

Abstract
Chemoresistance to gemcitabine (GEM) - a frontline chemotherapeutic, resulting from its dysfunctional uptake and metabolism in cancer cells, is a major contributing factor for failed therapy in pancreatic cancer (PanC) patients. Therefore, there is an urgent need for agents that could reverse GEM resistance and allow continued chemosensitivity to the drug. We employed natural non-toxic agent (with anti-PanC potential) bitter melon juice (BMJ) and GEM to examine their combinatorial benefits against tumorigenesis of PanC patient derived xenograft (PDX) - pancreatic ductal adenocarcinomas explants PDX272 (wild-type KRAS), PDX271 (mutant KRAS and SMAD4), and PDX266 (mutant KRAS). Anti-PanC efficacy of single agents versus combination in the three tumor explants, both at the end of active dosing regimen and following a drug-washout phase were compared. In animal studies, GEM alone treatment significantly inhibited PDX tumor growth, but effects were not sustained, as GEM-treated tumors exhibited regrowth post-treatment termination. However, combination-regimen displayed enhanced and sustained efficacy. Mechanistic assessments revealed that overcoming GEM resistance by co- administration with BMJ was possibly due to modulation of GEM transport/ metabolism pathway molecules [Ribonucleotide reductase regulatory subunit M1 (RRM1), human equilibrative nucleoside transporter 1 (hENT1) and deoxycytidine kinase (dCK)]. Study outcomes, highlighting significantly higher and sustained efficacy of GEM in combination with BMJ, make a compelling case for a clinical trial in PanC patients wherein BMJ could be combined with GEM to target and overcome GEM resistance. Additionally, given their specific effectiveness against KRAS- mutant tumors, this combination could be potentially beneficial to a broader PanC patient population.
Keywords: Pancreatic cancer, patient derived xenograft, bitter melon juice, gemcitabine, chemoresistance
1. INTRODUCTION
Pancreatic cancer (PanC) has the lowest 5-year survival rate (<3%) of all cancer types and is projected to be the second leading cause of cancer-associated mortalities by year 2030 in the United States.1,2 Pancreatic ductal adenocarcinoma (PDAC) is the most frequently encountered subtype of PanC (~95%), with a symptomless progression to metastasis and invasion.3 Surgical resection is the main curative option; however, only 15–20% of PanC patients qualify for this procedure owing to its highly aggressive nature, complex spatial location and the advanced stage of the disease at clinical diagnosis.4 Surgery alone fails to suffice in majority of cases; the disease relapses in 70–80% of patients and is fatal without additional therapy.5–7
Currently, of all the available treatment modalities for PanC, gemcitabine (GEM) is the standard of care frontline drug, with a moderate success rate in improving the median overall PanC patient survival by about 5.7–6.8 months.8,9 Moreover, chemoresistance to GEM is a major contributing factor for PanC patient’ morbidity and mortality resulting from dysfunctional uptake and metabolism of GEM in cancer cells.10,11 Therefore, there is an urgent need for agents that could reverse GEM resistance and allow continued chemosensitivity in PanC.
On another front, a lack of efficient, efficacy-predictive, preclinical cancer models is also one of the foremost reasons due to which the drugs previously exhibiting remarkable anticancer potential in preclinical animal models eventually fail in real-life clinical settings.12 Although the availability of athymic-immunodeficient mice models eases tumor xenograft establishment using human cancer cell lines, the generated xenograft tumors cannot recapitulate the tumorigenesis in its entirety, including the tumor associated microenvironment. Additionally, these tumors harbor little resemblance with the true nature of patient tumor complexity and heterogeneity, thus resulting in differential treatment outcomes between preclinical and clinical scenario.12 In recent times, patient derived xenograft (PDX) tumors are being generated by engrafting tumor tissue derived straight from the patient into an immunodeficient mouse and passaged in vivo for efficacy studies of various anti-cancer drugs.13 PDXs have an inherent high reliability owing to the preservation of overall patient genomic profile, including tumor histology and stromal component despite serial passages and thus recapitulate the genetic heterogeneity of PanC.14,15 To this end, a patient-derived pancreatic adenocarcinoma explant xenograft model bank has been established at UC Denver-AMC. Because these pancreatic carcinomas are maintained in vivo, their utilization in PDX-based efficacy studies simulates the first stage of a phase II clinical trial. 16,17
In the light of the above background, herein, we employed PanC-PDX- pancreatic ductal adenocarcinomas explants in immunocompromised mice as a relevant model system to examine the combinatorial effects of a natural agent bitter melon juice (BMJ) and GEM against PanC tumorigenesis [harboring wild-type (WT) KRAS or mutant (MT) KRAS and SMAD4 states]. The selection of BMJ as a combination agent, is based on our recent studies, where we have reported in vitro and in vivo anti-PanC efficacy of BMJ,18,19 together with its potential to enhance GEM sensitivity in resistant PanC spheroids, in addition to targeting self-renewal and kinetics of PanC stem cell pool expansion.18,20–22 Thus, it is very likely that BMJ intake has potential benefits against PanC including those in GEM-resistant disease.
2. MATERIALS AND METHODS
2.1. Cell lines and reagents
PANC1, AsPC1, and BxPC3 were obtained from ATCC (Manassas, VA) during past 4–6 years, and aliquots frozen and cultured as needed. All these cell lines were tested and authenticated by DNA profiling for polymorphic short tandem repeat markers at our Molecular Biology Core Facility. Last authentication was performed in 12/ 2018 (during the time when this study was performed and completed). Cells were grown under standard culture conditions with 10% FBS and 1% Penicillin-Streptomycin [PANC1 in DMEM with high glucose while BxPC3 and AsPC1 cells in RPMI 1640 (1X)]; AsPC1 media was also supplemented with essential amino acids. GEM used was from Zydus Hospira. Primary antibodies: Ki67 (ab16667, RRID:AB_302459), PECAM-1/CD31 (ab28364, RRID:AB_726362), RRM1 (ab81085, RRID:AB_1640848) and hENT1 (ab135756) were from Abcam (Cambridge, MA); VEGF (sc-152, RRID:AB_2212984) and dCK (sc-393099) from Santa Cruz (Dallas, TX), and cleaved (C)-caspase3 (9661, RRID:AB_2341188) from Cell Signaling (Danvers, MA). BMJ preparation, characterization and standardization have recently been reported by us.19 For cell culture studies, lyophilized BMJ was dissolved in DMSO/water, centrifuged at 3000 rpm and added to the media.
2.2. Cell viability assay
Cells were seeded for 24 hours, and then treated with BMJ (0.5–2% v/v), GEM (2–25 µM), or their combination (where cells were exposed to BMJ for 1 hour and then GEM added to same media); controls were exposed to media containing DMSO (≤0.1%). Cell viability was determined at 72 hours by Trypan Blue dye exclusion assay. All experiments were performed in triplicate.
2.3. Patient derived xenograft study
Female athymic nude mice (Hsd: athymic nude Foxn1nu; model 069 from Harlan/Envigo) aged ~8–10 weeks were housed in animal facility (UCDenver-AMC) and switched to AIN76A pellet diet (Envigo) a week prior to tumor study initiation (for acclimatization). Utilizing a trocar, the mice were subcutaneously implanted with (Matrigel immersed) ~3mm3 of PanC patient tumor tissue (expanded as explant tumors in nude mice) designated PDX272 (KRASWT), PDX266 (KRASMT, SMAD4MT) and PDX271 (KRASMT) [as previously described].16,17 Animals were implanted with tumor tissue in both flanks, enough to generate at least ~4–10 tumors per treatment cohort for each PDX explant. The difference in sample size between different PDX explants was due to differential tumor take rate in these explants (which also limited explant availability for grafting) and was not based on power analysis. Once tumors reached 200 mm3 size (day 1), they were randomized into four groups and treatment initiated: control (untreated); BMJ oral gavage in water (200mg/kg body wt.; 5 days a week); GEM(i.p. 50mg/kg biweekly in saline); and BMJ and GEM combination (Combo). The animals were maintained on this active drug dosing regimen for 32 (PDX272 and PDX271) and 35 (PDX266) days. Following completion of the active dosing (32 or 35 days), a subgroup of the animals was sacrificed (sacrifice time point 1), and their tumors harvested. [Since tumor take rate for PDX271 explants was low, we only recorded tumor volume at the end of dosing period (32 days) but did not sacrifice any mice to collect explants]. Post day 32 or 35, in remaining mice, all treatments were discontinued to analyze sustained effects (till day 64, sacrifice time point 2) of the drugs alone or in combination, post-treatment stoppage. In all study end points, harvested tumors were flash frozen in liquid nitrogen and a portion fixed in formalin. Throughout the study, body weight and tumor volume were recorded biweekly. Tumor volume was measured using a digital caliper and calculated using the formula 0.5236 L1 (L2)2, where L1 is the long axis and L2 is the short axis of the tumor. All animal studies were conducted in accordance with Institutional Animal Care and Use Committee under approved animal protocol [# 90316 (03)1E (UC Denver-AMC)].
2.4. Immunohistochemical (IHC) and statistical analyses
IHC analysis using paraffin-embedded tumor (5 μm-thick) sections was performed as described previously 23; percentage of (brown stained) positive cells counted in 5–8 arbitrarily selected fields/section. For cytoplasmic staining, arbitrary immunoreactivity scores were allotted based on intensity of brown staining as 0 (no), 1 (weak), 2 (moderate), 3 (strong), 4 (very strong) staining. Images were captured by AxioCam MrC5 camera. H&E and Picrosirius red (PSR) staining was performed at Histology Shared Resource, UCD Dept. of Pathology and analyzed/ quantified by co-author- histopathologist (Dr. Orlicky)-who was blinded to the study; images were captured using Olympus BX51, 17mp high-definition camera employing Olympus CellSens (RRID:SCR_014551) software (Olympus, Waltham, MA). Polarized light images of PSR staining were quantified using SlideBook V.6.0 (RRID:SCR_014300) (Intelligent Imaging Innovations, Denver, CO). Statistical analyses were performed using Sigma Stat software (version 3.5, Jandel Scientific) (RRID:SCR_010285). Quantitative data are presented as mean±SEM. Statistical significance of difference between treatment groups and specific controls was determined by unpaired t-test or one-way analysis of variance (ANOVA) followed by Tukey’s test for multiple comparisons. P ≤ 0.05 was considered significant.
3. RESULTS
3.1. Combinatorial in vitro effects of BMJ and GEM against human PanC cells
Human PanC cells BxPC3, AsPC1 and PANC1 were exposed to different concentrations of BMJ (0.5–2% v/v) and GEM (2–25 µM) alone or a combination of BMJ+GEM for 72 hours, and the drug/s effects on cell viability was determined by Trypan Blue dye exclusion assay. BxPC3 cells displayed the most significant decrease in total cell number after BMJ+GEM (1 – 2% BMJ with 2µM GEM) exposure compared to both drugs alone, together with a significant increase in percentage (%) of dead cells; a ≥50% BxPC3 cell death was observed with a combination of BMJ (2% v/v) + GEM (25 µM) compared to only ~25% cell death with GEM (25µM) alone (Fig. 1A). AsPC1 cells also showed similar increase in anti-cancer effects with BMJ+GEM compared to GEM alone-where a pronounced effect of ~30% cell death was observed with a combination of BMJ (2% v/v) + GEM (25 µM) (Fig. 1B). Interestingly, BMJ + GEM combinations failed to elicit any improved response over effects of either drug alone in PANC1 cells (Fig. 1C).
Figure 1. Combinatorial effects of BMJ and GEM against human PanC cells.
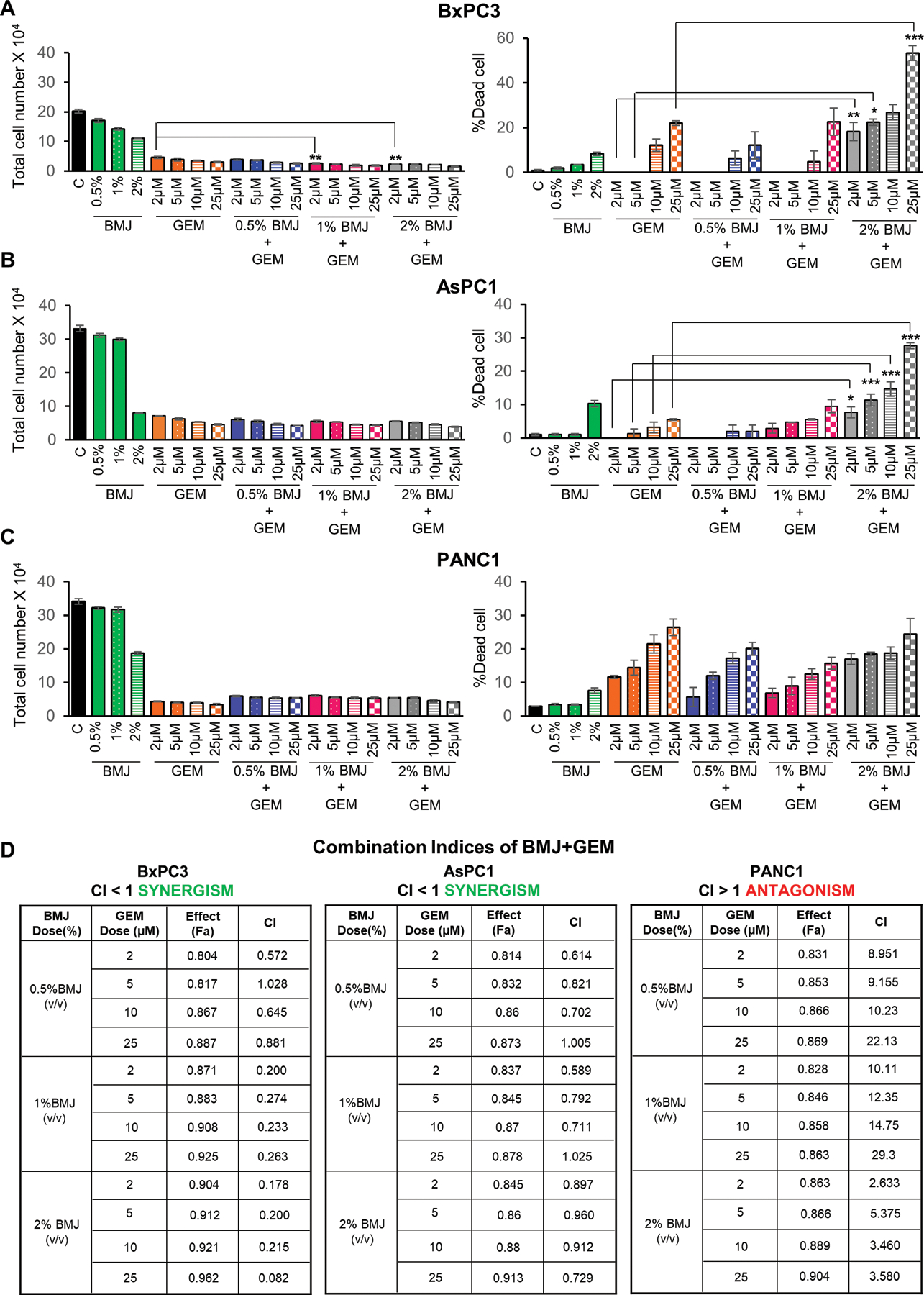
Effect on total cell number and percent dead cells of (A) BxPC3, (B) AsPC1 and (C) PANC1 cells with varying concentrations of BMJ, GEM and a combination of BMJ+GEM for 72 hours. (D) Represents the combination indices for BMJ+GEM for BxPC3, AsPC1 and PANC1 cells (as determined by data from viability assays using non-constant combination ratios with Chou-Talalay method employing CompuSyn software program) after 72 hrs of drug exposure. Fa represents the fraction of cells that is growth-inhibited in response to drug treatments and is calculated as 1-fraction of surviving cells. ***p≤0.001, **p≤0.01 and *p≤0.05.
Use of the CompuSyn software, employing Chou Talalay method,24 to determine the nature of drug combination effects in PanC cells, revealed a highly synergistic effect in BxPC3 cells with BMJ+GEM [combination indices (CIs) in the range of ~0.02–1.00]. The combinatorial effect in AsPC1 cells was comparable to that seen in BxPC3 cells (with a CI range of ~0.6–1.0). However, analysis of PANC1 cells viability data, revealed an antagonistic effect resulting from BMJ+GEM combinations where the CIs ranged between ~2.6–22.1, thereby supporting the cell-count results in the Trypan Blue assay (Fig. 1D).
3.2. Histological characterization of PanC- PDX272, PDX266 and PDX271 explants
As shown in Figure 2A, pathological analysis identified PDX272 as classical PDAC while PDX266 and PDX271 had a mucinous appearance, where PDX271 had the most mucinous morphology. A summary is provided in Figure 2B corresponding to the criteria for PDX classification, including differences based on PanC grade/stage, extent of metastasis, inflammation, epithelial hyperplasia, and stromal changes.
Figure 2. Comparative histopathological characterization of PanC-PDX272, PDX266, and PDX271 explants.
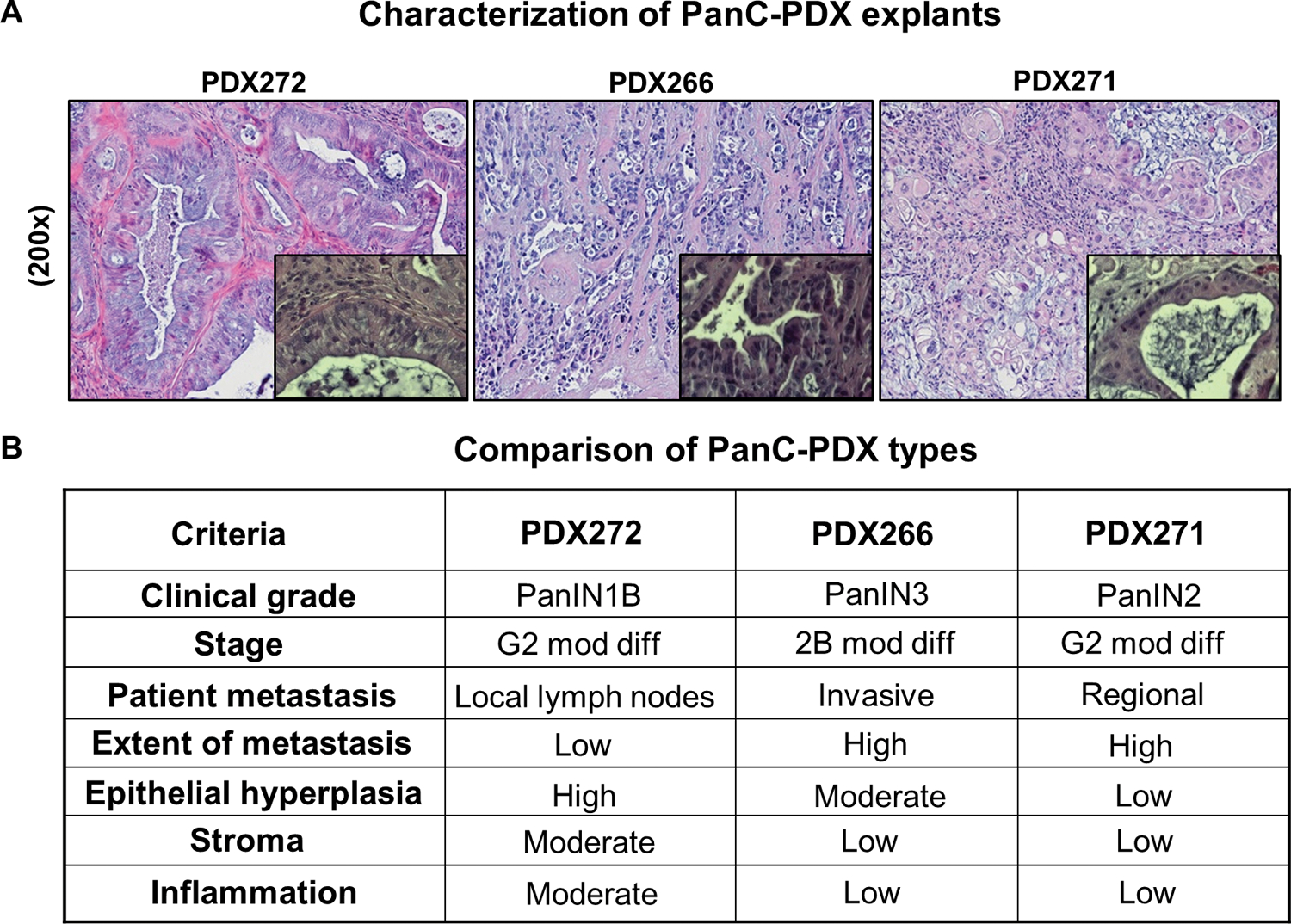
For our PDX study, excess tumor tissues not needed for diagnosis from Whipple resection of human pancreatic tumor specimens (patients without preoperative chemo- or radiation therapy) were used to propagate the PDX-explants in vivo in the PanC-PDX bank (UC Denver-AMC). (A) Representative pictographs (x200) of hematoxylin and eosin (H&E) stained PDX explants; magnified insets depicted at x400 magnification highlight the progression/clinical stage of PanC as seen by the ductal morphology. (B) A summary of the histological features of these explants provides details on PanC grade/stage, extent of metastasis, inflammation, epithelial hyperplasia, and stromal pattern. Moderately differentiated adenocarcinoma (mod diff).
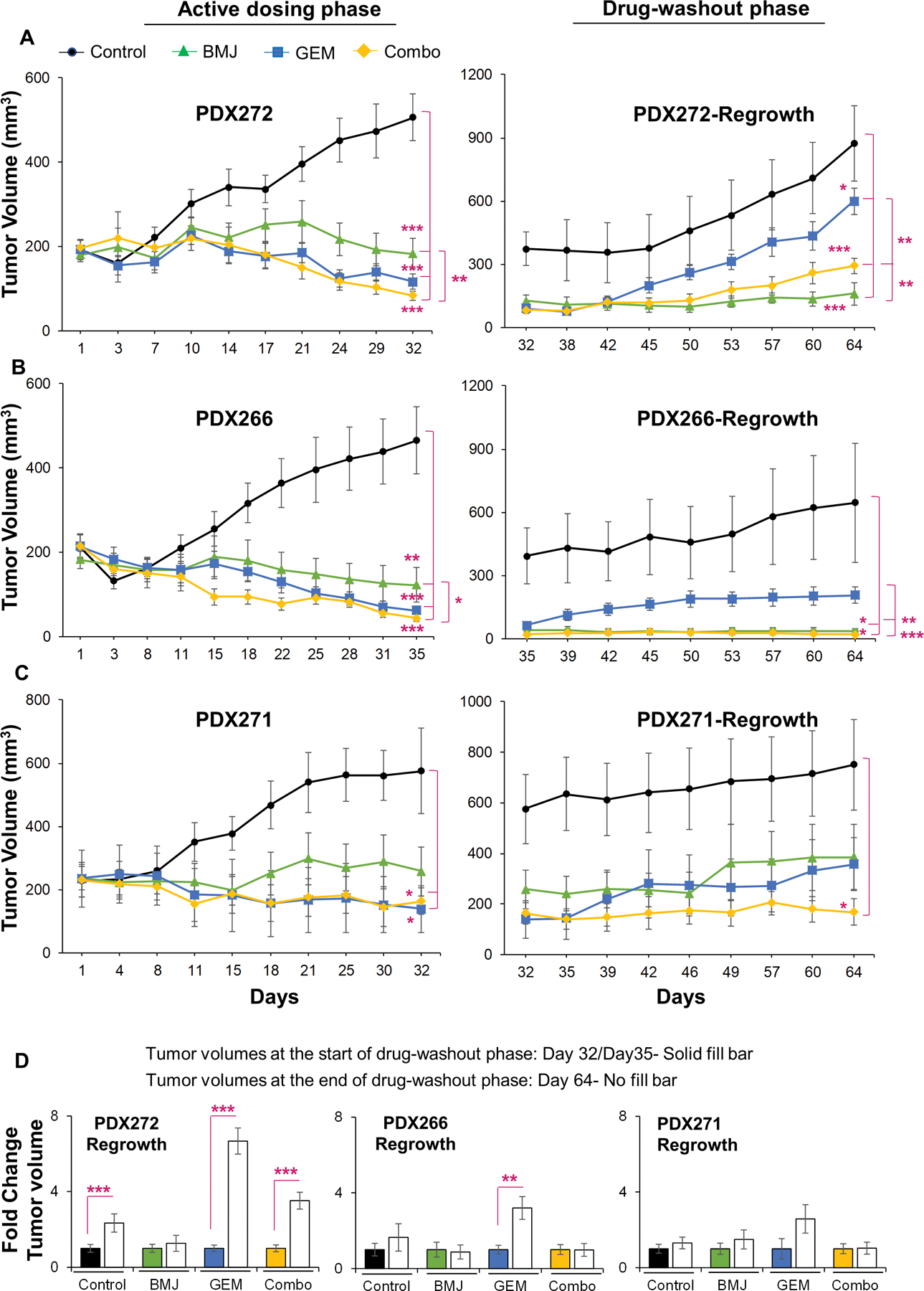
3.3. Combinatorial in vivo effects of BMJ and GEM against PanC-PDX explants
Next, to determine whether the differential response of PanC cells to BMJ+GEM combination was reflective of combinatorial effects of these drugs in a clinical scenario, we performed efficacy studies with these drugs (alone or in combination) against a number of PDX- explants. Figure 3 presents the tumor growth data obtained from PanC-PDX explants following treatment with BMJ or GEM alone or their combination (Combo), as well as their effect thereafter once the treatments were stopped. In general, longitudinal assessments of PDX tumor volumes, as a function of time, during the course of study indicated that irrespective of explant type, the drug efficacy trends of both drugs (either alone or in combination) were highly comparable in all three explants (Fig. 3A–3C). Dosing with BMJ displayed a significant inhibition in tumor growth in all three explants (∼55–74% decrease in PDX272, PDX266, and PDX271 tumor volumes). On the other hand, dosing with GEM or Combo showed relatively better anti-tumor effects compared to BMJ. Specifically, with respect to untreated controls, dosing with GEM reduced tumor volumes by ∼77% (PDX272), ∼87% (PDX266), and ∼76% (PDX271); while dosing with Combo reduced tumor volumes by ∼84% (PDX272), ∼91% (PDX266), and ∼72% (PDX271).
Figure 3. Combinatorial in vivo effects of BMJ and GEM against growth of PanC-PDX tumor explants.
Right and left panel depict longitudinal assessment of tumor volumes (mm3) as a function of time (measured biweekly). (A-C, left panel) depicts tumor volumes during active dosing with drugs (either alone or as combination ‘Combo’)-until day 32 for both PDX272 and PDX271, and day 35 for PDX266 tumors; (A-C, right panel) depicts tumor volumes during drug-washout phase (tumor regrowth was measured post-treatment termination until day 64). Values are expressed as mean ± S.E.M. Tumor trends: black (untreated controls) green (BMJ-exposed), blue (GEM-exposed), and yellow (Combo-exposed) groups. Drug treatments were initiated at day 1: control (untreated); BMJ oral gavage in water (200mg/kg body wt.; 5 days a week); GEM (i.p. 50mg/kg biweekly in saline); and BMJ and GEM combination (Combo). No. of explants per group in active dosing phase: PDX272 [control (n=12), BMJ (n=11), GEM (n=12), Combo (n=12)]; PDX266 [control (n=10), BMJ (n=9), GEM (n=10), Combo (n=11)] and PDX271 [control (n=5), BMJ (n=5), GEM (n=4), Combo (n=4). (D) Comparison of PDX-tumor volumes (fold change) at the start of drug-washout phase [(day 32 or day 35) represented by solid fill bars] to the tumor volumes at the end of drug-washout phase [day 64 represented by no fill bars]. No. of explants per group evaluated in drug-wash out phase: PDX272 [control (n=7), BMJ (n=3), GEM (n=6), Combo (n=6)]; PDX266 [control (n=4), BMJ (n=5), GEM (n=5), Combo (n=5)] and PDX266 [control (n=5), BMJ (n=5), GEM (n=4), Combo (n=4)]. ***p≤0.001, **p≤0.01 and *p≤0.05.
Interestingly, during drug-washout phase a completely different trend in tumor volumes was observed. Notably, a sharp tumor regrowth was observed in GEM-washout tumors (Fig. 3A–3C); however, BMJ and Combo exposures displayed extended efficacy with sustained decrease in tumor volumes post-treatment termination in the drug-washout phase (Fig. 3A–3C). Overall, comparing the tumor volumes (within a specific treatment group) taken at the start of drug-washout phase (day 32 or 35) and at the end of drug-washout phase (day 64) yielded that there was a ∼7-fold increase in PDX272 tumor volume, and a ∼3-fold increase in tumor volumes of PDX266 and PDX271 explants in GEM-exposed groups. Notably, these fold-changes in tumor volumes in GEM-exposed PDX explants were more profound than untreated control tumors (Fig. 3D). Importantly, negligible fold changes in tumor volumes were observed in all three PDX explants exposed to BMJ alone, while Combo-exposed PDX272 tumors did show a 4-fold increase in tumor volumes at the end of drug-wash out phase (Fig. 3D).
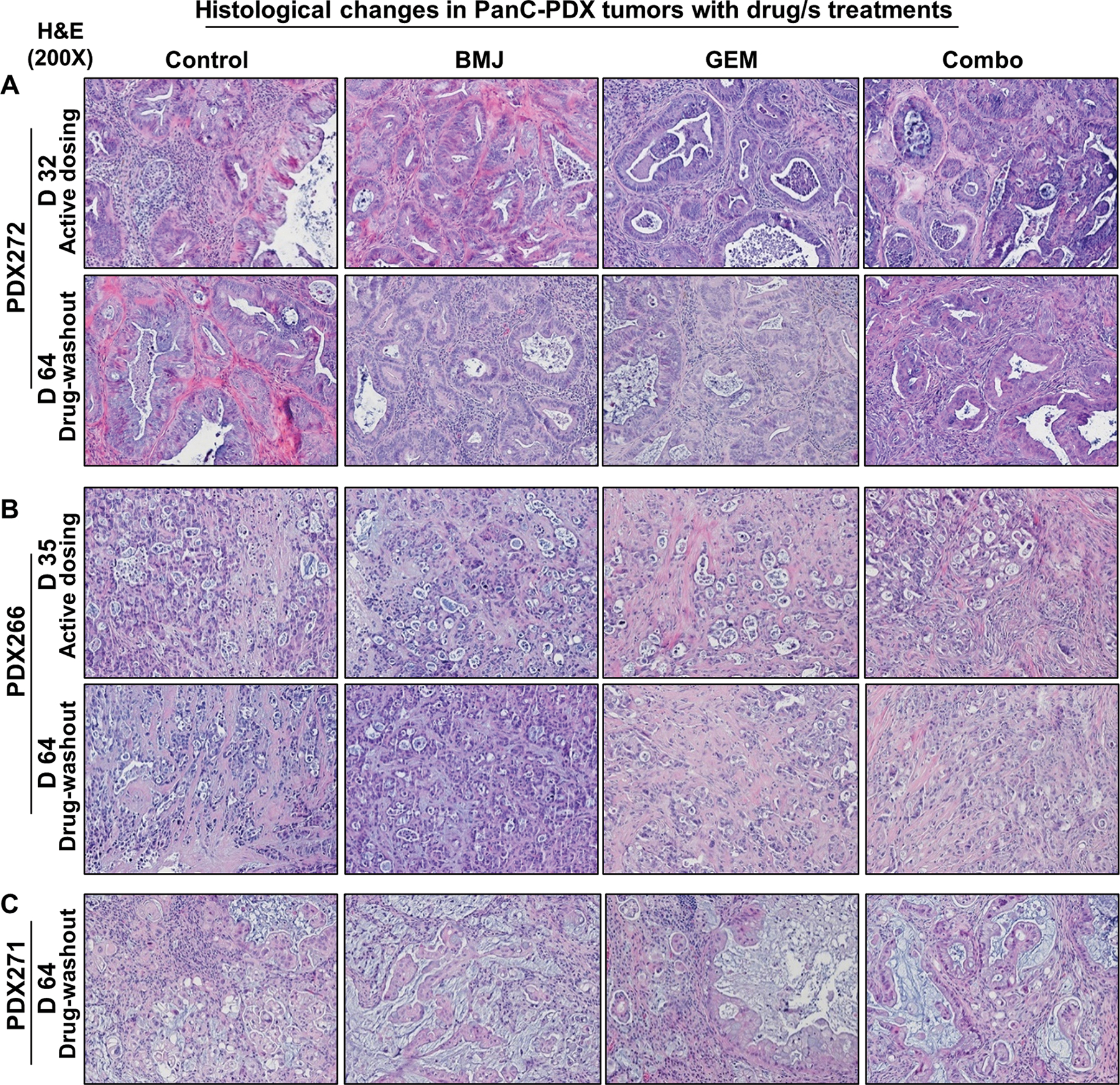
3.4. Phenotypic diversity induced by BMJ and GEM combinatorial effects in PanC-PDX explants
Next, we assessed the tumor histology of the treated PDX explants to determine whether the induced-phenotypic changes could be correlated to the in vivo anti-tumor effect of the drug/s (alone or as Combo).
PDX272 (Fig. 4A):
Figure 4. Comparative phenotypic changes induced in PanC-PDX tumor explants after treating with BMJ or GEM alone or as combination.
Representative pictographs (x200) of hematoxylin and eosin stained (A) PDX272, (B) PDX266, and (C) PDX271 explant tumors harvested after active dosing with the drug/s (day 32 or day 35) or after drug-washout phase (day 64). Descriptive details of the histopathological changes induced by drug/s is elaborated in the Results section.
During the course of the xenograft study, the harvested untreated classical PDAC tumors displayed presence of lesions having a PanIN-1B morphology, basally located nuclei with minimal changes and abundant supranuclear mucin. There was little evidence of local invasion either at day 32 or 64. Histopathological assessment of BMJ-exposed tumors indicated that dosing with BMJ for 32 days induced considerable cell death in the tumor cells and sloughing into the gland lumens with inflammatory cells present outside of the tumor mass. In the BMJ-washout tumors (at day 64), abundant inflammatory cells [lymphocytes, polymorphonuclear neutrophils (PMNs) and macrophages] were present throughout the tumor mass, dead tumor cells were visible, but overall, there was less mucin secretion and thicker glandular epithelium. Dosing with GEM also induced cell death & sloughing into gland lumens with visible inflammation, but there was no increase in thickness of glandular epithelium. In GEM-washout PDX272 explants, mucin production was reduced and there was presence of glandular epithelial thickening (few glands were also filled with necrotic debris) and inflammatory cells were present throughout the tumor mass. On the other hand, PDX272 explants dosed with Combo for 32 days displayed greatly reduced mucin and decreased epithelium thickening; notably, there was increased collagen & connective tissue deposition around the glands. By day 64 in Combo-wash out PDX272 explants, mucin secretion had further decreased, but the epithelium was piled up with increased dead cells and sloughing into gland lumens. In addition, in the Combo-washout tumors there was an increase in the number of inflammatory cells, pigmented macrophages and non-degranulated mast cells, together with increased collagen deposition in the connective tissue around glands (Fig. 4A).
PDX266 (Fig. 4B):
The harvested untreated tumors displayed lesions with PanIN-3 morphology; there were small acini with a low cuboidal cancer cells (pleomorphic in appearance), and increased invasive cells in the surrounding connective tissue with many cells pinching off into the gland lumens. Dosing of PDX266 explants with BMJ resulted in large patches of necrosis; the size of such necrotic patches increased in the BMJ-washout tumors by day 64. On the other hand, dosing with GEM caused an increase in the number of smaller glands, with lots of fibrotic connective tissue deposition and lymphocytes around the cancer mass (some calcification of materials in the gland lumens was also observed). At the end of GEM-washout phase, a further increase in the number of smaller glands (with no calcification) was observed with increased collagen connective tissue between the glands. Also, very evident invasive cell groups migrating out through the outer rim of connective tissue around the cancer mass were also present. PDX266 explants dosed with Combo also displayed an increase in the number of small glands as well as the surrounding connective tissue. There was also an increase in the number of dead tumor cells with reduced presence of lymphocytes around the cancer mass. However, in the Combo-washout PDX266 explants, way fewer glands were present or were replaced by small masses of single/few cells and there was also a marked increase in collagen deposition; more PMNs and non-degranulated mast cells were also observed in the connective tissue surrounding the cancer mass (Fig. 4B).
PDX271 (Fig. 4C):
The harvested untreated tumors displayed presence of lesions with PanIN-2 morphology and lots of larger glands with pseudostratified as well as columnar epithelium and abundant mucin (presumably even some goblet cells). There were signs of epithelial cells budding off into the gland lumen, and also signs of local invasion in surrounding tissue. Histopathological assessments in (day 64) tumors revealed that both BMJ and GEM-washout tumors had similar characteristics; a highly mucinous looking simple columnar epithelium and basal nuclei flattened against the basement membrane. On the other hand, Combo-washout PDX271 explants showed presence of two main cell populations: one with abundant mucin as evidenced in BMJ or GEM groups and the other population of cells containing much less mucin. Also, the glands were dense with increased number of inflammatory cells including pigmented macrophages and showed traces of local invasion in the surrounding tissue (Fig. 4C).
3.5. Strong fibrogenic effects of GEM, alone and in combination with BMJ, dominate the anti-tumorigenic pattern induced by these treatments in PanC-PDX explants
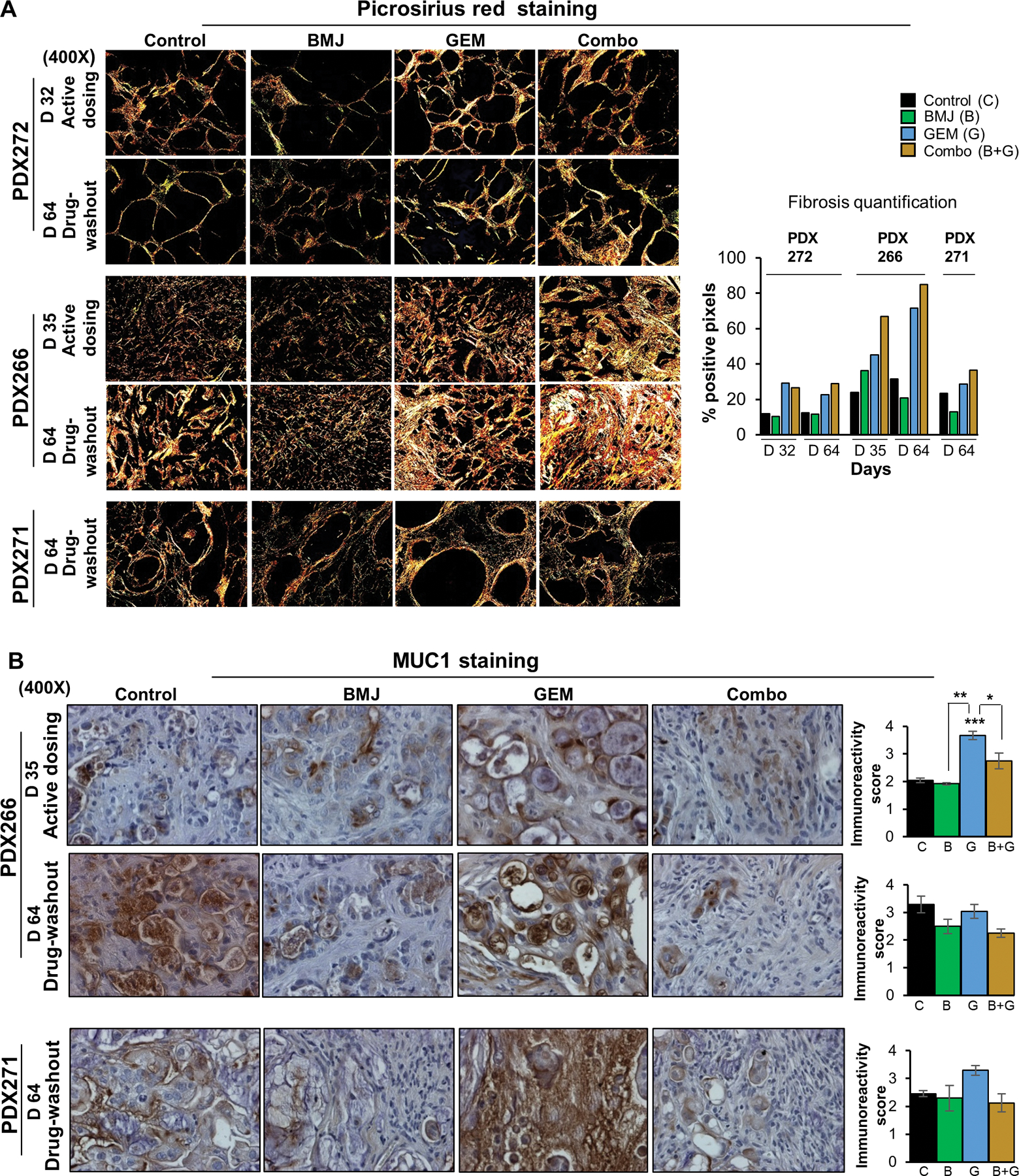
Given that in the GEM and Combo-exposed tumors (either dosed with the drugs or in the drug-washout phase) there was an increase in fibrotic connective tissue surrounding the tumor mass, we performed Picrosirius red (PSR) staining to determine the extent of fibrosis/collagen deposition in these PanC-PDXs. As shown in Fig. 5A, in PDX272 explants, fibrosis was markedly increased in dosed or drug-washout tumors in both GEM and Combo groups compared to untreated control and BMJ groups (Fig. 5A). In PDX266 explants, at the end of dosing regimen, fibrotic deposition was least in untreated control, followed by BMJ and then GEM; notably, the maximum fibrotic content was observed in dosed Combo tumors. By the end of drug-washout phase (day 64), there was a significant increase in fibrosis in GEM and Combo-washout PDX266 explants, while BMJ group had the least deposition (Fig. 5A). PDX271 explants also exhibited a similar trend as observed for PDX266 explants; the GEM and Combo-washout tumors had copious amount of fibrotic deposition while BMJ had the least (Fig. 5A). Overall, PDX266 explants had the highest fibrotic deposition in GEM and Combo groups, as determined by the percent positive pixels (Fig. 5A, right panel).
Figure 5. Combinatorial effect of BMJ and GEM on fibrosis and MUC1 expression in PanC-PDX tumors explants.
(A, left panel) Representative polarized light images (x400) from Picrosirius red (PSR) staining of PDX explant tumors after active dosing with the drug/s (day 32 or day 35) or after drug-washout phase (day 64). (A, right panel) Fibrosis quantification represented as percent positive pixels using SlideBook V.6.0. Treatment cohorts represented by C (untreated controls), B (BMJ), G (GEM) and B+G (Combo). (B, left panel) Representative pictographs (x400) of MUC1 expression in the mucinous PDX266 and PDX271 tumors after immunohistochemistry, and (B, right panel) quantification of the MUC1 immunoreactivity score (arbitrary values) in PDX tumor explants, as detailed in Methods section. Values are expressed as mean ± S.E.M. ***p≤0.001, **p≤0.01 and *p≤0.05.
Next, based on the histopathological assessments of the PDX explants which indicated that the untreated controls and the drug-exposed tumors varied in their mucin content, we evaluated the differential mucinous content of PDX266 and PDX271 explants by staining the tumors for MUC1 (Fig. 5B). Importantly, MUC1- a marker for mucin is also a marker for PanC aggressiveness and GEM resistance.25,26 IHC analysis of MUC1 staining in PDX266 explants indicated that dosing with GEM caused a significant increase in MUC1 expression. After the drug-washout phase, while GEM-washout tumors had higher MUC1 levels than BMJ and Combo-washout tumors, the BMJ and Combo groups displayed lower MUC1 levels than those of untreated controls. Similarly, in PDX271 explants, MUC1 levels were elevated in GEM-washout tumors, while the MUC1 expression in BMJ and Combo tumors was lower than the untreated controls (Fig. 5B).
3.6. Molecular effects of BMJ and GEM combinatorial treatments in PanC-PDX explants
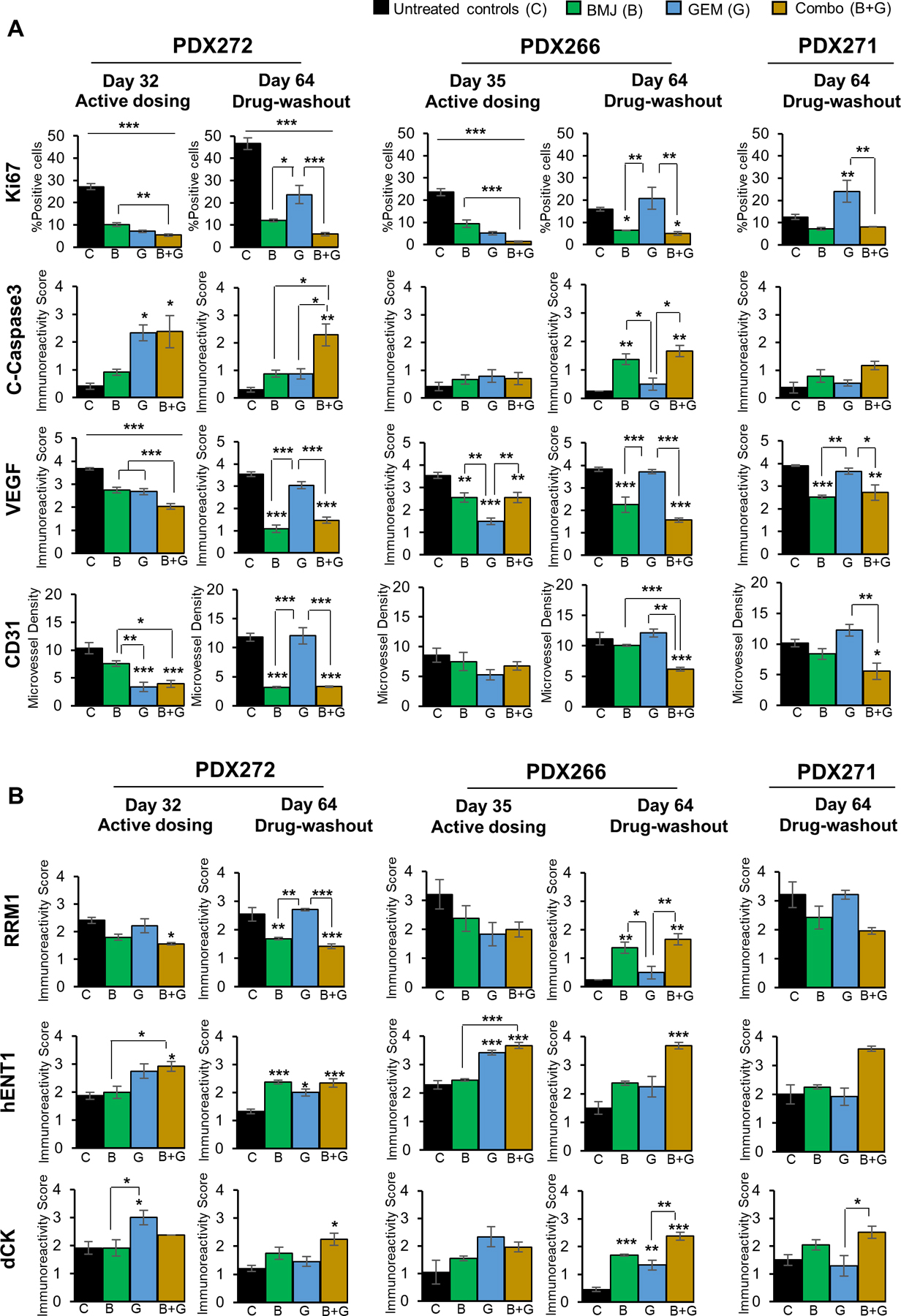
The PDX tumors generated above, either at the end of the dosing regimen or following a post treatment-washout period (no drug administration), were next subjected to molecular assessments by IHC staining to determine effects on proliferation, apoptosis, and angiogenesis. While qualitative IHC staining for these markers is depicted in Supplementary Fig. S1A, the quantitative assessments are detailed in Fig 6. IHC analysis (Fig. 6A) of cell proliferation marker Ki67 revealed a significant decrease in proliferative cells in tumors subjected to dosing with BMJ, GEM and Combo treatments. Specifically, the proliferative index was decreased by ~65%, ∼72% and ∼79% in PDX272, and by ~58%, ∼75% and ∼91% in PDX266 tumors treated with BMJ, GEM and Combo, respectively. However, this trend shifted in post treatment-washout tumors (by day 64) where GEM-washout tumors displayed a reversal in proliferative index, with significant increase in proliferative (Ki67 +ve) cells, while BMJ and Combo-washout tumors continued to have less proliferative capacity (Fig. 6A). Similar effect was observed in PDX271 post treatment-washout explants at day 64.
Figure 6.
Molecular effects of BMJ and GEM combinatorial treatments in PanC-PDX tumor explants. Immunohistochemical staining of tumors for markers involved in (A) proliferation (Ki67), apoptosis (cleaved-caspase 3), angiogenesis (CD31 (microvessel density, VEGF), and (B) GEM transport/ metabolism pathway (RRM1, hENT1 and dCK). Data details the comparative outcomes after active dosing with the drug/s (day 32 or day 35) or after drug-washout phase (day 64). Values are expressed as % positive cells or as immunoreactivity score (arbitrary values) as detailed in Methods section. Treatment cohorts represented by C-black (untreated controls), B-green (BMJ), G-blue (GEM), and B+G-yellow (Combo). Values are expressed as mean ± S.E.M. ***p≤0.001, **p≤0.01 and *p≤0.05.
Next, the extent of apoptosis induced by the drugs alone or in combination was analyzed by assessing the PDX explants for cleaved (C)-caspase 3 staining. While PDX272 explants subjected to dosing with the drugs showed a significant increase in apoptosis with GEM and Combo treatment, by the end of the study (at day 64), Combo-washout tumors were the only PDX272 explants that continued to display increased apoptosis. Interestingly, no apoptotic effects with any of the drug treatments were visible in PDX266 explants during the dosing phase (Fig. 6A); however, at day 64, both BMJ and Combo-washout tumors displayed significant increase in c-caspase 3 staining. On the other hand, no significant apoptotic signs with regards to c-caspase 3 staining were observed in PDX271 explants (Fig. 6A, Supplementary Fig. S1).
Next, we assessed whether the anti-tumor effects of the drugs alone or their combination were associated with modulation of tumor angiogenesis. IHC staining of the tumors was performed for quantifying microvessel density using PECAM-1/ CD31 as the identifying marker (Fig. 6A, Supplementary Fig. S1A). Results indicated that there was a significant decrease in microvessel density in PDX272 explants subjected to dosing with BMJ, GEM and Combo treatments. Also, GEM and Combo treatments displayed comparatively more significant anti-angiogenic effects than BMJ alone during dosing. Importantly, the effect of GEM was lost during treatment-washout phase while the BMJ and Combo-washout tumors continued to have decreased microvessel density. While anti-angiogenic effects of both drugs alone and their combination were observed during dosing in PDX266 explants, only the BMJ alone and Combo treatments caused significant decrease in the microvessel density in the washout phase at day 64, and GEM alone effect was lost. Similar protective effects of BMJ alone and Combo against angiogenesis were also observed in PDX271 washout explants. To determine whether the anti-angiogenic effect was caused due to the modulation of vascular endothelial growth factor (VEGF) levels, we next assessed the tumors for the effect on VEGF expression levels (Fig. 6A, Supplementary Fig. S1A). Importantly, while the differential expression of VEGF molecule in the tumor tissues was in line with the observed inhibitory effect (if any) of the drugs or their combination on angiogenesis, it was observed that the decrease in VEGF expression preceded the decrease in microvessel density. Another interesting observation was that during the drug-washout phase if the VEGF expression levels were restored, it negatively impacted the inhibitory effect on angiogenesis, i.e., the microvessel density was increased too.
Overall, comparing single agents versus combination in three explants PDX272, PDX266, and PDX271, both at the end of the dosing regimen as well as following a post treatment-washout period (no drug administration), revealed enhanced efficacy of the combination treatment over failed GEM, as seen in the post treatment termination cohort. Thus, to determine whether combination with BMJ was overcoming GEM resistance possibly due to the modulation of GEM uptake/metabolism pathway molecules, we performed IHC assessments for the expression of these molecules in the explants (Fig. 6B, Supplementary Fig. S1B).
Ribonucleotide reductase regulatory subunit M1 (RRM1, one of the key enzymes in GEM metabolism) is mainly known as a marker of poor survival in PanC patients undergoing GEM-based chemotherapy.27 IHC staining for RRM1 indicated that all drug exposures (either alone or in combination) led to a modest decrease in RRM1 expression during dosing regimens in PanC-explants; however, at the end of treatment-washout phase, PDX272 explants displayed restored RRM1 levels in GEM-washout group, and a sustained significant decrease in both BMJ and Combo-washout groups. Similar trend in RRM1 levels (though not significant) was observed in GEM-wash out PDX271 explants at day 64. Interestingly, in PDX266 explants, the RRM1 expression levels in untreated controls and GEM-washout tumors were both decreased at day 64 compared to the expression levels observed at day 35 (dosing phase).
Investigation into hENT1 (human equilibrative nucleoside transporter 1) expression, responsible for bidirectional GEM trafficking,28 revealed increased transporter levels in GEM and Combo groups during dosing regimens for both PDX272 and PDX266 explants. However, at the end of the washout phase, Combo tumors continued to express high levels of hENT1 for all three explants compared to GEM-washout tumors which showed poor hENT1 expression with respect to dosing tumors. Furthermore, IHC analysis for deoxycytidine kinase (dCK) expression (the enzyme required in the rate-limiting step for production of active GEM metabolites), revealed increased levels of dCK in PDX explants during dosing with GEM, while the expression was decreased during GEM-washout phase. Notably, the Combo-washout tumors had the most augmented dCK expression levels by study end at day 64 (Fig. 6B, Supplementary Fig. S1).
In summary, the expression of molecules/ transporters/ enzymes involved in GEM metabolism were significantly altered in GEM-washout tumors compared to when the tumors were dosed with the drug, thereby conferring a loss of drug sensitivity in tumor cells towards GEM. However, BMJ (partly) and mainly Combo-dosed tumors displayed sustained efficacies, possibly by overcoming the GEM resistance and conferring extended drug sensitivity in these PanC-PDX explants even in drug-washout phase.
4. DISCUSSION
GEM has been the most widely utilized first-line chemotherapeutic for PanC patients, regardless of its use resulting in only a nominal increase in patient survival.9 While patient survival and increased toxicity due to repeated high dosages with GEM treatments still remain a challenge, the rising cases of inherited or acquired GEM resistance worsen the current state of PanC therapeutics and pose serious concerns.10,11 Based on the synergistic effects of GEM in treatment combinations with other chemotherapeutics, recent strategies expanded the treatment horizon by using these combinatorial treatment regimens in PDAC patients. However, this strategy availed no significant benefits; rather it increased the rate of toxicities, thereby limiting the use of these approaches.29,30 In the light of this background, combination approaches of mainstream chemotherapeutics and non-toxic natural agents (with anti-PanC efficacy) can be explored to determine their synergistic efficacy against PanC with minimal toxic side-effects. 31,32
Previous research outcomes from our lab have indicated that the anticancer effect of BMJ in PanC18–22 is mediated via activation of cellular energy sensor AMP-activated protein kinase, targeting both CSC and bulk tumor cells, and contextually more relevant, by imparting sensitivity to GEM resistant PanC cells and spheroids.18–20 Thus, in the present study, we focused our approach on determining the combinatorial potential of BMJ and GEM to increase potency of GEM efficacy (with minimal side effects) in clinically-relevant PanC-PDX tumorigenesis model, and to further understand and establish the underlying mechanisms associated with such effects.
Even though, under in vitro conditions, the combination of BMJ+GEM showed a differential response in different PanC cell lines, under in vivo scenario, it was very evident that the combination effects were more potent. While dosing of PDX explants with GEM alone showed significant reduction in tumor size, the effects were not sustained; GEM-washout tumors exhibited tumor regrowth implying a loss of GEM sensitivity post treatment stoppage. On the other hand, Combo and BMJ (to a greater extent) demonstrated sustained anti-tumor effects even after drug-washout phase. Overall, in all the three PDX explants, the anti-tumor trends were consistent, and Combo emerged as the preferred treatment regimen with the most significant anticancer potential. Notably, the sustained effect of BMJ was observed in all the washout tumors irrespective of the mutational status of KRAS, while the sustained anti-tumor effects of Combo were more prominent in tumors harboring mutant KRAS (PDX266 and PDX271) compared to when the tumors possessed wild-type KRAS. This finding has great clinical significance given that mutant/ oncogenic KRAS is present in more than 90% of the PDACs33; importantly, KRAS mutation is considered as the initiating genetic event as well as tumor progression (metastasis) driver in PDAC.33
Furthermore, GEM exposures of the PDX explants had a fibrogenic effect in the tumors which was significantly increased in the Combo tumors. Surprisingly, the more significant the reduction in tumor size, the higher the fibrogenic content. Even though GEM has been reported to induce interstitial lung disease (associated with increased fibrosis)34 during treatment regimens, it was interesting to see this phenomenon unfold in the PDX explants itself. Although stromal remodeling and collagen degradation have been associated with improved drug delivery, certain studies also report these elements to act by restraining PDAC growth,35,36 a possible explanation of the outcomes in the present study. Hence, this phenomenon needs to be explored further and is a possible subject for deeper investigation, because contrary to the popular belief associating higher fibrotic content with PDAC aggressiveness, GEM and Combo treatments indeed resulted in increased fibrosis, albeit accompanying the most significant anti-tumor effects in Combo group.
IHC analysis of PDX explants revealed inhibition of tumor cell proliferation and angiogenesis alongside apoptosis induction, decreased MUC1 expression, and sustained efficacy post treatment termination in BMJ and Combo groups. The subtle differences between treatment responses in PDX explants warrant further histological analysis and interpretation of our findings but can be partly attributed to the different tumor histological grade/type of each of the three PDX explants. The switch in GEM efficacy, from being highly effective during dosing regimen to lost drug sensitivity during washout phase, also pointed towards aberrant GEM transport /metabolism. This was verified by IHC staining for relevant molecules which indicated differential levels of hENT1, dCK, and RRM1 molecules correlating with GEM resistance and dismal patient survival.27,37,38 Interestingly, in our other continuing study on BMJ-associated molecular mechanisms 39 we have observed that BMJ-exposure of PanC cells results in the modulation of PanC cell metabolome, where BMJ caused an intracellular lactate accumulation in PanC cells, thereby decreasing the acidity of extracellular environment, which in turn could possibly result in improved GEM uptake in PanC cells by BMJ,40,41 thus explaining the enhanced efficacy of Combo compared to GEM alone. However, additional studies focused on GEM uptake in combination with BMJ are warranted to validate these observations.
Overall, these studies provide compelling evidence regarding significant combinatorial efficacy of BMJ and GEM in a high fidelity preclinical PDX mouse model of PanC, thereby providing a promising approach for future clinical investigations targeting mutant/oncogenic KRAS harboring PDAC tumors.
Supplementary Material
Supplementary Figure S1. Representative pictographs (x400) of immunohistochemical staining of PanC-PDX tumor explants for markers involved in (A) proliferation (Ki67), apoptosis (cleaved-caspase 3), angiogenesis (CD31 (microvessel density, VEGF), and (B) GEM transport/ metabolism pathway (RRM1, hENT1 and dCK). PanC-PDX272, PDX266 and PDX271 tumor explants were either treated with BMJ or GEM alone or their combination, tumors were harvested after active dosing with the drug/s (day 32 or day 35) or after drug-washout phase (day 64).
Acknowledgements and Funding source:
This work was supported by National Institute of Health/ National Cancer Institute grant R01CA195708 (RA). The authors acknowledge the assistance of E. Erin Smith, TL(ASCP)CMQIHC; Jenna Van Der Volgen, HT(ASCP)CM; Allison Quador, HTL(ASCP)CM; and Jessica Arnold, HTL(ASCP)CM at University of Colorado Denver Histology Shared Resource, which is supported in part by the University of Colorado Cancer Center Shared Resources Grant (P30CA046934).
Abbreviations:
- PanC
pancreatic cancer
- BMJ
bitter melon juice
- GEM
gemcitabine
- PDX
patient derived xenograft
- IHC
immunohistochemistry
- PanIN
pancreatic intraepithelial neoplasia
- PDAC
pancreatic ductal adenocarcinoma
- AMPK
adenosine monophosphate activated protein kinase
- RRM1
Ribonucleotide reductase regulatory subunit M1
- hENT1
human equilibrative nucleoside transporter 1
- dCK
deoxycytidine kinase
- MUC1
Mucin 1
- VEGF
vascular endothelial growth factor
- PMNs
polymorphonuclear neutrophils
Footnotes
Disclosure of Potential Conflict of Interest: The authors declare no potential conflicts of interest.
Supplementary Data-description: Supplementary Fig. S1 is available as an addendum (representative pictographs of IHC staining of PanC-PDX tumor explants) to the quantitative data shown in Figure 6.
Data Availability:
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin 2018;68(1):7–30. [DOI] [PubMed] [Google Scholar]
- 2.Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res 2014;74(11):2913–2921. [DOI] [PubMed] [Google Scholar]
- 3.Zhang L, Sanagapalli S, Stoita A. Challenges in diagnosis of pancreatic cancer. World J Gastroenterol 2018;24(19):2047–2060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rossi ML, Rehman AA, Gondi CS. Therapeutic options for the management of pancreatic cancer. World J Gastroenterol 2014;20(32):11142–11159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sener SF, Fremgen A, Menck HR, Winchester DP. Pancreatic cancer: a report of treatment and survival trends for 100,313 patients diagnosed from 1985–1995, using the National Cancer Database. J Am Coll Surg 1999;189(1):1–7. [DOI] [PubMed] [Google Scholar]
- 6.Kleeff J, Korc M, Apte M, et al. Pancreatic cancer. Nat Rev Dis Primers 2016;2:16022. [DOI] [PubMed] [Google Scholar]
- 7.Takai E, Yachida S. Genomic alterations in pancreatic cancer and their relevance to therapy. World J Gastrointest Oncol 2015;7(10):250–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Von Hoff DD, Ervin T, Arena FP, et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med 2013;369(18):1691–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Burris HA 3rd, Moore MJ, Andersen J, et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J Clin Oncol 1997;15(6):2403–2413. [DOI] [PubMed] [Google Scholar]
- 10.Wang Y, Hu GF, Zhang QQ, et al. Efficacy and safety of gemcitabine plus erlotinib for locally advanced or metastatic pancreatic cancer: a systematic review and meta-analysis. Drug Des Devel Ther 2016;10:1961–1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Binenbaum Y, Na’ara S, Gil Z. Gemcitabine resistance in pancreatic ductal adenocarcinoma. Drug Resist Updat 2015;23:55–68. [DOI] [PubMed] [Google Scholar]
- 12.Hutchinson L, Kirk R. High drug attrition rates--where are we going wrong? Nat Rev Clin Oncol 2011;8(4):189–190. [DOI] [PubMed] [Google Scholar]
- 13.Tentler JJ, Tan AC, Weekes CD, et al. Patient-derived tumour xenografts as models for oncology drug development. Nat Rev Clin Oncol 2012;9(6):338–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nunes M, Vrignaud P, Vacher S, et al. Evaluating patient-derived colorectal cancer xenografts as preclinical models by comparison with patient clinical data. Cancer Res 2015;75(8):1560–1566. [DOI] [PubMed] [Google Scholar]
- 15.Topp MD, Hartley L, Cook M, et al. Molecular correlates of platinum response in human high-grade serous ovarian cancer patient-derived xenografts. Mol Oncol 2014;8(3):656–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pitts TM, Tan AC, Kulikowski GN, et al. Development of an integrated genomic classifier for a novel agent in colorectal cancer: approach to individualized therapy in early development. Clin Cancer Res 2010;16(12):3193–3204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rubio-Viqueira B, Jimeno A, Cusatis G, et al. An in vivo platform for translational drug development in pancreatic cancer. Clin Cancer Res 2006;12(15):4652–4661. [DOI] [PubMed] [Google Scholar]
- 18.Dhar D, Deep G, Kumar S, et al. Bitter melon juice exerts its efficacy against pancreatic cancer via targeting both bulk and cancer stem cells. Mol Carcinog 2018. doi 10.1002/mc.22833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kaur M, Deep G, Jain AK, et al. Bitter melon juice activates cellular energy sensor AMP-activated protein kinase causing apoptotic death of human pancreatic carcinoma cells. Carcinogenesis 2013;34(7):1585–1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Somasagara RR, Deep G, Shrotriya S, Patel M, Agarwal C, Agarwal R. Bitter melon juice targets molecular mechanisms underlying gemcitabine resistance in pancreatic cancer cells. Int J Oncol 2015;46(4):1849–1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dhar D, Raina K, Agarwal R. Mechanisms and Drug Targets for Pancreatic Cancer Chemoprevention. Current medicinal chemistry 2018;25(22):2545–2565. [DOI] [PubMed] [Google Scholar]
- 22.Raina K, Kumar D, Agarwal R. Promise of bitter melon (Momordica charantia) bioactives in cancer prevention and therapy. Seminars in cancer biology 2016;40–41:116–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.da Silva RF, Dhar D, Raina K, et al. Nintedanib inhibits growth of human prostate carcinoma cells by modulating both cell cycle and angiogenesis regulators. Scientific reports 2018;8(1):9540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chou TC. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol Rev 2006;58(3):621–681. [DOI] [PubMed] [Google Scholar]
- 25.Nath S, Daneshvar K, Roy LD, et al. MUC1 induces drug resistance in pancreatic cancer cells via upregulation of multidrug resistance genes. Oncogenesis 2013;2:e51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shukla SK, Purohit V, Mehla K, et al. MUC1 and HIF-1alpha Signaling Crosstalk Induces Anabolic Glucose Metabolism to Impart Gemcitabine Resistance to Pancreatic Cancer. Cancer Cell 2017;32(1):71–87 e77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nakahira S, Nakamori S, Tsujie M, et al. Involvement of ribonucleotide reductase M1 subunit overexpression in gemcitabine resistance of human pancreatic cancer. Int J Cancer 2007;120(6):1355–1363. [DOI] [PubMed] [Google Scholar]
- 28.Spratlin J, Sangha R, Glubrecht D, et al. The absence of human equilibrative nucleoside transporter 1 is associated with reduced survival in patients with gemcitabine-treated pancreas adenocarcinoma. Clin Cancer Res 2004;10(20):6956–6961. [DOI] [PubMed] [Google Scholar]
- 29.Jin SF, Fan ZK, Pan L, Jin LM. Gemcitabine-based combination therapy compared with gemcitabine alone for advanced pancreatic cancer: a meta-analysis of nine randomized controlled trials. Hepatobiliary Pancreat Dis Int 2017;16(3):236–244. [DOI] [PubMed] [Google Scholar]
- 30.Mizusawa J, Fukutomi A, Katayama H, et al. Protocol digest of randomized phase II study of modified FOLFIRINOX versus gemcitabine plus nab-paclitaxel combination therapy for locally advanced pancreatic cancer: Japan clinical oncology group study (JCOG1407). Pancreatology 2018. doi 10.1016/j.pan.2018.07.007. [DOI] [PubMed] [Google Scholar]
- 31.Marasini B, Sahu RP. Natural Anti-Cancer Agents: Implications in Gemcitabine-Resistant Pancreatic Cancer Treatment. Mini Rev Med Chem 2017;17(11):920–927. [DOI] [PubMed] [Google Scholar]
- 32.Yue Q, Gao G, Zou G, Yu H, Zheng X. Natural Products as Adjunctive Treatment for Pancreatic Cancer: Recent Trends and Advancements. Biomed Res Int 2017. doi 10.1155/2017/8412508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Waters AM, Der CJ. KRAS: The Critical Driver and Therapeutic Target for Pancreatic Cancer. Cold Spring Harb Perspect Med 2018;8(9):a031435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Umemura S, Yamane H, Suwaki T, et al. Interstitial lung disease associated with gemcitabine treatment in patients with non-small-cell lung cancer and pancreatic cancer. Journal of cancer research and clinical oncology 2011;137(10):1469–1475. [DOI] [PubMed] [Google Scholar]
- 35.Rhim AD, Oberstein PE, Thomas DH, et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell 2014;25(6):735–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ozdemir BC, Pentcheva-Hoang T, Carstens JL, et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 2014;25(6):719–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Farrell JJ, Elsaleh H, Garcia M, et al. Human equilibrative nucleoside transporter 1 levels predict response to gemcitabine in patients with pancreatic cancer. Gastroenterology 2009;136(1):187–195. [DOI] [PubMed] [Google Scholar]
- 38.Ohhashi S, Ohuchida K, Mizumoto K, et al. Down-regulation of deoxycytidine kinase enhances acquired resistance to gemcitabine in pancreatic cancer. Anticancer Res 2008;28(4B):2205–2212. [PubMed] [Google Scholar]
- 39.Dhar D, Raina K, Kant R, et al. Bitter melon juice-intake modulates glucose metabolism and lactate efflux in tumors in its efficacy against pancreatic cancer. Carcinogenesis 2019. doi 10.1093/carcin/bgz114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Phua LC, Goh S, Tai DWM, et al. Metabolomic prediction of treatment outcome in pancreatic ductal adenocarcinoma patients receiving gemcitabine. Cancer Chemother Pharmacol 2018;81(2):277–289. [DOI] [PubMed] [Google Scholar]
- 41.Wike-Hooley JL, Haveman J, Reinhold HS. The relevance of tumour pH to the treatment of malignant disease. Radiother Oncol 1984;2(4):343–366. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1. Representative pictographs (x400) of immunohistochemical staining of PanC-PDX tumor explants for markers involved in (A) proliferation (Ki67), apoptosis (cleaved-caspase 3), angiogenesis (CD31 (microvessel density, VEGF), and (B) GEM transport/ metabolism pathway (RRM1, hENT1 and dCK). PanC-PDX272, PDX266 and PDX271 tumor explants were either treated with BMJ or GEM alone or their combination, tumors were harvested after active dosing with the drug/s (day 32 or day 35) or after drug-washout phase (day 64).
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.