

Abstract
Introduction
Repeated incursions of highly pathogenic avian influenza virus (HPAIV) H5 subtype of Gs/GD lineage pose a serious threat to poultry worldwide. We provide a detailed analysis of the spatio-temporal spread and genetic characteristics of HPAIV Gs/GD H5N8 from the 2019/20 epidemic in Poland.
Material and methods
Samples from poultry and free-living birds were tested by real-time RT-PCR. Whole genome sequences from 24 (out of 35) outbreaks were generated and genetic relatedness was established. The clinical status of birds and possible pathways of spread were analysed based on the information provided by veterinary inspections combined with the results of phylogenetic studies.
Results
Between 31 December 2019 and 31 March 2020, 35 outbreaks in commercial and backyard poultry holdings and 1 case in a wild bird were confirmed in nine provinces of Poland. Most of the outbreaks were detected in meat turkeys and ducks. All characterised viruses were closely related and belonged to a previously unrecognised genotype of HPAIV H5N8 clade 2.3.4.4b. Wild birds and human activity were identified as the major modes of HPAIV spread.
Conclusion
The unprecedentedly late introduction of the HPAI virus urges for re-evaluation of current risk assessments. Continuous vigilance, strengthening biosecurity and intensifying surveillance in wild birds are needed to better manage the risk of HPAI occurrence in the future.
Keywords: highly pathogenic avian influenza, H5N8, epidemic
Introduction
Highly pathogenic avian influenza (HPAI) is an infectious and highly contagious viral disease of birds (2, 5). The HPAI viruses (HPAIV) emerge as a result of mutation from low-virulence progenitors (low pathogenic avian influenza viruses, LPAIV), for which wild aquatic birds are the natural reservoir (2, 30). Influenza viruses belong to the Orthomyxoviridae family that encompasses negative-sense single-stranded RNA viruses with segmented genomes (12). Based on the antigenic structure of the surface glycoproteins (haemagglutinin and neuraminidase), influenza viruses prevalent in birds have been classified into 16 haemagglutinin subtypes (H1–H16) and 9 neuraminidase subtypes (N1–N9) that form different combinations. However, only the H5 and H7 LPAIV subtypes (irrespective of the N subtype) have the capacity to mutate to HPAIV and it is believed that the transformation occurs upon transmission from wild birds and adaptation of an LPAIV to gallinaceous poultry (2, 18). The HPAIV which have emerged pose a significant risk to poultry worldwide due to high mortality and impact on trade. Additionally, HPAIV can be re-introduced to wild migratory birds and quickly spread to new geographic areas (3).
HPAI as a global problem is mostly associated with the A/goose/Guangdong/1/1996 (Gs/GD) lineage of HPAIV H5 viruses (H5 Gs/GD), which were first reported in 1996 in China as an H5N1 subtype (8). In subsequent years, the H5 Gs/GD viruses evolved into multiple genetic clades and genotypes through genetic drift (i.e. accumulation of point mutations over time) or reassortment (i.e. switching of viral RNA segments between different influenza viruses co-infecting the same avian host) (10, 18). More than a decade ago, H5 Gs/GD viruses of clade 2.3.4 started to evolve into lower-order sub-clades and it has been shown that the sub-clade 2.3.4.4 viruses (further differentiated into four subgroups unofficially designated 2.3.4.4a to 2.3.4.4d) were unusually prone to reassortment (18, 25). Since 2014, a rapid global expansion of H5 Gs/GD clade 2.3.4.4 has been observed, and frequent reassortment events with LPAIV prevailing in local populations of Eurasian wild birds resulted in the generation of novel genotypes bearing different neuraminidase subtypes, e.g. H5N2, H5N5, H5N6 or H5N8 (15, 18). The viruses spread from Asia to Europe, North America and Africa but the magnitude of the epidemics varied greatly (1, 13, 15).
Poland was among the countries affected by the 2016–17 HPAI epidemic of the H5 Gs/GD clade 2.3.4.4b viruses, which so far has been the largest in Europe (1). On 31 December 2019, a novel HPAIV H5N8 clade 2.3.4.4b genotype was confirmed in Poland (27) and quickly spread across the country. This article provides details on the epidemic with a special focus on the clinical outcome of infections in different poultry species, possible pathways of spread, preventive measures undertaken in response to the situation and molecular characterisation of the representative HPAIV H5N8 isolates.
Material and Methods
Sample collection. Since the detection of the first outbreak on 31 December 2019, passive surveillance in poultry was enhanced and entailed sampling suspected cases (those presenting clinical signs indicative of HPAI) and a “testing to exclude” (TTE) scheme (sampling birds presenting clinical signs not suggestive of HPAI). In response to the confirmation of the HPAI outbreak in poultry, laboratory testing of birds in contact holdings (whether with sick or healthy birds), clinically healthy flocks before shipment to slaughterhouses or movement within the restriction zone, and clinically healthy flocks after re-population was carried out in compliance with the Directive 2005/94/EC (6). A standard set of organ samples or oropharyngeal or cloacal swabs were collected from dead wild birds.
Between 31 December 2019 and 16 July 2020, a total of 428 poultry flocks were tested, including laying hens, broiler chickens, fattening turkeys, geese, and ducks, breeding geese and duck and guinea fowl (Table 1). Additionally, 84 wild birds mainly comprising swans, ducks and pigeons, were examined in the same period. Descriptive data on the clinical course as well as morbidity and mortality were collected by official veterinarians. Epidemiological investigation was performed to determine the most likely route of virus introduction into the flock.
Table 1.
Number and type of tested flocks per species and production category
| Type of poultry holding tested | Laying hens | Broiler chickens | Fattening turkeys | Fattening geese | Breeding geese | Fattening ducks | Breeding ducks | Guinea fowl |
|---|---|---|---|---|---|---|---|---|
| Flocks suspected of being infected and TTE (passive surveillance)1 | 24 | 25 | 34 | 3 | 2 | 58 | 4 | 3 |
| Contact (sick1 or holdings healthy birds2) | 1 | 0 | 8 | 0 | 0 | 4 | 0 | 0 |
| Flocks assumed free of slaughterhouses HPAI before shipment or within to restriction zones2 | 1 | 146 | 18 | 0 | 0 | 40 | 0 | 1 |
| Flocks of HPAI assumed after re-free population3 | 3 | 5 | 27 | 5 | 1 | 9 | 6 | 0 |
| TOTAL | 29 | 176 | 87 | 8 | 3 | 111 | 10 | 4 |
TTE – testing to exclude
HPAI – highly pathogenic avian influenza
organs from at least 5 birds (dead or sick) as well as oropharyngeal and cloacal swabs from at least 20 birds per flock (dead or sick)
oropharyngeal swabs from 60 randomly selected birds per flock
organs from dead poultry or swabs taken from their carcasses from up to 10 birds per week during the 21-day period
Virus detection and subtyping. Cloacal and oropharyngeal/tracheal swabs were pooled separately: 5 swabs from the same flock in the case of suspicion/TTE and 10 swabs in the case of healthy flocks. Swabs were immersed in 3 ml of phosphate buffered saline (PBS) (Biomed, Lublin, Poland), shaken and centrifuged for 10 minutes at 3,000 × g. Organ samples (pools from a maximum of 5 birds) were prepared in two ways: 10% w/v suspension for intestines and 20% w/v suspension for other tissues. Samples were homogenized and centrifuged for 10 minutes at 3,000 × g. Total RNA was extracted from 0.2 mL of supernatant using an RNeasy Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s protocol. A real-time RT-PCR test with primers and probe targeting the M gene (26) was performed, and in the case of positive samples, real-time RT-PCRs for the H5 and N8 genes were carried out (11, 22). The QuantiTect Probe RT-PCR Kit (Qiagen) was used for all real time RT-PCR assays in an ABI 7500 instrument (Applied Biosystems, Foster City, CA, USA).
Virus sequencing and phylogenetic analysis. Whole genome sequences were generated by the Sanger method as previously described (28) or by high-throughput sequencing (HTS). Briefly, for Sanger sequencing the virus genome was amplified in an RT-PCR with primer pairs specific to each viral segment. The RT-PCR products were sequenced using a BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems) in a 3500 Genetic Analyzer (Applied Biosystems). Sequences were assembled and analysed in SeqScape v2.7 (Applied Biosystems).
For HTS, all gene segments were amplified using universal primers and a SuperScript III One-Step RT-PCR System with Platinum Taq High Fidelity DNA Polymerase (Invitrogen, Waltham, MA, USA) (29, 31). The libraries were prepared from purified PCR products using a Nextera XT DNA Library Preparation Kit following the manufacturer’s instructions and sequenced in MiSeq (Illumina, San Diego, CA, USA) using a MiSeq Reagent Kit v3 (Illumina). Raw reads were filtered and trimmed with Trimmomatic (4) and mapped to a reference sequence (A/turkey/Poland/23/2020(H5N8)) using the Burrows–Wheeler Alignment tool (BWA) (16). Samtools (17) was used to obtain consensus sequences. Sequences were submitted to the GISAID EpiFlu database with the following isolate IDs: EPI_ISL_405813, EPI_ISL_525439–EPI_ISL_525455, EPI_ISL_525459, and EPI_ISL_525461– EPI_ISL_525465. Phylogenetic analysis was performed using Bayesian inference in BEAST v.1.8.4 software (7). A time-resolved tree was constructed using the HKY+SRD06 substitution model and lognormal relaxed clock with Bayesian skyline tree prior. Markov chain Monte Carlo analysis was performed with a chain length of 50 million and sampling frequency of once every 5,000 generations. Tracer v1.7 (19) was used to analyse the quality of the obtained data. Maximum clade credibility trees were constructed with TreeAnnotator v1.8.4 (7). FigTree v1.4.2 (20) was used to visualise the trees and parse the divergence times for the time of the most recent common ancestor (tMRCA).
Results
Chronology of events. The first outbreak of HPAI H5N8 was confirmed on a farm of meat turkeys on 31 December 2019 in the morning. Two more outbreaks were detected on neighbouring meat turkey holdings on the same day in the evening. They were all located in a densely populated region of poultry production (mostly turkeys) in the Lubartowski district in Lubelskie Province (Fig. 1). Up to 5 January 2020, three more outbreaks were registered in meat turkeys (n=2) and guinea fowl (n=1) in the same district and two additional outbreaks in backyard poultry in another district of the same province. On 3 January, an outbreak of HPAI H5N8 was detected in laying hens in Wielkopolskie Province, more than 350 km from the index case. On 7 January, the presence of HPAIV H5N8 virus was confirmed in the carcass of a goshawk found dead approximately 2 km from the index case holding. Three days later, a positive result was obtained in meat turkeys on a farm in Zachodniopomorskie Province, 570 km away. Ongoing to the end of January, new outbreaks were detected in Wielkopolskie Province in breeding geese (n=1), breeding ducks (n=1), fattening ducks (n=1), meat turkeys (n=2) and backyard holdings (n=2); in Warmińsko-Mazurskie in meat turkeys (n=1); and in backyard poultry in the Dolnośląskie (n=1) and Śląskie (n=1) provinces. On 8 February, another outbreak of HPAI H5N8 in Warmińsko-Mazurskie Province was confirmed in meat turkeys. Starting on 22 February, numerous outbreaks in ducks (mostly in young fattening ducklings) were detected almost simultaneously in different regions of Poland, particularly in Łódzkie (fattening ducks, n=5; breeding ducks, n=1), Wielkopolskie (fattening ducks, n=2), Śląskie (fattening ducks and breeding geese, n=2) and Opolskie (fattening ducks, n=1). Three HPAI H5N8-positive establishments were confirmed in March 2020: one backyard holding in Dolnośląskie Province on 6 March and two meat turkey flocks in Lubuskie Province on 24 and 31 March. In summary, a total of 35 H5N8 HPAI outbreaks in poultry (Table 2) and one case in a wild bird were reported, mainly in eastern and central Poland (Fig. 1).
Fig. 1.
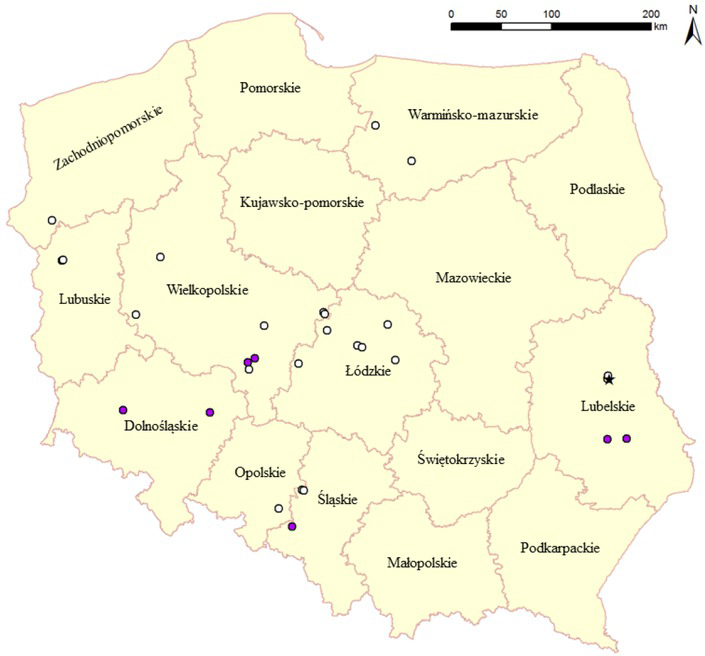
Location of HPAI H5N8 outbreaks in Poland between 31 December 2019 and 31 March, 2020 (white dots: commercial farms; purple dots: backyard holdings; asterisk: wild bird)
Table 2.
A summary of HPAI outbreaks in poultry in Poland reported between 31 December 2019 and 31 March 2020
| Production type | Province | Number of outbreaks | Total |
|---|---|---|---|
| fattening turkeys | Lubelskie Zachodniopomorskie Wielkopolskie Warmińsko-Mazurskie Lubuskie |
5 1 2 2 2 |
12 |
| fattening ducks | Wielkopolskie Opolskie Łódzkie Śląskie |
3 1 5 1 |
10 |
| breeding ducks | Wielkopolskie Łódzkie |
1 1 |
2 |
| breeding geese | Wielkopolskie Śląskie |
1 1 |
2 |
| laying hens | Wielkopolskie | 1 | 1 |
| guinea fowl | Lubelskie | 1 | 1 |
| backyard holdings (mostly chickens) | Lubelskie Dolnośląskie Wielkopolskie Śląskie |
2 2 2 1 |
7 |
| Total | 35 | ||
All positive results were obtained for samples collected in the frame of passive surveillance (on grounds of suspicion or as TTE). No virus was detected in clinically healthy birds tested before movement to slaughterhouses or after re-population.
Clinical outcome of HPAI H5N8 in poultry. Clinical signs of HPAI observed in the field varied depending on the species. The most severe clinical manifestation was observed in turkeys, in which sudden and high mortality was always noted, and in some cases (the outbreaks in the Lubartowski district) it reached 100% within 24-72 hours of the onset of clinical signs. The mortality rate was not so high and peaked over a longer period in other cases. The disease was characterised by depression, reduction in vocalisation, decreased feed and water intake and nervous signs such as tremors, incoordination, paralysis of the wings and fast alternate movements of the legs. Lethargy, ataxia, bloody nasal discharge, diarrhoea and higher mortality were observed in chickens. In caged laying hens, mortality increased slowly and reached 3% during the first three days after the onset of clinical signs. In breeding geese, depression, a drop in food consumption, tremors, movements of the neck and head, sinusitis and nasal discharge were noted and mortality ranged between 2 and 13%. No clear clinical signs were seen in guinea fowl except for an increased mortality. Fattening ducklings exhibited violent clinical manifestations that included neurological disorders such as tremors, incoordination, lying on the back and making pedalling movements of the legs, opisthotonus, and circling movements of the body. Mortality often exceeded 20% and could reach a maximum of 65%. The mildest clinical course of HPAI infection was observed in breeding ducks, in which only single deaths were noted, and the total mortality in two positive flocks at the time of official intervention was 0.27 to 0.4%. However, in both flocks the veterinary inspectors reported drop in food and water consumption as well as a significant (by 90%) decrease in egg production.
Phylogenetic analysis. The previous study showed that the first outbreak confirmed on 31 December 2019 was caused by a novel genotype of H5N8 HPAIV clade 2.3.4.4b (27). Phylogenetic analysis of whole genome sequences of additional 23 strains from Polish outbreaks selected as representative revealed that all of them belonged to the same genotype, generated by the reassortment of African-origin H5N8 HPAIV and Eurasian LPAIV. Two clusters of the 2019/2020 viruses could be distinguished for most gene segments, with Polish sequences belonging to both of them (Fig. 2). The tMRCA of all genome segments of the European viruses ranged from June to September 2019. The comparison of sequences from the index case and the two outbreaks in backyard holdings in Lubelskie Province confirmed that one of them was a secondary outbreak caused by human-mediated spread of the virus while the second one was an independent event. Similarly, it was shown that the first outbreak outside the Lubelskie Province (in Wielkopolskie) was a primary outbreak, as its sequence grouped in a different cluster than that of the index case sequence (Fig. 2). In contrast, the identity of sequences from outbreaks in young fattening ducks reported in February corroborated the hypothesis of the common origin of the virus, while their association with an outbreak in breeding ducks detected in the same period and region was excluded.
Fig. 2.
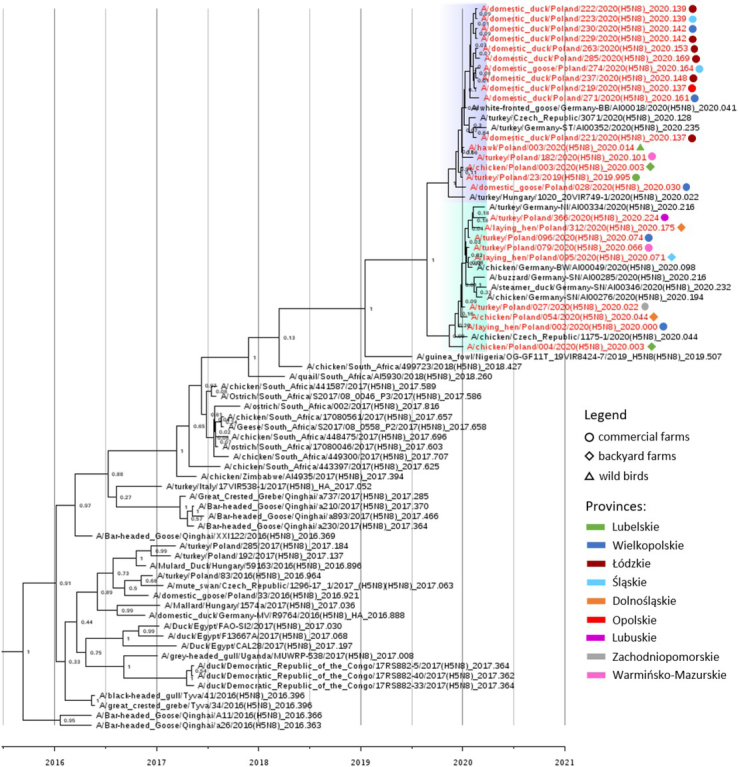
Maximum clade credibility phylogenetic tree of the HA gene segment of H5N8 viruses sequenced in the present study and sequences available in the EpiFlu GISAID database. The European H5N8 viruses from the 2018/2020 epidemic are highlighted with blue and green. The names of Polish strains are coloured red and are labelled according to the legend. The posterior probability values are indicated next to the nodes
Discussion
So far, there have been four recorded HPAI epidemics in Poland, all caused by H5 Gs/GD lineage viruses (23, 24, 27, 28). The causative agent of the first two HPAI epidemics (in 2006 and 2007) was H5N1 clade 2.2 virus, but the genetic analysis confirmed that the outbreaks in 2006 and 2007 were caused by separate incursions of genetically distinguishable viruses (23). The highly pathogenic H5 Gs/GD clade 2.3.4.4b viruses were first introduced into Poland in 2016 (28). Between November 2016 and March 2017, 65 outbreaks in poultry and 68 detections in wild birds were caused by two subtypes (mostly H5N8 and H5N5) and at least four different genotypes of the H5 Gs/GD clade 2.3.4.4b virus (27).
The latest 2019/20 HPAIV epidemic in Poland was caused by a novel genotype of H5 Gs/GD clade 2.3.4.4b that has African ancestry and was generated through reassortment between sub-Saharan Africa H5N8 Gs/GD clade 2.3.4.4b viruses (six gene segments) and Eurasian LPAIV (two segments) (27). All the analysed outbreaks in Poland were caused by the same reassortant virus and relatively high homology was noted for all genome segments of Polish strains (>99.3%), suggesting their recent divergence from a common ancestor. This observation was confirmed by phylogenetic analysis employing estimation of divergence times, as the MRCAs for all genome segments of the European viruses from the current epidemic dated back to summer/early autumn 2019.
The role of wild birds in the introduction and spread of the virus was initially questioned, as there were no HPAIV detections in the period preceding the occurrence of the H5N8 virus in Europe and during the epidemic positive cases in wild avifauna were extremely rare (9). However, almost simultaneous detection of genetically similar viruses in Poland, Hungary, Slovakia, Romania, Germany, the Czech Republic and Ukraine (9, 14) pointed indirectly at wild birds as the most probable disseminators of the virus. The possible reasons for the late incursion of the HPAI virus in 2019/20 into Europe compared to the previous epidemics can be explained by the moderate temperatures reported in the moulting areas in Russia in November/December 2019, different routes of virus spread or undetected circulation of the HPAIV H5 virus in wild birds (9). Presently purely speculative reasons for virus detections in wild avian species being infrequent are insufficient surveillance, pre-existing herd immunity elicited by exposures to antigenically similar viruses of H5 Gs/GD clade 2.3.4.4 in previous seasons and/or reduced pathogenicity of the new H5N8 genotype for certain “sentinel” bird species.
Despite the limited sequence variation, the phylogenetic analysis could assist in the investigation of the pathways of virus spread by complementing the findings of classic epidemiological investigations. The first outbreak in Poland was detected on a farm in a region with high turkey density and the abundant water bodies of the Łęczna-Włodawa Lakeland, where wild migratory waterfowl had gathered in large flocks at that time. Indirect contact with wild birds was therefore suggested as the most plausible source of virus introduction onto the farm. The HPAIV-positive holdings detected in the following days on the neighbouring farms were likely caused by human-mediated spread of the virus, owing to the close proximity of the poultry establishments to the location of the index case. However, airborne transmission cannot be ruled out and there are more evidence-based studies to support the role of wind in the dissemination of the virus over short distances (21). The detection of an HPAI H5N8 outbreak in a flock of laying hens on a farm located in Wielkopolskie Province more than 350 km from the index case in Lubelskie Province raised questions about the origin of the pathogen. The phylogenetic studies ruled out the possibility of a direct connection between the first outbreaks in Lubelskie and Wielkopolskie, and so far the only explanation is indirect contact with wild birds. On the other hand, the index case turkey farm was suggested as the source of the outbreak in backyard poultry in Lubelskie, 60 km away. In the days preceding the appearance of signs arousing suspicion of HPAI at the farm where the index case was diagnosed, animal by-products were sold. Epidemiological investigation revealed that they were to feed foxes kept on the same premises where on a small scale, poultry were kept which subsequently tested positive for HPAI. It was later confirmed by whole genome sequencing, which showed that the two viruses were identical. Interestingly, the viruses in question clustered separately from the virus detected at the same time in a nearby (~20 km) backyard flock and thus direct interconnections were excluded. Most of the outbreaks in young fattening ducks detected in February had a common source, i.e. one specific transport company that delivered one-day-old birds to farms in different, sometimes distant locations. It was also corroborated by phylogenetic studies, as the viruses from the outbreaks in ducklings were either identical or differed only at single nucleotide positions. As the risk of vertical transmission of HPAIV is negligible and laboratory examinations excluded the possibility of infection at hatcheries, the young ducklings were most probably infected during transport in a contaminated vehicle.
Control measures applied during the epidemic were in agreement with the Regulation of the Minister of Agriculture and Rural Development of December 18, 2007 on eradication of avian influenza (implementation of Council Directive 2005/94/EC (6)) and included stamping-out, zoning, movement restrictions, cleansing and disinfection, post-outbreak surveillance, information campaigns, housing orders and strengthening other biosecurity measures (9). Although preventive culling can be implemented in a protection zone (i.e. 3 km around the HPAI outbreak), it was not applied during the recent epidemic. Similarly to previous epidemics in Poland (2006-2017), vaccination in poultry and zoo birds was prohibited. In most cases, samples from clinically healthy broiler/meat poultry flocks from restriction zones were tested in a laboratory before shipment to a slaughterhouse. Despite the large number of tested consignments (>200), no positive results were found. Additionally, in spite of the introduction of the virus into regions with intensive poultry production (e.g. Wielkopolskie, Lubuskie, and Lubelskie provinces), secondary spread was rather limited and the outbreaks were quickly contained.
In summary, repeated incursions of HPAI viruses into new countries or territories raise serious concerns for poultry producers. Scientific evidence collected in recent years highlights the predominant role of wild birds in disseminating the HPAI virus to previously disease-free areas (1, 3, 15). However, the 2019/20 HPAI epidemic revealed the ineffectiveness of wild bird surveillance as an early warning tool for disease detection, and the reasons for that failure should be carefully addressed to improve future detectability of the virus in wild avifauna. As the movement of wild birds is beyond human control and most European countries apply a non-vaccination policy, the only means of preventing virus introduction into poultry flocks is strict adherence to biosecurity practices. Furthermore, timely notification of health problems suggestive of HPAI and fast laboratory diagnosis minimise the risk of secondary spread. Based on the experience gathered during the recent epidemic in Poland, passive surveillance in poultry can be very effective. The most common clinical signs predictive of HPAI included higher mortality, drop in food and water consumption, decreased egg production, and neurological signs. However, it is also very important to note that in breeding ducks the mortality was extremely low and neurological symptoms were absent, thus the stereotypical approach to clinical manifestation in the flock engenders the risk of erroneous suspicion and prolonged time to diagnosis. As HPAI will continue to pose a risk in the future, better preparedness including constant vigilance (demonstrably necessary by the late onset of the recent epidemic), increased surveillance activity in wild birds, awareness campaigns among stakeholders, timely diagnosis, and notification of and response to outbreaks in poultry are needed to reduce the risk of occurrence and minimise the losses posed by HPAI.
Acknowledgements
The authors wish to thank Justyna Opolska, Anna Lisowska, Anna Sawicka, and Joanna Sajewicz-Krukowska for their excellent assistance with laboratory diagnosis, Karolina Piekarska and Karolina Adamska for support with the necropsies, and members of the Department of Epidemiology and Risk Assessment: Agnieszka Stolarek, Anna Ziętek-Barszcz and Łukasz Bocian for creating and updating the map. We are also grateful to the Department of Omics Analyses for performing high throughput sequencing. District veterinary officers deserve our gratitude for providing all necessary information about the outbreaks. We also acknowledge the originating and submitting laboratories of the sequences from the GISAID EpiFlu database which were used for phylogenetic analysis.
Footnotes
Conflict of Interest
Conflict of Interests Statement: The authors declare that there is no conflict of interests regarding the publication of this article.
Financial Disclosure Statement: This study was supported by the reference activity of the National Veterinary Research Institute, Puławy, Poland.
Animal Rights Statement: Not applicable.
References
- 1.Alarcon P., Brouwer A., Venkatesh D., Duncan D., Dovas C.I., Georgiades G., Monne I., Fusaro A., Dan A., Śmietanka K., Ragias V., Breed A.C., Chassalevris T., Goujgoulova G., Hjulsager C.K., Ryan E., Sánchez A., Niqueux E., Tammiranta N., Zohari S., Stroud D.A., Savić V., Lewis N.S., Brown I.H.. Comparison of 2016-17 and previous epizootics of highly pathogenic avian influenza H5 Guangdong lineage in Europe. Emerg Infect Dis. 2018;24:2270–2283. doi: 10.3201/eid2412.171860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alexander D. J.. An overview of the epidemiology of avian influenza. Vaccine. 2007;25:5637–5644. doi: 10.1016/j.vaccine.2006.10.051. [DOI] [PubMed] [Google Scholar]
- 3.Bodewes R., Kuiken T.. Changing role of wild birds in the epidemiology of avian influenza A viruses. Adv Virus Res. 2018;100:279–307. doi: 10.1016/bs.aivir.2017.10.007. [DOI] [PubMed] [Google Scholar]
- 4.Bolger A.M., Lohse M., Usadel B.. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Capua I., Alexander D. J.. Avian influenza infection in birds: A challenge and opportunity for the poultry veterinarian. Poultry Sci. 2009;88:842–846. doi: 10.3382/ps.2008-00289. [DOI] [PubMed] [Google Scholar]
- 6.Council Directive 2005/94/EC of 20 December 2005 on Community measures for the control of avian influenza and repealing Directive 92/40/EEC.
- 7.Drummond A.J., Suchard M.A., Xie D., Rambaut A.. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol. 2012;29:1969–1973. doi: 10.1093/molbev/mss075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Duan L., Campitelli L., Fan X.H., Leung Y.H.C., Vijaykrishna D., Zhang J. X., Donatelli I., Delogu M., Li K. S., Foni E., Chiapponi C., Wu W. L., Kai H., Webster R. G., Shortridge K. F., Peiris J. S. M., Smith G. J. D., Chen H., Guan Y.. Characterization of low-pathogenic H5 subtype influenza viruses from Eurasia: Implications for the origin of highly pathogenic H5N1 viruses. J Virol. 2007;81:7529–7539. doi: 10.1128/JVI.00327-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.EFSA (European Food Safety Authority), European Centre for Disease Prevention and Control, EURL (European Reference Laboratory for Avian Influenza); Adlhoch C., Fusaro A., Kuiken T., Niqueux É., Terregino C., Staubach C., Muñoz Guajardo I., Baldinelli F.. Scientific report: Avian influenza overview November 2019 – February 2020. EFSA Journal. 2020;18:6096. [Google Scholar]
- 10.Guan Y., Smith G. J. D., Webby R., Webster R. G.. Molecular epidemiology of H5N1 avian influenza. Rev Sci Tech. 2009;28:39–47. doi: 10.20506/rst.28.1.1868. [DOI] [PubMed] [Google Scholar]
- 11.Hoffmann B., Hoffmann D., Henritzi D., Beer M., Harder T.C.. Riems influenza a typing array (RITA): An RT-qPCR-based low density array for subtyping avian and mammalian influenza a viruses. Sci Rep. 2016;6:27211. doi: 10.1038/srep27211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.ICTV Taxonomy 2019 release. https://talk.ictvonline.org/taxonomy/ available at.
- 13.Ip H.S., Torchetti M.K., Crespo R., Kohrs P., DeBruyn P., Mansfield K.G., Baszler T., Badcoe L., Bodenstein B., Shearn-Bochsler V., Killian M.L., Pedersen J.C., Hines N., Gidlewski T., DeLiberto T., Sleeman J.M.. Novel Eurasian highly pathogenic avian influenza A H5 viruses in wild birds, Washington, USA, 2014. Emerg Infect Dis. 2015;21:886–890. doi: 10.3201/eid2105.142020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.King J., Schulze C., Engelhardt A., Hlinak A., Lennermann S.L., Rigbers K., Skuballa J., Staubach C., Mettenleiter T. C., Harder T., Beer M., Pohlmann A.. Novel HPAIV H5N8 Reassortant (Clade 2.3.4.4b) Detected in Germany. Viruses. 2020;12:281. doi: 10.3390/v12030281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee D.H., Bertran K., Kwon J.H., Swayne D.E.. Evolution, global spread, and pathogenicity of highly pathogenic avian influenza H5Nx clade 2.3.4.4. J Vet Sci. 2017;18:269–280. doi: 10.4142/jvs.2017.18.S1.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li H., Durbin R.. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R.. 1000 Genome Project Data Processing Subgroup: The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lycett S.J., Duchatel F., Digard P.. A brief history of bird flu. Philos T R Soc B. 2019;374:20180257. doi: 10.1098/rstb.2018.0257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rambaut A., Drummond A.J., Xie D., Baele G., Suchard M.A.. Posterior summarisation in Bayesian phylogenetics using Tracer 1.7. Systematic Biology. 2018;67:901–904. doi: 10.1093/sysbio/syy032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rambaut A. FigTree, a Graphical Viewer of Phylogenetic Trees. Institute of Evolutionary Biology University of Edinburgh; 2009. http://tree.bio.ed.ac.uk/software/figtree [Google Scholar]
- 21.Scoizec A., Niqueux E., Thomas R., Daniel P., Schmitz A., Le Bouquin S.. Airborne Detection of H5N8 Highly Pathogenic Avian Influenza Virus Genome in Poultry Farms, France. Front Vet Sci. 2018;5:15. doi: 10.3389/fvets.2018.00015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Slomka M.J., Pavlidis T., Banks J., Shell W., McNally A., Essen S., Brown I.H.. Validated H5 Eurasian real-time reverse transcriptase-polymerase chain reaction and its application in H5N1 outbreaks in 2005-2006. Avian Dis. 2007;51:373377. doi: 10.1637/7664-060906R1.1. [DOI] [PubMed] [Google Scholar]
- 23.Smietanka K., Fusaro A., Domanska-Blicharz K., Salviato A., Monne I., Dundon W.G., Cattoli G., Minta Z.. Full-Length Genome Sequencing of the Polish HPAI H5N1 Viruses Suggests Separate Introductions in 2006 and 2007. Avian Diseases. 2010;54:335–339. doi: 10.1637/8782-040109-ResNote.1. [DOI] [PubMed] [Google Scholar]
- 24.Smietanka K., Minta Z.. Avian influenza in Poland. Acta Biochim Pol. 2014;61:453–457. [PubMed] [Google Scholar]
- 25.Smith G.J., Donis R.O.. World Health Organization/World Organisation for Animal Health/ Food and Agriculture Organization (WHO/OIE/FAO) H5 Evolution Working Group. Nomenclature updates resulting from the evolution of avian influenza A(H5) virus clades 2.1.3.2a, 2.2.1, and 2.3.4 during 2013-2014. Influenza Other Respir Viruses. 2015;9:271–276. doi: 10.1111/irv.12324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Spackman E., Senne D.A., Myers T.J., Bulaga L.L., Garber L.P., Perdue M.L., Lohman K., Daum L.T., Suarez D.L.. Development of a real-time reverse transcriptase PCR assay for type A influenza virus and the avian H5 and H7 hemagglutinin subtypes. J Clin Microbiol. 2002;40:3256–3260. doi: 10.1128/JCM.40.9.3256-3260.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Swieton E., Fusaro A., Shittu I., Niemczuk K., Zecchin B., Joannis T., Bonfante F., Smietanka K., Terregino C.. Sub-Saharan Africa and Eurasia Ancestry of Reassortant Highly Pathogenic Avian Influenza A(H5N8) Virus, Europe, December 2019. Emerg Infect Dis. 2020;26:1557–1561. doi: 10.3201/eid2607.200165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Swieton E., Smietanka K.. Phylogenetic and molecular analysis of highly pathogenic avian influenza H5N8 and H5N5 viruses detected in Poland in 2016-2017. Transbound Emerg Dis. 2018;65:1664–1670. doi: 10.1111/tbed.12924. [DOI] [PubMed] [Google Scholar]
- 29.Watson S.J., Welkers M.R., Depledge D.P., Coulter E., Breuer J.M., de Jong M.D., Kellam P.. Viral population analysis and minority-variant detection using short read next-generation sequencing. Philos Trans R Soc Lond B Biol Sci. 2013;368:20120205. doi: 10.1098/rstb.2012.0205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Webster R.G., Bean W.J., Gorman O.T., Chambers T.M., Kawaoka Y.. Evolution and Ecology of Influenza-a Viruses. Microbiol Rev. 1992;56:152–179. doi: 10.1128/mr.56.1.152-179.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhou B., Donnelly M.E., Scholes D.T., St George K., Hatta M., Kawaoka Y., Wentworth D.E.. Single-reaction genomic amplification accelerates sequencing and vaccine production for classical and Swine origin human influenza a viruses. J Virol. 2009;83:10309–10313. doi: 10.1128/JVI.01109-09. [DOI] [PMC free article] [PubMed] [Google Scholar]