

Abstract
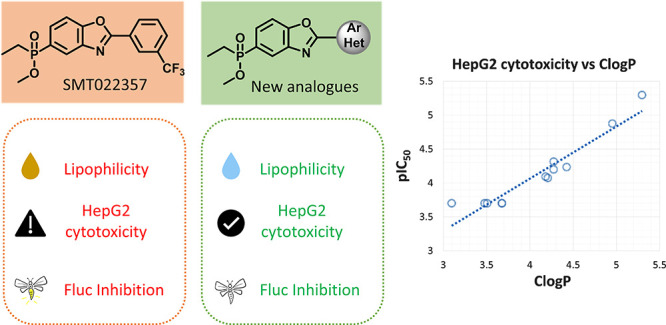
Utrophin modulation is a disease-modifying therapeutic strategy for Duchenne muscular dystrophy that would be applicable to all patient populations. To improve the suboptimal profile of ezutromid, the first-in-class clinical candidate, a second generation of utrophin modulators bearing a phosphinate ester moiety was developed. This modification significantly improved the physicochemical and ADME properties, but one of the main lead molecules was found to have dose-limiting hepatotoxicity. In this work we describe how less lipophilic analogues retained utrophin modulatory activity in a reporter gene assay, upregulated utrophin protein in dystrophic mouse muscle cells, but also had improved physicochemical and ADME properties. Notably, ClogP was found to directly correlate with pIC50 in HepG2 cells, hence leading to a potentially safer toxicological profiles in this series. Compound 21 showed a balanced profile (H2K EC50: 4.17 μM, solubility: 477 μM, mouse hepatocyte T1/2 > 240 min) and increased utrophin protein 1.6-fold in a Western blot assay.
Keywords: Duchenne muscular dystrophy, utrophin upregulation, hepatotoxicity, phosphinate esters
Duchenne muscular dystrophy (DMD) is an X-linked neuromuscular disorder caused by loss of function mutations on the dystrophin gene.1 Though classified as a rare disease, it is one of the most common fatal genetic diseases affecting children.1 Dystrophin is a critical component of the dystrophin-associated protein complex (DAPC) connecting the internal cytoskeleton to the surrounding extracellular matrix. In the absence of functional dystrophin, the muscles gradually degenerate, leading to inflammation, fibrosis, and failed cycles of regeneration.2,3
More than 30 years after the discovery of dystrophin,4 there is still a paucity of treatment options for the disease, as the only disease-modifying therapeutics are targeting specific subpopulations and the standard of care is aimed mostly to alleviate the inflammation and other secondary effects. A promising therapeutic strategy that would be both disease modifying and could target all patient subpopulations is functional replacement of dystrophin with its structural paralogue utrophin.2 Data from transgenic animal models5−7 led to the initiation of drug discovery programs, the most advanced of which progressed to Phase 2 proof of concept clinical trials.
Ezutromid (Figure 1, 1), formerly SMT C1100, a utrophin modulator initially discovered via a HTS using a firefly luciferase (Fluc) reporter gene assay,8,9 led to promising results in animal models of DMD10 and a safe profile in multiple Phase 1 clinical trials.11−13 Encouraging interim 24-week data in a Phase 2 trial (NCT02858362, Summit Therapeutics PLC, PhaseOut DMD) showed increase of utrophin protein and reduction of muscle fiber damage. However, the primary and secondary clinical end points were not met at the end of the trial and development of ezutromid was discontinued.14 The lack of sustained efficacy has been attributed to extensive metabolism of ezutromid and induction of CYP1A (ezutromid’s main metabolic pathway),15 which is further supported by reduction in ezutromid’s exposure after repeated dosing in both healthy volunteers and DMD patients.11−13
Figure 1.

Structures of ezutromid (1), SMT022357 (2), and SMT022332 ((+) enantiomer, 3).
A medicinal chemistry program was launched to improve ezutromid’s suboptimal properties and led to a second generation of utrophin modulators.16−18 Therein, a dual strategy was implemented to improve solubility by replacement of the sulfone moiety and to improve metabolic stability by replacement of naphthalene with halo- and heteroaryls. A promising lead molecule from this series (Figure 1, SMT022357, 2) with an improved ADME profile increased levels of utrophin protein in skeletal, respiratory, and cardiac muscles, reduced regeneration, and improved muscle function when administered orally to mdx mice.16 Further progression of SMT022357 (2) was halted, due to dose-limiting hepatotoxicity observed during subsequent maximum tolerated dose studies in rats. A structurally similar, but less lipophilic, analogue SMT022332 (Figure 1, (+)-enantiomer 3) was instead progressed because of its encouraging activity and efficacy in mdx mice and its improved safety profile.18
During these studies, the relationship between lipophilicity and hepatotoxicity within this compound series was investigated. Several empirical studies have correlated lipophilicity with increased nonspecific binding and off-target effects, and a decreased chance of clinical success.19,20 However, to our knowledge, there are few published studies that directly correlate lipophilicity with hepatotoxicity within a compound series.21−25 Lipophilic compounds are more prone to extensive metabolism and thus may result more frequently in drug-induced liver injury (DILI).26 As we observed extensive metabolism and in vivo hepatotoxicity in rodents with 2, which translated in in vitro cellular hepatotoxicity for 2 and other related examples,18 we felt that reducing lipophilicity would be a good strategy for optimizing the series. To achieve this, analogues that bear the -CHF2 and -OCHF2 groups and their pyridyl equivalents were synthesized as they are predicted to have lower lipophilicity than 2.27
To access the phosphinate esters of the benzo[d]oxazoles, two main synthetic routes were devised (Scheme 1). In General Route 1, the phosphinate esters were synthesized first, followed by cyclization to the benzoxazole, and finally attachment of the C(2)-aryl via C–H activation. Bromo 2-nitrophenols were O-benzylated (4–5) and substituted with phosphinic acid (6–7), and the resulting acids were esterified to their corresponding ethyl phosphinate esters (8–9). Subsequent hydrogenation led to 2-aminophenols 10 and 11 in a one-step reduction/deprotection, which in turn gave the benzo[d]oxazoles 12 and 13 in a reaction with diethoxymethoxyethane in the presence of PPTS. The final products (14–27) were obtained with C–H activation using Pd(PPh3)2Cl2 or Pd(dppf)Cl2 and CuI as catalysts. In another variation Pd(OAc)2, Xantphos, and 1 M LiHMDS, in DME, were used (14, 15). This led to hydrolysis of the ester in the case of 12, and the resulting phosphinic acid (14a) was reacted with MeOH to give the final product 14.
Scheme 1. General Synthetic Routes to Phosphinate Esters.
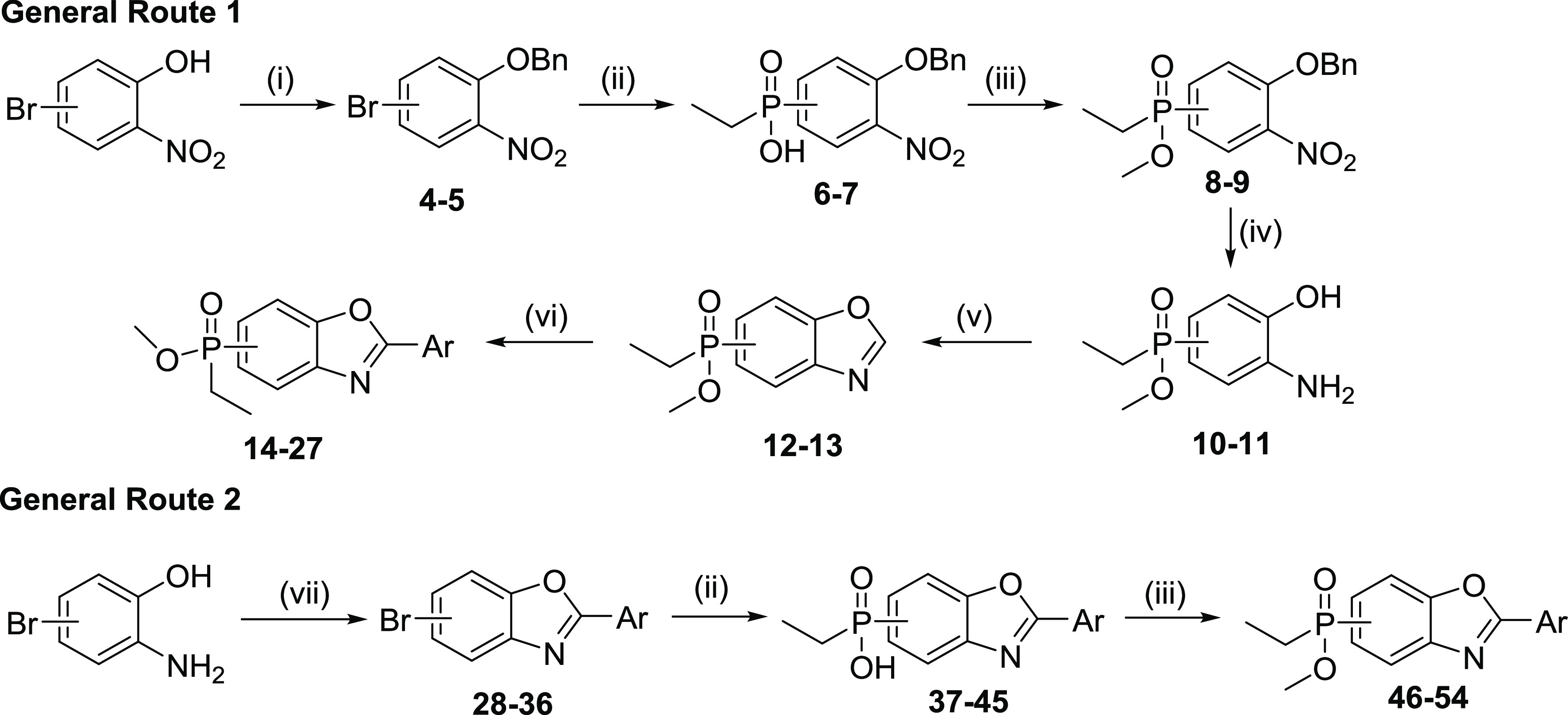
Reagents and conditions: (i) BnBr, K2CO3, acetone, 60 °C, 6 h, (yields 97% and 74% for 4 and 5, respectively); (ii) EtPH(O)OH, Pd(OAc)2, Xantphos, DIPEA, DME/toluene, 90 °C, 2 h, (yields 71–98%); (iii) (1) SOCl2, DMF, 70 °C, 1–2 h; (2) DIPEA, MeOH, DMAP, CH2Cl2, rt, 30 min (yields 41–85%) or MeOH, rt, 15 min for 9 (yield 81%); (iv) 10% Pd/C, EtOH/CH2Cl2, H2, rt, 3 h (yields 94–96%); (v) CH(OEt)3, PPTS, xylenes, 140 °C, 3 h (yields 91–95%); (vi) bromoaryl, Pd(PPh3)2Cl2 or Pd(dppf)Cl2, CuI, Cs2CO3, dioxane, 110 °C, 12 h (yields 7–40%) or bromoaryl, Pd(OAc)2, N-Xantphos, 1 M LiHMDS, DME, 85 °C, 1 h, for 14a, 15 (yields 22 and 12%, respectively) or chloroaryl, In(OTf)3, dioxane, 100 °C, 24 h, (yield 26%); (vii) (1) ArCOCl, xylenes, 155 °C, 1 h; (2) MsOH, 155 °C, 2–3 h (yields 12–81%).
General route 2 was described in detail in our previous work.18 In brief, intermediate 2-arylbromobenzo[d]oxazoles (28–36) were derived from the condensation of 2-aminophenols with the respective acid chloride, followed by acid catalyzed dehydration. The final products were obtained from the bromobenzo[d]oxazoles in a Pd-catalyzed cross-coupling with ethyl phosphinic acid (37–45) and were then converted to the methyl phosphinate esters through formation of the phosphinic chloride and reaction with MeOH (46–54).
Utrophin modulation activity was assessed using a reporter gene assay in the previously reported cell line H2K-mdx utrnA-luc (H2K), a murine myoblast cell line that contains a stably integrated 8.4 kb fragment of the human utroA promoter linked to a firefly luciferase gene.15,16,18 In the reporter gene assay (Table 1), most of the o-substituted analogues appeared to be inactive (18, 23, 50, 54); however, the o-difluoromethyl analogues 14, 16, and 51 were active. Similarly, analogues bearing the phosphinate ester substituent in position 6 were inactive (23–27, 53–54), apart from the o- and p-difluoromethyl analogues (51–52). Finally, pyridyl analogue 49 was also found to be inactive.
Table 1. Structure Activity Relationships (SARs) of C2 (Hetero)aryl Analogues.


Firefly luciferase reporter gene assay reported as EC50 ± SD (μM) of 3 biological replicates (3 technical replicates per experiment).
Firefly luciferase enzymatic assay reported as IC50 ± SD (μM) of 2 biological replicates (3 technical replicates per experiment).
logD is calculated with the shaking flask method at pH = 7.4 from the partition between n-octanol/water.
Reference: Tetrahedron2020, 76, 130819.
IA, inactive.
ND, not determined.
In our previous work,18 confirmation of utrophin upregulation activity with an orthogonal assay was accomplished with labor-intensive Western blots. As we had previously demonstrated that ezutromid is a noncompetitive inhibitor of the firefly luciferase enzyme (Fluc),28 and recognizing the complications Fluc inhibition can cause in the interpretation of reporter gene assays,29 we sought a more facile counter-screen to include in our assay cascade. Quantification of utrophin protein using Western blots was still used for confirmation of utrophin upregulation activity, but the inclusion of a biochemical assay to directly measure inhibition of recombinant Fluc enzyme activity in our screening workflow allowed an early detection of compounds which exhibited interference in the H2K reporter assay.
Interestingly, despite their structural similarity with ezutromid,29 several of the new analogues showed little or no inhibitory activity on Fluc (Table 1), with the exception of methoxy analogues 17 and 47. Two difluoromethyl analogues (14, 15) inhibited Fluc in the low micromolar range, and 19 was even less potent (IC50 of 23.6 μM). The previously reported analogue 3 was found to inhibit Fluc with an IC50 in the nanomolar range, while 2 was less potent with a low micromolar IC50. None of the pyridyl-containing analogues inhibited Fluc. Interestingly, we observed a trend that less lipophilic compounds (logD7.4 < 2.5) showed reduced or no activity in the Fluc assay, while several examples still showed activity in the H2K reporter assay. This finding is consistent with the work of others who have shown that this Fluc has a preference for binding lipophilic molecules.30
Decreasing lipophilicity significantly improved aqueous solubility, as was expected (Table 2). All the newly synthesized analogues were equally or more water-soluble than 2 and 3. The extent of plasma protein binding varied between different analogues. Pyridyl derivatives were found to bind <90%, with the exception of 25. All the new molecules were highly permeable in Caco-2 cells, and more importantly no efflux was noted for any of the new examples ([B-A]/[A-B] < 1). Finally, the metabolic stability of the compounds was found to vary considerably. Analogues 14, 16–18, 50, 51, and 54 were cleared very quickly in mouse hepatocytes. Interestingly, most of these analogues bear electron withdrawing groups in the ortho position, which may suggest a potential opening of the more electrophilic benzoxazole ring in these examples, enabling further CYP oxidation. On the other hand, 20, 21, 25–27, and 49, m-substituted pyridyl analogues, were the most metabolically robust. Apart from o-substituted analogues 50 and 54, all pyridyl analogues had improved metabolic stability compared to their phenyl counterparts.
Table 2. Structure Property Relationships (SPR) of the Synthesized Analogues.
|
Caco-2c |
mHep Stability |
|||||
|---|---|---|---|---|---|---|
| # | Sola | mPPBb | A-B | B-A | Clintd | T1/2e |
| 2 | 468 | 97 | 18.0 | 16.4 | 11.9 | 58 |
| 3 | 378 | 95 | 18.0 | 15.7 | 9.12 | 76 |
| 14 | >250 | 97 | 16.2 | 11.2 | >250 | <3 |
| 15 | >250 | 94 | 14.2 | 10.1 | 34.5 | 20 |
| 46 | 530 | 98 | 8.99 | 77 | ||
| 16 | 518 | 97 | 53.0 | 13 | ||
| 17 | >250 | 96 | 15.7 | 12.6 | >250 | <3 |
| 18 | 366 | 97 | 18.8 | 15.7 | >250 | <3 |
| 19 | 576 | 98 | 12.7 | 11.7 | 4.49 | 154 |
| 20 | 733 | 86 | 22.3 | 20.6 | <2.89 | >240 |
| 21 | 477 | 75 | <2.89 | >240 | ||
| 22 | 440 | 74 | <2.89 | >240 | ||
| 23 | 469 | 97 | 12.3 | 11.5 | 18.2 | 38 |
| 24 | 574 | 98 | 17.2 | 16.2 | 4.49 | 154 |
| 25 | 834 | 93 | 21.8 | 16.9 | <2.89 | >240 |
| 26 | 603 | 80 | <2.89 | >240 | ||
| 27 | 487 | 77 | <2.89 | >240 | ||
| 47 | 265 | 13.3 | 10.7 | 12.4 | 56 | |
| 48 | 13.3 | 9.37 | 54.4 | 13 | ||
| 49 | 12.3 | 11.0 | <2.89 | >240 | ||
| 50 | 854 | 89 | 184 | 4 | ||
| 51 | 446 | 97 | >250 | <3 | ||
| 52 | 556 | 96 | 8.77 | 79 | ||
| 53 | 8.4 | 96 | 18.1 | 15.9 | 19.0 | 37 |
| 54 | 699 | 76 | 142 | 5 | ||
Aqueous kinetic solubility (μM).
mPPB, murine plasma protein binding (% bound).
Caco-2 permeability assay (Papp (10–6 cm/s)).
Murine hepatocyte intrinsic clearance (μL/min/106 cells).
Murine hepatocyte half-life (min).
Confirmation of utrophin modulation activity for representative analogues (20, 21, and 52) was determined using Western blot to quantify the increase of the utrophin protein levels in dystrophic mouse muscle cells (LUmdx), as before.18 Pyridyl analogues 20 and 21 were selected because of their combination of potency in the H2K assay, lack of Fluc inhibition, and optimal ADME properties. Analogue 52 was selected as a representative haloaryl derivative, since it had a well-balanced overall profile. All three analogues were found to increase utrophin by approximately 1.3–1.6-fold (Figure 2). The modest increase in utrophin levels in combination with the variability and low sensitivity of this assay are likely responsible for the apparent lack of a dose–response; however, these results demonstrate utrophin modulation in a cellular context, confirming the activity in this series of new analogues.
Figure 2.

Treatment with 20, 21, and 52 increases utrophin protein in LUmdx cells. *p < 0.05, **p < 0.01, ***p < 0.001. pos ctrl = positive control.
In an attempt to visualize the move from a suboptimal physicochemical property space, we plotted selected physicochemical properties of the previously synthesized phosphinate ester analogues against the new ones (Figure S1, Table S1). At a glance, it was observed that the optimal chemical space proposed by Gleeson (Figure S1a, gray area defined with clogP < 4 and MW < 400)31 is mostly occupied by new analogues. These “not large/not greasy” molecules were found to have the best drug developability profiles according to a separate analysis by Ritchie et al.32 Considering the more polar nature of some of the analogues, we went on to experimentally measure the logD of selected analogues and observed that indeed they move almost entirely into the desired area (Figure S1b).
The discovery that 2 was hepatotoxic during the maximum tolerated dose study in rats,18 and the observation that 2 and related compounds showed cytotoxicity in vitro in liver cells, led us to probe the new analogues for their cytotoxic potential in HepG2 cells (Table 3) as an early indication of hepatotoxicity. Cytotoxicity was determined by quantification of cellular ATP levels, using an evolved luciferase known to have substantially reduced promiscuity.33 Increased lipophilicity has been known to contribute to compound promiscuity and further toxicological outcomes.19,20,34,35 From the results in Table 3 it was apparent that the most toxic compounds are those with high lipophilicity and low TPSA, in good agreement with other literature reports.19
Table 3. Cytotoxicity of Selected Compounds in HepG2 Cells.
Next, we sought to quantify the relationship between HepG2 cytotoxicity with key physicochemical properties. As our sample size described herein was rather small, we also expanded this analysis to include all synthesized compounds in this series (Table S2, Figures 3 and S2).
Figure 3.
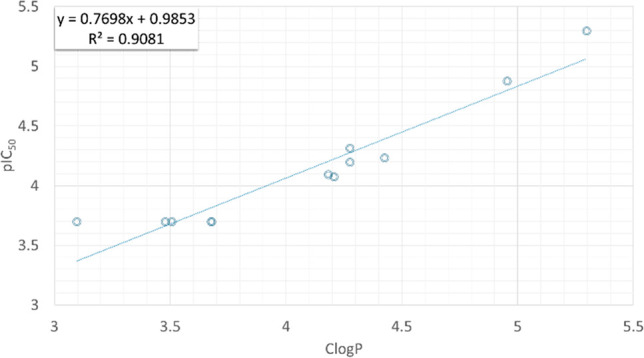
Correlation between cytotoxicity in HepG2 cells and lipophilicity as expressed from their pIC50 and clogP plot after the removal of two outliers (n = 12). Predicted clogP (a) was generated with OSIRIS Datawarrior version 5.0.0 © 2002–2019 Idorsia Pharmaceuticals Ltd.
Plotting pIC50 against TPSA did not give a statistically significant correlation (data not shown); however, pIC50 against ClogP derived equation S1 (n = 14, R2 = 0.46, Figure S2a). Examining the graph, we observed two compounds that diverted from the group and considered removing them as potential outliers. Indeed, after removal of the two, the correlation between clogP and cytotoxicity appeared to improve as shown in equation S2 (n = 12, R2 = 0.91, Figure 3).
The aberrant behavior of the two outliers may partly be explained by gaps in the ability of software to predict clogP successfully. Analogue 24 was predicted to be the most lipophilic compound with Datawarrior; however, it was almost one log unit less polar by the two other calculators (Table S3). Similarly, analogue 55 was significantly more lipophilic when other calculators were used. However, this analysis does not rule out the participation of other mechanisms contributing to the significantly higher cytotoxicity of this compound in HepG2 cells. Importantly utrophin modulation activity in H2K cells did not increase with clogP; plotting the ratio of EC50 in H2K cells and the IC50 in HepG2 cells showed only a weak but negative correlation (Table S2, Figure S2).
Aiming to improve the ADMET profile of existing benzo[d]oxazole phosphinate utrophin modulators, we implemented a strategy of reducing lipophilicity in the design of new analogues. A facile synthetic route was devised that allows faster access to analogues involving C–H activation of the intermediate benzoxazole phosphinate ester. The new analogues were shown to be in a more optimal chemical space and have better drug developability potential. The decrease in lipophilicity gave inactive or only weakly active analogues as Fluc inhibitors, while maintaining utrophin modulatory activity. We were able to show that cytotoxicity in HepG2 cells is directly proportional to lipophilicity and that reducing lipophilicity led to analogues that were only mildly or not cytotoxic at all in HepG2 cells.
Acknowledgments
The authors kindly acknowledge Evotec (UK) for the physicochemical and ADME characterization and Evotec (France) for the biological evaluation. We further acknowledge G.E. Morris (Oswestry, UK) for the MANCHO3 antibody.
Glossary
Abbreviations
- ADMET
administration distribution metabolism excretion toxicity
- CYP
cytochrome P450; DAPC, dystrophin associated complex
- DIPEA
N,N-diisopropylethylamine
- DMAP
4-dimethylaminopyridine
- DMD
Duchenne muscular dystrophy
- DME
dimethoxyethane
- dppf
1,1′-bis(diphenylphosphino)ferrocene
- Fluc
firefly luciferase
- H2K
reporter cell line H2K-mdx utrnA-luc
- HMDS
hexamethyldisilazane
- LUmdx
dystrophic mouse cells
- PPB
plasma protein binding
- PPTS
pyridinium p-toluenesulfonate
- SAR
structure–activity relationship
- SPR
structure–property relationship
- TPSA
topological polar surface area.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.0c00405.
Experimental details describing the biological assays and the synthesis and characterization of compounds. Computational and experimental calculations of physicochemical properties, graphs and equations. NMR and mass spectroscopy spectra of the synthesized compounds. (PDF)
Author Contributions
M.C. and A.J.R. collated the data and wrote the manuscript. All authors have given approval to the final version of the manuscript.
The authors wish to thank Summit Therapeutics plc (M.C., E.E., J.A.R, L.M.) and the Medical Research Council (1501AV003/CA2, K.E.D., S.E.S.) for financial support.
The authors declare the following competing financial interest(s): S.H. and F.X.W. are or were Summit Therapeutics plc employees or consultants at the time that the work was conducted. K.E.D., S.G.D. and A.J.R. are minor shareholders of Summit Therapeutics plc.
Supplementary Material
References
- Wynne G. M.; Russell A. J.. Drug discovery approaches for rare neuromuscular diseases. Orphan Drugs and Rare Diseases; The Royal Society of Chemistry: 2014; pp 257–343. [Google Scholar]
- Guiraud S.; Davies K. E. Pharmacological advances for treatment in Duchenne muscular dystrophy. Curr. Opin. Pharmacol. 2017, 34, 36–48. 10.1016/j.coph.2017.04.002. [DOI] [PubMed] [Google Scholar]
- Guiraud S.; Roblin D.; Kay D. E. The potential of utrophin modulators for the treatment of Duchenne muscular dystrophy. Expert Opin. Orphan Drugs 2018, 6 (3), 179–192. 10.1080/21678707.2018.1438261. [DOI] [Google Scholar]
- Monaco A. P.; Neve R. L.; Colletti-Feener C.; Bertelson C. J.; Kurnit D. M.; Kunkel L. M. Isolation of candidate cDNAs for portions of the Duchenne muscular dystrophy gene. Nature 1986, 323 (6089), 646–50. 10.1038/323646a0. [DOI] [PubMed] [Google Scholar]
- Deconinck N.; Tinsley J.; De Backer F.; Fisher R.; Kahn D.; Phelps S.; Davies K.; Gillis J. M. Expression of truncated utrophin leads to major functional improvements in dystrophin-deficient muscles of mice. Nat. Med. 1997, 3 (11), 1216–21. 10.1038/nm1197-1216. [DOI] [PubMed] [Google Scholar]
- Tinsley J.; Deconinck N.; Fisher R.; Kahn D.; Phelps S.; Gillis J. M.; Davies K. Expression of full-length utrophin prevents muscular dystrophy in mdx mice. Nat. Med. 1998, 4 (12), 1441–4. 10.1038/4033. [DOI] [PubMed] [Google Scholar]
- Cerletti M.; Negri T.; Cozzi F.; Colpo R.; Andreetta F.; Croci D.; Davies K. E.; Cornelio F.; Pozza O.; Karpati G.; Gilbert R.; Mora M. Dystrophic phenotype of canine X-linked muscular dystrophy is mitigated by adenovirus-mediated utrophin gene transfer. Gene Ther. 2003, 10 (9), 750–757. 10.1038/sj.gt.3301941. [DOI] [PubMed] [Google Scholar]
- Wynne G. M.; Wren S. P.; Johnson P. D.; Price D.; De Moor O.; Nugent G.; Tinsley J. M.; Storer R.; Mulvaney A.; Pye R. J.; Dorgan C. R.. Preparation of benzimidazoles, benzoxazoles, benzothiazoles, indoles and their analogs for the treatment of muscular dystrophy and cachexia. WO2007091106A2, 2007.
- Chancellor D. R.; Davies K. E.; De Moor O.; Dorgan C. R.; Johnson P. D.; Lambert A. G.; Lawrence D.; Lecci C.; Maillol C.; Middleton P. J.; Nugent G.; Poignant S. D.; Potter A. C.; Price P. D.; Pye R. J.; Storer R.; Tinsley J. M.; van Well R.; Vickers R.; Vile J.; Wilkes F. J.; Wilson F. X.; Wren S. P.; Wynne G. M. Discovery of 2-arylbenzoxazoles as upregulators of utrophin production for the treatment of Duchenne muscular dystrophy. J. Med. Chem. 2011, 54 (9), 3241–50. 10.1021/jm200135z. [DOI] [PubMed] [Google Scholar]
- Tinsley J. M.; Fairclough R. J.; Storer R.; Wilkes F. J.; Potter A. C.; Squire S. E.; Powell D. S.; Cozzoli A.; Capogrosso R. F.; Lambert A.; Wilson F. X.; Wren S. P.; De Luca A.; Davies K. E. Daily treatment with SMTC1100, a novel small molecule utrophin upregulator, dramatically reduces the dystrophic symptoms in the mdx mouse. PLoS One 2011, 6 (5), e19189 10.1371/journal.pone.0019189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tinsley J.; Robinson N.; Davies K. E. Safety, tolerability, and pharmacokinetics of SMT C1100, a 2-arylbenzoxazole utrophin modulator, following single- and multiple-dose administration to healthy male adult volunteers. J. Clin. Pharmacol. 2015, 55 (6), 698–707. 10.1002/jcph.468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricotti V.; Spinty S.; Roper H.; Hughes I.; Tejura B.; Robinson N.; Layton G.; Davies K.; Muntoni F.; Tinsley J. Safety, tolerability, and pharmacokinetics of SMT C1100, a 2-arylbenzoxazole utrophin modulator, following single- and multiple-dose administration to pediatric patients with Duchenne muscular dystrophy. PLoS One 2016, 11 (4), e0152840 10.1371/journal.pone.0152840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muntoni F.; Tejura B.; Spinty S.; Roper H.; Hughes I.; Layton G.; Davies K. E.; Harriman S.; Tinsley J. A phase 1b trial to assess the pharmacokinetics of ezutromid in pediatric Duchenne muscular dystrophy patients on a balanced diet. Clin. Pharmacol. Drug Dev. 2019, 8 (7), 922–933. 10.1002/cpdd.642. [DOI] [PubMed] [Google Scholar]
- https://musculardystrophynews.com/2018/06/29/summit-therapeutics-ends-ezutromid-development-trail-failure/; accessed 2020-08-31.
- Chatzopoulou M.; Claridge T. D. W.; Davies K. E.; Davies S. G.; Elsey D. J.; Emer E.; Fletcher A. M.; Harriman S.; Robinson N.; Rowley J. A.; Russell A. J.; Tinsley J. M.; Weaver R.; Wilkinson I. V. L.; Willis N. J.; Wilson F. X.; Wynne G. M. Isolation, structural identification, synthesis, and pharmacological profiling of 1,2-trans-dihydro-1,2-diol metabolites of the utrophin modulator ezutromid. J. Med. Chem. 2020, 63 (5), 2547–2556. 10.1021/acs.jmedchem.9b01547. [DOI] [PubMed] [Google Scholar]
- Guiraud S.; Squire S. E.; Edwards B.; Chen H.; Burns D. T.; Shah N.; Babbs A.; Davies S. G.; Wynne G. M.; Russell A. J.; Elsey D.; Wilson F. X.; Tinsley J. M.; Davies K. E. Second-generation compound for the modulation of utrophin in the therapy of DMD. Hum. Mol. Genet. 2015, 24 (15), 4212–24. 10.1093/hmg/ddv154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babbs A.; Berg A.; Chatzopoulou M.; Davies K. E.; Davies S. G.; Edwards B.; Elsey D. J.; Emer E.; Figuccia A. L. A.; Fletcher A. M.; Guiraud S.; Harriman S.; Moir L.; Robinson N.; Rowley J. A.; Russell A. J.; Squire S. E.; Thomson J. E.; Tinsley J. M.; Wilson F. X.; Wynne G. M. Synthesis of SMT022357 enantiomers and in vivo evaluation in a Duchenne muscular dystrophy mouse model. Tetrahedron 2020, 76 (2), 130819. 10.1016/j.tet.2019.130819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babbs A.; Berg A.; Chatzopoulou M.; Davies K. E.; Davies S. G.; Edwards B.; Elsey D.; Emer E.; Guiraud S.; Harriman S.; Lecci C.; Moir L.; Peters D.; Robinson N.; Rowley J.; Russell A. J.; Squire S.; Tinsley J.; Wilson F.; Wynne G. M. 2-Arylbenzo[d]oxazole phosphinate esters as second-generation modulators of utrophin for the treatment of Duchenne muscular dystrophy. J. Med. Chem. 2020, 63, 7880. 10.1021/acs.jmedchem.0c00807. [DOI] [PubMed] [Google Scholar]
- Meanwell N. A. Improving drug candidates by design: a focus on physicochemical properties as a means of improving compound disposition and safety. Chem. Res. Toxicol. 2011, 24 (9), 1420–1456. 10.1021/tx200211v. [DOI] [PubMed] [Google Scholar]
- Tarcsay A.; Keseru G. M. Contributions of molecular properties to drug promiscuity. J. Med. Chem. 2013, 56 (5), 1789–1795. 10.1021/jm301514n. [DOI] [PubMed] [Google Scholar]
- Fratello G.; Marchini S.; Zucco F.; Sapora O.; Stammati A. Cytotoxicity of halogenated benzenes and its relationship with logP. Toxicol. In Vitro 1997, 11 (5), 673–677. 10.1016/S0887-2333(97)00086-6. [DOI] [PubMed] [Google Scholar]
- Moridani M. Y.; Siraki A.; O’Brien P. J. Quantitative structure toxicity relationships for phenols in isolated rat hepatocytes. Chem.-Biol. Interact. 2003, 145 (2), 213–223. 10.1016/S0009-2797(02)00258-2. [DOI] [PubMed] [Google Scholar]
- Moridani M. Y.; Siraki A.; Chevaldina T.; Scobie H.; O’Brien P. J. Quantitative structure toxicity relationships for catechols in isolated rat hepatocytes. Chem.-Biol. Interact. 2004, 147 (3), 297–307. 10.1016/j.cbi.2004.02.001. [DOI] [PubMed] [Google Scholar]
- Bartalis J.; Halaweish F. T. Relationship between cucurbitacins reversed-phase high-performance liquid chromatography hydrophobicity index and basal cytotoxicity on HepG2 cells. J. Chromatogr. B: Anal. Technol. Biomed. Life Sci. 2005, 818 (2), 159–166. 10.1016/j.jchromb.2004.12.020. [DOI] [PubMed] [Google Scholar]
- Chan K.; Jensen N. S.; Silber P. M.; O’Brien P. J. Structure–activity relationships for halobenzene induced cytotoxicity in rat and human hepatoctyes. Chem.-Biol. Interact. 2007, 165 (3), 165–174. 10.1016/j.cbi.2006.12.004. [DOI] [PubMed] [Google Scholar]
- Chan R.; Benet L. Z. Evaluation of the relevance of DILI predictive hypotheses in early drug development: review of in vitro methodologies vs. BDDCS classification. Toxicol. Res. 2018, 7 (3), 358–370. 10.1039/C8TX00016F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smart B. E. Fluorine substituent effects (on bioactivity). J. Fluorine Chem. 2001, 109 (1), 3–11. 10.1016/S0022-1139(01)00375-X. [DOI] [Google Scholar]
- Wilkinson I. V. L.; Reynolds J. K.; Galan S. R. G.; Vuorinen A.; Sills A. J.; Pires E.; Wynne G. M.; Wilson F. X.; Russell A. J.. Characterisation of utrophin modulator SMT C1100 as a non-competitive inhibitor of firefly luciferase. Bioorg. Chem. 2020, 94, 103395. 10.1016/j.bioorg.2019.103395. [DOI] [PubMed] [Google Scholar]
- Thorne N.; Shen M.; Lea W. A.; Simeonov A.; Lovell S.; Auld D. S.; Inglese J. Firefly luciferase in chemical biology: a compendium of inhibitors, mechanistic evaluation of chemotypes, and suggested use as a reporter. Chem. Biol. 2012, 19 (8), 1060–1072. 10.1016/j.chembiol.2012.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auld D. S.; Southall N. T.; Jadhav A.; Johnson R. L.; Diller D. J.; Simeonov A.; Austin C. P.; Inglese J. Characterization of chemical libraries for luciferase inhibitory activity. J. Med. Chem. 2008, 51 (8), 2372–2386. 10.1021/jm701302v. [DOI] [PubMed] [Google Scholar]
- Gleeson M. P. Generation of a set of simple, interpretable ADMET rules of thumb. J. Med. Chem. 2008, 51 (4), 817–834. 10.1021/jm701122q. [DOI] [PubMed] [Google Scholar]
- Ritchie T. J.; Macdonald S. J. F.; Peace S.; Pickett S. D.; Luscombe C. N. Increasing small molecule drug developability in sub-optimal chemical space. MedChemComm 2013, 4 (4), 673–680. 10.1039/c3md00003f. [DOI] [Google Scholar]
- Auld D. S.; Zhang Y.-Q.; Southall N. T.; Rai G.; Landsman M.; MacLure J.; Langevin D.; Thomas C. J.; Austin C. P.; Inglese J. A basis for reduced chemical library inhibition of firefly luciferase obtained from directed evolution. J. Med. Chem. 2009, 52 (5), 1450–1458. 10.1021/jm8014525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu S. Y.; Jessen B.; Strock C.; Will Y. The contribution of physicochemical properties to multiple in vitro cytotoxicity endpoints. Toxicol. In Vitro 2012, 26 (4), 613–620. 10.1016/j.tiv.2012.01.025. [DOI] [PubMed] [Google Scholar]
- Hughes J. D.; Blagg J.; Price D. A.; Bailey S.; DeCrescenzo G. A.; Devraj R. V.; Ellsworth E.; Fobian Y. M.; Gibbs M. E.; Gilles R. W.; Greene N.; Huang E.; Krieger-Burke T.; Loesel J.; Wager T.; Whiteley L.; Zhang Y. Physiochemical drug properties associated with in vivo toxicological outcomes. Bioorg. Med. Chem. Lett. 2008, 18 (17), 4872–4875. 10.1016/j.bmcl.2008.07.071. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.