

Abstract

We designed and synthesized a series of inhibitors of the bacterial enzymes DNA gyrase and DNA topoisomerase IV, based on our recently published benzothiazole-based inhibitor bearing an oxalyl moiety. To improve the antibacterial activity and retain potent enzymatic activity, we systematically explored the chemical space. Several strategies of modification were followed: varying substituents on the pyrrole carboxamide moiety, alteration of the central scaffold, including variation of substitution position and, most importantly, modification of the oxalyl moiety. Compounds with acidic, basic, and neutral properties were synthesized. To understand the mechanism of action and binding mode, we have obtained a crystal structure of compound 16a, bearing a primary amino group, in complex with the N-terminal domain of E. coli gyrase B (24 kDa) (PDB: 6YD9). Compound 15a, with a low molecular weight of 383 Da, potent inhibitory activity on E. coli gyrase (IC50 = 9.5 nM), potent antibacterial activity on E. faecalis (MIC = 3.13 μM), and efflux impaired E. coli strain (MIC = 0.78 μM), is an important contribution for the development of novel gyrase and topoisomerase IV inhibitors in Gram-negative bacteria.
Keywords: DNA gyrase, topoisomerase IV, GyrB, ParE, antibacterial, benzothiazole
The treatment of infections has advanced beyond recognition since the first discoveries of antimicrobial compounds. Widespread distribution, low price, and high safety profile have established antimicrobial drugs as a cornerstone of modern medicine. However, due to rising resistance issues, the efficacy of existing infection treatments is rapidly decreasing. The dearth of new anti-infectives introduced to the market over recent years, and no discovery of novel classes, has deepened the problem.1−4 Antimicrobial resistance is now a serious global health concern; infections that are currently readily treatable could become untreatable in the near future.5
DNA topoisomerases are enzymes that alter DNA topology, being involved in important biological processes in the cell (replication, transcription, chromosome condensation, etc.).6−11 Bacterial DNA gyrase and DNA topoisomerase IV (topo IV) are attractive targets for antibacterial drug discovery due to their well-described structure and mechanism, as well as their absence from eukaryotes. Both enzymes are type II topoisomerases, sharing high functional and structural similarity. They change the topology of DNA by cleaving both strands of the double-helix. Gyrase has the unique function of introducing negative supercoiling ahead of the replication fork, while topo IV is involved in chromosome decatenation. The enzymes consist of two pairs of subunits with different functions, forming heterotetrameric structures: A2B2 (gyrase) or C2E2 (topo IV). The main role of the GyrA and ParC subunits is to release torsional stress by breaking and rejoining strands of the DNA molecule. GyrB and ParE contain the ATP-binding site and provide the energy required for the enzyme function by ATP hydrolysis.12,13
DNA gyrase inhibitors can influence the enzyme action at different levels.14 Inhibition of the GyrA subunit can cause stabilization of the DNA–enzyme complex, and thus broken strands of DNA cannot be rejoined. GyrA inhibitors, so-called gyrase poisons, are represented by fluoroquinolones, which are widely in clinical use, although struggling with side effects and growing resistance concerns.6 These problems have encouraged further research on inhibitors with different mechanisms of action. GyrB inhibitors are not currently in clinical use; novobiocin was withdrawn from the market in the 1960s due to significant side effects. Since then, no other GyrB inhibitor has passed clinical trials.7 Low cross-resistance between potential GyrB inhibitors and fluoroquinolones is a promising and motivating factor for further research. Novel gyrase inhibitors based on various chemical structures were discovered in recent years.8
Our research group has recently published several articles focusing on ATP-competitive inhibitors of the DNA gyrase B protein (GyrB).15−17 The compounds share a common pyrrole-2-carboxamide moiety, originating from marine alkaloids such as oroidin.18 Compounds are potent inhibitors of gyrase; however, they lack potent antimicrobial activity. The mode of action for this series of compounds was confirmed by solving the crystal structure of a complex of E. coli GyrB with a benzothiazole-type inhibitor 1 (PDB 5L3J).15
Our goal was to explore the chemical space of pyrrole-benzothiazole GyrB inhibitors by selecting those that retain potent enzyme inhibition while optimizing their physicochemical properties. Several strategies of modification illustrated in Figure 1 were followed: alteration of the pyrrole carboxamide moiety, alteration of the central scaffold, including variation of the substitution position, and most importantly modification of the oxalyl moiety.
Figure 1.
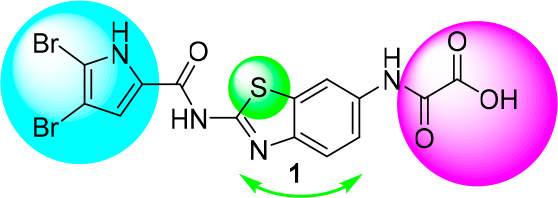
Structure of previously discovered compound 1 (PDB 5L3J) with highlighted positions selected for structure modifications.
The 4,5-dibromo-1H-pyrrole-2-carboxamide interacting with the protein through hydrogen bonding interactions of the pyrrole nitrogen and adjacent carbonyl with Asp73, either directly or via a network of hydrogen bonds involving conserved water molecule, respectively does not permit substantial changes. Modification to 3,4-dichloro-5-methyl-1H-pyrrole-2-carboxamide has several potential beneficial properties. The key role of the pyrrole nitrogen and nonsubstituted carboxamide moiety remains unchanged. Smaller chlorine atoms (compared to bromine) are likely to fit better into the E. coli topo IV hydrophobic pocket and the Staphylococcus aureus gyrase hydrophobic pocket, which has a smaller volume than the E. coli gyrase hydrophobic pocket.19 The 3,4-dichloro-5-methyl-1H-pyrrole-carboxamide moiety is present in the natural antibiotics kibdelomycin20−22 and amycolamicin23,24 and was used in the Astra Zeneca pyrrolamide series of gyrase B inhibitors.25−27
To explore the influence of the central scaffold we interchanged the substituents on positions 2 and 6 of benzothiazole, resulting in interchanging the position of the benzene ring and thiazole ring of the benzothiazole moiety28 in the binding site and its π–cation interactions with Arg76, as well as influencing the acidity of the pyrrole amide proton. An obvious bioisosteric approach was the replacement of the ring sulfur (benzothiazole) with nitrogen to give a benzimidazole central scaffold. Indeed, benzimidazole-ureas were presented as potent dual inhibitors of bacterial topoisomerases in a recent study by Vertex Pharmaceuticals.29 In this manner, crucial cation−π interactions between the core benzene ring and Arg76 and the Glu50-Arg76 salt bridge interactions were preserved. Additional interactions might have been introduced by enlarging the central scaffold, but we did not plan to increase the scaffold MW.
The focus of the optimization strategy was the replacement of the oxalyl moiety, which is likely too acidic and might impair permeation into bacteria and thus diminish antimicrobial activity. As confirmed by the crystal structure of 1, the oxalyl moiety extending out of the binding pocket and making only a single direct hydrogen bond to the protein, via the carbonyl group to Arg136, can be replaced without loss of inhibitory activity. We systematically changed the acidic oxalyl moiety to (i) a neutral small aliphatic acyl moiety (acetamide group), (ii) a neutral aromatic moiety (isonicotinamide moiety), (iii) a neutral polar group with H-bond donor/acceptor potential (urea derivative), and (iv) an aliphatic primary amino group (derivatives of glycine and beta alanine).
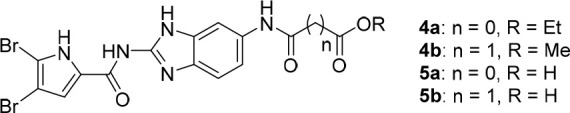
Synthesis of benzimidazole compounds, illustrated in Scheme 1, starts with coupling of 6-nitro-1H-benzo[d]imidazol-2-amine with 2,2,2-trichloro-1-(4,5-dibromo-1H-pyrrol-2-yl)ethan-1-one and a weak base in DMF. The nitro group of intermediate 2 is reduced using tin(II) chloride dihydrate to give amino-derivative 3, further acylated with respective acyl chloride to obtain esters 4a–b. Corresponding carboxylic acids 5a–b were obtained by alkaline hydrolysis.
Scheme 1.
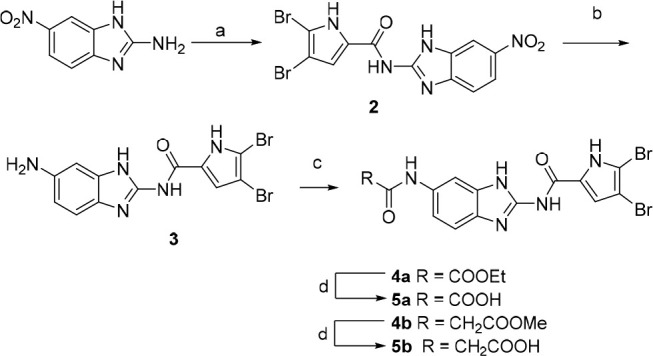
Reagents and conditions: (a) 2,2,2-trichloro-1-(4,5-dibromo-1H-pyrrol-2-yl)ethan-1-one, Na2CO3, DMF, 80 °C, 16 h; (b) SnCl2·2H2O, EtOH, reflux, 12 h; (c) ethyl oxalyl chloride (for 4a) or methyl malonyl chloride (for 4b), Et3N, 1,4-dioxane, rt, 12 h; (d) 1 M NaOH, 1,4-dioxane, rt, 16 h.
Synthesis of benzothiazole-based compounds is illustrated in Scheme 2. Acylation of 6-nitro-benzo[d]thiazol-2-amine (6) with ethyl oxalyl chloride/methyl malonyl chloride/acetyl chloride gives intermediates 7a, 7b, and 7c respectively.
Scheme 2.
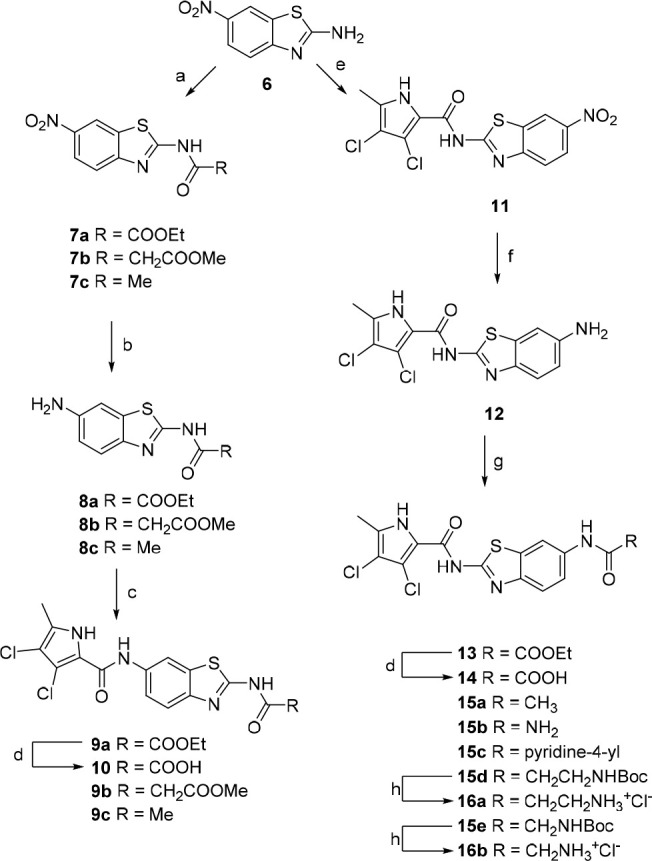
Reagents and conditions: (a) corresponding acyl chloride, Et3N, 1,4-dioxane, rt, 4 h; (b) H2, 10% Pd/C, EtOH (for 8a and 8c) or MeOH (for 8b), rt, 24 h; (c) 3,4-dichloro-5-methyl-1H-pyrrole-2-carbonyl chloride, pyridine, DCM; (d) 1 M NaOH, MeOH, rt, 24 h,; (e) 3,4-dichloro-5-methyl-1H-pyrrole-2-carbonyl chloride, toluene, reflux, 16 h; (f) SnCl2·2H2O, EtOH, reflux, 12 h; (g) ethyl oxalyl chloride, Et3N, 1,4-dioxane, rt, 8 h (for the synthesis of 13); (g) Ac2O, Et3N, DCM, rt, 2 h (for the synthesis of 15a); or CDI, DMF, rt, 3 h; NH3, rt, 16 h (for the synthesis of 15b); or nicotinic acid, EDC, NMM, HOBt, DMF, rt, 12 h (for the synthesis of 15c); or corresponding Boc-amino acid, EDC, NMM, HOBt, DMF, rt, 12 h (for the synthesis of 15d–e); (h) 4 M HCl, 1,4-dioxane, rt, 5 h.
The nitro group in position 6 of the benzothiazole was then reduced using hydrogen and palladium catalyst to obtain compounds 8a–c. Coupling with 3,4-dichloro-5-methyl-1H-pyrrole-2-carboxylic acid (activated in situ with oxalyl chloride) in DCM in the presence of pyridine as base gave compounds 9a–c. The ethyl oxalyl derivative 9a was further hydrolyzed to carboxylic acid 10. Synthesis of the reversed isomers with the pyrrole moiety at position 2 is more demanding. The coupling of benzothiazole 6 with the pyrrole-2-carbonyl chloride moiety was done in refluxing toluene without any base, to obtain intermediate 11. The nitro group is reduced using tin(II) chloride dihydrate to give key amino derivative 12, which was further derivatized. Acylation with oxalyl chloride moiety gave ester 13. The corresponding carboxylic acid 14 is isolated after hydrolysis with 1 M sodium hydroxide. Reaction of amino derivative 12 with acetic anhydride in the presence of triethylamine in DCM gave target compound 15a. To obtain the urea derivative 15b, aminobenzothiazole 12 was first stirred at RT with CDI in DMF for 3 h and heated to 50 °C overnight after addition of ammonia gas. Compounds 15c–e were prepared by coupling of 12 with nicotinic acid (15c), Boc-β-alanine (15d), or Boc-glycine (15e) in the presence of coupling agents EDC, HOBt, or NMM, respectively, in DMF. The Boc protective group of 15d–e was finally removed with HCl in dioxane, to obtain 16a–b.
Compounds were evaluated in E. coli and S. aureus gyrase supercoiling assays as well as in E. coli and S. aureus topo IV relaxation assays. The results are presented in Tables 1–3 as IC50 values or residual activity (RA) of the enzyme in a concentration of 10 μM of the inhibitor.
Table 1. Inhibitory Activity of Series I of Compounds with Benzimidazole Central Scaffold.
| IC50 (μM) or RA (%)a |
||||||
|---|---|---|---|---|---|---|
| Cmpd | n | R | E. coli gyrase | E. coli topo IV | S. aureus gyrase | S. aureus topo IV |
| 4a | 0 | Et | 4.0 ± 1.6 μM | 100% | 100% | 100% |
| 4b | 1 | Me | 7.0 ± 3.4 μM | 100% | 100% | 100% |
| 5a | 0 | H | 0.60 ± 0.32 μM | 12 ± 2 μM | 80 ± 23 μM | 31 ± 1 μM |
| 5b | 1 | H | 1.5 ± 0.2 μM | 100% | 100% | 100% |
| 1 | 0.058 μM | 13 μM | >100 μM | 10 μM | ||
| novobiocin | 0.17 μM | 11 μM | 0.041 μM | 27 μM | ||
Residual activity of the enzyme at 10 μM concentration of the compound.
Table 3. Inhibitory Activity of Benzothiazole Compounds with Neutral or Basic Terminal Functional Groups.

| IC50 (nM) or RA (%)a |
|||||
|---|---|---|---|---|---|
| Cmpd | R | E. coli gyrase | E. coli topo IV | S. aureus gyrase | S. aureus topo IV |
| 9c | 66 ± 8 nM | 100% | 35 400 nM | 100% | |
| 12 | 16 000 ± 4 000 nM | 100% | 60% | 100% | |
| 15a | -CH3 | 9.5 ± 2.5 nM | 4 600 ± 100 nM | 400 ± 120 nM | 1 600 ± 300 nM |
| 15b | -NH2 | 26 ± 8 nM | 5 200 ± 2 700 nM | 1 300 ± 600 nM | 5 800 ± 3 100 nM |
| 15c | -pyridine-4-yl | 2 500 ± 1 500 nM | 100% | 100% | 100% |
| 15d | -CH2CH2NHBoc | 110 ± 20 nM | 100% | 780 nM | 100% |
| 15e | -CH2NHBoc | 29 ± 15 nM | 60% | 260 ± 120 nM | 100% |
| 16a | -CH2CH2NH3+Cl– | 110 ± 50 nM | 10 000 ± 2 000 nM | 1 500 ± 600 nM | 290 ± 180 nM |
| 16b | -CH2NH3+Cl– | 280 ± 10 nM | 100% | 380 nM | 100% |
Residual activity of the enzyme at 10 μM concentration of the compound.
Table 2. Inhibitory Activity of Benzothiazole Compounds with Acidic Terminal Functional Groups.

| IC50 (nM) or RA (%)a |
||||||
|---|---|---|---|---|---|---|
| Cmpd | n | R | E. coli gyrase | E. coli topo IV | S. aureus gyrase | S. aureus topo IV |
| 9a | 0 | Et | 290 ± 170 nM | 100% | 69% | 100% |
| 9b | 1 | Me | 200 ± 180 nM | 100% | 59% | 48% |
| 10 | 0 | H | 29 ± 16 nM | 6 400 ± 3 000 nM | 250 ± 130 nM | 910 ± 340 nM |
| 13 | 0 | Et | 48 ± 12 nM | 100% | 100% | 100% |
| 14 | 0 | H | 4.8 ± 2.1 nM | 75 ± 28 nM | 38 ± 16 nM | 290 ± 180 nM |
| 1 | 58 nM | 13 000 nM | >100 μM | 10 000 nM | ||
| novobiocin | 168 nM | 11 000 nM | 41 nM | 27 000 nM | ||
Residual activity of the enzyme at 10 μM concentration of the compound.
The benzimidazole-based series was shown to be only weakly active (Table 1), with inhibitory activity against E. coli gyrase in the micromolar range. Carboxylic acids 5a–b were approximately 10-fold more active than the corresponding esters 4a–b, due to possible ionic interactions additional to hydrogen bonds with Arg136. The most potent compound 5a (IC50 = 0.60 μM) showed weak activity against S. aureus gyrase and E. coli/S. aureus topo IV). Direct comparison of benzothiazole 1 with benzimidazole 5a shows that replacement of sulfur with nitrogen resulted in 10-fold lower enzymatic inhibition against E. coli gyrase. Poor activity results and poor solubility of benzimidazoles pointed us back to the benzothiazole central scaffold, and the benzimidazole series was not further extended. Replacement of 4,5-dibromo-1H-pyrrole with 3,4-dichloro-5-methyl-1H-pyrrole indeed resulted in good inhibitory activity against E. coli gyrase, but even more importantly it introduced good inhibitory activity against E. coli topo IV (14: IC50 topo IV = 75 nM) and potent inhibitory activity against S. aureus gyrase and topo IV, which was completely absent in the case of the dibromo analogue 1. When comparing compounds with the pyrrole attached to position 2 (compound 14) to a regioisomer with the pyrrole attached to position 6 (compound 10), the inhibitory activity on E. coli gyrase is favorable for compound 10 and even more favorable regarding E. coli topo IV inhibitory activity as well as S. aureus gyrase and topo IV. Overall, compound 14 has superior enzymatic activity against all four tested enzymes compared to novobiocin. Benzothiazole with 3,4-dichloro-5-methyl-1H-pyrrole on position 2 was thus selected to explore the possible replacements of the unfavorable oxalyl moiety. The results of the E. coli gyrase inhibitory activity assays reveal that the anionic center is not required for potent inhibitory activity. Compounds with the acetyl moiety, the urea derivative, and the glycine derivative with a free primary amino group all possess E. coli gyrase inhibitory activity in the low nanomolar range (10–25 nM). Having an aromatic moiety (15c) pointed to the water environment (and possibly having π–cationic interactions with Arg136) is clearly not optimal for this series of compounds, although such an approach was successful in tricyclic inhibitors of GyrB (PDB: 4KFG).30 Investigation of the Boc-protected amino acid derivatives 15d and 16a reveals that the bulky lipophilic moiety can have favorable binding to E. coli GyrB. Although this might seem contradictory, it is known from thermodynamic evaluations that the binding of compound with “unfavorable” lipophilic moieties extending into a water environment can be beneficial as more polar/ionized groups can pay a high desolvation penalty, which contributes to net unfavorable binding.31 The amino compound 12, lacking the carbonyl group, is a very weak binder, which indicates that a carbonyl moiety is a prerequisite for potent enzyme binding. Acetyl derivative 15a with lowest molecular weight in the series and single digit nanomolar binding with IC50 = 9.5 nM seemed very interesting; therefore, regioisomer 9c with a pyrrole attached to position 6 and acetyl to position 2 of benzothiazole was prepared. The trend observed already from previous compounds was the same in this series: the isomer 15a with pyrrole attached to position 2 of benzothiazole was 7-fold more potent than regioisomer with pyrrole attached to position 6.
To confirm binding to the ATP-pocket of GyrB and to gain insight into molecular interactions, crystallization of selected ligands with the 24 kDa fragment (N-terminal subdomain) of GyrB was attempted. Among crystallization attempts with 15a, 16a, and 9c, cocrystals of 16a with E. coli GyrB24 were obtained and the structure was solved to a resolution of 1.60 Å (Figure 2). The 3,4-dichloro-5-methyl-1H-pyrrole moiety of 16a is bound to the adenine-binding pocket, making a hydrogen bond between the pyrrole NH group and Asp73 side chain, while interaction of the Asp73 side chain and pyrrolamide carbonyl oxygen is bridged by a coordinated water molecule (NHCO–H2O H-bond distance is 2.61 Å). Comparison with the GyrB crystal structure of kibdelomycin (PDB: 4URM)32 and Astra Zeneca’s pyrrolamide (PDB: 3TTZ)33 reveals identical interactions of the 3,4-dichloro-5-methyl-1H-pyrrole moiety. There is a strong π–cation interaction (3.28 Å) between the aromatic ring and Arg76. The carbonyl moiety of β-alanine interacts with Arg136 through a H-bond (2.87 Å), while the primary amino group of β-alanine is pointed toward the water environment, making no interactions with the protein. While interactions of the oxalyl moiety of 1 with Arg136 were not apparent (PDB code 5L3J, Resolution: 2.83 Å), improved resolution now offers clear insight into the binding mode. Hydrogen bonding of Arg136 with the carbonyl oxygen of β-alanine is evident, thus demonstrating that a carboxylic acid moiety is not prerequisite for binding, and explains strong enzymatic inhibitory activity of compounds having a neutral side chain.
Figure 2.
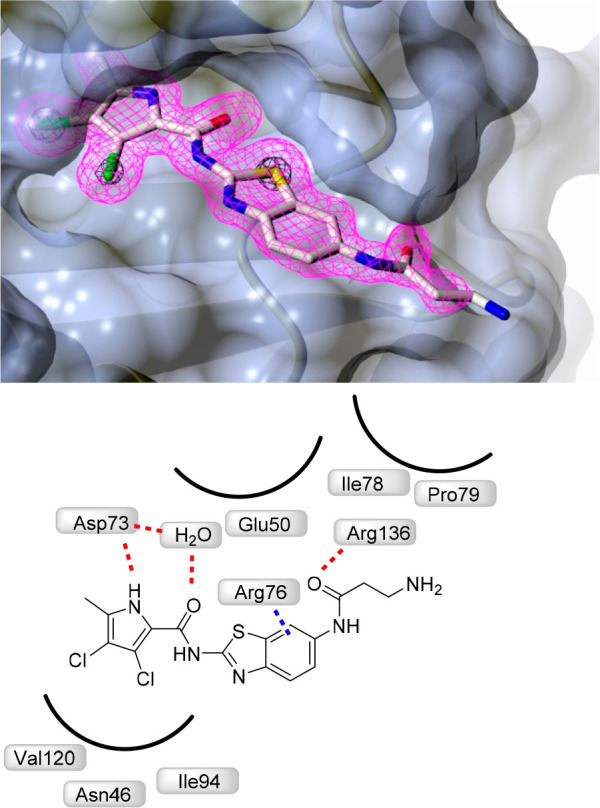
Crystal structure of the complex formed between 16a and E. coli GyrB 24 kDa fragment (PDB: 6YD9). The protein is depicted in cartoon representation covered by a semitransparent molecular surface. Omit mFobs-DFcalc positive difference electron density for the ligand at 1.6 Å resolution is depicted at two contour levels: 3σ (magenta mesh) and 12σ (black mesh), with the latter highlighting the locations of the electron-dense chlorine and sulfur atoms. Shown below are the interactions between amino acid residues with ligand (red line: hydrogen bond, blue line: π–cation interactions).
Compounds were assayed on 4 wild type bacterial strains to determine their antibacterial activity (E. coli, S. aureus, Enterococcus faecalis, Pseudomonas aeruginosa) (Table 4). Additionally, we included two other E. coli strains, an E. coli strain with impaired outer membrane (lpxC deletion mutant) and an E. coli strain with a defective efflux pump (tolC deletion mutant), to evaluate if weak antibacterial activity on wild type could be due to weak permeability and/or strong efflux. Both benzimidazole and benzothiazole inhibitors with the oxalyl moiety (5a, 10, and 14) were inactive in all bacterial strains (MIC > 50 μM). Ester derivative 9b which had good enzymatic inhibitory activity (IC50 = 200 nM) showed also a good MIC (1.56 μM) in the E. coli strain with a defective efflux pump, indicating that the ester moiety is beneficial for entry into bacteria; however, such compounds are often the substrates for efflux pumps. Ester derivative 13 which had very good enzymatic inhibitory activity (IC50 = 48 nM) showed only weak MIC (50 μM) in the E. coli strain with a defective efflux pump.
Table 4. Antibacterial Activity of Selected Compounds.
| MIC
(μM)a |
||||||
|---|---|---|---|---|---|---|
| Cmpd | E. coli (ATCC 25922) | S. aureus (ATCC 29213) | E. faecalis (ATCC 29212) | P. aeruginosa (ATCC 27853) | E. coli (JD17464)b | E. coli (JW5503)c |
| 4a, 4b, 5a, 5b, 9a, 9c, 10, 14, 15c, 15d | >50 μM | >50 μM | >50 μM | >50 μM | >50 μM | >50 μM |
| 9b | >50 μM | >50 μM | >50 μM | >50 μM | >50 μM | 1.56 μM |
| 13 | >50 μM | >50 μM | >50 μM | >50 μM | >50 μM | 50 μM |
| 15a | >50 μM | >50 μM | 3.13 μM | >50 μM | >50 μM | 0.78 μM |
| 15b | >50 μM | >50 μM | >50 μM | >50 μM | >50 μM | 1.56 μM |
| 15e | >50 μM | >50 μM | 3.13 μM | >50 μM | >50 μM | 0.78 μM |
| 16a | >50 μM | >50 μM | 12.5 μM. | >50 μM | >50 μM | 12.5 μM |
| 16b | >50 μM | 25 μM | 6.25 μM | >50 μM | >50 μM | 3.13 μM |
| ciprofloxacin | 0.05 μM | 1.51 μM | 3.02 μM | 3.02 μM | 0.121 μM | 0.015 μM |
MIC (minimum inhibitory concentration that inhibits the growth of bacteria by ≥90%) values against E. coli and S. aureus.
E. coli strain with impaired outer membrane, lpxC deletion mutant.
E. coli strain with defective efflux pump, tolC deletion mutant.
A significant improvement in MIC value was expected with the compound lacking the acidic oxalyl moiety. Compound 16b, having a glycine moiety with a free primary amino group, was the only compound showing some antibacterial activity on S. aureus wild type (MIC = 25 μM), confirming that good enzymatic inhibitory activity (S. aureus gyrase IC50 = 380 nM) with suitable physicochemical properties is needed for antibacterial activity. When looking at Gram-positive E. faecalis, the results were more encouraging, as four compounds showed good antibacterial activity. Both compounds with a free amino group (16a MIC = 12.5 μM and 16b MIC = 6.25 μM) had good antibacterial activity. Boc-protected analogue 15e and compound 15a with an acetyl moiety had even better antibacterial activity with MIC = 3.13 μM, which also correlated with enzymatic inhibitory potency. When looking at more challenging Gram-negative E. coli and P. aeruginosa, the results were less encouraging, as none of the compounds showed antibacterial activity.
Testing on the E. coli strain with impaired outer membrane (lpxC deletion mutant), revealed that permeability is not a main issue as tested compounds did not show any antibacterial activity. On the contrary, the compounds were very active when tested on the E. coli strain with a defective efflux pump. The most potent compounds were 15e and 15a with submicromolar MIC values (MIC = 0.78 μM). Both are neutral molecules with lipophilic moieties (acetyl and Boc). Compounds with free amino group were also potent antibacterial compounds (16a and 16b) but were not superior to the neutral compounds (15a and 15e). This is somehow contrary to the recent proposal that the introduction of a primary amino group improves entry and causes accumulation in Gram-negative bacteria.34 To confirm the mechanism of uptake for the reported compounds (porin pathway or direct diffusion),35 additional studies should be performed in order to guide optimization.36,37 Urea derivative 15b was also a potent antibacterial compound on the efflux impaired E. coli strain (MIC = 1.56 μM), while it was inactive on all other strains.
In summary, three series of GyrB/ParE inhibitors were designed, synthesized, and evaluated in enzymatic and antibacterial assays. Chemical space was thoroughly explored; compounds with acidic, basic, and neutral properties were synthesized, possessing very potent inhibitory activity on E. coli and/or S. aureus gyrase and/or topo IV. The best compounds were active on Gram-positive bacterium E. faecalis with the best compound having MIC= 3.13 μM. The compounds were inactive on Gram-negative bacteria because they are good substrates for bacterial efflux pumps, but 15a and 15e showed potent antibacterial activity on the efflux impaired E. coli strain (MIC = 0.78 μM). Compound 15a with low molecular weight 383 Da displayed potent inhibitory activity on E. coli gyrase (IC50 = 9.5 nM) and potent antibacterial activity on E. faecalis (MIC = 3.13 μM) and on the efflux impaired E. coli strain (MIC = 0.78 μM) and thus makes an important contribution for the development of novel gyrase and topoisomerase inhibitors in Gram-negative bacteria.
Acknowledgments
We acknowledge Diamond Light Source for access to beamline I04 under proposal MX18565 and Sara Henderson for providing the protein sample.
Glossary
Abbreviations
- MIC
minimal inhibitory concentration
- RA
residual activity
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.0c00416.
(i) Experimental conditions for biochemical and microbiological assays, (ii) experimental conditions for cocrystallization, collection, and refinement statistics for compound 16a, (iii) experimental procedures for the synthesis of all the compounds described in this manuscript, and (iv) representative analytical data (PDF)
Author Contributions
⊥ Ž.S. and M.B. contributed equally. The manuscript was written with contributions from all of the authors.
The work was funded by the EU H2020 ITN-ETN Project INTEGRATE (Project Reference: 642620), EU FP7 Integrated Project MAREX (Project No. FP7-KBBE-2009-3-245137), Academy of Finland (Grant No. 277001), and Slovenian Research Agency (Grant No. P1-0208). Work in A.M.’s lab was supported by the Biotechnology and Biosciences Research Council (UK) Institute Strategic Programme Grant BB/P012523/1, and the Wellcome Trust (Investigator Award 110072/Z/15/Z).
The authors declare no competing financial interest.
Supplementary Material
References
- Aminov R. I. A brief history of the antibiotic era: lessons learned and challenges for the future. Front. Microbiol. 2010, 1, 134. 10.3389/fmicb.2010.00134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathan C.; Cars O. Antibiotic resistance--problems, progress, and prospects. N. Engl. J. Med. 2014, 371, 1761–1763. 10.1056/NEJMp1408040. [DOI] [PubMed] [Google Scholar]
- Payne D. J.; Gwynn M. N.; Holmes D. J.; Pompliano D. L. Drugs for bad bugs: confronting the challenges of antibacterial discovery. Nat. Rev. Drug Discovery 2007, 6, 29–40. 10.1038/nrd2201. [DOI] [PubMed] [Google Scholar]
- Silver L. L. Challenges of antibacterial discovery. Clin Microbiol Rev. 2011, 24, 71–109. 10.1128/CMR.00030-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown E. D.; Wright G. D. Antibacterial drug discovery in the resistance era. Nature 2016, 529, 336–343. 10.1038/nature17042. [DOI] [PubMed] [Google Scholar]
- Emmerson A. M.; Jones A. M. The quinolones: decades of development and use. J. Antimicrob. Chemother. 2003, 51 (Suppl 1), 13–20. 10.1093/jac/dkg208. [DOI] [PubMed] [Google Scholar]
- Bisacchi G. S.; Manchester J. I. A New-Class Antibacterial-Almost. Lessons in Drug Discovery and Development: A Critical Analysis of More than 50 Years of Effort toward ATPase Inhibitors of DNA Gyrase and Topoisomerase IV. ACS Infect. Dis. 2015, 1, 4–41. 10.1021/id500013t. [DOI] [PubMed] [Google Scholar]
- Barančokova M.; Kikelj D.; Ilaš J. Recent progress in the discovery and development of DNA gyrase B inhibitors. Future Med. Chem. 2018, 10, 1207–1227. 10.4155/fmc-2017-0257. [DOI] [PubMed] [Google Scholar]
- Bates A.D.; Maxwell A.. DNA Topology; Oxford University Press, Oxford, 2005. [Google Scholar]
- Chen S. H.; Chan N. L.; Hsieh T. S. New mechanistic and functional insights into DNA topoisomerases. Annu. Rev. Biochem. 2013, 82, 139–170. 10.1146/annurev-biochem-061809-100002. [DOI] [PubMed] [Google Scholar]
- Vos S. M.; Tretter E. M.; Schmidt B. H.; Berger J. M. All tangled up: how cells direct, manage and exploit topoisomerase function. Nat. Rev. Mol. Cell Biol. 2011, 12, 827–841. 10.1038/nrm3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bush N.G.; Evans-Roberts K.; Maxwell A.. DNA Topoisomerases. EcoSal Plus 2015, 6, 10.1128/ecosalplus.ESP-0010-2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoeffler A. J.; Berger J. M. DNA topoisomerases: harnessing and constraining energy to govern chromosome topology. Q. Rev. Biophys. 2008, 41, 41–101. 10.1017/S003358350800468X. [DOI] [PubMed] [Google Scholar]
- Collin F.; Karkare S.; Maxwell A. Exploiting bacterial DNA gyrase as a drug target: current state and perspectives. Appl. Microbiol. Biotechnol. 2011, 92, 479–497. 10.1007/s00253-011-3557-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gjorgjieva M.; Tomašič T.; Barančokova M.; Katsamakas S.; Ilaš J.; Tammela P.; Mašič L. P.; Kikelj D. Discovery of Benzothiazole Scaffold-Based DNA Gyrase B Inhibitors. J. Med. Chem. 2016, 59, 8941–8954. 10.1021/acs.jmedchem.6b00864. [DOI] [PubMed] [Google Scholar]
- Zidar N.; Macut H.; Tomašič T.; Brvar M.; Montalvao S.; Tammela P.; Šolmajer T.; Mašič L. P.; Ilaš J.; Kikelj D. N-Phenyl-4,5-dibromopyrrolamides and N-Phenylindolamides as ATP Competitive DNA Gyrase B Inhibitors: Design, Synthesis, and Evaluation. J. Med. Chem. 2015, 58, 6179–6194. 10.1021/acs.jmedchem.5b00775. [DOI] [PubMed] [Google Scholar]
- Zidar N.; Tomasič T.; Macut H.; Sirc A.; Brvar M.; Montalvao S.; Tammela P.; Ilaš J.; Kikelj D. New N-phenyl-4,5-dibromopyrrolamides and N-Phenylindolamides as ATPase inhibitors of DNA gyrase. Eur. J. Med. Chem. 2016, 117, 197–211. 10.1016/j.ejmech.2016.03.079. [DOI] [PubMed] [Google Scholar]
- Zidar N.; Montalvão S.; Hodnik Ž.; Nawrot D. A.; Žula A.; Ilaš J.; Kikelj D.; Tammela P.; Peterlin Mašič L. Antimicrobial Activity of the Marine Alkaloids, Clathrodin and Oroidin, and Their Synthetic Analogues. Mar. Drugs 2014, 12, 940–963. 10.3390/md12020940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamut A.; Skok Ž.; Barančoková M.; Gutierrez L. J.; Cruz C. D.; Tammela P.; Draskovits G.; Szili P. É.; Nyerges Á.; Pál C.; Molek P.; Bratkovič T.; Ilaš J.; Zidar N.; Zega A.; Enriz R. D.; Kikelj D.; Tomašič T. Second-generation 4,5,6,7-tetrahydrobenzo[d]thiazoles as novel DNA gyrase inhibitors. Future Med. Chem. 2020, 12 (4), 277–297. 10.4155/fmc-2019-0127. [DOI] [PubMed] [Google Scholar]
- Phillips J. W.; Goetz M. A.; Smith S. K.; Zink D. L.; Polishook J.; Onishi R.; Salowe S.; Wiltsie J.; Allocco J.; Sigmund J.; Dorso K.; Lee S.; Skwish S.; de la Cruz M.; Martin J.; Vicente F.; Genilloud O.; Lu J.; Painter R. E.; Young K.; Overbye K.; Donald R. G.; Singh S. B. Discovery of kibdelomycin, a potent new class of bacterial type II topoisomerase inhibitor by chemical-genetic profiling in Staphylococcus aureus. Chem. Biol. 2011, 18, 955–965. 10.1016/j.chembiol.2011.06.011. [DOI] [PubMed] [Google Scholar]
- Singh S. B.; Goetz M. A.; Smith S. K.; Zink D. L.; Polishook J.; Onishi R.; Salowe S.; Wiltsie J.; Allocco J.; Sigmund J.; Dorso K.; de la Cruz M.; Martin J.; Vicente F.; Genilloud O.; Donald R. G.; Phillips J. W. Kibdelomycin A, a congener of kibdelomycin, derivatives and their antibacterial activities. Bioorg. Med. Chem. Lett. 2012, 22, 7127–7130. 10.1016/j.bmcl.2012.09.071. [DOI] [PubMed] [Google Scholar]
- Lu J.; Patel S.; Sharma N.; Soisson S. M.; Kishii R.; Takei M.; Fukuda Y.; Lumb K. J.; Singh S. B. Structures of kibdelomycin bound to Staphylococcus aureus GyrB and ParE showed a novel U-shaped binding mode. ACS Chem. Biol. 2014, 9, 2023–2031. 10.1021/cb5001197. [DOI] [PubMed] [Google Scholar]
- Tohyama S.; Takahashi Y.; Akamatsu Y. Biosynthesis of amycolamicin: the biosynthetic origin of a branched alpha-aminoethyl moiety in the unusual sugar amycolose. J. Antibiot. 2010, 63, 147–149. 10.1038/ja.2010.1. [DOI] [PubMed] [Google Scholar]
- Sawa R.; Takahashi Y.; Hashizume H.; Sasaki K.; Ishizaki Y.; Umekita M.; Hatano M.; Abe H.; Watanabe T.; Kinoshita N.; Homma Y.; Hayashi C.; Inoue K.; Ohba S.; Masuda T.; Arakawa M.; Kobayashi Y.; Hamada M.; Igarashi M.; Adachi H.; Nishimura Y.; Akamatsu Y. Amycolamicin: a novel broad-spectrum antibiotic inhibiting bacterial topoisomerase. Chem. - Eur. J. 2012, 18, 15772–15781. 10.1002/chem.201202645. [DOI] [PubMed] [Google Scholar]
- Sherer B. A.; Hull K.; Green O.; Basarab G.; Hauck S.; Hill P.; Loch J. T.; Mullen G.; Bist S.; Bryant J.; Boriack-Sjodin A.; Read J.; DeGrace N.; Uria-Nickelsen M.; Illingworth R. N.; Eakin A. E. Pyrrolamide DNA gyrase inhibitors: Optimization of antibacterial activity and efficacy. Bioorg. Med. Chem. Lett. 2011, 21, 7416–7420. 10.1016/j.bmcl.2011.10.010. [DOI] [PubMed] [Google Scholar]
- Eakin A. E.; Green O.; Hales N.; Walkup G. K.; Bist S.; Singh A.; Mullen G.; Bryant J.; Embrey K.; Gao N.; Breeze A.; Timms D.; Andrews B.; Uria-Nickelsen M.; Demeritt J.; Loch J. T. 3rd; Hull K.; Blodgett A.; Illingworth R. N.; Prince B.; Boriack-Sjodin P. A.; Hauck S.; MacPherson L. J.; Ni H.; Sherer B. Pyrrolamide DNA gyrase inhibitors: fragment-based nuclear magnetic resonance screening to identify antibacterial agents. Antimicrob. Agents Chemother. 2012, 56, 1240–1246. 10.1128/AAC.05485-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uria-Nickelsen M.; Blodgett A.; Kamp H.; Eakin A.; Sherer B.; Green O. Novel DNA gyrase inhibitors. Int. J. Antimicrob. Agents 2013, 41, 28–35. 10.1016/j.ijantimicag.2012.08.017. [DOI] [PubMed] [Google Scholar]
- Cascioferro S.; Parrino B.; Carbone D.; Schillaci D.; Giovannetti E.; Cirrincione G.; Diana P. Thiazoles, Their Benzofused Systems, and Thiazolidinone Derivatives: Versatile and Promising Tools to Combat Antibiotic Resistance. J. Med. Chem. 2020, 63, 7923–7956. 10.1021/acs.jmedchem.9b01245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charifson P. S.; Grillot A.-L.; Grossman T. H.; Parsons J. D.; Badia M.; Bellon S.; Deininger D. D.; Drumm J. E.; Gross C. H.; LeTiran A.; Liao Y.; Mani N.; Nicolau D. P.; Perola E.; Ronkin S.; Shannon D.; Swenson L. L.; Tang Q.; Tessier P. R.; Tian S.-K.; Trudeau M.; Wang T.; Wei Y.; Zhang H.; Stamos D. Novel Dual-Targeting Benzimidazole Urea Inhibitors of DNA Gyrase and Topoisomerase IV Possessing Potent Antibacterial Activity: Intelligent Design and Evolution through the Judicious Use of Structure-Guided Design and Structure–Activity Relationships. J. Med. Chem. 2008, 51, 5243–5263. 10.1021/jm800318d. [DOI] [PubMed] [Google Scholar]
- Tari L. W.; Li X.; Trzoss M.; Bensen D. C.; Chen Z.; Lam T.; Zhang J.; Lee S. J.; Hough G.; Phillipson D.; Akers-Rodriguez S.; Cunningham M. L.; Kwan B. P.; Nelson K. J.; Castellano A.; Locke J. B.; Brown-Driver V.; Murphy T. M.; Ong V. S.; Pillar C. M.; Finn J. Tricyclic GyrB/ParE (TriBE) Inhibitors: A New Class of Broad-Spectrum Dual-Targeting Antibacterial Agents. PLoS One 2013, 8 (12), e84409. 10.1371/journal.pone.0084409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cramer J.; Krimmer S. G.; Heine A.; Klebe G. Paying the Price of Desolvation in Solvent-Exposed Protein Pockets: Impact of Distal Solubilizing Groups on Affinity and Binding Thermodynamics in a Series of Thermolysin Inhibitors. J. Med. Chem. 2017, 60, 5791–5799. 10.1021/acs.jmedchem.7b00490. [DOI] [PubMed] [Google Scholar]
- Lu J.; Patel S.; Sharma N.; Soisson S. M.; Kishii R.; Takei M.; Fukuda Y.; Lumb K. J.; Singh S. B. Structures of Kibdelomycin Bound to Staphylococcus Aureus Gyrb and Pare Showed a Novel U-Shaped Binding Mode. ACS Chem. Biol. 2014, 9, 2023. 10.1021/cb5001197. [DOI] [PubMed] [Google Scholar]
- Sherer B. A.; Hull K.; Green O.; Basarab G.; Hauck S.; Hill P.; Loch J. T.; Mullen G.; Bist S.; Bryant J.; Boriack-Sjodin A.; Read J.; Degrace N.; Uria-Nickelsen M.; Illingworth R. N.; Eakin A. E. Pyrrolamide DNA gyrase inhibitors: Optimization of antibacterial activity and efficacy. Bioorg. Med. Chem. Lett. 2011, 21, 7416–7420. 10.1016/j.bmcl.2011.10.010. [DOI] [PubMed] [Google Scholar]
- Richter M. F.; Drown B. S.; Riley A. P.; Garcia A.; Shirai T.; Svec R. L.; Hergenrother P. J. Predictive rules for compound accumulation yield a broad-spectrum antibiotic. Nature 2017, 545 (7654), 299–304. 10.1038/nature22308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergalli J.; Bodrenko I. V.; Masi M.; Moynié L.; Acosta-Gutiérrez S.; Naismith J. H.; Davin-Regli A.; Ceccarelli M.; van den Berg B.; Winterhalter M.; Pagès J. M. Porins and small-molecule translocation across the outer membrane of Gram-negative bacteria. Nat. Rev. Microbiol. 2020, 18 (3), 164–176. 10.1038/s41579-019-0294-2. [DOI] [PubMed] [Google Scholar]
- Winterhalter M.; Ceccarelli M. Physical methods to quantify small antibiotic molecules uptake into Gram-negative bacteria. Eur. J. Pharm. Biopharm. 2015, 95, 63–7. 10.1016/j.ejpb.2015.05.006. [DOI] [PubMed] [Google Scholar]
- Vergalli J.; Dumont E.; Pajović J.; Cinquin B.; Maigre L.; Masi M.; Réfrégiers M.; Pagés J. M. Spectrofluorimetric quantification of antibiotic drug concentration in bacterial cells for the characterization of translocation across bacterial membranes. Nat. Protoc. 2018, 13 (6), 1348–1361. 10.1038/nprot.2018.036. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.