimental measurements of 24‐h fractional survival after varying durations of transient or sustained (24 h) TNF stimulation (mean of three independent experiments ± standard deviation; two‐tailed Student's t‐test ***P < 0.001, or no statistically significant difference, n.s., P > 0.05).
Source data are available online for this figure.
Figure EV3. Mathematical model quantitatively accounts for RelA‐A20 controlled necroptosis kinetics.
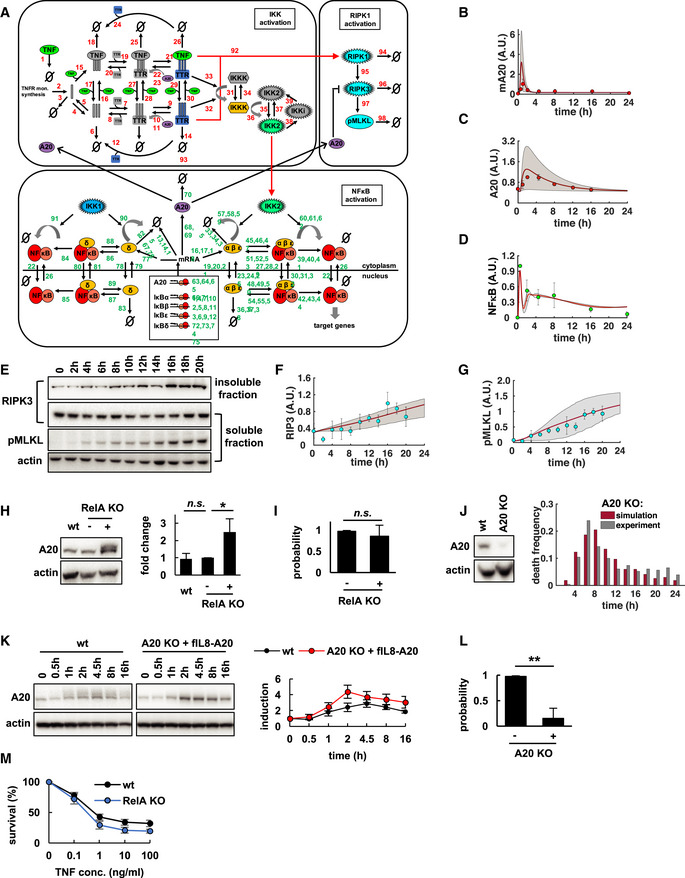
-
ADetailed schematic diagram of model reaction network (for reactions and parameters see Tables in Appendix).
-
B–DModel simulations (smoothed line represents population average, and shaded area the 20th percentile around the median) and experimental measurements (data points, mean of three independent experiments ± standard deviation) of indicated species over the time course of TNF treatment in L929 wild‐type cells. A.U., arbitrary units.
-
EImmunoblot for pMLKL and RIPK3 in detergent‐soluble and ‐insoluble fractions of wild‐type cells as a biochemical correlate of the active necrosome (representative data of three independent experiments).
-
F, GSimulations (smoothed line represents population average, and shaded area the 20th percentile around the median) and relative quantification of experimental measurements of necrosome activity in (E) (mean of three independent experiments ± standard deviation).
-
HOne representative of three immunoblots of A20 in wild‐type (wt), parental RelA‐knockout (KO) cells (−) or RelA KO cells expressing A20 from a constitutive transgene (pBabe‐A20, +). Relative quantification across three independent experiments (mean ± standard deviation; two‐tailed Student's t‐test *P < 0.05, or no statistically significant difference, n.s., P > 0.05).
-
IProbability of unimodal distributions of death times calculated by Hartigan's dip significance (mean of three independent experiments ± standard deviation; two‐sample t‐test revealed no statistical significance, n.s., P > 0.05).
-
JA20 immunoblot in wt or A20 KO cells (left). Simulated and measured distribution of death times in A20 KO cells (representative data of three independent experiments).
-
KImmunoblot of A20 in wt and A20 KO cells reconstituted with an NFκB‐inducible transgene (fIL8‐A20) and relative quantification (mean of three independent experiments ± standard deviation).
-
LProbability of unimodal distributions of death times calculated by Hartigan's dip significance in parental A20 KO cells (−) or A20 KO cells expressing fIL8‐A20 (+). Mean of three independent experiments ± standard deviation; two‐sample t‐test **P < 0.01).
-
M24‐h fractional survival in response to varying concentrations of TNF (mean of three independent experiments ± standard deviation).
Source data are available online for this figure.
Previous work demonstrated that while A20 plays a key role in modulating NFκB dynamics, its NFκB‐inducible expression does not (Werner et al, 2008). We wondered if the inducibility of A20 was instead required to provide proper dynamic regulation of TNF‐induced cell death decisions. We employed the mathematical model, set the inducible expression of A20 to zero, and simulated 24 h of TNF treatment with different levels of only constitutive A20 expression. While constitutively elevated A20 expression (twofold or fourfold) protected from death, two‐phased necroptosis dynamics as characteristic in wild‐type cells were not predicted (Fig 3D). To test this experimentally, we expressed A20 from a constitutive transgene in RelA‐knockout cells, which led to 2.5‐fold increased basal expression (Fig EV3H). Indeed, death dynamics remained unimodal (Figs 3E and EV3I), although cells were protected from necroptosis compared with RelA‐knockout cells (Fig 3F). However, when we reconstituted L929 A20‐knockout cells (Fig EV3J) with an NFκB‐inducible transgene (Fig EV3K), two‐phased necroptosis dynamics were restored (Figs 3G and H, and EV3L). These results indicated that the TNF‐inducible RelA‐A20‐RIPK3 circuit motif plays a critical role in shaping necroptotic death kinetics.
While in physiological settings TNF is typically secreted in transient bursts to coordinate and resolve inflammation, prolonged secretion and elevated levels of TNF are associated with autoimmune and chronic inflammatory diseases (Beutler et al, 1985b; Agbanoma et al, 2012; Wallach & Kovalenko, 2016). We hypothesized that the inducible RelA‐A20‐RIPK3 circuit may determine the fractional survival of cells in response to transient TNF stimulation, while leaving cells sensitive to long‐lasting TNF exposures. Model simulations testing a range of different temporal TNF doses (3–12 h, Appendix) predicted that L929 wild‐type cells would indeed be better protected from transient TNF exposures than RelA‐knockout cells, while remaining sensitive when exposed to long‐lasting TNF stimulation (Fig 3I). Experiments confirmed that wild‐type, but not RelA‐knockout populations, were able to discriminate short‐term exposures of up to 12 h from sustained 24‐h TNF treatment (Fig 3J). However, wild‐type and RelA‐knockout cells responded similarly to different TNF concentration doses (Fig EV3M), suggesting that the primary role of the RelA‐A20‐RIPK3 circuit motif is to discriminate between transient and sustained TNF, rather than concentration doses.
Dysregulated NFκB dynamics diminish the cellular discrimination of TNF exposures
As dysregulated NFκB activity is often associated with disease (Hanahan & Weinberg, 2011; Taniguchi & Karin, 2018), we utilized our mathematical model to explore necroptosis fate decisions as a function of altered RelA dynamics. To this end, we defined NFκB dynamics with an extrinsic pulse function rather than the normal IκB‐circuit (Appendix). The model predicted that prolonged NFκB dynamics and A20 expression (Fig 4A) led to increased fractional survival in 24‐h simulations of TNF treatment (Fig 4B). To experimentally test this scenario, we targeted the IκB regulatory system via CRISPR/Cas9‐mediated gene knockout (Fig 4C), resulting in significantly prolonged TNF‐induced RelA activity (Figs 4D, and EV4A and B), as well as prolonged expression of A20 mRNA and protein, while basal expression was unchanged (Fig 4E and F). As expected, IκBα/IκBε‐knockout cells were more resistant to TNF‐induced necroptosis with significantly increased fractional survival of 67% (Fig 4G) similar to the model prediction (56%, Fig 4B), and overall decreased death rates in response to 24 h of sustained TNF treatment (Fig 4H). This effect was even more pronounced in a clonal population selected for IκBα/IκBε‐knockout and CRISPR/Cas9‐induced heterozygosity for p100 to compensate for upregulated IκB inhibitory activity (O'Dea & Hoffmann, 2010), while maintaining wildtype‐like basal A20 expression (Figs 4H and EV4C). Finally, siRNA‐mediated knockdown targeting A20 in IκBα/IκBε‐knockout cells confirmed that the protective effect was largely due to A20, as death rates now resembled those of wild‐type cells treated with siA20 treatment (Fig 4I). Together, these data implicate that in conditions of dysregulated NFκB dynamics and prolonged expression of A20, cells are more likely to resist even long‐lasting TNF exposures.
Figure 4. Dysregulated NFκB dynamics diminish the cellular discrimination of TNF exposures.
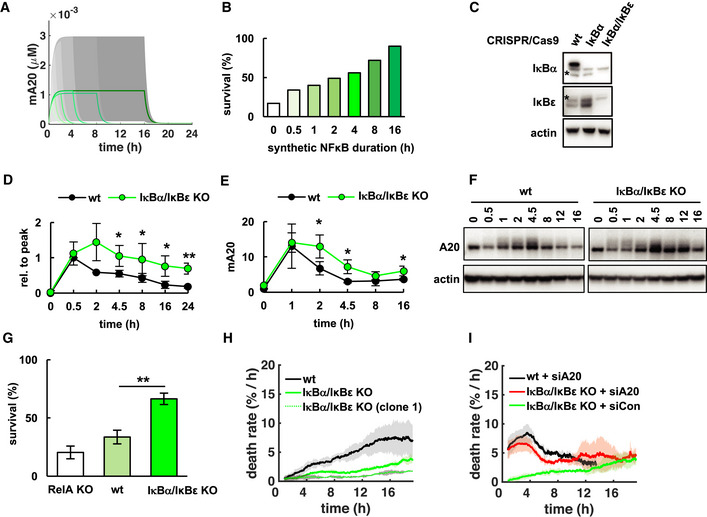
- Simulations of A20 mRNA concentrations in versions of the NFκB‐necroptosis model where expression is under the control of synthetic NFκB activity following step functions of 0.5, 1, 2, 4, 8, or 16 h duration (smoothed line is population average, and shaded area the 30th percentile around the median).
- Fractional survival that results from simulations in (A).
- Immunoblot for IκBα and IκBε in L929 wild‐type (wt) and CRISPR/Cas9‐knockout cell lines. Asterisks depict unspecific bands.
- Normalized RelA activity dynamics after TNF treatment quantified via EMSA (mean of three independent experiments ± standard deviation; two‐tailed Student's t‐test *P < 0.05, **P < 0.01; corresponding images of representative experiment in Fig EV4A).
- A20 mRNA quantified via qPCR (mean of three independent experiments ± standard deviation; two‐tailed Student's t‐test *P < 0.05).
- Immunoblot for A20 (representative data of three independent experiments).
- Fractional survival after 24 h of TNF treatment in indicated cell lines (mean of three independent experiments ± standard deviation; two‐tailed Student's t‐test **P < 0.01).
- Death rates in TNF‐treated indicated cell lines including isogenic IκBα/IκBε‐knockout population (mean of three independent experiments ± standard deviation).
- Death rates in TNF‐treated cell lines treated with targeting (siA20) or non‐targeting (siCon) siRNA (mean of three independent experiments ± standard deviation).
Source data are available online for this figure.
Figure EV4. Dysregulated NFκB dynamics in IκB‐knockout cells.
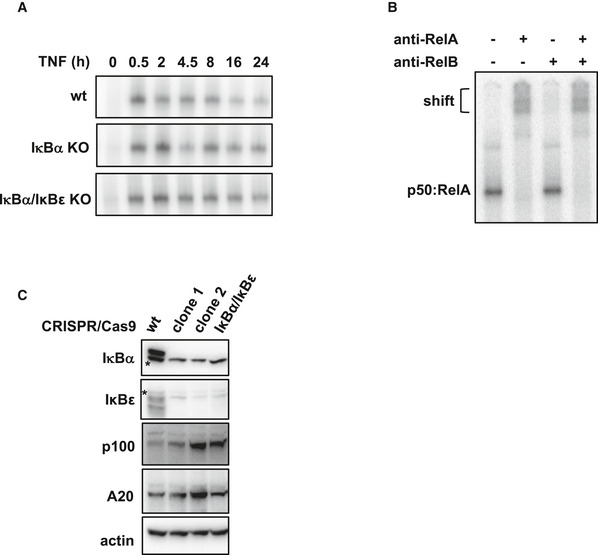
- TNF‐induced NFκB activity in indicated cell lines measured via EMSA (representative data of three independent experiments).
- Antibody for RelA, but not RelB induces shift of bands, revealing they largely consist of p50:RelA (representative data of three independent experiments).
- Immunoblot for indicated proteins; clonal population 1 (clone 1) was selected for maintaining basal expression of p100 and A20 despite of IκBα/IκBε‐knockout. Asterisks depict unspecific bands.
Source data are available online for this figure.
Discussion
In this study, we have addressed the regulatory mechanisms that determine TNF's dual roles in inflammation, namely whether TNF elicits a cellular response that includes coordination and resolution of the inflammatory condition, or necroptotic cell death that may further amplify inflammation. Using time‐lapse microscopy, we identified an incoherent feedforward loop involving TNF‐induced NFκB/RelA activity and de novo expressed A20 protein, which provides potent, though transient protection to RIPK3‐mediated necroptosis. We demonstrated that this molecular circuit ensures that a majority of cells survive transient TNF exposures, but, because of the transience of A20 expression, does not protect from long‐lasting TNF exposure.
While a potential role of NFκB in inhibiting necroptosis was previously suggested (Thapa et al, 2011; Shindo et al, 2013; Xu et al, 2018), the molecular regulatory circuits and its significance for necroptosis decision‐making remained unknown. Although the anti‐inflammatory protein A20 is a prominent NFκB‐response gene, its robust TNF‐inducibility is not required for inhibiting NFκB (Werner et al, 2008), prompting the question of why A20 expression is so highly TNF‐inducible. Here, we demonstrate that TNF‐inducible A20 is in fact key to linking NFκB and the regulation of necroptosis decisions. Even under conditions of exceptional TNF sensitivity as demonstrated in the L929 cell model system, NFκB‐responsive A20 provides potent, though transient protection from necroptosis, which is critically determined by the duration of induced A20 expression and TNF exposure times. The A20 expression time course is controlled by NFκB dynamics, which is in turn a function of stimulus duration and IκB feedback regulation (Werner et al, 2008; Lane et al, 2017). We found that when cells are deprived of negative feedback mechanisms that ensure physiological NFκB dynamics, subsequent prolonged expression of A20 will diminish TNF‐induced necroptosis.
Previous studies established A20 as an inhibitor of TNF‐induced cell death (Lee et al, 2000; Draber et al, 2015; Onizawa et al, 2015; Polykratis et al, 2019; Martens et al, 2020; Razani et al, 2020). Via its ubiquitin‐binding domain ZnF7, A20 is believed to stabilize M1‐linked ubiquitin chains in TNFR1‐induced complex I, which may restrict complex II formation and thereby apoptosis and/or necroptosis as shown in MEFs (Draber et al, 2015), macrophages (Polykratis et al, 2019), and intestinal epithelial cells (Martens et al, 2020). In addition, previous work in T cells and MEFs suggested that A20 binds to the necrosome, which may also be mediated via its ZnF7 domain, to enable ubiquitin editing and disruption of RIPK1‐RIPK3‐complexes (Onizawa et al, 2015; Dondelinger et al, 2016). Our iterative approach of mathematical modeling and experiments provides a refined, quantitative, and dynamic picture of A20's roles in determining TNF‐mediated fate decisions. We show that inducible A20 expression kinetics shape the dynamics of TNF‐induced necroptosis decisions. As upstream complex I is activated rapidly within minutes of TNF stimulation (Micheau & Tschopp, 2003), we reason that constitutively (but not induced) expressed A20 may integrate and limit the rate of transitioning into death‐inducing complex II (Priem et al, 2019) to determine both the apoptotic and necroptotic propensity. The active necrosome, however, forms only within hours (Vanlangenakker et al, 2011a; Vanlangenakker et al, 2011b) and is therefore more susceptible to inducibly expressed A20, which is why NFκB activity dynamics and the duration of the stimulus may be more critical in shaping necroptosis decisions. Indeed, our biochemical analyses showed that A20 expression kinetics coincided with its dynamic integration into RIPK1‐RIPK3‐complexes. Further work may address how the distinct molecular mechanisms ascribed to A20 in complex I/II contribute to necroptotic and apoptotic death decisions. The regulatory principles identified here put A20 into a position to limit necroptosis: When RIPK3 activation is slowed, but not blocked by low Caspase 8 activity, there is sufficient time for induced A20 to provide transient protection to cells from necroptotic death. In this scenario, the dynamics (transience/duration) of the two branches of the incoherent feedforward loop determine the cell fate decision of necroptosis vs. survival. This insight provides a guide on how to interpret studies on necroptosis vs. survival decisions in other cell types or experimental systems, which require inhibition of caspases to undergo necroptosis.
NFκB also protects cells from apoptosis, as genetic and pharmacologic perturbation studies revealed (Beg et al, 1995; Micheau & Tschopp, 2003) by contributing to the expression of anti‐apoptotic target genes such as caspase inhibitor cFLIP (Micheau et al, 2001). However, these studies do not demonstrate that apoptosis decisions are regulated by TNF‐induced gene expression (Beg et al, 1995; Micheau & Tschopp, 2003). While protein synthesis inhibitors sensitize cells to TNF‐induced apoptosis (Micheau et al, 2001), they also block constitutive protein expression, including key anti‐apoptotic target genes such as cFLIP, whose short half‐life requires continuous constitutive synthesis (Kreuz et al, 2001; Poukkula et al, 2005). Of note, cFLIP is only weakly induced by TNF (Kreuz et al, 2001; Micheau et al, 2001). The fact that TNF pulses as short as 30 s may be as effective as continuous exposure in eliciting apoptotic responses (Lee et al, 2016) may suggest that the stimulus itself merely sorts cells by a pre‐existing apoptotic propensity, which may in turn be affected by the level of tonic NFκB activity (Lin et al, 1998; Loriaux & Hoffmann, 2009; Gaudet et al, 2012).
What might be the physiological consequences of the differential regulatory strategies by which NFκB controls apoptosis and necroptosis? A cell's decision to undergo apoptosis appears to be inherent, depending on the general health of a cell, tonic NFκB, and hence its history of having responded appropriately to prior inflammatory conditions. If cells are unhealthy, they will be weeded out via apoptosis without causing much inflammation. In contrast, whether cells that express the necroptosis machinery will die of necroptosis is also a function of the dynamics of NFκB and duration of the TNF signal (Fig EV5A). Healthy NFκB activity dynamics in response to physiological TNF doses will ensure these cells participate in immune modulatory tissue processes rather than die. However, if TNF doses last longer, as they may in persistent infections, sepsis or chronic inflammatory diseases, cells may die via necroptosis and thereby release DAMPs to fuel an overwhelming inflammatory response (Fig EV5A). Indeed, loss of Caspase‐8 (Gunther et al, 2011) or FADD (Welz et al, 2011) induces TNF‐mediated necroptosis and inflammatory lesions in murine intestinal epithelium, resembling the pathology of inflammatory bowel disease. In turn, loss of MLKL or RIPK3 protected mice from TNF‐induced systemic inflammatory response syndrome (SIRS) (Newton et al, 2016). Two recent reports further pointed out that A20's anti‐inflammatory properties are not solely reliant on inhibiting IKK/NFκB, but depend on the prevention of TNF‐induced cell death (Polykratis et al, 2019; Martens et al, 2020). In this context, our work establishes physiological NFκB dynamics as a safeguard against overwhelming inflammation, namely by securing physiological expression of A20 and therefore protecting from TNF‐induced necroptosis.
Figure EV5. Pathophysiological consequences of the NFκB‐A20‐RIPK3 circuit.
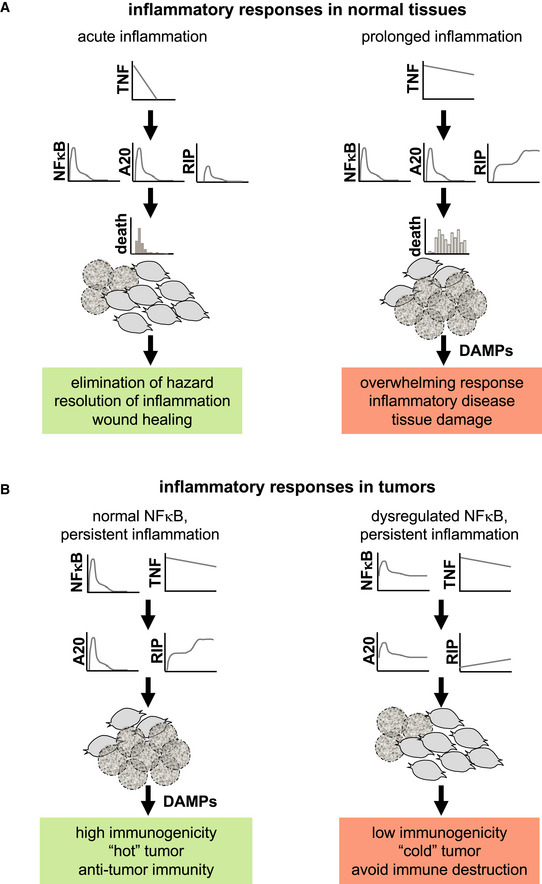
- In normal tissues, the NFκB‐A20‐RIPK3 circuit protects the majority of fibroblasts from death induced by transient signals of TNF to participate in the coordination and resolution of acute inflammation (left). In conditions of prolonged inflammation, sustained TNF signals overcome the protective NFκB‐A20‐RIPK3 circuit, leading to massive necroptosis, the release of DAMPs, and overwhelming inflammation, which may contribute to inflammatory diseases and tissue damage (right).
- In tumors with normal NFκB activity, sustained TNF signals may induce sufficient levels of necroptotic cell death to establish immunogenicity and an effective anti‐tumor response (left). In contrast, in tumors with dysregulated NFκB activity, prolonged expression of A20 may contribute to low immunogenicity and avoidance of immune destruction (right).
In contrast, in tumors amplifying inflammatory responses via necroptotic cell death may have beneficial effects, increasing immunogenicity and helping to establish effective anti‐tumor immunity (Fig EV5B). In this context, sensitizing cells to TNF or other necroptotic stimuli by counteracting inducible NFκB or the protective functions of A20 may have potential therapeutic value to enhance anti‐tumor immunity.
Materials and Methods
Reagents and Tools table
| Reagent/resource | Reference or source | Identifier or catalog number |
|---|---|---|
| Experimental Models | ||
| NCTC clone 929 [L cell, L‐929, derivative of Strain L] CCL‐1 (M. musculus) | ATCC | |
| Recombinant DNA | ||
| lentiCRISPR v2 | Addgene | Cat # 52961 |
| pBabe‐A20 | Werner et al (2008) | |
| fIL8‐A20 | Lois et al (2002), Werner et al (2008) | |
| Antibodies | ||
| Rabbit monoclonal [EPR9515(2)] to MLKL (phospho S345) | Abcam | Cat # ab196436 |
| Mouse anti‐RIP | BD Biosciences | Cat # 610459 |
| Rabbit Phospho‐RIP (Ser166) Antibody | Cell Signaling | Cat # 31122S |
| Rabbit anti‐RIP3 | Sigma Aldrich | Cat # PRS2283 |
| Rabbit NFκB p65 Antibody (C‐20) | Santa Cruz | Cat # sc‐372 |
| Rabbit RelB Antibody (C‐19) | Santa Cruz | Cat # sc‐226 |
| Rabbit IκB‐α Antibody (C‐21) | Santa Cruz | Cat # sc‐371 |
| Rabbit IκB‐β Antibody (C‐20) | Santa Cruz | Cat # sc‐945 |
| Rabbit IκB‐ε Antibody (M‐121) | Santa Cruz | Cat # sc‐7156 |
| Rabbit p52/100 (NR‐145) | generous gift from Nancy Rice | |
| Mouse A20 Antibody (A‐12) | Santa Cruz | Cat # sc‐166692 |
| Rat FLIP Antibody [Dave‐2] | ProSci | Cat # XA‐1008 |
| Mouse cIAP Pan‐specific Antibody | R&D | Cat # MAB3400 |
| Rat c‐IAP1 monoclonal antibody (1E1‐1‐10) | Enzo Life Sciences | Cat # ALX‐803‐335 |
| Oligonucleotides and sequence‐based reagents | ||
| qPCR primers | This study | Table EV1 |
| Oigonucleotides for smFISH | This study | Table EV2 |
| gRNAs for CRISPR/Cas9 | This study | Table EV3 |
| non‐targeting siRNA control | Dharmacon | Cat # D‐001206‐13 |
| A20 siRNA | Dharmacon | Cat # M‐058907 |
| pBabe‐A20 | Werner et al (2008) | |
| fIL8‐A20 | Lois et al (2002), Werner et al (2008) | |
| Chemicals, enzymes and other reagents | ||
| Mouse recombinant TNF | R&D | Cat # 410‐MT‐10 |
| Propidium iodide (PI) | Sigma Aldrich | Cat # P4864 |
| Hoechst 33342 | Thermo Fisher Scientific | Cat # H21492 |
| ZVAD‐fmk | Enzo Life Sciences | Cat # BML‐P416‐0001 |
| Necrostatin‐1 | Enzo Life Sciences | Cat # BML‐AP309 |
| Butylated hydroxyanisole (BHA, ROS inhibitor) | Sigma Aldrich | Cat # B1253 |
| SP600125 (JNK inhibitor) | Sigma Aldrich | Cat # S5567 |
| Direct‐zol RNA Miniprep Plus Kit | Zymogen | Cat # R2071 |
| iScript cDNA Synthesis Kit | Bio‐Rad | Cat # 1708890 |
| SYBR Green PCR Master Mix | Bio‐Rad | Cat # 1725150 |
| Quick‐DNA Miniprep Plus | Zymogen | Cat # D4068 |
| DharmaFECT1 | Dharmacon | Cat # T‐2001‐01 |
| Software | ||
| MATLAB R2015b (Version 8.6.0.267246) | ||
| Image Lab (Version 5.2 build 14) | Bio‐Rad | |
| Zen2 (blue edition, Version 2.0.0.0) | Carl Zeiss Microscopy GmbH | |
| Gen5 (Version 1.11.5) | Bio‐Tek Instruments | |
| CFX Maestro 1.1 (Version 4.1.2433.1219) | Bio‐Rad | |
| ImageQuant TL, 1D (Version 7.0) | GE Healthcare | |
| Other | ||
| AxioObserver | Carl Zeiss Microscopy GmbH | |
| CoolSnap HQ2 camera | Photometrics | |
| Epoch microplate reader | BioTek | |
| ChemiDoc MP imaging system | BioRad | |
Methods and Protocols
Cell culture
L929 cells were maintained in DMEM (Corning) containing 10% FBS (Omega Scientific), 1% l‐Glutamine, and 1% penicillin/streptomycin (Thermo Fisher Scientific) at 5% CO2 and 37°C. Isogenic L929 wild‐type or CRISPR/Cas9‐modified cell lines (RelA‐ or IκBα/IκBε‐knockout) were established by single‐cell sorting and clonal expansion as indicated.
Live‐cell microscopy and image analysis
Seed L929 cells (3 × 105) into eight‐well μ‐slides (ibidi) and grow for 24 h.
Immediately before experiment, add Hoechst (15 ng/ml) to culture medium and stain cells in incubator for 20 min.
Remove Hoechst containing medium and add culture medium containing propidium iodide (1 μg/ml), and TNF (10 ng/ml).
Transfer dish to Zeiss AxioObserver equipped with an incubation chamber, a 20× objective, LED (light‐emitting diode) fluorescence excitation, and CoolSnap HQ2 camera. Equilibrate for 30 min at 5% CO2 and 37°C.
Set imaging positions and image differential interference contrast (DIC), Hoechst (excitation at 365 nm, Zeiss Filter Set 49), and PI (excitation at 587 nm, Semrock mCherry B‐000) every 1.5 min for 24 h.
Export TIFF images for automated analysis using MATLAB.
Our automated image analysis tool NECtrack identifies, segments and tracks individual cells based on DIC and Hoechst images (Selimkhanov et al, 2014), and measures their mean nuclear PI intensity over time. Cells were declared dead when numerical threshold of nuclear PI was crossed for at least six consecutive time frames, and first frame was stored to generate histogram of death times. The status of each cell per frame was then integrated into a single binary matrix with cells and time points in rows and columns, respectively, with 0 representing “alive” and 1 indicating death had occurred. Absolute numbers of death events were converted to a rate of cell death proportional to the number of cells alive at a time point by using a 5‐h sliding window, in which the number of new death events within a window was divided by the number of cells alive at the beginning of the window. The death rate at a time point is therefore the rate of death that will occur over the following 5 h for cells alive at that time point; this accounts for continuous numerical changes in the population, e.g., by cell death or division. Average death rates per hour, i.e., the probability for individual cells to die within a given time window, is reported as the mean percentage of three independent experiments ± standard deviations until remaining alive cell population drops under one third of the starting population (approx. 250–300 cells per experiment and condition). To obtain proliferative index, cell divisions were manually counted per 4‐h time window and normalized to the number of alive cells present at the beginning of each window.
Statistical analysis of distributions of death times
All death times were placed into a single‐ordered array and passed to MATLAB's “histcounts” function with “Normalization” set to “pdf” and histogram bins defined every 2 h from 30 min (first time point) to 24.5 h (final time point). The calculated probability distribution of cell death was plotted as a histogram. The null hypothesis that the sorted array of death times was drawn from a unimodal distribution was tested by Hartigan's dip significance test for unimodality, which calculates a probability of unimodality (Hartigan, 1985). This was repeated for three independent experiments for TNF‐treated wild‐type or RelA‐knockout cells. The probabilities of unimodality in each condition were compared with a two‐sample t‐test.
Cell viability endpoint assays
Crystal violet assay:
Grow 1 × 104 cells per well in 96‐well plates for 24 h.
Add drugs (ZVAD: 30 μM, BHA: 50 μM, or JNK inhibitor: 10 μM) for 1 h as indicated, prior to TNF (10 ng/ml) treatment.
For pulse stimulation experiments, treat cells with TNF for the indicated durations, wash, and then incubate in culture medium before taking 24‐h endpoint measurements.
At endpoint, stain cells with crystal violet staining solution (0.5% in 20% methanol) for 10 min.
Wash plates with water, add sodium citrate solution (0.1 M in 50% ethanol) to dry wells and measure absorption using a microplate reader. Normalize data to mock‐treated controls.
Manual cell counting:
Grow 6 × 105 cells per well were grown in six‐well plates for 24 h.
Treat with TNF (10 ng/ml) for indicated durations.
Detach cells with trypsin resuspend in culture medium and count alive cells using a hemocytometer and Trypan blue exclusion staining.
Immunoblotting
Grow 2–3 × 106 cells in 10‐cm plates for 24 h and add treatments as indicated.
Remove dead cells by thoroughly washing with PBS, harvest remaining adherent cells, and lyse using RIPA buffer containing 1% Triton X‐100 supplemented with PMSF, DTT, and phosphatase inhibitors.
Normalize samples for total protein amounts using a Bradford assay (Bio‐Rad).
Boil detergent insoluble fractions for 10 min in 3× SDS sample buffer and subject to gel electrophoresis and immunoblotting.
Incubate with respective antibodies and develop signal using chemiluminescent substrate (SuperSignal West Pico Plus, Thermo Fisher Scientific). Visualize and quantify bands using ChemiDoc MP imaging system (Bio‐Rad).
Co‐immunoprecipitation (Co‐IP)
Grow 6 × 106 cells in 15‐cm plates for 24 h.
Wash with PBS and lyse on ice in 30 mM Tris–HCL pH 7.4, 150 mM NaCl, 10% glycerol, 2 mM EDTA, 0.5% Triton, 0.5% NP‐40, 1 mM DTT containing de‐ubiquitinase inhibitor PR‐619, protease and phosphatase inhibitors.
Normalize lysates for total protein amounts and incubate with anti‐RIPK3 (1 μg antibody per 1 mg protein) for 4 h at 4°C.
Add protein G beads (Dynabeads, Thermo Fisher Scientific) for 1 h to isolate complexes and wash five times in Co‐IP lysis buffer.
If indicated, subject flow through to secondary co‐immunoprecipitation with anti‐RIPK1 (1 μg antibody per 1 mg protein; incubate overnight at 4°C).
Boil IP fraction, flow through, and input lysates in 1× or 3× SDS sample buffer and subject to immunoblotting.
Electrophoretic mobility shift assay (EMSA)
For gel‐shift assays, nuclear extracts (Basak et al, 2007; Schrofelbauer et al, 2012) were incubated with 32P‐labeled, double‐stranded DNA probes containing kB‐binding sites in the presence or absence of anti‐RelA or anti‐RelB, prior to nondenaturing acrylamide gel electrophoresis. Bands were visualized by autoradiography and quantified using ImageQuant software.
Quantitative real‐time PCR (qRT–PCR)
RNA was purified using Direct‐zol RNA Miniprep Plus Kit (Zymogen), and cDNA synthesized with iScript cDNA Synthesis Kit (Bio‐Rad). qRT–PCR was performed with SYBR Green PCR Master Mix reagent using the D(DCt) method with RPL as normalization control, relative to unstimulated and stimulated signals in L929 cells to derive fold induction (Table EV1).
Single‐molecule fluorescence in situ hybridization (smFISH) and image analysis
Design oligos (50 bp) using custom software developed by the Zhuang laboratory for MERFISH (Moffitt et al, 2016). Oligos in this study were comprised of a 30‐bp region complementary to A20 or IκBα, and a 20‐bp mouse orthogonal sequence that binds to dye‐labeled “readout oligo”. 52 different gene targeting regions were selected to tile along the length of the transcripts of interest (Table EV2). Dye‐labeled oligos used Cy5 and Atto 565, respectively, to image labeled transcripts as diffraction limited spots.
Plate cells (3 × 105) in eight‐well μ‐slides (ibidi), grow for 24 h, and stimulate with 10 ng/ml TNF for different durations.
Remove medium and fix with 4% paraformaldehyde in PBS buffer.
Rinse with PBS and permeabilize cells with 0.5% v/v Triton X‐100 in PBS, followed by 3× rinse with Tris‐buffered 300mM NaCl supplemented with 0.1% Tween‐20 (TBS2Xtw).
Equilibrate for 10 min in TBS2Xtw supplemented with 30% formamide (MW), aspirate liquid, and add TBS2Xtw supplemented with 30% formamide and 10% dextran sulfate.
Hybridize with 50 nM oligo mixture for both A20 and IκBα overnight (16 h) at 37°C.
Wash 2× with MW at 47°C for 30 min, and hybridize with a PER amplifier oligo at 25 nM concentration in MH for 30 min at room temperature (Kishi et al, 2019)
Wash and stain for nuclei with DAPI, and stain with readout oligo in TB2XStw supplemented with 10% ethylene carbonate for 30 min, before washing with the same solution without readout oligo.
Add imaging buffer (4 mM PCA and 0.3 U/ml of rPCO oxygen scavenging system in TBS2Xtw supplemented with 0.1 v/v murine RNAase inhibitor) before imaging on a custom configured Zeiss Axio Observer Z1 with 63× planapo objective, Zyla 4.2 sCMOS, and custom LED light engine built for MERFISH (Foreman & Wollman, 2020).
Images were background subtracted by difference of Gaussian filtering with a high pass filter of 2.5 pixel sigma and 0.8 pixel low pass blurring filter. Local maxima were detected, and a 3 × 3 pixel region around local maxima intensity averaged. Number of spots found as a function of spot intensity was used to hysteresis threshold background spots from mRNA spots (Tsanov et al, 2016). Nuclei were segmented using watershed seed with smoothed local maxima in valleys of the negative smoothed intensity of the DAPI channel. Cells were imaged in a Green channel that produces smooth autofluorescence around the cell cytoplasm, and cytoplasm was segmented using the nuclei segmentation as seeds in a valley of negative autofluorescence intensity. RNA spots were assigned to cells if the cellular segmentation mask contained a particular spot. Cellular areas were calculated for each cell and used in volume normalization of RNA counts. Raw counts were normalized by dividing the count by the area of each cell and multiplying by the average area of all cells to rescale counts back to the same means as before normalization. These volume normalized counts were log2 transformed with a pseudo‐count of 1. Fraction of responders was calculated by finding the fraction of cells with an A20 count > 1 transcript per cell at each timepoint.
CRISPR/Cas9‐gene editing
Guide RNAs (gRNAs, Table EV3) were cloned into lentiCRISPR v2 (52961, Addgene) (Sanjana et al, 2014) and used for lentivirus production in HEK293T cells. Infected L929 cells were selected with Puromycin (8 µg/ml) until cell death subsided, used for experiments or subjected to single‐cell sorting and clonal expansion as indicated. Knockout was confirmed by Western blot or—in the case of cIAP2—by HRM analysis. To this end, genomic DNA was isolated using Quick‐DNA Miniprep Plus (Zymogen) and subjected to PCR amplification (5′ – ACAGTCCCATGGAGAAGCAC – 3′, and 5′ – CTTGTGCTCAAAGCAGGACA – 3′), and subsequent melt curve analysis using SYBR Green (Bio‐Rad) and temperature increments (0.2°C steps).
Transfection of short interference (si) RNA
Reverse transfection of L929 cells (Metzig et al, 2011a; Metzig et al, 2011b) was performed using transfection reagent (DharmaFECT1) and siRNAs (Dharmacon) at a final concentration of 50 nM. After 24 h, knockdown efficiencies were tested prior to and in response to TNF treatment on mRNA and protein level as indicated.
Expression of A20 transgenes
pBabe‐A20 containing a human A20 ORF was used for retroviral production (Werner et al, 2008) and infection of L929 RelA‐knockout cells. In L929 A20‐knockout cells, inducible A20 was reconstituted from transgene fIL8‐A20 containing A20 ORF under the control of the IL8‐promoter (Lois et al, 2002; Werner et al, 2008).
Data reproducibility and statistical analysis
Each experiment was repeated at least three independent times and performed in different weeks. All measurements were taken from distinct cellular samples, rather than measuring the same sample repeatedly. Microscopy analysis and all other data or images presented in the same figure panel were taken from measuring the respective conditions side‐by‐side in the same independent experiment. Statistical analysis was performed on means of these three individual experiments using two‐tailed Student’s t‐test with P values noted in respective figure legends.
Author contributions
MOM initiated, and MOM and AH designed the study. MOM performed the experiments and analyzed the data, with assistance from RF for smFISH experiments. MOM, SM, and BT developed the automated image analysis tools for NECTrack. YT performed the mathematical modeling. MOM and AH interpreted the data and wrote the paper with valuable contributions by RW.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Movie EV1
Movie EV2
Source Data for Expanded View
Source Data for Figures
Review Process File
Acknowledgements
We are grateful to Tracy Johnson, Shubhamoy Ghosh, and Sho Ohta for providing IκB‐targeting CRISPR constructs. We thank Zhang Cheng for preparatory work on the NFκB module for the mathematical model and Carlos Lopez for critical reading of the manuscript. This work was supported by NIH grants R01AI127867 and R01AI132731 to AH, and with a Research Fellowship to MOM by the Deutsche Forschungsgemeinschaft (DFG).
Mol Syst Biol. (2020) 00: e9677
Data availability
The datasets and computer code produced in this study are available in the following databases:
NFκB‐necroptosis modeling code: Dryad (https://doi.org/10.5068/D1W09.)
Live‐cell imaging: Dryad (https://doi.org/10.5068/D1C38J.)
NECtrack package: Dryad (https://doi.org/10.5068/D1W09J.)
smFISH code and source images: Figshare (accessory files: https://doi.org/10.6084/m9.figshare.12769667; Dataset 1: https://doi.org/10.6084/m9.figshare.12769658; Dataset 2: https://doi.org/10.6084/m9.figshare.12769655; Dataset 3: https://doi.org/10.6084/m9.figshare.12769661
References
- Aaes TL, Kaczmarek A, Delvaeye T, De Craene B, De Koker S, Heyndrickx L, Delrue I, Taminau J, Wiernicki B, De Groote P et al (2016) Vaccination with necroptotic cancer cells induces efficient anti‐tumor immunity. Cell Rep 15: 274–287 [DOI] [PubMed] [Google Scholar]
- Agbanoma G, Li C, Ennis D, Palfreeman AC, Williams LM, Brennan FM (2012) Production of TNF‐alpha in macrophages activated by T cells, compared with lipopolysaccharide, uses distinct IL‐10‐dependent regulatory mechanism. J Immunol 188: 1307–1317 [DOI] [PubMed] [Google Scholar]
- Albeck JG, Burke JM, Aldridge BB, Zhang M, Lauffenburger DA, Sorger PK (2008) Quantitative analysis of pathways controlling extrinsic apoptosis in single cells. Mol Cell 30: 11–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alon U (2007) Network motifs: theory and experimental approaches. Nat Rev Genet 8: 450–461 [DOI] [PubMed] [Google Scholar]
- Annibaldi A, Meier P (2018) Checkpoints in TNF‐induced cell death: implications in inflammation and cancer. Trends Mol Med 24: 49–65 [DOI] [PubMed] [Google Scholar]
- Basak S, Kim H, Kearns JD, Tergaonkar V, O'Dea E, Werner SL, Benedict CA, Ware CF, Ghosh G, Verma IM et al (2007) A fourth IkappaB protein within the NF‐kappaB signaling module. Cell 128: 369–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beg AA, Sha WC, Bronson RT, Ghosh S, Baltimore D (1995) Embryonic lethality and liver degeneration in mice lacking the RelA component of NF‐kappa B. Nature 376: 167–170 [DOI] [PubMed] [Google Scholar]
- Beutler B, Greenwald D, Hulmes JD, Chang M, Pan YC, Mathison J, Ulevitch R, Cerami A (1985a) Identity of tumour necrosis factor and the macrophage‐secreted factor cachectin. Nature 316: 552–554 [DOI] [PubMed] [Google Scholar]
- Beutler BA, Milsark IW, Cerami A (1985b) Cachectin/tumor necrosis factor: production, distribution, and metabolic fate in vivo . J Immunol 135: 3972–3977 [PubMed] [Google Scholar]
- Brumatti G, Ma C, Lalaoui N, Nguyen NY, Navarro M, Tanzer MC, Richmond J, Ghisi M, Salmon JM, Silke N et al (2016) The caspase‐8 inhibitor emricasan combines with the SMAC mimetic birinapant to induce necroptosis and treat acute myeloid leukemia. Sci Transl Med 8: 339ra69 [DOI] [PubMed] [Google Scholar]
- Carswell EA, Old LJ, Kassel RL, Green S, Fiore N, Williamson B (1975) An endotoxin‐induced serum factor that causes necrosis of tumors. Proc Natl Acad Sci USA 72: 3666–3670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng CS, Behar MS, Suryawanshi GW, Feldman KE, Spreafico R, Hoffmann A (2017) Iterative modeling reveals evidence of sequential transcriptional control mechanisms. Cell Syst 4: 330–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, Cuny GD, Mitchison TJ, Moskowitz MA, Yuan J (2005) Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol 1: 112–119 [DOI] [PubMed] [Google Scholar]
- Degterev A, Ofengeim D, Yuan J (2019) Targeting RIPK1 for the treatment of human diseases. Proc Natl Acad Sci USA 116: 9714–9722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dondelinger Y, Jouan‐Lanhouet S, Divert T, Theatre E, Bertin J, Gough PJ, Giansanti P, Heck AJ, Dejardin E, Vandenabeele P et al (2015) NF‐kappaB‐independent role of IKKalpha/IKKbeta in preventing RIPK1 kinase‐dependent apoptotic and necroptotic cell death during TNF signaling. Mol Cell 60: 63–76 [DOI] [PubMed] [Google Scholar]
- Dondelinger Y, Darding M, Bertrand MJ, Walczak H (2016) Poly‐ubiquitination in TNFR1‐mediated necroptosis. Cell Mol Life Sci 73: 2165–2176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dondelinger Y, Delanghe T, Rojas‐Rivera D, Priem D, Delvaeye T, Bruggeman I, Van Herreweghe F, Vandenabeele P, Bertrand MJM (2017) MK2 phosphorylation of RIPK1 regulates TNF‐mediated cell death. Nat Cell Biol 19: 1237–1247 [DOI] [PubMed] [Google Scholar]
- Draber P, Kupka S, Reichert M, Draberova H, Lafont E, de Miguel D, Spilgies L, Surinova S, Taraborrelli L, Hartwig T et al (2015) LUBAC‐recruited CYLD and A20 regulate gene activation and cell death by exerting opposing effects on linear ubiquitin in signaling complexes. Cell Rep 13: 2258–2272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foreman R, Wollman R (2020) Mammalian gene expression variability is explained by underlying cell state. Mol Syst Biol 16: e9146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman N, Cai L, Xie XS (2006) Linking stochastic dynamics to population distribution: an analytical framework of gene expression. Phys Rev Lett 97: 168302 [DOI] [PubMed] [Google Scholar]
- Fulda S (2015) Promises and challenges of Smac mimetics as cancer therapeutics. Clin Cancer Res 21: 5030–5036 [DOI] [PubMed] [Google Scholar]
- Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, Alnemri ES, Altucci L, Amelio I, Andrews DW et al (2018) Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ 25: 486–541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaudet S, Spencer SL, Chen WW, Sorger PK (2012) Exploring the contextual sensitivity of factors that determine cell‐to‐cell variability in receptor‐mediated apoptosis. PLoS Comput Biol 8: e1002482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong YN, Guy C, Olauson H, Becker JU, Yang M, Fitzgerald P, Linkermann A, Green DR (2017) ESCRT‐III acts downstream of MLKL to regulate necroptotic cell death and its consequences. Cell 169: 286–300.e16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green DR (2019) The coming decade of cell death research: five riddles. Cell 177: 1094–1107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunther C, Martini E, Wittkopf N, Amann K, Weigmann B, Neumann H, Waldner MJ, Hedrick SM, Tenzer S, Neurath MF et al (2011) Caspase‐8 regulates TNF‐alpha‐induced epithelial necroptosis and terminal ileitis. Nature 477: 335–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144: 646–667 [DOI] [PubMed] [Google Scholar]
- Harhaj EW, Dixit VM (2012) Regulation of NF‐kappaB by deubiquitinases. Immunol Rev 246: 107–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris LA, Frick PL, Garbett SP, Hardeman KN, Paudel BB, Lopez CF, Quaranta V, Tyson DR (2016) An unbiased metric of antiproliferative drug effect in vitro . Nat Methods 13: 497–500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartigan JA (1985) The dip test of unimodality. Ann Stat 13: 70–84 [Google Scholar]
- He S, Wang L, Miao L, Wang T, Du F, Zhao L, Wang X (2009) Receptor interacting protein kinase‐3 determines cellular necrotic response to TNF‐alpha. Cell 137: 1100–1111 [DOI] [PubMed] [Google Scholar]
- Hoffmann A, Baltimore D (2006) Circuitry of nuclear factor kappaB signaling. Immunol Rev 210: 171–186 [DOI] [PubMed] [Google Scholar]
- Hughes MA, Powley IR, Jukes‐Jones R, Horn S, Feoktistova M, Fairall L, Schwabe JW, Leverkus M, Cain K, MacFarlane M (2016) Co‐operative and hierarchical binding of c‐FLIP and caspase‐8: a unified model defines how c‐FLIP isoforms differentially control cell fate. Mol Cell 61: 834–849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito Y, Ofengeim D, Najafov A, Das S, Saberi S, Li Y, Hitomi J, Zhu H, Chen H, Mayo L et al (2016) RIPK1 mediates axonal degeneration by promoting inflammation and necroptosis in ALS. Science 353: 603–608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaern M, Blake WJ, Collins JJ (2003) The engineering of gene regulatory networks. Annu Rev Biomed Eng 5: 179–206 [DOI] [PubMed] [Google Scholar]
- Kamata H, Honda S, Maeda S, Chang L, Hirata H, Karin M (2005) Reactive oxygen species promote TNFalpha‐induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell 120: 649–661 [DOI] [PubMed] [Google Scholar]
- Kishi JY, Lapan SW, Beliveau BJ, West ER, Zhu A, Sasaki HM, Saka SK, Wang Y, Cepko CL, Yin P (2019) SABER amplifies FISH: enhanced multiplexed imaging of RNA and DNA in cells and tissues. Nat Methods 16: 533–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreuz S, Siegmund D, Scheurich P, Wajant H (2001) NF‐kappaB inducers upregulate cFLIP, a cycloheximide‐sensitive inhibitor of death receptor signaling. Mol Cell Biol 21: 3964–3973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krysko O, Aaes TL, Kagan VE, D'Herde K, Bachert C, Leybaert L, Vandenabeele P, Krysko DV (2017) Necroptotic cell death in anti‐cancer therapy. Immunol Rev 280: 207–219 [DOI] [PubMed] [Google Scholar]
- Lane K, Van Valen D, DeFelice MM, Macklin DN, Kudo T, Jaimovich A, Carr A, Meyer T, Pe'er D, Boutet SC et al (2017) Measuring signaling and RNA‐Seq in the same cell links gene expression to dynamic patterns of NF‐kappaB activation. Cell Syst 4: 458–469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurien L, Nagata M, Schunke H, Delanghe T, Wiederstein JL, Kumari S, Schwarzer R, Corona T, Kruger M, Bertrand MJM et al (2020) Autophosphorylation at serine 166 regulates RIP kinase 1‐mediated cell death and inflammation. Nat Commun 11: 1747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee EG, Boone DL, Chai S, Libby SL, Chien M, Lodolce JP, Ma A (2000) Failure to regulate TNF‐induced NF‐kappaB and cell death responses in A20‐deficient mice. Science 289: 2350–2354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee RE, Qasaimeh MA, Xia X, Juncker D, Gaudet S (2016) NF‐kappaB signalling and cell fate decisions in response to a short pulse of tumour necrosis factor. Sci Rep 6: 39519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, McQuade T, Siemer AB, Napetschnig J, Moriwaki K, Hsiao YS, Damko E, Moquin D, Walz T, McDermott A et al (2012) The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis. Cell 150: 339–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin KI, DiDonato JA, Hoffmann A, Hardwick JM, Ratan RR (1998) Suppression of steady‐state, but not stimulus‐induced NF‐kappaB activity inhibits alphavirus‐induced apoptosis. J Cell Biol 141: 1479–1487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lois C, Hong EJ, Pease S, Brown EJ, Baltimore D (2002) Germline transmission and tissue‐specific expression of transgenes delivered by lentiviral vectors. Science 295: 868–872 [DOI] [PubMed] [Google Scholar]
- Loriaux P, Hoffmann A (2009) Of elections and cell‐death decisions. Mol Cell 34: 257–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martens A, Priem D, Hoste E, Vetters J, Rennen S, Catrysse L, Voet S, Deelen L, Sze M, Vikkula H et al (2020) Two distinct ubiquitin‐binding motifs in A20 mediate its anti‐inflammatory and cell‐protective activities. Nat Immunol 21: 381–387 [DOI] [PubMed] [Google Scholar]
- Metzig M, Nickles D, Boutros M (2011a) Large‐scale RNAi screens to dissect TNF and NF‐kappaB signaling pathways. Adv Exp Med Biol 691: 131–139 [DOI] [PubMed] [Google Scholar]
- Metzig M, Nickles D, Falschlehner C, Lehmann‐Koch J, Straub BK, Roth W, Boutros M (2011b) An RNAi screen identifies USP2 as a factor required for TNF‐alpha‐induced NF‐kappaB signaling. Int J Cancer 129: 607–618 [DOI] [PubMed] [Google Scholar]
- Micheau O, Lens S, Gaide O, Alevizopoulos K, Tschopp J (2001) NF‐kappaB signals induce the expression of c‐FLIP. Mol Cell Biol 21: 5299–5305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micheau O, Tschopp J (2003) Induction of TNF receptor I‐mediated apoptosis via two sequential signaling complexes. Cell 114: 181–190 [DOI] [PubMed] [Google Scholar]
- Mitchell S, Roy K, Zangle TA, Hoffmann A (2018) Nongenetic origins of cell‐to‐cell variability in B lymphocyte proliferation. Proc Natl Acad Sci USA 115: E2888–E2897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moffitt JR, Hao J, Wang G, Chen KH, Babcock HP, Zhuang X (2016) High‐throughput single‐cell gene‐expression profiling with multiplexed error‐robust fluorescence in situ hybridization. Proc Natl Acad Sci USA 113: 11046–11051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Najafov A, Chen H, Yuan J (2017) Necroptosis and cancer. Trends Cancer 3: 294–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton K, Dixit VM (2012) Signaling in innate immunity and inflammation. Cold Spring Harb Perspect Biol 4: a006049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton K, Dugger DL, Maltzman A, Greve JM, Hedehus M, Martin‐McNulty B, Carano RA, Cao TC, van Bruggen N, Bernstein L et al (2016) RIPK3 deficiency or catalytically inactive RIPK1 provides greater benefit than MLKL deficiency in mouse models of inflammation and tissue injury. Cell Death Differ 23: 1565–1576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton K, Wickliffe KE, Dugger DL, Maltzman A, Roose‐Girma M, Dohse M, Komuves L, Webster JD, Dixit VM (2019) Cleavage of RIPK1 by caspase‐8 is crucial for limiting apoptosis and necroptosis. Nature 574: 428–431 [DOI] [PubMed] [Google Scholar]
- Oberst A, Dillon CP, Weinlich R, McCormick LL, Fitzgerald P, Pop C, Hakem R, Salvesen GS, Green DR (2011) Catalytic activity of the caspase‐8‐FLIP(L) complex inhibits RIPK3‐dependent necrosis. Nature 471: 363–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Dea E, Hoffmann A (2010) The regulatory logic of the NF‐kappaB signaling system. Cold Spring Harb Perspect Biol 2: a000216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ofengeim D, Yuan J (2013) Regulation of RIP1 kinase signalling at the crossroads of inflammation and cell death. Nat Rev Mol Cell Biol 14: 727–736 [DOI] [PubMed] [Google Scholar]
- Oliver Metzig M, Fuchs D, Tagscherer KE, Grone HJ, Schirmacher P, Roth W (2016) Inhibition of caspases primes colon cancer cells for 5‐fluorouracil‐induced TNF‐alpha‐dependent necroptosis driven by RIP1 kinase and NF‐kappaB. Oncogene 35: 3399–3409 [DOI] [PubMed] [Google Scholar]
- Onizawa M, Oshima S, Schulze‐Topphoff U, Oses‐Prieto JA, Lu T, Tavares R, Prodhomme T, Duong B, Whang MI, Advincula R et al (2015) The ubiquitin‐modifying enzyme A20 restricts ubiquitination of the kinase RIPK3 and protects cells from necroptosis. Nat Immunol 16: 618–627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paek AL, Liu JC, Loewer A, Forrester WC, Lahav G (2016) Cell‐to‐cell variation in p53 dynamics leads to fractional killing. Cell 165: 631–642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasparakis M, Vandenabeele P (2015) Necroptosis and its role in inflammation. Nature 517: 311–320 [DOI] [PubMed] [Google Scholar]
- Polykratis A, Martens A, Eren RO, Shirasaki Y, Yamagishi M, Yamaguchi Y, Uemura S, Miura M, Holzmann B, Kollias G et al (2019) A20 prevents inflammasome‐dependent arthritis by inhibiting macrophage necroptosis through its ZnF7 ubiquitin‐binding domain. Nat Cell Biol 21: 731–742 [DOI] [PubMed] [Google Scholar]
- Poukkula M, Kaunisto A, Hietakangas V, Denessiouk K, Katajamaki T, Johnson MS, Sistonen L, Eriksson JE (2005) Rapid turnover of c‐FLIPshort is determined by its unique C‐terminal tail. J Biol Chem 280: 27345–27355 [DOI] [PubMed] [Google Scholar]
- Priem D, Devos M, Druwe S, Martens A, Slowicka K, Ting AT, Pasparakis M, Declercq W, Vandenabeele P, van Loo G et al (2019) A20 protects cells from TNF‐induced apoptosis through linear ubiquitin‐dependent and ‐independent mechanisms. Cell Death Dis 10: 692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Razani B, Whang MI, Kim FS, Nakamura MC, Sun X, Advincula R, Turnbaugh JA, Pendse M, Tanbun P, Achacoso P et al (2020) Non‐catalytic ubiquitin binding by A20 prevents psoriatic arthritis‐like disease and inflammation. Nat Immunol 21: 422–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakon S, Xue X, Takekawa M, Sasazuki T, Okazaki T, Kojima Y, Piao JH, Yagita H, Okumura K, Doi T et al (2003) NF‐kappaB inhibits TNF‐induced accumulation of ROS that mediate prolonged MAPK activation and necrotic cell death. EMBO J 22: 3898–3909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samson AL, Zhang Y, Geoghegan ND, Gavin XJ, Davies KA, Mlodzianoski MJ, Whitehead LW, Frank D, Garnish SE, Fitzgibbon C et al (2020) MLKL trafficking and accumulation at the plasma membrane control the kinetics and threshold for necroptosis. Nat Commun 11: 3151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjana NE, Shalem O, Zhang F (2014) Improved vectors and genome‐wide libraries for CRISPR screening. Nat Methods 11: 783–784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrofelbauer B, Polley S, Behar M, Ghosh G, Hoffmann A (2012) NEMO ensures signaling specificity of the pleiotropic IKKbeta by directing its kinase activity toward IkappaBalpha. Mol Cell 47: 111–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selimkhanov J, Taylor B, Yao J, Pilko A, Albeck J, Hoffmann A, Tsimring L, Wollman R (2014) Systems biology. Accurate information transmission through dynamic biochemical signaling networks. Science 346: 1370–1373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalek AK, Satija R, Adiconis X, Gertner RS, Gaublomme JT, Raychowdhury R, Schwartz S, Yosef N, Malboeuf C, Lu D et al (2013) Single‐cell transcriptomics reveals bimodality in expression and splicing in immune cells. Nature 498: 236–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan B, Pan H, Najafov A, Yuan J (2018) Necroptosis in development and diseases. Genes Dev 32: 327–340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih VF, Kearns JD, Basak S, Savinova OV, Ghosh G, Hoffmann A (2009) Kinetic control of negative feedback regulators of NF‐kappaB/RelA determines their pathogen‐ and cytokine‐receptor signaling specificity. Proc Natl Acad Sci USA 106: 9619–9624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shindo R, Kakehashi H, Okumura K, Kumagai Y, Nakano H (2013) Critical contribution of oxidative stress to TNFalpha‐induced necroptosis downstream of RIPK1 activation. Biochem Biophys Res Commun 436: 212–216 [DOI] [PubMed] [Google Scholar]
- Spencer SL, Gaudet S, Albeck JG, Burke JM, Sorger PK (2009) Non‐genetic origins of cell‐to‐cell variability in TRAIL‐induced apoptosis. Nature 459: 428–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun SC (2020) A20 restricts inflammation via ubiquitin binding. Nat Immunol 21: 362–364 [DOI] [PubMed] [Google Scholar]
- Tang D, Kang R, Berghe TV, Vandenabeele P, Kroemer G (2019) The molecular machinery of regulated cell death. Cell Res 29: 347–364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi K, Karin M (2018) NF‐kappaB, inflammation, immunity and cancer: coming of age. Nat Rev Immunol 18: 309–324 [DOI] [PubMed] [Google Scholar]
- Thapa RJ, Basagoudanavar SH, Nogusa S, Irrinki K, Mallilankaraman K, Slifker MJ, Beg AA, Madesh M, Balachandran S (2011) NF‐kappaB protects cells from gamma interferon‐induced RIP1‐dependent necroptosis. Mol Cell Biol 31: 2934–2946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsanov N, Samacoits A, Chouaib R, Traboulsi AM, Gostan T, Weber C, Zimmer C, Zibara K, Walter T, Peter M et al (2016) smiFISH and FISH‐quant ‐ a flexible single RNA detection approach with super‐resolution capability. Nucleic Acids Res 44: e165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuchiya Y, Nakabayashi O, Nakano H (2015) FLIP the switch: regulation of apoptosis and necroptosis by cFLIP. Int J Mol Sci 16: 30321–30341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyson JJ, Novak B (2010) Functional motifs in biochemical reaction networks. Annu Rev Phys Chem 61: 219–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanhaesebroeck B, Decoster E, Van Ostade X, Van Bladel S, Lenaerts A, Van Roy F, Fiers W (1992) Expression of an exogenous tumor necrosis factor (TNF) gene in TNF‐sensitive cell lines confers resistance to TNF‐mediated cell lysis. J Immunol 148: 2785–2794 [PubMed] [Google Scholar]
- Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G (2010) Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol 11: 700–714 [DOI] [PubMed] [Google Scholar]
- Vanlangenakker N, Bertrand MJ, Bogaert P, Vandenabeele P, Vanden Berghe T (2011a) TNF‐induced necroptosis in L929 cells is tightly regulated by multiple TNFR1 complex I and II members. Cell Death Dis 2: e230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanlangenakker N, Vanden Berghe T, Bogaert P, Laukens B, Zobel K, Deshayes K, Vucic D, Fulda S, Vandenabeele P, Bertrand MJ (2011b) cIAP1 and TAK1 protect cells from TNF‐induced necrosis by preventing RIP1/RIP3‐dependent reactive oxygen species production. Cell Death Differ 18: 656–665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallach D, Varfolomeev EE, Malinin NL, Goltsev YV, Kovalenko AV, Boldin MP (1999) Tumor necrosis factor receptor and Fas signaling mechanisms. Annu Rev Immunol 17: 331–367 [DOI] [PubMed] [Google Scholar]
- Wallach D, Kang TB, Dillon CP, Green DR (2016) Programmed necrosis in inflammation: toward identification of the effector molecules. Science 352: aaf2154 [DOI] [PubMed] [Google Scholar]
- Wallach D, Kovalenko AV (2016) Tumor necrosis factor alpha (TNFalpha) In Compendium of Inflammatory Diseases, Parnham MJ. (ed). Basel: Springer; [Google Scholar]
- Welz PS, Wullaert A, Vlantis K, Kondylis V, Fernandez‐Majada V, Ermolaeva M, Kirsch P, Sterner‐Kock A, van Loo G, Pasparakis M (2011) FADD prevents RIP3‐mediated epithelial cell necrosis and chronic intestinal inflammation. Nature 477: 330–334 [DOI] [PubMed] [Google Scholar]
- Werner SL, Kearns JD, Zadorozhnaya V, Lynch C, O'Dea E, Boldin MP, Ma A, Baltimore D, Hoffmann A (2008) Encoding NF‐kappaB temporal control in response to TNF: distinct roles for the negative regulators IkappaBalpha and A20. Genes Dev 22: 2093–2101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C, Wu X, Zhang X, Xie Q, Fan C, Zhang H (2018) Embryonic lethality and host immunity of RelA‐deficient mice are mediated by both apoptosis and necroptosis. J Immunol 200: 271–285 [DOI] [PubMed] [Google Scholar]
- Yang Z, Wang Y, Zhang Y, He X, Zhong CQ, Ni H, Chen X, Liang Y, Wu J, Zhao S et al (2018) RIP3 targets pyruvate dehydrogenase complex to increase aerobic respiration in TNF‐induced necroptosis. Nat Cell Biol 20: 186–197 [DOI] [PubMed] [Google Scholar]
- Yu JW, Jeffrey PD, Shi Y (2009) Mechanism of procaspase‐8 activation by c‐FLIPL. Proc Natl Acad Sci USA 106: 8169–8174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Su SS, Zhao S, Yang Z, Zhong CQ, Chen X, Cai Q, Yang ZH, Huang D, Wu R et al (2017) RIP1 autophosphorylation is promoted by mitochondrial ROS and is essential for RIP3 recruitment into necrosome. Nat Commun 8: 14329 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Movie EV1
Movie EV2
Source Data for Expanded View
Source Data for Figures
Review Process File
Data Availability Statement
The datasets and computer code produced in this study are available in the following databases:
NFκB‐necroptosis modeling code: Dryad (https://doi.org/10.5068/D1W09.)
Live‐cell imaging: Dryad (https://doi.org/10.5068/D1C38J.)
NECtrack package: Dryad (https://doi.org/10.5068/D1W09J.)
smFISH code and source images: Figshare (accessory files: https://doi.org/10.6084/m9.figshare.12769667; Dataset 1: https://doi.org/10.6084/m9.figshare.12769658; Dataset 2: https://doi.org/10.6084/m9.figshare.12769655; Dataset 3: https://doi.org/10.6084/m9.figshare.12769661