

Abstract
CRISPR/Cas gene editing and messenger RNA (mRNA)-based protein replacement therapy hold tremendous potential to effectively treat disease-causing mutations with diverse cellular origin. However, it is currently impossible to rationally design nanoparticles that selectively target specific tissues. Here, we report a strategy termed Selective ORgan Targeting (SORT) wherein multiple classes of lipid nanoparticles (LNPs) are systematically engineered to exclusively edit extrahepatic tissues via addition of a supplemental SORT molecule. Lung-, spleen-, and liver-targeted SORT LNPs were designed to selectively edit therapeutically relevant cell types including epithelial cells, endothelial cells, B cells, T cells, and hepatocytes. SORT is compatible with multiple gene editing techniques, including mRNA, Cas9 mRNA / sgRNA, and Cas9 ribonucleoprotein (RNP) complexes, and is envisioned to aid development of protein replacement and gene correction therapeutics in targeted tissues.
Reporting summary.
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
The development of CRISPR/Cas-based gene editing1–3 and mRNA-based gene replacement technologies4, 5 have ushered in a hopeful era that dreams of new therapies for currently untreatable genetic diseases.6–8 Because mutated proteins are produced in specific cells, there is a critical need to develop organ specific delivery strategies to reach the full potential of genomic medicines. Non-viral synthetic nanoparticles represent a safe and efficacious approach that allows repeated administrations. Among carriers, lipid nanoparticles (LNPs) represent a broad class of materials that can deliver therapeutic nucleic acids to the liver,2, 4, 9 including a recently FDA-approved siRNA LNP therapy for transthyretin-mediated amyloidosis called Onpattro.10 Despite these advances, it is currently impossible to predictably and rationally design nanoparticles for delivery to targeted tissues beyond the liver.
We report a strategy termed Selective ORgan Targeting (SORT) that allows nanoparticles to be systematically engineered for accurate delivery of diverse cargoes including mRNA, Cas9 mRNA / sgRNA, and Cas9 ribonucleoprotein (RNP) complexes to the lungs, spleens, and livers of mice following intravenous (IV) administration (Fig. 1a). Traditional LNPs are composed of ionizable cationic lipids, amphipathic phospholipids, cholesterol, and poly(ethylene glycol) (PEG) lipids. Here we show that addition of a supplemental component (termed a SORT molecule) precisely alters the in vivo RNA delivery profile and mediates tissue-specific gene delivery and editing as a function of the percentage and biophysical property of the added SORT molecule. In this work, we provide evidences for tissue-specific delivery, establish that this methodology is applicable to various nanoparticle systems, and provide a new method for predictable LNP design to target therapeutically relevant cells.
Fig. 1 |. Selective ORgan Targeting (SORT) allows lipid nanoparticles (LNPs) to be systematically and predictably engineered to accurately deliver mRNA into specific organs.
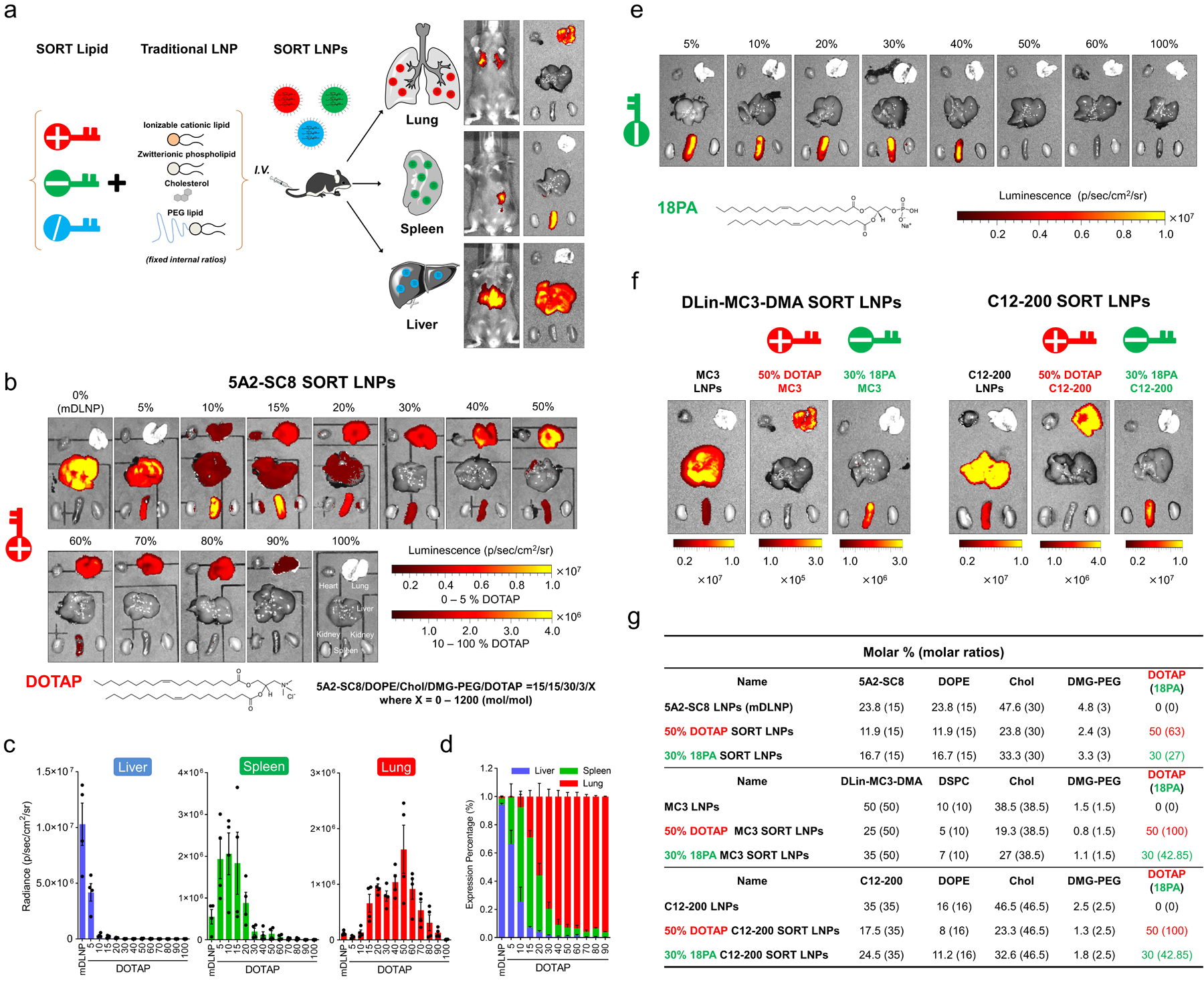
a, Addition of a supplemental component (termed a SORT molecule) to traditional LNPs systematically alters the in vivo delivery profile and mediates tissue specific delivery as a function of the percentage and biophysical property of the SORT molecule. This methodology successfully redirected multiple classes of nanoparticles. b, 5A2-SC8 SORT LNPs were formulated as indicated to make series of LNPs with 0% to 100% SORT lipid (fraction of total lipids). Here, inclusion of a permanently cationic lipid (DOTAP) systematically shifted luciferase protein expression from the liver to spleen to lung as a function of DOTAP percentage. c, Quantification data demonstrated that SORT molecule percentage is the most important factor for tissue-specific delivery. Data were shown as mean±s.e.m. (n=4 biologically independent animals). d, Relative luciferase expression in each organ demonstrated that fractional expression could be predictable tuned (0.1 mg/kg Luc mRNA, IV, 6h). Data are shown as mean±s.e.m. (n=4 biologically independent animals). e, Inclusion of an anionic SORT molecule enabled selective mRNA delivery to the spleen. Luciferase expression was observed only in spleen when introducing 18PA lipid into mDLNPs up to 40%. f, Ex vivo images of luminescence in major organs of DLin-MC3-DMA SORT LNPs and C12–200 SORT LNPs (0.1 Luc mRNA mg/kg, IV, 6h). g, Details of selected SORT molecule formulations.
Effective intracellular delivery materials have conventionally relied on an optimal balance of ionizable amines to bind and release RNAs (pKa between 6.0 – 6.5) and nanoparticle stabilizing hydrophobicity.9, 11–13 This exhaustive focus on ionizable cationic lipids has produced highly effective carriers for liver hepatocytes, but has not yielded effective carriers capable of reaching other organs. Stimulated by our work on charge unbalanced lipids with multi-organ tropism,14 demonstration that surface charge-adjusted mRNA lipoplexes could promote delivery to dendritic cells15 of the immune mononuclear phagocyte system (MPS) system,16, 17 and other reports on non-selective delivery,18–22 we speculated that internal and/or external charge may be a key factor for tuning tissue tropism.23, 24 In this paper, we therefore studied if one could augment established LNP molar compositions with supplemental molecules to tune the internal charge, thereby altering cell fate in vivo. Indeed, intravenous administration of the developed SORT LNPs enabled high levels of mRNA delivery and tissue-specific gene editing.
SORT is compatible with various methods of deploying gene editing machinery, including mRNA, Cas9 mRNA / sgRNA, and Cas9 RNPs. Lung, spleen, and liver enhanced SORT LNP delivery of Cre mRNA to tdTom mice, resulting in organ selective transfection of 40% of epithelial cells and 65% of endothelial cells; 12% of B cells and 10% of T cells; and 93% of hepatocytes, respectively. Lung, spleen, and liver SORT LNP delivered mRNAs to produce therapeutically relevant levels of proteins including IL-10, EPO, and Klotho. In addition, co-delivery of Cas9 mRNA and sgPCSK9 by liver SORT LNPs enabled complete ~100% knockout of serum and protein levels of PCSK9, a therapeutically attractive target for treatment of cardiovascular disease.25
Discovery and development of SORT
To examine the hypothesis that internal charge adjustment could mediate tissue-specific delivery, we conceived a strategy to add a 5th molecule to established LNP compositions with validated efficacy in liver hepatocytes. The rationale was to tune efficacious LNP formulations without destroying the core 4-component ratios that are essential for mediating RNA encapsulation and endosomal escape.26, 27
We first examined the effect of adding a permanently cationic lipid (defined as positively charged without pKa or pKa >8) to a degradable dendrimer ionizable (pKa <8) cationic lipid 5A2-SC8 formulation named mDLNPs,28–30 which effectively delivered fumarylacetoacetate hydrolase (FAH) mRNA to liver hepatocytes and extended survival in FAH knockout mice.27 This initial base mDLNP formulation consisted of 5A2-SC8, 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE), cholesterol, DMG-PEG (15/15/30/3, mol/mol), and mRNA (5A2-SC8/mRNA, 20/1, wt/wt) (Supplementary Fig. 1). We then formed a series of LNPs by systematically increasing the percentage of additional permanently cationic lipid from 5 to 100% of total lipids (Fig. 1b and Supplementary Fig. 1). We focused herein on lipids as SORT molecules to aid self-assembly, and initially selected 1,2-dioleoyl-3-trimethylammonium-propane (DOTAP) because it is a well-known quaternary amino lipid and prepared a titrated series of 5-component formulations (Supplementary Fig. 1).
We then evaluated the effects of SORT modification by delivering Luciferase (Luc) mRNA intravenously (IV) at dose of 0.1 mg/kg. Surprisingly, with increasing molar percentage of DOTAP, resulting luciferase protein expression moved progressively from liver to spleen, and then to lung, demonstrating a clear and precise organ-specific delivery trend with a threshold that allowed exclusive lung delivery (Fig. 1b). DOTAP percentage was the key factor that tuned tissue specificity. Base mDLNPs (0% DOTAP) were optimal for liver delivery, which was anticipated since they had been previously optimized for hepatocyte delivery,27 10–15% aided spleen delivery, and 50% was optimal for lung delivery albeit with some reduction in activity (Fig. 1c). We note that in vitro and in vivo delivery efficacy did not correlate (Supplementary Fig. 2), demonstrating that LNPs with high in vivo efficacy may be missed through traditional in vitro assays that do not capture the organism-level biology of specific cells in various organs. This suggests one explanation why the discovery of predictable organ-specific delivery has been so elusive. Calculating the relative expression in each organ, SORT completely altered delivery from liver to lungs (Fig. 1d and Supplementary Fig. 3).
With the functional role of the permanently cationic SORT lipid elucidated, we then hypothesized that inclusion of other molecules may also alter tissue tropism. To explore this potential, negatively charged 1,2-dioleoyl-sn-glycero-3-phosphate (18PA) was incorporated as a SORT molecule in a similar manner as DOTAP (Supplementary Fig. 1). At 10–40% 18PA incorporation, SORT LNPs now mediated completely selective delivery to the spleen with no luciferase expression in any other organs (Fig. 1e). Thus, negatively charged SORT lipids allow for explicit delivery to the spleen. These results indicated that SORT molecule chemistry and percentage can be tailored for tissue-specific delivery via IV injection.
SORT is generalizable to LNP types and molecular classes
We next asked if the SORT methodology could be applied to other classes of established 4-component LNPs. First, DLin-MC3-DMA was formulated with DSPC, cholesterol, and PEG-DMG with the same molar composition of lipids used in the FDA-approved Onpattro (Patisiran) formulation31 (Supplementary Fig. 4). This formulation has been established as a benchmark for both siRNA11 and mRNA32–34 delivery. To date, DLin-MC3-DMA LNPs have only been shown to deliver to the liver following IV administration, which we confirmed (Fig. 1f). As expected, supplementing DLin-MC3-DMA LNPs with DOTAP altered the protein expression profile from the liver to spleen to lung, mirroring the results of 5A2-SC8 SORT LNPs. To study this further, we included DOTAP into C12–200 LLNPs (Fig. 1g and Supplementary Fig. 4), which are also well validated for RNA delivery to the liver,35, 36 and observed the identical trend (Fig. 1f, and Supplementary Fig. 4). Additionally, inclusion of 18PA as a SORT molecule mirrored our results with 5A2-SC8 and mediated exclusive delivery of Luc mRNA to the spleen for both DLin-MC3-DMA and C12–200 LNPs (Fig. 1f,g). While DLin-MC3-DMA is a two-tailed lipid with a single dimethylamine headgroup that forms stable nucleic acid lipid nanoparticles (SNALPs), C12–200 is a representative lipidoid that forms lipid-like LNPs (LLNPs). Thus, we show that the SORT methodology is generalizable to other classes of ionizable cationic lipid LNPs, which will allow existing liver-targeting LNPs to be tuned to deliver mRNA to the spleen or lungs. Specifically, the SORT technology may allow pre-clinical and clinical liver-targeting LNPs to be quickly redeveloped for treatment of diseases in the lung and spleen.
To understand whether the tissue tropism profiles observed were specific to exact chemical structures or generalizable to defined chemical classes, we evaluated multiple permanently cationic, anionic, zwitterionic, and ionizable cationic SORT lipids (Fig. 2 and Supplementary Table 1). First, we generated 5A2-SC8 SORT LNPs with two additional permanently cationic lipids: DDAB and EPC. These lipids all contain quaternary amino groups, but there are major chemical differences in the polar head group, linker region, and hydrophobic domain (e.g. degree of saturation). LNPs containing 5, 15, 40, 50, and 100% DDAB or EPC were formulated and characterized. Importantly, the in vivo luciferase expression profile matched that of the DOTAP LNPs, where the luminescence activity systematically shifted from the liver to spleen, then to lung with increasing DDAB or EPC percentages (Fig. 2a). We next examined 14PA and 18BMP as representative anionic lipids with very different structures. All anionic SORT lipids promoted exclusive delivery to the spleen (Fig. 2b). This design flexibility provides a path to optimize future SORT molecules to balance potency, selectivity, and tolerability.
Fig. 2 |. SORT relies on general biophysical properties and not exact chemical structures to deliver mRNAs encoding for therapeutically relevant proteins.
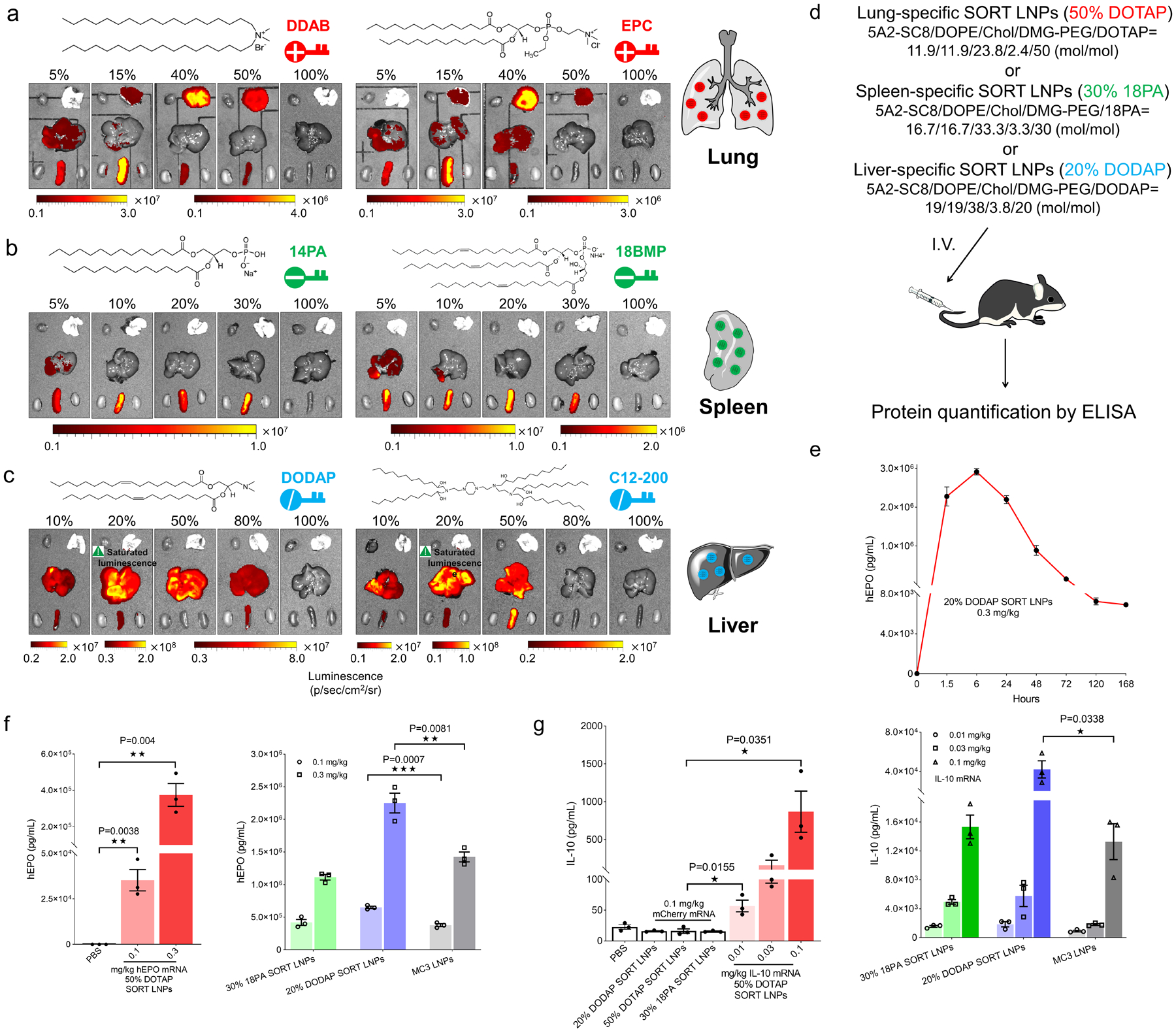
a, SORT molecules could be divided into specific groups with defined biophysical properties. Permanently cationic SORT lipids (DDAB, EPC, and DOTAP) all resulted in the same mRNA delivery profile. b, Anionic SORT lipids (14PA, 18BMP, 18PA) all resulted in the same mRNA delivery profile. c, Ionizable cationic SORT lipids with tertiary amino groups (DODAP, C12–200) enhanced liver delivery without luciferase expression in the lungs (0.1 mg/kg, 6h). d, Scheme for mRNA delivery of secreted proteins. e, High levels of hEPO expression persisted for > one week following administration of 0.3 mg/kg hEPO mRNA in 20% DODAP SORT LNPs. Data are presented as mean±s.e.m. (n=3 biologically independent animals). f, hEPO was quantified by ELISA in serum following IV administration of hEPO mRNA in lung-, spleen-, and liver-specific SORT LNPs, and MC3 LNPs. Data are presented as mean±s.e.m. (n=3 biologically independent animals). g, IL-10 was quantified by ELISA in serum following IV administration of mouse IL-10 mRNA in lung-, spleen-, and liver-specific SORT LNPs, and MC3 LNPs. mCherry mRNA SORT formulations and PBS were used as controls. Data are presented as mean±s.e.m. (n=3 biologically independent animals). A two-tailed unpaired t-test was used to determine the significance of the indicated comparisons of data from f and g. (*P < 0.05; **P<0.01; ***P < 0.001; ****P<0.0001).
Inspired by these findings, we then asked what would happen if other ionizable cationic lipids were added to established formulations. As expected, addition of DODAP or C12–200 to 5A2-SC8 mDLNPs did not significantly alter tissue tropism, but surprisingly did enhance liver delivery >10-fold at 20% incorporation (Fig. 2c). Interestingly, simply supplementing already established mDLNPs with additional 5A2-SC8 as a SORT molecule dramatically improved liver mRNA delivery, producing 107 photons/sec/cm2 at the low dose of 0.05 mg/kg. SORT thus offers a new strategy to further improve liver targeting LNP systems (Supplementary Fig. 5). We also evaluated the effect of using zwitterionic lipids (DOCPe and DSPC) as SORT molecules. While we found that the tissue tropism moved from liver to spleen, it was not as purely selective compared to use of cationic or anionic SORT lipids (Supplementary Fig. 6).
To test the limits of the SORT methodology, we examined if SORT could “activate” otherwise inactive formulations. Indeed, supplementing a completely inactive formulation with DODAP or DOTAP resulted in tissue-specific delivery to the spleen and lung (Supplementary Fig. 7). We note that all SORT LNPs still contain ionizable cationic lipids, which are considered essential for endosomal escape26 due to their ability to acquire charge. We performed control experiments and confirmed that inclusion of an ionizable cationic lipid was needed for efficacy (Supplementary Fig. 8). Taking these results together, we conclude that SORT is a modular and generalizable strategy to achieve tissue-targeted delivery.
This work led to selection of lung-, spleen-, and liver-specific 5A2-SC8 SORT LNPs (Fig. 2d). We confirmed that Luc delivery was dose responsive (Supplementary Fig. 9). We also verified that SORT-enabled tissue specificity occurs quickly and is not dependent on time (Supplementary Fig. 10). We further analyzed the tissue specific mRNA delivery more precisely by homogenizing isolated tissues to quantify luminescence normalized to the tissue weight and total protein for each organ (Supplementary Fig. 11). Considering potential therapeutic applications, formulation stability was monitored over time, which revealed that SORT LNPs maintained physiochemical properties and in vivo delivery efficacy after storage at 4 °C (Supplementary Fig. 12).
We next evaluated the ability of these formulations to deliver mRNAs encoding secreted therapeutic proteins: human Erythropoietin (hEPO), mouse Interleukin-10 (IL-10), and mouse Klotho (KL). This approach allowed us to quantify protein concentrations in serum to directly test all organ-specific SORT LNPs (Fig. 2d). hEPO production following 20% DODAP Liver SORT LNP IV administration peaked at 6 hours and was maintained for >1 week (Fig. 2e and Supplementary Fig. 13). All three tissue-specific formulations mediated high levels of protein production at low mRNA doses for both hEPO mRNA (0.1 and 0.3 mg/kg) and IL-10 mRNA (0.01 mg/kg to 0.1 mg/kg) in a dose-responsive manner (Fig. 2f,g). Similar delivery trends were observed for Klotho (Supplementary Fig. 13). We next evaluated in vivo toxicity using a dose higher than needed to produce therapeutically relevant protein levels in the blood. SORT LNPs with single or multiple dosing did not alter kidney and liver function and serum cytokines (Supplementary Fig. 14), and no adverse signs of injury in tissue histology (Supplementary Fig. 15) were observed at the tested dose (1 mg/kg mRNA). These results suggest that SORT LNPs are well tolerated and mediate therapeutically relevant levels of protein production.
SORT enables delivery to therapeutically relevant cell types
Given the ability of SORT LNPs to target specific organs, we next applied our finding to tissue-specific gene editing via IV injection. The CRISPR/Cas (clustered regularly interspaced short palindromic repeat / CRISPR-associated protein (Cas)) technology6–8 can edit the genome in a precise and sequence dependent manner and has rapidly developed for use in diverse applications, including for potential correction of disease-causing mutations.37–41 Gene editing can be achieved by local administration.42–44 However, many serious genetic disorders arise from mutations in cells deep in organs, where correction of specific cells will be required to cure disease. Such correction may be best achieved by systemic administration. We, and others, recently reported that IV co-delivery of Cas9 mRNA and sgRNA is a safe and effective strategy to enable gene editing.14, 39, 45 To date, however, there have been no reports to our knowledge of LNPs rationally engineered to edit cells in organs outside of the liver.
To examine and quantify the ability of SORT LNPs to mediate organ-specific gene editing, we utilized genetically engineered tdTomato (tdTom) reporter mice containing a LoxP flanked stop cassette46 that prevents expression of the tdTom protein.44 Once the stop cassette is deleted, tdTom fluorescence is turned on, allowing detection of gene edited cells (Fig. 3a). We initially delivered Cre recombinase mRNA (Cre mRNA), producing Cre protein that deletes the stop to activate tdTom in edited cells. Fluorescent tissues were readily apparent (Fig. 3b) in selected organs treated with liver, lung, and spleen selective SORT LNPs. We note that these mice have weaker background organ autofluorecense in the spleen compared to other organs, which required the use of separate controls (Fig. 3c and Supplementary Fig. 16). This makes detection of spleen specificity more challenging; nevertheless, tdTom positive cells were easily seen by confocal imaging of tissue sections (Fig. 3d).
Fig. 3 |. SORT LNPs enabled tissue-specific Td-Tomato activation by Cre mRNA delivery.

a, Schematic illustration shows that delivery of Cre mRNA activates Td-Tom expression in Td-Tom transgenic mice via Cre-mediated genetic deletion of the stop cassette. b, mDLNP and liver SORT LNPs (20% DODAP) induced Td-Tom fluorescence specifically in the liver and lung SORT LNPs (50% DOTAP) selectively edited the lung. Td-Tom fluorescence of main organs was detected 2 days following IV injection of Cre mRNA-loaded LNPs (n=3 biologically independent animals). c, Spleen SORT LNPs (30% 18-PA) induced gene editing in the spleen (note high liver background fluorescence in PBS injected mice). Data are presented as mean±s.e.m. (n=3 biologically independent animals). d, Confocal microscopy was employed to further verify effective tissue editing (n=3 biologically independent animals). Scale bars = 20 μm and 100 μm. e, FACS was used to quantify the percentage of TdTom+ cells within defined cell type populations of the liver, lung, and spleen (0.3 mg/kg, day 2). Data are presented as mean±s.e.m. (n=3 biologically independent animals).
To initially estimate cell types amenable to delivery by SORT LNPs, we quantified delivery to specific cell types within the liver, lung, and spleen using flow cytometry of cells extracted from edited organs following delivery of Cre mRNA (Fig. 3e). Liver-specific SORT LNPs (20% DODAP) delivered Cre mRNA to ~93% of all hepatocytes in the liver following a single injection of 0.3 mg/kg Cre mRNA (Fig. 3e and Supplementary Fig. 17). Lung-specific SORT LNPs (50% DOTAP) transfected ~40% of all epithelial cells, ~65% of all endothelial cells, and ~20% of immune cells in the lungs at the same dose (Fig. 3e and Supplementary Fig. 18). Given that epithelial cells are a primary target for correction of mutations in CFTR that cause Cystic Fibrosis, this result establishes lung-specific SORT LNPs as a compelling delivery system with immediate application for correcting CFTR mutations. Finally, spleen-specific SORT LNPs (30% 18PA) transfected ~12% of all B cells, ~10% of all T cells, and ~20% of all macrophages (Fig. 3e and Supplementary Fig. 19). Due to the improved selectivity, spleen-specific SORT LNPs could be applicable to treat non-Hodgkin’s B cell lymphoma, and other immune disorders. Although our initial focus was on single, low dose injection quantification, we note that higher levels of transfection are achievable by administering higher doses or multiple injections.
SORT allows tissue-specific gene editing
We next examined the ability of SORT LNPs to achieve tissue-specific CRISPR/Cas gene editing via IV co-delivery of Cas9 mRNA and sgRNA in a single nanoparticle (Fig. 4a, Supplementary Fig. 20 and Supplementary Table 2). We injected liver and lung targeting SORT LNPs at a dose of 2.5 mg/kg total RNA (4:1 mRNA:sgRNA, wt:wt) and quantified gene editing 10 days following a single IV injection. As shown in Fig. 4b, strong tdTom fluorescence in the liver was observed for both mDLNPs and 20% DODAP SORT LNP treated mice, and strong fluorescence in the lung of 50% DOTAP SORT LNP treated mice. The fluorescence was then confirmed by imaging tissue sections with confocal microscopy (Fig. 4c). All results were consistent with the Luc mRNA delivery results. Due to fast turnover of splenic immune cells in mice,47 we optimized the weight ratio of Cas9/sgRNA to be 2/1 (Supplementary Fig. 21) and tested the spleen editing two days after injection. Accounting for background autofluorescence, bright tdTom fluorescence was observed in the spleen of 30% 18PA-treated mice (Supplementary Fig. 22). Next, we focused on direct delivery of Cas9 RNPs, which is the most challenging strategy for synthetic carriers. The use of permanently cationic SORT lipids enabled Cas9 protein / sgTom1 complexes to be encapsulated with control over tissue tropism. IV injection of 7% DOTAP SORT LNPs enabled liver editing, while 55% DOTAP SORT LNPs enabled exclusive lung editing (Fig. 4f). These data indicate that the described methodology enables liver-, lung-, and spleen-specific CRISPR/Cas gene editing.
Fig. 4 |. SORT LNPs mediated tissue-specific CRISPR/Cas gene editing of Td-Tom transgenic mice and C57/BL6 wild type mice by co-delivering Cas9 mRNA and sgRNA and by delivering Cas9 RNPs.
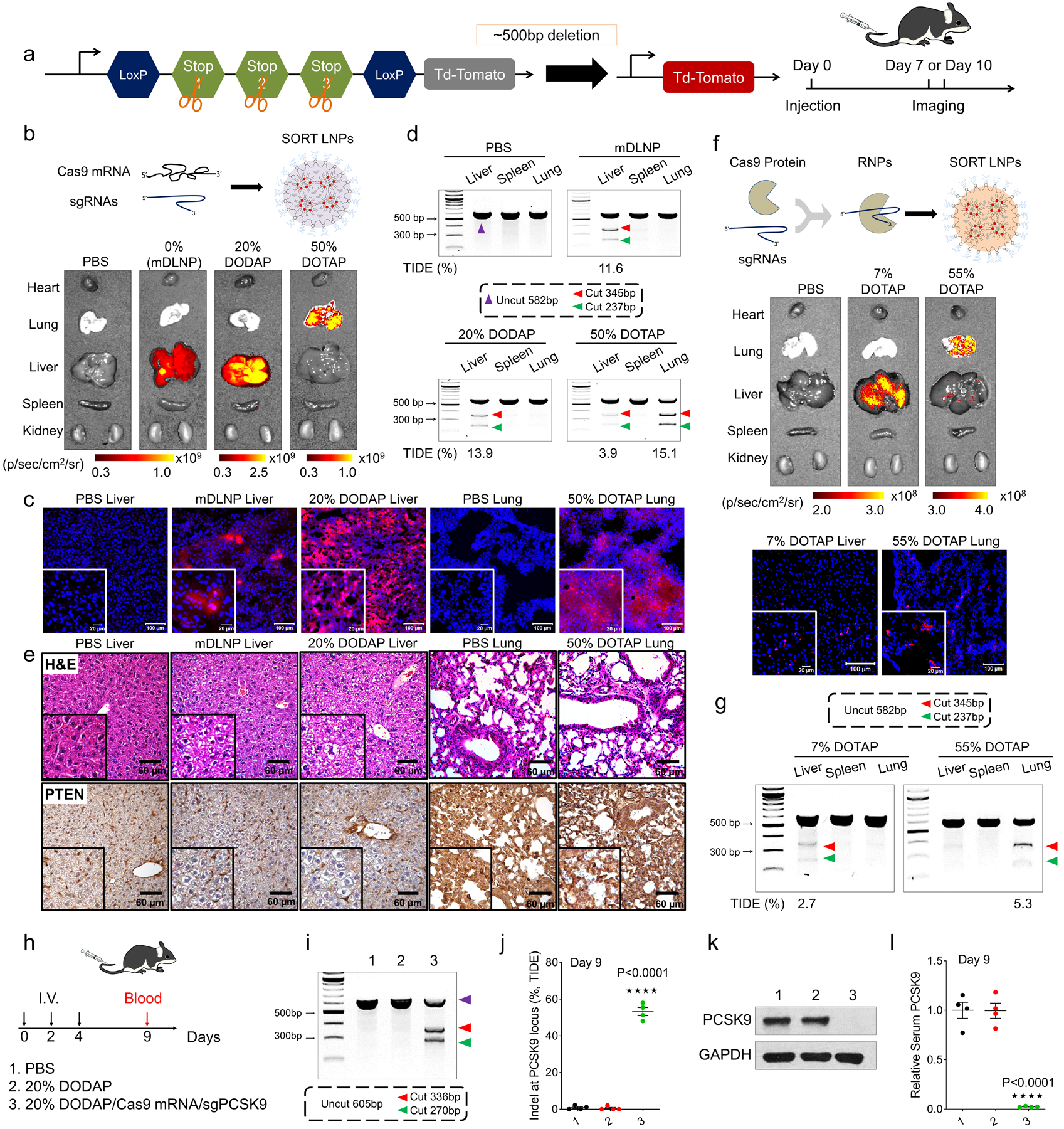
a, Schematic illustration shows that co-delivery of Cas9 mRNA (or Cas9 protein) and sgTom1 activates Td-Tom expression in Td-Tom transgenic mice. b, mDLNP and SORT LNPs (20% DODAP) induced Td-Tom fluorescence specifically in the liver and SORT LNPs (50% DOTAP) selectively edited the lung with 2.5 mg/kg dose (Cas9 mRNA/sgTom1, 4/1, wt/wt; measured day 10, n=3 biologically independent animals). c, tdTom expression was confirmed by confocal imaging of tissue sections (n=3 biologically independent animals). Scale bars = 20 μm and 100 μm. d, Cas9 mRNA and sgPTEN were co-delivered in SORT LNPs to selectively edit the liver, lung, and spleen of C57/BL6 mice with 2.5 mg/kg total mRNA (Cas9 mRNA/sgPTEN, 4/1, wt/wt; measured day 10, n = 3 biologically independent animals). The T7E1 assay indicated that tissue-specific PTEN editing was achieved. Editing was quantified by DNA sequencing and TIDE analysis. e, H&E sections and IHC further confirmed successful PTEN editing (n=3 biologically independent animals). Clear cytoplasm indicated lipid accumulation in H&E sections and PTEN loss in IHC images. Scale bar = 60 μm. f, Delivery of Cas9/sgTom1 ribonucleoprotein (RNP) complexes in 7% DOTAP or 55% DOTAP SORT LNPs induced Td-Tom fluorescence specifically in the liver and lungs, respectively (1.5 mg/kg sgTom1, day 7, n=3 biologically independent animals). tdTom expression was confirmed by confocal imaging of tissue sections. Scale bars = 20 μm and 100 μm. g, Liver- and lung-tropic SORT LNPs also delivered Cas9/sgPTEN RNPs to selectively edit the liver and lungs C57/BL6 mice (1.5 mg/kg sgPTEN; day 7, n = 3 biologically independent animals). The T7E1 and TIDE assays indicated that tissue-specific PTEN editing was achieved. h, C57BL/6 mice were IV injected three times (days 0, 2, 4) by co-delivery of Cas9 mRNA and modified sgPCSK9 with 20% DODAP SORT LNPs (n = 4 biologically independent animals). At day 9, ~60% Indel at PCSK9 locus of liver tissue was quantified by (i) T7E1 assay and (j) TIDE analysis, which resulted to ~100% PCSK9 protein reduction in (k) liver tissue (western blot) and (l) serum (ELISA), respectively. The results of h-l were given from 4 biologically independent animals. Data of j and l are presented as mean±s.e.m. A two-tailed unpaired t-test was used to determine the significance of the indicated comparisons of data from j and l. (*P < 0.05; **P<0.01; ***P < 0.001; ****P<0.0001).
To expand beyond reporter mice, we tested the ability of tissue-specific LNPs to edit an endogenous target. We selected PTEN because it is a well-established tumor suppressor expressed in most cells. Wild type C57BL/6 mice were injected with SORT LNPs co-loaded with Cas9 mRNA and sgPTEN (2.5 mg/kg total RNA). Generation of insertions and deletions (indels) was quantified 10 days following a single IV injection. As shown in Fig. 4d, clear DNA cleavage bands were observed in specific tissues by T7E1 assay which demonstrated that both base mDLNPs (11.6% indels by TIDE analysis) and 20% DODAP SORT LNPs (13.9%) mediated effective PTEN editing in liver, but not at all in lung or spleen. Remarkably, 50% DOTAP SORT LNPs (15.1%) showed PTEN editing exclusively in the lungs. To further confirm PTEN editing, H&E staining and immunohistochemistry (IHC) of tissue sections was performed. As shown in Fig. 4e, cells in tissue sections obviously displayed clear cytoplasm, which is a known phenotype of PTEN loss due to lipid accumulation.48 Moreover, negative staining of PTEN was observed in IHC sections in both liver and lung tissues. Although spleen-specific 18PA SORT LNP editing was more challenging to distinguish in the tdTom mouse model, clear spleen PTEN editing could be observed in wild type mice (Supplementary Fig. 22). Next, we applied SORT to Cas9 RNPs and examined endogenous editing of PTEN. As before, 7% and 55% DOTAP SORT LNPs containing Cas9 protein / sgPTEN enabled liver (2.7%) and lung (5.3%) specific editing, respectively (Fig. 4g). We also evaluated off target effects for PTEN and did not detect any off target DNA editing (Supplementary Fig. 23 and Supplementary Fig. 24).
Finally, to establish that SORT LNPs can edit therapeutically relevant targets at significant levels, we co-delivered Cas9 mRNA and an sgRNA against PCSK9 (Fig. 4h), a highly attractive target for Familial Hypercholesterolemia (FH) and Atherosclerotic Cardiovascular Disease (ASCVD).25 Indeed, 20% DODAP SORT LNPs were able to significantly induce Indels at the PCSK9 locus (~60% by TIDE) (Fig. 4i,j and Supplementary Fig. 25), leading to ~100% liver (Fig. 4k and Supplementary Fig. 25) and serum (Fig. 4l and Supplementary Fig. 25) PCSK9 protein reduction. We also observed higher liver to body weight ratio, which demonstrated a clear phenotypic change due to lipid accumulation in the liver after PCSK9 knockout (Supplementary Fig. 25). These results, targeting endogenous genes, demonstrate rationally guided tissue-selective gene editing achieved by synthetic carriers.
Conclusions
The discovery of Selective ORgan Targeting (SORT), which allows predictable nanoparticle delivery of RNA to specific organs, is anticipated to aid development of protein replacement and gene correction therapeutics. Because delivery efficacy correlates strongly to modular molecular classes, we believe this methodology can be widely applied to existing LNPs and other nanoparticle systems. We speculate that SORT may allow rational design of carriers for a variety of cargoes (including proteins and other drugs) and organs. Current efforts are focused on determining precise mechanisms that explain how SORT enables tissue targeting. Although more work remains, initial data indicates that inclusion of SORT molecules alters the biodistribution of SORT LNPs to different tissues, changes the global apparent pKas,11 and endows distinct protein coronas.49, 50 These investigations will be published in a future manuscript. Nevertheless, the current data disclosing the high degree of editing in specific cells positions the discovery of SORT LNPs to treat an array of diseases in a highly accurate manner. It is thus anticipated that SORT may open new avenues of development for gene correction therapeutics.
Methods
Materials
5A2-SC828 and C12–20035 were synthesized and purified by following published protocols. DLin-MC3-DMA11 was purchased from MedKoo Biosciences. 1,2-dioleoyl-3-trimethylammonium-propane (DOTAP), dimethyldioctadecylammonium (DDAB), 1,2-dimyristoyl-sn-glycero-3-ethylphosphocholine (EPC), 1,2-dioleoyl-sn-glycero-3-phosphate (sodium salt) (18PA), 1,2-dimyristoyl-sn-glycero-3-phosphate (sodium salt) (14PA), sn-(3-oleoyl-2-hydroxy)-glycerol-1-phospho-sn-3’-(1’,2’-dioleoyl)-glycerol (ammonium salt) (18:1 Hemi BMP, 18BMP), 1,2-dioleoyl-3-dimethylammonium-propane (DODAP), 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC), 2-((2,3-bis(oleoyloxy)propyl)dimethylammonio)ethyl ethyl phosphate (DOCPe) and 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE) were purchased from Avanti Polar Lipids. Cholesterol was purchased from Sigma-Aldrich. 1,2-Dimyristoyl-sn-glycerol-methoxy(poly((ethylene glycol) MW 2000) (DMG-PEG2000) was purchased from NOF America Corporation. Cas9 protein was purchased from Thermo Fisher. Sucrose and Sodium Dodecyl Sulfate (SDS) was purchased from Sigma Aldrich. The ONE-Glo + Tox Luciferase Reporter assay kit was purchased from Promega Corporation. Pur-A-Lyzer Midi Dialysis Kits (WMCO, 3.5kDa) were purchased from Sigma-Aldrich. 4’,6-Diamidino-2-phenylindole dihydrochloride (DAPI) was purchased from Thermo Fisher Scientific. Cas9 mRNA was produced by in vitro translation (IVT). Firefly luciferase mRNA (Luc mRNA) and mCherry mRNA were purchased from TriLink BioTechnologies. D-Luciferin (Sodium Salt) was purchased from Gold Biotechnology. Modified sgTom1, sgPTEN, and sgPCSK9 (Supplementary Table 2) were purchased from Synthego.
Nanoparticle formation
RNA-loaded LNP formulations were formed using the ethanol dilution method28. The liver-targeted mRNA formulation (mDLNP) was developed and reported in our previous paper27, and the base formulations were prepared as previously described35, 11. Unless otherwise stated, all lipids with specified molar ratios were dissolved in ethanol and RNA was dissolved in 10 mM citrate buffer (pH 4.0). The two solutions were rapidly mixed at an aqueous to ethanol ratio of 3:1 by volume (3:1, aq.:ethanol, vol:vol) to satisfy a final weight ratio of 40:1 (total lipids:mRNA), then incubated for 10 min at room temperature. To prepare SORT LNP formulations containing anionic SORT lipids (such as 18PA, 14PA and 18BMP), the anionic lipids were dissolved in tetrahydrofuran (THF) first then mixed with other lipid components in ethanol, finally yielding formulations with mRNA buffer (10 mM, pH 3.0) as described above. All formulations were named based on the additional lipids. Taking DOTAP mDLNP as an example, the internal molar ratio of mDLNP was fixed as reported in our published paper with 5A2-SC8/DOPE/Cholesterol/DMG-PEG of 15/15/30/327. DOTAP, as the additional lipid, was dissolved into the above ethanol lipid mixture with specified amount, making the molar ratio of 5A2-SC8/DOPE/Cholesterol/DMG-PEG/DOTAP equal to 15/15/30/3/X, then rapidly mixed with aq. mRNA solutions following the above standard protocol, finally producing SORT LNPs named Y% DOTAP, where Y means the molar percent of DOTAP in total lipids. Formulations with other additional lipids were formed similarly with the above methods (Supplementary Fig. 1 and Table 1). As a representative example, liver targeted SORT LNPs (20% DODAP) could be prepared as follows. A solution of lipids in ethanol was prepared consisting of 7.59 mM 5A2-SC8, 7.59 mM DOPE, 15.18 mM Cholesterol, 1.52 mM DMG-PEG2000, and 7.97 mM DODAP to make the final molar ratio of 19.05/19.05/38.1/3.81/20. To reach a final 40/1 (wt/wt) of total lipids to total RNAs ratio, 1.16 μL lipid solution could be used per μg RNA. For example, to make a final 5 μg RNA formulation, 5.8 μL lipid mixture and 9.2 μL ethanol were mixed first (total 15 μL), then 45 μL mRNA solution was prepared consisting of 5 μg RNA in citrate buffer (10 mM, pH 4.0). The 45 μL mRNA solution was rapidly combined into 15 μL of the ethanol lipid solution to form 20% DODAP SORT LNPs. For Cas9/sgRNA ribonucleoprotein (RNP) encapsulation, 1X PBS was used for formulation and the molar ratio of Cas9 and sgRNA was fixed at 1:3. After SORT LNP formation, the fresh LNP formulations were diluted with 1X PBS to 0.5 ng/μL mRNA (with final ethanol concentration < 5%) for in vitro assays and size detection by Dynamic Light Scattering (DLS, Malvern MicroV model; He-Ne laser, λ = 632 nm). For in vivo experiments, the formulations were dialyzed (Pur-A-Lyzer Midi Dialysis Kits, WMCO 3.5kDa, Sigma-Aldrich) against 1X PBS for 2h, and diluted with PBS to 15 μL/g for intravenous (IV) injections.
In vivo Luc mRNA delivery
C57BL/6 mice with weight of 18–20g, were IV injected by various Luc mRNA formulations. n = 3–4 per group. At the stated detection time points, mice were injected with D-Luciferin (150 mg/kg, intraperitoneal (IP)) and imaged by an IVIS Lumina system (Perkin Elmer). In this work, BLI imaging was performed at various time-points (3h, 6h, 8h, and 24h) following injection of various Luc mRNA doses (0.02 mg/kg to 0.5 mg/kg, intravenous (IV)). To further quantify the luciferase mRNA delivery in vivo, we normalized the relative luciferase expression to per mg tissue and per μg total proteins. Briefly, LNPs were formed as described above and mice were I.V. injected with dose of 0.1 mg/kg luc mRNA. After 6 hours, tissues were collected and cut by scissors, then a known weight of tissue (20–30 mg) was homogenized by T-PER Tissue Protein Extraction Reagent (Thermo Fisher). 20 μL supernatant was used to measure the luciferase expression by ONE-Glo kits based on Promega’s standard protocol, and 10 μL supernatant was used to measure the total proteins by BCA kit (Thermo Fisher). Finally, relative luciferase expression was normalized by tissue weight and total protein.
LNP stability testing
To study LNP stability, the sizes, PDI, and encapsulation efficacy (EE) were monitored for one week during storage in PBS at 4 °C. mDLNPs, 50% DOTAP SORT LNPs (lung), 30% 18PA SORT LNPs (spleen), and 20% DODAP SORT LNPs (liver) were formed as described above and dialyzed with 1X PBS, thendiluted to 5 ng/μL luciferase mRNA in 1X PBS (n=3). 250 μL was pipetted into DLS Ultramicro cuvettes for size and PDI monitoring and 250 μL was pipetted into a 1.5 mL tube for EE evaluation by the Ribogreen assay for one week. Additionally, to evaluate mRNA delivery efficacy in vivo, mice were I.V. injected with LNPs that had been stored for one week in 4 °C, then luminescence was measured 6h after injection.
mRNA synthesis
Optimized mouse IL-10, human EPO, mouse Klotho ECD, Cre recombinase, and Cas9 mRNAs were produced by in vitro transcription (IVT). Briefly, the coding fragments of each protein were prepared by PCR program (Supplementary Table 3). Then, the these fragments were cloned into pCS2+MT vector with optimized 5’(3’)-untranslated regions (UTR) and poly A sequences. IVT reactions were performed following standard protocols but with N1-methylpseudouridine-5’-triphosphate replacing the typical UTP. Finally the mRNA was capped (Cap-1) by Vaccinia Capping Enzyme and 2’-O-methyltransferase (NEB). The coding sequences for these proteins are detailed in the Supplementary Information.
Western blot
The quality of IVT Cas9 mRNA was analyzed by western blot. 293T cells were seeded into 12-well plate with the density of 1×105 cells per well the day before transfection. Cells were treated with various formulations in 600 μL total volume for another 24h, including mCherry mDLNP (0.5 μg mRNA per well), mCherry mDLNP (1.0 μg mRNA per well), IVT Cas9 mDLNP (0.5 μg mRNA per well), IVT Cas9 mDLNP (1.0 μg mRNA per well) and Lipofectamine2000/Cas9 pDNA (0.5 μg pDNA per well). After washing three times with 1X PBS, 100 μL lysis buffer (50 mM Tris HCl, pH 7.4, with 150 mM NaCl, 1 mM EDTA and 1% TRITON X-100) and 1 μL protein inhibitor cocktail (100X, Thermo Fisher) were added into each well and rocked for 20 min at RT. Cell lysates were collected into 1.6 mL tubes and centrifuged for 10 min (13,000 g) at 4 °C. Supernatants were collected into new tubes and stored in −80 °C if not used immediately. Before executing a western blot, protein concentrations were measured using a BCA assay kit (Thermo Fisher). Fifteen microgram total proteins were loaded and separated by 4–20% polyacrylamide gel (Thermo Fisher). Separated proteins were then transferred into polyvinylidene membrane (BioRad) and blocked by 5% BSA (dissolved in PBST) for 1h at RT. Primary antibodies were applied overnight at 4 °C. After washing four times using PBST, the membrane was incubated by secondary antibody for 1h at RT then imaged with ECL substrate after washing four times by PBST (Thermo Fisher). For PCSK9 detection of liver tissue, total protein was extracted by RIPA Lysis and Extraction Buffer (Thermo Fisher) using standard protocols, then western blot was executed as described above.
In vivo toxicity evaluation
Male C57BL/6 mice, weight of 18g to 20g, were randomly divided into 5 groups, n = 4. To maximally expose toxicity in vivo, we here selected high dose of mRNA (1 mg/kg mCherry mRNA) for I.V. injection, and three-tissue targeting formulations were used. The 50% DOTAP SORT LNPs for lung, 30% 18PA SORT LNPs for spleen, and 20% DODAP SORT LNPs for liver. Lipopolysaccharide (LPS, 5 mg/kg) was intraperitoneal (I.P.) injected for positive control and PBS (I.V.) for negative control, respectively. After 24h, and 48h, whole blood was collected and serum was separated. Then liver function (AST and ALT) and renal function (BUN and CREA) were measured by UT Southwestern Metabolic Phenotyping Core, and tissue (heart, liver, spleen, lung, and kidney) sections were analyzed with H&E staining by UT Southwestern Tissue Management Shared Resource. To further study potential toxicity in vivo, we performed I.V. injections twice with the 1 mg/kg mCherry mRNA doses. Briefly, mice were I.V. injected by SORT LNPs at day 0 and day 3, serum was separated at 24h and 48h after the second injection, then liver function and renal function were measured as described above. Meanwhile, serum cytokines (IL-1β and TNF-α) were measured by the UT Southwestern Genomics Sequencing & Microarray Core Facility.
In vivo therapeutic mRNA delivery
Male C57BL/6 mice, weight of 18g to 20g, were randomly divided into 5 groups (n = 3). For therapeutic mRNAs delivery, we prepared three different mRNAs: mouse Interleukin 10 (mIL-10), human Erythropoietin (hEPO), and mouse Klotho (mKL). For mIL-10 and hEPO mRNAs delivery, we applied different doses to target all three organs, lung (50% DOTAP SORT LNPs), spleen (30% 18PA SORT LNPs), and liver (20% DODAP SORT LNPs). At the same time, we included Onpattro (DLin-MC3-DMA LNPs)31 for efficacy comparison of liver-targeted formulations. At 6h after I.V. injection, serum was separated from whole blood, then concentrations were detected by ELISA kits (IL-10 Mouse ELISA Kit and Erythropoietin Human ELISA Kit, Thermo Fisher). Furthermore, we evaluated the hEPO protein kinetics in serum with 20% DODAP SORT LNPs (0.3 mg/kg), we collected serum from 0h to day 7 after I.V. injection, and then quantified the hEPO by ELISA. For mKL assay, the quantification of serum was analyzed by UT Southwestern Physiology Core (O’Brien Kidney Center).
Gene editing (Cre mRNA) in Td-Tomato mice model
Cre mRNA formulations were prepared as described above and performed IV injections (0.3 mg/kg Cre mRNA). After two days, mice (n = 3 per group) were sacrificed and major organs were imaged by IVIS Lumina system (Perkin Elmer).
Cell isolation and staining for flow cytometry
To test the Td-Tomato+ cells in cell types of each organ, cell isolation and staining was performed after 2 days of treatment with Cre mRNA formulations (0.3 mg/kg), then analyzed by flow cytometry.
For hepatocyte isolation, two-step collagenase perfusion was executed as described before27. Briefly, mice were anesthetized by isofluorane and fixed. Perfusion was started with liver perfusion medium (Thermo Fisher Scientific, 17701038) for 7–10 min, then switched to liver digestion medium (Thermo Fisher Scientific, 17703034) for another 7–10 min. The liver was collected into a plate containing 10 mL of liver digestion medium and cut to release the hepatocytes. Then the released hepatocytes were collected and washed with ice-cold hepatocyte wash medium (Thermo Fisher Scientific, 17704024) via low speed centrifugation (50xg) for 5 minutes. The supernatant was decanted and the pellet was re-suspended with ice hold hepatocyte wash medium. The cell suspension was passed through 100 μM filter into a new tube. The hepatocyte-cell suspension was washed twice with ice cold hepatocyte wash medium and and once with 1X PBS via centrifugation (50xg) for 5 minutes. Afterwards, the hepatocytes were further isolated by straining through a 100μm filter and using low speed (50xg) centrifugation for 5 minutes, the supernatant was removed and the re-suspended hepatocytes were analyzed by FACS Aria II SORP machine (BD Biosciences).
For isolation and staining of spleen cell types, the removed spleen was minced up by a sterile blade and homogenized in 250 μL of 1X digestion medium (45 units/μL Collagenase I, 25 units/μL DNAse I and 30 units/μL Hyaluronidase). The spleen solution was transferred into a 15 mL tube that contained 5–10 mL of 1X digestion medium. Next, the spleen solution was filtered using a 70 μm filter and washed once with 1X PBS. A cell pellet was obtained by centrifuging for 5 min at the speed of 300xg at 4°C. The supernatant was removed and the cell pellet was resuspended in 2 mL of 1X RBC lysis buffer (BioLegend, 420301) and incubated on ice for 5 min. After incubation, 4 mL of cell staining buffer (BioLegend) was added to stop RBC lysis. The solution was centrifuged again at 300xg for 5 min to obtain a cell pellet. The single cells were resuspended in cell staining buffer and added into flow tubes that contained antibodies (100 μL total volume). The cells were incubated with antibodies for 20 min in the dark at 4 °C. The stained cells were washed twice with 1 mL 1X PBS, then resuspended in 500 μL 1X PBS for flow cytometry analysis. The antibodies used were Pacific Blue anti-mouse CD45 (BioLegend, 103126), Alexa Fluor 488 anti-mouse/human CD11b (BioLegend, 101217), Alexa Fluor 647 anti-mouse CD19 (BioLegend, 115522) and PerCP-Cyanine5.5 Anti-Mouse CD3e (145–2C11) (Tonbo Biosciences, 65–0031). Ghost Dye Red 780 (Tonbo Biosciences, 13–0865-T500) was used to discriminate live cells. The spleen cells were analyzed by LSRForessa SORP machine (BD Biosciences).
For isolation and staining of lung cell types, isolated lungs were minced up by a sterile blade and then transferred into 15 mL tube that contained 10 mL 2X digestion medium (90 units/μL Collagenase I, 50 units/μL DNAse I and 60 units/μL Hyaluronidase) and incubated at 37 °C for 1h with shaking. After incubation, any remaining lung tissue was homogenized. The following steps were similar to the spleen protocol described above. The antibodies here used were Pacific Blue anti-mouse CD45 (BioLegend, 103126), Alexa Fluor 488 anti-mouse CD31 (BioLegend, 102414) and Alexa Fluor 647 anti-mouse CD326 (Ep-CAM) (BioLegend, 118212). Ghost Dye Red 780 (Tonbo Biosciences, 13–0865-T500) was used to discriminate live cells. The lung cells were analyzed by the LSRForessa SORP machine (BD Biosciences).
Gene editing (Cas9 mRNA/sgRNA and Cas9/sgRNA RNPs) in Td-Tomato mice model
To evaluate in vivo gene editing, Td-Tom mice were selected for comparable weight and same sex. We co-delivered Cas9 mRNA and sgRNA (Supplementary Table 2) to tdTomato (td-Tom) mice. Cas9 mRNA/sgTom1 (4/1, wt/wt) were co-delivered by various formulations with the total RNA dose equal to 2.5 mg/kg. 10 days following IV injection, the main organs were removed and imaged on an IVIS Lumina system. For spleen-targeted formulations, the total RNA dose was 4 mg/kg and the weight ratio of Cas9 mRNA to sgTom1 was 2/1, with a detection time was 2 days. For RNP delivery, the mole ratio of Cas9 protein to sgRNA was fixed at 1:3, the injection dose was 1.5 mg/kg RNA, and the detection time was day 7 after injection (n = 3 per group). To confirm Td-Tom expression, tissue sections were further prepared and imaged by confocal microscopy. Briefly, tissue blocks were embedded into optimal cutting temperature compound (OCT) (Sakura Finetek) and cyro-sectioned (8 μm) on a Cryostat instrument (Leica Biosystems). Mounted tissue slices were stained with 4,6-diamidino-2-phenylindole (DAPI, Vector Laboratories) before imaging by confocal microscopy on a Zeiss LSM 700.
Gene editing (Cas9 mRNA/sgPTEN and Cas9/sgRNA RNPs) in C57BL/6 mice
To examine endogenous gene editing in vivo, PTEN was selected. Wild type C57BL/6 mice were IV injected with various carriers by co-delivery of Cas9 mRNA and modified sgPTEN (Supplementary Table 2) at a total dose of 2.5 mg/kg (4/1, mRNA/sgRNA, wt/wt) (n = 3 per group). After 10 days, tissues were collected, and genomic DNA was extracted using a PureLink Genomic DNA Mini Kit (Thermo Fisher). For spleen-targeted formulations, the total RNA dose was 4 mg/kg, Cas9 mRNA/sgTom1 was 2/1 (wt/wt), and the detection time was 2 days after injection. For RNP delivery, the mole ratio of Cas9 protein to sgRNA was fixed at 1:3, the injection dose was 1.5 mg/kg RNA, and the detection time was day 7 after injection (n = 3 per group). After obtaining PTEN PCR products, the T7E1 assay51, 52 was performed to confirm gene editing efficacy by the established protocol from the manufacturer (NEB) (Supplementary Table 3). Detailed instructions including an interactive protocol are available online53. Furthermore, evaluation of PTEN editing was executed on tissue sections by H&E staining and immunohistochemistry (IHC). Briefly, paraformaldehyde (PFA) fixed tissues were embedded in paraffin, sectioned and H&E stained by the Molecular Pathology Core at UTSW. The 4 μm sections were performed in the standard fashion and detected with Elite ABC Kit and DAB Substrate (Vector Laboratories) for IHC. For off-target prediction of sgPTEN, the Cas-OFFinder webtool was employed54. Eight potential targeted positions were amplified by PCR, then analyzed by T7E1 assay and Sanger sequencing.
Gene editing (Cas9 mRNA/sgPCSK9) in C57BL/6 mice
To perform liver PCSK9 gene knock out in vivo. Wild type C57BL/6 mice were IV injected three times (day 0, day 2 and day 4) by co-delivery of Cas9 mRNA and modified sgPCSK9 (Supplementary Table 2) at a total dose of 2.5 mg/kg (1/1, mRNA/sgRNA, wt/wt) (n = 4 per group). After 7 and 9 days, whole blood was collected and serum was separated for serum PCSK9 protein detection by ELISA kit (Abcam). And PCSK9 protein expression of liver tissue was detected by western blot (day 9). For gene knock out validation in genomic level, liver tissues were collected at day 9, genome DNA was extracted using a PureLink Genomic DNA Mini Kit (Thermo Fisher). After examining PCSK9 PCR process, the Indel of PCSK9 was analyzed by T7E1 assay (described above) and TIDE webtool (https://tide.deskgen.com/) (Supplementary Table 3).
Supplementary Material
Acknowledgments
D.J.S. acknowledges financial support from the National Institutes of Health (NIH) National Institute of Biomedical Imaging and Bioengineering (NIBIB) (R01 EB025192-01A1), the American Cancer Society (ACS) (RSG-17-012-01), the Welch Foundation (I-1855), and the Cystic Fibrosis Foundation (CFF) (SIEGWA18XX0). We acknowledge the UTSW Tissue Resource, supported in part by the National Cancer Institute (5P30CA142543), the Moody Foundation Flow Cytometry Facility, and the UTSW Proteomics Core. We thank Yuemeng Jia, Yu-Hsuan Lin, Dr. Yonglong Wei and Prof. Hao Zhu for assistance with tissue processing and analyses.
Footnotes
Competing interests
D.J.S., Q.C., T.W., and the Reagents of the University of Texas System have filed patent applications on SORT and related technologies. D.J.S. is a co-founder of ReCode Therapeutics, which has licensed intellectual property from UT Southwestern.
Supplementary information is available in the online version of the paper.
Please see Supplementary Information for Additional Supplementary Methods, Supplementary Figures, Supplementary Tables, and Additional Supplementary References.
Data availability
The data that support the plots within this paper and other findings of this study are available from the corresponding author upon reasonable request.
References
- 1.Doudna JA & Charpentier E Genome editing: The new frontier of genome engineering with CRISPR-Cas9. Science 346, 1258096 (2014). [DOI] [PubMed] [Google Scholar]
- 2.Wang HX et al. CRISPR/Cas9-based genome editing for disease modeling and therapy: Challenges and opportunities for nonviral delivery. Chem. Rev 117, 9874–9906 (2017). [DOI] [PubMed] [Google Scholar]
- 3.Sander JD & Joung JK CRISPR-Cas systems for editing, regulating and targeting genomes. Nat. Biotechnol 32, 347–355 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hajj KA & Whitehead KA Tools for translation: Non-viral materials for therapeutic mRNA delivery. Nat. Rev. Mater 2, 17056 (2017). [Google Scholar]
- 5.Sahin U, Kariko K & Tureci O mRNA-based therapeutics: Developing a new class of drugs. Nat. Rev. Drug Discovery 13, 759–780 (2014). [DOI] [PubMed] [Google Scholar]
- 6.Jinek M et al. A Programmable Dual-RNA-Guided DNA Endonuclease in Adaptive Bacterial Immunity. Science 337, 816–821 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cong L et al. Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mali P et al. RNA-guided human genome engineering via Cas9. Science 339, 823–826 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kanasty R, Dorkin JR, Vegas A & Anderson D Delivery materials for siRNA therapeutics. Nat. Mater 12, 967–977 (2013). [DOI] [PubMed] [Google Scholar]
- 10.Wood H FDA approves Patisiran to treat hereditary transthyretin amyloidosis. Nat. Rev. Neurol 14, 570 (2018). [DOI] [PubMed] [Google Scholar]
- 11.Jayaraman M et al. Maximizing the potency of siRNA lipid nanoparticles for hepatic gene silencing in vivo. Angew. Chem. Int. Ed 51, 8529–8533 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nelson CE et al. Balancing cationic and hydrophobic content of PEGylated siRNA polyplexes enhances endosome escape, stability, blood circulation time, and bioactivity in vivo. ACS Nano 7, 8870–8880 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hao J et al. Rapid synthesis of a lipocationic polyester library via ring-opening polymerization of functional valerolactones for efficacious siRNA delivery. J. Am. Chem. Soc 137, 9206–9209 (2015). [DOI] [PubMed] [Google Scholar]
- 14.Miller JB et al. Non-viral CRISPR/Cas gene editing in vitro and in vivo enabled by synthetic nanoparticle co-delivery of Cas9 mRNA and sgRNA. Angew. Chem. Int. Ed 56, 1059–1063 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kranz LM et al. Systemic RNA delivery to dendritic cells exploits antiviral defence for cancer immunotherapy. Nature 534, 396–401 (2016). [DOI] [PubMed] [Google Scholar]
- 16.Wilhelm S et al. Analysis of nanoparticle delivery to tumours. Nat. Rev. Mater 1, 16014 (2016). [Google Scholar]
- 17.Gustafson HH, Holt-Casper D, Grainger DW & Ghandehari H Nanoparticle uptake: The phagocyte problem. Nano Today 10, 487–510 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fehring V et al. Delivery of therapeutic siRNA to the lung endothelium via novel lipoplex formulation DACC. Mol. Ther 22, 811–820 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dahlman JE et al. In vivo endothelial siRNA delivery using polymeric nanoparticles with low molecular weight. Nat. Nanotechnol 9, 648–655 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fenton OS et al. Synthesis and biological evaluation of ionizable lipid materials for the in vivo delivery of messenger RNA to B lymphocytes. Adv. Mater 29, 1606944 (2017). [DOI] [PubMed] [Google Scholar]
- 21.Kowalski PS et al. Ionizable amino‐polyesters synthesized via ring opening polymerization of tertiary amino‐alcohols for tissue selective mRNA delivery. Adv. Mater, 1801151 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sago CD et al. High-throughput in vivo screen of functional mRNA delivery identifies nanoparticles for endothelial cell gene editing. Proc. Natl. Acad. Sci. U.S.A 115, E9944–E9952 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kaczmarek JC et al. Polymer-lipid nanoparticles for systemic delivery of mRNA to the lungs. Angew. Chem. Int. Ed 55, 13808–13812 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yan Y, Xiong H, Zhang X, Cheng Q & Siegwart DJ Systemic mRNA delivery to the lungs by functional polyester-based carriers. Biomacromolecules 18, 4307–4315 (2017). [DOI] [PubMed] [Google Scholar]
- 25.Kazi DS et al. Cost-effectiveness of PCSK9 inhibitor therapy in patients with heterozygous familial hypercholesterolemia or atherosclerotic cardiovascular disease. JAMA 316, 743–753 (2016). [DOI] [PubMed] [Google Scholar]
- 26.Wittrup A et al. Visualizing lipid-formulated siRNA release from endosomes and target gene knockdown. Nat. Biotechnol 33, 870–976 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cheng Q et al. Dendrimer-based lipid nanoparticles deliver therapeutic FAH mRNA to normalize liver function and extend survival in a mouse model of hepatorenal tyrosinemia type I. Adv. Mater 30, e1805308 (2018). [DOI] [PubMed] [Google Scholar]
- 28.Zhou K et al. Modular degradable dendrimers enable small RNAs to extend survival in an aggressive liver cancer model. Proc. Natl. Acad. Sci. U.S.A 113, 520–525 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang S et al. Knockdown of anillin actin binding protein blocks cytokinesis in hepatocytes and reduces liver tumor development in mice without affecting regeneration. Gastroenterology 154, 1421–1434 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang S et al. The polyploid state plays a tumor suppressive role in the liver Dev. Cell 44, 447–459 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.ONPATTRO Prescribing Information Label. https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/210922s000lbl.pdf.
- 32.Sabnis S et al. A novel amino lipid series for mRNA delivery: Improved endosomal escape and sustained pharmacology and safety in non-human primates. Mol. Ther 26, 1509–1519 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hassett KJ et al. Optimization of lipid nanoparticles for intramuscular administration of mRNA vaccines. Mol. Ther. - Nucl. Acids 15, 1–11 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ramaswamy S et al. Systemic delivery of factor IX messenger RNA for protein replacement therapy. Proc. Natl. Acad. Sci. U.S.A 114, E1941–E1950 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Love K et al. Lipid-like materials for low-dose, in vivo gene silencing. Proc. Natl. Acad. Sci. U.S.A 107, 1864–1869 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kauffman KJ et al. Optimization of lipid nanoparticle formulations for mRNA delivery in vivo with fractional factorial and definitive screening designs. Nano Lett. 15, 7300–7306 (2015). [DOI] [PubMed] [Google Scholar]
- 37.Hendel A et al. Chemically modified guide RNAs enhance CRISPR-Cas genome editing in human primary cells. Nat. Biotechnol 33, 985–989 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yin H et al. Therapeutic genome editing by combined viral and non-viral delivery of CRISPR system components in vivo. Nat. Biotechnol 34, 328–333 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yin H et al. Structure-guided chemical modification of guide RNA enables potent non-viral in vivo genome editing. Nat. Biotechnol 35, 1179 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang L et al. Meganuclease targeting of PCSK9 in macaque liver leads to stable reduction in serum cholesterol. Nat. Biotechnol (2018). [DOI] [PubMed] [Google Scholar]
- 41.Amoasii L et al. Gene editing restores dystrophin expression in a canine model of Duchenne muscular dystrophy. Science 362, 86–91 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zuris JA et al. Cationic lipid-mediated delivery of proteins enables efficient protein-based genome editing in vitro and in vivo. Nat. Biotechnol 33, 73–80 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sun WJ et al. Self-assembled DNA nanoclews for the efficient delivery of CRISPR-Cas9 for genome editing. Angew. Chem. Int. Ed 54, 12029–12033 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Staahl BT et al. Efficient genome editing in the mouse brain by local delivery of engineered Cas9 ribonucleoprotein complexes. Nat. Biotechnol 35, 431–434 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Finn JD et al. A single administration of CRISPR/Cas9 lipid nanoparticles achieves robust and persistent in vivo genome editing. Cell Rep. 22, 2227–2235 (2018). [DOI] [PubMed] [Google Scholar]
- 46.Tabebordbar M et al. In vivo gene editing in dystrophic mouse muscle and muscle stem cells. Science 351, 407–411 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kamath AT et al. The development, maturation, and turnover rate of mouse spleen dendritic cell populations. J Immunol 165, 6762–6770 (2000). [DOI] [PubMed] [Google Scholar]
- 48.Xue W et al. CRISPR-mediated direct mutation of cancer genes in the mouse liver. Nature 514, 380–384 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Monopoli MP, Aberg C, Salvati A & Dawson KA Biomolecular coronas provide the biological identity of nanosized materials. Nat. Nanotechnol 7, 779–786 (2012). [DOI] [PubMed] [Google Scholar]
- 50.Akinc A et al. Targeted delivery of RNAi therapeutics with endogenous and exogenous ligand-based mechanisms. Mol. Ther 18, 1357–1364 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu JJ et al. CasX enzymes comprise a distinct family of RNA-guided genome editors. Nature 566, 218–223 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nihongaki Y, Kawano F, Nakajima T & Sato M Photoactivatable CRISPR-Cas9 for optogenetic genome editing. Nat. Biotechnol 33, 755–760 (2015). [DOI] [PubMed] [Google Scholar]
- 53.Determining Genome Targeting Efficiency using T7 Endonuclease I. https://www.neb.com/protocols/2014/08/11/determining-genome-targeting-efficiency-using-t7-endonuclease-i
- 54.Cas-OFFinder. http://www.rgenome.net/cas-offinder/.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.