

Abstract
In-cell NMR can investigate protein conformational changes at atomic resolution, such as those changes induced by drug binding or chemical modifications, directly in living human cells, and therefore has great potential in the context of drug development as it can provide an early assessment of drug potency. NMR bioreactors can greatly improve the cell sample stability over time and, more importantly, allow for recording in-cell NMR data in real time to monitor the evolution of intracellular processes, thus providing unique insights into the kinetics of drug-target interactions. However, current implementations are limited by low cell viability at >24 h times, the reduced sensitivity compared to “static” experiments and the lack of protocols for automated and quantitative analysis of large amounts of data. Here, we report an improved bioreactor design which maintains human cells alive and metabolically active for up to 72 h, and a semiautomated workflow for quantitative analysis of real-time in-cell NMR data relying on Multivariate Curve Resolution. We apply this setup to monitor protein–ligand interactions and protein oxidation in real time. High-quality concentration profiles can be obtained from noisy 1D and 2D NMR data with high temporal resolution, allowing further analysis by fitting with kinetic models. This unique approach can therefore be applied to investigate complex kinetic behaviors of macromolecules in a cellular setting, and could be extended in principle to any real-time NMR application in live cells.
A complete characterization of the three-dimensional structure of a disease-related protein and its dynamic behavior in the native cellular environment are critical to fully understand its function within the cell and to develop molecules that efficiently and selectively interfere with its function. The first steps of a structure-based drug design involve the screening of ligand libraries, followed by further optimization of lead compounds. Classically, these steps are performed in vitro on the isolated target protein/domain, where the most promising molecules are selected solely on the basis of their binding affinity toward the target. This initial design phase does not, by definition, take into account the interactions that potentially occur between the newly designed molecules and cellular structures such as the plasma membrane or the intracellular milieu. Often, this leads to a high attrition rate when drugs move from preclinical to clinical trials. To provide this crucial information, cellular assays are therefore necessary further down the drug development pipeline to select those among the compounds most active toward the isolated protein that are able to exert the desired effect in a cell culture model.
In recent years, in-cell Nuclear Magnetic Resonance (NMR) spectroscopy has emerged as a powerful approach to investigate structural features of macromolecules in their native cellular context.1−6 Thanks to the high sensitivity of the chemical shift of each nucleus to chemical or conformational changes in the immediate surroundings, in-cell NMR has made it possible to investigate functionally relevant processes such as protein folding/misfolding,7−9 metal binding,7,10 disulfide bond formation,11,12 and protein–protein interactions13 in living human cells. Recent developments in methodology have focused on the screening and characterization of the interactions between small ligands and proteins/nucleic acids.14,15 These uses naturally lend themselves to using in-cell NMR for drug candidate screening. Indeed, a recent study in human cells used this approach to examine different binding modalities of trial compounds to their intracellular targets, their ability to be taken up by target cells, and their binding selectivity.16 Each of these parameters are important for predicting and improving the efficacy of the potential drugs, and in-cell NMR provides unique advantages by allowing them to be studied simultaneously.16
Two main factors limiting the applicability of in-cell NMR to drug development are the intrinsically low sensitivity of NMR and the short lifetime of the cells during the experiment. A common workaround to this problem is to densely pack cells in a closed NMR tube for a short period of time, which provides sufficient NMR signal intensity without sacrificing cell viability. However, such a trade-off imposes severe time constraints and prevents the study of biological processes that take more than minutes/hours to occur. Furthermore, even the investigation of stationary states may suffer from artifacts caused by the cellular response to hypoxia, media acidification, and starvation.17 A more appropriate solution involves the use of NMR bioreactors, which apply a constant flow of growth medium to the sample tube within the NMR spectrometer to provide fresh nutrients and remove the byproducts of cellular metabolism. Several examples of NMR bioreactors have been developed for both bacteria and mammalian cells, where cells are either encapsulated in biocompatible hydrogels or kept in suspension through the use of a high MW cutoff membrane.18−22 NMR bioreactors improve sample stability by maintaining cell viability at acceptable levels for longer periods of time, ranging from a few hours to up to ∼24 h, allowing longer NMR experiments to be recorded and consequently boosting the signal-to-noise ratio (S/N). In addition to enabling long acquisition times for single, time-averaged experiments, NMR bioreactors can also be used to record cellular processes in real time.21,22 This application provides unprecedented levels of information on the kinetics of functional processes within living cells, due to the high chemical and conformational sensitivity of NMR.
To date, factors limiting the use of bioreactors for real-time in-cell NMR include the lack of detailed data on the viability of cells in different sample regions and the reduction of S/N due to a lower cell density compared to the “static” in-cell NMR setups. Moreover, real-time NMR imposes a trade-off between a higher S/N obtained with longer experiments, that translates into a lower error in the time-dependent measurements, and a higher temporal resolution obtained by sampling at a higher rate with shorter NMR experiments. Protocols for automated data processing and quantitative analysis are also lacking, which are even more necessary in high-temporal-resolution regimes, due to the high number of NMR spectra recorded. Finally, further optimization of the bioreactor setup is required to enable monitoring intracellular events for periods of time longer than 24 h with sufficiently high cell viability.
In this work, we report an improved bioreactor setup, obtained by adapting the modular bioreactor design previously reported,22 that maintains human cells encapsulated in agarose gel threads highly viable and metabolically active for up to 72 h of real-time in-cell NMR experiments. To demonstrate the utility of the NMR bioreactor design for the study of cellular processes in real time, we studied the kinetics of interaction of small molecules, some of which are already used as drugs, with two clinically relevant proteins, carbonic anhydrase and copper, zinc superoxide dismutase. Quantification of the relative concentrations of protein species was performed using the Multivariate Curve Resolution-Alternating Least Squares (MCR-ALS) algorithm.23 MCR-ALS is a mathematical approach that has been applied to various analytical and spectroscopic techniques to recover pure response profiles of different chemical species contained in the sample (here, the NMR spectra and the relative concentration profiles of each state of the protein) with minimal prior information on the composition and the spectral features of the analyzed mixture (here, the cell sample).23−26 Recently, MCR-ALS has been applied to deconvolute NMR spectra of complex metabolic mixtures27 and to improve the S/N of 2D Solid-State NMR spectra.28 To evaluate the applicability of MCR-ALS to real-time in-cell NMR data, the improved bioreactor setup was employed to monitor conformational and chemical modifications of proteins directly in human cells, in response to cell treatment with exogenous molecules.
First, we monitored as a function of time the binding of inhibitors of the second isoform of human carbonic anhydrase (CA2) by observing the unlabeled intracellular protein with 1D 1H NMR spectra. CA2 is one of 15 isoforms of human carbonic anhydrase, which catalyze the reversible hydration of carbon dioxide to bicarbonate ion and proton. Carbonic anhydrases are responsible for intracellular pH balance and regulate many physiological processes, and have been associated with several human diseases, including epilepsy, obesity, and cancer.29,30 As such, they have been extensively investigated as drug targets.31 The approved antiglaucoma drugs acetazolamide (AAZ) and methazolamide (MZA) are often used as reference compounds when developing novel, isoform-selective CA inhibitors. Both compounds have been extensively characterized in vitro, where they inhibit CA2 with nanomolar KI.29,31 The binding of AAZ and MZA to intracellular CA2 has been recently observed by in-cell NMR without the use of a bioreactor, revealing that both compounds quantitatively bind intracellular CA2 but exhibit remarkably different rates of diffusion through the plasma membrane.16 Therefore, these experiments served to validate the performance of our NMR bioreactor on different time scales.
We then monitored the formation of the native intramolecular disulfide bond of the copper-depleted, zinc-bound form of human superoxide dismutase 1 (SOD1) catalyzed by ebselen, a redox-active organoselenium drug,32 by observing 15N-labeled intracellular protein with 2D 1H–15N correlation NMR spectra. SOD1 is a ubiquitous enzyme responsible for the elimination of the cytotoxic superoxide anion produced during cellular respiration. The misfolding and aggregation of human SOD1 are correlated to the onset of Amyotrophic Lateral Sclerosis (ALS), a fatal neurodegenerative disease.33 The maturation of human SOD1 and its ALS-related mutants has been extensively characterized by NMR both in vitro34−36 and in human cells.7,8,37 In its mature active state, human SOD1 is a stably folded homodimer where each monomer binds one zinc ion and one copper ion, and harbors an intramolecular disulfide bond between C57 and C146 that provides structural stability (Cu,Zn-SOD1SS). When overexpressed in human cells, apo-SOD1 spontaneously binds zinc and forms a stable, disulfide-reduced homodimer (E,Zn-SOD1SH), while copper binding and cysteine oxidation can only occur upon interaction with the specific copper chaperone for SOD1, CCS.7,38 However, it has been shown that premature disulfide bond formation can be induced by external factors, e.g., in response to cadmium-induced oxidative stress39 or by treatment with ebselen.40 Specifically, ebselen reacts directly with the cysteines of E,Zn-SOD1SH, forming a selenylsulfide intermediate. Subsequent intramolecular disulfide bond formation then releases ebselenol, which is then oxidized back to ebselen by reacting with H2O2.41 The catalytic action of ebselen makes it a promising lead compound for potential drugs that could prevent SOD1 misfolding and aggregation by modulating its intracellular redox state. Previous NMR applications did not investigate the effect of ebselen over time, therefore crucial information on the kinetics of its activity within living cells is currently lacking.
Time-dependent concentration curves were obtained for both ligand binding and protein oxidation data sets, with a time resolution as high as ∼7 min over a period of time up to 60 h, during which the cells remained viable. The comparably good quality of the concentration curves obtained by MCR-ALS, in spite of the low S/N of each single NMR spectrum, suggests that the experimental setup combining bioreactor and automated quantitative spectral analysis will provide, with minimal additional inputs, reliable data to model complex kinetic behaviors of macromolecules in living cells at atomic resolution.
Experimental Section
Human Cell Culture and Transfection
HEK293T cells (ATCC CRL-3216) were maintained in Dulbecco-modified Eagle medium (DMEM) high-glucose (Gibco) supplemented with 2 mM l-glutamine, antibiotics (100 U/mL penicillin and 100 μg/mL streptomycin) and 10% fetal bovine serum (FBS, Gibco) in uncoated 75 cm2 plastic flasks and incubated at 37 °C, 5% CO2 in a humidified atmosphere. Cells were transiently transfected following a previously reported protocol.42 Briefly, the pHLsec plasmid containing the gene of CA2 or SOD1, which were cloned by removing the secretion sequence from the original vector as previously described,16 was mixed to polyethylenimine (PEI), with a DNA/PEI ratio of 1:2 (25 μg/flask DNA, 50 μg/flask PEI) and, after 20 min, added to the cells. Protein expression was carried out for 48 h in either 20 mL DMEM for unlabeled cell samples or 20 mL [U–15N]-BioExpress6000 medium (Cambridge Isotope Laboratories) for uniform 15N-labeled cell samples. Both expression media were supplemented with 2% FBS, antibiotics, and 10 μM ZnSO4. After protein expression, cells were collected by trypsinization, washed once with phosphate-buffered saline (PBS, pH 7.4, Gibco), and spun down in Eppendorf tubes at 500g prior to the NMR sample preparation.
Production of Agarose Threads
Low-gelling agarose (Sigma-Aldrich, A4018) was dissolved at 1.5% (w/v) in PBS at 85 °C, sterilized by filtration with a 0.22 μm filter, aliquoted in Eppendorf tubes and stored at 4 °C. For sample preparation, one aliquot of solidified agarose was melted at 85 °C in a water bath and subsequently kept in solution at 37 °C in a thermoblock. A pellet of cells overexpressing the desired protein, collected from one 75 cm2 flask (∼3 × 107 cells), was heated up at 37 °C for 15–20 s in the thermoblock. Cells were then resuspended in 450 μL of agarose solution, carefully avoiding the formation of bubbles. The cell–agarose suspension was aspirated into a chromatography PEEK tubing (outer diameter, o.d. = 1/16”, inner diameter, i.d. as specified in the Results section) connected to a 1 mL syringe and was cooled down at RT for 2 min. Threads were then cast into the flow unit NMR tube, which contained an ∼5 mm-high bottom plug of 1.5% agarose gel (to place the cell sample within the active volume of the 1H NMR coil) and was prefilled with with 100 μL PBS.
Bioreactor Setup
The NMR bioreactor used in this study is a variant of a setup described earlier,22 which was based on the InsightMR system (Bruker),43 and consists of a flow NMR tube (o.d. = 5 mm, i.d. = 4.2 mm) connected to a watertight sealing that sets the total sample height to 38 mm, for a total sample volume of 526 μL. The inlet consists of a PEEK capillary tubing (o.d. = 1/32”, i.d. = 0.5 mm) inserted in the NMR tube down to ∼6 mm from the bottom, while the outlet is a PTFE tubing (o.d. = 1/32”, i.d. = 0.5 mm) attached at the top of the tube holder (Figure S1 of the Supporting Information, SI). The transfer line was temperature-controlled through a water bath set at 37 °C. The bioreactor was attached to an FPLC pump with reciprocating piston heads (P-920 module from ÄKTA FPLC, GE Healthcare) through PEEK tubing (o.d. = 1/16”, i.d. = 0.5 mm). During the in-cell NMR experiments, unlabeled DMEM (Sigma-Aldrich D5648, powder, reconstituted in sterile-filtered Milli-Q H2O and supplemented with 2% FBS, 10 mM NaHCO3, antibiotics, and 2% D2O, pH 7.4) was supplied at a flow rate of 0.1 mL/min. The medium was contained in a 250 or 500 mL reservoir bottle kept at 37 °C in the water bath. The bottle was sealed with a steel headpiece with two hose nozzles, one connected to the pump through a FEP tubing (o.d. = 1/8”, i.d. = 1.6 mm), the other connected to a 0.22 μm PTFE syringe filter for air intake.
After each run, the bioreactor was rinsed with water and cleaned-in-place by flowing at 1 mL/min the following solutions: 0.2 M NaOH, 3 M citric acid, and 0.2 M NaOH, for at least 30 min each, followed by sterile-filtered Milli-Q water for 2 h. The reservoir assembly and tubing were washed and autoclaved after each run.
Cell Treatment with External Molecules
Cells overexpressing unlabeled CA2 were treated with either 25 μM AAZ or 10 μM MZA; cells overexpressing 15N-labeled SOD1 were treated with 20 μM ebselen. These concentrations were chosen on the basis of different rates of cell permeability previously estimated and do not affect cell viability, as determined from dose-dependent cellular vitality tests previously reported.16,40 The molecules were supplemented by injecting concentrated stock solutions directly in the medium reservoir.
Trypan Blue Test
Viability measurements were performed by Trypan blue exclusion test on thin slices obtained from threads extracted from the NMR tube either immediately after casting (t = 0) or after a given amount of time either under flow conditions (in the NMR bioreactor at 37 °C, 0.1 mL/min flow) or under static conditions (in conventional NMR tubes kept at 37 °C in an incubator). The NMR tube content was recovered with a Pasteur pipet, and the agarose threads were transferred to an Eppendorf tube. The residual medium was removed by rinsing the threads with 600 μL PBS, followed by centrifugation (4000g, 1 min, RT). The supernatant was discarded, and 250 μL of PBS and 50 μL of 0.4% Trypan blue were added. After 2 min of incubation with continuous pipetting, two washes with 600 μL of PBS were performed. The threads were chopped with razor blades to create small slices of gel, which were transferred into a self-made chamber consisting of two glass slides spaced by three layers of parafilm on each side. Images of cells inside the agarose were acquired on a Zeiss SteREO Discovery V8 microscope using AxioVision 4.7 software. Cell counting was performed manually using ImageJ software. Viability in each slice of agarose thread was calculated as (blue cells–white cells)/total cells. Intrasample variability (reported as standard deviation) was assessed by measuring cell staining in slices of agarose threads from different regions of the NMR tube.
NMR Experiments
1D 1H and 2D 1H–15N NMR spectra were collected at 310 K at a 950 MHz Bruker Avance III spectrometer equipped with a TCI CryoProbe. Series of 1D 1H WATERGATE 3–9–19 spectra44 were recorded on unlabeled cell samples (256 scans, 6 min 49 s each for cells treated with AAZ; 512 scans 13 min 32 s each for cells treated with MZA); 2D 1H–15N SOFAST-HMQC spectra45 were recorded on the 15N-labeled cell sample (16 scans × 128 increments, 12 min 37 s each). 1D 1H excitation sculpting spectra (32 scans, 1 min 7 s each) were alternated with the others for monitoring the general state of each sample. 31P NMR spectra were collected at 310 K at a 400 Bruker Avance III spectrometer equipped with a BBO probe. Series of 1D 31P spectra were recorded on a sample of untransfected, unlabeled cells (1760 scans, 60 min each). NMR spectra were processed with Bruker Topspin 4.0, by applying zero filling and exponential line broadening (to the 1D spectra, LB = 20 Hz) or square sine bell (to the 2D spectra, SSB = 2) window functions. 31P NMR spectra were analyzed using Bruker Dynamics Center 2.5.5. ATP levels were obtained by integrating the β-ATP signal at −19 ppm, and inorganic phosphate levels were obtained by integrating the signal at 0.5–1.8 ppm.
MCR-ALS Analysis
MCR-ALS analysis was performed in MATLAB, using the MCR-ALS 2.0 graphical user interface (GUI),46 on a portable computer equipped with an 8th gen. Intel Core i5 microprocessor and 24 GB RAM. For 1D spectra, the spectral region of interest was exported in ASCII format from Topspin, imported and stacked in a 2D array (time points × 1H spectral intensities). 2D spectra were imported using the Read_Bruker_2D script provided by NMRFAM, Univ. of Wisconsin-Madison (http://pine.nmrfam.wisc.edu/download_scripts.html), 2D arrays (1H × 15N spectral intensities) of the regions of interests were cut, converted to row vectors for each time point, and stacked in a 2D array (time points × spectral intensities). MCR-ALS 2.0 was run in GUI mode. The number of components (n = 2 for all experiments) was evaluated by Singular Value Decomposition (SVD), and the initial estimation of pure spectra was made by either purest variable detection47 or Evolving Factor Analysis (EFA).48 Fast non-negativity-constrained least-squares49 was used as the non-negativity constraint for the pure components, both in rows (concentrations) and columns (spectra). As a closure constraint, a sum of concentrations equal to 1 was applied to obtain concentration profiles of each species normalized with respect to the total protein. The fitting was run with 0.01 convergence criterion and reached convergence after <30 iterations.
Curve Fitting
Nonlinear curve fitting and linear regression were performed with OriginPro. Single exponential and biexponential fittings were performed with the following equations:
Fitting of CA2 binding curves was performed with membrane diffusion-limited binding kinetics, approximated with the following equation:16
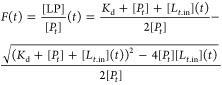 |
where [Pt] is the total intracellular protein concentration, [LP] is the bound protein concentration, Kd is the dissociation constant, and [Lt.in](t) is the total intracellular ligand concentration, defined as follows:
where Lt = Lo + Li + LP are the total moles of ligand (Lo and Li are the extracellular and intracellular ligand, respectively), Vt is the total external volume, Kp is the permeability coefficient, and A is the total area of the membrane.
Values of Kp·A were obtained from linear regression by considering that in the initial phase of the binding [Li] in steady-state and at negligible concentration, so that [LP](t) is small and linearly dependent on the membrane diffusion rate:
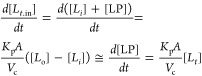 |
where Vc is the intracellular volume. Therefore,
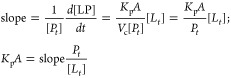 |
Results and Discussion
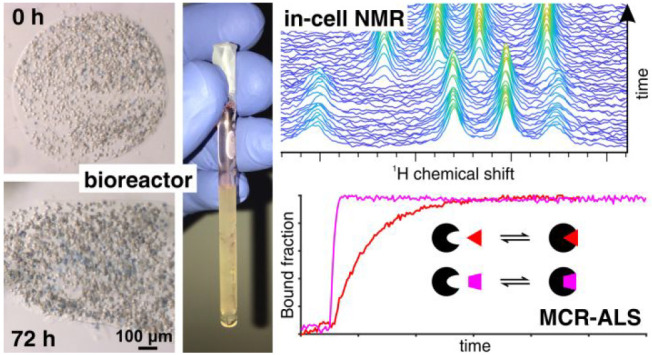
The NMR bioreactor setup was optimized to maximize cell viability for long periods of time, while simultaneously allowing high cell numbers in the NMR spectrometer. To this aim, cells were encapsulated in threads of hydrogel, a strategy that has been successfully employed before with other mammalian cell lines.19,21,50 In our implementation, HEK293T cells were suspended in a 1.5% solution of low-gelling agarose, cooled down in FPLC capillary tubing to form threads, which were then packed into the flow unit NMR tube, where growth medium was supplemented at a constant flow rate through an inlet placed at the bottom of the tube (see Experimental Section and Figure 1a). To optimize the protocol, the viability of cells within the agarose threads was measured by Trypan blue exclusion test as a function of different parameters. The effect of the thread thickness was evaluated by comparing viability of cells cast in capillaries with internal diameters of 0.75 mm and 0.5 mm (Figure 1b). After 24 h in the bioreactor, cell viability decreased slightly in both samples (3–4%) with respect to the t = 0 reference samples. A minor improvement was observed in 0.75 mm threads, which were chosen for the subsequent experiments. We then assessed whether cells in the center of the agarose threads survived as well as those closer to the surface and found only a minor decrease in cell viability (by ∼2%, lower than the experimental error) at increasing distances from the surface of the thread after 24 h (Figure 1c).
Figure 1.

Cell viability in the NMR bioreactor. (a) Annotated images of the flow NMR tube filled with agarose threads (left) and of the empty flow unit assembly (right). (b–d) Viability of HEK293T cells in agarose threads in the NMR bioreactor measured by Trypan blue staining (b) for different thread diameters (i.d. = 0.75 mm and 0.5 mm) at 0 and 24 h; (c) at different distances from the surface of the thread (from left to right: outer, middle, inner cells) at 24 h; (d) at different heights in the NMR tube (from left to right: average at 0 h; average at 72 h; top, middle, and bottom fractions at 72 h). Values are reported as mean ± SD (n = 3) of different slices of agarose thread within the same sample. (e) Cell viability in agarose threads (i.d. = 0.75 mm) as a function of time in the NMR bioreactor at 0.1 mL/min flow (black) and under static conditions (red). Values are reported as mean ± SD of different experiments (n = 3 at 0 h; n = 2 at 24 and 48 h); at 72 h (n = 1) SD is not reported. (f) Representative images of agarose threads sliced and stained with Trypan blue immediately after casting (0 h, left) and after 72 h in the bioreactor at 0.1 mL/min flow (72 h, right).
To study the kinetics of cellular processes via NMR, which provides values of intensity and chemical shift averaged over all cells within the active volume, it is important to ensure that the cells are experiencing a uniform microenvironment. To address this concern, the homogeneity of the cell population at different heights of the NMR tube, i.e., at different distances from the flow of fresh medium at the bottom, was evaluated after 72 h (Figure 1d). On average, viability decreased by ∼5% from the value at t = 0, while cells at the bottom were slightly more viable than those at the top (by ∼2%, lower than the experimental error). Overall, these data indicate that cell viability is marginally influenced by the location within the thread and within the NMR tube. These effects are intuitively explained by considering that (1) fresh nutrients reach the superficial layers of the thread first and take longer to diffuse to its center, and (2) the medium composition changes slightly as it moves within the NMR tube, so that cells at the bottom will receive more nutrients than those at the top. The differences in viability caused by the above effects are minor when compared to the high intrinsic variability that is observed within each viability measurement (see error bars in Figure 1), possibly due to other sources of inhomogeneity of the cell population, and therefore are not expected to introduce artifacts when recording population-averaged data such as the in-cell NMR spectra. Finally, cell viability as a function of time measured with the final bioreactor setup showed that the bioreactor could preserve ∼90% cell viability after 72 h of operation (Figure 1e,f), suggesting that this setup could enable even longer experimental times, whereas control samples of cells in agarose threads incubated in the absence of flow showed a marked decrease in viability, dropping to ∼50% after 24 h and ∼30% after 72 h (Figure 1e).
Cell metabolic activity in the NMR bioreactor was assessed by real-time 31P NMR measurement of the intracellular levels of ATP and inorganic phosphate (Pi). In highly active cells such as HEK293T, ATP levels mostly depend on the rate of glycolysis, both anaerobic and aerobic, and therefore provide a measure of the cellular metabolic activity, whereas Pi levels increase mainly as a consequence of ATP hydrolysis and therefore provide a measure of nutrient starvation. As expected, cell metabolic activity was sustained by the bioreactor over the course of 72 h, as shown by the ATP levels (Figures 2a and S2a). An increase in ATP was actually observed during the first 24 h, possibly due to the recovery of cells which were deprived of nutrients during sample preparation or to cell growth within the bioreactor. Consistently, ATP was completely lost within 1 h after the cells were placed in the bioreactor in the absence of flow, or within 2 h when the flow was switched off after 24 h (Figures 2a and S2b,c). Pi showed the opposite trend, as it was detected at low and constant levels under active flow but steadily increased after the flow was switched off (Figures 2b and S2). Notably, the increase in Pi occurred at a slower rate with respect to the ATP decrease, and the corresponding signal reached a much higher intensity compared to that of the initial ATP, suggesting that other enzymatic reactions could contribute to the total Pi increase as a consequence of starvation, such as diphosphate breakdown and protein dephosphorylation.
Figure 2.

Cell metabolic activity in the NMR bioreactor. Metabolic activity of HEK293T cells in agarose threads (i.d. = 0.75 mm) in the NMR bioreactor assessed by 31P NMR of (a) ATP and (b) inorganic phosphate (Pi) in separate experiments with constant flow for 72 h (black), in the absence of flow for 24 h (red) and for 42 h with flow switched off after 24 h (magenta). Values are reported as integrals of (a) β-ATP and (b) Pi ± SD from spectral noise.
The bioreactor setup was applied to investigate in real time intracellular processes, where an intracellular protein of interest undergoes conformational or chemical changes, by 1D and 2D in-cell NMR spectroscopy. In both series of experiments, signals arising from different protein species were simultaneously present in the spectra, so that it was not straightforward to obtain the relative concentration of each species by signal integration, due to spectral overlap (especially true for 1D NMR spectra) and to the low S/N of single signals (especially true for 2D NMR spectra). To overcome the above limits, MCR-ALS was used to fit the data to retrieve both the NMR spectra of the pure components and the relative concentration profiles.
The binding of the two inhibitors, AAZ and MZA, to human CA2 overexpressed in the cytosol was monitored by 1D 1H NMR spectra. In the previous work, “static” in-cell NMR spectra were collected on a series of samples treated for different times and were manually fitted to obtain time-dependent binding curves.16 Here, binding curves were obtained by MCR-ALS analysis of the data from a single sample of cells treated with either 25 μM AAZ or 10 μM MZA in the NMR bioreactor. Different ligand concentrations were chosen based on the expected difference of diffusion rates. Raw NMR spectra recorded in ∼7 min (AAZ) or ∼14 min (MZA) exhibited sufficient S/N in the region between 11 and 16 ppm (Figure 3a). This spectral region is almost background-free and contains signals arising from aromatic protons of side chains located in the ligand binding site. These signals shift upon ligand binding in a slow exchange regime, giving rise to separate sets of signals from free and bound protein (Figures 3a and S3). MCR-ALS analysis separated almost completely the two sets of signals, as shown in the reconstructed spectra of the pure species, i.e., free and bound CA2 (Figure 3b), and simultaneously provided the relative concentration profiles of the two species (Figure 3c). For comparison with the MZA data, the AAZ data were also analyzed after 2× binning (i.e., spectra were added two-by-two to simulate 2×-long spectra), resulting in a binding curve with lower noise and lower time resolution (Figure 3c). Considering that the binding kinetics of both drugs follow a membrane diffusion-limited model,16 the initial slope of the curve is directly proportional to the membrane permeability coefficient, to the total area of the membrane and to the external drug concentration (see the Experimental Section). Comparison of the values obtained by linear regression (Figure S4) reveals that in the bioreactor MZA permeates the cell membrane ∼15 times faster than AAZ (KpAAAZ = 3.5 × 10–8 dm3/s, KpAMZA = 5.1 × 10–7 dm3/s), confirming previous observations on adherent cells (∼12-fold difference16). Notably, however, the absolute values obtained are ∼60% lower than those previously reported (KpAAAZ = 1.0 × 10–7 dm3/s, KpAMZA = 1.2 × 10–6 dm3/s), possibly due to a decrease in the cell surface-to-volume ratio when transitioning from a flat shape (cells attached to the culture flask) to a spherical shape (cells detached and embedded in agarose).
Figure 3.

Real-time NMR analysis of ligand binding to intracellular CA2. (a) Representative 1H NMR spectra (region between 15.6 and 11.1 ppm) of intracellular CA2 in the absence of ligands (black) and after quantitative binding of AAZ (red) or MZA (magenta) in the NMR bioreactor; (b) 1H NMR spectra of the pure components reconstructed by MCR-ALS, color-coded as in (a); (c) Relative concentration profiles obtained by MCR-ALS of intracellular free (black) and bound (red/magenta) CA2 as a function of time upon addition of AAZ (red), from original spectra (top) and 2× binned spectra (middle), and MZA (magenta, bottom). Times of ligand treatment are marked with arrows.
The effect of ebselen on the redox state of zinc-bound human SOD1 was monitored by 2D 1H–15N correlation NMR spectra. Cysteine oxidation causes a conformational change of E,Zn-SOD1, which is clearly reflected in the 2D 1H–15N spectrum of the backbone,34,39,40 whereas 1D spectra suffer from severe signal overlap and do not allow the two species to be separated (Figure S5a). MCR-ALS was therefore applied on 2D spectra recorded on cells expressing SOD1 and treated with 20 μM ebselen in the NMR bioreactor (Figure S5b–e). In addition to the signals from SOD1, the 2D spectra contained other signals from 15N-labeled background cellular components, each evolving differently as a function of time, thus complicating the correct reconstruction of the pure spectra for each independent component. Therefore, MCR-ALS analysis was carried out on selected spectral regions, which only contained signals from either reduced or oxidized E,Zn-SOD1 (Figure 4a). Notably, while the separate fitting of each spectral region provided similar concentration curves (Figure 4b), global fitting resulted in an improved curve (Figure 4c).
Figure 4.

Real-time NMR analysis of SOD1 oxidation catalyzed by ebselen. (a) 2D 1H–15N NMR spectral regions analyzed by MCR-ALS. Reconstructed spectra of disulfide-reduced SOD1 (RED = E,Zn-SOD1SH) and disulfide-oxidized SOD1 (OX = E,Zn-SOD1SS) are shown in black and red, respectively. Crosspeaks from residues perturbed by SOD1 oxidation are labeled. (b) Relative concentration profiles of reduced (RED, black) and oxidized SOD1 (OX, red) as a function of time upon addition of ebselen obtained by MCR-ALS by separately fitting the single spectral regions (I–IV) shown in (a). (c) Relative concentration profiles as in (b) obtained by MCR-ALS by global fitting of the spectral regions (I–IV). The time of ebselen addition is marked with an arrow.
To assess the robustness of the MCR-ALS algorithm, the two investigated systems, i.e., CA2-ligand binding experiments and SOD1 oxidation by ebselen, were analyzed again with different methods for the initial estimation of pure components. Overall, for CA2-ligand binding experiments, similar relative concentration curves were obtained, which differed only slightly in the temporal regions where only one species is present, i.e., either pure free CA2 before ligand addition, or pure bound CA2 at the end of the runs (Figure S6a–c). Somewhat larger variation was observed in the curves of disulfide-reduced and oxidized SOD1 in the temporal region after ebselen addition (Figure S6d). Notably, the shape of the actual curves was mostly unaffected, suggesting that the initial choice of pure components does not introduce bias in the fitting and therefore the shape of the curve can be reliably used to test different kinetic models. Interestingly, the CA2-ligand binding curves are best fitted by different equations (Figure 5). Specifically, the slow binding of AAZ to intracellular CA2 is approximated by a single exponential function better than the fast binding of MZA and the ebselen-dependent SOD1 oxidation (Figure 5a). Both CA2 binding curves can be fitted with the kinetic model described previously,16 in which the bound species initially increases at a constant rate due to the membrane diffusion being the rate-determining step. However, proper fitting requires apparent dissociation constants several orders of magnitude larger than those reported for AAZ and MZA, suggesting the occurrence of a pharmacodynamic response still unaccounted for (Figure 5b). Intracellular SOD1 oxidation appears to follow yet another mechanism, which is best modeled with a biexponential equation, suggesting that ebselen may promote SOD1 disulfide bond formation through different coexisting pathways (Figure 5c). These observations show that, with properly designed sets of experiments, real-time in-cell NMR can provide detailed information on the mechanism of the observed biological processes.
Figure 5.

Nonlinear curve fitting of real-time NMR data. (a) Fraction of CA2 bound to AAZ (left) and MZA (center), and disulfide-oxidized SOD1 (right) fitted with single exponential build-up curves. (b) Fraction of CA2 bound to AAZ (left) and MZA (right) fitted with a membrane diffusion-limited model.16 (c) Fraction of disulfide-oxidized SOD1 fitted with a biexponential build-up curve. The exponential time constants (t1, t2), the permeability coefficients × membrane area (KpA) and the apparent dissociation constants (KdAPP) are reported in each panel. Raw data and residuals are shown as gray and red dots, respectively. Best-fit curves are shown as continuous red lines. In (b), diffusion-limited binding curves with values of KdAPP fixed to the KI determined in vitro (KIAAZ = 12 × 10–9 M; KIMZA = 14 × 10–9 M) are shown as dashed red lines.
Conclusions
NMR bioreactors are fundamental tools for the investigation of biological processes by real-time in-cell NMR. However, their successful implementation critically depends on their capability of maintaining cells viable and metabolically stable for long periods of time, coupled with a robust, automated approach to analyze the large amounts of output NMR data. Our results show that the optimized bioreactor setup allows real-time in-cell NMR experiments on human cells for prolonged periods of time, up to 72 h. During the acquisition, the cell samples experienced minimal loss of viability and remained metabolically active as shown by the constant production of ATP. The MCR-ALS algorithm is an ideal approach for the quantitative analysis of time-dependent data, as it provides concentration curves and reconstructed NMR spectra of the pure components in a semiautomated workflow. The concentration profiles obtained had relatively low noise and could be further analyzed by nonlinear fitting with simple models or in principle with more complex kinetic equations.
The straightforward implementation of the MCR-ALS 2.0 GUI allowed high flexibility of the input data: here it was applied to analyze 1D spectra and linearized 2D spectral regions (analyzed either separately or concatenated), but in principle it can accept >2D NMR data as input, provided that the nD matrices of each time point are linearized to 1D vectors. Importantly, the effectiveness of MCR-ALS in reconstructing the pure spectral components correlates with the total NMR acquisition time, but not with the S/N of the single time points, i.e., it performs equally well with a high number of low S/N spectra and with a low number of high S/N spectra. Therefore, short NMR experiments can be preferred to get maximum time resolution without the drawbacks from the lower S/N. While the algorithm may be less reliable in situations where the pure species do not span the full relative concentration range (0%–100%, see the SOD1 data in Figure S6d), this in principle can be resolved by performing global fitting on concatenated multiple data sets from different runs (e.g., including positive/negative control samples), to cover the full range of each species.
Concerning the general applicability to NMR, the automated workflow described here will be applicable in principle not only to macromolecular in-cell NMR, but also to real-time metabolic analysis on live cells, for which NMR has been shown to be a powerful approach.51 Specifically, MCR-ALS should perform best in cases where the line shapes and chemical shifts of each species in the mixtures are constant in time, i.e., when all the NMR signals are in the slow exchange regime, while it may be not ideal in cases where signals are changing shape or chemical shift as a function of time, i.e., in the presence of fast/intermediate exchange regime.
Further improvements of the workflow will focus on the approaches for cell immobilization. Comparing the above results with those previously obtained from cell treated in the CO2 incubator, prior to collection for NMR analysis,16 suggests that the cell behavior might be affected by the detachment from the monolayer and by the embedding in agarose hydrogel. Indeed, it has recently been shown that the choice of detachment protocol and type of hydrogel matrix can largely affect membrane receptor internalization,52 which in turn may change membrane properties such as drug permeability. Despite this, appropriate control experiments can be designed to allow the comparison between flow-in-cell NMR, static-in-cell NMR, and in vitro NMR conditions, even if agarose-based hydrogels are still far from a truly physiological cell culture environment. Eventually, understanding how different hydrogels affect the cell phenotype will allow the development of more physiological matrices. Methylcellulose, which has been recently employed for live-cell NMR analysis51,52 to provide a more physiological environment, could be combined with other scaffolds that provide the mechanical strength required for the bioreactor operation.
In conclusion, the experimental setup described here combines an optimized NMR bioreactor design and automated data analysis and can be successfully applied to quantitatively analyze time-dependent processes involving intracellular molecules at atomic resolution, providing a unique way to investigate the kinetic behavior of complex molecular mechanisms in the cellular environment.
Acknowledgments
This work has been supported by iNEXT-Discovery, grant number 871037, funded by the Horizon 2020 programme of the European Commission, by Instruct-ULTRA, grant number 731005, an EU H2020 project to further develop the services of Instruct-ERIC, and by Ministero dell’Istruzione, dell’Università e della Ricerca PRIN grant 20177XJCHX. The authors acknowledge the support of Instruct-ERIC, a Landmark ESFRI project, through the JRA Award number 815 and the use of resources of the CERM/CIRMMP Italy Centre. T.F.C. was supported by a U.S. −Italy Fulbright Scholarship. We thank Matteo Pennestri (Bruker, U.K.) for providing support for the InsightMR flow unit operation.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.analchem.0c01677.
Figure S1, Cross-section view of the empty InsightMR flow unit; Figure S2, real-time 31P NMR spectra; Figure S3, real-time in-cell NMR spectra of CA2; Figure S4, permeability coefficients from linear regression of CA2 ligand binding; Figure S5, real-time in-cell NMR spectra of SOD1; and Figure S6, MCR-ALS with different choice of initial components (PDF)
Author Present Address
⊥ (T.F.C.) DuPage lab, University of California, Berkeley, 142 Life Sciences Addition # 3200, Berkeley, California 94720–3200, United States.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Inomata K.; Ohno A.; Tochio H.; Isogai S.; Tenno T.; Nakase I.; Takeuchi T.; Futaki S.; Ito Y.; Hiroaki H.; Shirakawa M. High-Resolution Multi-Dimensional NMR Spectroscopy of Proteins in Human Cells. Nature 2009, 458 (7234), 106–109. 10.1038/nature07839. [DOI] [PubMed] [Google Scholar]
- Luchinat E.; Banci L. In-Cell NMR: A Topical Review. IUCrJ 2017, 4 (2), 108–118. 10.1107/S2052252516020625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dzatko S.; Krafcikova M.; Hänsel-Hertsch R.; Fessl T.; Fiala R.; Loja T.; Krafcik D.; Mergny J.-L.; Foldynova-Trantirkova S.; Trantirek L. Evaluation of the Stability of DNA I-Motifs in the Nuclei of Living Mammalian Cells. Angew. Chem., Int. Ed. 2018, 57 (8), 2165–2169. 10.1002/anie.201712284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luchinat E.; Banci L. In-Cell NMR in Human Cells: Direct Protein Expression Allows Structural Studies of Protein Folding and Maturation. Acc. Chem. Res. 2018, 51 (6), 1550–1557. 10.1021/acs.accounts.8b00147. [DOI] [PubMed] [Google Scholar]
- Tanaka T.; Ikeya T.; Kamoshida H.; Suemoto Y.; Mishima M.; Shirakawa M.; Güntert P.; Ito Y. High-Resolution Protein 3D Structure Determination in Living Eukaryotic Cells. Angew. Chem., Int. Ed. 2019, 58 (22), 7284–7288. 10.1002/anie.201900840. [DOI] [PubMed] [Google Scholar]
- Siegal G.; Selenko P. Cells, Drugs and NMR. J. Magn. Reson. 2019, 306, 202–212. 10.1016/j.jmr.2019.07.018. [DOI] [PubMed] [Google Scholar]
- Banci L.; Barbieri L.; Bertini I.; Luchinat E.; Secci E.; Zhao Y.; Aricescu A. R. Atomic-Resolution Monitoring of Protein Maturation in Live Human Cells by NMR. Nat. Chem. Biol. 2013, 9 (5), 297–299. 10.1038/nchembio.1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luchinat E.; Barbieri L.; Rubino J. T.; Kozyreva T.; Cantini F.; Banci L. In-Cell NMR Reveals Potential Precursor of Toxic Species from SOD1 FALS Mutants. Nat. Commun. 2014, 5, 5502. 10.1038/ncomms6502. [DOI] [PubMed] [Google Scholar]
- Theillet F.-X.; Binolfi A.; Bekei B.; Martorana A.; Rose H. M.; Stuiver M.; Verzini S.; Lorenz D.; van Rossum M.; Goldfarb D.; Selenko P. Structural Disorder of Monomeric α-Synuclein Persists in Mammalian Cells. Nature 2016, 530 (7588), 45–50. 10.1038/nature16531. [DOI] [PubMed] [Google Scholar]
- Barbieri L.; Luchinat E.; Banci L. Intracellular Metal Binding and Redox Behavior of Human DJ-1. JBIC, J. Biol. Inorg. Chem. 2018, 23 (1), 61–69. 10.1007/s00775-017-1509-5. [DOI] [PubMed] [Google Scholar]
- Banci L.; Barbieri L.; Luchinat E.; Secci E. Visualization of Redox-Controlled Protein Fold in Living Cells. Chem. Biol. 2013, 20 (6), 747–752. 10.1016/j.chembiol.2013.05.007. [DOI] [PubMed] [Google Scholar]
- Mercatelli E.; Barbieri L.; Luchinat E.; Banci L. Direct Structural Evidence of Protein Redox Regulation Obtained by In-Cell NMR. Biochim. Biophys. Acta, Mol. Cell Res. 2016, 1863 (2), 198–204. 10.1016/j.bbamcr.2015.11.009. [DOI] [PubMed] [Google Scholar]
- Barbieri L.; Luchinat E.; Banci L. Protein Interaction Patterns in Different Cellular Environments Are Revealed by In-Cell NMR. Sci. Rep. 2015, 5, 14456. 10.1038/srep14456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeMott C. M.; Girardin R.; Cobbert J.; Reverdatto S.; Burz D. S.; McDonough K.; Shekhtman A. Potent Inhibitors of Mycobacterium Tuberculosis Growth Identified by Using In-Cell NMR-Based Screening. ACS Chem. Biol. 2018, 13 (3), 733–741. 10.1021/acschembio.7b00879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krafcikova M.; Dzatko S.; Caron C.; Granzhan A.; Fiala R.; Loja T.; Teulade-Fichou M.-P.; Fessl T.; Hänsel-Hertsch R.; Mergny J.-L.; Foldynova-Trantirkova S.; Trantirek L. Monitoring DNA-Ligand Interactions in Living Human Cells Using NMR Spectroscopy. J. Am. Chem. Soc. 2019, 141 (34), 13281–13285. 10.1021/jacs.9b03031. [DOI] [PubMed] [Google Scholar]
- Luchinat E.; Barbieri L.; Cremonini M.; Nocentini A.; Supuran C. T.; Banci L. Drug Screening in Human Cells by NMR Spectroscopy Allows the Early Assessment of Drug Potency. Angew. Chem., Int. Ed. 2020, 59 (16), 6535–6539. 10.1002/anie.201913436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hembram D. S. S.; Haremaki T.; Hamatsu J.; Inoue J.; Kamoshida H.; Ikeya T.; Mishima M.; Mikawa T.; Hayashi N.; Shirakawa M.; Ito Y. An In-Cell NMR Study of Monitoring Stress-Induced Increase of Cytosolic Ca2+ Concentration in HeLa Cells. Biochem. Biophys. Res. Commun. 2013, 438 (4), 653–659. 10.1016/j.bbrc.2013.07.127. [DOI] [PubMed] [Google Scholar]
- Sharaf N. G.; Barnes C. O.; Charlton L. M.; Young G. B.; Pielak G. J. A Bioreactor for In-Cell Protein NMR. J. Magn. Reson. 2010, 202 (2), 140–146. 10.1016/j.jmr.2009.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubo S.; Nishida N.; Udagawa Y.; Takarada O.; Ogino S.; Shimada I. A Gel-Encapsulated Bioreactor System for NMR Studies of Protein-Protein Interactions in Living Mammalian Cells. Angew. Chem., Int. Ed. 2013, 52 (4), 1208–1211. 10.1002/anie.201207243. [DOI] [PubMed] [Google Scholar]
- Inomata K.; Kamoshida H.; Ikari M.; Ito Y.; Kigawa T. Impact of Cellular Health Conditions on the Protein Folding State in Mammalian Cells. Chem. Commun. 2017, 53 (81), 11245–11248. 10.1039/C7CC06004A. [DOI] [PubMed] [Google Scholar]
- Breindel L.; DeMott C.; Burz D. S.; Shekhtman A. Real-Time In-Cell Nuclear Magnetic Resonance: Ribosome-Targeted Antibiotics Modulate Quinary Protein Interactions. Biochemistry 2018, 57 (5), 540–546. 10.1021/acs.biochem.7b00938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerofolini L.; Giuntini S.; Barbieri L.; Pennestri M.; Codina A.; Fragai M.; Banci L.; Luchinat E.; Ravera E. Real-Time Insights into Biological Events: In-Cell Processes and Protein-Ligand Interactions. Biophys. J. 2019, 116 (2), 239–247. 10.1016/j.bpj.2018.11.3132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tauler R. Multivariate Curve Resolution Applied to Second Order Data. Chemom. Intell. Lab. Syst. 1995, 30 (1), 133–146. 10.1016/0169-7439(95)00047-X. [DOI] [Google Scholar]
- de Juan A.; Tauler R. Multivariate Curve Resolution (MCR) from 2000: Progress in Concepts and Applications. Crit. Rev. Anal. Chem. 2006, 36 (3–4), 163–176. 10.1080/10408340600970005. [DOI] [Google Scholar]
- Garrido M.; Rius F. X.; Larrechi M. S. Multivariate Curve Resolution–Alternating Least Squares (MCR-ALS) Applied to Spectroscopic Data from Monitoring Chemical Reactions Processes. Anal. Bioanal. Chem. 2008, 390 (8), 2059–2066. 10.1007/s00216-008-1955-6. [DOI] [PubMed] [Google Scholar]
- Ruckebusch C.; Blanchet L. Multivariate Curve Resolution: A Review of Advanced and Tailored Applications and Challenges. Anal. Chim. Acta 2013, 765, 28–36. 10.1016/j.aca.2012.12.028. [DOI] [PubMed] [Google Scholar]
- Puig-Castellví F.; Alfonso I.; Tauler R. Untargeted Assignment and Automatic Integration of 1H NMR Metabolomic Datasets Using a Multivariate Curve Resolution Approach. Anal. Chim. Acta 2017, 964, 55–66. 10.1016/j.aca.2017.02.010. [DOI] [PubMed] [Google Scholar]
- Bruno F.; Francischello R.; Bellomo G.; Gigli L.; Flori A.; Menichetti L.; Tenori L.; Luchinat C.; Ravera E. Multivariate Curve Resolution for 2D Solid-State NMR Spectra. Anal. Chem. 2020, 92 (6), 4451–4458. 10.1021/acs.analchem.9b05420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supuran C. T. Carbonic Anhydrases: Novel Therapeutic Applications for Inhibitors and Activators. Nat. Rev. Drug Discovery 2008, 7 (2), 168–181. 10.1038/nrd2467. [DOI] [PubMed] [Google Scholar]
- Neri D.; Supuran C. T. Interfering with PH Regulation in Tumours as a Therapeutic Strategy. Nat. Rev. Drug Discovery 2011, 10 (10), 767–777. 10.1038/nrd3554. [DOI] [PubMed] [Google Scholar]
- Alterio V.; Di Fiore A.; D’Ambrosio K.; Supuran C. T.; De Simone G. Multiple Binding Modes of Inhibitors to Carbonic Anhydrases: How to Design Specific Drugs Targeting 15 Different Isoforms?. Chem. Rev. 2012, 112 (8), 4421–4468. 10.1021/cr200176r. [DOI] [PubMed] [Google Scholar]
- Azad G. K.; Tomar R. S. Ebselen, a Promising Antioxidant Drug: Mechanisms of Action and Targets of Biological Pathways. Mol. Biol. Rep. 2014, 41 (8), 4865–4879. 10.1007/s11033-014-3417-x. [DOI] [PubMed] [Google Scholar]
- Trist B.; Hilton J. B.; Crouch P. J.; Hare D. J.; Double K. L.. Superoxide Dismutase 1 in Health and Disease: How a Front-Line Antioxidant Becomes Neurotoxic. Angew. Chem., Int. Ed. 2020 10.1002/anie.202000451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banci L.; Bertini I.; Cantini F.; Kozyreva T.; Massagni C.; Palumaa P.; Rubino J. T.; Zovo K. Human Superoxide Dismutase 1 (HSOD1) Maturation through Interaction with Human Copper Chaperone for SOD1 (HCCS). Proc. Natl. Acad. Sci. U. S. A. 2012, 109 (34), 13555–13560. 10.1073/pnas.1207493109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fetherolf M. M.; Boyd S. D.; Taylor A. B.; Kim H. J.; Wohlschlegel J. A.; Blackburn N. J.; Hart P. J.; Winge D. R.; Winkler D. D. Copper-Zinc Superoxide Dismutase Is Activated through a Sulfenic Acid Intermediate at a Copper Ion Entry Site. J. Biol. Chem. 2017, 292 (29), 12025–12040. 10.1074/jbc.M117.775981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sala F. A.; Wright G. S. A.; Antonyuk S. V.; Garratt R. C.; Hasnain S. S. Molecular Recognition and Maturation of SOD1 by Its Evolutionarily Destabilised Cognate Chaperone HCCS. PLoS Biol. 2019, 17 (2), e3000141 10.1371/journal.pbio.3000141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danielsson J.; Mu X.; Lang L.; Wang H.; Binolfi A.; Theillet F.-X.; Bekei B.; Logan D. T.; Selenko P.; Wennerström H.; Oliveberg M. Thermodynamics of Protein Destabilization in Live Cells. Proc. Natl. Acad. Sci. U. S. A. 2015, 112 (40), 12402–12407. 10.1073/pnas.1511308112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luchinat E.; Barbieri L.; Banci L. A Molecular Chaperone Activity of CCS Restores the Maturation of SOD1 FALS Mutants. Sci. Rep. 2017, 7 (1), 17433. 10.1038/s41598-017-17815-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polykretis P.; Cencetti F.; Donati C.; Luchinat E.; Banci L. Cadmium Effects on Superoxide Dismutase 1 in Human Cells Revealed by NMR. Redox Biol. 2019, 21, 101102. 10.1016/j.redox.2019.101102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capper M. J.; Wright G. S. A.; Barbieri L.; Luchinat E.; Mercatelli E.; McAlary L.; Yerbury J. J.; O’Neill P. M.; Antonyuk S. V.; Banci L.; Hasnain S. S. The Cysteine-Reactive Small Molecule Ebselen Facilitates Effective SOD1Maturation. Nat. Commun. 2018, 9 (1), 1693. 10.1038/s41467-018-04114-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao R.; Holmgren A. A Novel Antioxidant Mechanism of Ebselen Involving Ebselen Diselenide, a Substrate of Mammalian Thioredoxin and Thioredoxin Reductase. J. Biol. Chem. 2002, 277 (42), 39456–39462. 10.1074/jbc.M206452200. [DOI] [PubMed] [Google Scholar]
- Barbieri L.; Luchinat E.; Banci L. Characterization of Proteins by In-Cell NMR Spectroscopy in Cultured Mammalian Cells. Nat. Protoc. 2016, 11 (6), 1101–1111. 10.1038/nprot.2016.061. [DOI] [PubMed] [Google Scholar]
- Foley D. A.; Bez E.; Codina A.; Colson K. L.; Fey M.; Krull R.; Piroli D.; Zell M. T.; Marquez B. L. NMR Flow Tube for Online NMR Reaction Monitoring. Anal. Chem. 2014, 86 (24), 12008–12013. 10.1021/ac502300q. [DOI] [PubMed] [Google Scholar]
- Piotto M.; Saudek V.; Sklenár V. Gradient-Tailored Excitation for Single-Quantum NMR Spectroscopy of Aqueous Solutions. J. Biomol. NMR 1992, 2 (6), 661–665. 10.1007/BF02192855. [DOI] [PubMed] [Google Scholar]
- Schanda P.; Brutscher B. Very Fast Two-Dimensional NMR Spectroscopy for Real-Time Investigation of Dynamic Events in Proteins on the Time Scale of Seconds. J. Am. Chem. Soc. 2005, 127 (22), 8014–8015. 10.1021/ja051306e. [DOI] [PubMed] [Google Scholar]
- Jaumot J.; de Juan A.; Tauler R. MCR-ALS GUI 2.0: New Features and Applications. Chemom. Intell. Lab. Syst. 2015, 140, 1–12. 10.1016/j.chemolab.2014.10.003. [DOI] [Google Scholar]
- Windig W.; Stephenson D. A. Self-Modeling Mixture Analysis of Second-Derivative near-Infrared Spectral Data Using the SIMPLISMA Approach. Anal. Chem. 1992, 64 (22), 2735–2742. 10.1021/ac00046a015. [DOI] [Google Scholar]
- Maeder M. Evolving Factor Analysis for the Resolution of Overlapping Chromatographic Peaks. Anal. Chem. 1987, 59 (3), 527–530. 10.1021/ac00130a035. [DOI] [Google Scholar]
- Bro R.; De Jong S. A Fast Non-Negativity-Constrained Least Squares Algorithm. J. Chemom. 1997, 11 (5), 393–401. . [DOI] [Google Scholar]
- Carvalho J.; Alves S.; Castro M. M. C. A.; Geraldes C. F. G. C.; Queiroz J. A.; Fonseca C. P.; Cruz C. Development of a Bioreactor System for Cytotoxic Evaluation of Pharmacological Compounds in Living Cells Using NMR Spectroscopy. J. Pharmacol. Toxicol. Methods 2019, 95, 70–78. 10.1016/j.vascn.2018.11.004. [DOI] [PubMed] [Google Scholar]
- Alshamleh I.; Krause N.; Richter C.; Kurrle N.; Serve H.; Günther U. L.; Schwalbe H. Real□Time NMR Spectroscopy for Studying Metabolism. Angew. Chem., Int. Ed. 2020, 59 (6), 2304–2308. 10.1002/anie.201912919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mateos B.; Sealey-Cardona M.; Balazs K.; Konrat J.; Staffler G.; Konrat R. NMR Characterization of Surface Receptor Protein Interactions in Live Cells Using Methylcellulose Hydrogels. Angew. Chem., Int. Ed. 2020, 59 (10), 3886–3890. 10.1002/anie.201913585. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.