

Abstract
Insulin gene mutation is the second most common cause of neonatal diabetes (NDM). It is also one of the genes involved in maturity-onset diabetes of the young (MODY). We aim to investigate molecular behaviors of different INS gene variants that may correlate with the clinical spectrum of diabetes phenotypes. In this study, we concentrated on two previously uncharacterized MODY-causing mutants, proinsulin-p.Gly44Arg [G(B20)R] and p.Pro52Leu [P(B28)L] (a novel mutant identified in one French family), and an NDM causing proinsulin-p.(Cys96Tyr) [C(A7)Y]. We find that these proinsulin mutants exhibit impaired oxidative folding in the endoplasmic reticulum (ER) with blocked ER export, ER stress, and apoptosis. Importantly, the proinsulin mutants formed abnormal intermolecular disulfide bonds that not only involved the mutant proinsulin, but also the co-expressed WT-proinsulin, forming misfolded disulfide-linked proinsulin complexes. This impaired the intracellular trafficking of WT-proinsulin and limited the production of bioactive mature insulin. Notably, although all three mutants presented with similar defects in folding, trafficking, and dominant negative behavior, the degrees of these defects appeared to be different. Specifically, compared to MODY mutants G(B20)R and P(B28)L that partially affected folding and trafficking of co-expressed WT-proinsulin, the NDM mutant C(A7)Y resulted in an almost complete blockade of the ER export of WT-proinsulin, decreasing insulin production, inducing more severe ER stress and apoptosis. We thus demonstrate that differences in cell biological behaviors among different proinsulin mutants correlate with the spectrum of diabetes phenotypes caused by the different INS gene mutations.
Keywords: Neonatal diabetes mellitus, Maturity onset diabetes of the young, Proinsulin misfolding, ER stress, Dominant negative effect, Insulin gene mutations
1. Introduction
Upon delivery into the oxidized endoplasmic reticulum (ER) lumen, proinsulin (the insulin precursor) undergoes rapid oxidative folding, forming three highly conserved disulfide bonds (B7-A7, B19-A20, and A6-A11) (Liu, Weiss, Arunagiri et al., 2018,Sun, Cui, He et al., 2015). Proinsulin bearing its native disulfide bonds exits the ER to travel through the Golgi complex and immature secretory granules, where proinsulin begins to be processed by prohormone convertase 1/2 (PC1/2) and carboxypeptidase E (CPE), forming mature insulin. It has been long believed that proinsulin folding, intracellular trafficking, and processing occur very efficiently (Dodson and Steiner, 1998,Steiner, Cunningham, Spigelman et al., 1967); however, more recent genetic and biological evidence indicate that proinsulin folding in the ER is not as efficient as was previously thought. Under normal physiological conditions, up to 10–15% of newly synthesized proinsulin may form mispaired intramolecular and/or intermolecular disulfide bonds (Guo, Xiong, Witkowski et al., 2014,Liu, Lara-Lemus, Shan et al., 2012,Liu, Li, Cavener et al., 2005,Liu, Ramos-Castañeda and Arvan, 2003,Schuit, In’t Veld and Pipeleers, 1988), and misfolded proinsulin may increase further when beta cells are forced to synthesize more proinsulin to compensate insulin resistance (Arunagiri, Haataja, Pottekat et al., 2019) or in beta cells with defective ER protein folding and export machinery (Zhu, Li, Xu et al., 2019,Jang, Pottekat, Poothong et al., 2019,Tsuchiya, Saito, Kadokura et al., 2018,Zito, Chin, Blais et al., 2010,Li, Itani, Haataja et al., 2019).
The pathological significance of misfolded proinsulin in the pathogenesis of diabetes has been highlighted by the discovery of new diabetogenic insulin gene mutations (Colombo, Porzio, Liu et al., 2008,Stoy, Edghill, Flanagan et al., 2007). To date, about 60 insulin gene variants have been identified in patients with monogenic diabetes (Liu, Sun, Cui et al., 2015,Liu, Hodish, Haataja et al., 2010,Weiss, 2009). More than half of these variants are predicted or experimentally confirmed to cause proinsulin misfolding in the ER (Liu et al., 2012,Park, Ye, Steiner et al., 2010,Liu, Haataja, Wright et al., 2010). Interestingly, the diabetes phenotypes caused by these variants range from severe insulin-deficient neonatal diabetes (NDM) to relative mild maturity onset diabetes of the young (MODY) (Stoy et al., 2007,Liu et al., 2015,Liu et al., 2010,Polak, Dechaume, Cavé et al., 2008,Edghill, Flanagan, Patch et al., 2008,Meur, Simon, Harun et al., 2010,Molven, Ringdal, Nordbø et al., 2008). The molecular mechanisms underlying the extent of this phenotypic spectrum of diabetes remain unclear.
In this study, we functionally characterized two new INS gene variants [G(B20)R and P(B28)L, identified in three French MODY patients with age of onset from 17 – 40 years old] in comparison to the NDM proinsulin mutant C(A7)Y. We found that MODY mutants showed partial defects in their oxidative folding and ER export compared with the NDM mutant. All of the mutants formed disulfide-linked proinsulin complexes (DLPC) with WT-proinsulin, thus impairing WT-proinsulin ER export and decreasing mature insulin production, but the severity of the phenotypes varied, with C(A7) being most severe. Thus, cell biological defects exhibited by mutant proinsulins appear to correlate with the clinical spectrum of diabetes phenotypes associated with different INS gene mutants.
2. Material and Methods
2.1. Patients
Three patients were recruited for genetic testing of monogenic diabetes. The biological collection of the Department of Genetics of Pitié-Salpêtrière Hospital has been declared to the Minister for research and the Director of the Regional Health Agency (biobank ID #DC2009–957). Patients signed an informed consent for the molecular diagnosis of their diabetes also indicating they approved to any research project performed in relation with their disease. Results of the genetic analyses are registered in a diagnosis database (CNIL certificate 16/02/2010-n°1412729). The study was done in agreement with the Declaration of Helsinky.
2.2. Genetic analyses
Targeted sequencing was performed based on a multiplex PCR assay (MODY-MASTRTM assay, Agilent) as previously described(Donath, Saint-Martin, Dubois-Laforgue et al., 2019). We used the sequence variant nomenclature recommendations (http://varnomen.hgvs.org/) for describing INS (NM_000207.2) variants and classified them following the American College of Medical Genetics and Genomics (ACMG) guidelines(Richards, Aziz, Bale et al., 2015).
2.3. Reagents and antibodies
Lipofectamine 2000 and 4–12% NuPage gel were purchased from Invitrogen (Carlsbad, CA, USA). Protein phosphatase inhibitor was purchased from Beyotime Biotechnology (Beijing, China). Protein A-Agarose was from Santa Cruz Biotechnology (Dallas, TX, USA). Guinea pig anti-insulin (dilution: 1:2000) was from Merck Millipore (Billerica, MA, USA) and mouse anti-proinsulin antibody was from Novus Biologicals (Littleton, CO, USA). Rabbit anti-Hsp90 (dilution: 1:2000) antibody was from Assay designs (Ann Arbor, MI, USA). Rabbit anti-cleaved caspase 3 antibody was from Cell Signalling Technology (Danvers, MA, USA). Annexin V with Alexa Fluor™ 555 conjugation, Goat anti-guinea pig IgG Alexa Fluor 555 and goat anti-rabbit IgG Alexa Fluor 435 was bought from Invitrogen (Carlsbad, CA, USA). Rabbit anti-Myc antibody was from Immunology Consultants Labs. Horseradish peroxidase–conjugated antibodies were from Jackson ImmunoResearch Laboratories (West Grove, PA, USA). Enhanced chemiluminescence Western blotting substrate was from Millipore (Billerica, MA, USA). Trans35S label and pure 35S-methionine were from PerkinElmer (Waltham, MA, USA).
2.4. Construction of plasmids encoding WT and mutant proinsulin
The plasmids encoding human WT preproinsulin with or without Myc-tag or GFP-tag in the C-peptide were described as previously (Liu et al., 2012,Liu et al., 2010,Guo, Sun, Li et al., 2018). G(B20)R or P(B28)L mutations was introduced into the constructs using the following primers: G(B20)R 5’-TCTACCTAGTGTGCAGGGAACGAGGCTTCTTC-3’, P(B28)L 5’-CTTCTTCTACACACTCAAGACCCGCCGG-3’, with the QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA, USA). All mutations were confirmed by DNA sequencing in using primer 5’-CTGTGGATGCGCCTCCTGC-3’.
2.5. Cell culture
Human embryonic kidney 293T (293T) cells and INS1 rat insulinoma cells were purchased from ATCC (Manassas, VA, USA). 293T cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM) with 10% Fetal bovine serum (FBS), penicillin (100 units/mL), and streptomycin (100 μg/mL). The INS1 cells were cultured in RPMI 1640 supplemented with 10% FBS, 1 mM sodium pyruvate, 10 mM Hepes, and 0.05 mM 2-mercaptoethanol (Sigma, St Louis, MO, USA). Mycoplasma detection and STR analysis were performed in these two cell lines.
2.6. Cells transfection, 35S-Met/Cys labeling, and immunoprecipitation
293T cells were seeded into 12-well plates 24 hours before transfection to achieve 70–90% cell confluent on the day of transfection. For each well, a total of 1ug plasmid DNA was transfected using Lipofectamine 2000. At 48 hours post transfection, the cells were cultured with 35S labeled amino acids and chased for the times indicated. The cells were washed once with PBS containing 20 mmol/L N-ethyl maleimide, then lysed in immunoprecipitation buffer [0.1M Tris-HCl 25mM (PH 7.0) , 20mM EDTA 5mM (PH 8.0), 1M NaCl 100mM, Triton×−100 0.1%]. A proteinase inhibitor cocktail was added to cell lysates and chase media. Trichloroacetic acid (TCA)-precipitable counts were used to quantify and normalize the amount of total protein among samples. The samples were immunoprecipitated with anti-insulin at 4°C overnight. Anti-insulin immunoprecipitates were washed twice with immunoprecipitation buffer and then boiled in SDS sample buffer with or without 100 mM DTT for 5 minutes, and analyzed in tris-tricine-urea-SDS-PAGE or 4–12% NuPage as indicated. Bands were quantified using ImageJ.
2.7. Fully /Partially-reduced two-dimensional electrophoresis (2-DE)
After 48 hours post transfection, 293T cells were lysed and resolved in 4–12% NuPage gel under non reducing condition. Fully-reduced 2-DE assay was carried out as following protocol: the gel was cut into six pieces, corresponding to the molecular weight of 6–14KD, 14–28KD, 28KD-38KD, 38KD-49KD, 49–98KD and 98–198KD. The gel pieces were boiled in loading buffer containing 100mM DTT for 5 minutes and run again in 4–12% NuPage gel. For Partially-reduced 2-DE assay, the gel was incubated with 1M Tris-HCl PH 6.8 with 50mM DTT at room temperature for 20 min. The gel was horizontally placed in a new stacking gel, re-run in 15% separating gel. Finally, the gel was then transferred to nitrocellulose and blotted by the anti-proinsulin antibody. The percentage of different molecular weight complexes was quantified using ImageJ.
2.8. BiP promoter-driven luciferase assay
INS1 cells were plated into 12-well plates 1 day before transfection. The cells were triple transfected with BiP promoter firefly-luciferase reporter plasmid (kindly provided by Dr. Randy J. Kaufman at Sanford Burnham Prebys Medical Discovery Institute), CMV-driven renilla-luciferase plasmid (Promega, Madison, WI, USA), and plasmid encoding human WT or mutant proinsulin at a DNA ratio of 1:2:5, respectively. At 48 h post-transfection, cells were collected and lysed. The severity of ER stress response was measured calculating the ratio of BiP-firefly-luciferase / Renilla luciferase activity, using Dual-Glo Luciferase assay.
2.9. Immunofluorescence and Annexin V staining
Immunofluorescence was employed in INS1 cells transfected with plasmid encoding sfGFP-tagged WT or mutant proinsulin. Briefly, transfected INS1 cells monolayer grown on coverslips were fixed with 4% paraformaldehyde for 30 min at room temperature, followed by permeabilization with 2% (wt/vol) saponin (Sigma, St Louis, MO, USA) for an additional 60 min and then blocking. The cell samples were incubated with primary antibodies followed by appropriate secondary antibodies conjugated with different fluor as indicated. Immunofluorescence images were acquired by using Axio Imager M2 (ZENISS, Baden-Württemberg, Germany).
For measuring apoptotsis in beta cells expressing proinsulin mutants, INS1 cells transfected with sfGFP-tagged proinsulin were fixed, permeabilized, blocked and incubated with cleaved caspase 3 antibody following the protocol described above. For surface Annexin V staining, adherent cells were incubated with Annexin V (detected by Alexa Fluor™ 555) directly at room tempture for 1 hour, followed by fixation.
2.10. Statistical analysis
All data were processed with GraphPad Prism 7 software and presented as means ± SD. Student’s t test and ANOVA was used to determine significance between groups. A p value < 0.05 was considered as statistically significant.
3. Results
3.1. Clinical characterization of three patients with INS variants in the B chain
A previously published heterozygous INS gene variant, c.130G>A p.Gly44Arg [G(B20)R], was identified in two unrelated female patients (Flannick, Beer, Bick et al., 2013). One of them was diagnosed with impaired fasting glucose (IFG)at the age of 17 years, without symptoms of diabetes. Currently, 18 years after the initial manifestation of diabetes, her blood glucose is well controlled [glycated hemoglobin A1c, (HbA1c), 5.4%] by diet alone. The other proband, who is now aged 33 years, was identified by an elevated HbA1c (11.0%) 4 years ago. Metformin therapy was started upon diagnosis of diabetes, and insulin injection was initiated several months ago because of pregnancy. Notably, the HbA1c decreased to 5.4% at the last visit. No relatives of either patient were affected.
A novel leucine substitution for proline at the 52nd residue, c.155C>T p.Pro52Leu [P(B28)L], was identified in a 40-year-old male who suffered from polyuria and unexplained weight loss, and presented with fasting hyperglycemia and elevated HbA1c (11.8%). He was treated with metformin and the HbA1c had decreased to 7.9% at the last visit, 2 years after diagnosis. The proband’s father and paternal grandfather were diagnosed with diabetes prior to the age of 40 years. The clinical characteristics of the three probands are listed in Table 1.
Table 1.
Clinical characterization of the three patients with INS variants in the B chain.
| Patient 1 | Patient 2 | Patient 3 | ||
|---|---|---|---|---|
| Gender | ||||
| At diagnosis | Age (y) | 17 | 29 | 40 |
| BMI (kg/m2) | 21.3 | 22.3 | 22.7 | |
| Fasting glycemia(mmol/l) | 6.44 | ND | 8.91 | |
| HbA1C (% - mmol/mol) | 5.5 – 37 | 11 – 97 | 11.8 – 105 | |
| Symptoms of diabetes | No | No | Polyuria, weight loss | |
| Treatment | Diet | Metformin | Metformin | |
| Family history | None | None | 3 affected generations | |
| Last visit | Age (y) | 35 | 33 | 42 |
| HbA1C (% - mmol/mol) | 5.4 – 36 | 5.4 – 36 | 7.9 – 63 | |
| Treatment | Diet | Insulin (pregnancy) | Metformin | |
| Arterial hypertension | No | ND | No | |
| Dyslipidemia | No | ND | No | |
| INS gene mutation | nuc. | c.130G>A | c.130G>A | c.155C>T |
| prot. | p.Gly44Arg G(B20)R |
p.Gly44Arg G(B20)R |
p.Pro52Leu P(B28)L |
|
ND: Not Determined
3.2. INS variants impair proinsulin oxidative folding and the ER export
Previous research has described that INS gene variants commonly cause severe insulin-deficient NDM (Stoy, Edghill, Flanagan et al., 2007,Colombo, Porzio, Liu et al., 2008) and also could be a rare cause of MODY (Boesgaard, Pruhova, Andersson et al., 2010). The three patients from this study presented with relatively mild diabetes phenotypes that could be managed with diet and/or oral hypoglycemic agents. We therefore investigated the extent to which MODY-INS variants affected proinsulin folding, trafficking, and maturation compared with variants associated with NDM. Preproinsulin consists sequentially of the signal peptide (SP), insulin B-chain, C peptide, and insulin A-chain. Three highly conserved disulfide bonds (B19-A20, A6-A11, and B7-A7) are critical for proper folding of proinsulin in the ER (Fig. 1). Amino acid sequence alignment of proinsulin B-chains from various species showed that the glycine at B20 and the proline at B28 are both highly conserved, suggesting that they may be important for proinsulin folding. To experimentally test this, we expressed WT, MODY mutants G(B20)R and P(B28)L, and NDM mutant C(A7)Y proinsulin in 293T cells. The folding of newly synthesized proinsulin was examined using tris-tricine-urea-SDS-PAGE under both reducing and non-reducing conditions (Guo et al., 2014,Liu et al., 2005). Although the total amount of newly synthesized proinsulin was comparable under reducing conditions (Fig. 2, right panel), the monomeric forms (including native, disulfide isomer, and reduced forms, Fig. 2, left panel) of mutant proinsulins were significantly decreased compared with that of WT-proinsulin (Fig. 2, left panel), suggesting that proinsulin mutants formed more mispaired disulfide isomers and disulfide-linked protein complexes that were not recovered as proinsulin monomers under non-reducing conditions.
Fig. 1. Highly conserved amino acids B20 and B28 in different species.
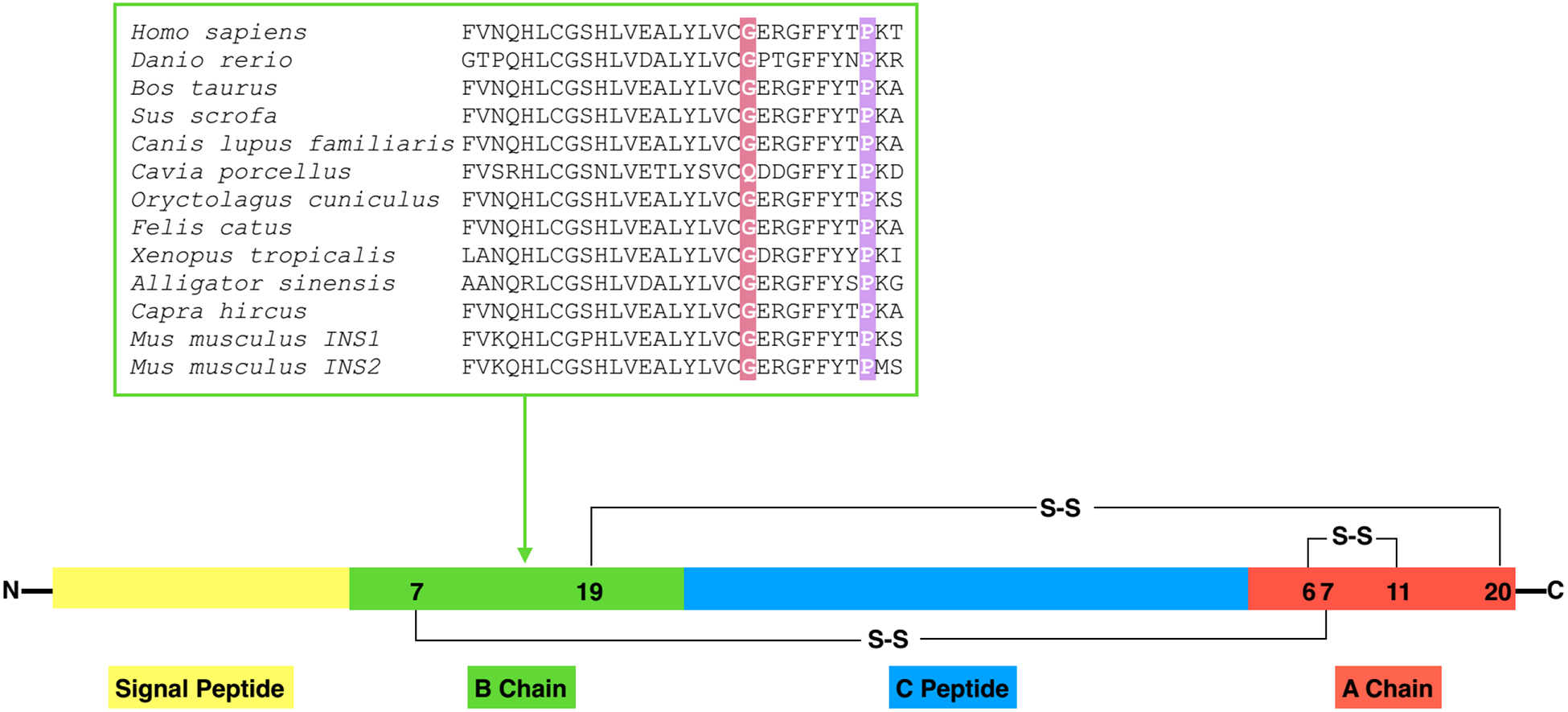
Structure of preproinsulin: The signal peptide (SP, yellow), insulin B chain (green), C peptide (blue), insulin A chain (red), S-S indicate the B19-A20, A6-A11, B7-A7 three different disulfide bonds. B chain alignment between human INS and orthologs. Amino acids B20 and B28 were marked with red and purple, respectively.
Fig. 2. INS variants impair proinsulin oxidative folding and ER export.
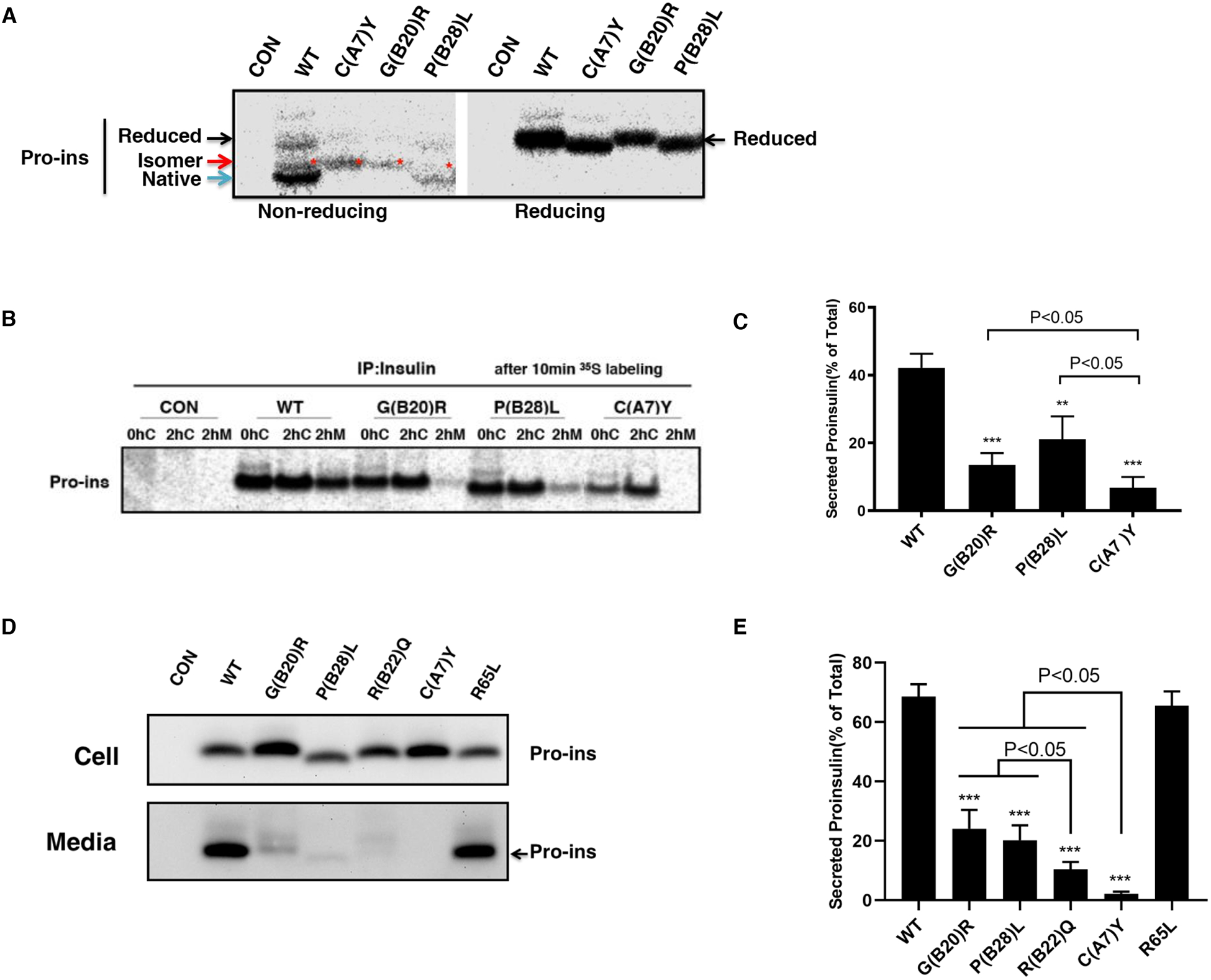
A. 293T cells were transfected with plasmids encoding wild-type (WT), or mutants C(A7)Y, G(B20)R, P(B28)L. At 48 h post-transfection, the cells were labeled with 35S-Met/Cys for 10 min. The newly synthesized proinsulin were precipitated with anti-insulin and analyzed by Tris–tricine–urea–SDS-PAGE under both non-reducing or reducing conditions. Reduced forms proinsulin marked by the black arrow, native forms marked by the blue arrow, disulfide isomers marked by the red arrow and star. B. 293T cells were transfected as Fig. 2A and pulse-labeled at 48 h with 35S-Met/Cys for 10 min followed by 0 or 2 h chase. Both cell lysates harvested after 0h (0hC) or 2h (2hC) chase and chase media (2hM) were immunoprecipitated with the anti-insulin and analyzed in 4–12% NuPage gel under reducing conditions with autoradiography. C. The secretion efficiency of WT or mutant proinsulin from at least three independent experiments shown in Fig. 2B was quantified using ImageJ. The results were shown as mean±SD, ** p <0.01 and *** p <0.001 comparing to WT (ANOVA test). D. 293T cells were transfected with plasmids encoding WT or mutant proinsulin as indicated. At 24 h post-transfection, the culture media were changed. After additional 24 hour incubation, the media were collected and cells were lysed. Both media and lysates were subjected to western blotting using anti-proinsulin antibody. E. The secretion efficiency of WT or mutant proinsulin under steady state from at least three independent experiments shown in Fig. 2D was quantified using ImageJ. The results were shown as mean±SD, *** p <0.001 comparing to WT (ANOVA test).
Misfolded proteins can be recognized and retained by the ER quality control system. We therefore asked the extent to which NDM and MODY mutations impair proinsulin ER export. We used two approaches: pulse-chase radiolabeling to follow newly-synthesized proinsulin, and Western blotting to evaluate the efficiency of proinsulin secretion at steady state. We found that compared with WT-Proinsulin, the secretion of MODY or NDM mutants were dramatically decreased for both the newly synthesized (Fig. 2B–C) and steady state proinsulin (Fig. 2D–E). The hyperproinsulinemia-inducing mutation proinsulin-R65L was well secreted. Importantly, although the NDM-causing C(A7)Y mutation almost abolished proinsulin secretion, up to 20% of the MODY mutations G(B20)R- or P(B28)L-proinsulin could indeed escape from the ER and be secreted from cells (Fig. 2B–E). Furthermore, another MODY-inducing mutation R(B22)Q, which usually causes diabetes during adolescence (Stoy, Olsen, Park et al., 2017), showed decreased secretion compared to G(B20)R or P(B28)L. These results indicate that the severity of the proinsulin secretion defect correlates with the diabetes phenotypes associated with these INS mutations.
3.3. Proinsulin mutants form misfolded disulfide-linked proinsulin complexes (DLPC) in the ER
As the monomeric mutant proinsulins exhibited dramatically decreased recovery under non-reducing conditions (Fig. 2A), we asked whether these mutants formed disulfide linked protein complexes. We performed Western blotting using anti-proinsulin monoclonal antibody that could detect misfolded proinsulin under nonreducing conditions (Arunagiri et al., 2019,Zhu et al., 2019), and found that the proinsulin mutants indeed formed more DLPCs (Fig. S1). To further confirm that proinsulin molecules were involved in formation of these DLPCs, we ran two-dimensional SDS-PAGE to allow intermolecular disulfide bonds present in the first dimension to be partially broken such that proinsulin monomers were released from the complexes. We found that some proinsulin molecules in the DLPCs were released as monomers in the presence of the reducing agent dithiothreitol (DTT) (Fig. S2). Next, we cut the first dimensional non-reducing gel into six pieces based on the molecular weight and boiled in SDS plus 100mM DTT, and re-ran the samples. Consistent with our previous reports (Liu et al., 2012,Liu, Li, Cavener et al., 2005,Arunagiri, Haataja, Pottekat et al., 2019), we found that more than 50% of WT-proinsulin was expressed as native monomer with a molecular weight ranging from 6–14 KD, followed by about 20% expressed as HMW complexes ranging from 49 to 198 KD, and < 10% WT-proinsulin expressed in the dimeric, trimeric, or tetrameric forms (Figs. 3A, E). However, C(A7)Y, G(B20)R, or P(B28)L proinsulin showed decreased monomer (< 40%) and increased dimer, trimer, tetramer, and high HMW DLPCs (Figs. 3B–E). Together, these data demonstrate that C(A7)Y, G(B20)R, and P(B28)L proinsulin were misfolded to an abnormally high degree.
Fig. 3. Proinsulin mutants form misfolded disulfide-linked proinsulin complexes (DLPC) in the ER.
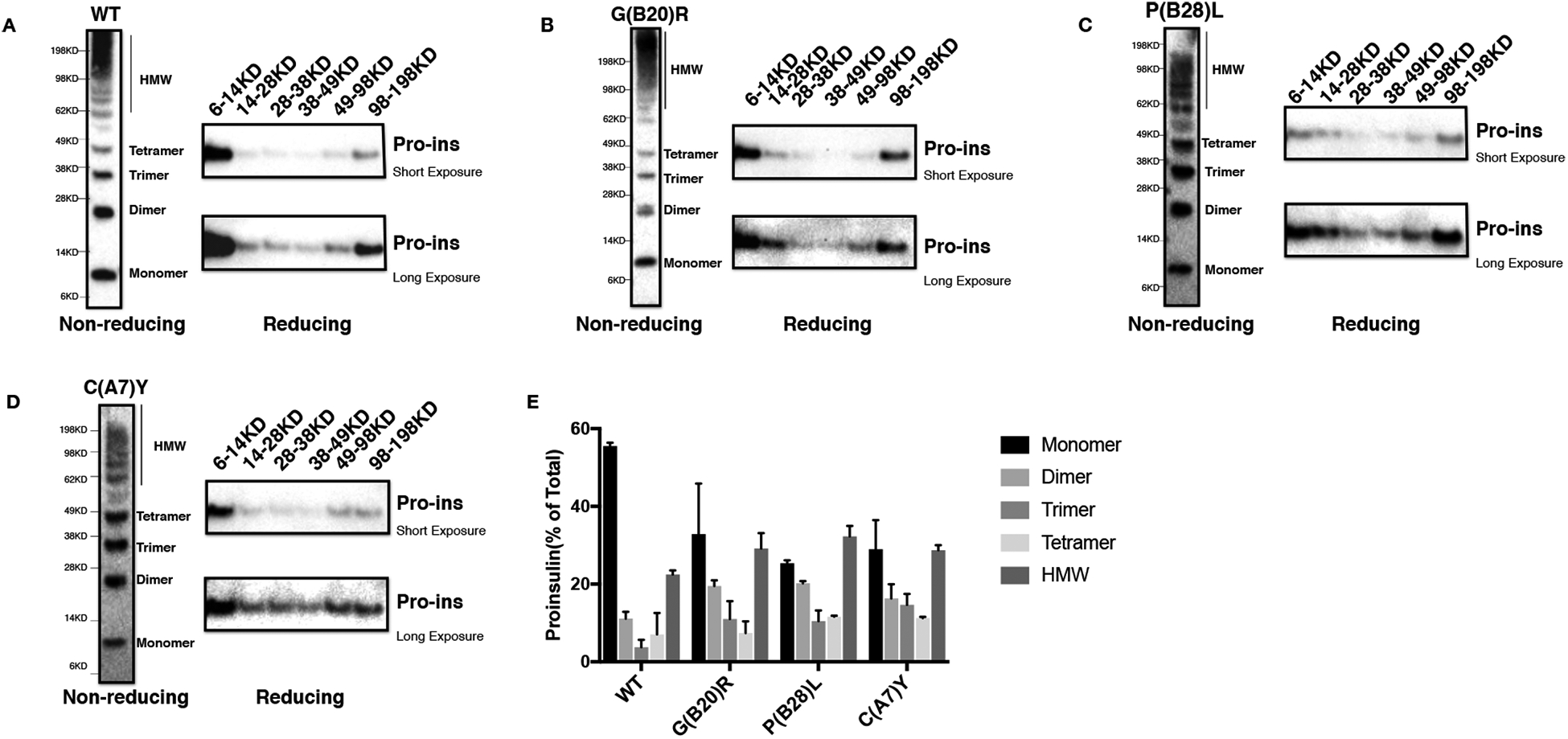
293T cells were transfected with Myc-tagged plasmids encoding WT or mutant proinsulin. At 48 h post-transfection, cell lysates were resolved in 4–12% NuPage under non-reducing condition (left panel). The gels were cut into 6 pieces corresponding to the molecular weight, then boiled in the sample buffer containing 100mM DTT followed by resolved again in 4–12% NuPage. The gels were transferred to nitrocellulose following by blotting with anti-proinsulin antibody. The same procedure was processed both for WT (A), G(B20)R (B), P(B28)L (C), and C(A7)Y (D). E. The 2-DE assay shown in Fig. 3A-D from at least three independent experiments was quantified using ImageJ. The percentages of fully reduced proinsulin monomer(6–14KD), dimer(14–28KD), trimer(28–38KD), tetramer(38–49KD) and high molecular weight (HMW) complexes (49–198KD) in total proinsulin molecules were calculated and shown as mean±SD.
3.4. Proinsulin mutants interact with co-expressed WT-proinsulin and impair the ER export of WT-proinsulin.
All NDM and MODY proinsulin mutants behave in a dominant fashion. We have previously shown that C(A7)Y mutant could interact with co-expressed WT-proinsulin and block its ER export (Liu, Hodish, Rhodes et al., 2007). To explore whether this dominant negative mechanism is also in play for MODY G(B20)R and P(B28)L mutants, we co-expressed Myc-tagged WT-proinsulin with untagged mutant (or WT) proinsulin in 293T cells. We found that although the mutants impaired secretion of co-expressed WT-proinsulin, a fraction of WT-proinsulin could still escape. Specifically, the dominant-negative effect of G(B20)R, P(B28)L and R(B22)Q was milder than that of C(A7)Y (Fig. 4A–B, Fig. S3). In contrast, the hyperproinsulinemia-inducing mutant R65L-proinsulin did not accumulate in the ER (Fig 2D), was well secreted, and failed to block secretion of co-expressed WT-proinsulin (Fig.4A–B). We then examined physical interactions between diabetes-causing proinsulin mutants and WT-proinsulin. As shown in the Fig. 4C–D, heterodimers and heterotrimers were clearly formed between untagged WT-proinsulin and Myc-tagged mutants. Co-immunoprecipitation experiments further confirmed interactions of WT-proinsulin and mutants (Fig. 4E–F). Since the mutants were misfolded and retained in the ER, the recruitment of WT-proinsulin into mixed disulfide-linked dimers/complexes was likely the underlying mechanism of the secretory blockade of co-expressed WT-proinsulin.
Fig.4. Proinsulin mutants interact with co-expressed WT-proinsulin and impair the ER export of WT-proinsulin.
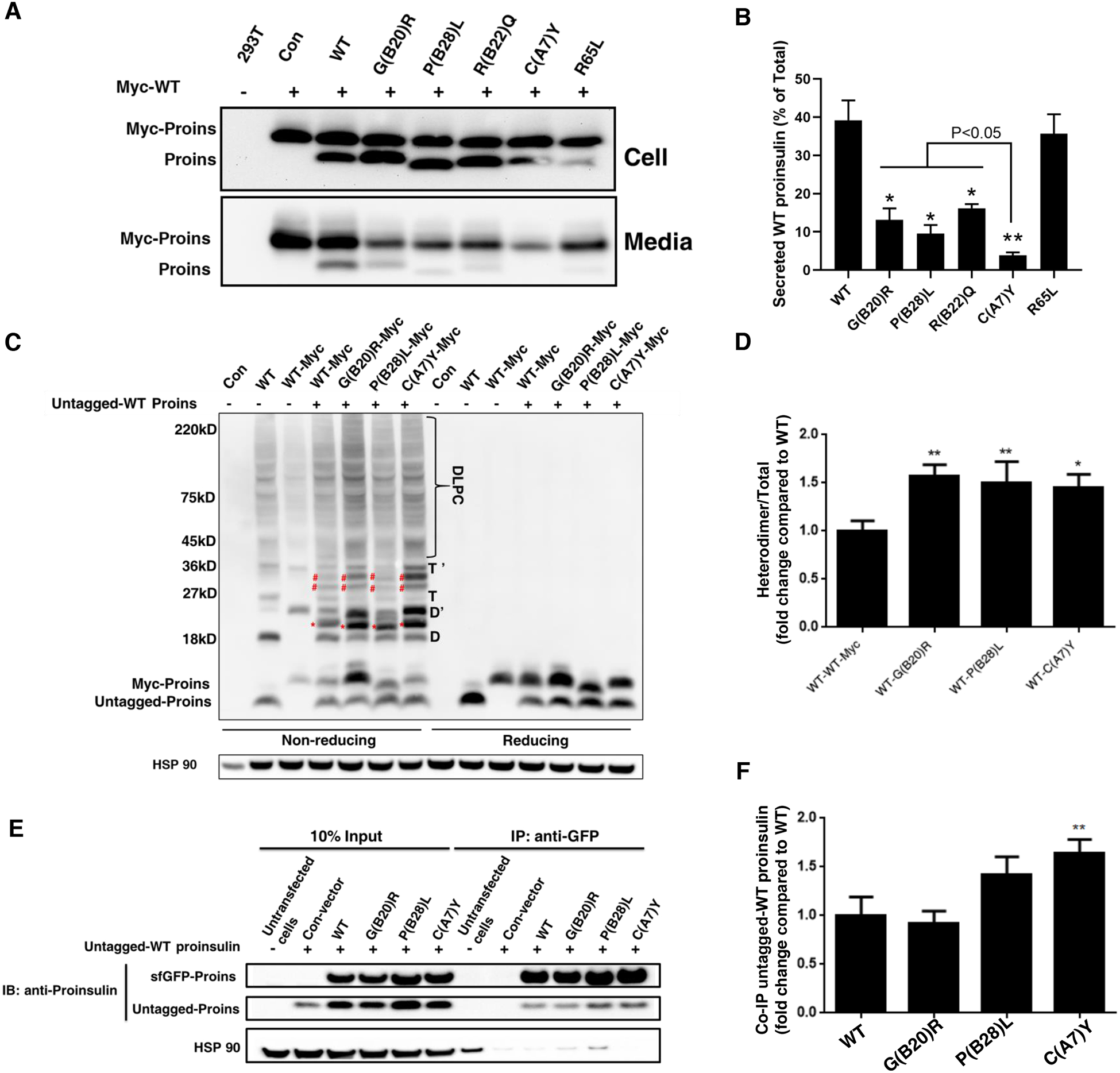
A-B. 293T cells were co-transfected with Myc-tagged WT-proinsulin (upper bands) and untagged WT-proinsulin or mutants (lower bands) as indicated. The secretion of Myc-tagged WT-proinsulin in the presence of untagged WT-proinsulin or mutants under 24h steady state was examined by immuno-blotting using anti-proinsulin. The percentages of secreted WT-proinsulin were quantified and calculated. * p <0.05 and ** p <0.01 comparing to WT (ANOVA test). C-D. 293T cells were co-transfected with untagged WT-proinsulin and Myc-tagged WT-proinsulin or mutants. The monomers, dimers (D refers to homodimers formed by untagged Proins, and D’ refers to homodimers formed by Myc-Proins, red star refers to heterodimers formed by untagged Proins and Myc-Proins), trimers (T refers to homotrimers formed by untagged Proins, and T’ refers to homotrimers formed by Myc-Proins, blue star refers to heterotrimers formed by untagged Proins and Myc-Proins), and higher-molecular weight disulfide-linked proinsulin complexes (DLPC) were analyzed under non reducing conditions. The total amount of untagged WT-proinsulin and Myc-tagged WT or mutants were analyzed under reducing condition. The percentages of heterodimer (red star marked) formed by untagged WT Proins and Myc-tagged proinsulin mutant were calculated. The percentage of heterodimer formed by untagged Proins-WT and Myc tagged Proins-WT was set to 1. * p <0.05 and ** p <0.01 comparing to WT (ANOVA test). E-F. 293T cells were co-transfected with untagged WT-proinsulin and super folder (sf) GFP-tagged WT-proinsulin or mutants. At 48 h post-transfection, cells were lysed and immunoprecipitated with the anti-GFP antibody, followed by immuno-blotting (IB) with anti-proinsulin antibody. The percentages of untagged WT-proinsulin pulled down by sfGFP-tagged proinsulin-WT or mutants were quantified and calculated. ** p <0.01 comparing to WT (ANOVA test).
3.5. Mutated proinsulin impairs endogenous insulin production and induces ER stress, leading to apoptosis in beta cells.
To further confirm that proinsulin mutants have dominant negative effects, we transfected rat insulinoma cell line (INS1E) with superfolder GFP (sfGFP)-tagged WT or mutant proinsulin and examined insulin content in the transfected cells (the cells with sfGFP signal). As expected, the endogenous insulin production of the cells transfected with sfGFP-tagged WT-proinsulin (white arrows) was comparable to that of neighboring non-transfected INS1E cells. By contrast, cells expressing sfGFP-tagged proinsulin mutants showed significantly decreased insulin production compared to non-transfected cells that served as an internal control (Fig. 5A), further confirming dominant-negative effects of the mutants.
Fig.5. Proinsulin mutants decrease endogenous insulin production and induce ER stress, leading to apoptosis in beta cells.
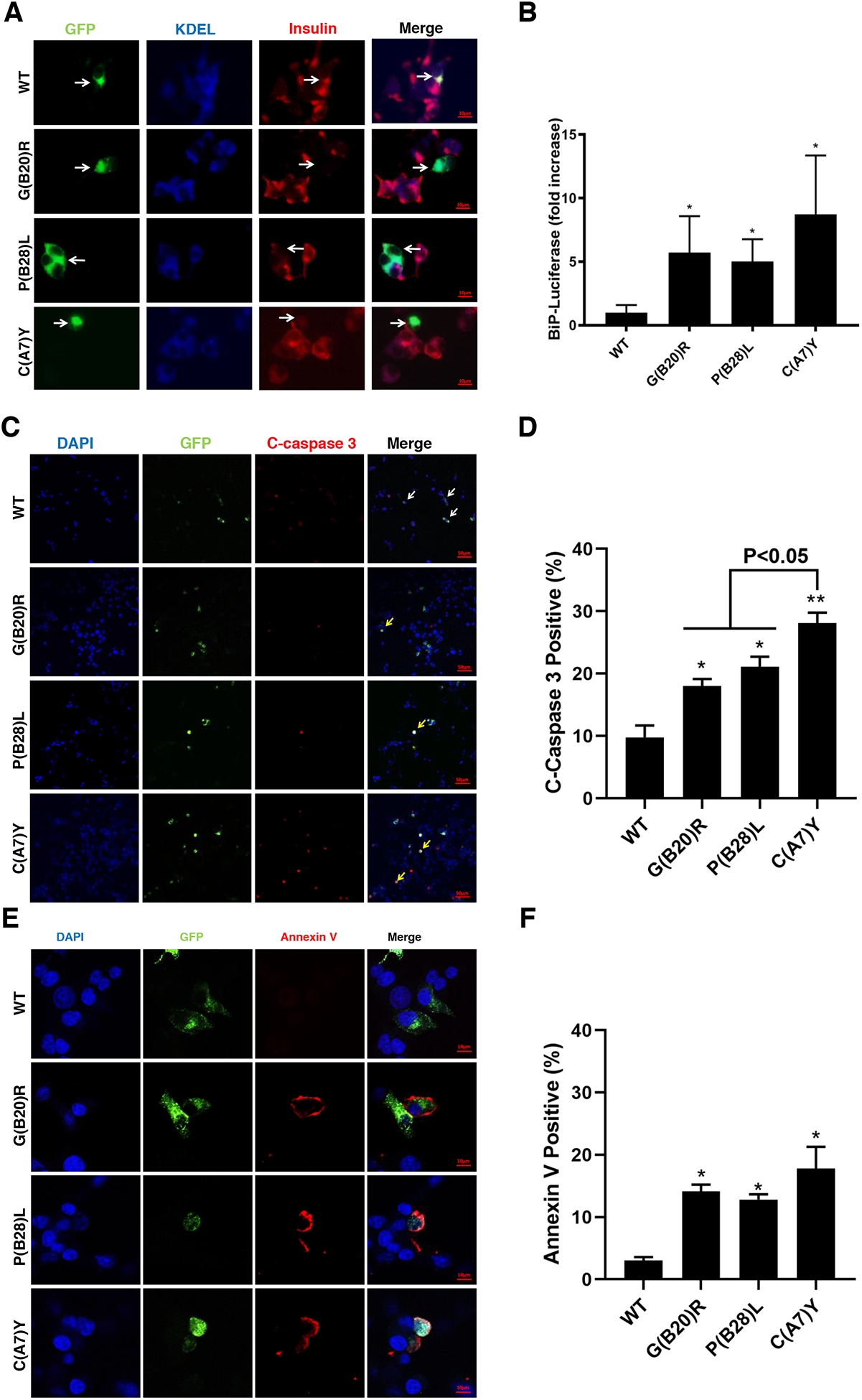
A. INS1 cells were transfected with plasmid encoding sfGFP-tagged WT, G(B20)R, P(B28)L or C(A7)Y proinsulin. At 48h post-transfection, the cells were permeabilized and immunoblotted with anti-insulin (red) and anti-KDEL (blue, ER marker). Arrows indicate the cells expressed exogenous sfGFP-tagged WT-proinsulin or mutants.
B. INS1 cells were transiently triple-transfected with the plasmids encoding BiP promoter-firefly luciferase, CMV-driven Renilla luciferase, and WT or mutant proinsulin at ratio 1 : 2 : 5 (This ratio helps ensure that BiP-luciferase serves as a reporter from cells synthesizing exogenously expressed proinsulins). At 48 h post-transfection, the cells were lysed and a ratio of firefly/renilla luciferase was measured. The relative activities of the BiP promoter in cells expressing proinsulin mutants were compared to that in cells expressing WT-proinsulin, which served as a control and set to 1. Results are from at least three independent experiments. *p <0.05 compared with WT-proinsulin (Student’s t-test). C. INS1 cells were transfected with sfGFP-tagged WT-proinsulin and variants as indicated. After 3 days post transfection, cells were fixed and stained with anti-cleaved caspase 3 antibody. Arrows indicate cells expressed exogenous proinsulin with (yellow arrow) or without (white arrow) apoptosis. D. Percentages of cleaved caspase 3 positive cell in transfected INS1 cells were quantified. * p <0.05 and ** p <0.01 comparing to WT (Student’s t-test). E. Representative images of INS1 cells expressing sfGFP-tagged proinsulin stained with Annexin V (red) and DAPI (blue) were shown. F. Proportion of Annexin V positive cells were quantitative analyzed. *p <0.05 compared with WT-proinsulin (Student’s t-test)
The accumulation of misfolded proinsulin (both mutant and WT) may result in ER stress, which has been implicated in pancreatic beta cell dysfunction (Sun, Cui, He et al., 2015). We explored whether these three mutants induced ER stress using the BiP-promoter firefly luciferase assay, as previously reported (Liu et al., 2012,Tirasophon, Welihinda and Kaufman, 1998). From this, it was apparent that the NDM C(A7)Y mutant induced a greater ER stress response compared with that of MODY G(B20)R and P(B28)L mutants in beta cells (Fig. 5B).
Persistent ER stress from NDM INS mutants can lead to apoptosis(Colombo et al., 2008). To verify whether MODY mutant proinsulins could induce apoptosis in beta cells, cleaved caspase-3 (an early apoptosis marker) was stained in INS1E cells expressing sfGFP-tagged proinsulins. Beta cells containing the C(A7)Y mutant induced apoptosis more strongly than the MODY mutants G(B20)R and P(B28)L (Fig.5C–D). We then assessed surface staining of annexin V, an additional apoptosis marker. As shown in Fig.5E–F, the percentage of Annexin V positive cells increased in beta cells 3 days after transfection with mutant proinsulins, and once again, proinsulin-C(A7)Y appeared to trigger a higher level of apoptosis than either of the two MODY mutants.
4. DISCUSSION
In this study, we studied two INS mutations G(B20)R and P(B28)L from patients with a MODY phenotype, in comparison to the NDM-causing C(A7)Y mutation. Our study showed that the G(B20)R, P(B28)L and C(A7)Y mutations each impaired proinsulin oxidative folding in the ER, causing proinsulin misfolding and DLPC formation, with impaired ER export (Figs. 2 and 3). Among the three mutants, C(A7)Y showed the most severe proinsulin misfolding and defect of ER export, but even the newly identified MODY mutation, P(B28)L appears to form more mispaired disulfide isomers than WT-proinsulin (Fig. 2A), suggesting that proinsulin misfolding underlies the disease in each of the three cases.
It is reported that one functional INS gene is sufficient to maintain normoglycemia, while in patients about 80% of all INS gene mutations are inherited in an autosomal dominant way (Liu, Sun, Cui et al., 2015). This strongly suggests a gain-of-toxic function from the mutant protein. Previous studies have reported that abnormal interactions between co-expressed mutant and proinsulin-WT in the ER can limit WT insulin production (Liu et al., 2012,Liu et al., 2010). In this paper, we confirmed all three mutants could form DLPC with co-expressed WT-proinsulin, which impaired intracellular trafficking of WT-proinsulin, limited mature insulin production, and induced ER stress and even cell apoptosis (Fig. 4–5). These findings extend those of previous reports (Colombo et al., 2008) to include mutations that elicit a MODY phenotype. On the one hand, we found that the NDM mutation C(A7)Y caused an almost complete blockade of co-expressed WT-proinsulin export, induced the most severe ER stress and apoptosis. In contrast, the MODY mutants G(B20)R and P(B28)L partially blocked the export of co-expressed WT-proinsulin (Fig. 4) and triggered milder ER stress response and less apoptosis (Fig. 5B–F). The milder degree of cell biological defect thus appears to correlate with the MODY phenotype rather than the NDM phenotype.
To date, around half of the autosomal dominant INS gene mutations have been predicted and/or experimentally confirmed to affect the folding process of proinsulin in the ER (Liu et al., 2010,Liu et al., 2015,Liu, Weiss, Arunagiri et al., 2018). The most well-studied INS gene mutation of this type is the C(A7)Y mutation, the severity of which may be due to the availability of an unpaired B7 cysteine to form abnormal disulfide linkages with other cysteine residues. INS gene mutations with unpaired cysteines are prone to interfering with disulfide maturation, leading to proinsulin misfolding (Liu et al., 2010,Liu et al., 2005) (Rajpal, Schuiki, Liu et al., 2012). However, the MODY mutants studied here bear all 6 native cysteine residues.
The B chain of insulin contains a type-II’ beta-turn (B7-B10) and a type-I beta-turn (B20-B23), both of which contain highly conserved glycines, including GlyB8 and GlyB20(Weiss, 2009); folding efficiency appears to depend to a much greater extent on the dihedral angle at GlyB8 (Nakagawa, Zhao, Hua et al., 2005).] Indeed, an alanine substitution at B20 actually results in an increased affinity for the insulin receptor (Kristensen, Kjeldsen, Wiberg et al., 1997). Conceivably, G(B20)R might also enhance insulin receptor binding affinity. Nevertheless, previous work has found that replacing GlyB20 (or ArgB22) with alanine produced poor yield in a yeast expression system, which may due to structure alteration of beta-turn B20–B23 (Kristensen et al., 1997). Especially, GlyB20 appears to be essential for the shift from the alpha-helix B8–B19 to the beta-turn B20–B23 and maintains a positive phi dihedral angle (“D-glycines”)(Nakagawa, Hua, Hu et al., 2006), which could be perturbed by any L-amino acid substitution.
Indeed, chain combination studies showed a reduced yield of insulin chain combination for L-AlaB20, but remarkably, yield could be rescued by chiral inversion D-AlaB20 (Nakagawa et al., 2006). This implies that the negative phi angle of G(B20)R is likely to enable folding, albeit with decreased efficiency. The G(B20)R proinsulin mutation does not directly generate any novel unpaired cysteine residues, yet structural analysis predicts it is highly possible that it might diminish the efficiency of (Cys)B19-(Cys)A20 disulfide bond formation (given that the 20th residue of the B-chain is adjacent to the disulfide bond B19-A20). Further investigation is still needed to verify if this is the case.
It should also be noted that two female probands carrying the same G(B20)R mutation showed different clinical features: one was diagnosed with mild fasting hyperglycemia controlled by diet alone, while the other presented with obviously increased HbAc1 requiring medical therapy. Indeed, a single mutation in the INS gene can be associated with a spectrum of phenotypes even within the same family (Edghill, Flanagan, Patch et al., 2008). One example comes from a proband carrying the p.Cys43Gly [C(B19)G] mutation, which disrupts one of the conserved disulfide bonds, leading to proinsulin misfolding. The proband developed very severe diabetes at 43 weeks after birth; however his father who carried the same mutation was diagnosed with type 2 diabetes at the age of 30 years.
Another example is the NDM-causing mutation p. Gly32Ser [G(B8)S], which has also been found to cause diabetes onset at the age of ~ 3 years (Bonfanti, Colombo, Nocerino et al., 2009). Almost certainly the variation in clinical presentation depends on additional genetic and environmental factors (Stoy et al., 2007). Notably, Weiss and colleagues have reported that proinsulin-G(B8)S could lead to an insulin that has a higher-than-WT affinity to the insulin receptor, yet proinsulin-G(B8)S displays impaired folding (Avital-Shmilovici, Whittaker, Weiss et al., 2014). Altogther these findings, and our present results, highlight diabetic phenotypes initiated by impaired proinsulin folding, followed thereafter by additional downstream consequences.
Supplementary Material
Funding
This work was supported by the National Natural Science Foundation of China (81700699, 81620108004, 81830025, 81870533); the Ministry of Science and Technology of China (2019YFA0802502); the Tianjin Municipal Science and Technology Bureau (17ZXMFSY00150 and 18JCYBJC93900) and The Second Hospital of Tianjin Medical University Youth Program (2017YDEY19). The work of L.H. and P.A. was supported by NIH DK48280.
Abbreviations
- DLPC
Disulfide-linked proinsulin complexes
- ER
Endoplasmic reticulum
- EV
Empty vector
- HMW
High molecular weight
- INS
Insulin
- MODY
Maturity onset diabetes of the young
- NDM
Neonatal diabetes mellitus
- WT
Wide type
Footnotes
Declaration of competing interest
None.
References
- Liu M, Weiss MA, Arunagiri A, Yong J, Rege N, Sun JH, Haataja L, Kaufman RJ and Arvan P, 2018. Biosynthesis, structure, and folding of the insulin precursor protein, Diabetes Obesity & Metabolism. 20, 28–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J, Cui J, He Q, Chen Z, Arvan P and Liu M, 2015. Proinsulin misfolding and endoplasmic reticulum stress during the development and progression of diabetes, Mol Aspects Med. 42, 105–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodson G and Steiner DF, 1998. The role of assembly in insulin’s biosynthesis., Curr Opin Struct Biol. 8, 189–94. [DOI] [PubMed] [Google Scholar]
- Steiner DF, Cunningham D, Spigelman L and Aten B, 1967. Insulin Biosynthesis: Evidence for a Precursor, Science. 157, 697–700. [DOI] [PubMed] [Google Scholar]
- Guo H, Xiong Y, Witkowski P, Cui J, Wang LJ, Sun J, Lara-Lemus R, Haataja L, Hutchison K, Shan SO, Arvan P and Liu M, 2014. Inefficient translocation of preproinsulin contributes to pancreatic beta cell failure and late-onset diabetes, J Biol Chem. 289, 16290–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Lara-Lemus R, Shan SO, Wright J, Haataja L, Barbetti F, Guo H, Larkin D and Arvan P, 2012. Impaired cleavage of preproinsulin signal peptide linked to autosomal-dominant diabetes, Diabetes. 61, 828–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Li Y, Cavener D and Arvan P, 2005. Proinsulin disulfide maturation and misfolding in the endoplasmic reticulum., J Biol Chem. 280, 13209–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Ramos-Castañeda J and Arvan P, 2003. Role of the Connecting Peptide in Insulin Biosynthesis, Journal of Biological Chemistry. 278, 14798–14805. [DOI] [PubMed] [Google Scholar]
- Schuit FC, In’t Veld PA and Pipeleers DG, 1988. Glucose stimulates proinsulin biosynthesis by a dose-dependent recruitment of pancreatic beta cells, Proceedings of the National Academy of Sciences of the United States of America. 85, 3865–3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arunagiri A, Haataja L, Pottekat A, Pamenan F, Kim S, Zeltser LM, Paton AW, Paton JC, Tsai B, Itkin-Ansari P, Kaufman RJ, Liu M and Arvan P, 2019. Proinsulin misfolding is an early event in the progression to type 2 diabetes, Elife. 8, e44532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu R, Li X, Xu J, Barrabi C, Kekulandara D, Woods J, Chen X and Liu M, 2019. Defective endoplasmic reticulum export causes proinsulin misfolding in pancreatic β cells, Molecular and Cellular Endocrinology. 110470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang I, Pottekat A, Poothong J, Yong J, Lagunas-Acosta J, Charbono A, Chen Z, Scheuner DL, Liu M, Itkin-Ansari P, Arvan P and Kaufman RJ, 2019. PDIA1/P4HB is required for efficient proinsulin maturation and ß cell health in response to diet induced obesity, eLife. 8, e44528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuchiya Y, Saito M, Kadokura H, Miyazaki JI, Tashiro F, Imagawa Y, Iwawaki T and Kohno K, 2018. IRE1-XBP1 pathway regulates oxidative proinsulin folding in pancreatic beta cells, J Cell Biol. 217, 1287–1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zito E, Chin KT, Blais J, Harding HP and Ron D, 2010. ERO1-beta, a pancreas-specific disulfide oxidase, promotes insulin biogenesis and glucose homeostasis, J Cell Biol. 188, 821–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Itani OA, Haataja L, Dumas KJ, Yang J, Cha J, Flibotte S, Shih HJ, Delaney CE, Xu J, Qi L, Arvan P, Liu M and Hu PJ, 2019. Requirement for translocon-associated protein (TRAP) alpha in insulin biogenesis, Sci Adv. 5, eaax0292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombo C, Porzio O, Liu M, Massa O, Vasta M, Salardi S, Beccaria L, Monciotti C, Toni S, Pedersen O, Hansen T, Federici L, Pesavento R, Cadario F, Federici G, Ghirri P, Arvan P, Lafusco D, Barbetti F and Diab ISPE, 2008. Seven mutations in the human insulin gene linked to permanent neonatal/infancy-onset diabetes mellitus, Journal of Clinical Investigation. 118, 2148–2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoy J, Edghill EL, Flanagan SE, Ye H, Paz VP, Pluzhnikov A, Below JE, Hayes MG, Cox NJ, Lipkind GM, Lipton RB, Greeley SAW, Patch A-M, Ellard S, Steiner DF, Hattersley AT, Philipson LH, Bell GI and Neonatal Diabetes International Collaborative Group, 2007. Insulin gene mutations as a cause of permanent neonatal diabetes, Proceedings of the National Academy of Sciences. 104, 15040–15044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Sun J, Cui J, Chen W, Guo H, Barbetti F and Arvan P, 2015. INS-gene mutations: From genetics and beta cell biology to clinical disease, Mol Aspects Med. 42, 3–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Hodish I, Haataja L, Lara-Lemus R, Rajpal G, Wright J and Arvan P, 2010. Proinsulin misfolding and diabetes: mutant INS gene-induced diabetes of youth, Trends in Endocrinology & Metabolism. 21, 652–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss MA, 2009. Proinsulin and the Genetics of Diabetes Mellitus, J. Biol. Chem 284, 19159–19163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S-Y, Ye H, Steiner DF and Bell GI, 2010. Mutant proinsulin proteins associated with neonatal diabetes are retained in the endoplasmic reticulum and not efficiently secreted, Biochemical and Biophysical Research Communications. 391, 1449–1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Haataja L, Wright J, Wickramasinghe NP, Hua QX, Phillips NF, Barbetti F, Weiss MA and Arvan P, 2010. Mutant INS-gene induced diabetes of youth: proinsulin cysteine residues impose dominant-negative inhibition on wild-type proinsulin transport, PLoS One. 5, e13333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polak M, Dechaume A, Cavé H, Nimri R, Crosnier H, Sulmont V, de Kerdanet M, Scharfmann R, Lebenthal Y, Froguel P and Vaxillaire M, 2008. Heterozygous Missense Mutations in the Insulin Gene Are Linked to Permanent Diabetes Appearing in the Neonatal Period or in Early Infancy, Diabetes. 57, 1115–1119. [DOI] [PubMed] [Google Scholar]
- Edghill EL, Flanagan SE, Patch A-M, Boustred C, Parrish A, Shields B, Shepherd MH, Hussain K, Kapoor RR, Malecki M, MacDonald MJ, Støy J, Steiner DF, Philipson LH, Bell GI, Hattersley AT and Ellard S, 2008. Insulin Mutation Screening in 1,044 Patients With Diabetes, Diabetes. 57, 1034–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meur G, Simon A, Harun N, Virally M, Dechaume A.l., Bonnefond A.l., Fetita S, Tarasov AI, Guillausseau P-J, Boesgaard T.W.v., Pedersen O, Hansen T, Polak M, Gautier J.-F.ß., Froguel P, Rutter GA and Vaxillaire M, 2010. Insulin Gene Mutations Resulting in Early-Onset Diabetes: Marked Differences in Clinical Presentation, Metabolic Status, and Pathogenic Effect Through Endoplasmic Reticulum Retention, Diabetes. 59, 653–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molven A, Ringdal M, Nordbà AM, Ræder H, Støy J, Lipkind GM, Steiner DF, Philipson LH, Bergmann I, Aarskog D, Undlien DE, Joner G, Søvik O, Bell GI and Njølstad P.l.R., 2008. Mutations in the Insulin Gene Can Cause MODY and Autoantibody-Negative Type 1 Diabetes, Diabetes. 57, 1131–1135. [DOI] [PubMed] [Google Scholar]
- Donath X, Saint-Martin C, Dubois-Laforgue D, Rajasingham R, Mifsud F, Ciangura C, Timsit J, Bellanne-Chantelot C and Monogenic Diabetes Study Group of the Societe Francophone du, D., 2019. Next-generation sequencing identifies monogenic diabetes in 16% of patients with late adolescence/adult-onset diabetes selected on a clinical basis: a cross-sectional analysis, BMC Med. 17, 132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL and Committee ALQA, 2015. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology, Genet Med. 17, 405–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo H, Sun J, Li X, Xiong Y, Wang H, Shu H, Zhu R, Liu Q, Huang Y, Madley R, Wang Y, Cui J, Arvan P and Liu M, 2018. Positive charge in the n-region of the signal peptide contributes to efficient post-translational translocation of small secretory preproteins, J Biol Chem. 293, 1899–1907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flannick J, Beer NL, Bick AG, Agarwala V, Molnes J, Gupta N, Burtt NP, Florez JC, Meigs JB, Taylor H, Lyssenko V, Irgens H, Fox E, Burslem F, Johansson S, Brosnan MJ, Trimmer JK, Newton-Cheh C, Tuomi T, Molven A, Wilson JG, O’Donnell CJ, Kathiresan S, Hirschhorn JN, Njolstad PR, Rolph T, Seidman JG, Gabriel S, Cox DR, Seidman CE, Groop L and Altshuler D, 2013. Assessing the phenotypic effects in the general population of rare variants in genes for a dominant Mendelian form of diabetes, Nat Genet. 45, 1380–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoy J, Edghill EL, Flanagan SE, Ye H, Paz VP, Pluzhnikov A, Below JE, Hayes MG, Cox NJ, Lipkind GM, Lipton RB, Greeley SA, Patch AM, Ellard S, Steiner DF, Hattersley AT, Philipson LH, Bell GI and Neonatal Diabetes International Collaborative, G., 2007. Insulin gene mutations as a cause of permanent neonatal diabetes, Proc Natl Acad Sci U S A. 104, 15040–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombo C, Porzio O, Liu M, Massa O, Vasta M, Salardi S, Beccaria L, Monciotti C, Toni S, Pedersen O, Hansen T, Federici L, Pesavento R, Cadario F, Federici G, Ghirri P, Arvan P, Iafusco D, Barbetti F, Early Onset Diabetes Study Group of the Italian Society of Pediatric, E. and Diabetes, 2008. Seven mutations in the human insulin gene linked to permanent neonatal/infancy-onset diabetes mellitus, J Clin Invest. 118, 2148–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boesgaard TW, Pruhova S, Andersson EA, Cinek O, Obermannova B, Lauenborg J, Damm P, Bergholdt R, Pociot F, Pisinger C, Barbetti F, Lebl J, Pedersen O and Hansen T, 2010. Further evidence that mutations in INS can be a rare cause of Maturity-Onset Diabetes of the Young (MODY), BMC Med Genet. 11, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoy J, Olsen J, Park SY, Gregersen S, Hjorringgaard CU and Bell GI, 2017. In vivo measurement and biological characterisation of the diabetes-associated mutant insulin p.R46Q (GlnB22-insulin), Diabetologia. 60, 1423–1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Li Y, Cavener D and Arvan P, 2005. Proinsulin disulfide maturation and misfolding in the endoplasmic reticulum, J Biol Chem. 280, 13209–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arunagiri A, Haataja L, Pottekat A, Pamenan F, Kim S, Zeltser LM, Paton AW, Paton JC, Tsai B, Itkin-Ansari P, Kaufman RJ, Liu M and Arvan P, 2019. Proinsulin misfolding is an early event in the progression to type 2 diabetes, Elife. 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Hodish I, Rhodes CJ and Arvan P, 2007. Proinsulin maturation, misfolding, and proteotoxicity, Proc Natl Acad Sci U S A. 104, 15841–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J, Cui J, He Q, Chen Z, Arvan P and Liu M, 2015. Proinsulin misfolding and endoplasmic reticulum stress during the development and progression of diabetes, Mol Aspects Med. 42, 105–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirasophon W, Welihinda AA and Kaufman RJ, 1998. A stress response pathway from the endoplasmic reticulum to the nucleus requires a novel bifunctional protein kinase/endoribonuclease (Ire1p) in mammalian cells, Genes & Development. 12, 1812–1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Sun J, Cui J, Chen W, Guo H, Barbetti F and Arvan P, 2015. INS-gene mutations: from genetics and beta cell biology to clinical disease, Mol Aspects Med. 42, 3–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Weiss MA, Arunagiri A, Yong J, Rege N, Sun J, Haataja L, Kaufman RJ and Arvan P, 2018. Biosynthesis, structure, and folding of the insulin precursor protein, Diabetes Obes Metab. 20 Suppl 2, 28–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajpal G, Schuiki I, Liu M, Volchuk A and Arvan P, 2012. Action of protein disulfide isomerase on proinsulin exit from endoplasmic reticulum of pancreatic beta-cells, J Biol Chem. 287, 43–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss MA, 2009. The structure and function of insulin: decoding the TR transition, Vitam Horm. 80, 33–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa SH, Zhao M, Hua QX, Hu SQ, Wan ZL, Jia W and Weiss MA, 2005. Chiral mutagenesis of insulin. Foldability and function are inversely regulated by a stereospecific switch in the B chain, Biochemistry. 44, 4984–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristensen C, Kjeldsen T, Wiberg FC, Schaffer L, Hach M, Havelund S, Bass J, Steiner DF and Andersen AS, 1997. Alanine scanning mutagenesis of insulin, J Biol Chem. 272, 12978–83. [DOI] [PubMed] [Google Scholar]
- Nakagawa SH, Hua QX, Hu SQ, Jia W, Wang S, Katsoyannis PG and Weiss MA, 2006. Chiral mutagenesis of insulin. Contribution of the B20-B23 beta-turn to activity and stability, J Biol Chem. 281, 22386–96. [DOI] [PubMed] [Google Scholar]
- Edghill EL, Flanagan SE, Patch AM, Boustred C, Parrish A, Shields B, Shepherd MH, Hussain K, Kapoor RR, Malecki M, MacDonald MJ, Stoy J, Steiner DF, Philipson LH, Bell GI, Neonatal Diabetes International Collaborative, G., Hattersley AT and Ellard S, 2008. Insulin mutation screening in 1,044 patients with diabetes: mutations in the INS gene are a common cause of neonatal diabetes but a rare cause of diabetes diagnosed in childhood or adulthood, Diabetes. 57, 1034–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonfanti R, Colombo C, Nocerino V, Massa O, Lampasona V, Iafusco D, Viscardi M, Chiumello G, Meschi F and Barbetti F, 2009. Insulin gene mutations as cause of diabetes in children negative for five type 1 diabetes autoantibodies, Diabetes Care. 32, 123–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avital-Shmilovici M, Whittaker J, Weiss MA and Kent SB, 2014. Deciphering a molecular mechanism of neonatal diabetes mellitus by the chemical synthesis of a protein diastereomer, [D-AlaB8]human proinsulin, J Biol Chem. 289, 23683–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.