

Abstract
The glycoslated macrocyclic antibiotic fidaxomicin (1, tiacumicin B, lipiarmycin A3) displays good to excellent activity against Gram-positive bacteria and was approved for the treatment of Clostridium difficile infections (CDI). Among the main limitations for this compound, its low water solubility impacts further clinical uses. We report on the synthesis of new fidaxomicin derivatives based on structural design and utilizing an operationally simple one-step protecting group-free preparative approach from the natural product. An increase in solubility of up to 25-fold with largely retained activity was observed. Furthermore, hybrid antibiotics were prepared that show improved antibiotic activities.
Keywords: Fidaxomicin, antibiotics, semisynthesis, homology modeling, water solubility
Fidaxomicin (1, tiacumicin B, lipiarmycin A3) is a glycosylated macrocyclic lactone produced by actinomycetes and has been isolated from four different soil bacteria.1−7 Fidaxomicin shows good antibiotic activity in vitro against many Gram-positive bacteria, with minimum inhibitory concentration (MIC) values between 0.012 μg/mL and 0.25 μg/mL for Clostridium difficile,8,9 a pathogen causing nosocomial diarrhea. Since 2011, fidaxomicin has been approved for the treatment of inflammations of the intestine caused by C. difficile.10−12 Furthermore, excellent activity against multiresistant Mycobacterium tuberculosis (MIC values <0.008–0.045 μg/mL) and Staphylococcus aureus (MIC values 2–16 μg/mL) has been reported.7,13 Due to the low water solubility of fidaxomicin (1) and in consequence its poor systemic absorption, its application for the treatment of systemic infections has not yet been achieved. The development of semisynthetic derivatives is therefore a promising approach to render this class of antibiotics available for such treatments of systemic infections, in particular by improving water solubility.
With regard to its aglycon ring system, fidaxomicin (1) structurally belongs to the macrocyclic lactone antibiotics (Figure 1), but features a larger 18-membered ring.14,15 The aglycon is connected to a modified noviose (isobutyl ester instead of a methoxy group in 4″-position) as well as to a rhamnose substituent, which is esterified to a dichlorohomoorsellinic acid subunit. The complex structure and the remarkable biological properties of fidaxomicin (1) aroused the interest of many research groups and led to several synthetic studies toward the preparation of the aglycon16−20 and finally the first total synthesis of this natural product reported by some of us21 and subsequently by others.22 Furthermore, investigations of the mechanism of action revealed that fidaxomicin, in contrast to other macrolide antibiotics, does not affect ribosomal protein synthesis, but rather inhibits the transcription process catalyzed by bacterial RNA polymerase (RNAP).8,23−30 In order to identify promising semisynthetic target molecules based on structural considerations, docking calculations of fidaxomicin on a homology model of M. tuberculosis RNAP as well as stability assays31 under various conditions were carried out first.
Figure 1.

Structure of fidaxomicin (tiacumicin B, lipiarmycin A3).
We started our work with the prediction of the binding mode of fidaxomicin in a structural model of M. tuberculosis RNAP. We applied multitemplate homology modeling and a multimodel docking approach.32,33 In the predicted complex, fidaxomicin forms one direct and three indirect interactions (mediated by water) with an RNAP residue whose mutation triggered resistance (Figure 2).7,26,34−36 The dichlorohomoorsellinic acid is located outside the binding pocket and its phenolic hydroxy groups do not take part in any of these interactions. This led to the hypothesis that modifications at these positions represent a promising strategy when aiming to improve water solubility. Furthermore, these hydroxy groups are in proximity to the binding pocket of the antibiotic rifampicin (Figure S1) and we used our binding mode prediction to calculate the optimal linker length (24 atoms of a polyethylene glycol chain) to covalently connect the two antibiotics in order to target both binding sites at the same time. The actual binding mode of fidaxomicin in M. tuberculosis RNAP was recently elucidated using cryo-electron microscopy (cryo-EM).29,30 A comparison with our binding mode prediction revealed that the macrocycles are overlapping, but the resorcinol moieties point in opposite directions (Figure S2). However, the cryo-EM structure also revealed a solvent pocket around the dichlorohomoorsellinic acid subunit, thus further corroborating the hypothesis that this part is a suitable site for modifications.
Figure 2.

Interaction diagram of the predicted fidaxomicin binding mode in M. tuberculosis RNAP. The protein subunits are given in Greek letters. Gray circles around atoms show exposition to solvent. Orange circles show residues that lead to resistance when mutated; indirectly affected residues are displayed as hexagons.
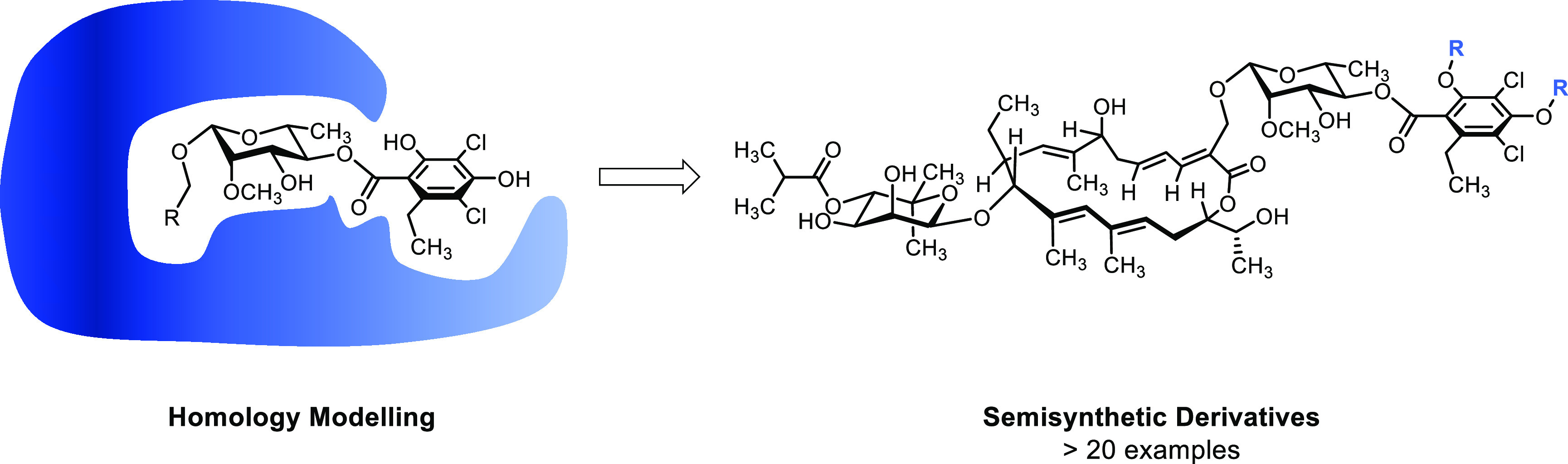
According to our binding mode prediction, 3′′′- and 5′′′-positions displayed promising sites for modifications as they are exposed to the solvent. Furthermore, functionalizations in these positions turned out to be synthetically feasible without the use of any protecting group; thus, analogs were accessible with only one preparative step.37 Therefore, fidaxomicin (1) was treated with different electrophiles under slightly basic conditions in DMF at elevated temperatures (Scheme 1). The products 2–10 were obtained as separable mixtures of mono- and disubstituted compounds (except 9b(38)). It is noteworthy that the monosubstitutions were exclusively observed at 5′′′-hydroxy group since electronic effects might render this hydroxy group more nucleophilic. Under basic conditions, formation of trace amounts of isomers, which proved to be the transacylated isobutyric ester, could not be prevented.31 After purification by preparative HPLC, the desired compounds were obtained in moderate to good yields (28–81%).
Scheme 1. Functionalization at Positions 3′′′ and 5′′′ of the Resorcinol Unit (Ns = Nosyl).
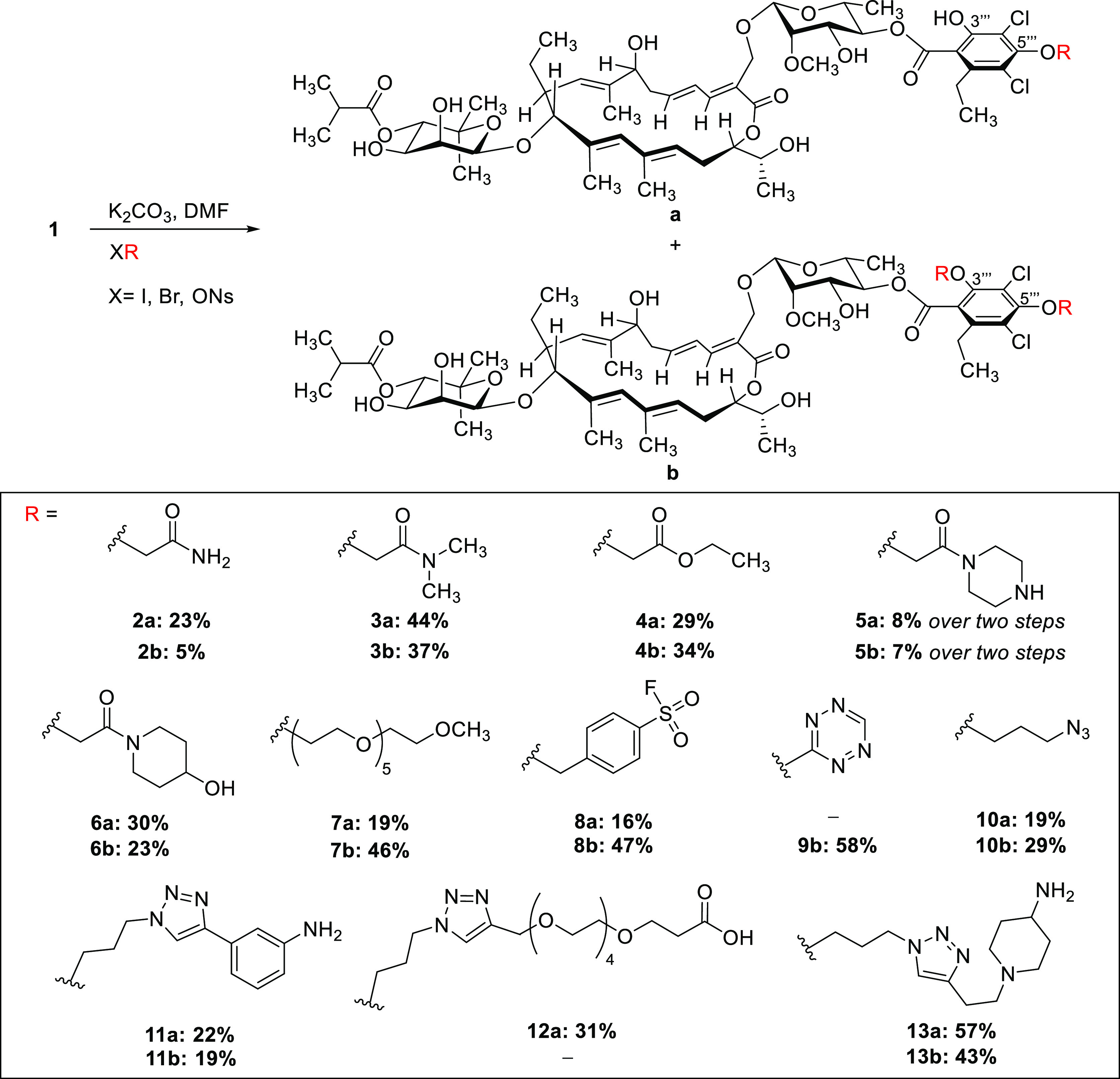
The substituents in compounds 2–7 were introduced for their high polarity and their common use in medicinal chemistry. The sulfonyl fluorides 8a/8b were synthesized for their ability to form covalent bonds with the enzyme of interest (SuFEx-Click chemistry),39 which might also be useful for MS-based studies on the binding site. In the context of a study on labeling of macromolecules with tetrazine moieties, we synthesized tetrazine-fidaxomicin 9b.38 Furthermore, we reacted the nosylated azidopropanol to obtain azide derivatives 10a/b, which were used as a platform to connect several structurally diverse alkynes to the resorcinol moiety using Click chemistry (CuAAC) with a dicopper catalyst developed by Straub and co-workers40 or commercially available Cu(I)OAc. Using azide 10a as starting material, anilines 11, PEG5-acid 12a, and piperidinamine 13a/b were obtained, which showed improved water solubility in comparison with fidaxomicin (see below).
In addition to the derivatives discussed so far, hybrids of fidaxomicin with other antibiotics were synthesized. These either have different targets or interact with different binding sites within a common target. Fluoroquinolones are common components of hybrid antibiotics due to their stability in a great variety of reaction conditions and their broad spectrum of activity.41,42 Modifications on the amine of the piperazine moiety were shown to barely influence their biological activity.43 Therefore, this position was chosen for covalently connecting ciprofloxacin to fidaxomicin, thus expecting only minor effects on both molecules’ biological properties. For the synthesis of the fidaxomicin-ciprofloxacin hybrid 15, ciprofloxacin was first transformed into the corresponding bromoacetyl-ciprofloxacin 14 using bromoacetyl bromide (Scheme 2). Subsequent exposure to basic conditions (K2CO3 in DMF) together with fidaxomicin resulted in the desired hybrid 15.
Scheme 2. Synthesis of a Fidaxomicin–Ciprofloxacin Hybrid.

As fidaxomicin and rifampicin share the same target enzyme (RNAP), but interact with different binding sites (Figure S1),8 we synthesized a fidaxomicin–rifampicin hybrid. Based on our calculations on the predicted binding mode, an octaethylene glycol linker was deemed suitable in order to covalently connect the two antibiotics while retaining their ability to interact with their respective binding sites at the same time. Starting from fidaxomicin (1) and nosylated octaethylene glycol azide 16, the octaethlyene glycol linker was introduced and azide 17 was then connected with literature-known alkynylated rifampicin 18(44) using a Cu(I)-catalyst40 to give the desired fidaxomicin-rifampicin hybrid 19 (Scheme 3).
Scheme 3. Synthesis of the Fidaxomicin–Rifampicin Hybrid 19.

We investigated the antibiotic activity of the synthesized derivatives on different bacterial strains such as Bacillus subtilis (DSM3256), S. aureus (ATCC29213), and M. tuberculosis by evaluation of the minimum inhibitory concentration (MIC) (see SI). Derivatives featuring promising antibiotic activity in these tests were further assessed for their antibiotic activity against several isolates of C. difficile (Table 1). Although some of the derivatives did not retain their antibiotic activity, it was demonstrated that large substituents on the resorcinol do not necessarily impair biological activity, which is shown by the good biological activities of derivatives 4a/b, 5a, 9b, 13a, and hybrids 15a and 19. Although derivatives 4a/b and 13a/b display decreased activity against C. difficile, some promising activity was observed against M. tuberculosis, S. aureus, and B. subtilis. Moreover, compounds 5a and 9b retained excellent activity against C. difficile.
Table 1. Summary of the Minimum Inhibitory Concentrations (MIC) of Selected Derivatives.
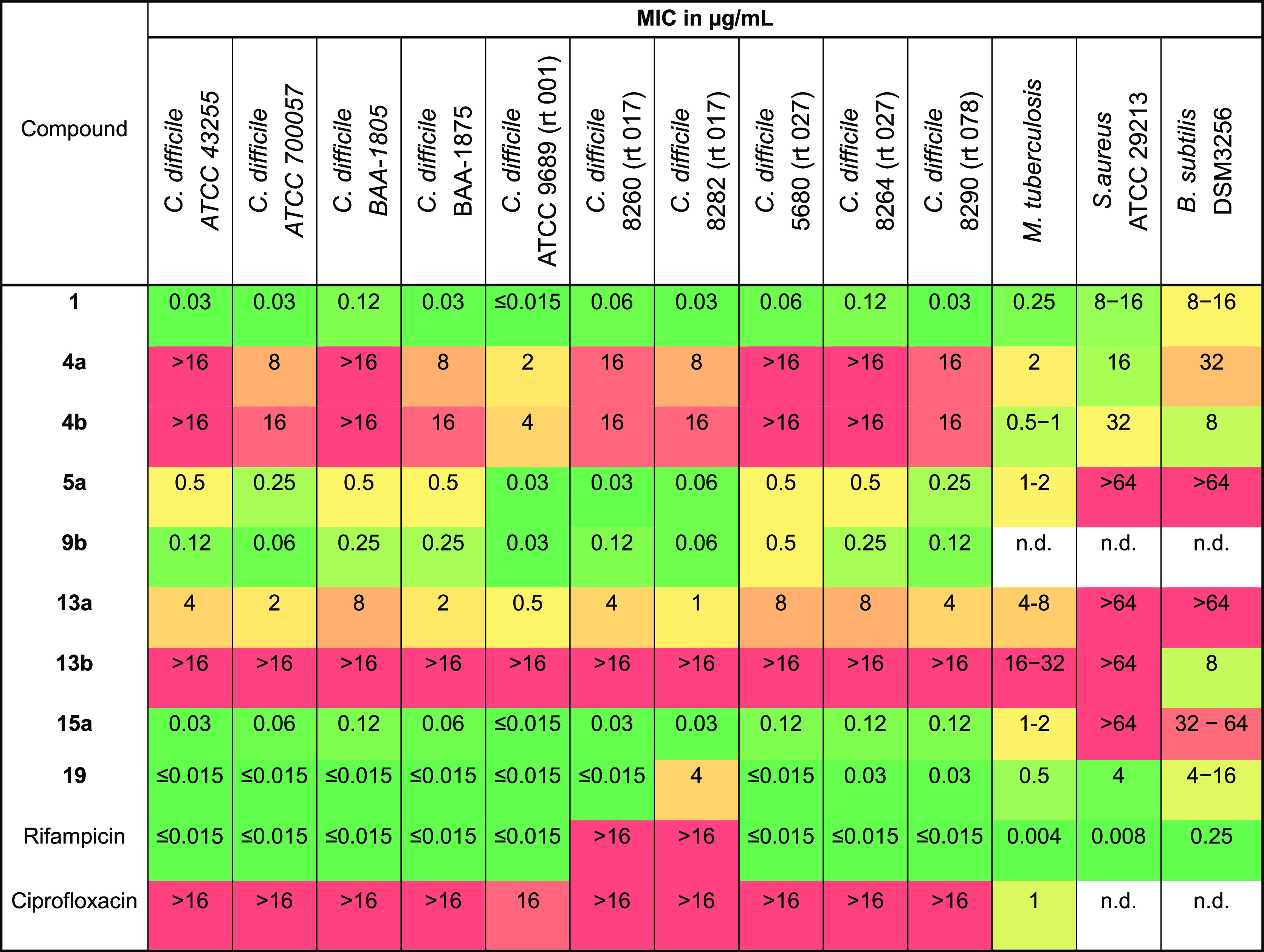
The fidaxomicin–rifampicin hybrid 19 shows an improved activity compared to fidaxomicin against all investigated strains and similar activity against C. difficile when compared to rifampicin. Interestingly, hybrid 19 retains its biological activity even against strains which are not susceptible to rifampicin (C. difficile 8260 and 8282). Though no antibiotic activity against S. aureus and B. subtilis has been observed, the fidaxomicin-ciprofloxacin hybrid 15a retains its excellent activity against all C. difficile strains even though ciprofloxacin itself is inactive against the latter.
Additionally, we investigated the water solubility of the obtained derivatives at pH = 7 by HPLC. For this purpose, saturated solutions of the derivatives in phosphate buffer were prepared and the concentration was determined after filtration of the resulting suspensions. The results (Figure 3) provide evidence for higher water solubility of PEG-derivatives 7b and 12a, piperidinol 6a/b, and piperidinamine 13b. Thus, PEG5-acid 12a displays 25-fold and amine 13b 5-fold increase in water solubility, though its antibiotic activity is reduced.
Figure 3.
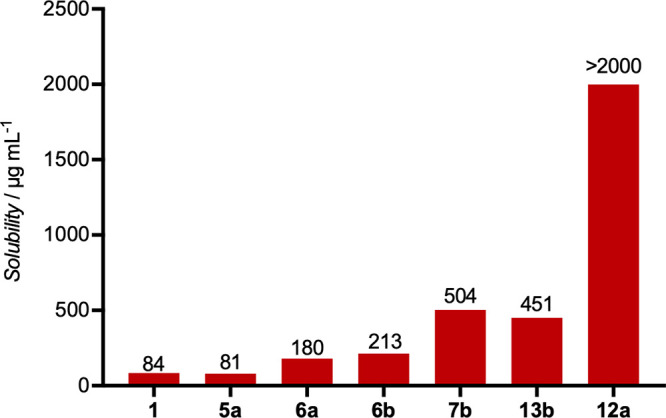
Solubility of selected derivatives in phosphate buffer (pH = 7).
In conclusion, apart from methylations45 and benzylations30 at the phenolic hydroxy groups, the new analogs presented here are, to our knowledge, the first examples of complex semisynthetic derivatives of fidaxomicin obtained via phenolic modifications. Other derivatives recently reported are obtained by semisynthesis,46−48 fermentation,49,50 and fermentation of knockout mutants.51−54 Based on our predictions of a binding mode of fidaxomicin in a homology model of M. tuberculosis RNAP, which suggested that modifications on the dichlorohomoorsellinic acid would be promising, several analogs have been synthesized and tested for their biological activity and water solubility. Some of these compounds showed improved water solubility with maintained antibiotic activity. The synthesized hybrid antibiotics 15 and 19 show improved activity compared to fidaxomicin, while also retaining activity against strains that show resistance against the attached antibiotic itself. Access to a great variety of derivatives of this complex natural product was achieved by using easy and reliable synthetic methods. These derivatives could provide an important contribution in ongoing efforts to reduce the rate of antibiotic resistance development in bacteria and broaden the scope of application of fidaxomicin.
Acknowledgments
We thank the Swiss National Science Foundation for financial support (200021_182043 (K.G.) and 31003A_153349/1 (P.S.)). Work in the laboratory of P.S. is supported by Swiss Lung Association (2018-02). We acknowledge partial financial support by the DFG (GRK 2158, P4 for H.G.). We gratefully acknowledge the NMR and MS services of the University of Zurich and F. Fazzi and G. Uzunidis for skillful laboratory assistance. We further thank A. Siegle and O. Trapp (LMU München) for using the HPLC in their research groups and R. Wehlauch and J. Michel for proofreading the manuscript.
Glossary
Abbreviations
- CDI
C. difficile infection
- MIC
minimum inhibitory concentration
- RNAP
RNA polymerase
- cryo-EM
cryo-electron microscopy
- HPLC
high performance liquid chromatography
- PEG
polyethylene glycol.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.0c00381.
Supplementary Figures S1 and S2, experimental procedures and methods, characterization data, 1H and 13C NMR spectra, supplementary MIC tables, and computational details (PDF)
Author Contributions
⊥ A.D. and R.B. contributed equally. The manuscript was written through contributions of all authors. A.D., R.B., and K.G. designed the study. A.D., R.B., and S.D.S. carried out the synthesis and characterization of the derivatives. C.G.W.G., and H.G. performed the homology modeling and computational studies. D.S. and P.S. developed the MIC tests for M. tuberculosis and performed the biological evaluation of the derivatives. K.Z. and M.G. performed the MIC tests against S. aureus and B. subtilis. A.D., R.B., and K.G. analyzed and discussed the results. A.D.,R.B., and K.G. wrote the manuscript. All authors have given approval to the final version of the manuscript.
The authors declare the following competing financial interest(s): A patent application (WO2019135010A1, EP18150671.8A) was filed Jan 8th, 2018 that includes antibiotics presented in this work.
Supplementary Material
References
- Parenti F.; Pagani H.; Beretta G. Lipiarmycin, A New Antibiotic From Actinoplanes I. Description of the Producer Strain and Fermentation Studies. J. Antibiot. 1975, 28, 247–252. 10.7164/antibiotics.28.247. [DOI] [PubMed] [Google Scholar]
- Coronelli C.; White R. J.; Lancini G. C.; Parenti F. Lipiarmycin, A New Antibiotic from Actinoplanes II. Isolation, Chemical, Biological and Biochemical Charaterization. J. Antibiot. 1975, 28, 253–259. 10.7164/antibiotics.28.253. [DOI] [PubMed] [Google Scholar]
- Õmura S.; Imamura N.; Õiwa R.; Kuga H.; Iwata R.; Masuma R.; Iwai Y. Clostomicins, New Antibiotics Produced by Micromonospora Echinospora Subsp. Armeniaca Subsp. Nov. I. Production, Isolation, and Physico-Chemical and Biological Properties. J. Antibiot. 1986, 39, 1407–1412. 10.7164/antibiotics.39.1407. [DOI] [PubMed] [Google Scholar]
- Takahashi Y.; Iwai Y.; Õmura S. Clostomicins, New Antibiotics Produced by Micromonospora Echinospora Subsp. Armeniaca Subsp. Nov. II. Taxonomic Study of the Producing Microorganism. J. Antibiot. 1986, 39, 1413–1418. 10.7164/antibiotics.39.1413. [DOI] [PubMed] [Google Scholar]
- Hochlowski J. E.; Swanson S. J.; Ranfranz L. M.; Whittern D. N.; Buko A. M.; McAlpine J. B. Tiacumicins, a Novel Complex of 18-Membered Macrolides II. Isolation and Structure Determination. J. Antibiot. 1987, 40, 575–588. 10.7164/antibiotics.40.575. [DOI] [PubMed] [Google Scholar]
- Theriault R. J.; Karwowski J. P.; Jackson M.; Girolami R. L.; Sunga G. N.; Vojtko C. M.; Coen L. J. Tiacumicins, a Novel Complex of 18-Membered Macrolide Antibiotics I. Taxonomy, Fermentation and Antibacterial Activity. J. Antibiot. 1987, 40, 567–574. 10.7164/antibiotics.40.567. [DOI] [PubMed] [Google Scholar]
- Kurabachew M.; Lu S. H. J.; Krastel P.; Schmitt E. K.; Suresh B. L.; Goh A.; Knox J. E.; Ma N. L.; Jiricek J.; Beer D.; Cynamon M.; Petersen F.; Dartois V.; Keller T.; Dick T.; Sambandamurthy V. K. Lipiarmycin Targets RNA Polymerase and Has Good Activity against Multidrug-Resistant Strains of Mycobacterium Tuberculosis. J. Antimicrob. Chemother. 2008, 62, 713–719. 10.1093/jac/dkn269. [DOI] [PubMed] [Google Scholar]
- Srivastava A.; Talaue M.; Liu S.; Degen D.; Ebright R. Y.; Sineva E.; Chakraborty A.; Druzhinin S. Y.; Chatterjee S.; Mukhopadhyay J.; Ebright Y. W.; Zozula A.; Shen J.; Sengupta S.; Niedfeldt R. R.; Xin C.; Kaneko T.; Irschik H.; Jansen R.; Donadio S.; Connell N.; Ebright R. H. New Target for Inhibition of Bacterial RNA Polymerase: Switch Region. Curr. Opin. Microbiol. 2011, 14, 532–543. 10.1016/j.mib.2011.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babakhani F.; Gomez A.; Robert N.; Sears P. Killing Kinetics of Fidaxomicin and Its Major Metabolite, OP-1118, against Clostridium Difficile. J. Med. Microbiol. 2011, 60, 1213–1217. 10.1099/jmm.0.029470-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erb W.; Zhu J. From Natural Product to Marketed Drug: The Tiacumicin Odyssey. Nat. Prod. Rep. 2013, 30, 161–174. 10.1039/C2NP20080E. [DOI] [PubMed] [Google Scholar]
- Dorst A.; Gademann K. Chemistry and Biology of the Clinically Used Macrolactone Antibiotic Fidaxomicin. Helv. Chim. Acta 2020, 103, e2000038 10.1002/hlca.202000038. [DOI] [Google Scholar]
- Dorst A.; Jung E.; Gademann K. Recent Advances in Mode of Action and Biosynthesis Studies of the Clinically Used Antibiotic Fidaxomicin. Chimia 2020, 74, 270–273. 10.2533/chimia.2020.270. [DOI] [PubMed] [Google Scholar]
- Biedenbach D. J.; Ross J. E.; Putnam S. D.; Jones R. N. In Vitro Activity of Fidaxomicin (OPT-80) Tested against Contemporary Clinical Isolates of Staphylococcus Spp. and Enterococcus Spp. Antimicrob. Agents Chemother. 2010, 54, 2273–2275. 10.1128/AAC.00090-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnone A.; Nasini G.; Cavalleri B. Structure Elucidation of the Macrocyclic Antibiotic Lipiarmycin. J. Chem. Soc., Perkin Trans. 1 1987, 1353–1359. 10.1039/p19870001353. [DOI] [Google Scholar]
- Serra S.; Malpezzi L.; Bedeschi A.; Fuganti C.; Fonte P. Final Demonstration of the Co-Identity of Lipiarmycin A3 and Tiacumicin B (Fidaxomicin) through Single Crystal X-Ray Analysis. Antibiotics 2017, 6, 7. 10.3390/antibiotics6010007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaus F.; Altmann K. H. Total Synthesis of the Tiacumicin B (Lipiarmycin A3/Fidaxomicin) Aglycone. Angew. Chem., Int. Ed. 2015, 54, 1937–1940. 10.1002/anie.201409510. [DOI] [PubMed] [Google Scholar]
- Miyatake-Ondozabal H.; Kaufmann E.; Gademann K. Total Synthesis of the Protected Aglycon of Fidaxomicin (Tiacumicin B, Lipiarmycin A3). Angew. Chem., Int. Ed. 2015, 54, 1933–1936. 10.1002/anie.201409464. [DOI] [PubMed] [Google Scholar]
- Erb W.; Grassot J. M.; Linder D.; Neuville L.; Zhu J. Enantioselective Synthesis of Putative Lipiarmycin Aglycon Related to Fidaxomicin/Tiacumicin B. Angew. Chem., Int. Ed. 2015, 54, 1929–1932. 10.1002/anie.201409475. [DOI] [PubMed] [Google Scholar]
- Jeanne-Julien L.; Masson G.; Astier E.; Genta-Jouve G.; Servajean V.; Beau J. M.; Norsikian S.; Roulland E. Synthesis of a Tiacumicin B Protected Aglycone. Org. Lett. 2017, 19, 4006–4009. 10.1021/acs.orglett.7b01744. [DOI] [PubMed] [Google Scholar]
- Jeanne-Julien L.; Masson G.; Astier E.; Genta-Jouve G.; Servajean V.; Beau J.-M.; Norsikian S.; Roulland E. Study of the Construction of the Tiacumicin B Aglycone. J. Org. Chem. 2018, 83, 921–929. 10.1021/acs.joc.7b02909. [DOI] [PubMed] [Google Scholar]
- Kaufmann E.; Hattori H.; Miyatake-Ondozabal H.; Gademann K. Total Synthesis of the Glycosylated Macrolide Antibiotic Fidaxomicin. Org. Lett. 2015, 17, 3514–3517. 10.1021/acs.orglett.5b01602. [DOI] [PubMed] [Google Scholar]
- Norsikian S.; Tresse C.; François-Eude M.; Jeanne-Julien L.; Masson G.; Servajean V.; Genta-Jouve G.; Beau J.-M.; Roulland E. Total Synthesis of Tiacumicin B: Implementing Hydrogen Bond Directed Acceptor Delivery for Highly Selective β-Glycosylations. Angew. Chem., Int. Ed. 2020, 59, 6612–6616. 10.1002/anie.202000231. [DOI] [PubMed] [Google Scholar]
- Talpaert M.; Campagnari F.; Clerici L. Lipiarmycin: An Antibiotic Inhibiting Nucleic Acid Polymerases. Biochem. Biophys. Res. Commun. 1975, 63, 328–334. 10.1016/S0006-291X(75)80047-7. [DOI] [PubMed] [Google Scholar]
- Sonenshein A. L.; Alexander H. B.; Rothstein D. M.; Fisher S. H. Lipiarmycin-Resistant Ribonucleic Acid Polymerase Mutants of Bacillus Subtilis. J. Bacteriol. 1977, 132, 73–79. 10.1128/JB.132.1.73-79.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonenshein A. L.; Alexander H. B. Initiation of Transcription in Vitro Is Inhibited by Lipiarmycin. J. Mol. Biol. 1979, 127, 55–72. 10.1016/0022-2836(79)90459-5. [DOI] [PubMed] [Google Scholar]
- Tupin A.; Gualtieri M.; Leonetti J. P.; Brodolin K. The Transcription Inhibitor Lipiarmycin Blocks DNA Fitting into the RNA Polymerase Catalytic Site. EMBO J. 2010, 29, 2527–2537. 10.1038/emboj.2010.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artsimovitch I.; Seddon J.; Sears P. Fidaxomicin Is an Inhibitor of the Initiation of Bacterial RNA Synthesis. Clin. Infect. Dis. 2012, 55, 127–131. 10.1093/cid/cis358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morichaud Z.; Chaloin L.; Brodolin K. Regions 1.2 and 3.2 of the RNA Polymerase σ Subunit Promote DNA Melting and Attenuate Action of the Antibiotic Lipiarmycin. J. Mol. Biol. 2016, 428, 463–476. 10.1016/j.jmb.2015.12.017. [DOI] [PubMed] [Google Scholar]
- Boyaci H.; Chen J.; Lilic M.; Palka M.; Mooney R. A.; Landick R.; Darst S. A.; Campbell E. A. Fidaxomicin Jams Mycobacterium Tuberculosis RNA Polymerase Motions Needed for Initiation via RbpA Contacts. eLife 2018, 7, 1–19. 10.7554/eLife.34823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin W.; Das K.; Degen D.; Zhang C.; Ebright R. H.; Lin W.; Das K.; Degen D.; Mazumder A.; Duchi D.; Wang D.; Ebright Y. W. Structural Basis of Transcription Inhibition by Fidaxomicin (Lipiarmycin A3). Mol. Cell 2018, 70, 60–71. 10.1016/j.molcel.2018.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hattori H.; Kaufmann E.; Miyatake-Ondozabal H.; Berg R.; Gademann K. Total Synthesis of Tiacumicin A. Total Synthesis, Relay Synthesis, and Degradation Studies of Fidaxomicin (Tiacumicin B, Lipiarmycin A3). J. Org. Chem. 2018, 83, 7180–7205. 10.1021/acs.joc.8b00101. [DOI] [PubMed] [Google Scholar]
- Gertzen C. G. W.; Spomer L.; Smits S. H. J.; Häussinger D.; Keitel V.; Gohlke H. Mutational Mapping of the Transmembrane Binding Site of the G-Protein Coupled Receptor TGR5 and Binding Mode Prediction of TGR5 Agonists. Eur. J. Med. Chem. 2015, 104, 57–72. 10.1016/j.ejmech.2015.09.024. [DOI] [PubMed] [Google Scholar]
- Milić D.; Dick M.; Mulnaes D.; Pfleger C.; Kinnen A.; Gohlke H.; Groth G. Recognition Motif and Mechanism of Ripening Inhibitory Peptides in Plant Hormone Receptor ETR1. Sci. Rep. 2018, 8, 3890. 10.1038/s41598-018-21952-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gualtieri M.; Tupin A.; Brodolin K.; Leonetti J. P. Frequency and Characterisation of Spontaneous Lipiarmycin-Resistant Enterococcus Faecalis Mutants Selected in Vitro. Int. J. Antimicrob. Agents 2009, 34, 605–616. 10.1016/j.ijantimicag.2009.07.003. [DOI] [PubMed] [Google Scholar]
- Ebright R. H.RNA-Exit Channel: Target and Method for Inhibition of Bacterial RNA. WO2005/001034, 2005.
- Babakhani F.; Seddon J.; Sears P. Comparative Microbiological Studies of Transcription Inhibitors Fidaxomicin and the Rifamycins in Clostridium Difficile. Antimicrob. Agents Chemother. 2014, 58, 2934–2937. 10.1128/AAC.02572-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gademann K.; Berg R.; Meier A.. Glycosylated Macrolides and Their Use as Antibiotics. WO 2019/135010 A1, 2019.
- Schnell S. D.; Hoff L. V.; Panchagnula A.; Wurzenberger M. H. H.; Klapötke T. M.; Sieber S.; Linden A.; Gademann K. 3-Bromotetrazine: Labelling of Macromolecules via Monosubstituted Bifunctional s-Tetrazines. Chem. Sci. 2020, 11, 3042–3047. 10.1039/C9SC06169J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z.; Li J.; Li S.; Li G.; Sharpless K. B.; Wu P. SuFEx Click Chemistry Enabled Late-Stage Drug Functionalization. J. Am. Chem. Soc. 2018, 140, 2919–2925. 10.1021/jacs.7b12788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg R.; Straub J.; Schreiner E.; Mader S.; Rominger F.; Straub B. F. Highly Active Dinuclear Copper Catalysts for Homogeneous Azide-Alkyne Cycloadditions. Adv. Synth. Catal. 2012, 354, 3445–3450. 10.1002/adsc.201200734. [DOI] [Google Scholar]
- Hu Y. Q.; Zhang S.; Xu Z.; Lv Z. S.; Liu M. L.; Feng L. S. 4-Quinolone Hybrids and Their Antibacterial Activities. Eur. J. Med. Chem. 2017, 141, 335–345. 10.1016/j.ejmech.2017.09.050. [DOI] [PubMed] [Google Scholar]
- Pokrovskaya V.; Baasov T. Dual-Acting Hybrid Antibiotics: A Promising Strategy to Combat Bacterial Resistance. Expert Opin. Drug Discovery 2010, 5, 883–902. 10.1517/17460441.2010.508069. [DOI] [PubMed] [Google Scholar]
- Gootz T. D.; Brighty K. E. Fluoroquinolone Antibacterials: SAR, Mechanism of Action, Resistance, and Clinical Aspects. Med. Res. Rev. 1996, 16, 433–486. 10.1002/(SICI)1098-1128(199609)16:5<433::AID-MED3>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- Cochrane S. A.; Li X.; He S.; Yu M.; Wu M.; Vederas J. C. Synthesis of Tridecaptin-Antibiotic Conjugates with in Vivo Activity against Gram-Negative Bacteria. J. Med. Chem. 2015, 58, 9779–9785. 10.1021/acs.jmedchem.5b01578. [DOI] [PubMed] [Google Scholar]
- McAlpine J. E.; Hochlowski J. E.. Dialkyltiacumicin Compounds. US E5,583,115, 1996.
- Wu M.-C.; Huang C.-C.; Lu Y.-C.; Fan W.-J.. Derivatives of Tiacumicin B as Anti-Cancer Agents. US 2009/0110718 A1, 2008.
- Dorst A.; Shchelik I. S.; Schäfle D.; Sander P.; Gademann K. Synthesis and Biological Evaluation of Iodinated Fidaxomicin Antibiotics. Helv. Chim. Acta 2020, 103, e2000130 10.1002/hlca.202000130. [DOI] [Google Scholar]
- Dailler D.; Dorst A.; Schäfle D.; Sander P.; Gademann K. Novel Fidaxomicin Antibiotics through Site-Selective Catalysis. ChemRxiv 2020, 10.26434/chemrxiv.12593429.v1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochlowski J. E.; Jackson M.; Rasmussen R. R.; Buko A. M.; Clement J. J.; Whittern D. N.; McAlpine J. B. Production of Brominated Tiacumicin Derivatives. J. Antibiot. 1997, 50, 201–205. 10.7164/antibiotics.50.201. [DOI] [PubMed] [Google Scholar]
- Hochlowski J. E.; Jackson M.; McAlpine J. B.; Rasmussen R. R.. Bromotiacumicin Compounds. US 5,767,096, 1998.
- Xiao Y.; Li S.; Niu S.; Ma L.; Zhang G.; Zhang H.; Zhang G.; Ju J.; Zhang C. Characterization of Tiacumicin B Biosynthetic Gene Cluster Affording Diversified Tiacumicin Analogues and Revealing a Tailoring Dihalogenase. J. Am. Chem. Soc. 2011, 133, 1092–1105. 10.1021/ja109445q. [DOI] [PubMed] [Google Scholar]
- Niu S.; Hu T.; Li S.; Xiao Y.; Ma L.; Zhang G.; Zhang H.; Yang X.; Ju J.; Zhang C. Characterization of a Sugar-O-Methyltransferase TiaS5 Affords New Tiacumicin Analogues with Improved Antibacterial Properties and Reveals Substrate Promiscuity. ChemBioChem 2011, 12, 1740–1748. 10.1002/cbic.201100129. [DOI] [PubMed] [Google Scholar]
- Zhang H.; Tian X.; Pu X.; Zhang Q.; Zhang W.; Zhang C. Tiacumicin Congeners with Improved Antibacterial Activity from a Halogenase-Inactivated Mutant. J. Nat. Prod. 2018, 81, 1219–1224. 10.1021/acs.jnatprod.7b00990. [DOI] [PubMed] [Google Scholar]
- Yu Z.; Zhang H.; Yuan C.; Zhang Q.; Khan I.; Zhu Y.; Zhang C. Characterizing Two Cytochrome P450s in Tiacumicin Biosynthesis Reveals Reaction Timing for Tailoring Modifications. Org. Lett. 2019, 21, 7679–7683. 10.1021/acs.orglett.9b03100. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.