

Publisher's Note: There is a Blood Commentary on this article in this issue.
Key Points
CD27/CD70 deficiencies are IEIs characterized by EBV-associated immune dysregulation including HLH, lymphoproliferation, and malignancy.
The excellent outcome following HSCT in cases with CD27/CD70 deficiency supports its timely use particularly in patients with lymphoma.
Abstract
Biallelic mutations in the genes encoding CD27 or its ligand CD70 underlie inborn errors of immunity (IEIs) characterized predominantly by Epstein-Barr virus (EBV)-associated immune dysregulation, such as chronic viremia, severe infectious mononucleosis, hemophagocytic lymphohistiocytosis (HLH), lymphoproliferation, and malignancy. A comprehensive understanding of the natural history, immune characteristics, and transplant outcomes has remained elusive. Here, in a multi-institutional global collaboration, we collected the clinical information of 49 patients from 29 families (CD27, n = 33; CD70, n = 16), including 24 previously unreported individuals and identified a total of 16 distinct mutations in CD27, and 8 in CD70, respectively. The majority of patients (90%) were EBV+ at diagnosis, but only ∼30% presented with infectious mononucleosis. Lymphoproliferation and lymphoma were the main clinical manifestations (70% and 43%, respectively), and 9 of the CD27-deficient patients developed HLH. Twenty-one patients (43%) developed autoinflammatory features including uveitis, arthritis, and periodic fever. Detailed immunological characterization revealed aberrant generation of memory B and T cells, including a paucity of EBV-specific T cells, and impaired effector function of CD8+ T cells, thereby providing mechanistic insight into cellular defects underpinning the clinical features of disrupted CD27/CD70 signaling. Nineteen patients underwent allogeneic hematopoietic stem cell transplantation (HSCT) prior to adulthood predominantly because of lymphoma, with 95% survival without disease recurrence. Our data highlight the marked predisposition to lymphoma of both CD27- and CD70-deficient patients. The excellent outcome after HSCT supports the timely implementation of this treatment modality particularly in patients presenting with malignant transformation to lymphoma.
Visual Abstract

Introduction
Epstein-Barr virus (EBV) is 1 of 9 human herpesviruses and infects up to 90% of the adult population.1 Upon primary exposure, EBV infects oropharyngeal epithelial cells and B cells, but acquires latency and persists predominantly in B cells.1-3 Host defense against EBV is largely mediated by CD8+ T cells and natural killer (NK) cells.2,4-6 In immunocompetent hosts, EBV exposure during early childhood is often asymptomatic, but causes infectious mononucleosis (IM) in ∼25% of infected adolescents.2,3 In contrast, EBV infection is associated with significant morbidity and mortality in immunocompromised individuals. Thus, when the host-virus balance is disrupted, a wide range of EBV-associated immunopathologic conditions may arise, including lymphoproliferative disorders (LPDs), hemophagocytic lymphohistiocytosis (HLH), and chronic active EBV infection (CAEBV). Severe EBV manifestations commonly occur in acquired T-cell immunodeficiencies, such as HIV infection or iatrogenic immunosuppression following organ or hematopoietic stem cell transplantation (HSCT).3,4,7,8 Importantly, recent discoveries of single-gene defects presenting as severe and often fatal EBV-induced disease have defined cellular networks essential for controlling acute EBV infection. For instance, patients may be highly vulnerable to the pathogenic consequences of EBV infection due to germline-inherited loss-of-function mutations in SH2D1A, XIAP, ITK, MAGT1, CTPS1, CORO1A, RASGRP1, STK4, CARMIL2, TNFRSF9, CD27, or CD70.5,6,9-14
CD27 is a member of the tumor necrosis factor (TNF) receptor superfamily expressed on a broad range of human lymphocytes including naive and central memory T (TCM) cells, germinal center and memory B cells, plasma cells, and some NK cells.15 By contrast, expression of its unique ligand CD70 is restricted to activated lymphoid and myeloid cells.15 CD27 engagement by CD70 provides costimulatory signals that enhance T-cell activation, survival, proliferation, and differentiation.15 In vivo studies have established that CD27 enhances generation and maintenance of antigen-specific CD4+ and CD8+ T cells. Furthermore, deletion of Cd27 in mice compromises cytotoxic CD8+ T-cell responses to viral and bacterial infections.15-19 CD27 also has a role on human B cells, promoting memory cell differentiation and plasma cell survival.20 The nonredundant role of the CD27-CD70 axis in humans has been revealed by the discovery of individuals with biallelic mutations in CD2721-25 or CD70,24,26,27 who typically present with chronic EBV viremia, severe EBV-induced HLH, EBV-associated LPD, Hodgkin (HL) and non-Hodgkin lymphoma (NHL), and/or hypogammaglobulinemia.21-27 Patients also suffer from recurrent bacterial and other viral infections, underlining a role for CD27-CD70 interactions in host defense beyond EBV.5,6,21-24,26,27
Due to the limited numbers of CD27- and CD70-deficient patients reported to date,21-27 no consensus on treatment strategies has been defined. Here, we report the largest cohort of genetically defined CD27- and CD70-deficient patients providing unprecedented insights into the immunological characteristics, mechanisms of disease pathogenesis, and clinical course of individual patients undergoing various treatments, including HSCT.
Methods
Patients and diagnosis
We performed a retrospective analysis of CD27- and CD70-deficient patients. Centers with patients were identified through the European Society for Immunodeficiencies (ESID)/European Society for Blood and Marrow Transplantation (EBMT) registries, published case reports, and communication with defined expert clinicians working in the field. Questionnaires regarding patient demographics, clinical and laboratory features, transplant characteristics, and outcome were distributed. Analysis was performed using data collected for 49 patients from 20 centers worldwide. Diagnosis of CD27 or CD70 deficiency was made based on molecular genetics, and flow cytometry or western blot in selected cases. Patients and families provided written informed consent in accordance with the Declaration of Helsinki. Study approval was granted by the institutional review boards, including the ethics committees of University of Duesseldorf (Duesseldorf, Germany; 3208), Royal Prince Alfred Hospital (Camperdown, Australia; X16-0210/LNR/16/RPAH/257), Royal Children’s Hospital (Melbourne, VIC, Australia; 33146A), and Medical University of Vienna, Austria (1796/2018), and the National Institute of Allergy and Infectious Diseases (NIAID) Institutional Review Board. Data from 25 patients (P1-17, P26, P31, P34-39) have been previously published,21-27 with additional information including longer-term follow-up collected for this study. A case report on patient P42 has been submitted in parallel to this manuscript.28
Lymphocyte phenotyping and function
Peripheral blood was collected from healthy blood donors and patients with CD27 or CD70 mutations (Tables 1 and 2).21-23 All samples were run at a single site (Garvan Institute of Medical Research, Sydney, NSW, Australia); shipping controls, heterozygous healthy family members and numerous age-matched controls were also included in the analysis. Proportions of CD3+, CD4+ T (CD3+CD4+), CD8+ T (CD3+CD8+), B cells (CD20+); naive (CD45RA+CCR7+), central memory (TCM; CD45RA−CCR7+), effector memory (TEM; CD45RA−CCR7−), CD45RA+ revertant memory (TEMRA; CD45RA+CCR7−) cells; αβ (CD3+TCRαβ+) and γδ (CD3+TCRγδ+) T cells; mucosal-associated invariant T (MAIT; CD3+TCRVα7.2+CD161+), NK (CD3−CD56+) and invariant NKT (iNKT) cells (CD3+TCRVα24+Vβ11+); transitional (CD20+CD10+CD27−), naive (CD20+CD10-CD27-), and memory (CD20+CD10-CD27+) B-cell subsets were determined by flow cytometry.26,29-32 EBV- and CMV-specific CD8+ T cells were detected using major histocompatibility complex (MHC) class I-specific tetramers.26,29 For in-depth phenotyping, peripheral blood mononuclear cells (PBMCs) were further stained with monoclonal antibodies (mAbs) against specific cell surface and intracellular molecules.26,29,30,32 Data were acquired on an LSRII SORP or LSR Fortessa (both from Becton Dickinson) and analyzed using FlowJo (Tree Star). PBMCs or sorted CD8+ T-cell subsets were isolated and cultured in vitro under various conditions. Proliferation, cytokine expression and secretion, apoptosis, and expression of FASLG were then determined.26,29,30
Table 1.
Clinical features and genetic variants of patients with CD27 deficiency
| Age at onset* | Age at diagnosis* | Sex | Ethnicity | Consanguinity† | Mutation NM_001242.5 | Infections | EBV-related symptoms | Max EBV load‡ | MALIGNANCY | Other symptoms | Treatment | Outcome | Center | Ref | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| P1 | 2 | 21 | M | Moroccan | + | c.G24A, p.W8* | No | IM | 3 000 | No | No | IgRT | Alive | Utrecht | 21, 23 |
| P2 | 3 | PM | M | Moroccan | + | c.G24A, p.W8* | Sepsis due to cytopenia | IM, possibly HLH (previously reported as aplastic anemia) | 2 050 | No | Uveitis | No | Died | Utrecht | 21, 23 |
| P3 | 1 | 1 | F | Turkish | + | c.G158A, p.C53Y | LRTI, sepsis, phlegmons | HLH, LPD | 2 000 000 | No | No | IgRT, RTX | Alive | Vienna | 22, 23 |
| P4 | NA | 12 | M | Turkish | + | c.G158A, p.C53Y | No | No | 0 | No | No | No | Alive | Vienna | 22, 23 |
| P5 | 1 | 0 | F | Turkish | + | c.G158A, p.C53Y | Phlegmons | IM | 4 400 | No | Arthritis | IgRT | Alive | Vienna | 22, 23 |
| P6 | 1 | 4 | M | Lebanese | − | c.G158A, p.C53Y | Recurrent infections | LPD, HLH | 5 000 000 | No | Oral ulcers | HLH2004, RTX, HSCT | Alive | Melbourne | 22, 23 |
| P7 | 1 | 1 | F | Lebanese | − | c.G158A, p.C53Y | URTI | Meningitis | 8 000 000 | No | Oral ulcers, uveitis | RTX, HSCT | Alive | Melbourne | 22, 23 |
| P8 | 15 | 19 | M | Lebanese | + | c.G158A, p.C53Y | No | CAEBV, LPD | 25 000 000 | No | Oral ulcers, uveitis | RTX, chemo, HSCT | Alive | Melbourne | 22, 23 |
| P9 | 2 | PM | F | Lebanese | + | c.G158A, p.C53Y | No | LPD | NA | DLBCL (EBV+) | Oral ulcers | chemo | Died | Melbourne | 22, 23 |
| P10 | 22 | PM | F | Lebanese | + | c.G158A, p.C53Y | No | CAEBV, LPD | 270 000 | No | No | Steroids | Died | Melbourne | 22, 23 |
| P11 | 4 | 9 | F | German | − | het c.C30A/ p.C10* | No | LPD, HLH | 1 000 000 | No | No | IgRT, HLH2004, RTX | Alive | Krefeld | 23 |
| P12 | 13 | 17 | F | German | − | c.G24A/ c.C319T p.W8*/ p.R107C | Ulcers | CAEBV | 320 000 | mc-HL | Oral ulcers, uveitis | IgRT, chemo | Alive | Krefeld | 23 |
| P13 | 7 | 7 | F | Turkish | + | c.G287A, p.C96Y | LRTI | LPD, HLH | 2 200 000 | No | No | RTX, HLH2004, HSCT | Alive | Leiden/ Rotterdam | 23 |
| P14 | 6 | 13 | F | Turkish | + | c.G287A, p.C96Y | No | IM | 0 | No | No | No | Alive | Leiden/ Rotterdam | 23 |
| P15 | 6 | PM | F | Iranian | + | c.G287A, p.C96Y | RTI, skin abscesses bronchiectasis, toxoplasmosis | IM | 3 700 | ns-HL | Recurrent fever, eczema | IgRT, chemo | Died | Tehran | 23 |
| P16 | 8 | 32 | M | Iranian | + | c.G287A, p.C96Y | URTI | IM | 930 | ns-HL | No | chemo | Alive | Tehran | 23 |
| P17 | 8 | PM | F | Iranian | + | c.C232T, p.R78W | Recurrent infections | IM, EBV pneumonia | 930 | No | Recurrent fever, stomatitis | No | Died | Tehran | 23 |
| P18 | 2 | 2 | F | Syrian | + | c.266_267del, p.S89Wfs*14 | No | HLH, LPD | 259 000 | DLBCL | Recurrent fever | RTX, IgRT, chemo, HSCT | Alive | Duesseldorf | 23 |
| P19 | NA | 2 | M | Syrian | + | c.266_267del, p.S89Wfs*14 | No | No | 626 | No | No | No | Alive | Duesseldorf | |
| P20 | 15 | 20 | F | Iraqi | NA | c.G158A, p.C53Y | No | Viremia | 12 00 | ns-HL | No | chemo, IgRT | Alive | Duesseldorf/Essen | |
| P21 | 13 | 13 | M | Iraqi | NA | c.G158A, p.C53Y | No | Viremia | 32 195 | mc-HL | No | chemo, IgRT | Alive | Essen | |
| P22 | NA | 30 | M | Bangladeshi | NA | c.C280T, p.R94C | No | No | 45 031 | No | Asthma, eczema | No | Alive | London | |
| P23 | NA | 8 | M | Bangladeshi | NA | c.C280T, p.R94C | No | Viremia | 16 652 | No | No | No | Alive | London | |
| P24 | 8 mo | 7 | M | Bangladeshi | NA | c.C280T, p.R94C | URTI | EBV encephalitis | 2 000 000 | No | Vasculitis, arthritis, | RTX, IgRT | Alive | London | |
| P25 | 1 | 3 | F | Turkish | + | c.G137A, p.G46Q | No | Viremia, LPD | 665 000 | No | No | HSCT | Alive | Kayseri | |
| P26 | 14 | 14 | M | English | − | c.251_252insT, p.C71fs*44 | No | Viremia | 45 000 000 | HL, DLBCL | Recurrent tonsillitis | HSCT | Alive | London | 33 |
| P27 | 5 | 5 | F | Tunisian | − | c.G329A, p.W110* | LRTI, skin infections | Viremia, LPD | 1 000 000 | No | Arthritis, uveitis | MTX, HSCT | Alive | Paris | |
| P28 | 5 | 7 | M | Iranian | + | c.T94C, p.Y32H | LRTI | IM, LPD, HLH | 7 250 | No | Recurrent fever, uveitis, arthritis | IgRT, RTX | Alive | Tehran | |
| P29 | 4 | 5 | F | Iranian | + | c.T94C, p.Y32H | LRTI | IM, LPD, HLH | 8 220 | No | Recurrent fever | IgRT, RTX | Alive | Tehran | |
| P30 | 3 | 4 | M | Turkish | + | c.G98A, p.W33* | No | No | 0 | HL | No | HSCT | Alive | Kayseri | |
| P31 | 3 | 10 | F | Indian | − | c.A95G, p.Y32C | LRTI | LPD | 474 188 | B-NHL | Oral ulcers, IBD | RTX, steroids | Died | Delhi | 25 |
| P32 | 8 | 8 | F | Turkish | + | c.18 del, p.W7G*44 | No | No | 85 | mc-HL | No | R-ABVD, HSCT | Alive | Ankara | |
| P33 | 3 | 3 | F | Turkish | + | c.18 del, p.W7G*44 | No | No | 2 500 | ns-HL, Burkitt | No | RTX, HSCT | Alive | Ankara |
B-NHL, B-cell non-Hodgkin lymphoma; chemo, chemotherapy; DLBCL, diffuse large B-cell lymphoma; F, female; IBD, inflammatory bowel disease; IgRT, immunoglobulin replacement therapy; LRTI, lower respiratory tract infection; M, male; Max, maximum; mc-HL, mixed-cellularity Hodgkin lymphoma; MTX, methotrexate; NA, not applicable; ns-HL, nodular sclerosis Hodgkin lymphoma; PM, postmortem; R-ABVD, rituximab with doxorubicin, bleomycin, vinblastine, and dacarbazine; Ref., reference for previous publications; RTI, respiratory tract infection; RTX, rituximab; URTI, upper respiratory tract infection.
Age in years unless stated otherwise. Please note that the asterisks used in the “Age” column headings have no relation to those used in the “Mutation” column.
+indicates positive; −, negative.
PCR copy numbers
Table 2.
Clinical features and genetic variants of patients with CD70 deficiency
| Age at onset, y* | Age at diagnosis, y* | Sex | Ethnicity | Consanguinity | Mutation NM_001242.5 | Bacterial infections | EBV-related symptoms | Max EBV load | Malignancy | Other symptoms | Treatment | Outcome | Center | Ref. | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| P34 | 3 | 7 | M | Egyptian | + | c.C535T, p.R179* | No | LPD | NA | ns-HL | Recurrent fever | HSCT | Alive | Paris | 24 |
| P35 | 5 | 9 | F | Iranian | + | c.250delT, p.S84Pfs27* | LRTI | IM | 6 910 | ns-HL | Behcet-like disease | No | Alive | Tehran | 26 |
| P36 | 0 | 36 | M | Iranian | + | c.250delT, p.S84Pfs27* | Sinusitis | Encephalitis | 322 | No | No | No | Alive | Tehran | 26 |
| P37 | 3 | 16 | M | Turkish | + | c.555_557del, p.F186del* | Otitis, diarrhea | Cervical LAP | 5 620 | mc-HL | Recurrent fever, chronic diarrhea | HSCT | Alive | Ankara | 26 |
| P38 | 2 | 8 | M | Turkish | + | c.555_557del, p.F186del* | URTI | Cervical LAP | NA | mc-HL | No | HSCT | Alive | Ankara | 26 |
| P39 | 1 | 6 | M | Caucasian | + | c.163-2A>G, p.W55Dfs*44 | No | IM, LPD | 27 700 | No | PFAPA-like periodic fever | HSCT | Alive | Genova | 27 |
| P40 | 7 | 16 | M | Turkish | + | c.G570A, p.Trp190* | URTI | LPD | 17 856 | HL | PFAPA-like periodic fever | HSCT | Alive | Ankara | |
| P41 | 9 | 11 | M | Turkish | + | c.C332T, p.T111M | No | Lymphadenitis | 411 000 | B-NHL | No | HSCT | Died | Istanbul | |
| P42 | 3 | 7 | F | Turkish | + | c.T2C, p.M1T | LRTI | IM LPD | NA | No | No | IgRT | Alive | Berlin | 28 |
| P43 | 4 | 6 | M | Turkish | + | c.C332T, p.T111M | LRTI | LPD | 233 | No | Chronic diarrhea | HSCT | Alive | Istanbul | |
| P44 | 3 | 3 | F | Turkish | + | c.C332T, p.T111M | LRTI | No | 309 402 | ns-HL | Chronic diarrhea | HSCT | Alive | Istanbul | |
| P45 | 1 | 1 | M | Turkish | + | c.C332T, p.T111M | LRTI | No | 24 000 | ns-HL | No | Planned for HSCT | Alive | Istanbul | |
| P46 | 3 | 10 | F | Iranian | + | c.G437T, p.S146I | LRTI, URTI | IM, LPD | 4 310 | No | Recurrent fever | No | Alive | Tehran | |
| P47 | 1 | 5 | F | Turkish | + | c.C332T, p.T111M | URTI | LPD | 67 500 | No | No | RTX, sirolimus, planned for HSCT | Alive | Ankara | |
| P48 | NA | 14 | M | Turkish | + | c.C332T, p.T111M | No | No | 0 | No | No | Planned for HSCT | Alive | Ankara | |
| P49 | 6 | 13 | M | Turkish | + | c.C332T, p.T111M | No | No | 0 | Burkitt lymphoma | No | Planned for HSCT | Alive | Ankara |
LAP, lymphadenopathy; PFAPA, periodic fever, aphthous stomatitis, pharyngitis, cervical adenitis. See Table 1 for expansion of other abbreviations.
Age in years unless stated otherwise. Please note that the asterisks used in the “Age” column headings have no relation to those used in the “Mutation” column.
Statistical analysis
For single comparisons of independent groups, a Mann-Whitney test was performed. For multiple comparisons, a 2-way analysis of variance (ANOVA) or multiple Student t tests were applied. Analyses were performed with the use of PRISM software (GraphPad Software Inc).
Results
Demographic characteristics of patients with inborn errors in CD27 or CD70
We reviewed clinical records of 49 patients from 29 unrelated families with a history of severe EBV-related diseases. Thirty-three patients from 19 families were diagnosed with CD27 deficiency, whereas CD70 mutations were detected in 16 patients from 10 families (Tables 1 and 2). Distribution of sex was balanced (25 male, 24 female). Mean age of patients was 18.8 years (range, 5-46 years) and 16.7 years (range, 5-40 years) for CD27- and CD70-deficient cohorts. Average age of disease manifestation was 7.3 years (8 months to 22 years) for CD27 deficiency and 3.4 years (6 months to 9 years) for CD70 deficiency (Tables 1 and 2). Seven of 49 patients (CD27, 6 of 33; CD70, 1 of 16) died of lymphoproliferation (n = 4, CD27 deficiency: P9 died of diffuse large B-cell lymphoma [DLBCL], P10 died of LPD [earlier interpreted as DLBCL]; P15 of HL, P31 of NHL) or infection (n = 3; CD27 deficiency: P2 succumbed to gram-positive sepsis during cytopenia, retrospectively interpreted as HLH; P17 to EBV pneumonia; 1 CD70-deficient patient (P41) succumbed to Pneumocystis jirovecii infection post-HSCT). Twenty allogeneic HSCT procedures were performed in 19 patients, with 95% overall survival (Table 3; supplemental Table 1, available on the Blood Web site). P14 and P36 suffered from severe IM-like presentation and encephalitis, respectively, but remained symptom-free thereafter. Five individuals in our cohort (P4, P19, P22, P23, P48) remained clinically asymptomatic at time of analysis (CD27, n = 4; CD70, n = 1; 5, 14, 17, 23, and 38 years of age, respectively) and were identified by familial screening after detection of an affected family member. Only 3 of 5 asymptomatic patients had documented antibody formation against EBV. Thus, based on this cohort, clinical penetrance of CD27 or CD70 deficiency is ∼90%
Table 3.
HSCT characteristics
| ID | Age of onset, y | Clinical manifestations | Age at HSCT, y | HLA match | Graft | Conditioning | Serotherapy | GVHD prophylaxis | Cell dose | ANC engraftment, day | GVHD | Complications | Last chimerism, % (time after transplant) | Last FU |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CD27 | ||||||||||||||
| P6 | 1 | HLH | 2 | MMUD, 9 of 10 | CB | Bu, Cy | ATG, 3 × 30 mg/kg | CSA, Pred | 0.5 × 108/kg, TNC | +34 | Acute IV (gut 3-4) | Infections, ADV, EBV, HHV6, cord colitis syndrome, poor growth | 96 (4.5 y) | 9.5 y |
| P7 | 1 | Recurrent URTI, skin abscesses, meningitis | 2 | MMUD, 9 of 10 | CB | Flu, Bu (MAC) | ATG, 3 × 2.5 mg/kg | CSA, MMF | 0.7 × 108/kg, TNC | +20 | Acute I (skin 1-2), chronic limited skin | Severe infections, CMV | 100 (5 y) | 6.5 y |
| P8 | 15 | CAEBV, EBV-LPD, oral ulcers, uveitis | 17 | MMUD, 8 of 10 | CB | Flu, Cy, TBI | None | CSA, MMF | NA | +5 | Acute I (skin 1-2) | HHV6 infection | 100 (4 y) | 9 y |
| P13 | 6 | Recurrent pneumonia, EBV viremia, HLH | 12 | MRD, 10 of 10 | BM | Eto + Dexa (HLH protocol) | Campath, 4 × 0.3 mg/kg | CSA, Pred | 9.1 × 106/kg, CD34 | +13 | Acute III (liver 3) | Severe infections, mucor, HLH relapse, EBV | 100 (2 y) | 2 y |
| P18 | 2 | Pneumonia, EBV-HLH/LPD, DLCBL | 2 | Haplo, 5 of 10 | PB TCRab/ CD19 depletion | Flu, Mel, TT | ATG, 3 × 10 mg/kg | MMF | 20.6 × 106/kg, CD34 | 20 | None | ADV viremia, CMV reactivation, CMV VST D+60 | 100 (2 y) | 2 y |
| P25 | 1 | EBV-LPD, EBV viremia | 4 | MSD, 10 0f 10 | BM | Flu, Bu (MAC) | None | CSA, MTX | 8.6 × 106/kg, CD34 | 16 | None | Pulmonary candidosis (cleared) | 96 (3 mo) | 1 y |
| P26 | 14 | Recurrent tonsillitis, EBV− ns-HL, EBV+ NHL, DLBCL | 18 | MMUD, 9 of 10 | PB | Flu, Mel | Campath, 5 × 20 mg total | CSA | 6.4 × 106/kg, CD34 | 12 | Acute I (skin 2) chronic limited skin | EBV, rhinovirus | 100 (1 y) | 4 y |
| P27 | 5 | Recurrent pneumonia, skin infections, arthritis, LPD | 6 | MMUD, 9 of 10 | PB CD45RA depletion | Flu, Bu, TT | ATG, 3 × 10 mg/kg | CSA | 13.2 × 106/kg, CD34 | None | None | Graft failure | ||
| P27 2nd | 6 | Same | BM | Flu, Cy | ATG, 3 × 2.5 mg/kg | CSA, MMF | 3.3 × 106/kg, CD34 | 28 | None | 100 (3 mo) | 3 y | |||
| P30 | 3 | HL | 5 | MUD, 10 of 10 | PB | Flu, Bu (MAC) | ATG, 3 × 10 mg/kg | CSA, MTX | 8 × 106/kg, CD34 | 12 | None | None | 100 (1 mo) | 10 mo |
| P32 | 8 | mc-HL | 8 | MRD, 10 of 10 | BM | Flu, Bu | ATG, 3 × 10 mg/kg | CSA, MTX | 2.3 × 106/kg, CD34 | 15 | None | 100 (1 y) | 2 y | |
| P33 | 3 | ns-HL | 3 | MRD, 10 of 10 | BM | Flu, Bu, TT | ATG, 3 × 10 mg/kg | CSA, MTX | 2.8 × 106/kg, CD34 | 17 | None | Infections, hemorrhage | 100 (2 y) | 2 y |
| CD70 | ||||||||||||||
| P34 | 3 | ns-HL stage 2, LPD, recurrent fever | 10 | Haplo, 3 of 6 | BM | Flu, Bu (MAC) | ATG,1 × 10 mg/kg; Campath, 1 × 0.5 mg/kg; RTX, 375 | CSA, MMF, Post-Cy | 4.9 × 106/kg, CD34 | 22 | Acute II (skin 2, gut 1) chronic limited skin | None | 100 (1 y) | 3.5 y |
| P37 | 3 | Recurrent otitis, recurrent fever, chronic enteritis, mc-EBV-HL, HL relapse | 18 | MUD, 10 of 10 | PB | Flu, Treo, RTX | ATG | CSA, MMF | 3.4 × 106/kg, CD34 | 20 | None | RSV pneumonia hemopericardium, acute renal failure, thrombocytopenia | 75 (6 mo) | 1 y |
| P38 | 2 | URTI, IM, mc-EBV-HL, HL relapse | 10 | MUD, 10 of 10 | PB | Flu, Treo, RTX | ATG | CSA, MMF | 3.7 × 106/kg, CD34 | 20 | Acute II (gut 1) | Acute renal failure, RSV pneumonia | 100 (3 mo) | 1 y |
| P39 | 1 | LAP, recurrent fever, PFAPA like, EBV PTLD, mosquito bite hypersensitivity | 5 | MUD, 10 of 10 | BM | Flu, Treo, TT | ATG, 3 × 10 mg/kg | CSA, MMF, MTX | 24 | Acute II (gut 1) | CMV reactivation | 100 (1 y) | 4 y | |
| P40 | 7 | Tonsillitis, URTI, EBV viremia, EBV-LPD, recurrent fever | 16 | MSD, 10 of 10 | PB | Flu, Treo, RTX | No | CSA, MMF | 7 × 106/kg, CD34 | 20 | Acute III (gut 2-3), chronic gut | CMV infection, thrombocytopenia | 83 (4 mo) | 1 y |
| P41 | 9 | LAP, EBV–B-NHL | 11 | Haplo, 5 of 10 | BM | Flu, Bu, TT | ATG | CSA, post-Cy | 15.2 × 106/kg, CD34 | 13 | None | Severe engraftment syndrome, day +166 PJP | 100 (3 mo) | Died day +166 |
| P43 | 4 | Recurrent pneumonia, chronic enteritis, EBV LAP | 7 | MRD, 10 of 10 | PB | Flu, Treo | No | CSA, MMF | 5.4 × 106/kg, CD34 | 22 | Acute II (skin, liver) | None | 99 (4 mo) | 1 y |
| P44 | 3 | LRTI, ns-HL | 6 | MRD. 10 of 10 | PB | Flu, Treo | No | CSA, MMF | Chronic skin and liver | None | 99 (4 mo) | 1 y |
ADV, adenovirus; ANC, absolute neutrophil count; ATG, antithymocyte globulin; BM, bone marrow; Bu, busulfan; CB, cord blood; CSA, cyclosporin A; Cy, cyclophosphamide; Dexa, dexamethasone; Eto, etoposide; Flu, fludarabine; FU, follow-up; Haplo, haploidentical; HHV6, human herpesvirus 6; MAC, myeloablative conditioning; Mel, melphalan; MMF, mycophenolate mofetil; MMUD, mismatched unrelated donor; MRD, matched related donor; MSD, matched sibling donor; MUD, matched unrelated donor; PB, peripheral blood; PJP, Pneumocystis jirovecii pneumonia; Pred, prednisolone; PTLD, posttransplant lymphoproliferative disorder; TBI, total-body irradiation; TCRab, T-cell receptor αβ; TNC, total nucleated cell; Treo, treosulfan; TT, thiotepa; VST, virus-specific T cells. See Table 1 for expansion of other abbreviations.
Genetic characteristics
Thirty-one patients from 17 families had homozygous mutations in CD27 (11 missense, 3 nonsense, 3 frameshift). One patient had compound heterozygous (1 nonsense, 1 missense) CD27 mutations; 1 further patient had only 1 identified heterozygous (1 nonsense) CD27 mutation.23 We documented 16 unique pathogenic variants in CD27; 10 of 16 were novel (Figure 1A; Table 1). All CD70-deficient patients (n = 16) carried homozygous mutations (10 families: 5 missense, 2 nonsense, 3 frameshift) leading to 8 different genetic lesions (4 of 8 novel) (Figure 1A; Table 2). The CD27 p.C53Y and the CD70 p.T111M variants were found in 4 and 3 unrelated families from the same geographic region, respectively, suggesting a founder effect. All mutations led to abolished or reduced expression of CD27 or CD70 protein (not shown). Twenty mutations localized to the extracellular domain of CD27 or CD70, whereas only 1 CD70 (p.M1T) and 3 CD27 mutations (p.W7G, p.W8*, p.C10*) localized to the intracellular domain (Figure 1A). The mutations impacting cysteine residues (p.Y32C, p.C53Y, p.C96Y, p.R107C, p.R94C) are detrimental to the tertiary structure of CD27, whereas the remainder of the mutations impair the CD27-CD70 protein interaction.34
Figure 1.
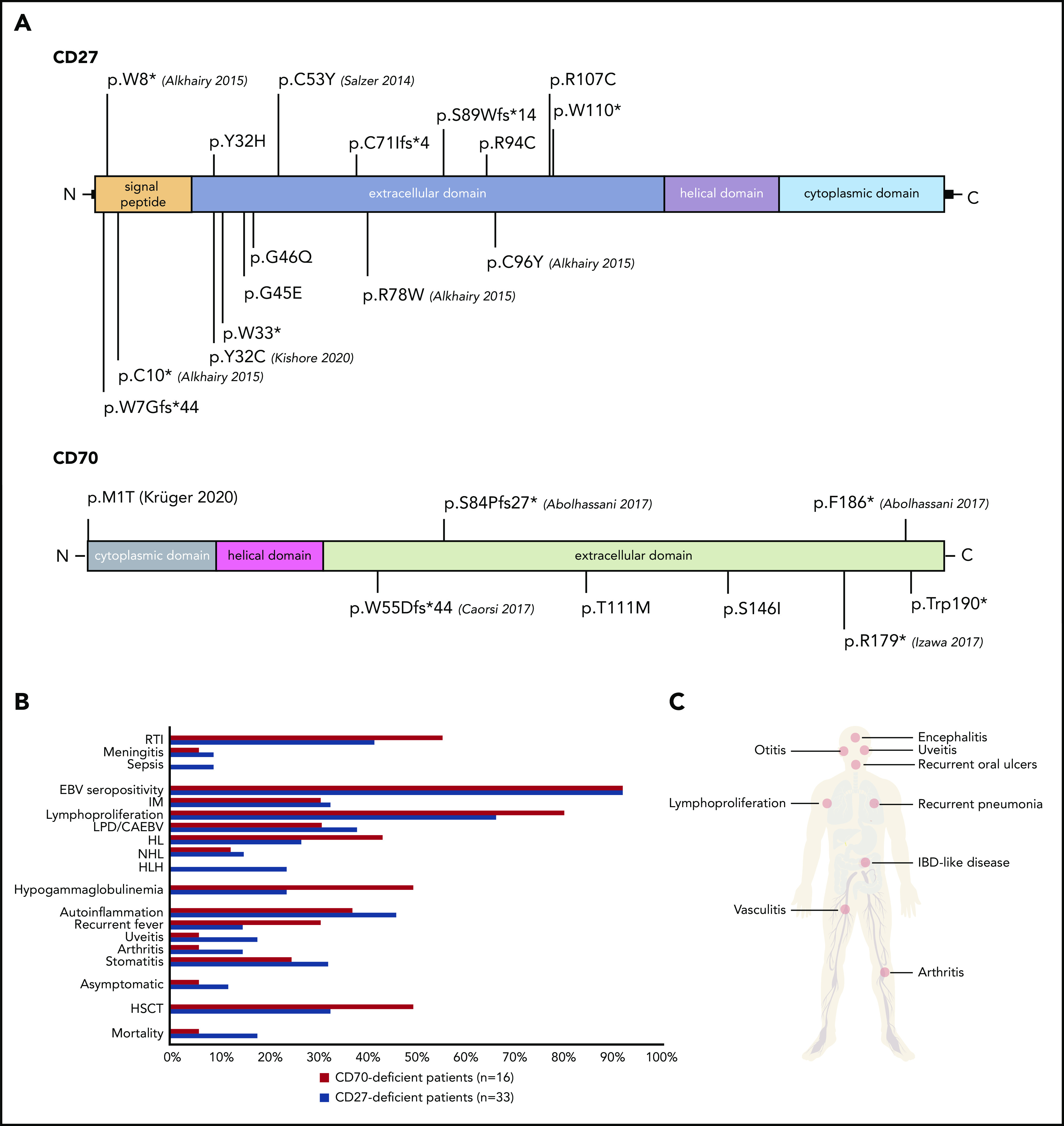
Clinical and genetic features of patients harboring mutations in CD27 and CD70. (A) Identified variants in CD27 (top) and CD70 (bottom). Corresponding publications for previously reported mutations are indicated in brackets. (B) Comparison of clinical findings and outcomes in CD27- and CD70-deficient patients. (C) Clinical manifestations of CD27 and CD70 deficiencies. IBD, inflammatory bowel disease; RTI, respiratory tract infection.
Clinical phenotype
Consistent with previous reports on CD27 and CD70 deficiency,21-27 the most common clinical features of the patients were EBV-related: IM (37%, 18 of 49 patients), LPD (37%, 18 of 49 patients) and/or lymphoma (43%, 21 of 49 patients), and HLH (18%, 9 of 49) (Figure 1B-C; supplemental Table 1). EBV positivity (evidenced by viremia, serology, or histology) at diagnosis was detected in 31 of 33 CD27-deficient and 15 of 16 CD70-deficient patients. EBV load was available for 46 patients (mean EBV load, 2.0 × 106 copies per milliliter; range, 0 to 4.5 × 107). Lymphoproliferation was the major manifestation in 71% of the cohort (35 of 49); lymphoma was diagnosed in 60% of patients (21 of 35) by biopsy, being the initial presenting symptom in 14 of 49. Interestingly, 2 patients developed EBV- HL. Thirty-six percent of CD27-deficient patients (12 of 33) and 56% of CD70-deficient patients (9 of 16) developed lymphoma at a median age of 8.5 years and 3 years, respectively. HL was the most prevalent malignancy (16 of 49), followed by NHL (n = 7 of 49 patients, 14%: 2 unclassified B-NHL, 2 Burkitt, 3 DLBCL) (Tables 1 and 2; Figure 1B). Five of 16 CD70-deficient patients suffered a relapse following initial remission after first treatment of HL, prompting allogeneic HSCT (Figure 2). All patients received a genetic diagnosis prior to HSCT.
Figure 2.

Clinical course of patients having CD27 and CD70 mutations. The scheme depicts the main clinical characteristics, therapeutic interventions, and outcome of CD27- and CD70-deficient patients within the follow-up time of each individual patient (P).
HLH occurred in 27% of CD27-deficient patients (9 of 33) (median age of onset, 4 years), and was the initial presenting symptom in 4 individuals (Figure 1B-C; supplemental Table 1). Notably, HLH progressed to lymphoproliferation within 1 year in 7 of 9 patients (Figure 2). Three of 9 CD27-deficient patients with HLH underwent HSCT, whereas 5 of 9 patients responded to conventional treatment. Eight of 9 patients who developed HLH are currently alive, at a median follow-up of 4 years since HLH; 1 patient succumbed (P2) to infection (see earlier in this section) (Figure 2). Interestingly, HLH was not observed in any CD70-deficient patients.
Various autoinflammatory features were reported in a total of 21 patients (CD27, n = 15; CD70, n = 5; 43% total) including periodic fever (n = 10), oral ulcers or stomatitis (n = 14), uveitis (n = 7), arthritis (n = 6), and vasculitis (n = 1), without documentation of a causative infectious agent and/or autoantibodies. In 4 cases (P27, P35, P39, P40), rheumatological symptoms (uveitis, arthritis) occurred initially and were treated before an EBV predisposition syndrome was suspected (Figure 1B-C; supplemental Table 1). The autoinflammatory complications resolved following HSCT.
Infectious profile
Beyond EBV-related symptoms, recurrent infections were common in our cohort, with viral infections (cytomegalovirus [CMV], herpes simplex virus, human herpes virus 6 [HHV6], varicella zoster virus, coxsackie virus) most frequent. Two patients suffered from viral encephalitis (P24, EBV and HHV6; P36, unknown etiology), causing intellectual disability in P36. P35 had severe varicella and P42 had herpes zoster. Recurrent respiratory tract infections occurred in 56% of patients (23 of 49). Although fungus (Candida, Aspergillus, Rhizopus; 4 patients) and parasites (Toxoplasma gondii, Giardia lamblia; 5 patients) were occasionally isolated, these infections were likely secondary to immunosuppressive treatment of HLH and lymphoma rather than resulting directly from CD27 or CD70 deficiency.
Effect of CD27 and CD70 deficiency on lymphocyte differentiation in vivo
To determine the impact of CD27 and CD70 mutations on lymphocyte differentiation in vivo, we performed detailed flow cytometric analyses on PBMCs from 10 CD27-deficient (average age, 13.5 years; range, 1.5-32 years) and 11 CD70-deficient (average age, 12.5 years; range, 4-36 years) patients. Although the healthy donors were older than some of the patients studied, proportions of lymphocyte subsets in peripheral blood are relatively stable between 5 years of age and adulthood.31,35-37 Proportions of T, B, and NK cells were unaffected by CD27 or CD70 mutations (Figure 3A). However, CD8+ T cells were significantly increased in CD27-deficient patients and a similar trend noted for CD70 deficiency (Figure 3A), resulting in a significantly decreased CD4/CD8 ratio in CD27-deficient individuals (Figure 3B). Although proportions of CD3+ T cells were intact for both genotypes, γδ T cells were modestly but significantly increased in CD70-deficient patients accompanied by reduced αβ T cells (Figure 3C). Proportions of MAIT, but not iNKT, cells were reduced in CD27/CD70 deficiency without reaching statistical significance (Figure 3D-E). NK-cell subsets, defined by differential expression of CD56, revealed increased proportions of CD56high and corresponding reductions in CD56dim cells in CD27/CD70 deficiency (Figure 3F).
Figure 3.
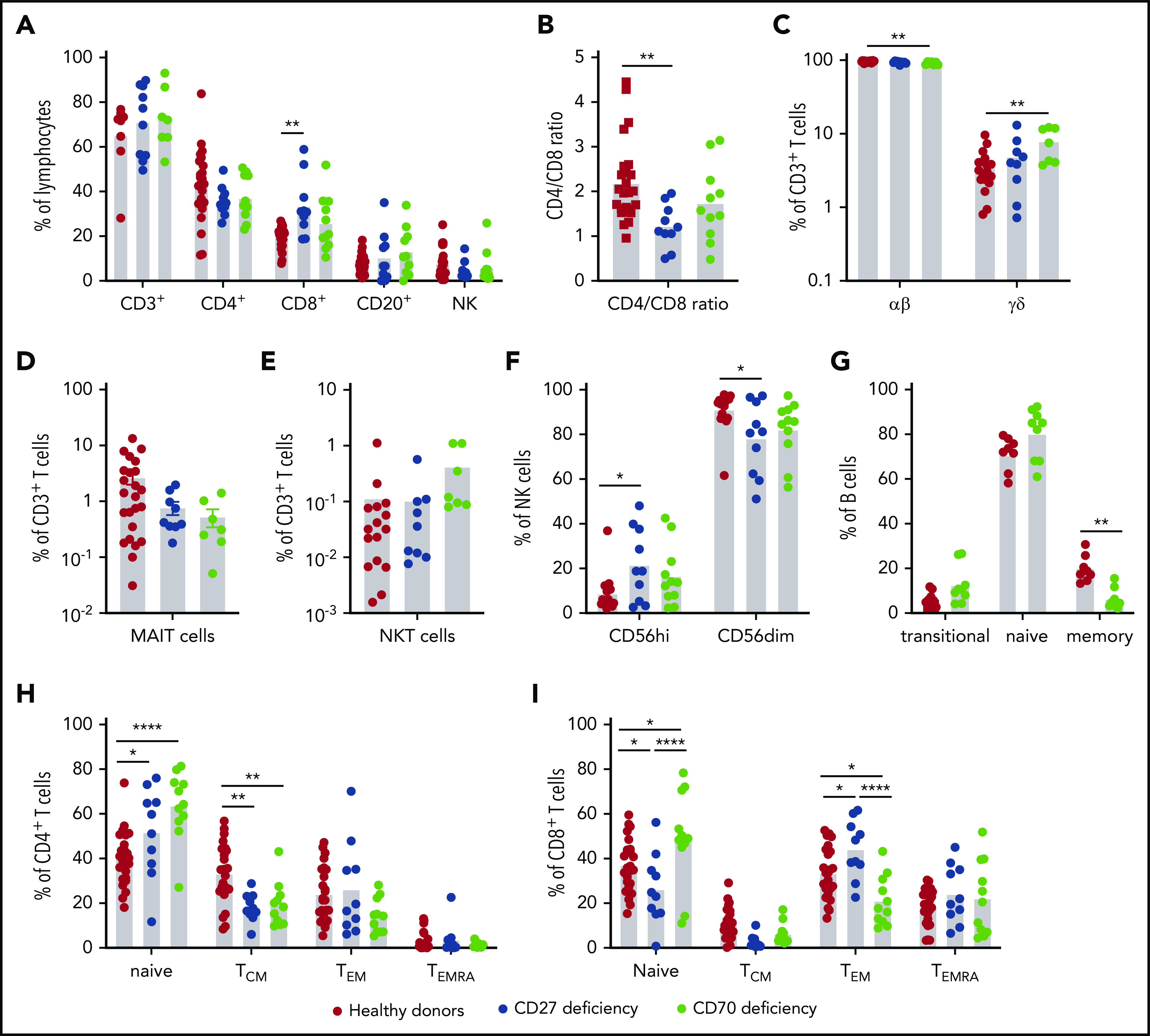
Impact of CD27 and CD70 mutations in lymphocyte differentiation in vivo: lymphocytes and subsets. PBMCs from healthy controls (n = 18-26), CD27-deficient patients (n = 10), or CD70-deficient patients (n = 7-11) were labeled with mAbs against CD3, CD4, CD8, CD56, CD20, CD10, CD27, CD161, TCR Vβ11, TCR Vα7-2, TCR Vα24, CCR7, and CD45RA. (A) Proportions of total (CD3+), CD4+ (CD3+CD4+CD8−), and CD8+ (CD3+CD4−CD8+) T cells, B cells (CD20+), and NK cells (CD3−CD56+) in peripheral lymphocytes of healthy donors and patients. (B) Ratio of CD4/CD8 T cells. (C) Proportions of CD3+ T cells expressing αβ or γδ TCR. (D-E) proportions of MAIT (D) or NKT (E) cells within the total CD3+ T-cell population. (F) Proportions of CD56high and CD56dim NK subsets within the total NK population. (G) Proportions of transitional, naive, and memory B cells within the total B-cell population. (H-I) Proportion of CD4+ (H) and CD8+ (I) cells with a naive, TCM, TEM, or TEMRA phenotype. Statistics performed using ANOVA. *P < .05; ** P < .01; ****P < .0001.
CD27/CD70 interactions are required for the generation of memory B and T cells
CD27 expression delineates memory B cells.33,38 Although this precludes defining distinct B-cell subsets in CD27 deficiency, flow cytometry revealed significantly reduced memory B-cell proportions in CD70 deficiency (Figure 3G). CD4+ and CD8+ T-cell subsets can be classified into 4 distinct populations: naive (CD45RA+CCR7+); TCM (CD45RA−CCR7+); TEM (CD45RA−CCR7−) and TEMRA (CD45RA+CCR7−).39 Proportions of naive CD4+ T cells were significantly increased and CD4+ TCM cells corresponding reduced in CD27/CD70-deficient patients (Figure 3H). Proportions of regulatory T cells and circulating T follicular helper cells were intact in all patients (not shown). The CD8+ T-cell compartment was also altered. In CD27 deficiency, naive CD8+ T cells were reduced and TEM cells increased, whereas CD70 deficiency yielded increased naive CD8+ T cells and fewer TEM cells. Despite these opposing observations, TCM cells tended to be reduced in both genotypes (Figure 3I).
Impaired cytokine production and cytolytic function of CD27-deficient CD8+ T cells
To identify mechanisms underlying impaired immune cell function and subsequent infectious susceptibility due to abolished CD27/CD70 signaling, we studied functional responses of CD8+ T cells from individuals with CD27 mutations. Consistent with CD70-deficient T cells,26 proliferation of CD4+ and CD8+ T cells in vitro was unaltered by CD27 deficiency (not shown). In contrast, expression of granzyme B and perforin was significantly reduced in CD27-deficient CD8+ TEMRA cells, and trended to be reduced in TEM cells (Figure 4A-B). Similarly, expression of CD107a, a correlate of degranulation, and IFNγ by CD8+ T cells following ex vivo stimulation was reduced by CD27 mutations (Figure 4C). To extend these findings, we examined sort-purified CD8+ T cells. Expression and/or production of cytolytic (granzyme A and B, perforin, CD107a) and effector (IFNγ, TNFα, IL-2) molecules were reduced for TCM/EM and TEMRA cells from CD27-deficient individuals (Figure 4D-E). Thus, production of cytokines and granzymes by, and activation of the lytic machinery in, memory CD8+ T cells is compromised by CD27 deficiency. As CD27 costimulation maintains effector cells, thereby providing host defense following pathogen exposure,15 we determined the impact of CD27 deficiency on T-cell survival. CD27-deficient phytohemaglutinin (PHA) blasts exhibited greater death (Figure 4F) and significantly increased expression of FASLG (Figure 4G) upon TCR engagement compared with healthy donors. Thus, CD27-deficient CD8+ T cells are intrinsically more susceptible to apoptosis than CD27-sufficient CD8+ T cells.
Figure 4.
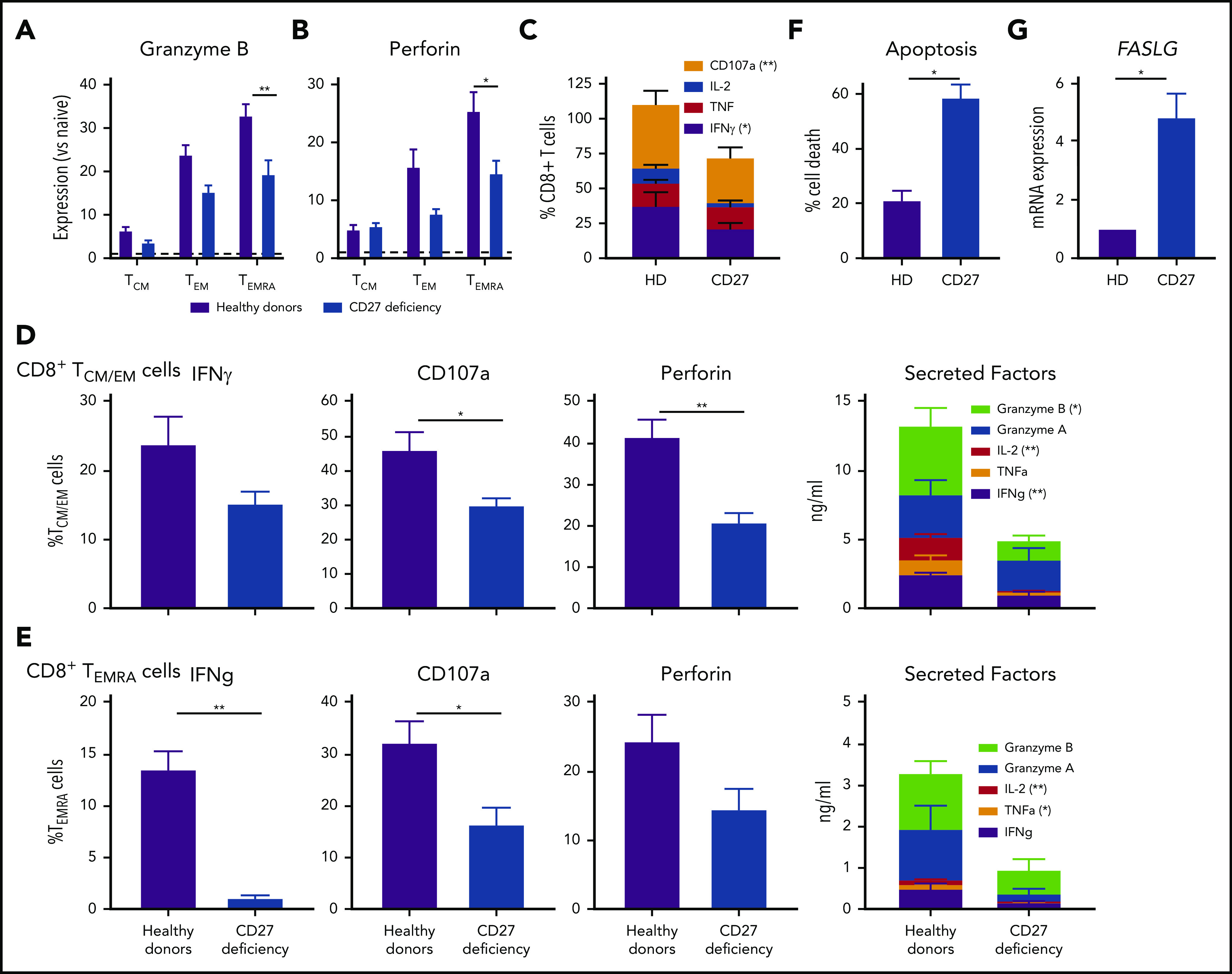
CD8 T-cell function. CD27 deficiency compromises effector function and survival of CD8+ T cells. (A-B) PBMCs were stained with mAbs to CD8, CCR7, CD45RA, granzyme B, and perforin. Expression levels of (A) granzyme B or (B) by CD8+ TCM, TEM, and TEMRA cells were determined relative to naive CD8+ T cells (normalized to 1.0). (C) PBMCs from healthy individuals (n = 5) and CD27-deficient individuals (n = 5) were stimulated for 14 hours (PMA/ionomycin) in the presence of brefeldin A and monensin. Percentage of cells expressing IFNγ, TNF, IL-2, or CD107a was determined by intracellular staining and flow cytometric analysis. (D-E) CD8+ memory (TCM/TEM; D) and TEMRA (E) cells were sort-purified from healthy individuals (n = 6) and CD27-deficient individuals (n = 4) and cultured with anti-CD2/CD3/CD28 mAbs for 5 days. Proportions of cells expressing IFNγ, CD107a, or perforin were determined by intracellular staining and flow cytometry; secretion of IFNγ, TNFα, IL-2, and granzyme A and B was determined by cytometric bead arrays. (F-G) PHA blasts were expanded from PBMCs from healthy donors (n = 5) and CD27-deficient patients (n = 3). After 5 to 7 days, the cells were restimulated with plate-bound α-CD3. (F) Percentage of apoptotic cells was determined after 24 hours. (G) Relative expression of FASLG expression was determined after 4 hours stimulation with α-CD3 mAb (normalized to PHA blasts from healthy donors). For all graphs, values represent mean plus or minus SEM. Statistics performed using Student t tests with Mann-Whitney tests. *P < .05; ** P < .01.
Altered phenotype of EBV-specific T cells in CD27-deficient individuals
EBV poses the greatest pathogenic threat to CD27/CD70-deficient individuals (Figure 1).21-27 Our previous studies found variable proportions of EBV-specific CD8+ T cells in CD70-deficient individuals.26 Furthermore, CD70-deficient memory CD8+ T cells have reduced expression of 2B4 and NKG2D,26 molecules critical for regulating CD8+ T- and NK-cell–mediated immunity against EBV-infected B cells.5,6 Hence, we used peptide/MHC class I tetramers to identify EBV-specific CD8+ T cells. Frequencies of these cells were in the normal range in 2 of 7 patients, but approximated levels detected in HLA-mismatched controls in 5 of 7 patients, suggesting that EBV-specific CD8+ T cells were negligible in these individuals (Figure 5A). CMV-specific CD8+ T cells were detected at comparable frequencies in CD27-deficient individuals and healthy donors (Figure 5B). The phenotype of EBV-specific CD8+ T cells in CD27-deficient patients was generally comparable to healthy donors (Figure 5C), with a predominance of TEM cells, and the remainder being TCM and TEMRA.40,41 However, similar to CD70 deficiency,26 expression of NKG2D and 2B4 was reduced (∼50%) on EBV-specific CD8+ T cells (Figure 5D-E), but not on CMV-specific CD8+ T cells (Figure 5E), from CD27-deficient patients. Expression of granzyme B and perforin by CD27-deficient EBV-specific CD8+ T cells was also reduced (Figure 5F). Overall, our results suggest that CD27 deficiency selectively impairs the generation of EBV-specific CD8+ T cells, hence the function of these cells is likely compromised by lack of expression of key cytotoxic receptors.
Figure 5.
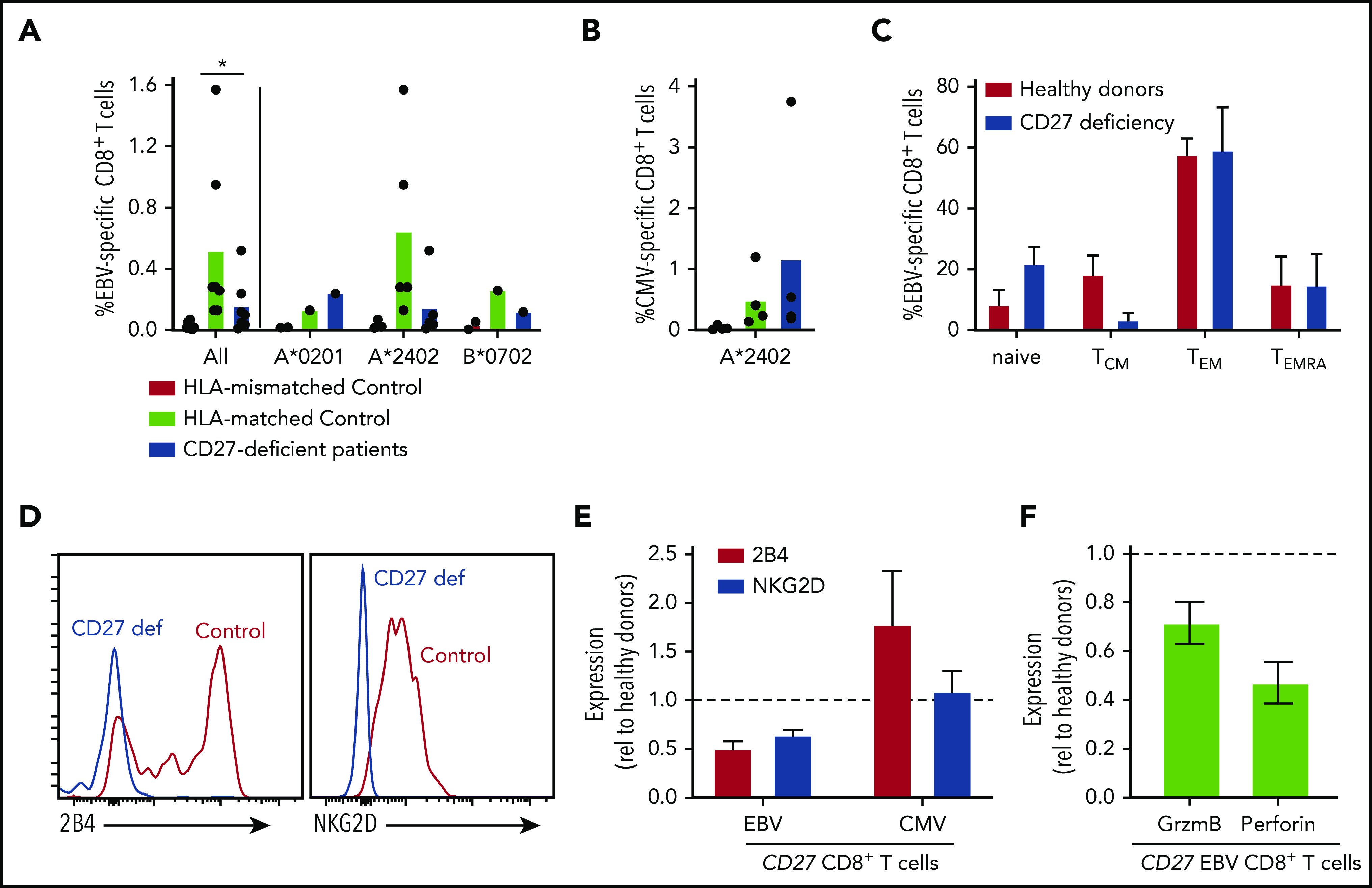
Impaired generation and function of EBV-specific CD8+ T cells in CD27-deficient individuals. (A-B) PBMCs from healthy HLA-mismatched, HLA-matched (n = 4-8), and CD27-deficient patients (n = 4-7) were stained with specific EBV- or CMV-peptide-MHC class I tetramers, mAbs to CD4, CD8, CCR7, CD45RA, CD57, CD95, PD-1, 2B4, and NKG2D. EBV-specific and CMV-specific CD8+ T cells quantified in HLA-mismatched or -matched controls and CD27-deficient patients, presented for all individuals as well as based on the specific HLA alleles (HLA-A*0201, HLA-A*2402, or HLA B*0702). Statistics were performed using ANOVA; *P < .05. (C) Distribution of EBV-specific CD8+ T cells in the naive, TCM, TEM, and TEMRA CD8+ T-cell populations in HLA-matched controls and CD27-deficient patients. (D) Expression of 2B4 and NKG2D on EBV-specific CD8+ T cells from healthy control and CD27-deficient patients. (E) Relative expression of 2B4 and NKG2D on EBV- and CMV-specific CD8+ T cells from CD27-deficient patients determined by calculating fold change relative to virus-specific CD8+ T cells from HLA-matched donors. (F) PBMCs were stained ex vivo with EBV-specific HLA tetramer, and mAbs to CD8, granzyme B, and perforin. Expression of granzyme B or perforin in EBV-specific CD8+ T cells from CD27-deficient patients was determined relative to that in EBV-specific CD8+ T cells from healthy donors.
Therapeutic interventions and outcome
Given the wide and variable phenotypic spectrum of disease, therapeutic approaches were also variable. Hypogammaglobulinemia was detected in 18 of 49 patients (only in P5 prior to EBV infection) and they received IgG substitution (n = 18 of 49) and antibiotic prophylaxis (n = 17/49), whereas patients with HLH received the HLH-2004/HLH-1994 protocol. Patients with HL or NHL were treated according to disease-specific protocols (cyclophosphamide, hydroxydaunorubicin, vincristine, prednisolone [CHOP]-based). Relapse protocols (doxorubicin, bleomycin, vinblastine, dacarbazine [ABVD] and ifosfamide, gemcitabine, vinorelbine, prednisolone [IGEV] regimen) with consecutive treatment with anti-CD30 (brentuximab), autologous HSCT, and radiotherapy were administered to patients with relapsed HL (P4, P5, P11, P12, P40).
HSCT and outcome
Eleven of 33 CD27-deficient patients (33%) and 8 of 16 CD70-deficient patients (50%) underwent HSCT (39% total). The median age at HSCT differs between the groups (CD27, 5.0 years [range, 2-18 years]; CD70, 10.0 years [range, 5-18 years]). Severe infection was the HSCT indication in only 1 of these patients. Indications for HSCT in the remaining 18 patients were persistent EBV viremia/LPD (n = 4), HL/NHL (n = 9), or a combination of these disease manifestations (LPD/viremia plus HLH [n = 2], lymphoma plus HLH [n = 1], lymphoma plus LPD [n = 2]). Median follow-up time in all our patients has been 2.0 years post-HSCT (range, 1 year to 9.5 years).
In total, 20 allogenic HSCT procedures (in 19 patients; Table 3) were performed; the majority of patients received unrelated donor transplants (12 of 19). Seven patients were transplanted with matched related donors, 4 with 10 of 10 HLA-matched unrelated donors, and 8 with mismatched unrelated donors, including 3 haploidentical transplants. Three of the unrelated donations used cord blood grafts. One patient received a TCRαβ/CD19-depleted graft; in 2 patients, haploidentical HSCT was performed with posttransplant cyclophosphamide prophylaxis.
The choice of conditioning regimen largely reflects the recommendation of the ESID-EBMT guidelines for inherited immune disorders and experience of the transplant community for patients with nonmalignant disorders. Most patients were treated with a fludarabine-busulfan (n = 8) or fludarabine-treosulfan–based reduced toxicity, but myeloablative regimen. Fludarabine-melphalan was used in 2 patients, and fludarabine-cyclophosphamide was used in 1 case prior to second transplant (P27). In 1 patient (P13), conditioning was discontinued after alemtuzumab and etoposide (as part of the HLH treatment) due to systemic toxicity and invasive fungal infection. Most patients received serotherapy with antithymocyte globulin (ATG; n = 13), or alemtuzumab (n = 3). For the 3 patients with haploidentical donors, 1 (P18) received a TCRαβ/CD19-depleted graft, and 2 (P34, P41) received posttransplant cyclophosphamide as GVHD prophylaxis. In other patients, GVHD prophylaxis consisted mostly of cyclosporine A (CSA; n = 17) and mycophenolate mofetil (MMF) or methotrexate
At 1 year posttransplant, donor chimerism (>90% donor in whole blood) was documented in all patients. Acute GVHD grade I/II and grade III-IV occurred in 7 of 19 patients (37%) and 3 of 19 patients (16%), respectively. Limited chronic skin GVHD was seen in 3 of 19 patients, and 2 patients (P39, P44) had chronic GVHD. One patient developed severe engraftment syndrome and 2 patients acute renal failure, with full recovery in both cases. No cases of sinusoidal obstruction syndrome or other toxicity were reported, even in the transplanted patients with HLH.
We observed infectious complications in 12 of 19 patients post-HSCT. Viremia resulted from reactivation of CMV (n = 4, 1 patient treated with virus-specific T cells at day +60), EBV (n = 3), adenovirus (n = 2), and HHV6 (n = 2). Two patients suffered from respiratory syncytial virus (RSV) pneumonia. It remains unclear whether viral reactivations other than EBV were already present prior to HSCT. One patient (P41) succumbed to P jirovecii pneumonia at day +166.
At a median follow-up time of 2 years (range, 1-9.5 years), overall survival was 95% (100% of CD27-deficient patients [11 of 11] and 88% of CD70-deficient patients [7 of 8]). Importantly, lymphoma relapse or secondary malignancies have not occurred in any transplanted patients and all remain in continuous remission. Hence, event-free survival in the combined group, including death and relapse, is also 95%. At 1 year post-HSCT, 16 of 18 surviving patients no longer require immunosuppression (Table 3). Within the untransplanted cohort (n = 31), 6 patients died (at 2, 4, 10, 20, 22, and 35 years of age), 4 during their first malignant event (P9 died of DLBCL, P10 of LPD; P15 of HL, P31 of NHL), and 2 due to infections.
Discussion
Combined immunodeficiencies due to germline biallelic mutations in CD2721 or CD70,24,26 characterized by increased susceptibility to bacterial and viral infections, impaired humoral immunity, and hypogammaglobulinemia, were first described in 2012 and 2017, respectively. The major pathogenic threat to these patients is EBV, causing chronic viremia and severe diseases including HLH, lymphoproliferation, and lymphoma. A growing number of experts emphasize the need to implement immunological and EBV screening in most cases of HL or NHL.42 However, there are no current definitions or “reference values” of immunological parameters or biomarkers in patients with malignancies prior to treatment. In our study, 61% of CD27- and 81% of CD70-deficient patients presented with EBV-associated lymphoproliferation or lymphoma often at a young age (median age: CD27, 11 years; CD70, 3 years), suggesting the inability to control EBV infection is a strong indication to raise attention among physicians taking care of these patients.
Besides lymphoma, a high number of CD27- and CD70-deficient patients experience autoinflammatory symptoms. In vivo analysis of gene-targeted mice established that CD27-CD70 costimulation inhibits Th17 differentiation, dampening Th17-mediated autoimmunity and inflammation.43 The accumulation of autoinflammatory symptoms suggest a regulatory effect of CD27-CD70 signaling on fine-tuning immune responses. As seen in our cohort, some patients (P39, P40) are followed as periodic fever, aphthous stomatitis, pharyngitis, cervical adenitis (PFAPA)-like disorders for several years before typical CD27-CD70 disease manifestations occur. So far, no specific disease biomarkers exist, so clinical awareness is of paramount importance in patients with signs of autoinflammation, especially with atypical presentation or unusual/lack of response where CD27/CD70 defects should be considered.
The spectrum of EBV-associated diseases in patients with inborn errors of immunity (IEIs) results from defective CD8+ T-cell activation, expansion and/or cytotoxicity that compromise immune-mediated control of EBV infection.5,6,9,11,12,14 Patients with CD27 or CD70 mutations have increased CD8+ T cells and naive CD4+ T cells, but reduced proportions of CD4+ and CD8+ TCM cells, MAIT cells, memory B cells, and EBV-specific CD8+ T cells. In contrast to the initial studies of CD27 deficiency,21,22 proportions of iNKT cells were intact in both CD27- and CD70-deficient patients. CD27-deficient memory CD8+ T cells have reduced production of cytokines and cytotoxic molecules, reduced expression of NKG2D and 2B4, and increased apoptosis in vitro.Similar functional defects have been reported for CD70-deficient CD8+ T cells.24,26 CD27-CD70 interactions are important for expansion of EBV-specific T cells, evidenced by lack of expansion of CD27-deficient T cells in response to CD70-expressing EBV+ B cells, and impaired T-cell expansion, including EBV-specific T cells, to CD70-deficient B cells.24 Collectively, these cellular defects likely manifest as impaired cytotoxic T-cell–mediated control of EBV-infected B cells, resulting in EBV-associated disease. These cellular and functional defects, together with a lack of detectable expression of CD27 or CD70 on patient immune cells, could be used as biomarkers for the early identification and putative diagnosis of individuals with inactivating mutations in CD27 or CD70. Interestingly, the spectrum of clinical manifestations varied between patients in the same family, including identification of asymptomatic individuals with biallelic CD27 mutations, indicating that additional mechanisms including environmental factors may contribute to the variable penetrance of this genetic disease.
CD27-CD70 signaling is unequivocally nonredundant for EBV immune surveillance. The clinical phenotype of CD27 and CD70 deficiency phenocopy each other with regard to disease presentation, yet there are notable differences. Age of onset is earlier in CD70 deficiency with greater incidence of hypogammaglobulinemia and lymphoma, whereas HLH is so far only seen in CD27 deficiency. Curiously, we recorded malignancies in 50% of family members carrying heterozygous CD70 mutations consistent with initial findings.26 However, only 21% of heterozygous CD27 mutation carriers reported malignant events. These differences might be due to relatively disproportional sizes of the 2 cohorts and selection bias resulting from the more recent discovery of CD70 deficiency. Targeting the CD27-CD70 pathway as a therapy for autoimmune diseases and cancers is being tested clinically.44-47 It remains unclear whether CD27/CD70 have cell-intrinsic roles in tumorigenesis which may explain the difference in lymphoma occurrence.48-50 Notably, CD70 ligation can induce apoptosis of EBV-transformed human B-cell lines.51 This may explain the excess of malignancies associated with CD70 mutations.
Following diagnosis, the decision on the therapeutic regimen remains a challenge. Although HSCT is the only cure for individuals with refractory or relapsed malignancies, treatment strategies in less severely affected patients differ broadly. Unlike patients with classical cytotoxicity defects, most patients in our cohort with HLH developed further lymphoproliferation. Therefore, persistent EBV viremia could be a biomarker to alert clinicians to the necessity of early curative interventions.42 The high mortality of CD27-deficient patients during their first malignant event, combined with excellent event-free survival post-HSCT, strongly supports consideration of curative HSCT, guided by relatively mild disease manifestations or positive family history, as demonstrated recently for other genetic forms of HLH.52 Recent studies on HSCT in IEIs have demonstrated encouraging results are not only obtained with matched but also alternative donor sources, including haploidentical.30,53-55 Based on our data that heterozygous carrier status of CD27/CD70 deficiency may be associated with increased risk for malignancy, we would preferably recommend usage of suitable nonrelated donors for allogenic HSCT, if available.
The aggressive clinical course and/or lack of a genetic diagnosis in most of these cases hindered the use of a rescue HSCT. Notably, a recent study demonstrates the feasibility and curative potential of HSCT in IEI patients with lymphoproliferative disorders even in those without complete remission at HSCT.56 A restrained attitude is often observed regarding preemptive HSCT, especially in adolescent and adult patients, even following a first malignant event explainable by the usually less favorable HSCT outcomes. However, recent HSCT studies in adolescent and young adult IEI patients have demonstrated encouraging results.57,58
In conclusion, we report the heterogeneous spectrum and clinical course in a large cohort of patients with CD27 and CD70 deficiency, which often manifest following EBV infection. These findings further highlight the critical role of the CD27-CD70 axis in regulating cellular immunity in humans, especially in the context of EBV control and lymphomagenesis. Importantly, this relates to both EBV+ and EBV− B-cell lymphomas, revealed by the key role that CD27-CD70 interaction plays in enabling antigen-presenting B cells to efficiently activate cytotoxic lymphocytes.5,6 The excellent outcome after HSCT in CD27- or CD70-deficient patients with severe disease manifestations emphasizes that HSCT needs timely consideration as a definitive treatment, especially in patients with malignant transformation to lymphoma.
Supplementary Material
The online version of this article contains a data supplement.
Acknowledgments
The authors thank the patients and their families for participating in the study, Helen Matthews for regulatory and logistical assistance, and Tatjana Hirschmugl for illustrating the graphical abstract.
This work was supported by the European Research Council (ERC Consolidator Grant 820074 [K. Boztug]), the Austrian Science Fund (FWF; Project P29951-B30 [K. Boztug]), The Susan and John Freeman Cancer Research Grant from Cancer Council NSW (Australia), and the National Health and Medical Research Council of Australia (NHMRC) (1127157 [S.G.T.]). D.A.P. was supported by a Wellcome Trust Senior Investigator Award (100326/Z/12/Z). C.S.M. was supported by a Mid-Career Research Fellowship awarded by the Office of Health and Medical Research of the New South Wales Government of Australia. S.G.T. was supported by a Principal Research Fellowship (104925) and a Peter Doherty Leadership Grant (176665) awarded from the NHMRC. F.H. received funding from the German Centre for Infection Research (DZIF; TTU 07.909), the Else Kröner-Fresenius Stiftung (EKFS; 2017_A110), and the German Federal Ministry of Education and Research (BMBF; 01GM1910C). A.W and E.G.D. are supported by the UK National Institutes of Health Research and the Great Ormond Street Hospital Biomedical Research Centre. Y.Z., H.C.S., and M.J.L. are supported by funds from the Intramural Research Program of the National Institute of Allergy and Infectious Diseases, National Institutes of Health.
Footnotes
Please contact the corresponding authors for patient data from the current study. Previously published patient data can be accessed via indicated references.21-28
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: S.G. and S.K.B. designed the standard questionnaires, and collected and analyzed the data; E.S.J.E., B.P., and G.R. designed, performed, and analyzed the experiments; R.J.H., B.E., and S. Ceylaner analyzed exome-sequencing data, performed the variant filtering and Sanger validation, and identified the mutations in P32 and P42, P43-45, and P47-49, respectively; Y.Z., A.J.O., and C.G.-J. analyzed exome-sequencing data and identified CD70 mutations for patients P37, P38, P40, and P41 supervised by M.J.L. and H.C.S.; H.A. analyzed exome-sequencing data and identified mutations in P28, P29, P46 supervised by Q.P.-H. and L.H. F.H. interpreted the clinical data and identified the mutations in P25 and P30; F.E.C., S.H., A.M., K. Baskin, G.D., S. Burns, H.A., and S. Choo provided patient samples; E.S., S.Z., T.M., A.M., I.I., S.H., C.I., K. Baskin, E.Y., E.U., M.K., D.B., T.C., A.K.G., A.I.M.H., S. Baris, E.K.-A., A.O., L.K., D.H., H.K., M.P., R.K., R.M., P.T.O., E.M., B.N., A.W., J.v.M., P.L.A.F., S. Choo, F.D., E.G.D., S. Burns, G.D., R.P.B., H.v.B., S.L., M.F., M.G., T.N., A.A., N.R., and A.I. followed the patients, and provided and interpreted the clinical data; E.G. and D.A.P. provided custom-designed HLA class I tetramers; C.S.M. supervised experimental design and data interpretation; K. Boztug, A.C.L. and S.G.T. conceptualized, initiated, supervised, and funded the study; and S.G., S.K.B., E.S.J.E., K. Boztug, A.C.L., and S.G.T. wrote the manuscript, which was reviewed and approved by all the authors.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
The current affiliation for E.S.J.E. is Department of Immunology and Pathology, Central Clinical School, Monash University, Melbourne, VIC, Australia.
Correspondence: Kaan Boztug, St. Anna Children’s Cancer Research Institute (CCRI) and Ludwig Boltzmann Institute for Rare and Undiagnosed Diseases (LBI-RUD), Zimmermannplatz 10, A-1090 Vienna, Austria; e-mail: kaan.boztug@ccri.at; or Arjan C. Lankester, Willem-Alexander Children’s Hospital, Department of Pediatrics, Leiden University Medical Center, 2300 RC Leiden, The Netherlands; e-mail: a.lankester@lumc.nl; and Stuart G. Tangye, Garvan Institute of Medical Research, 384 Victoria St, Darlinghurst, NSW 2010 Australia; e-mail: s.tangye@garvan.org.au.
REFERENCES
- 1.Zamora MR. DNA viruses (CMV, EBV, and the herpesviruses). Semin Respir Crit Care Med. 2011;32(4):454-470. [DOI] [PubMed] [Google Scholar]
- 2.Rickinson AB, Long HM, Palendira U, Münz C, Hislop AD. Cellular immune controls over Epstein-Barr virus infection: new lessons from the clinic and the laboratory. Trends Immunol. 2014;35(4):159-169. [DOI] [PubMed] [Google Scholar]
- 3.Thorley-Lawson DA, Gross A. Persistence of the Epstein-Barr virus and the origins of associated lymphomas. N Engl J Med. 2004;350(13):1328-1337. [DOI] [PubMed] [Google Scholar]
- 4.Taylor GS, Long HM, Brooks JM, Rickinson AB, Hislop AD. The immunology of Epstein-Barr virus-induced disease. Annu Rev Immunol. 2015;33:787-821. [DOI] [PubMed] [Google Scholar]
- 5.Tangye SG. Genetic susceptibility to EBV infection: insights from inborn errors of immunity. Hum Genet. 2020;139(6-7):885-901. [DOI] [PubMed] [Google Scholar]
- 6.Tangye SG, Latour S. Primary immunodeficiencies reveal the molecular requirements for effective host defense against EBV infection. Blood. 2020;135(9):644-655. [DOI] [PubMed] [Google Scholar]
- 7.Bibas M, Antinori A. EBV and HIV-related lymphoma. Mediterr J Hematol Infect Dis. 2009;1(2):e2009032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Young LS, Rickinson AB. Epstein-Barr virus: 40 years on. Nat Rev Cancer. 2004;4(10):757-768. [DOI] [PubMed] [Google Scholar]
- 9.Tangye SG. XLP: clinical features and molecular etiology due to mutations in SH2D1A encoding SAP. J Clin Immunol. 2014;34(7):772-779. [DOI] [PubMed] [Google Scholar]
- 10.Li FY, Chaigne-Delalande B, Su H, Uzel G, Matthews H, Lenardo MJ. XMEN disease: a new primary immunodeficiency affecting Mg2+ regulation of immunity against Epstein-Barr virus. Blood. 2014;123(14):2148-2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tangye SG, Palendira U, Edwards ES. Human immunity against EBV-lessons from the clinic. J Exp Med. 2017;214(2):269-283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Latour S, Fischer A. Signaling pathways involved in the T-cell-mediated immunity against Epstein-Barr virus: lessons from genetic diseases. Immunol Rev. 2019;291(1):174-189. [DOI] [PubMed] [Google Scholar]
- 13.Alosaimi MF, Hoenig M, Jaber F, et al. Immunodeficiency and EBV-induced lymphoproliferation caused by 4-1BB deficiency. J Allergy Clin Immunol. 2019;144(2):574-583.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Latour S, Winter S. Inherited immunodeficiencies with high predisposition to Epstein-Barr virus-driven lymphoproliferative diseases. Front Immunol. 2018;9:1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nolte MA, van Olffen RW, van Gisbergen KP, van Lier RA. Timing and tuning of CD27-CD70 interactions: the impact of signal strength in setting the balance between adaptive responses and immunopathology. Immunol Rev. 2009;229(1):216-231. [DOI] [PubMed] [Google Scholar]
- 16.Borst J, Hendriks J, Xiao Y. CD27 and CD70 in T cell and B cell activation. Curr Opin Immunol. 2005;17(3):275-281. [DOI] [PubMed] [Google Scholar]
- 17.Hendriks J, Gravestein LA, Tesselaar K, van Lier RA, Schumacher TN, Borst J. CD27 is required for generation and long-term maintenance of T cell immunity. Nat Immunol. 2000;1(5):433-440. [DOI] [PubMed] [Google Scholar]
- 18.Hendriks J, Xiao Y, Borst J. CD27 promotes survival of activated T cells and complements CD28 in generation and establishment of the effector T cell pool. J Exp Med. 2003;198(9):1369-1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Munitic I, Kuka M, Allam A, Scoville JP, Ashwell JD. CD70 deficiency impairs effector CD8 T cell generation and viral clearance but is dispensable for the recall response to lymphocytic choriomeningitis virus. J Immunol. 2013;190(3):1169-1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Avery DT, Ellyard JI, Mackay F, Corcoran LM, Hodgkin PD, Tangye SG. Increased expression of CD27 on activated human memory B cells correlates with their commitment to the plasma cell lineage. J Immunol. 2005;174(7):4034-4042. [DOI] [PubMed] [Google Scholar]
- 21.van Montfrans JM, Hoepelman AI, Otto S, et al. CD27 deficiency is associated with combined immunodeficiency and persistent symptomatic EBV viremia. J Allergy Clin Immunol. 2012;129(3):787-793.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Salzer E, Daschkey S, Choo S, et al. Combined immunodeficiency with life-threatening EBV-associated lymphoproliferative disorder in patients lacking functional CD27. Haematologica. 2013;98(3):473-478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Alkhairy OK, Perez-Becker R, Driessen GJ, et al. Novel mutations in TNFRSF7/CD27: Clinical, immunologic, and genetic characterization of human CD27 deficiency. J Allergy Clin Immunol. 2015;136(3):703-712.e10. [DOI] [PubMed] [Google Scholar]
- 24.Izawa K, Martin E, Soudais C, et al. Inherited CD70 deficiency in humans reveals a critical role for the CD70-CD27 pathway in immunity to Epstein-Barr virus infection. J Exp Med. 2017;214(1):73-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kishore R, Gupta A, Gupta AK, Kabra SK. Novel mutation in the CD27 gene in a patient presenting with hypogammaglobulinemia, bronchiectasis and EBV-driven lymphoproliferative disease. BMJ Case Rep. 2020;13(2):e233482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Abolhassani H, Edwards ES, Ikinciogullari A, et al. Combined immunodeficiency and Epstein-Barr virus-induced B cell malignancy in humans with inherited CD70 deficiency. J Exp Med. 2017;214(1):91-106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Caorsi R, Rusmini M, Volpi S, et al. CD70 deficiency due to a novel mutation in a patient with severe chronic EBV infection presenting as a periodic fever. Front Immunol. 2018;8:2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kruger Renate, Martin Emmanuelle, Dmytrus Jasmin, et al. CD70 deficiency associated with chronic Epstein-Barr virus infection, recurrent airway infections and severe gingivitis in a 24-year-old woman. Front Immunol. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Edwards ESJ, Bier J, Cole TS, et al. Activating PIK3CD mutations impair human cytotoxic lymphocyte differentiation and function and EBV immunity. J Allergy Clin Immunol. 2019;143(1):276-291.e6. [DOI] [PubMed] [Google Scholar]
- 30.Pillay BA, Avery DT, Smart JM, et al. Hematopoietic stem cell transplant effectively rescues lymphocyte differentiation and function in DOCK8-deficient patients. JCI Insight. 2019;5:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Avery DT, Kane A, Nguyen T, et al. Germline-activating mutations in PIK3CD compromise B cell development and function. J Exp Med. 2018;215(8):2073-2095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Payne K, Li W, Salomon R, Ma CS. OMIP-063: 28-color flow cytometry panel for broad human immunophenotyping [published online ahead of print 16 April 2020]. Cytometry A. doi:10.1002/cyto.a.24018. [DOI] [PubMed] [Google Scholar]
- 33.Tangye SG, Liu YJ, Aversa G, Phillips JH, de Vries JE. Identification of functional human splenic memory B cells by expression of CD148 and CD27. J Exp Med. 1998;188(9):1691-1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Teplyakov A, Obmolova G, Malia TJ, Gilliland GL. Crystal structure of CD27 in complex with a neutralizing noncompeting antibody. Acta Crystallogr F Struct Biol Commun. 2017;73(Pt 5):294-299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tosato F, Bucciol G, Pantano G, et al. Lymphocytes subsets reference values in childhood. Cytometry A. 2015;87(1):81-85. [DOI] [PubMed] [Google Scholar]
- 36.Schatorjé EJ, Gemen EF, Driessen GJ, Leuvenink J, van Hout RW, de Vries E. Paediatric reference values for the peripheral T cell compartment. Scand J Immunol. 2012;75(4):436-444. [DOI] [PubMed] [Google Scholar]
- 37.Garcia-Prat M, Álvarez-Sierra D, Aguiló-Cucurull A, et al. Extended immunophenotyping reference values in a healthy pediatric population. 2019;96(3):223-233. [DOI] [PubMed] [Google Scholar]
- 38.Klein U, Rajewsky K, Küppers R. Human immunoglobulin (Ig)M+IgD+ peripheral blood B cells expressing the CD27 cell surface antigen carry somatically mutated variable region genes: CD27 as a general marker for somatically mutated (memory) B cells. J Exp Med. 1998;188(9):1679-1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sallusto F, Lenig D, Förster R, Lipp M, Lanzavecchia A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature. 1999;401(6754):708-712. [DOI] [PubMed] [Google Scholar]
- 40.Callan MF, Tan L, Annels N, et al. Direct visualization of antigen-specific CD8+ T cells during the primary immune response to Epstein-Barr virus In vivo. J Exp Med. 1998;187(9):1395-1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hislop AD, Annels NE, Gudgeon NH, Leese AM, Rickinson AB. Epitope-specific evolution of human CD8(+) T cell responses from primary to persistent phases of Epstein-Barr virus infection. J Exp Med. 2002;195(7):893-905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bomken S, van der Werff Ten Bosch J, Attarbaschi A, et al. Current understanding and future research priorities in malignancy associated with inborn errors of immunity and DNA repair disorders: the perspective of an interdisciplinary working group. Front Immunol. 2018;9:2912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Coquet JM, Middendorp S, van der Horst G, et al. The CD27 and CD70 costimulatory pathway inhibits effector function of T helper 17 cells and attenuates associated autoimmunity. Immunity. 2013;38(1):53-65. [DOI] [PubMed] [Google Scholar]
- 44.Jacobs J, Deschoolmeester V, Zwaenepoel K, et al. CD70: An emerging target in cancer immunotherapy. Pharmacol Ther. 2015;155:1-10. [DOI] [PubMed] [Google Scholar]
- 45.Wajant H. Therapeutic targeting of CD70 and CD27. Expert Opin Ther Targets. 2016;20(8):959-973. [DOI] [PubMed] [Google Scholar]
- 46.Boleto G, Allanore Y, Avouac J. Targeting costimulatory pathways in systemic sclerosis. Front Immunol. 2018;9:2998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Starzer AM, Berghoff AS. New emerging targets in cancer immunotherapy: CD27 (TNFRSF7). ESMO Open. 2020;4(suppl 3):e000629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.de Miranda NF, Georgiou K, Chen L, et al. Exome sequencing reveals novel mutation targets in diffuse large B-cell lymphomas derived from Chinese patients. Blood. 2014;124(16):2544-2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Riether C, Schürch CM, Bührer ED, et al. CD70/CD27 signaling promotes blast stemness and is a viable therapeutic target in acute myeloid leukemia. J Exp Med. 2017;214(2):359-380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Scholtysik R, Nagel I, Kreuz M, et al. Recurrent deletions of the TNFSF7 and TNFSF9 genes in 19p13.3 in diffuse large B-cell and Burkitt lymphomas. Int J Cancer. 2012;131(5):E830-E835. [DOI] [PubMed] [Google Scholar]
- 51.Park GB, Kim YS, Lee HK, et al. Endoplasmic reticulum stress-mediated apoptosis of EBV-transformed B cells by cross-linking of CD70 is dependent upon generation of reactive oxygen species and activation of p38 MAPK and JNK pathway. J Immunol. 2010;185(12):7274-7284. [DOI] [PubMed] [Google Scholar]
- 52.Lucchini G, Marsh R, Gilmour K, et al. Treatment dilemmas in asymptomatic children with primary hemophagocytic lymphohistiocytosis. Blood. 2018;132(19):2088-2096. [DOI] [PubMed] [Google Scholar]
- 53.Balashov D, Shcherbina A, Maschan M, et al. Single-center experience of unrelated and haploidentical stem cell transplantation with TCRαβ and CD19 depletion in children with primary immunodeficiency syndromes. Biol Blood Marrow Transplant. 2015;21(11):1955-1962. [DOI] [PubMed] [Google Scholar]
- 54.Bertaina A, Merli P, Rutella S, et al. HLA-haploidentical stem cell transplantation after removal of αβ+ T and B cells in children with nonmalignant disorders. Blood. 2014;124(5):822-826. [DOI] [PubMed] [Google Scholar]
- 55.Shah RM, Elfeky R, Nademi Z, et al. T-cell receptor alphabeta(+) and CD19(+) cell-depleted haploidentical and mismatched hematopoietic stem cell transplantation in primary immune deficiency. J Allergy Clin Immunol. 2018;141(4):1417-1426.e1. [DOI] [PubMed] [Google Scholar]
- 56.Prunotto G, Offor UT, Samarasinghe S, et al. HSCT provides effective treatment for lymphoproliferative disorders in children with primary immunodeficiency [published online ahead of print 1 May 2020]. J Allergy Clin Immunol. doi:10.1016/j.jaci.2020.03.043. [DOI] [PubMed] [Google Scholar]
- 57.Fox TA, Chakraverty R, Burns S, et al. Successful outcome following allogeneic hematopoietic stem cell transplantation in adults with primary immunodeficiency. Blood. 2018;131(8):917-931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Morris EC, Albert MH. Allogeneic HSCT in adolescents and young adults with primary immunodeficiencies. Front Pediatr. 2019;7:437. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.