

Abstract
The idiosyncratic activation of mast cells (MCs) in response to the administration of nonselective cyclooxygenase (COX) inhibitors is a cardinal feature of aspirin exacerbated respiratory disease (AERD). Older studies using mast cell stabilizing drugs support a critical role for MCs and their products in driving the severe eosinophilic inflammation and respiratory dysfunction that is typical of AERD. Since patients with AERD react to all nonselective COX inhibitors regardless of their chemical structure, the mechanism of MC activation is not due to classical, antigen-induced cross-linking of IgE receptors. Recent studies in both human subjects and animal models have revealed a complex and multifactorial process culminating in dysregulation of MC function, and an aberrant dependency on COX-1-derived prostaglandin E2 to maintain a tenuous homeostasis. This article reviews the factors most likely to contribute to MC dysregulation in AERD, and the potential diagnostic and therapeutic implications.
Keywords: Aspirin, asthma, leukotrienes, mast cells, prostaglandins
Introduction
Mast cells abound in all barrier tissues, including the respiratory mucosa. Although their physiologic functions in healthy humans are unknown, animal studies strongly support a key role in the initiation and amplification of innate immune responses to microbial pathogens (1). The functions of MCs in type 1 hypersensitivity responses that underlie anaphylaxis, urticaria, and acute allergen-driven episodes of rhinitis and atopic asthma is well-established. MC-derived products such as histamine, cysteinyl leukotrienes (cysLTs), prostaglandin D2 (PGD2), cytokines and chemokines have functions in allergic diseases that are validated or strongly supported by experimental evidence (2). The ability of blocking antibodies against the Fc portion of IgE to block allergen-induced early and late phase responses in the airways of atopic asthmatics (3) further validates the importance of this classical activation pathway.
Aspirin exacerbated respiratory disease (AERD) is a distinctive syndrome, typically of adult onset, that is characterized by severe eosinophilic sinonasal disease with recurrent nasal polyposis, asthma (which is often but not uniformly severe), and pathognomonic clinical reactions to the ingestion of aspirin or any other drug that inhibits the enzymatic function of cyclooxygenase (COX)-1 (4). It affects ~7% of all asthmatic subjects in the United States and as many as 14% of subjects with severe asthma (5). The pathophysiology of AERD involves dysregulated MC function, and MC-derived products contribute substantially to the baseline immunopathology of the disease (6). Moreover, the pathognomonic clinical reactions to COX-1 active drugs are associated with marked activation of MCs by mechanisms that are poorly understood but are not due to IgE-dependent immune recognition of the drug (7–9). This review summarizes the potential significance of MCs in AERD, and the recent studies that highlight potential mechanisms responsible for their dysregulation and activation in this context.
Evidence to support a key role for MCs in AERD.
When activated, MCs generate LTC4, the parent of the cysLTs, through the 5-lipoxygenase (5-LO)/LTC4 synthase (LTC4S) pathway (10). This pathway is shared with eosinophils, basophils, and monocytes. MCs also metabolize arachidonic acid through the COX pathway to generate especially large quantities of PGD2 (11). CysLTs and PGD2 synergize to activate human innate group 2 lymphoid cells (ILC2s) and T helper type 2 (Th2) cells. CysLTs are the most potent known constrictors of human airways (12), elicit mucous secretion (13), and activate IL-25-expressing airway brush cells (14). The terminal cysLT metabolite, LTE4, can be measured in the urine, as can stable metabolites (PGD-M) of PGD2, as time-weighted reflections of LTC4 and PGD2 generation, respectively. In AERD, urinary levels of both LTE4 and PGD-M exceed those in aspirin tolerant controls by several fold (15, 16), consistent with ongoing MC activation. The administration of aspirin induces sharp additional increases in both mediators, accompanied by increases in plasma histamine (which may also reflect concomitant basophil activation) and (in some individuals) serum tryptase, confirming MC activation. The numbers of MCs are increased in the bronchial biopsies and sinonasal mucosa of patients with AERD compared with aspirin tolerant controls (17, 18). Thus, AERD involves relative MC hyperplasia, persistent steady-state MC activation (“leaky MCs”), and marked unbraking of MC activation with COX-1 blockade. Importantly, the unbraking phenomenon occurs with all drugs (regardless of structure) that interfere with COX-1, indicating that reactions are not manifestations of IgE-dependent “drug allergy”, but rather reflect an idiosyncratic dependency on one or more COX products to maintain a tenuous homeostasis over MCs and other effector cells.
Several older studies using classical MC-stabilizing cromone drugs strongly suggest that both the steady-state MC “leak” and the incremental activation with COX-1 inhibition contribute substantially to AERD pathophysiology. Although the administration of cromolyn has no acute effect on baseline lung function measurements in aspirin tolerant asthmatic control subjects, it increased FEV1 in subjects with AERD within minutes of administration (19). In another study, treatment with cromolyn for one week significantly decreased the numbers of sputum eosinophils and sputum levels of eosinophilic cationic protein in subjects with AERD (20). The administration of cromolyn or nedocromil to subjects with AERD blocks the reduction in FEV1 that occurs with aspirin challenges (21), as well as the accompanying increase in urinary LTE4 levels (22). Collectively, these studies strongly support the importance of MCs and their mediators in both the steady-state respiratory dysfunction and the clinical reactions to COX-1 inhibition in AERD, emphasizing the importance of understanding the idiosyncratic mechanisms responsible for the dysregulated function of MCs in this disease.
Factors contributing to MC activation in AERD.
1. Dysregulation of the prostaglandin E2/EP2 receptor system.
PGE2 is a ubiquitous COX product with MC-stabilizing properties. It is generated constitutively at steady-state and inducibly at very high levels during inflammatory responses. Epithelial cells, fibroblasts, and macrophages all generate substantial quantities of PGE2 when stimulated ex vivo with IL-1β or LPS. This response reflects inducible expression of COX-2 and microsomal PGE2 synthase-1 (mPGES-1), the principal terminal synthase responsible for generating PGE2 during inflammatory responses. Compared with constitutively expressed COX-1, COX-2 is much less sensitive to inhibition by aspirin (23). Consequently, PGE2 derived from COX-2/mPGES-1 during inflammatory responses may be maintained even in the face of low dose aspirin. PGE2 signaling through the E prostanoid 2 (EP2) receptor blocks MC degranulation in response to IgE receptor cross-linking (24), and dampens leukotriene production by inducing protein kinase A-dependent phosphorylation of 5-LO (25). Thus, PGE2 may function to restrain MC activation and cysLT generation during inflammatory responses.
Impaired function of the COX-2/mPGES-1 system may potentiate the risk of AERD (Fig. 1). Nasal polyp levels of PGE2 are markedly lower than in non-polypoid sinonasal tissue (26), and fibroblasts from nasal polyps show weak induction of COX-2 and mPGES-1 expression compared with cells from nonpolyp controls (27). These levels tend to be lowest in samples from subjects with AERD (28). Impaired expression of COX-2 and/or mPGES-1 could render the maintenance of local PGE2 generation disproportionately dependent on COX-1, predisposing to depletion below a critical threshold when aspirin is administered. Indeed, clinical reactions generally occur at low threshold doses (80-160 mg) of aspirin that are typically selective for COX-1. The administration of a single dose of PGE2 by inhalation blocks aspirin-induced changes in lung function and increases in urinary LTE4 in subjects with AERD (29), consistent with a major role for endogenous PGE2 as the critical COX-1-derived brake. The effect of depleting PGE2 with COX-1 inhibition may be potentiated by reduced levels of EP2 receptor expression by MCs (and other 5-LO-expressing leukocytes) in subjects with AERD, as suggested by immunohistochemical staining of sinonasal and lung tissues (30, 31). Mice lacking either mPGES-1 or EP2 receptors (Ptges−/− and Ptger2−/− mice, respectively) display AERD-like features, including aspirin-induced mast cell activation and increases in cysLTs, supporting the likely importance of perturbations in these systems as disease-causing lesions (32). Some studies support an epigenetic basis for these perturbations (33, 34), consistent with the acquired nature and persistence of AERD.
Figure 1. Biosynthesis of PGE2 and its potential dysregulation in AERD.
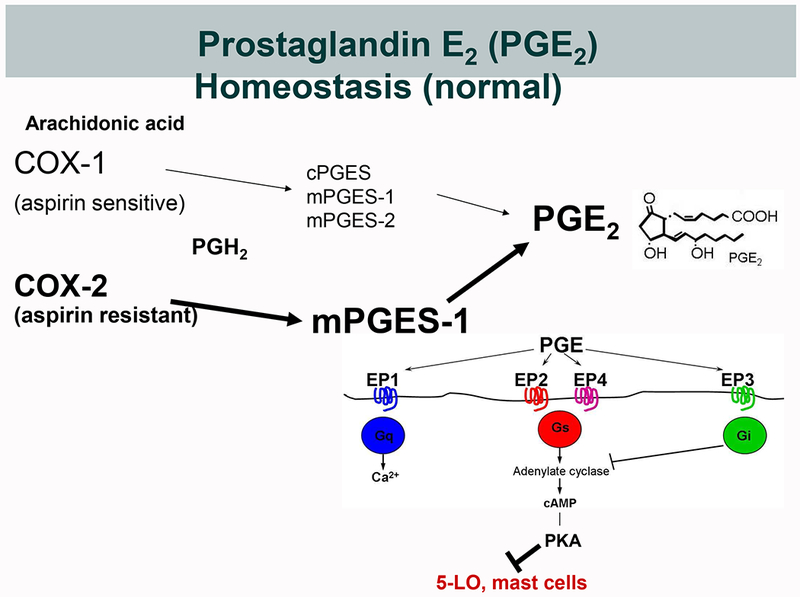
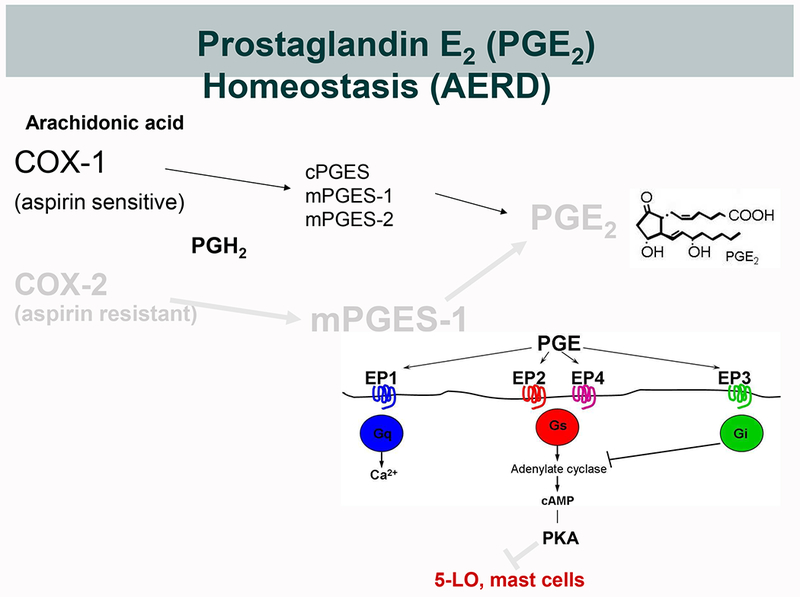
A. Under physiological conditions, PGE2 derives from either constitutively expressed COX-1 or inducibly expressed COX-2. COX-derived PGH2 can be converted to PGE2 by three different terminal PGE2 synthase (PGES) enzymes; cytosolic (c)PGES, and microsomal (m)PGES-1 and -2. During inflammation, the concomitantly induced expressions of COX-2 and mPGES-1 markedly increase the production of PGE2. Because COX-2 is much less sensitive to aspirin than is COX-1, this increase is relatively resistant to low dose aspirin. PGE2 elicits signaling through EP receptors 1-4. EP2 and EP4 activate the cAMP/PKA pathway that stabilizes MCs and blocks leukotriene formation by phosphorylating 5-LO. B. In AERD, the impaired expression and function of the COX-2/mPGES-1 system renders PGE2 production disproportionately dependent on COX-1 and susceptible to inhibition by low dose aspirin. The effects of PGE2 depletion is potentiated by diminished EP2 receptor expression, leading to unbraking of MCs and 5-LO.
Whereas low-dose aspirin-induced depletion of COX-1-derived PGE2 likely permits the triggering of MC activation during provocative challenges, high dose aspirin (325-650 mg twice daily, a dose sufficient to block COX-2) following desensitization can paradoxically improve sinonasal function and reduce the frequency of polyp recurrent in patients with AERD (35). Cahill and colleagues (36) demonstrated that patients with AERD on high dose aspirin display significantly increased total serum tryptase levels, as well as elevated urinary levels of LTE4, compared with their pre-challenge baselines, suggesting paradoxical amplification of MC “leak” even in the face of substantial clinical improvement on aspirin. This increase in MC activation markers was paralleled by sharply reduced levels of PGE2 metabolites in the urine, again consistent with the dependence of MCs on PGE2 to maintain a tenuous homeostasis in AERD. Since the tryptase was not fractionated in this study, it is not currently possible to ascertain whether ongoing MC degranulation is increased, or whether depletion of PGE2 dysregulates transcriptional control of tryptase synthesis. Notably, while PGD2 production increases during reactions, the desensitized state on high dose aspirin is accompanied by sharp reductions in urinary PGD-M levels (7). Nasal polyp MCs express markedly higher levels of mRNA encoding COX-2 than COX-1 (16), potentially explaining why PGD2 production resists inhibition during provocative challenges until the aspirin dose is escalated. The suppression of PGD2 production (with its attendant activating and chemotactic effects for eosinophils, basophils, group 2 innate lymphoid cells (ILC2s) and Th2 cells) could contribute to the therapeutic benefit of aspirin (7).
2. Dysregulation of cysLT synthesis and cysLT receptor function.
CysLTs contribute substantially to the baseline pathophysiology of AERD (37), and are essential to the pathognomonic reactions to aspirin (38). The dysregulated synthesis of the cysLTs in AERD (15) is paralleled by strongly upregulated expression of the type 1 cysLT receptor (CysLT1R) on hematopoietic cells, including MCs, in the airways (39), suggesting a mechanism driving a state of end-organ hyperreactivity to endogenous cysLTs. Indeed, subjects with AERD display a markedly higher sensitivity to bronchoconstriction induced by inhaled LTE4 than do aspirin tolerant asthmatic controls (40). Both hyperreactivity to LTE4 and overexpression of CysLT1R are reduced toward levels seen in aspirin tolerant controls after treatment with high dose aspirin (39, 40). A study by Fischer and colleagues strongly suggested that endogenous cysLTs are required for the idiosyncratic MC activation mechanism that characterizes AERD. In this study, the administration of the 5-LO inhibitor zileuton not only blocked incremental sinonasal symptoms in response to the challenge, but prevented the increase in nasal lavage fluid levels of tryptase (41). Although endogenous cysLTs are not required for IgE-dependent degranulation of MCs in ex vivo systems (42), exogenous cysLTs are sufficient to elicit calcium flux, cytokine production, and PGD2 generation from human cord blood MCs (42, 43). Moreover, a recent study demonstrated that inhalation of LTE4 by asthmatic (mostly aspirin-tolerant) subjects induced markedly increased urinary levels of PGD2, indicative of MC activation in vivo. This response was blocked by the CysLT1R antagonist montelukast (44). While cysLTs are very likely to signal directly to MCs to induce their activation, animal models suggest additional effects related to cysLT-dependent control of MC-active cytokines (see below).
3. The role of IgE and FcεRI.
While the rates of atopy in subjects with AERD vary depending on the study (45, 46), the disease frequently occurs in subjects who lack IgE with specificity to inhalant allergens (some of whom nonetheless have total IgE above the normal range)(47). A study by Taniguchi and colleagues demonstrated that treatment of patients with AERD with recombinant humanized anti-IgE (Omalizumab) in an open label protocol markedly improved symptomatic control of both asthma and sinonasal disease, while decreasing the urinary levels of LTE4 and PGD-M by >75% each (48). In a small placebo-controlled trial, Lang and colleagues demonstrated that omalizumab treatment for 16 weeks prevented clinical symptoms and increases in urinary LTE4 in 5 of 7 subjects challenged with aspirin (49), whereas all four placebo-treated subjects experienced reactions. Thus, despite the lack of evidence implicating classical allergen-induced MC activation in AERD physiology, these studies suggest that ongoing signaling through MC-associated IgE receptors permit and amplify activation responses to depletion of PGE2. The nature of the antigens driving the IgE production remains to be determined, though local production of antibodies against Staphylococcal antigens (50) and autoantigens (51) have been reported in nasal polyps. It is unknown whether the levels of these antibodies distinguish AERD subjects from aspirin tolerant controls.
4. The role of innate cytokines.
MCs express receptors for both IL-33 and thymic stromal lymphopoietin (TSLP). Both cytokines are abundant in nasal polyps (52), with mRNA for each localizing principally to epithelial basal cells (53). IL-33 potently induces the production of type 2 cytokines and chemokines by MCs ex vivo (54), and strongly induces COX-2-dependent production of PGD2 (16, 55). TSLP strongly potentiates these IL-33-induced effects (16, 54), suggesting that these resident tissue-derived cytokines may synergize to influence MC function in vivo. In AERD, the levels of TSLP expression in the sinonasal tissue correlate strongly with urinary levels of PGD-M (and with MC-specific transcripts such as tryptase, carboxypeptidase A3, and hematopoietic PGD2 synthase), suggesting that TSLP conditions MCs for augmented function in this disease (16). One study reported that the quantities of IL-33 protein in nasal polyp extracts from patients with AERD substantially exceeded those in non-AERD control samples (56).
IL-33 may be especially important in regulating MC function in AERD. In AERD-like Ptges−/− mice, antibody-mediated blockade of IL-33 or blockade of suppressor of tumorigenicity 2 (ST2, the IL-33 receptor) with a soluble Fc fusion protein completely prevents MC activation (with attendant release of proteases, production of cysLTs and PGD2) as well as the increases in airway resistance occurring in response to aspirin challenges (56). These mice display high levels of lung IL-33 protein following the induction of airway inflammation by house dust mite allergen. This induced expression of IL-33 in Ptges−/− mice requires endogenous cysLTs and signaling through the type 2 cysLT receptor (CysLT2R) (57). Moreover, cysLTs not only drive IL-33 expression by alveolar type 2 cells, but also induce the recruitment and activation of IL-33+ platelets, the latter of which are necessary for the MC response to aspirin challenge. Platelets release substantial amounts of pre-formed, cytosolic IL-33 in response to stimulation by LTC4 (58). Notably, large numbers of extravasated platelets are present in nasal polyps from patients with AERD compared with aspirin tolerant controls (59), and stimulation of freshly excised human nasal polyp tissue with LTC4 ex vivo results in the release of substantial amounts of IL-33 (58). It is tempting to speculate that the high levels of IL-33 protein found in AERD polyps may partially reflect this cysLT-driven platelet store.
Notably, while IL-33 alone is insufficient to elicit degranulation of MCs ex vivo, it does cause degranulation of mouse MCs that are passively sensitized with IgE, and can cause anaphylaxis in mice when IgE is present (60). This finding suggests a potential interaction between IL-33/ST2 and IgE-driven pathways that are permissive for the leaky MC phenotype in AERD, and may help to explain both the incremental degranulation of MCs with aspirin challenge and therapeutic responses to omalizumab.
Pathophysiologic and therapeutic implications
MC activation in AERD appears to be driven by complementary dysregulation of systems that respectively induce stabilization (PGE2/EP2) or activation (IgE, IL-33, cysLTs) of MCs (Fig. 2). Blockade of 5-LO with zileuton provides incremental improvement in sinonasal function for patients already treated with glucocorticoids (37). It seems possible that the marked effect of zileuton on signs, symptoms, and biomarkers of reactions to aspirin may reflect inhibition of both direct (CysLT1R-mediated) (44) and indirect (CysLT2R-driven, IL-33-mediated) cysLT-dependent pathways that facilitate MC activation. The potential efficacy of dual-specific antagonists of CysLT1R and CysLT2R could exceed that of the existing CysLT1R-selective drugs. Stable PGE2 analogues that selectively stimulate the EP2 receptor could inhibit the activation of MCs (and other EP2 receptor-bearing effector cells) and limit cysLT production without the cough-inducing effects that preclude the use of PGE2 itself, though reduced expression of EP2 may limit efficacy in some individuals. The encouraging results of omalizumab in small trials (48, 49) suggests a need for larger studies, and compels further study of the antigens potentially responsible for driving IgE synthesis. The high levels of PGD2 production in AERD suggests that antagonists of the type 2 PGD2 receptor (DP2, also known as CRTH2) may have efficacy by suppressing tissue eosinophilia. Biologics under development that target IL-33 and ST2 should provide opportunities to validate potential causative pathways implicated in experimental models, as well as potential efficacy. Lastly, the MC-depleting tyrosine kinase inhibitor imatinib was recently shown to improve lung function and airway reactivity in a population of refractory asthmatics not selected for the presence or absence of AERD (61). While imatinib has other potential targets (e.g., platelet-derived growth factor receptors) that may account for its benefits independent of MCs, it is possible that it may offer efficacy in AERD given the strong MC activation signatures associated with the disease.
Figure 2. Hypothetical factors contributing to dysregulation of MC activation in AERD.
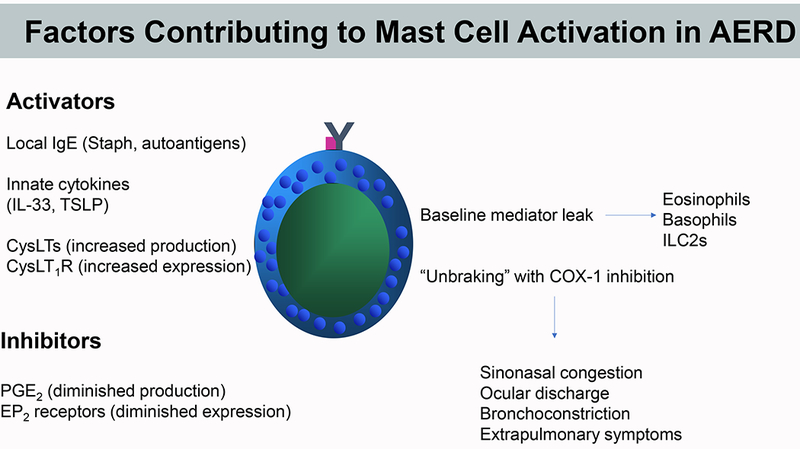
Occupancy of FcεRI, increased signaling through CysLT1R, and locally derived innate cytokines (TSLP and IL-33) may conspire to promote an ongoing “leak” of PGD2 and other MC products that facilitate eosinophilic pathology and baseline respiratory tissue dysfunction in AERD. PGE2 -dependent maintenance of homeostasis is compromised by poor COX-2/mPGES-1 and EP2 receptor function. Aspirin and other COX-1 inhibitors remove the brake provided by residual PGE2, markedly destabilizing the system to permit marked MC activation and associated end-organ responses during pathognomonic reactions.
Bullet points:
Diminished levels of COX-2 and mPGES-1 expression likely impair PGE2 generation in AERD and render residual PGE2 production highly dependent on COX-1
Reduced levels of EP2 receptor expression on mast cells and other effector cells may exacerbate the effects of depleting PGE2 with aspirin and other COX-1 active drugs.
The high levels of cysLT production in AERD are accompanied by increased levels of CysLT1R expression on hematopoietic cells.
Desensitization to aspirin downregulates CysLT1R expression and decreases end-organ reactivity to cysLTs, which may help explain clinical improvement despite substantial further increases in cysLT generation.
Local IgE produced against Staph or autoantigens in nasal polyps may synergize with IL-33 to promote ongoing MC activation, which is likely exaggerated in AERD due to reduced PGE2/EP2 signaling and enhanced cysLT generation.
What we do know:
Mast cells contribute substantially to both the steady-state respiratory dysfunction and to the pathognomonic reactions to aspirin and other COX-1-active drugs in AERD
PGE2 maintains an essential brake on mast cell activation
Local IgE and endogenous cysteinyl leukotrienes contribute to the idiosyncratic activation mechanism in AERD
What we do not yet know:
The role of innate type 2 cytokines (IL-33, TSLP) in conditioning mast cells for activation in AERD
The molecular basis of the impaired function of the endogenous PGE2/EP2 receptor system
Acknowledgments
Disclosure:
Dr. Boyce serves on the scientific advisory boards of Sanofi-Aventis and Siolta Therapeutics. He receives research funding from the National Institutes of Health.
Abbreviations used in this article:
- AERD
aspirin exacerbated respiratory disease
- COX
cyclooxygenase
- cysLTs
cysteinyl leukotrienes
- CysLT1R
type 1 cysLT receptor
- CysLT2R
type 2 cysLT receptor
- DP2
type 2 receptor for PGD2
- EP2
E prostanoid 2 receptor
- FcεRI
high affinity receptor for immunoglobulin E
- FEV1
forced expiratory volume in 1 second
- IL-1β
interleukin 1 β
- IL
interleukin
- ILC2
group 2 innate lymphoid cell
- LT
leukotriene
- LTC4S
leukotriene C4 synthase
- MCs
mast cells
- mPGES-1
microsomal PGE2 synthase-1
- PG
prostaglandin
- PGD-M
stable urinary PGD2 metabolite
- ST2
suppressor of tumorigenicity 2 (IL-33 receptor)
- Th2
T helper type 2
- TSLP
thymic stromal lymphopoietin
- 5-LO
5-lipoxygenase
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Malaviya R, Ikeda T, Ross E, and Abraham SN. Mast cell modulation of neutrophil influx and bacterial clearance at sites of infection through TNF-alpha. Nature. 1996;381(6577):77–80. [DOI] [PubMed] [Google Scholar]
- 2.Krystel-Whittemore M, Dileepan KN, and Wood JG. Mast Cell: A Multi-Functional Master Cell. Front Immunol. 2015;6:620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fahy JV, Fleming HE, Wong HH, Liu JT, Su JQ, Reimann J, et al. The effect of an anti-IgE monoclonal antibody on the early- and late-phase responses to allergen inhalation in asthmatic subjects. Am J Respir Crit Care Med. 1997;155(6):1828–34. [DOI] [PubMed] [Google Scholar]
- 4.Laidlaw TM, and Boyce JA. Aspirin-Exacerbated Respiratory Disease - New Prime Suspects. N Engl J Med. 2016;374(5):484–8. [DOI] [PubMed] [Google Scholar]
- 5.Rajan JP, Wineinger NE, Stevenson DD, and White AA. Prevalence of aspirin-exacerbated respiratory disease among asthmatic patients: A meta-analysis of the literature. J Allergy Clin Immunol. 2015;135(3):676–81. [DOI] [PubMed] [Google Scholar]
- 6.Yoshida S, Nakagawa H, Yamawaki Y, Sakamoto H, Akahori K, Nakabayashi M, et al. Bronchial hyperresponsiveness, hypersensitivity to analgesics and urinary leukotriene E4 excretion in patients with aspirin-intolerant asthma. Int Arch Allergy Immunol. 1998;117(2):146–51. [DOI] [PubMed] [Google Scholar]
- 7.Cahill KN, Bensko JC, Boyce JA, and Laidlaw TM. Prostaglandin D2: A dominant mediator of aspirin-exacerbated respiratory disease. J Allergy Clin Immunol. 2015;135(1):245–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cahill KN, Murphy K, Singer J, Israel E, Boyce JA, and Laidlaw TM. Plasma tryptase elevation during aspirin-induced reactions in aspirin-exacerbated respiratory disease. J Allergy Clin Immunol. 2019;143(2):799–803 e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sladek K, and Szczeklik A. Cysteinyl leukotrienes overproduction and mast cell activation in aspirin-provoked bronchospasm in asthma. Eur Respir J. 1993;6(3):391–9. [PubMed] [Google Scholar]
- 10.Hsieh FH, Lam BK, Penrose JF, Austen KF, and Boyce JA. T helper cell type 2 cytokines coordinately regulate immunoglobulin E-dependent cysteinyl leukotriene production by human cord blood-derived mast cells: profound induction of leukotriene C(4) synthase expression by interleukin 4. J Exp Med. 2001;193(1):123–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lewis RA, Holgate ST, Roberts LJ, Oates JA, and Austen KF. Preferential generation of prostaglandin D2 by rat and human mast cells. Kroc Found Ser. 1981;14:239–54. [PubMed] [Google Scholar]
- 12.Weiss JW, Drazen JM, McFadden ER Jr., Weller PF, Corey EJ, Lewis RA, et al. Comparative bronchoconstrictor effects of histamine, leukotriene C, and leukotriene D in normal human volunteers. Trans Assoc Am Physicians. 1982;95:30–5. [PubMed] [Google Scholar]
- 13.Marom Z, Shelhamer JH, Bach MK, Morton DR, and Kaliner M. Slow-reacting substances, leukotrienes C4 and D4, increase the release of mucus from human airways in vitro. Am Rev Respir Dis. 1982;126(3):449–51. [DOI] [PubMed] [Google Scholar]
- 14.Bankova LG, Dwyer DF, Yoshimoto E, Ualiyeva S, McGinty JW, Raff H, et al. The cysteinyl leukotriene 3 receptor regulates expansion of IL-25-producing airway brush cells leading to type 2 inflammation. Sci Immunol. 2018;3(28). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Christie PE, Tagari P, Ford-Hutchinson AW, Charlesson S, Chee P, Arm JP, et al. Urinary leukotriene E4 concentrations increase after aspirin challenge in aspirin-sensitive asthmatic subjects. Am Rev Respir Dis. 1991;143(5 Pt 1):1025–9. [DOI] [PubMed] [Google Scholar]
- 16.Buchheit KM, Cahill KN, Katz HR, Murphy KC, Feng C, Lee-Sarwar K, et al. Thymic stromal lymphopoietin controls prostaglandin D2 generation in patients with aspirin-exacerbated respiratory disease. J Allergy Clin Immunol. 2016;137(5):1566–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nasser SM, Pfister R, Christie PE, Sousa AR, Barker J, Schmitz-Schumann M, et al. Inflammatory cell populations in bronchial biopsies from aspirin-sensitive asthmatic subjects. Am J Respir Crit Care Med. 1996;153(1):90–6. [DOI] [PubMed] [Google Scholar]
- 18.Adamjee J, Suh YJ, Park HS, Choi JH, Penrose JF, Lam BK, et al. Expression of 5-lipoxygenase and cyclooxygenase pathway enzymes in nasal polyps of patients with aspirin-intolerant asthma. J Pathol. 2006;209(3):392–9. [DOI] [PubMed] [Google Scholar]
- 19.Imokawa S, Sato A, Taniguchi M, Toyoshima M, Nakazawa K, Hayakawa H, et al. [Sodium cromoglycate nebulized solution has an acute bronchodilative effect in patients with aspirin-intolerant asthma (AIA)]. Arerugi = [Allergy]. 1992;41(10):1515–20. [PubMed] [Google Scholar]
- 20.Amayasu H, Nakabayashi M, Akahori K, Ishizaki Y, Shoji T, Nakagawa H, et al. Cromolyn sodium suppresses eosinophilic inflammation in patients with aspirin-intolerant asthma. Ann Allergy Asthma Immunol. 2001;87(2):146–50. [DOI] [PubMed] [Google Scholar]
- 21.Robuschi M, Gambaro G, Sestini P, Pieroni MG, Refini RM, Vaghi A, et al. Attenuation of aspirin-induced bronchoconstriction by sodium cromoglycate and nedocromil sodium. Am J Respir Crit Care Med. 1997;155(4):1461–4. [DOI] [PubMed] [Google Scholar]
- 22.Yoshida S, Amayasu H, Sakamoto H, Onuma K, Shoji T, Nakagawa H, et al. Cromolyn sodium prevents bronchoconstriction and urinary LTE4 excretion in aspirin-induced asthma. Ann Allergy Asthma Immunol. 1998;80(2):171–6. [DOI] [PubMed] [Google Scholar]
- 23.Smith WL, Meade EA, and Dewitt DL. Interaction of PGH synthase isozymes-1 and -2 with nonsteroidal anti-inflammatory drugs. Adv Exp Med Biol. 1997;400A:189–96. [DOI] [PubMed] [Google Scholar]
- 24.Kay LJ, Yeo WW, and Peachell PT. Prostaglandin E2 activates EP2 receptors to inhibit human lung mast cell degranulation. Br J Pharmacol. 2006;147(7):707–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Feng C, Beller EM, Bagga S, and Boyce JA. Human mast cells express multiple EP receptors for prostaglandin E2 that differentially modulate activation responses. Blood. 2006;107(8):3243–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yoshimura T, Yoshikawa M, Otori N, Haruna S, and Moriyama H. Correlation between the prostaglandin D(2)/E(2) ratio in nasal polyps and the recalcitrant pathophysiology of chronic rhinosinusitis associated with bronchial asthma. Allergol Int. 2008;57(4):429–36. [DOI] [PubMed] [Google Scholar]
- 27.Machado-Carvalho L, Martin M, Torres R, Gabasa M, Alobid I, Mullol J, et al. Low E-prostanoid 2 receptor levels and deficient induction of the IL-1beta/IL-1 type I receptor/COX-2 pathway: Vicious circle in patients with aspirin-exacerbated respiratory disease. J Allergy Clin Immunol. 2016;137(1):99–107. [DOI] [PubMed] [Google Scholar]
- 28.Roca-Ferrer J, Perez-Gonzalez M, Garcia-Garcia FJ, Pereda J, Pujols L, Alobid I, et al. Low Prostaglandin E(2) and Cyclooxygenase Expression in Nasal Mucosa Fibroblasts of Aspirin-Intolerant Asthmatics. Respirology. 2013. [DOI] [PubMed] [Google Scholar]
- 29.Sestini P, Armetti L, Gambaro G, Pieroni MG, Refini RM, Sala A, et al. Inhaled PGE2 prevents aspirin-induced bronchoconstriction and urinary LTE4 excretion in aspirin-sensitive asthma. Am J Respir Crit Care Med. 1996;153(2):572–5. [DOI] [PubMed] [Google Scholar]
- 30.Ying S, Meng Q, Scadding G, Parikh A, Corrigan CJ, and Lee TH. Aspirin-sensitive rhinosinusitis is associated with reduced E-prostanoid 2 receptor expression on nasal mucosal inflammatory cells. J Allergy Clin Immunol. 2006;117(2):312–8. [DOI] [PubMed] [Google Scholar]
- 31.Corrigan CJ, Napoli RL, Meng Q, Fang C, Wu H, Tochiki K, et al. Reduced expression of the prostaglandin E2 receptor E-prostanoid 2 on bronchial mucosal leukocytes in patients with aspirin-sensitive asthma. J Allergy Clin Immunol. 2012;129(6):1636–46. [DOI] [PubMed] [Google Scholar]
- 32.Liu T, Laidlaw TM, Katz HR, and Boyce JA. Prostaglandin E2 deficiency causes a phenotype of aspirin sensitivity that depends on platelets and cysteinyl leukotrienes. Proc Natl Acad Sci U S A. 2013;110(42):16987–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cheong HS, Park SM, Kim MO, Park JS, Lee JY, Byun JY, et al. Genome-wide methylation profile of nasal polyps: relation to aspirin hypersensitivity in asthmatics. Allergy. 2011;66(5):637–44. [DOI] [PubMed] [Google Scholar]
- 34.Cahill KN, Raby BA, Zhou X, Guo F, Thibault D, Baccarelli A, et al. Impaired E Prostanoid2 Expression and Resistance to Prostaglandin E2 in Nasal Polyp Fibroblasts from Subjects with Aspirin-Exacerbated Respiratory Disease. Am J Respir Cell Mol Biol. 2016;54(1):34–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Berges-Gimeno MP, Simon RA, and Stevenson DD. Long-term treatment with aspirin desensitization in asthmatic patients with aspirin-exacerbated respiratory disease. J Allergy Clin Immunol. 2003;111(1):180–6. [DOI] [PubMed] [Google Scholar]
- 36.Cahill KN, Cui J, Kothari P, Murphy K, Raby BA, Singer J, et al. Unique Effect of Aspirin Therapy on Biomarkers in Aspirin-Exacerbated Respiratory Disease: A Prospective Trial. Am J Respir Crit Care Med. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dahlen B, Nizankowska E, Szczeklik A, Zetterstrom O, Bochenek G, Kumlin M, et al. Benefits from adding the 5-lipoxygenase inhibitor zileuton to conventional therapy in aspirin-intolerant asthmatics. Am J Respir Crit Care Med. 1998;157(4 Pt 1):1187–94. [DOI] [PubMed] [Google Scholar]
- 38.Israel E, Fischer AR, Rosenberg MA, Lilly CM, Callery JC, Shapiro J, et al. The pivotal role of 5-lipoxygenase products in the reaction of aspirin-sensitive asthmatics to aspirin. Am Rev Respir Dis. 1993;148(6 Pt 1):1447–51. [DOI] [PubMed] [Google Scholar]
- 39.Sousa AR, Parikh A, Scadding G, Corrigan CJ, and Lee TH. Leukotriene-receptor expression on nasal mucosal inflammatory cells in aspirin-sensitive rhinosinusitis. N Engl J Med. 2002;347(19):1493–9. [DOI] [PubMed] [Google Scholar]
- 40.Arm JP, O’Hickey SP, Spur BW, and Lee TH. Airway responsiveness to histamine and leukotriene E4 in subjects with aspirin-induced asthma. Am Rev Respir Dis. 1989;140(1):148–53. [DOI] [PubMed] [Google Scholar]
- 41.Fischer AR, Rosenberg MA, Lilly CM, Callery JC, Rubin P, Cohn J, et al. Direct evidence for a role of the mast cell in the nasal response to aspirin in aspirin-sensitive asthma. J Allergy Clin Immunol. 1994;94(6 Pt 1):1046–56. [DOI] [PubMed] [Google Scholar]
- 42.Mellor EA, Austen KF, and Boyce JA. Cysteinyl leukotrienes and uridine diphosphate induce cytokine generation by human mast cells through an interleukin 4-regulated pathway that is inhibited by leukotriene receptor antagonists. J Exp Med. 2002;195(5):583–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Paruchuri S, Jiang Y, Feng C, Francis SA, Plutzky J, and Boyce JA. Leukotriene E4 activates peroxisome proliferator-activated receptor gamma and induces prostaglandin D2 generation by human mast cells. J Biol Chem. 2008;283(24):16477–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lazarinis N, Bood J, Gomez C, Kolmert J, Lantz AS, Gyllfors P, et al. Leukotriene E4 induces airflow obstruction and mast cell activation through the cysteinyl leukotriene type 1 receptor. J Allergy Clin Immunol. 2018;142(4):1080–9. [DOI] [PubMed] [Google Scholar]
- 45.Bochenek G, Nizankowska E, and Szczeklik A. The atopy trait in hypersensitivity to nonsteroidal anti-inflammatory drugs. Allergy. 1996;51(1):16–23. [DOI] [PubMed] [Google Scholar]
- 46.Bochenek G, Kuschill-Dziurda J, Szafraniec K, Plutecka H, Szczeklik A, and Nizankowska-Mogilnicka E. Certain subphenotypes of aspirin-exacerbated respiratory disease distinguished by latent class analysis. J Allergy Clin Immunol. 2014;133(1):98–103 e1–6. [DOI] [PubMed] [Google Scholar]
- 47.Johns CB, and Laidlaw TM. Elevated total serum IgE in nonatopic patients with aspirin-exacerbated respiratory disease. Am J Rhinol Allergy. 2014;28(4):287–9. [DOI] [PubMed] [Google Scholar]
- 48.Hayashi H, Mitsui C, Nakatani E, Fukutomi Y, Kajiwara K, Watai K, et al. Omalizumab reduces cysteinyl leukotriene and 9alpha,11beta-prostaglandin F2 overproduction in aspirin-exacerbated respiratory disease. J Allergy Clin Immunol. 2016;137(5):1585–7.e4. [DOI] [PubMed] [Google Scholar]
- 49.Lang DM, Aronica MA, Maierson ES, Wang XF, Vasas DC, and Hazen SL. Omalizumab can inhibit respiratory reaction during aspirin desensitization. Annals of allergy, asthma & immunology: official publication of the American College of Allergy, Asthma, & Immunology. 2018;121(1):98–104. [DOI] [PubMed] [Google Scholar]
- 50.Gevaert P, Holtappels G, Johansson SG, Cuvelier C, Cauwenberge P, and Bachert C. Organization of secondary lymphoid tissue and local IgE formation to Staphylococcus aureus enterotoxins in nasal polyp tissue. Allergy. 2005;60(1):71–9. [DOI] [PubMed] [Google Scholar]
- 51.Tan BK, Li QZ, Suh L, Kato A, Conley DB, Chandra RK, et al. Evidence for intranasal antinuclear autoantibodies in patients with chronic rhinosinusitis with nasal polyps. J Allergy Clin Immunol. 2011;128(6):1198–206.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nagarkar DR, Poposki JA, Tan BK, Comeau MR, Peters AT, Hulse KE, et al. Thymic stromal lymphopoietin activity is increased in nasal polyps of patients with chronic rhinosinusitis. J Allergy Clin Immunol. 2013;132(3):593–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ordovas-Montanes J, Dwyer DF, Nyquist SK, Buchheit KM, Vukovic M, Deb C, et al. Allergic inflammatory memory in human respiratory epithelial progenitor cells. Nature. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Allakhverdi Z, Smith DE, Comeau MR, and Delespesse G. Cutting edge: The ST2 ligand IL-33 potently activates and drives maturation of human mast cells. Journal of immunology (Baltimore, Md: 1950). 2007;179(4):2051–4. [DOI] [PubMed] [Google Scholar]
- 55.Pan D, Buchheit KM, Samuchiwal SK, Liu T, Cirka H, Raff H, et al. Cyclooxygenase 1 mediates IL-33-induced extracellular signal regulated kinase activation in mast cells; Implications for aspirin sensitivity. J Allergy Clin Immunol. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liu T, Kanaoka Y, Barrett NA, Feng C, Garofalo D, Lai J, et al. Aspirin-Exacerbated Respiratory Disease Involves a Cysteinyl Leukotriene-Driven IL-33-Mediated Mast Cell Activation Pathway. J Immunol. 2015;195(8):3537–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liu T, Barrett NA, Kanaoka Y, Yoshimoto E, Garofalo D, Cirka H, et al. Type 2 Cysteinyl Leukotriene Receptors Drive IL-33-Dependent Type 2 Immunopathology and Aspirin Sensitivity. Journal of immunology (Baltimore, Md: 1950). 2018;200(3):915–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Liu T, Barrett NA, Kanaoka Y, Buchheit K, Laidlaw TM, Garofalo D, et al. Cysteinyl leukotriene receptor 2 drives lung immunopathology through a platelet and high mobility box 1-dependent mechanism. Mucosal immunology. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Laidlaw TM, Kidder MS, Bhattacharyya N, Xing W, Shen S, Milne GL, et al. Cysteinyl leukotriene overproduction in aspirin-exacerbated respiratory disease is driven by platelet-adherent leukocytes. Blood. 2012;119(16):3790–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pushparaj PN, Tay HK, H’ng SC, Pitman N, Xu D, McKenzie A, et al. The cytokine interleukin-33 mediates anaphylactic shock. Proceedings of the National Academy of Sciences. 2009;106(24):9773–8. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 61.Cahill KN, Katz HR, Cui J, Lai J, Kazani S, Crosby-Thompson A, et al. KIT Inhibition by Imatinib in Patients with Severe Refractory Asthma. N Engl J Med. 2017;376(20):1911–20. [DOI] [PMC free article] [PubMed] [Google Scholar]