

Abstract
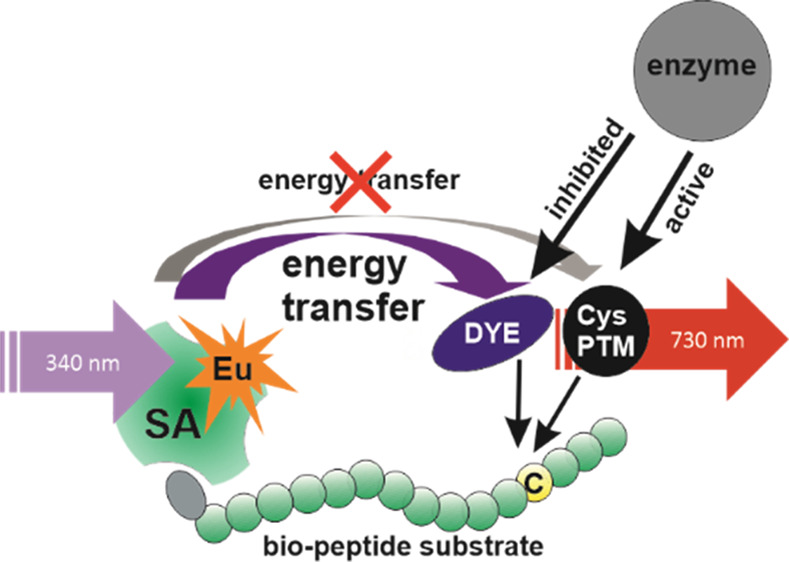
Post-translational modifications (PTMs) are one of the most important regulatory mechanisms in cells, and they play key roles in cell signaling both in health and disease. PTM catalyzing enzymes have become significant drug targets, and therefore, tremendous interest has been focused on the development of broad-scale assays to monitor several different PTMs with a single detection platform. Most of the current methodologies suffer from low throughput or rely on antibody recognition, increasing the assay costs, and decreasing the multifunctionality of the assay. Thus, we have developed a sensitive time-resolved Förster resonance energy transfer (TR-FRET) detection method for PTMs of cysteine residues using a single-peptide approach performed in a 384-well format. In the developed assay, the enzyme-specific biotinylated substrate peptide is post-translationally modified at the cysteine residue, preventing the subsequent thiol coupling with a reactive AlexaFluor 680 acceptor dye. In the absence of enzymatic activity, increase in the TR-FRET signal between the biotin-bound Eu(III)-labeled streptavidin donor and the cysteine-coupled AlexaFluor 680 acceptor dye is observed. We demonstrate the detection concept with cysteine modifying S-nitrosylation and ADP-ribosylation reactions using a chemical nitric oxide donor S-nitrosoglutathione and enzymatic ADP-ribosyltransferase PtxS1-subunit of pertussis toxin, respectively. As a proof of concept, three peptide substrates derived from the small GTPase K-Ras and the inhibitory α-subunit of the heterotrimeric G-protein Gαi showed expected functionality in both chemical and enzymatic assays. Measurements yielded signal-to-background ratios of 28.7, 33.0, and 8.7 between the modified and the nonmodified substrates for the three peptides in the S-nitrosylation assay, 5.8 in the NAD+ hydrolysis assay, and 6.8 in the enzymatic ADP-ribosyltransferase inhibitor dose–response assay. The developed antibody-free assay for cysteine-modifying enzymes provides a detection platform with low nanomolar peptide substrate consumption, and the assay is potentially applicable to investigate various cysteine-modifying enzymes in a high throughput compatible format.
Introduction
Post-translational protein-modifying enzymes have become one of the most studied drug targets.1,2 The main task of the post-translational modifications (PTMs) is to increase the protein diversity in cells by the addition or cleavage of chemical groups, such as phosphate, ADP-ribose, or acetyl.1 Because of these modifications, cell signaling and cellular functions are altered, and a malfunction in the regulation of these PTMs can lead to various disease conditions. For example, more than 40 PTMs have been linked to cancer and neurological disorders.3 The most common and targeted PTM is phosphorylation of tyrosine, serine, and threonine residues.4 Also glycosylation and acetylation are of high importance because of their prevalence.5,6 Cysteine is less frequently modified than, for example, lysine, as free reactive cysteines are often buried inside the folded protein structure or cysteine residues are bound to adjacent cysteine residues.7,8 However, cysteine modifications are highly important for cellular functions and cysteine is modified by many targets with a number of different types of modifications, such as nitrosylation, sulfhydrylation, glutathionylation, prenylation, palmitoylation, Michael adducts, and ADP-ribosylation, occur in cysteine residues.7−9 This abundance and variety of the PTMs sets challenges for the detection methods available today.
Even though some widely used established methods exist for PTM detection, new methods with improved functionalities are constantly developed. These current methods can be roughly divided as luminescence-based and biophysical methods. Most of the luminescence-based methods available are in a high throughput screening (HTS) compatible format and designed directly for one specific PTM or a group of PTMs. These technologies often rely on expensive antibodies, which make assays highly specific to a single PTM, but these methods are difficult to revise for other PTMs.9 Another widely used luminescence-based method relies on the monitoring of nucleotides, for example, ATP, ADP, and NAD (e.g., ATP assay kit, ADP Glo, and NAD/NADH Glo). The biophysical methods, mainly mass spectrometry (MS), are also widely utilized for PTM monitoring. MS is a universal method and has the ability to detect many of the PTMs occurring in their natural environment.10,11 Unfortunately, most of the MS methods are still relatively slow and reach only a moderate throughput, although positive development has been witnessed in recent years.12,13
Time-resolved luminescence (TRL) has been for years one of the most powerful techniques available for PTM detection. The long-lived emission of lanthanide chelates at a millisecond time scale allows time-gated of the detection, leading to enhanced assay sensitivity because of reduced background luminescence. This is because of decreased short-lived autofluorescence and scattering from the sample and used materials from which conventional fluorescence measurements often suffer. TRL has also been utilized to FRET (Förster resonance energy transfer) and time-resolved (TR)-FRET is one of the most used techniques to monitor biological interactions. In these assays, lanthanide chelate or cryptate acts as the energy transfer donor, whereas the monitored acceptor molecule can be a conventional organic luminophore. The advantages of TR-FRET to FRET are because of time-gated measurement. Using lanthanide donor, higher sensitivity, and robustness in biological matrixes is obtained, and thus multiple commercial assays for several analytes utilize the method.14 Previously, we have introduced sensitive antibody-free methods for the detection of several PTMs utilizing the TRL-signal readout.15−19 These methods were based on two interacting peptides leading to a TRL-signal shielded from soluble quencher molecules as a result of the paired peptide structure. Consequently, modification of the substrate peptide disintegrates this interaction reducing the monitored TRL-signal.15,17−19 These methods based on the peptide pairing via coiled-coil leucine-zipper or charged amino acid residues were suitable for different types of PTMs, but there are limitations in using natural substrates. Therefore, we aimed to develop a concept where structural and sequence constrains are less pronounced and the substrate sequence from the natural source of the PTM can be more freely selected. Here, we reported a single-peptide HTS-compatible TR-FRET detection platform for cysteine specific PTMs and demonstrated the assay functionality with the chemical cysteine-modifier, S-nitrosoglutathione (SNOG), and the ADP-ribosyltransferase PtxS1-subunit of pertussis toxin.
Experimental Section
Reagents and Instrumentation
Streptavidin (SA) was purchased from Biospa (Milan, Italy) and conjugated with {2,2′,2″,2‴-{[4-[(4-isothiocyanatophenyl)ethynyl]pyridine-2,6-diyl]-bis(methylenenitrilo)}tetrakis(acetato)}-europium(III) chelate20 obtained from QRET Technologies Ltd. (Turku, Finland). Alexa Fluor 680 C2 maleimide, nicotinamide adenine dinucleotide NAD+, nicotinamide, and peptide substrates, PC10 (bio-KKNNLKECGLY),26 PC15 (bio-KEEDVIIKKNNLKECGLY),26 and PR16 (bio-KDGKKKKKKSKTKCVIM),27 were from Invitrogen (CA, USA), Sigma Aldrich (MI, USA), and Pepmic Co., Ltd (Suzhou, China), respectively. The NAD/NADH-Glo Assay Kit was from Promega (WI, USA) and NAP 5 column Sephadex G-25 DNA Grade was purchased from GE Healthcare (IL, USA). Black low volume, round bottom, 384 assay plates were purchased from Corning (Corning, USA) and white 384 Optiplates were purchased from PerkinElmer (Groningen, Netherlands). Nitrocellulose membranes were from Santa Cruz Biotechnology (DA, USA). Synthetic DNA fragments, pNH-TrxT plasmid, oligonucleotide primers, BL21(DE3), IPTG, kanamycin monosulphate, lysozyme from chicken egg white, Pierce Protease inhibitor mini tablets, nickel-nitrilotriacetic acid (Ni-NTA) agarose beads, mouse monoclonal anti-HIS, mouse monoclonal anti-GST, and mouse IgG kappa binding protein conjugated to horseradish peroxidase were from Eurofins Genomics (Luxembourg, Luxembourg), Addgene (MA, USA), Novagen (TX, USA), Thermo Fisher Scientific (MA, USA), formedium (Norfolk, UK), Sigma Aldrich (MI, USA), Thermo Fisher Scientific (MA, USA), Macherey-Nagel (Düren, Germany), Structural Genomics Consortium, R&D Systems (MN, USA), and Santa Cruz Biotechnology (DA, USA), respectively. Basic buffer components and analytical grade solvents were from Sigma-Aldrich.
All nitrocellulose membranes were developed with WesternBright ECL from Advansta (CA, USA) and imaged on ImageQuant LAS 4000 from GE Healthcare (IL, USA). Size-exclusion chromatography was performed using a Superdex75 16/600 Hiload Superdex column from GE Healthcare (IL, USA). TR-FRET and TRL-signals were measured with a standard plate reader from Labrox Ltd. (Turku, Finland), using 340 ± 40 nm excitation and 733 ± 4 nm emission wavelengths, and 75 μs delay and 400 μs decay times for TR-FRET, and 340 ± 40 nm excitation and 616 ± 4 nm emission, with 600 μs delay and 400 μs decay times for the TRL-signal. Luminometry measurements for NAD/NADH Glo were performed using a 500 ms integration time using a Tecan Spark M20 plate reader (Männedorf, Switzerland). Tecan Spark was also used to record the excitation and emission spectra for EuSA, AlexaFluor 680 maleimide, and TR-FRET emission spectrum with AlexaFluor blocked or conjugated to the PC15. EuSA and TR-FRET luminescence emission lifetimes were recorded with a Varian Cary Eclipse fluorescence spectrophotometer (Agilent Technologies, Mulgrave, Australia).
SA Conjugation with 7-Dentate Eu-ITC
SA (200 μg) was diluted in 0.5 M carbonate buffer [Na2CO3/NaHCO3 (pH 9.8)] and labeled with 113 nmol of Eu-chelate (30-times excess of chelate). The reaction was incubated for 60 min at room temperature (RT), covered from light, and transferred to +4 °C overnight (o/n). The reaction was purified using a Sephadex NAP-5 column with an elution buffer [1 mM Hepes (pH 8)]. The collection was performed under UV-light to collect the right Eu(III)-labeled SA (EuSA) fractions, and the protein and Eu(III) concentrations were determined yielding a labeling degree of 3.2 (data not shown).
EuSA, AlexaFluor 680, and TR-FRET Spectral Characterization and Lifetime Measurements
EuSA (10 nM) and AlexaFluor 680 (500 nM) spectra were recorded in 20 μL of 384-well microtiter wells. Recording the TR-FRET spectra, PC15 (1 μM) was conjugated with AlexaFluor (1 μM) or blocked with 1 mM SNOG (incubated for 60 min in RT) before AlexaFluor (1 μM) addition. Labeling was incubated for 60 min before the addition of EuSA (250 nM) recording the spectra in 20 μL (1 nm steps). The EuSA excitation spectrum (300–500 nm) was recorded with 615 nm emission and emission spectra (550–800 nm) was recorded with 340 nm emission with 100 μs delay and 100 μs decay times. The AlexaFluor 680 excitation spectrum (550–725 nm) was recorded with a 750 nm emission and the emission spectrum (625–800 nm) was recorded with 600 nm excitation with 600 μs decay time without delay. The TR-FRET emission spectrum (550–800 nm) was recorded with 340 nm excitation using 75 μs delay and 400 μs decay times. EuSA and TR-FRET lifetimes were recorded in a quartz cuvette at 20 μL. EuSA emission lifetime was recorded at 400 nM. The TR-FRET lifetime sample was prepared incubating 1.6 μM PC15 with 25 μM AlexaFluor 680 for 60 min in RT. Thereafter, EuSA (400 nM) was added followed by 5 min incubation in RT. EuSA luminescence emission at a 615 nm lifetime was recorded with 340 nm excitation using 150 μs delay. The EuSA emission lifetime from the TR-FRET pair was measured using 75 μs delay. TR-FRET emission at 730 nm lifetime was recorded with 340 excitation using 50 μs delay.
Cloning of HIS-GST-Af1521 Expression Plasmid
A synthetic DNA fragment encoding for glutathione S-transferase (GST) was cloned in the place of thioredoxin (Trx)-encoding gene in pNH-TrxT with NdeI and NcoI to acquire pNH-HIS-GST. Next, a synthetic DNA fragment encoding for Archaeoglobus fulgidus Af1521 macrodomain21 (UniProt_O28751) was used in PCR to amplify a ligation independent cloning (LIC)-compatible fragment utilizing oligonucleotide primers prAPV-326 (TACTTCCAATCCATGGAACGGCGTACTTTAATCA, LIC nucleotides underlined) and prAPV-327 (TATCCACCTTTACTGTCAAAGACTCCTCTCAAAGACCT). The PCR product was then cloned into pNH-HIS-GST using the LIC method to acquire pNH-HIS-GST-Af1521 which allows the expression of an N terminally HIS- and GST-tagged Af1521 in Escherichia coli. Furthermore, the pNH-HIS-GST-Af1521 plasmid was verified by sequencing.
Protein Expression and Purification
Expression and purification of recombinant PtxS1 (wt and Q127D/E129D mutant) and Gαi proteins, here after called rPtxS1-wt, rPtxS1-Q127D/E129D and rGαi, have been described previously.22 For rHIS-GST-Af1521, the expression plasmid pNH-HIS-GST-Af1521 was transformed into BL21(DE3) and selected o/n at 37 °C on Luria–Bertani (LB) agar with appropriate antibiotics (kanamycin, 50 μg/mL). Next day, a single colony was transferred into LB media with appropriate antibiotics and cultured at 37 °C, 250 rpm, o/n. Thereafter, the culture was diluted (1:100) in LB with appropriate antibiotics and grown at 37 °C until optical density at 600 nm reached 0.5–0.6, before being induced with 0.5 mM IPTG. After 5–6 h of culturing at 37 °C, cultures were harvested by centrifugation and stored at −20 °C. Frozen bacteria were thawed and resuspended in lysis buffer [100 mM Hepes (pH 7.5), 500 mM NaCl, 10% (w/v) glycerol, 2 mM dithiothreitol, 10 mM imidazole, and 0.3 mg/mL lysozyme] supplemented with Pierce Protease inhibitor mini tablets [40 μL/mL of stock solution (one tablet/2 mL of H2O)]. Samples were sonicated and clarified by centrifugation. The supernatant was collected and loaded onto nickel-nitrilotriacetic acid (Ni-NTA) agarose column equilibrated in buffer [100 mM Hepes (pH 7.5), 500 mM NaCl, 10% glycerol, and 10 mM imidazole]. The unbound material was removed by extensive washing with the same buffer supplemented with 25–50 mM imidazole. The protein was eluted in buffer [100 mM Hepes (pH 7.0), 500 mM NaCl, and 500 mM imidazole] and subjected to size exclusion chromatography (SEC) on a Superdex75 16/600 Hiload Superdex column, using SEC buffer [100 mM Hepes (pH 7.5), 500 mM NaCl, and 10% (w/v) glycerol]. Protein fractions were pooled, flash frozen, and stored at −80 °C.
Single-Peptide TR-FRET Assay Optimization and Nonenzymatic S-Nitrosylation
The substrate peptide ratio to Eu3+-chelate-labeled SA (EuSA) was optimized with PC15 (1–40 nM). 5 μL of the peptide added in buffer 1 [20 mM Hepes (pH 7.5), 5 mM NaCl, and 0.01% Triton X-100] was mixed with 0.5 μL of AlexaFluor 680 maleimide ester (500 nM) added in buffer 2 [500 mM Hepes (pH 8.5)], and the conjugation reaction was incubated at RT for 90 min. Thereafter, 10 μL of EuSA (1–10 nM) was added in buffer 3 [50 mM Hepes (pH 7), and 0.01% Triton X-100] and the reaction was further incubated for 5 min, before being monitored at 730 nm (AlexaFluor 680 TR-FRET) and 615 nm (Eu TRL). Next, the optimal concentration for AlexaFluor 680 was determined in titration (0–11 μM) using the previously described protocol and concentrations for PC15 and EuSA. In all assays, AlexaFluor 680 conjugations were performed using light protection and triplicate reactions.
The nonenzymatic S-nitrosylation assay was used as a proof-of-concept. The peptide substrate (2.5 μL, 300 nM) (PC10, PC15, or PR16) was incubated with an equal volume of freshly prepared SNOG (0–1.67 mM), in buffer 1. Reactions were incubated for 60 min at RT, before the addition of 0.5 μL of 100 nM AlexaFluor 680 maleimide ester in buffer 2. The labeling reaction was incubated for 90 min, before 10 μL of EuSA (10 nM) was added in buffer 3. The reaction was further incubated for 5 min before TR-FRET- and TRL-signals were monitored, as previously.
ADP-Ribosylation and NAD+ Hydrolase Activities of rPtxS1-wt Using Various Substrates
First, we studied the ADP-ribosylation activity of rPtxS1-wt and rPtxS1-Q127D/E129D. In a 100 μL reaction performed in buffer 4 [100 mM Hepes (pH 7.5), 500 mM NaCl, and 10% (w/v) glycerol], 30 μM NAD+ was incubated with 1 μM rGαi and 0.3–10 μM rPtxS1 enzymes. ADP-ribosylation reactions were performed at RT for 60 min with shaking at 500 rpm. Reactions were stopped by the addition of Laemmli loading dye to 1× and heating for 10 min at 95 °C. The samples were separated on sodium dodecyl sulphate-polyacrylamide gel electrophoresis and transferred to nitrocellulose membranes, followed by blocking with 5% (w/v) fat free milk in TBST-buffer [10 mM Tris-HCl (pH 7.5), 150 mM NaCl, and 0.05% Tween 20]. After blocking, membranes were probed in TBST containing 5% (w/v) milk (24 h at 4 °C in rotation). For ADP-ribose, the probing was performed with a combination of rHIS-GST-Af1521 (10 μg/mL) and mouse monoclonal anti-GST antibody (1:1000), and for rPtxS1-wt, rPtxS1-Q127D/E129D, and rGαi proteins with mouse monoclonal anti-HIS antibody (1:1000). Membranes were washed thrice with TBST containing 5% (w/v) milk for 10 min at 4 °C in rotation. Membranes were subsequently incubated with a mouse IgG kappa binding protein conjugated to horseradish peroxidase (1:2500) for 3 h at 4 °C in rotation, before TBST washing was repeated. All membranes were subsequently developed with WesternBright ECL and imaged on ImageQuant LAS4000.
The NAD+ hydrolase activity of rPtxS1-wt was studied with peptides PC10 and PC15, both derived from human Gαi (Uniprot_P63096), which was also used as a comparison. The assay was performed using a commercial NAD/NADH-Glo assay according to the manufacturer’s instructions. Enzyme reactions containing 200 nM rPtxS1 (wt or Q127D/E129D mutant), 1 μM substrate (PC10, PC15 or rGαi), and 400 nM NAD+ were performed in assay buffer 1 and incubated for 60 min at RT. Duplicate reactions (20 μL) were mixed with an equal volume of a NAD/NADH Glo reagent in a white 384-well plate. The total luminescence signal intensity was measured and the signal values were compared to the linear part of the NAD+ standard curve monitored alongside the enzyme reactions.
ADP-Ribosylation Detection with a Single-Peptide TR-FRET System
The rPtxS1-wt enzyme was first titrated from 0 to 1 μM by mixing 1.5 μL of the enzyme with 1.5 μL of PC15 (40 nM) and 2 μL of NAD+ (10 μM) in buffer 1, and incubating the enzymatic reaction for 60 min, RT. Thereafter, 0.5 μL of AlexaFluor 680 maleimide ester (100 nM) was added in buffer 2 and further incubated for 90 min, RT. Then, 10 μL of EuSA (10 nM) was added and TR-FRET- and TRL-signals were monitored after 5 min incubation. In NAD+ titration, 0–30 μM NAD+ (2 μL) was mixed with 1.5 μL of PC15 (10 nM) and 1.5 μL of rPtxS1 (wt or Q127D/E129D mutant) (333 nM) in assay buffer 1. In inhibitor titrations, 1 μL of nicotinamide (0–0.5 M) was mixed with 1.5 μL of PC15 (10 nM) and 1.5 μL of rPtxS1-wt (333 nM) in assay buffer 1. Reactions were incubated for 5 min, before the addition of NAD+ (5 μM) in 1 μL volume. NAD+ and inhibitor titration reactions were incubated for 60 min at RT. Thereafter, 0.5 μL of AlexaFluor 680 maleimide ester (100 nM) was added in buffer 2 and incubation was continued for 90 min at RT. For the detection, 10 μL of EuSA (2.5 nM) was added and TR-FRET- and TRL-signals were monitored after 5 min of incubation.
Data Analysis
In all assays, the signal-to-background ratio (S/B) was calculated to be μmax/μmin and coefficient of variation (CV %) (σ/μ) × 100. That said, μmax corresponds to high TR-FRET-signal emission monitored at 730 nm when the substrate is conjugated with AlexaFluor 680 dye and μmin to a low TR-FRET-signal emission at 730 nm when the conjugation is blocked by a modification. In all formulas μ is the mean value, and σ is the standard deviation (SD). Enzyme rPtxS1-wt limit of detection was calculated to be 3 × σmax/slope (enzymatic titration). Data were analyzed using Origin 8 software and the half maximal inhibitory concentration (IC50) and the half maximal effective concentration (EC50) values were obtained using standard sigmoidal fitting functions. Data were analyzed using Origin 8 software (OriginLab, Northampton, MA).
Results and Discussion
Here, we report a homogeneous single-peptide technology for cysteine PTM detection based on TR-FRET. The simplified principle of the method is depicted in Figure 1. The method utilizes peptide substrates containing the target cysteine and biotinylated N-terminus for the binding of EuSA used as a TR-FRET donor. Nonmodified cysteine provides the basis for the TR-FRET pair formation as the cysteine containing peptide is labeled with a thiol-reactive AlexaFluor 680 acceptor. Upon chemical or enzymatic modification of the cysteine residue, the AlexaFluor 680 conjugation is prevented and, therefore, the label remains free in the solution leading to a low TR-FRET signal at 730 nm. When seeking for novel binders, for example, inhibitors of cysteine-modifying enzymes, inhibition of cysteine modifying enzymatic activity is monitored as an increase in the TR-FRET-signal at the acceptor emission at 730 nm. Therefore, the method is highly applicable to inhibitor screening of various cysteine-modifying enzymes.
Figure 1.

Single-peptide TR-FRET detection method for cysteine-specific PTMs. Using biotinylated substrate peptide containing PTM specific target sequence, chemically or enzymatically generated cysteine modification results conjugation blockage of the thiol-reactive AlexaFluor 680 acceptor dye monitored as a low TR-FRET-signal in the presence of the EuSA donor excited at 340 nm (right). If the cysteine modification is prevented, free cysteine allows the conjugation of the AlexaFluor 680 acceptor dye, resulting in the formation of short distance energy transfer pair monitored as a high TR-FRET-signal at 730 nm (AlexaFluor 680 emission) upon 340 nm excitation (left). In the used detection protocol, the PTM-addition (1) takes place prior the AlexaFluor conjugation (2) and thereafter, EuSA is added to form an efficient energy transfer pair (3).
Single-Peptide TR-FRET Assay Optimization and Nonenzymatic S-Nitrosylation
Many crucial cellular processes, for example, DNA repair23 and gene regulation,24 are regulated through cysteine modifying enzymes such as ADP-ribosyltransferases23 and farnesyltransferases.24 Moreover, inhibitors against these enzymes have been studied intensively for, for example, their potential use in cancer therapy.24,25 Because of our interest for these important potential drug target groups, we selected three terminal cysteine containing peptides as models to the single-peptide TR-FRET method. Our main target of interest was Gαi ADP-ribosylation, and thus, two Gαi derived peptides, PC10 and PC15, were selected.26 PR16, which was selected from the K-Ras C-terminus, had no resemblance in sequence to Gαi, and it was selected as an alternative sequence. It also has potential for further prenylation studies.
First, we screened the assay conditions step-by-step. In the overall assay protocol, peptide cysteine modification was first allowed to progress, following 90 min AlexaFluor cysteine coupling. Thereafter, EuSA was added and its binding to the N-terminal biotin of the peptide provided the proximity pair for TR-FRET (Figure 1). For the peptide modification step, that is, nonenzymatic or enzymatic modifications, the buffer conditions were selected to enable ADP-ribosylation under neutral conditions. Subsequently, pH was increased up to 8.5 to enhance the reactivity of AlexaFluor 680 maleimide ester in the coupling reaction. These buffers were also used in the assay optimization. The compatibility of AlexaFluor 680 as the acceptor luminophore in TR-FRET with EuSA was first studied by recording the excitation and emission spectra for AlexaFluor and emission spectra for EuSA (Figure 2A). The emission maximum of EuSA at 615 nm clearly overlaps with the AlexaFluor 680 excitation spectrum in which the maximum was monitored at 652 nm. Furthermore, the minor emission peak of EuSA at 675–700 nm, prevents us from using the AlexaFluor 680 emission maximum at 681 nm, and therefore, TR-FRET was recorded at 730 nm to prevent the measurement of Eu emission. Next, we evaluated the energy transfer efficiency and distance by monitoring spectra’s and luminescence lifetimes from the labeled or nonlabeled peptide and EuSA only (Figure 2B,C). Even the exact distance evaluation of the TR-FRET pair is not possible, thus tetrameric EuSA contains approximately three labels and we cannot determine the exact location of the label or position of the bound peptide, we found energy transfer to be efficient upon AlexaFluor 680 conjugation. No major TR-FRET signal was monitored from the blocked reaction, as a significant signal could be seen upon efficient acceptor conjugation (Figure 2B). Monitored lifetimes for EuSA emission monitored alone at 615 nm or in TR-FRET complex (615 nm), and AlexaFluor 680 emission monitored in the TR-FRET complex at 730 nm were 375, 188, and 37 μs, respectively. The EuSA lifetime fitted well to the value reported previously,27 and we decided to utilize 75 μs delay for TR-FRET assays because of TR-FRET emission is at 57% from maximum at 75 μs from the excitation (Figure 2C, black).
Figure 2.

Luminescence spectra and lifetimes for of AlexaFluor 680 and EuSA. (A) Excitation (black) and emission (red) spectra of AlexaFluor 680 and the emission spectra of EuSA (blue) showed maximums at 652, 681, and 615 nm, respectively. Based on the spectra, EuSA can efficiently transfer energy to AlexaFluor 680, and the TR-FRET can be monitored from the AlexaFluor 680 emission at 730 nm. (B) Significant TR-FRET signal change was monitored upon 340 nm excitation through EuSA when the AlexaFluor 680 was either conjugated to PC15 (black) or the conjugation was blocked by S-nitrosylation (red). The EuSA alone (blue) showed no interfering signal at the selected measurement wavelength at 730 nm. (C) Lifetimes for the TR-FRET reaction monitored through AlexaFluor 680 emission (black) or EuSA emission from the TR-FRET complex (red), and EuSA emission alone (blue), 37, 188, and 375 μs, respectively.
As the AlexaFluor-labeling reaction is performed using a significantly lower concentration than recommended by the manufacturer, concentrations of the multiple reactants play an important role in the method functionality. Therefore, we performed a series of experiments to understand the dynamics of the method. Based on the literature, PC15 is a highly suitable substrate for ADP-ribosylation and, therefore, it was selected for method optimization.26 First, the optimal ratio of PC15 and EuSA was studied. It was hypothesized that the optimal ratio is 4:1 for PC15 to EuSA, as SA is a tetrameric protein containing four binding sites for biotin. The ratio determination was performed using 1:1, 2:1, and 4:1 ratios with the PC15 peptide and three EuSA concentrations (1–10 nM) (Figure 3A). We decided not to investigate higher peptide excess ratios as it was anticipated to lower the assay functionality in the dye coupling step and to increase the TR-FRET background signal. In all experiments, a PC15/EuSA ratio of 4:1 was found to be optimal. The highest signal-to-background (S/B) ratio of 11.4, calculated from assays with or without PC15, was obtained with the highest EuSA concentration (10 nM).
Figure 3.
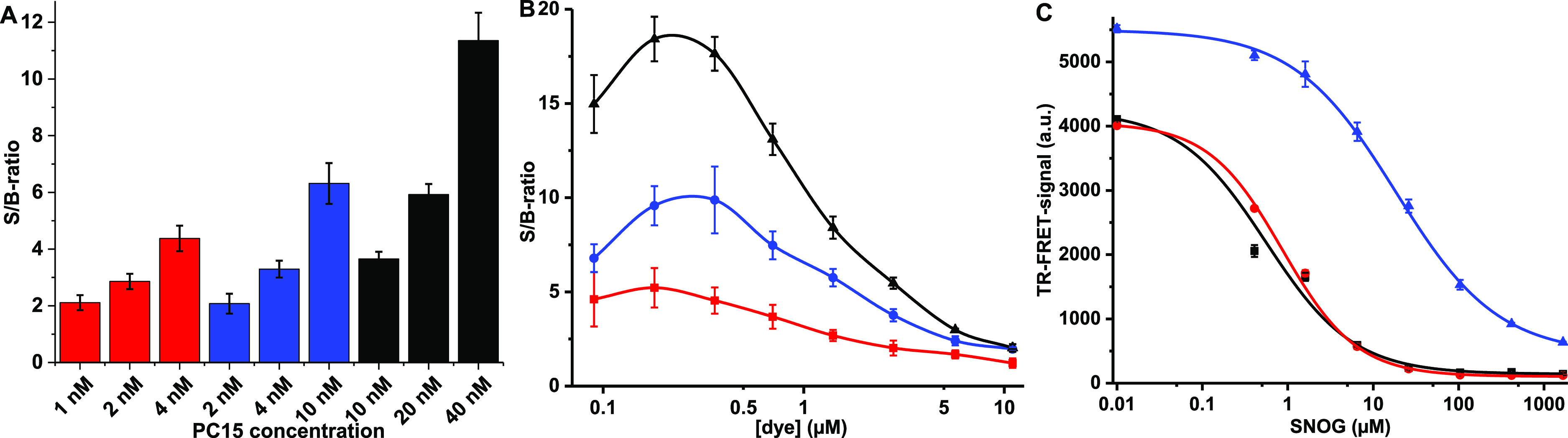
Single-peptide TR-FRET assay optimization and functionality. Data represent mean values ± SD (n = 3). (A) The optimal ratio of the peptide substrate PC15 (1–40 nM) to EuSA was determined in the presence of constant AlexaFluor 680 concentration (0.5 μM) and using 1 (red), 2.5 (blue) or 10 nM (black) EuSA, respectively. In all cases the 4:1 ration of PC15 and EuSA was found optimal and the S/B ratio increases upon PC15 concentration increase. (B) AlexaFluor titration was conducted using 4:1 PC15/EuSA ratio using 1 (red), 2.5 (blue) or 10 nM (black) EuSA, respectively. The most effective labeling was found between 0.1 and 0.5 μM AlexaFluor 680 with all used PC15/EuSA concentrations. (C) Assay functionality was further tested in the chemical S-nitrosylation assay with three peptide substrates PC10 (black), PC15 (red), and PR16 (blue). In SNOG titration (0–1.67 mM), EuSA (10 nM), and AlexaFluor (100 nM) concentration were kept constant. Sequentially, similar PC10 and PC15 gave similar EC50 values, as significantly weaker S-nitrosylation was monitored with K-Ras-derived PR16.
In the previous experiments, the PC15/EuSA ratio was optimized at a single 0.5 μM AlexaFluor concentration. Next, the assay was further studied by titrating AlexaFluor concentration up to 11 μM, and using the optimal PC15/EuSA ratio (4:1) with 1–10 nM EuSA (Figure 3B). The data suggest that the AlexaFluor concentration from 0.1 to 0.5 μM provides the highest S/B ratio, calculated from the TR-FRET signals with or without PC15, with all EuSA concentrations. Based on these results, already 2.5 nM EuSA provided a sufficiently high S/B ratio of 10 for the TR-FRET measurement and it was selected for further assays, while the highest S/B ratio of 18 was measured using 10 nM EuSA. Because of the very low TR-FRET background signal in the used concentrations, the S/B ratio was found to change slightly from assay to assay but not within the one assay. This small change was calculated as insignificant, as it was found not to affect assay result. As we aimed to detect ADP-ribosylation, the assay was also performed in the presence of the rPtxS1-wt enzyme but without any significant change in optimal AlexaFluor concentration or coupling efficiency (data not shown). This finding was significant, as any free cysteine, even rarely found in the protein, might also be labeled and thus the assay and/or enzyme functionality might be compromised.7,8
After the PC15/EuSA ratio optimization, the single-peptide TR-FRET system was tested for the chemical S-nitrosylation of cysteine as a proof-of-principle. In S-nitrosylation, the cysteine residue of the peptide is modified by nitric oxide (NO), spontaneously produced by SNOG in aqueous solution. SNOG is a sequence-independent cysteine modifier, and thus, it was an ideal reaction to test the functionality of the assay platform. The assays were performed with PC10 and PC15 derived from Gαi and selected for the ADP-ribosylation tests. As a third peptide, K-Ras-derived PR16 with a high positive charge because of lysine residues, was selected and it has a significantly different sequence compared to the other two peptides. The assay was performed using high 10 nM EuSA concentrations to provide a more efficient TR-FRET pair as with 2.5 nM EuSA already found optimal. S-nitrosylation was studied by performing SNOG titration up to 1.67 mM, and based on this data, SNOG is not entirely independent of the substrate sequence as the Gαi peptides PC10 and PC15 were more efficiently modified compared to the PR16 (Figure 3C). EC50 values for PC10, PC15, and PR16 were 0.56 ± 0.20, 0.86 ± 0.07, and 18.4 ± 1.5 μM and the S/B-ratios 28.7, 33.0, and 8.7, respectively. The reason for a more than 10-fold difference in the EC50 value was not further studied, but it can be speculated that the high positive charge of PR16 interferes with the S-nitrosylation. However, the data demonstrate the principle and potential of the TR-FRET for the detection of cysteine modifications.
ADP-Ribosylation and NAD+ Hydrolase Activities of rPtxS1-wt Using Various Substrates
As the nonenzymatic cysteine TR-FRET assay was found to be functional, and the rPtxS1-wt did not interfere with the AlexaFluor-labeling, we next established the assay for the enzymatic ADP-ribosylation. ADP-ribosylation of Gαi by the ADP-ribosyltransferase PtxS1-subunit of pertussis toxin was selected as the model.22,26,28 Before the TR-FRET ADP-ribosylation assay, we studied the activity of rPtxS1-wt. First, we conducted the ADP-ribosylation assay to verify and compare rPtxS1-wt and mutant rPtxS1-Q127D/E129D activity toward rGαi. The protein-conjugated ADP-ribose was detected in a western-based method using the bacterial Af1521-macrodomain, which has a nanomolar affinity to ADP-ribose.21 As shown in Figure 4A, ADP-ribosylated protein that migrated between 35 and 55 kDa markers was detected in the presence of NAD+, rGαi, and rPtxS1-wt, but not with rPtxS1-Q127D/E129D. This ADP-ribose signal is in accordance with the theoretical size of rGαi, for example, 40.4 kDa for isoform 1 of human rGαi. No ADP-ribose signal was detected between 35 and 55 kDa markers if the reaction conditions lacked NAD+. As previously reported,22 rPtxS1-wt and to lesser extent rPtxS1-Q127D/E129D had already become auto-ADP-ribosylated in the E. coli expression host as seen by the appearance of an ADP-ribose signal between 15 and 25 kDa markers. Based on these results, we concluded that the rPtxS1-wt ADP-ribosylated the rGαi target in the studied concentrations in the presence of NAD+. These results demonstrate that rPtxS1-wt is a prominent choice for the TR-FRET assay development, and the rPtxS1-Q127D/E129D mutant can be used as an inactive control.
Figure 4.

rPtxS1-wt ADP-ribosylation and NAD+ hydrolase activities with rGαi and peptides PC10 and PC15. (A) ADP-ribosylation assay was performed to estimate the ADP-ribosylation activity of HIS-tagged rPtxS1-wt and rPtxS1-Q127D/E129D. Reaction was performed with 1 μM N-terminally HIS-tagged rGαi in reactions with or without 30 μM NAD+. The protein-conjugated ADP-ribose was detected using bacterial Af1521-macrodomain. Protein loading is visualized with the anti-HIS antibody. Efficient ADP-ribosylation, migrating between 35 and 55 kDa markers, was detected only in the presence of NAD+, rGαi, and rPtxS1-wt. (B) PC10, PC15, and rGαi were further evaluated in the NAD/NADH Glo assay to demonstrate their suitability as rPtxS1-wt substrates. Assays were performed under a constant NAD+ concentration (400 nM) and in the presence of buffer (black), 200 nM rPtxS1-wt (red), or 200 nM rPtxS1-Q127D/E129D (blue). Based on the data, only rPtxS1-wt showed clear NAD+ hydrolase activity favoring rGαi followed by PC15 and PC10 as a substrate. Data represent mean values ± SD (n = 2).
We next investigated the functionality of the selected Gαi-derived peptides, PC10 and PC15,26 to act as substrates of rPtxS1-wt. We used the NAD/NADH Glo assay to measure the NAD+ consumption during catalysis. Of note, rPtxS1 is an ADP-ribosyltransferase, which consumes NAD+ also in the absence of its Gαi substrate, although to a much lower quantities.22 In all assays, the NAD+ concentration was fixed to 400 nM to ensure that the assay functionality is in the linear range determined by the manufacturer. The enzyme reaction was performed using 1 μM of substrates (PC10, PC15, and rGαi) and 200 nM of rPtxS1-wt. As expected, NAD+ consumption was negligible without rPtxS1-wt and with the rPtxS1-Q127D/E129D enzyme using either peptide substrates or rGαi (Figure 4B). However, a significant decrease in the concentration of NAD+ was apparent with rPtxS1-wt reflecting efficient ADP-ribosylation. The largest response was monitored with rGαi and the lowest with short PC10 peptide substrates, which is in line with reports by others.26 Thus, we selected PC15 for the further TR-FRET assays to study the ADP-ribosylation with the single-peptide TR-FRET system.
ADP-Ribosylation Detection with a Single-Peptide TR-FRET System
Based on the NAD/NADH Glo assay (Figure 4B), PC15 was selected as the substrate, and we investigated functionality of the single-peptide TR-FRET assay platform in the detection of enzymatic cysteine modification. The first step was to determine an efficient enzyme concentration to be used in further assays. The enzyme rPtxS1-wt was titrated from 0 to 1 μM and based on the results, 333 nM rPtxS1-wt was selected to be used in further assays (data not shown). The limit of detection, 37 nM, was determined based on the data obtained. In the NAD/NADH Glo assay, hydrolysis of NAD+ was monitored in a limited NAD+ concentration, and thus, limiting the enzymatic capacity of rPtxS1-wt. Quite opposite, the single-peptide TR-FRET assay is limited by the low peptide concentration. Therefore, a high NAD+ concentration is probably needed to ensure the assay functionality, and thus, we decided to optimize the NAD+ concentration next. The expected NAD+ concentration dependency was clearly observed in the NAD+ titration (0–30 μM) when assayed with the PtxS1 enzymes (Figure 5A). As previously, rPtxS1-Q127D/E129D did not show any activity even at high NAD+ concentrations. On the contrary, clear ADP-ribosylation activity using rPtxS1-wt was monitored from the reduced TR-FRET signal in the presence of the increasing NAD+ concentration. The calculated EC50 value for NAD+ was 3.6 ± 0.2 μM, which is in the expected concentration range based on the reported NAD+ affinity to PtxS1, Kd = 24 μM29 (Figure 5A).
Figure 5.
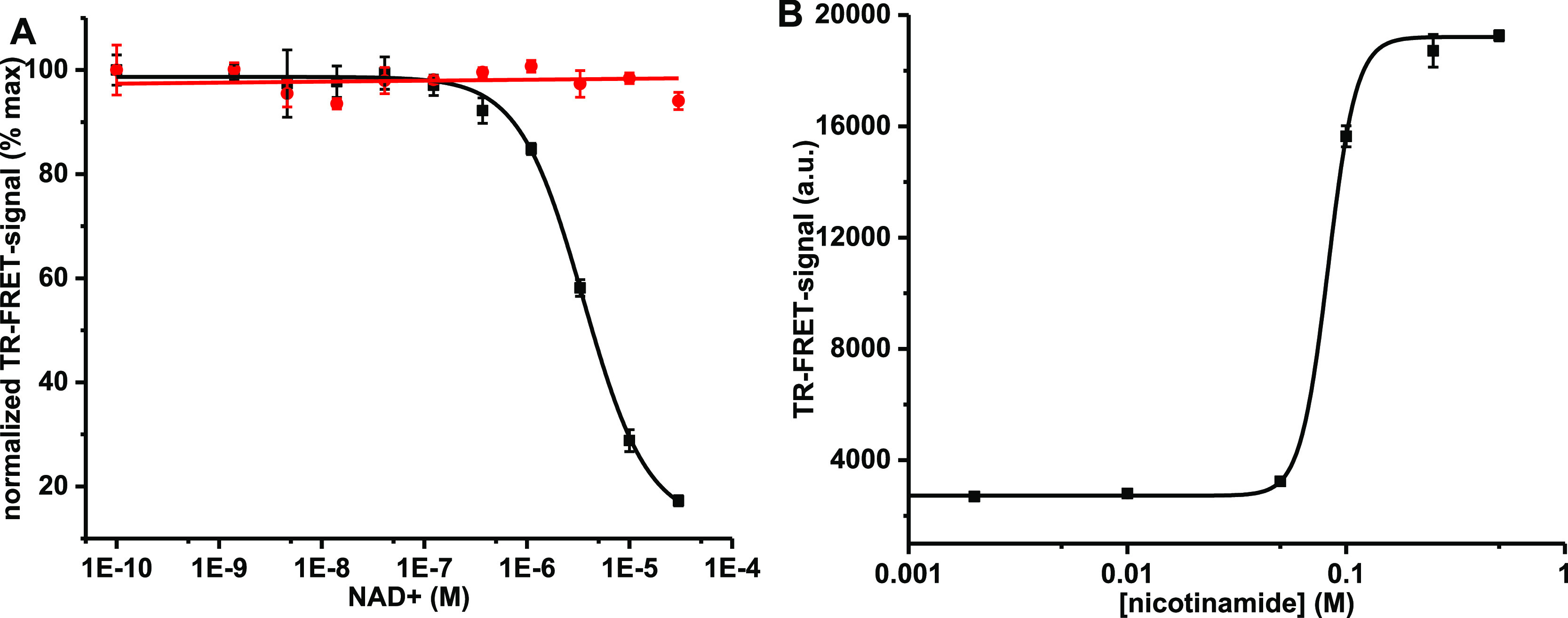
Single-peptide TR-FRET detection for ADP-ribosylation in NAD+ and NA titrations. Data represent mean values ± SD (n = 3). (A) Functionality of the ADP-ribosylation assay was analyzed with 333 nM rPtxS1-wt (black) or rPtxS1-Q127D/E129D (red) in a NAD+ titration (0–30 μM). Clear ADP-ribosylation was observed with rPtxS1-wt with a calculated EC50 value of 3.6 ± 0.2 μM for NAD+. Mutant rPtxS1-Q127D/E129D showed no response to NAD+ concentration increase. (B) ADP-ribosylation can be inhibited by NA. Inhibition assay was performed with 333 nM rPtxS1-wt and 5 μM NAD+ by titrating NA from 0 to 0.5 M. The determined IC50 value for NA was 82.8 ± 1.4 mM.
As the single-peptide TR-FRET method is potentially applicable for HTS, we next monitored the inhibition efficiency of ADP-ribosylation using the NAD+ precursor, nicotinamide (NA). Based on the NAD+ titration, 30 μM NAD+ provided maximal ADP-ribosylation with rPtxS1-wt under the given assay conditions. However, when the assay was performed using this NAD+ concentration, even high NA concentrations (500 mM) gave only a minor TR-FRET signal change (data not shown). This indicates that NA is able to compete with NAD+, but it is a very weak competitor. This led us to lower the NAD+ concentration to 5 μM for potentially improved inhibitory competition (Figure 5B). Based on the titration, NA inhibits NAD+ binding to rPtxS1-wt but with a relatively poor IC50 value of 82.8 ± 1.4 mM. Even at these conditions, the assay provided a sufficient S/B ratio of 6.8 at the highest 500 mM NA concentration.
Based on the data presented, the developed homogeneous single-peptide TR-FRET assay can be used to detect cysteine modifications at the low-nanomolar peptide substrate concentration as shown with nonenzymatic (S-nitrosylation) and enzymatic (ADP-ribosylation) reactions. ADP-ribosylation was performed with the rPtxS1-rGαi enzyme substrate pair and compared to the western blot-based assay and NAD/NADH Glo assays, which were additionally used to optimize the assay conditions. In comparison to the time-consuming western blot assay, the developed single-peptide TR-FRET assay significantly simplified the protocol and reduced the assay time. On the other hand, the NAD+ concentration is limiting the NAD/NADH Glo assay, which potentially also suffers from false positives related to NAD+ degradation and ADP-ribosyltransferase auto-ADP-ribosylation. This is because of the detection principle of the NAD/NADH Glo assay relying on the measurement of NAD+ consumption and not the actual ADP-ribosylation reaction. On the contrary, the single-peptide TR-FRET assay is not limited by the NAD+ concentration, although in the current assay setup the NAD+ concentration was reduced for the inhibitor assay because of the poor inhibition propensity of NA. In addition, TR-FRET assay concept directly monitors the ADP-ribosylation of the substrate peptide. However, at the current format, the method is limited to the use of protein fragments, that is, short peptides allowing the formation of an efficient FRET-pair. However, the use of peptides instead of complete protein structures reduces the costs and simplifies the assay conversion to other targets by only changing the targeted sequence. Potentially, the assay can be converted not only to all cysteine modified enzymes but also to the detection of lysine modifications with an amine reactive dye coupling chemistry. However, this is expected to be more challenging because of the higher number of protein surface lysines compared to cysteines. Based on the presented data, we expect that the introduced method works as a multifunctional tool for several different cysteine PTMs and has high potential also for HTS use.
Conclusions
We have demonstrated a homogeneous HTS-compatible cysteine-specific PTM detection platform based on single-peptide concept and sensitive TR-FRET signal detection. The aim was to construct a single-peptide platform to monitor multiple different cysteine PTMs. As a main target reaction, we selected rPtxS1-wt ADP-ribosylation of Gαi and derived peptides used to demonstrate the assay functionality. The developed homogeneous assay showed improved assay functionality and reduced assay time compared to the used reference methods. We were able show that ADP-ribosylation can be performed at a relatively high NAD+ concentration on the contrary to the NAD/NADH Glo assay, and rPtxS1-wt ADP-ribosylation activity can be inhibited with a structural analogue and precursor of NAD+ nicotinamide. The developed detection platform is potentially applicable to measuring different cysteine PTMs, opening new possibilities such as prenylation studies with K-Ras-derived PR16. Moreover, lysine PTMs, such as acetylation and methylation, can be potentially studied with the developed single-peptide TR-FRET system using, for example, NHS ester dyes. This further increases the applicability of the platform as a multifunctional tool for a variety of PTMs in a simple and sensitive HTS compatible format.
Acknowledgments
This work was supported by the Finnish Academy of Science and Letters (foundation of Vilho, Yrjö and Kalle Väisälä), Drug Research Doctoral Programme (University of Turku Graduate School), Academy of Finland (296225/K.K., 329012/K.K., 323433/K.K., 296093/H.H., and 295296/A.T.P.), Sigrid Jusélius Foundation (A.T.P.), and University of Turku.
Author Present Address
∥ Radiometer Turku Oy, Biolinja 12, FI-20750 Turku, Finland.
The authors declare the following competing financial interest(s): Kari Kopra and Harri Hrm have commercial interest through QRET Technologies Ltd.
References
- Mostaqul Huq M. D.; Wei L. N.. Protein Posttranslational Modification: A Potential Target in Pharmaceutical Development. Handbook of Pharmaceutical Biotechnology; John Wiley & Sons, Inc., 2006; pp 417–441. [Google Scholar]
- Su M.-G.; Weng J. T.-Y.; Hsu J. B.-K.; Huang K.-Y.; Chi Y.-H.; Lee T.-Y. Investigation and Identification of Functional Post-Translational Modification Sites Associated with Drug Binding and Protein-Protein Interactions. BMC Syst. Biol. 2017, 11, 132. 10.1186/s12918-017-0506-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saraswathy N.; Ramalingam P.. Concepts and Techniques in Genomics and Proteomics; Elsevier Science, 2011. [Google Scholar]
- Khoury G. A.; Baliban R. C.; Floudas C. A. Proteome-Wide Post-Translational Modification Statistics: Frequency Analysis and Curation of the Swiss-Prot Database. Sci. Rep. 2011, 1, 1–5. 10.1038/srep00090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selvi B. R.; Mohankrishna D. V.; Ostwal Y. B.; Kundu T. K. Small Molecule Modulators of Histone Acetylation and Methylation: A Disease Perspective. Biochim. Biophys. Acta, Gene Regul. Mech. 2010, 1799, 810–828. 10.1016/j.bbagrm.2010.09.005. [DOI] [PubMed] [Google Scholar]
- Hülsmeier A. J.; Tobler M.; Burda P.; Hennet T. Glycosylation Site Occupancy in Health, Congenital Disorder of Glycosylation and Fatty Liver Disease. Sci. Rep. 2016, 6, 1–12. 10.1038/srep33927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornton J. M. Disulphide Bridges in Globular Proteins. J. Mol. Biol. 1981, 151, 261–287. 10.1016/0022-2836(81)90515-5. [DOI] [PubMed] [Google Scholar]
- Wong J. W. H.; Ho S. Y. W.; Hogg P. J. Disulfide Bond Acquisition through Eukaryotic Protein Evolution. Mol. Biol. Evol. 2011, 28, 327–334. 10.1093/molbev/msq194. [DOI] [PubMed] [Google Scholar]
- Levilliers N.; Fleury A.; Hill A. M. Monoclonal and Polyclonal Antibodies Detect a New Type of Post-Translational Modification of Axonemal Tubulin. J. Cell Sci. 1995, 108, 3013–3028. [DOI] [PubMed] [Google Scholar]
- Mann M.; Jensen O. N. Proteomic Analysis of Post-Translational Modifications. Nat. Biotechnol. 2003, 21, 255–261. 10.1038/nbt0303-255. [DOI] [PubMed] [Google Scholar]
- Jensen O. N. Interpreting the Protein Language Using Proteomics. Nat. Rev. Mol. Cell Biol. 2006, 7, 391–403. 10.1038/nrm1939. [DOI] [PubMed] [Google Scholar]
- Kristensen B. K.; Askerlund P.; Bykova N. V.; Egsgaard H.; Møller I. M. Identification of Oxidised Proteins in the Matrix of Rice Leaf Mitochondria by Immunoprecipitation and Two-Dimensional Liquid Chromatography-Tandem Mass Spectrometry. Phytochemistry 2004, 65, 1839–1851. 10.1016/j.phytochem.2004.04.007. [DOI] [PubMed] [Google Scholar]
- Techner J.-M.; Kightlinger W.; Lin L.; Hershewe J.; Ramesh A.; Delisa M. P.; Jewett M. C.; Mrksich M. High-Throughput Synthesis and Analysis of Intact Glycoproteins Using SAMDI-MS. Anal. Chem. 2020, 92, 1963–1971. 10.1021/acs.analchem.9b04334. [DOI] [PubMed] [Google Scholar]
- Zwier J. M.; Hildebrandt N.. Time-Gated FRET Detection for Multiplex Biosensing. Reviews in Fluorescence; Springer: 2017, pp 17−43. [Google Scholar]
- Härmä H.; Tong-ochoa N.; Van Adrichem A. J.; Jelesarov I.; Wennerberg K.; Kopra K. Toward Universal Protein Post-Translational Modification Detection in High Throughput Format. Chem. Commun. 2018, 54, 2910–2913. 10.1039/C7CC09575A. [DOI] [PubMed] [Google Scholar]
- Kopra K.; Härmä H. Quenching Resonance Energy Transfer (QRET): A Single-Label Technique for Inhibitor Screening and Interaction Studies. New Biotechnol. 2015, 32, 575–580. 10.1016/j.nbt.2015.02.007. [DOI] [PubMed] [Google Scholar]
- Kopra K.; Tong-Ochoa N.; Laine M.; Eskonen V.; Koskinen P. J.; Härmä H. Homogeneous Peptide-Break Assay for Luminescent Detection of Enzymatic Protein Post-Translational Modification Activity Utilizing Charged Peptides. Anal. Chim. Acta 2019, 1055, 126–132. 10.1016/j.aca.2018.12.041. [DOI] [PubMed] [Google Scholar]
- Kopra K.; Eskonen V.; Seppälä T.; Jakovleva J.; Huttunen R.; Härmä H. Peptic Fluorescent “Signal-On” and “Signal-Off” Sensors Utilized for the Detection Protein Post-Translational Modifications. ACS Omega 2019, 4, 4269–4275. 10.1021/acsomega.8b03672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eskonen V.; Tong-Ochoa N.; Valtonen S.; Kopra K.; Härmä H. Thermal Dissociation Assay for Time-Resolved Fluorescence Detection of Protein Post-Translational Modifications. ACS Omega 2019, 4, 16501–16507. 10.1021/acsomega.9b02134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Von Lode P.; Rosenberg J.; Pettersson K.; Takalo H. A Europium Chelate for Quantitative Point-of-Care Immunoassays Using Direct Surface Measurement. Anal. Chem. 2003, 75, 3193–3201. 10.1021/ac0340051. [DOI] [PubMed] [Google Scholar]
- Karras G. I.; Kustatscher G.; Buhecha H. R.; Allen M. D.; Pugieux C.; Sait F.; Bycroft M.; Ladurner A. G. The Macro Domain Is an ADP-Ribose Binding Module. EMBO J. 2005, 24, 1911–1920. 10.1038/sj.emboj.7600664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashok Y.; Miettinen M.; de Oliveira D. K. H.; Tamirat M. Z.; Näreoja K.; Tiwari A.; Hottiger M. O.; Johnson M. S.; Lehtiö L.; Pulliainen A. T. Discovery of Compounds Inhibiting the ADP-Ribosyltransferase Activity of Pertussis Toxin. ACS Infect. Dis. 2020, 6, 588. 10.1021/acsinfecdis.9b00412. [DOI] [PubMed] [Google Scholar]
- Lüscher B.; Bütepage M.; Eckei L.; Krieg S.; Verheugd P.; Shilton B. H. ADP-Ribosylation, a Multifaceted Posttranslational Modification Involved in the Control of Cell Physiology in Health and Disease. Chem. Rev. 2018, 118, 1092–1136. 10.1021/acs.chemrev.7b00122. [DOI] [PubMed] [Google Scholar]
- Palsuledesai C. C.; Distefano M. D. Protein Prenylation: Enzymes, Therapeutics, and Biotechnology Applications. ACS Chem. Biol. 2015, 10, 51–62. 10.1021/cb500791f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slade D. PARP and PARG Inhibitors in Cancer Treatment. Genes Dev. 2020, 34, 360–394. 10.1101/gad.334516.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graf R.; Codina J.; Birnbaumer L. Peptide Inhibitors of ADP-Ribosylation by Pertussis Toxin Are Substrates with Affinities Comparable to Those of the Trimeric GTP-Binding Proteins. Mol. Pharmacol. 1992, 42, 760–764. [PubMed] [Google Scholar]
- Wang Q.; Nchimi Nono K.; Syrjänpää M.; Charbonnière L. J.; Hovinen J.; Härmä H. Stable and Highly Fluorescent Europium(III) Chelates for Time-Resolved Immunoassays. Inorg. Chem. 2013, 52, 8461–8466. 10.1021/ic400384f. [DOI] [PubMed] [Google Scholar]
- Kopra K.; Vuorinen E.; Abreu-Blanco M.; Wang Q.; Eskonen V.; Gillette W.; Pulliainen A. T.; Holderfield M.; Härmä H. Homogeneous Dual-Parametric Coupled Assay for Simultaneous Nucleotide Exchange and KRAS/RAF-RBD Interaction Monitoring. Anal. Chem. 2020, 92, 4971–4979. 10.1021/acs.analchem.9b05126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobban M. D.; Irons L. I.; van Heyningen S. Binding of NAD+ to Pertussis Toxin. Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol. 1991, 1078, 155–160. 10.1016/0167-4838(91)99004-c. [DOI] [PubMed] [Google Scholar]