

Abstract
Disruptions in the ubiquitin protein ligase E3A (UBE3A) gene cause Angelman syndrome (AS). Whereas AS model mice have associated synaptic dysfunction and altered plasticity with abnormal behavior, whether similar or other mechanisms contribute to network hyperactivity and epilepsy susceptibility in AS patients remains unclear. Using human neurons and brain organoids, we demonstrate that UBE3A suppresses neuronal hyperexcitability via ubiquitin-mediated degradation of calcium- and voltage-dependent big potassium (BK) channels. We provide evidence that augmented BK channel activity manifests as increased intrinsic excitability in individual neurons and subsequent network synchronization. BK antagonists normalized neuronal excitability in both human and mouse neurons and ameliorated seizure susceptibility in an AS mouse model. Our findings suggest that BK channelopathy underlies epilepsy in AS and support the use of human cells to model human developmental diseases.
Angelman syndrome (AS) is a neurodevelopmental disorder characterized by delayed development, intellectual disability, and seizures (1). Approximately 90% of AS cases are caused by a loss-of-function mutation of the ubiquitin protein ligase E3A (UBE3A) gene, which encodes a HECT E3 ubiquitin ligase (2). Loss of the UBE3A protein could result in accumulation of AS-relevant substrate proteins and contribute to disease pathogenesis (3). Studies using an AS mouse model have demonstrated impaired synaptic connectivity, an imbalance between network excitation and inhibition, and delayed neurodevelopmental processes (4–8). Studies using neurons differentiated from AS patient-derived induced pluripotent stem cells (iPSCs) have suggested disrupted maturation (9, 10), but neither the pathological mechanisms underlying seizures in AS nor the biological substrate(s) of UBE3A has been established.
Altered excitability and fAHP in UBE3A-null human neurons
To understand the cellular and functional consequences of the loss of UBE3A in human neurons, we used the CRISPR-Cas9 system to generate UBE3A knockout (KO) cells in the human embryonic stem cell line (hESC) H9 (Fig. 1A). One clone (KO) harbored a homozygous 5-bp deletion in exon 6 that resulted in a frame shift and early translational termination (Fig. 1A). This clone lacked the UBE3A protein (Fig. 1A). We found no off-target effects from CRISPR-Cas9 on the top 10 predicted off-target sites of our single guide RNA sequence (table S1). UBE3A KO hESCs were karyotypically normal (fig. S1A), expressed key pluripotency markers, and proliferated normally (fig. S1, B and C).
Fig. 1. Altered functional properties of UBE3A-deficient human neurons.
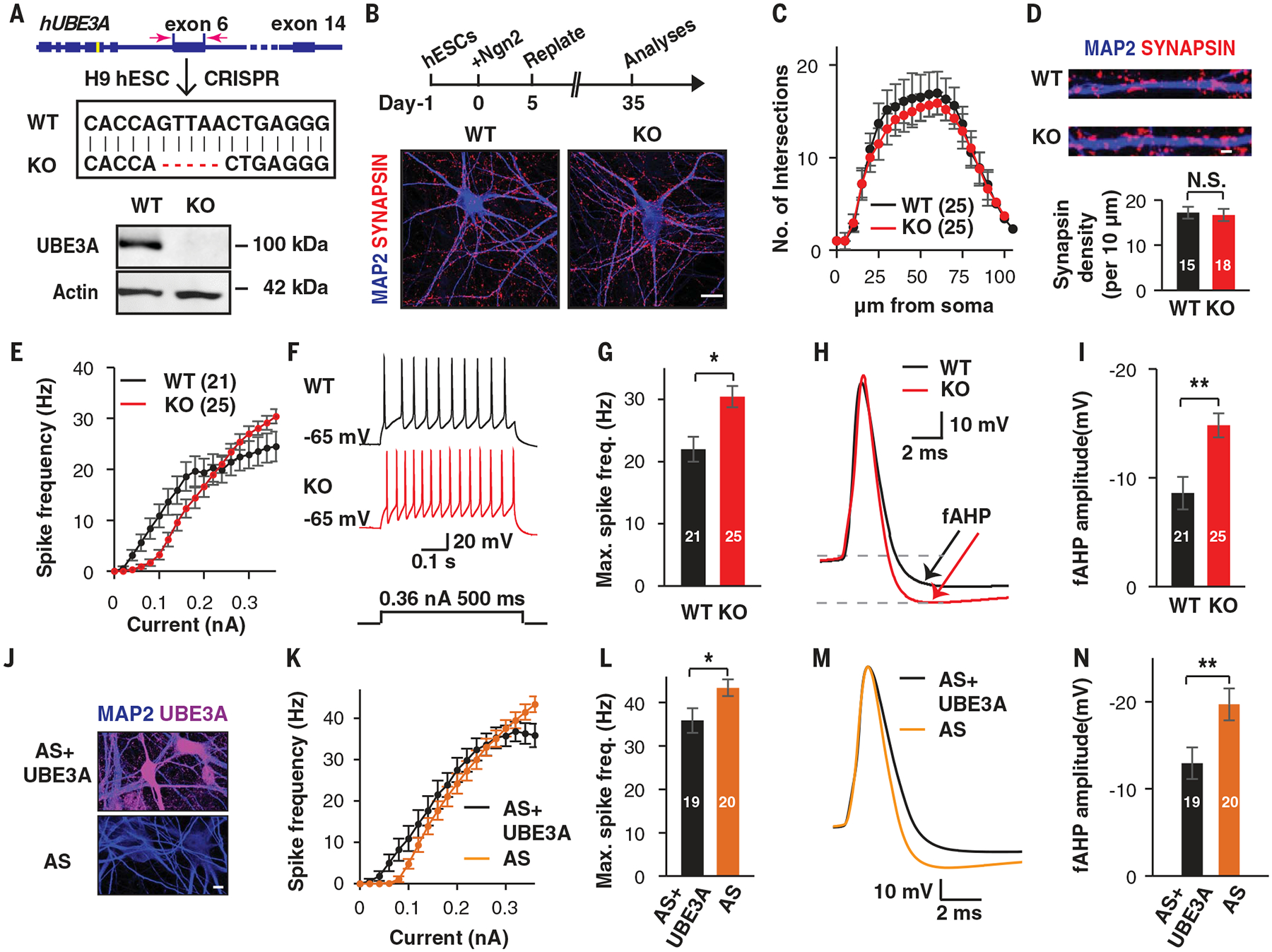
(A to D) Generation of human induced neurons from WT and UBE3A KO hESCs. (A) Schematic illustrating the CRISPR-Cas9-mediated gene editing approach used to knock out UBE3A in hESCs (top) and immunoblot showing the absence of UBE3A protein in UBE3A KO hESCs (bottom). (B) Schematic illustrating the protocol used to generate the human neurons used in this study (top) and representative morphological images of day-35 WT and KO neurons immunostained for MAP2 (blue) and synapsin (magenta) (bottom). Scale bar, 20 μm. (C) Sholl analysis of WT and KO induced neurons at day 35. (D) Representative images and quantification of synaptic puncta densities, calculated from neurons immunostained for synapsin. Scale bar, 1 μm. (E to I) Altered excitability in KO neurons (H9 derived). (E) Frequency-current (F-I) curves showing spike frequency versus current injections in WT and KO induced neurons. (F and G) Representative traces and quantification of maximal spike frequencies by current injection in induced WT and KO neurons. (H and I) Representative traces and quantification of spike fAHP amplitudes of induced WT and KO neurons. (J to N) Induced neurons derived from AS iPSCs showed reproduced excitability and fAHP changes. (J) Immunostaining for UBE3A in AS neurons, with and without ectopic expression of UBE3A. Scale bar, 10 μm. (K) F-I curves showing spike frequencies versus current injection in induced neurons derived from AS iPSCs. (L) Quantification of maximal spike frequencies in the current injections. (M and N) Representative traces and amplitude quantification of spike fAHP. Data represent means ± SEM. The two-tailed unpaired Student’s t test was used for all analyses. The numerals in all bars indicate the number of analyzed neurons. **P < 0.01; *P < 0.05; N.S., not significant.
Next, we induced neuronal differentiation from these isogenic hESCs by ectopically expressing Ngn2 (11). This protocol generates homogenous populations of electrically mature cortical neurons in a time frame shorter than that achieved with morphogen-guided differentiation protocols (12). Both UBE3A wild-type (WT) and KO hESCs were converted to neurons with similar efficiency (fig. S2A). At 35 days post-induction, we observed morphologically mature neurons with dendritic arborizations decorated with synaptic markers (Fig. 1B). There were no significant differences between WT and KO neurons in terms of dendritic complexity or the number of morphological synapses (Fig. 1, C and D). This lack of morphological alterations in KO neurons prompted us to investigate functional changes using whole-cell patch clamp recordings. By injecting stepped currents into neurons to trigger action potentials (APs), we observed that currents higher than 300 pA resulted in increased AP firing frequencies in KO neurons (Fig. 1, E to G). There were no differences in membrane capacitance, input resistance, AP threshold, and AP amplitudes between WT and KO neurons (fig. S2, B to E). Therefore, we monitored changes in voltage-gated sodium (V-Na+) and potassium (V-K+) channels, which are essential for AP generation (13). Current-voltage curves induced by stepped depolarization were indistinguishable between WT and KO neurons (fig. S2F). However, when we examined the fast components of after-hyperpolarization (fAHPs) (fig. S2G), which can affect neuronal excitability in a bidirectional manner (14–16), we observed that fAHPs were significantly augmented in UBE3A KO neurons [WT: −8.6 ± 1.5 mV (mean ± SEM); KO: −14.8 ± 1.1 mV; P = 0.0014] (Fig. 1, H and I). The changes in AP firing and fAHP observed in KO neurons were restored by the ectopic expression of WT UBE3A (fig. S2, H to L), confirming that the lack of UBE3A was responsible for these functional changes. To verify that these differences are not specific to this particular hESC line (H9), we generated independent pairs of isogenic UBE3A WT and KO clones in another hESC line (H1) (fig. S2M) and were able to reproduce similar functional changes (fig. S2, N to S). In humans, AS is associated with loss of the maternal UBE3A allele, and this could not be recapitulated in our homozygous KO clones. Thus, we sought to determine whether neurons from AS patient-derived iPSCs exhibit a similar increase in excitability with augmented fAHP. To this end, we generated neurons from an AS iPSC line that carried a microdeletion in chromosome 15q11.2-q13.1 (which includes the UBE3A gene) and confirmed that the UBE3A protein was not observed in induced neurons (Fig. 1J). The introduction of WT UBE3A into these AS iPSC-derived neurons similarly rescued functional changes in AP firing and fAHPs (Fig. 1, K to N). Taken together, our results established that the loss of UBE3A in human neurons results in altered neuronal excitability associated with enhanced fAHP.
BK augmentation underlies increased fAHP
fAHPs are primarily mediated by calcium- and voltage-dependent big potassium (BK) channels in neurons (17). To determine whether the enhanced fAHPs observed in KO neurons resulted from increased BK channel activity, we pharmacologically isolated BK currents (fig. S3A). Compared with WT-derived neurons, UBE3A KO neurons exhibited larger BK currents (Fig. 2A and fig. S3B). Although a previous report indicated that SK2 (small-conductance Ca2+-activated K+ channel 2) is a substrate of UBE3A (18), we did not observe any significant difference in SK2-mediated medium AHPs (fig. S3, C and D). The higher BK currents observed in KO neurons might simply be due to increased BK protein expression. Indeed, Western blot analyses showed that the level of the obligatory subunit of BK (BKα) was higher in KO neurons (fig. S3, E and F). We independently confirmed this finding by employing single-molecule, atomic force microscopy (AFM) to assess the densities of BK proteins on cellular membranes by analyzing the physical interactions between BK proteins and BK-specific antibodies conjugated to the AFM tip (Fig. 2B and fig. S4) (19). Sequential AFM force mapping was conducted by spatially moving the functionalized tip on the soma membranes of neurons. Figure 2C shows an example of a heatmap in which each colored block indicates the presence of BK channels at that location. The results revealed that the BK channel density was higher on the membranes of KO cells (Fig. 2, C and D). If increased BK levels lead to enhanced fAHP and elevated excitability in KO neurons, we hypothesized that treatment with a BK antagonist would normalize these differences. Indeed, application of BK antagonists (either 5 μM paxilline or 100 nM IBTX) (Fig. 2, E to H, and fig. S5, A to C), but not of an SK antagonist, apamin (200 nM) (fig. S5D), normalized the differences in fAHP amplitude and AP firing frequency between KO and WT neurons, demonstrating that BK channels mediate the augmented fAHP observed in KO neurons.
Fig. 2. UBE3A deletions increase BK channel function in human neurons.
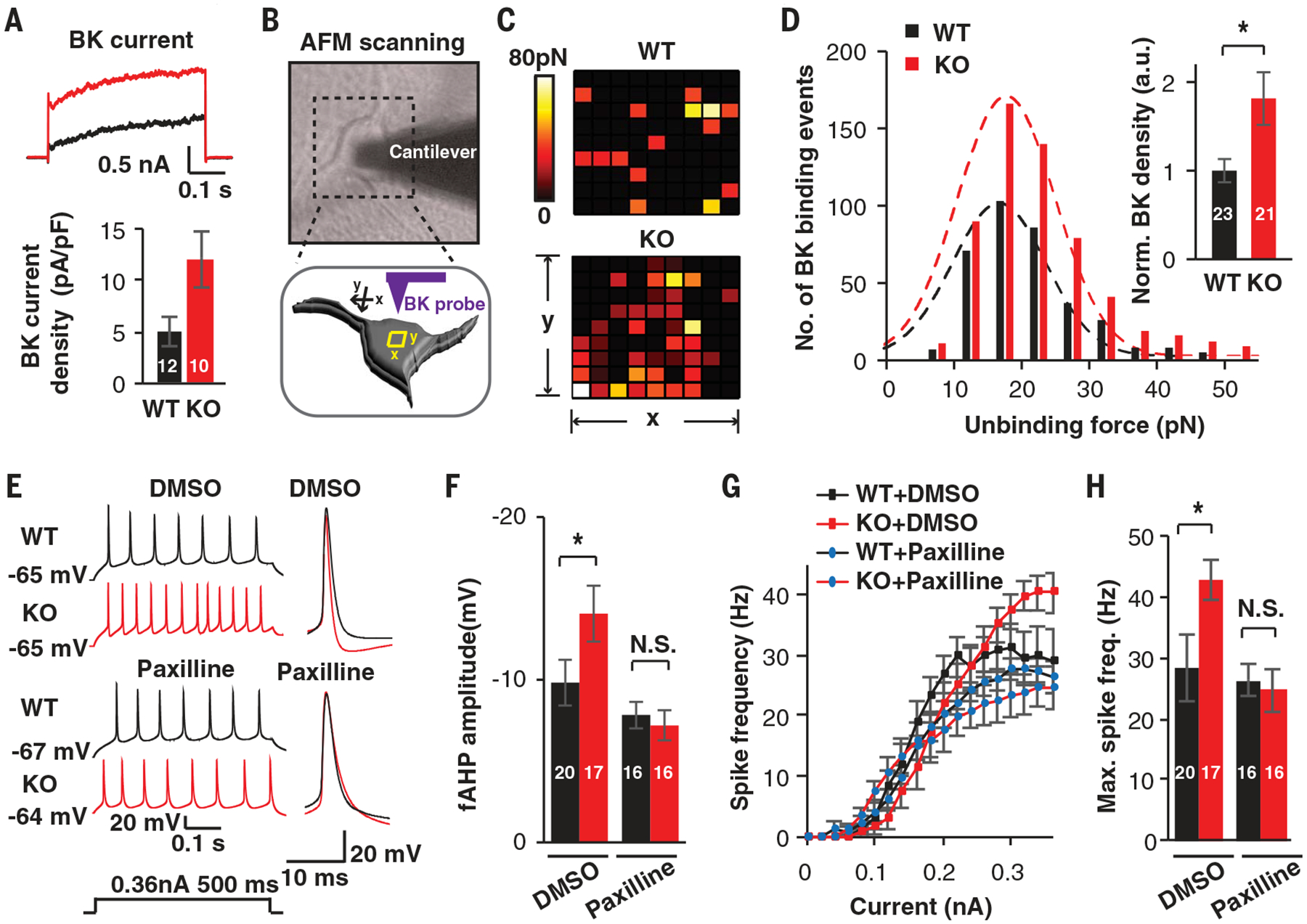
(A) Representative traces and quantification of BK currents isolated from WT and KO neurons treated with paxilline (5 μM). (B) Diagrams illustrating the detection of BK channels using a functionalized probe with AFM. (C) Representative heatmaps of specific BK probe binding events. Force-distance curves (10 data points by 10 data points) were obtained over 1-μm2 areas. Colors indicate the measured force of specific binding events. (D) Unbinding force distribution and BK channel density on the surface of WT and KO neurons. (E to H) Pharmacological rescue by the BK antagonist paxilline. (E and F) Representative traces and quantification of fAHP with and without paxilline (5 μM). DMSO, dimethyl sulfoxide. (G and H) F-I curves showing spike frequencies versus current injections and related quantification in induced neurons. Data represent means ± SEM. In all bars, the values indicate the number of analyzed neurons. The two-tailed unpaired Student’s t test was used. *P < 0.05; N.S., not significant.
UBE3A mediates ubiquitination and proteasomal degradation of BK
Next, we sought to determine how the loss of UBE3A led to BK augmentation. As UBE3A is an E3 ligase, we hypothesized that BK is one of the substrates of UBE3A-mediated ubiquitination and proteasomal degradation (3). To test this hypothesis, we first performed in vitro coimmunoprecipitation (IP) in heterologous cells and confirmed that there is an interaction between UBE3A and BK (fig. S6A). Second, when FLAG-tagged BK was coexpressed with hemagglutinin (HA)-tagged ubiquitin, we observed more BK ubiquitination when WT UBE3A was present than when a catalytically dead mutant (MT; C885A) was used (fig. S6B). BK ubiquitination by UBE3A was further confirmed in an in vitro ubiquitination assay using recombinant proteins (fig. S6C) and by assessing endogenous BK ubiquitination using mouse cortical neurons derived from either WT or Ube3am−/p+ mice (fig. S6, D and E). Further-more, coexpressing BK cDNA with increasing amounts of UBE3A cDNA resulted in a corresponding decline in the BK protein level, and this decrease in the BK level was abolished by the proteasome inhibitor MG312 (fig. S6, F to H). Collectively, these data demonstrate that BK is one of the targeted substrates of UBE3A-mediated ubiquitination and proteasomal degradation. A small subset of people with AS do not carry genetic deletions in UBE3A but instead possess missense mutations in UBE3A (2). Ectopically expressing AS-associated UBE3A missense mutants (Glu550→Leu, Leu502→Pro) (20), but not an autism-associated mutant (Thr485→Ala) (21), failed to down-regulate BK protein levels (fig. S6I), indicating that increased neuronal BK levels may also be a pathophysiological phenotype in AS patients with missense mutations in UBE3A. Although our data thus far suggest that UBE3A affects BK protein abundance via its E3 ligase activity, UBE3A could regulate BK levels via transcriptional modulation (22). However, quantitative real-time reverse transcription polymerase chain reaction (PCR) analysis revealed that there was no significant difference in BKα subunit expression between WT and KO neurons (fig. S7A). To identify which transcripts are altered by UBE3A deficiency at the genome-wide level, we performed mRNA sequencing on WT and KO neurons (fig. S7, B to D, and tables S2 to S4). Differential expression analysis showed a slight perturbation in the transcriptome overall: in total, 129 differentially expressed genes (adjusted P < 0.05) (figs. S7D and S8A and table S2) were identified, and all of these genes showed minor changes, as indicated by the small range of log2(fold change) values. Consistent with our quantitative PCR results, we found no significant differences in the expression levels of BK channel genes (fig. S8B and table S2). Taken together, our results support the notion that, in human neurons, UBE3A primarily regulates BK levels via a posttranslational mechanism.
A BK antagonist restores neuronal excitability and network activity in 3D organoids
One advantage of using our single-step neuronal induction protocol instead of morphogen-guided differentiation protocols is that our protocol can produce electrically mature neurons (11). However, neurons obtained using this protocol do not follow a normal developmental trajectory, in that they bypass the progenitor stage, raising the question of whether these functional deficits are relevant to AS disease progression. To address this issue, we generated three-dimensional (3D) cortical organoids and used this system to investigate functional changes in UBE3A-deficient neurons that follow a normal developmental maturation sequence (23). These brain organoids have been shown to better recapitulate the developmental timelines of human brains (24). After 20 days of differentiation, both WT and KO cells produced spheroids that were indistinguishable in size. They were similarly composed of polarized, proliferating neuroepithelial cells that expressed canonical neuronal progenitor markers, including PAX6, SOX2, and NESTIN (fig. S9, A and B). These brain organoids continued to grow over the next 100 days to ~1 to 2 mm (Fig. 3A). After 120 days, immunostaining revealed the presence of a substantial number of NeuN-positive (NeuN+) neurons. Consistent with previous reports, multiple cortical layer markers were also observed, including BRN2/CTIP2/CUX1, and these segregated the neurons into upper and deep layers (Fig. 3A). Quantification analyses showed that there was no significant difference in the number of NeuN+ neurons or the expression of the cortical layer markers between WT and KO organoids (fig. S9C). We detected a few γ-aminobutyric acid-positive (GABA+) neurons, indicating the presence of inhibitory neurons, within the organoids. Glial fibrillary acidic protein-positive cells, which are indicative of mature astrocytes, were also observed in both WT and KO organoids (fig. S9D). Collectively, these data demonstrated that both the WT and KO organoids contain similar compositions of mature cortical cell types when grown in long-term cultures.
Fig. 3. Altered functional properties of neurons and enhanced network activity in UBE3A-deficient organoids.
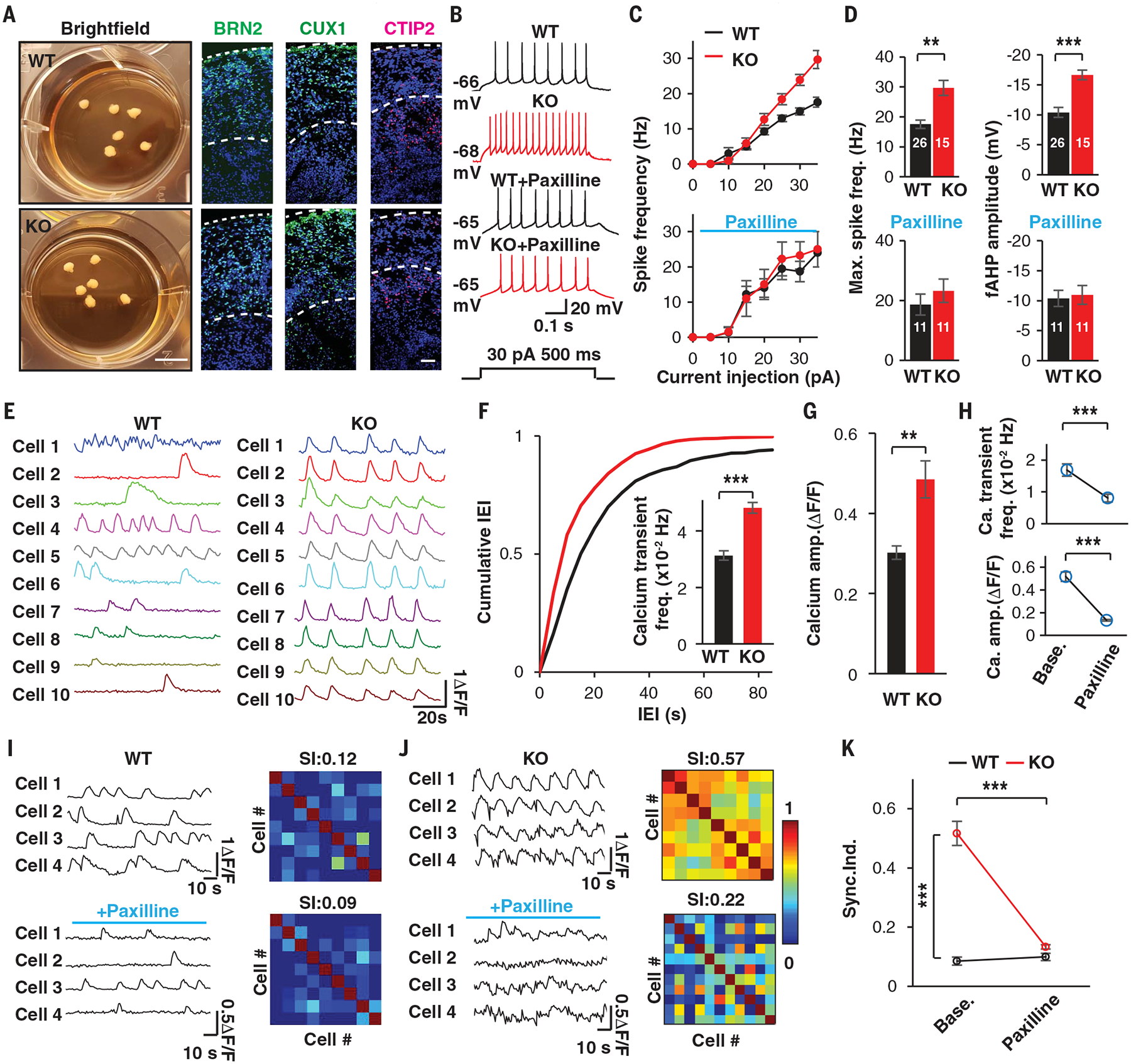
(A) Bright-field images of organoids and immunostaining of cortical layer markers in WT and KO organoids at day 120. Scale bars, 5 mm (bright-field images) or 50 μm (immunostaining). (B to D) Altered electrophysiological properties in neurons from KO organoids, as illustrated with representative traces (B), F-I curves evoked by current injection (C), and quantifications (D), with and without paxilline (10 μM). a.u., arbitrary units. (E to K) Two-photon (2P) live calcium imaging of WT and KO organoids. (E) Calcium transient traces extracted from individual neurons of WT and KO organoids. (F) Cumulative distribution of interevent intervals (IEI) in time bins for calcium transients recorded in WT and KO organoids. The inserted bar graph shows the quantification. n = 131 and 236 neurons from N = 12 and 17 organoids, respectively, for WT and KO organoids. (G) Quantification of the amplitudes of calcium transients recorded in WT and KO organoids. (H) Quantification of the frequencies and amplitudes of calcium transients before and after paxilline treatment (10 μM). N = 10 and 11 organoids for WT and KO, respectively. (I and J) Representative traces of calcium transients in individual neurons (left) and correlation heatmaps (right) obtained from WT (I) and KO (J) organoids. SI, synchronization index. (K) Summary of the synchronization index recorded in WT and KO organoids upon paxilline treatment (10 μM). N = 10 and 11 organoids for WT and KO, respectively. Data represent means ± SEM. Numerals in bars indicate the number of analyzed neurons. The two-tailed paired Student’s t test was used for analysis of data in (H) and (K); the two-tailed unpaired Student’s t test was used for analysis of data in all other panels. **P < 0.01; ***P < 0.001.
Next, we performed whole-cell patch-clamp recordings to evaluate the intrinsic properties of neurons in WT and KO organoids after 120 to 150 days in culture. Similar to the results obtained in 2D-induced neurons, pyramidal-shaped neurons within UBE3A KO organoids exhibited augmented excitability and elevated fAHPs (Fig. 3, B to D), whereas other electrophysiological properties remained the same, indicating that the overall developmental maturation is similar between WT and KO organoids (fig. S10, A to C). Immunoblots of BK proteins confirmed that BK levels were higher in KO organoids (fig. S10D). Paxilline treatment normalized the augmented neuronal excitability and fAHP changes observed in neurons of KO organoids (Fig. 3, B to D). We obtained similar results with cortical organoids generated from AS iPSCs (fig. S10, E to G). Thus, our recordings performed in long-term cultured 3D human cortical organoids reproduced the phenotypes observed in 2D human neurons.
Because 3D organoids are capable of assembling more sophisticated neuronal networks than can be achieved in 2D neuronal cultures, we sought to use calcium (Ca2+) imaging to monitor differences in neuronal network dynamics in large populations of cells in WT and KO cortical organoids. Incubating cortical organoids with Fluo-4AM (2 μM) resulted in labeled cells located up to 170 μm from the surface (movie S1). In WT organoids, bath application of TTX (2 μM) abolished spontaneous calcium transients (fig. S10H and movie S2). Whereas neurons in day-120 to day-150 WT organoids displayed prominent spontaneous Ca2+ transients, at the network level synchronization of transients was rare, and most neurons fired in a stochastic manner (Fig. 3E and movie S3) (25). In contrast, neurons in UBE3A KO organoids exhibited more frequent firing with augmented dynamic ranges of Fluo-4 fluorescence, and neurons in some KO organoids seemed to discharge regularly and synchronously (Fig. 3, E to G, and movie S3). Soma calcium transients were correlated with intrinsic neuronal activity, as indicated by the finding that the intensity of somatic Fluo-4 fluorescence gradually increased along with graded electrical train stimulations in individual neurons (fig. S10I), demonstrating that regular fluorescence fluctuations were related to intrinsic neuronal bursting, as shown in previous reports (26, 27). In addition, perforated patch recordings revealed that neurons exhibit spontaneous burst firing in UBE3A KO organoids (fig. S10J). Paxilline treatment completely converted spontaneous burst firing into single AP firing in these neurons (fig. S10, J and K) and decreased both the frequencies and the amplitudes of calcium transients in the organoids (Fig. 3H), suggesting that augmented BK activity promoted burst firing in individual neurons and subsequent hyperactivity in the entire network. Finally, we investigated network-level synchronization in cortical organoids. We found that KO organoids and AS iPSC-derived organoids exhibited higher levels of synchronization and that this effect was abolished by treatment with paxilline (Fig. 3, I to K, and movies S3 and S4). Taken together, our data suggest that aberrant intrinsic firing of UBE3A-null neurons collectively elicits network-level hyperactivity and synchronization that resemble the seizure activity observed in AS patients (28).
A BK antagonist ameliorates epileptic susceptibility in a mouse model of AS
Our data thus far demonstrate that changes in intrinsic excitability are mediated by augmented BK channel activities in neurons derived from both isogenic UBE3A KO hESCs and AS iPSCs. Because none of these phenotypes had been previously reported in AS mouse models, we sought to determine whether they are also present in neurons obtained from mice with a maternal Ube3a deletion (Ube3am−/p+) (fig. S11A), which has been shown to recapitulate many of the symptoms associated with AS in humans (29). At postnatal day 28, the BK currents were significantly larger in hippocampal neurons obtained from Ube3am−/p+ mice than in those obtained from WT mice (fig. S11B). Similar to human neurons, changes in excitability and comparatively higher fAHP were observed in neurons obtained from Ube3am−/p+ mice (fig. S11, C and D) (fAHP; WT: −4.4 ± 1.4 mV, n = 26; Ube3am−/p+: −6.4 ± 1.2 mV, n = 29; p < 0.001). Paxilline normalized these changes (fig. S11, C and D). These results indicate the presence of evolutionally conserved BK augmentation in mouse neurons that lack Ube3a, and they prompted us to test whether the enhanced seizure susceptibility observed in Ube3am−/p+ mice (7) could be normalized upon BK antagonist treatment. First, we exposed mice to the putative GABA type A receptor (GABAAR) antagonist flurothyl (100 μl/min), and we observed that Ube3am−/p+ mice exhibited a significant reduction in the latency to myoclonic seizure, and this effect could be normalized by paxilline pretreatment [0.35 mg/kg, intraperitoneal (i.p.) injection] (Fig. 4A). Similarly, another GABAAR antagonist, picrotoxin (3 mg/kg, i.p.), induced a grade 1 to 2 seizure phenotype based on Racine’s scale (30) in WT mice, whereas the same treatment induced a much more severe seizure phenotype (grade 5) in Ube3am−/p+ mice (Fig. 4B). Paxilline pretreatment was able to normalize the seizure grade of Ube3am−/p+ mice to a level comparable to that observed in WT mice (Fig. 4B). Next, to investigate the AS-like electroencephalogram (EEG) abnormalities observed in these mice, we recorded resting-state local field potentials (LFPs), which are analogous to intracortical EEG changes, in freely moving mice (fig. S12A) (31). As previously reported, compared to the WT mice, Ube3am−/p+ mice exhibited a strong trend toward total spectral power being increased in the delta rhythmicity (1 to 4 Hz) (Fig. 4, C to E, P = 0.0044) (31). We also observed increased power in other bands, including theta (4 to 8 Hz), beta (12 to 30 Hz), and gamma (30 to 80 Hz) bands, in Ube3am−/p+ mice, but these differences were not statistically significant (fig. S12B). Because increased delta rhythmicity has been linked to seizure susceptibility of Ube3am−/p+ mice (7), we sought to determine whether paxilline treatment could normalize the delta rhythmicity. Indeed, paxilline treatment normalized the enhancement observed in the delta band in Ube3am−/p+ mice (P = 0.85) (Fig. 4, F to H). Taken together, we conclude that BK augmentation similar to that observed in human UBE3A KO neurons is observed in neurons of Ube3am−/p+ mice, and paxilline treatment ameliorates the seizure threshold as well as the high delta oscillation observed in these mice.
Fig. 4. BK modulation ameliorates seizure susceptibility in a mouse model of AS.
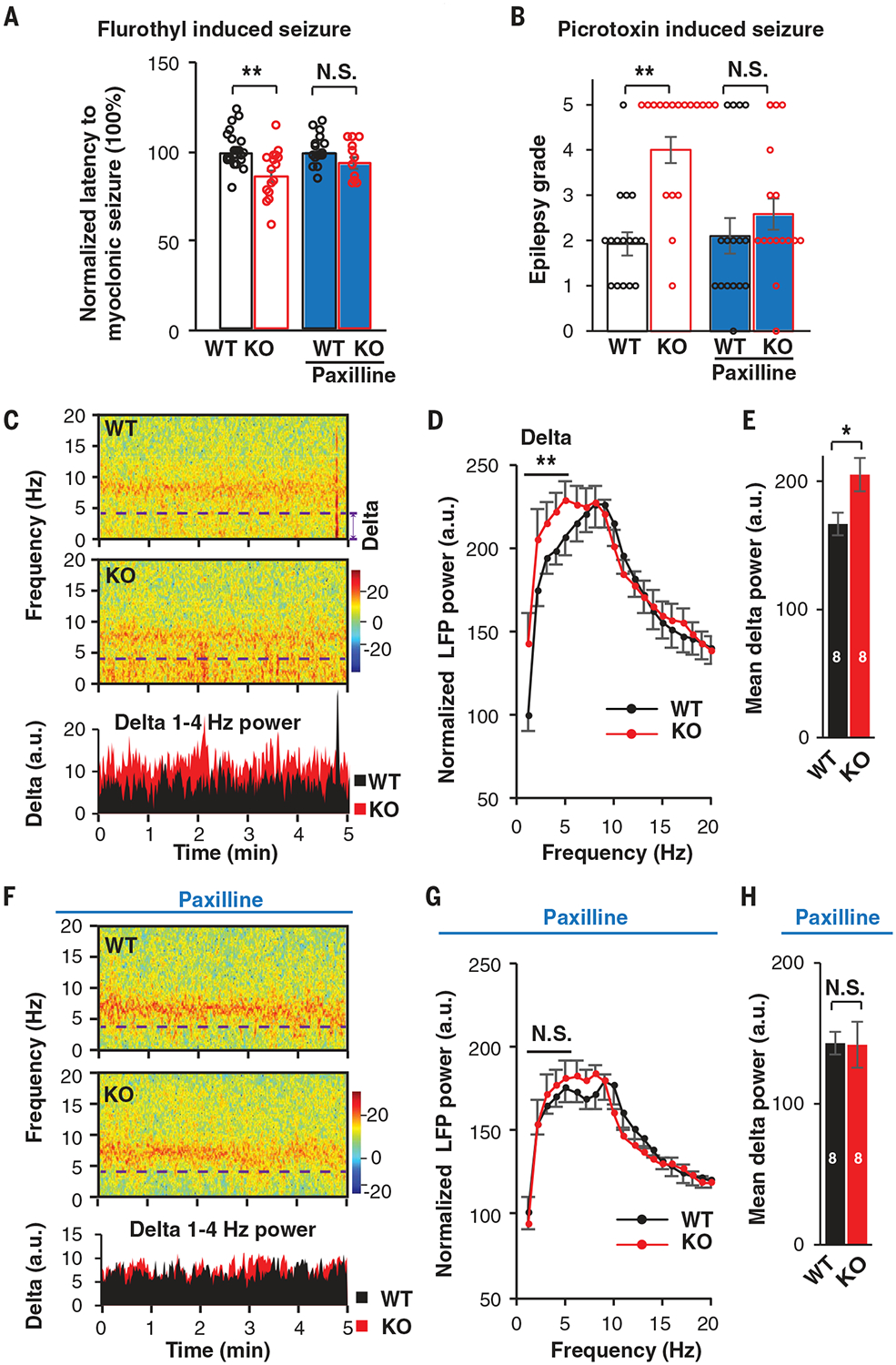
(A) Latency to myoclonic seizure induced by flurothyl in WT and KO (Ube3am−/p+) mice. WT without paxilline, N = 23; KO without paxilline, N = 17; WT with paxilline, N = 16; KO with paxilline, N = 12. (B) Epilepsy grade induced by picrotoxin in WT and KO mice. WT without paxilline, N = 16; KO without paxilline, N = 18; WT with paxilline, N = 16; KO with paxilline, N = 16. (C) Samples of spectrogram and averaged delta power data for LFP from BIC of WT and KO mice with sound stimuli. Delta rhythmicity below the dashed line was averaged for analysis. (D) Summary of power spectral density (PSD) of LFP recorded from BIC of WT and KO mice with sound stimuli. N = 8 mice from two batches. Normalization was performed to remove the baseline difference due to batch effect. A two-way analysis of variance (ANOVA) with the Bonferroni post hoc test was used for frequencies of 1 to 4 Hz to analyze differences between WT and KO mice. (E) Mean delta (1 to 4 Hz) power of WT and KO mice LFPs. N = 8 mice from two batches. (F) Sample spectrogram and averaged delta power of LFPs in BIC of WT and KO mice with paxilline treatment (0.35 mg/kg; four times, at 1-hour intervals). (G) Summary of PSD of LFPs in BIC recorded in WT and KO mice with paxilline treatment. Two-way ANOVA with the Bonferroni post hoc test was used for frequencies of 1 to 4 Hz to analyze differences between WT and KO mice. (H) Mean delta power (1 to 4 Hz) of WT and KO mice LFPs with paxilline treatment. N = 8 mice from two batches. Numerals in bars indicate the number of animals analyzed. Data represent means ± SEM. The two-tailed unpaired Student’s t test was used to analyze data for all panels except for (D) and (G). *P < 0.05; **P < 0.01; N.S., not significant.
Outlook
By characterizing the functional properties of 2D human neurons and 3D cortical organoids derived from UBE3A KO hESCs and AS iPSCs, we demonstrated an evolutionally conserved channelopathy that contributes to network dysfunction and hyperactivity in AS. Our results suggest that BK channels are substrates for UBE3A and that BK might thus be a therapeutic target for treatment of patients with AS.
Supplementary Material
ACKNOWLEDGMENTS
We thank E. Chua and N. Meenubharathi for their assistance in cell culture, molecular cloning, and Western blotting and N. Harmston for help with RNA sequencing (RNA-seq) data processing. We thank C. Yong (ZJU) for sharing the BK plasmid. We thank G. Augustine, J. Lee, and W.-K. Ho for valuable discussions. We thank S.T. Li (SJTU) for help with animal epilepsy experiments. We thank the Advanced Bioimaging Core of Duke-NUS/SingHealth for their technical support with multiphoton microscopy.
Funding: This work was supported by the Singapore Ministry of Education (MOE) Academic Research Fund (MOE2014-T2-2-071 to H.S.J.), a National Medical Research Council Collaborative Research Grant (NMRC/OFIRG/0050/2017 to A.X.S. and H.S.J.), a KHOO Bridge funding (Duke-NUS-KBrFA/2017/0004 to H.S.J.), a Duke-NUS Signature Research Program block grant (to H.S.J. and K.I.), a Khoo Postdoctoral Fellowship Award (Duke-NUS-KPFA/2016/0007 to M.F.), the National Parkinson’s Disease Translational Clinical Research Program fund (NMRC/TCR/013-NNI/2014 to E.K.T.), a Singapore Translational Research Investigator Award (NMRC/STaR/014/2013 to E.K.T.), and an NMRC Young Investigator Research Grant (NMRC/OFYIRG/0022/2016) to O.J.L.R.
Footnotes
Competing interests: The authors declare no competing interests.
Data and materials availability: RNA sequencing raw data and counts were deposited in the Gene Expression Omnibus with accession number GSE120225.
REFERENCES AND NOTES
- 1.Buiting K, Williams C, Horsthemke B, Nat. Rev. Neurol 12, 584–593 (2016). [DOI] [PubMed] [Google Scholar]
- 2.Mabb AM, Judson MC, Zylka MJ, Philpot BD, Trends Neurosci. 34, 293–303 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sell GL, Margolis SS, Front. Neurosci 9, 322 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jiang YH et al. , Neuron 21, 799–811 (1998). [DOI] [PubMed] [Google Scholar]
- 5.Wallace ML, Burette AC, Weinberg RJ, Philpot BD, Neuron 74, 793–800 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wallace ML, van Woerden GM, Elgersma Y, Smith SL, Philpot BD, J. Neurophysiol 118, 634–646 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Judson MC et al. , Neuron 90, 56–69 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Judson MC et al. , J. Neurosci 37, 7347–7361 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chamberlain SJ et al. , Proc. Natl. Acad. Sci. U.S.A 107, 17668–17673 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fink JJ et al. , Nat. Commun 8, 15038 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang Y et al. , Neuron 78, 785–798 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yi F et al. , Science 352, aaf2669 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bean BP, Nat. Rev. Neurosci 8, 451–465 (2007). [DOI] [PubMed] [Google Scholar]
- 14.Gu N, Vervaeke K, Storm JF, J. Physiol 580, 859–882 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Montgomery JR, Meredith AL, Proc. Natl. Acad. Sci. U.S.A 109, 18997–19002 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kimm T, Khaliq ZM, Bean BP, J. Neurosci 35, 16404–16417 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Storm JF, J. Physiol 385, 733–759 (1987). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sun J et al. , Cell Rep. 12, 449–461 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maciaszek JL, Soh H, Walikonis RS, Tzingounis AV, Lykotrafitis G, J. Neurosci 32, 11435–11440 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sadikovic B et al. , Hum. Mutat 35, 1407–1417 (2014). [DOI] [PubMed] [Google Scholar]
- 21.Yi JJ et al. , J. Biol. Chem 292, 12503–12515 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ramamoorthy S, Nawaz Z, Nucl. Recept. Signal 6, e006 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Paşca AM et al. , Nat. Methods 12, 671–678 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kelava I, Lancaster MA, Cell Stem Cell 18, 736–748 (2016). [DOI] [PubMed] [Google Scholar]
- 25.Kirwan P et al. , Development 142, 3178–3187 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kerr JN, Greenberg D, Helmchen F, Proc. Natl. Acad. Sci. U.S.A 102, 14063–14068 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Emiliani V, Cohen AE, Deisseroth K, Häusser M, J. Neurosci 35, 13917–13926 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pelc K, Boyd SG, Cheron G, Dan B, Seizure 17, 211–217 (2008). [DOI] [PubMed] [Google Scholar]
- 29.Jiang Y, Tsai TF, Bressler J, Beaudet AL, Curr. Opin. Genet. Dev 8, 334–342 (1998). [DOI] [PubMed] [Google Scholar]
- 30.Racine RJ, Electroencephalogr. Clin. Neurophysiol 32, 281–294 (1972). [DOI] [PubMed] [Google Scholar]
- 31.Sidorov MS et al. , J. Neurodev. Disord 9, 17 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.