

Abstract
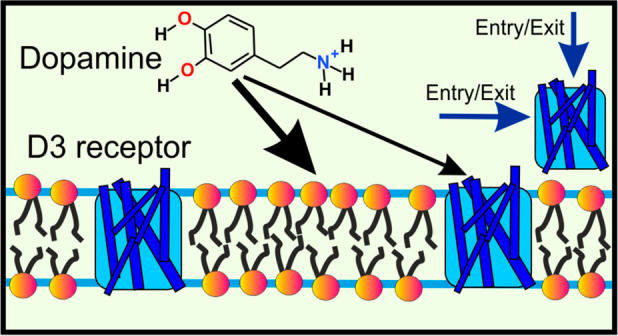
Synaptic neurotransmission has recently been proposed to function via either a membrane-independent or a membrane-dependent mechanism, depending on the neurotransmitter type. In the membrane-dependent mechanism, amphipathic neurotransmitters first partition to the lipid headgroup region and then diffuse along the membrane plane to their membrane-buried receptors. However, to date, this mechanism has not been demonstrated for any neurotransmitter–receptor complex. Here, we combined isothermal calorimetry measurements with a diverse set of molecular dynamics simulation methods to investigate the partitioning of an amphipathic neurotransmitter (dopamine) and the mechanism of its entry into the ligand-binding site. Our results show that the binding of dopamine to its receptor is consistent with the membrane-dependent binding and entry mechanism. Both experimental and simulation results showed that dopamine favors binding to lipid membranes especially in the headgroup region. Moreover, our simulations revealed a ligand-entry pathway from the membrane to the binding site. This pathway passes through a lateral gate between transmembrane alpha-helices 5 and 6 on the membrane-facing side of the protein. All in all, our results demonstrate that dopamine binds to its receptor by a membrane-dependent mechanism, and this is complemented by the more traditional binding mechanism directly through the aqueous phase. The results suggest that the membrane-dependent mechanism is common in other synaptic receptors, too.
Keywords: synaptic neurotransmission, ligand entry pathway prediction, lipid membrane, molecular dynamics, random acceleration molecular dynamics, umbrella sampling
Introduction
Synaptic receptors that bind to nonpeptidic neurotransmitters (NTs) belong to two structurally different groups: ionotropic receptors (IR; Figure 1A) or G protein-coupled receptors (GPCRs; Figure 1B). This classification correlates with the chemical properties of NTs, i.e., those that bind to IRs are predominantly hydrophilic, whereas those that bind to GPCRs are rather amphipathic. There are also NTs, e.g., acetylcholine, glutamate, and serotonin, which have receptor subtypes in both categories. As expected, the degree of hydrophilicity or hydrophobicity of NTs, reflected in their representative octanol/water partition coefficients (log P), also affects how strongly they interact with lipids in the synaptic membranes.1,2
Figure 1.
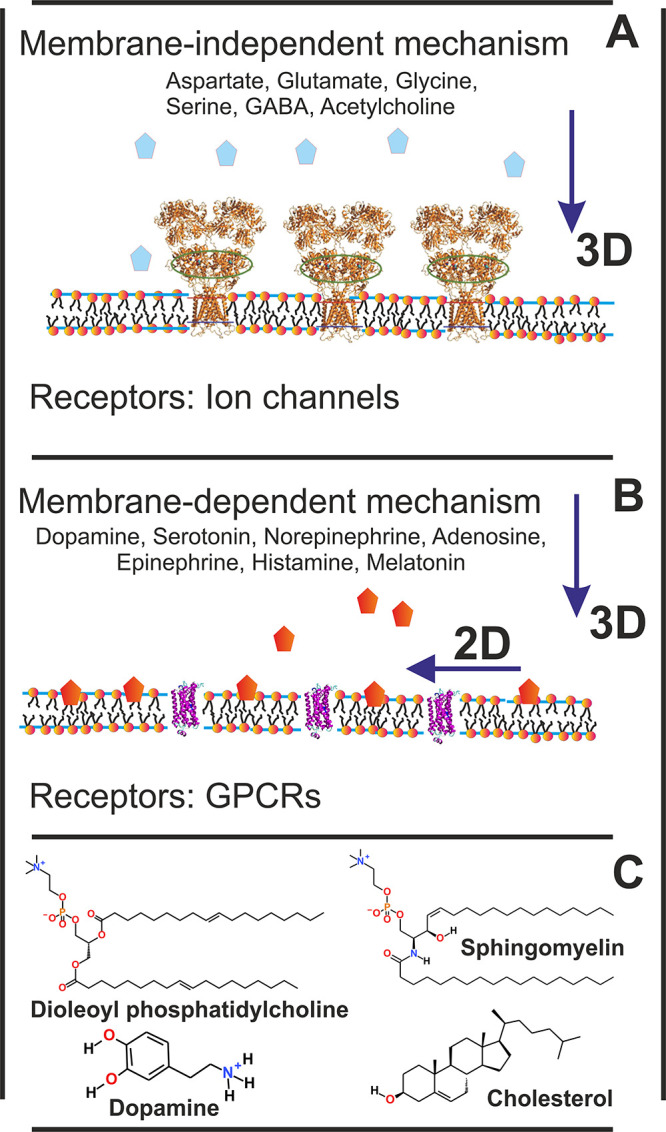
Synaptic neurotransmission models: (A) membrane-independent mechanism; (B) membrane-dependent mechanism.1 Chemical structures of compounds used in this study (C).
Recently, atomistic molecular dynamics (MD) simulations showed in various lipid membrane models that amphipathic NTs, including dopamine, serotonin, norepinephrine, adenosine, epinephrine, histamine, and melatonin (endogenous ligands of GPCRs), bind preferentially to the lipid headgroup region, although the barrier for translocation of NTs through a bilayer was high.1 In vitro studies have confirmed for dopamine the lipophilicity and partitioning into membranes using monolayer experiments,3 calorimetry,4 nuclear magnetic spectroscopy,5 and fluorescence lifetime imaging microscopy.6 Similar conclusions have been observed for serotonin7 and melatonin.8−10 On the other hand, according to MD simulations and experiments,1,11 hydrophilic NTs, such as aspartate, glutamate, glycine, serine, γ-aminobutyric acid (GABA), and acetylcholine (endogenous ligands of IRs), do not adhere to the membrane surface with comparable strength. Acetylcholine, however, has an affinity for lipid bilayers, especially in the presence of negatively charged lipids.11,12
Due to their low level of membrane association, hydrophilic NTs likely bind to their receptors via the membrane-independent mechanism (Figure 1A). On the other hand, amphipathic NTs released into the synaptic cleft are likely to associate with the postsynaptic membrane before binding to membrane-embedded receptor proteins. If NT entry is mediated or assisted by the membrane, then neurotransmission is considered to follow the membrane-dependent mechanism (Figure 1B): the random three-dimensional diffusion of NTs in the water phase is reduced to two dimensions at the membrane surface. The transition from 3D to 2D diffusion along the membrane plane is likely to accelerate NT binding. This picture, in which NT binding is classified into membrane-independent and membrane-dependent mechanisms, is appealing, but there is currently no solid evidence of any NT binding to its receptor via the membrane-dependent mechanism.
In this study, we used the binding of nonpeptidic NT dopamine to the dopamine D3 receptor as a case study to unravel the neurotransmission mechanism of amphipathic NTs (Figure 1B). Dopamine was chosen because of its considerable biological significance, since it is involved in functions such as learning, reward-motivated behavior, and motor control. Moreover, the dysfunction of dopamine-based signaling is linked to pathologies such as Parkinson’s disease.13−18 To characterize dopamine partitioning into physiologically relevant membranes and the mechanism of its entry into the ligand-binding site, we combined isothermal calorimetry experiments with a diverse set of MD-based methods.
Our isothermal titration calorimetry measurements showed that dopamine has a high affinity for liposomes composed of phosphatidylcholine, sphingomyelin, and cholesterol. These lipids are found in the extracellular leaflet of the cell membrane,19 where they are involved in the formation of ordered domains.20 Further, based on our measurements, the affinity of dopamine for bilayers composed of negatively charged phosphatidylserine was even higher. This finding is in agreement with previous experimental studies of dopamine’s interaction with charged and neutral lipids.4,5
To investigate the mechanism of ligand binding, we probed the ligand entry pathways into the binding pocket using unbiased MD simulations, random acceleration MD simulations, and umbrella sampling free energy calculations. The simulation results revealed that the entry of dopamine into the binding site in its receptor takes place through the membrane-dependent mechanism. This is complemented by direct entry from the water phase from the water-exposed face of the receptor.
Taken together, our results provide substantial support for a mechanism in which dopamine first partitions into the headgroup region of a synaptic membrane and then enters the binding pocket through a gate on the membrane-facing side of the receptor.
Results and Discussion
Dopamine Associates with POPC/SM/Chol and POPS Liposomes: Isothermal Titration Calorimetry Measurements
We used isothermal titration calorimetry (ITC) to characterize and quantify the adsorption of dopamine on the POPC/SM/Chol 1:1:1 and POPS liposomes. In general, ITC measures the heat released over time after injecting an aliquot of dopamine into a liposome suspension relative to a reference cell filled with water. Raw ITC data are then integrated over time to obtain incremental heat as a function of a dopamine/lipid molar ratio. After correction for dilution heats, for each injection the total heat change is fitted to an appropriate equilibrium binding model to determine the enthalpy of association (ΔHDA), association constant (KA), and the stoichiometry of association (or the number of binding sites, n).21 Since temperature (T) is held constant, related thermodynamic parameters such as the Gibbs free energy (ΔG) and the change in entropy (ΔS) can be calculated according to the standard relations:
| 1 |
First, we determined the enthalpy of dopamine binding to lipid membranes. Calorimetric titrations conducted under a large excess of lipid relative to dopamine (lipid/dopamine ratio >100:1) can be used to directly determine the enthalpy of the association process. If the association constant and lipid concentration are high enough, essentially the total amount of injected dopamine immediately associates with the lipid membrane. Therefore, consecutive injections of small aliquots of dopamine should yield identical heat values. The association enthalpy of dopamine to liposomes, ΔHDA, was determined by adding a small number of dopamine moles, dnDA, to the calorimeter cell containing the lipid dispersion, which produces a heat of association, dqi (corrected for dilution effects).22 For the POPS liposomes, the lipid/dopamine molar ratio ranged during titration from 1214:1 to 64:1, while for the POPC/SM/Chol liposomes this ratio varied at the end of titration from 1286:1 to 68:1. Thermograms of the dopamine-to-lipid titrations are shown in Figure 2, which reveal that the heats of association are very similar, especially for the first several consecutive injections. Thus, the association enthalpy was calculated using the equation
| 2 |
The ΔHDA values are given in Table 1. The association of dopamine to both types of liposomes is an exothermic process.
Figure 2.
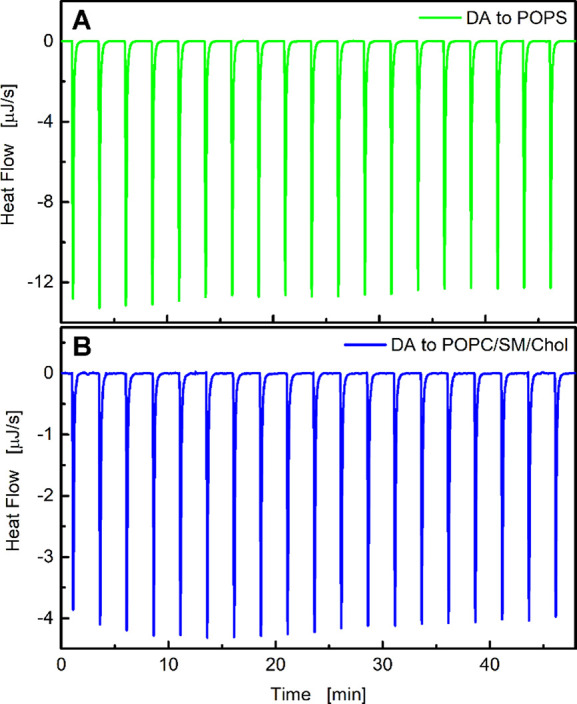
Titration of (A) POPS (51 mM) and (B) POPC/SM/Chol liposomes (54 mM) with a dopamine solution (4.2 mM) in 1 mM PBS at 25 °C. Each peak corresponds to a 2 μL injection. In the case of POPS, the lipid/dopamine ratio ranged from 1214:1 to 64:1, while in POPC/SM/Chol liposomes this ratio ranged at the end of titration from 1286:1 to 68:1.
Table 1. Thermodynamic Parameters of the Interactions between Dopamine and Liposomes Determined by ITC Experimentsa.
| system | ΔHDA (kJ mol–1) | KA (M–1) | ΔG (kJ mol–1) | TΔS (kJ mol–1) | ΔS (J K–1 mol–1) |
|---|---|---|---|---|---|
| POPS | –12.8 ± 0.1 | 1752 ± 95 | –18.5 ± 0.2 | 5.73 ± 0.14 | 19.2 ± 0.5 |
| POPC/SM/Chol | –4.66 ± 0.04 | 305.6 ± 15.0 | –14.2 ± 0.2 | 9.59 ± 0.12 | 32.2 ± 0.4 |
Results are given for the association enthalpy of dopamine to liposomes (ΔHDA); association constant (KA); Gibbs free energy (ΔG); entropic component in the free energy; and a change in entropy (ΔS).
Next, we performed ITC experiments at lower lipid/dopamine molar ratios (typically well below 100:1) to determine the association constant KA and the number of binding sites n on the surface of liposomes. MD simulations have previously indicated that dopamine associates to lipid headgroups predominantly through H-bonds, but it does not penetrate the membrane hydrophobic core.2,3Figure 3 depicts typical ITC results for titrations of the POPS and POPC/SM/Chol liposomes with a dopamine solution. Each injection of the dopamine solution into lipid dispersion led to a rapid exothermic heat flow (Figure 2A and C). The observed thermal effects result from establishing an equilibrium between dopamine molecules that interact with membrane headgroups and the dopamine molecules remaining in the aqueous phase. The dopamine fraction interacting with the lipid membrane after each injection can be calculated from the heat released, based on eq 2. Figure 2 shows that the injection of aliquots of the dopamine solution causes a series of decreasing heat releases, indicating that the number of molecules adsorbed on the liposomes decreases with each injection.
Figure 3.

ITC plots for dopamine association with (A, B) POPS and (C, D) POPC/SM/Chol liposomes. Experiments were performed at 25 °C in 1 mM PBS. Panels (A) and (C) show the heat flow for consecutive injections of the dopamine solution into POPS (3.2 mM) or POPC/SM/Chol liposomes (6.8 mM). Panels (B) and (D) present the corresponding integrated heats of each injection dqi (corrected for dilution effects) versus the dopamine/lipid molar ratio. The solid red lines correspond to the best fit made using a MicroCal PEAQ-ITC analysis software based on the one-site binding model.
Titration curves of kilojoules per mole vs molar ratio (dopamine/lipid) were generated by integrating the heat pulse of each injection with respect to time and dividing it by the number of moles of the injectant (Figure 3C and D). The resulting isotherms were fitted using a single set of the independent binding sites (SSIS)23 model that was sufficient to achieve a good fit to the calorimetric data. This model is based on the Langmuir adsorption isotherm and combines the heat signal from an ITC experiment with the affinity (KA) and the maximum number of bound dopamine molecules (i.e., the binding sites of the liposomes, n) assuming that all sites have the same KA and ΔHDA.23 Positively charged dopamine does not penetrate lipid membranes. Therefore, when injected into lipid vesicles, it can interact only with the outer leaflet of a lipid membrane, i.e., with the γ = 0.5 lipid fraction (γ is the asymmetry parameter).24 For this reason, the lipid concentration multiplied by the asymmetry parameter was used in the fitting routine. The calculated thermodynamic properties of the dopamine association with the POPS and POPC/SM/Chol liposomes, including the affinity constant (KA) and change in entropy (ΔS), are listed in Table 1.
Dopamine Is More Likely to Initially Bind to the Postsynaptic Membrane than to Its Receptor
To estimate the propensity of dopamine’s initial encounter with the components of the postsynaptic membrane, we performed a pool of 1000 short (20 ns long) unbiased simulations (spontaneous dopamine binding simulations), in each of which a dopamine molecule was allowed to diffuse freely, starting from a random position and orientation in the water phase. During these simulations, dopamine molecules translocated from the water phase (Figure 4A) to the water–membrane–protein interface (Figure 4B–F) (Table 2). In 736 cases, after 20 ns, dopamine molecules were in contact only with the lipids (a contact was defined as a pairing of heavy atoms at the distance ≤0.6 nm) (Figure 4B). In 62 cases, dopamine molecules were positioned at the protein surface (Figure 4C), and in 180 cases dopamine molecules were positioned at the protein–lipid interface (Figure 4D). Only 22 simulations captured a direct entry of a dopamine molecule from the water phase into the ligand-binding cavity (Figure 4E, F). While at very long times dopamine molecules are expected to partition based on their free energy of binding between the membrane and the ligand binding pocket, we wish to stress that the aim of our short simulations was to clarify whether there is a strong force driving dopamine toward the protein through the membrane phase, from which dopamine would eventually enter the ligand binding site.
Figure 4.
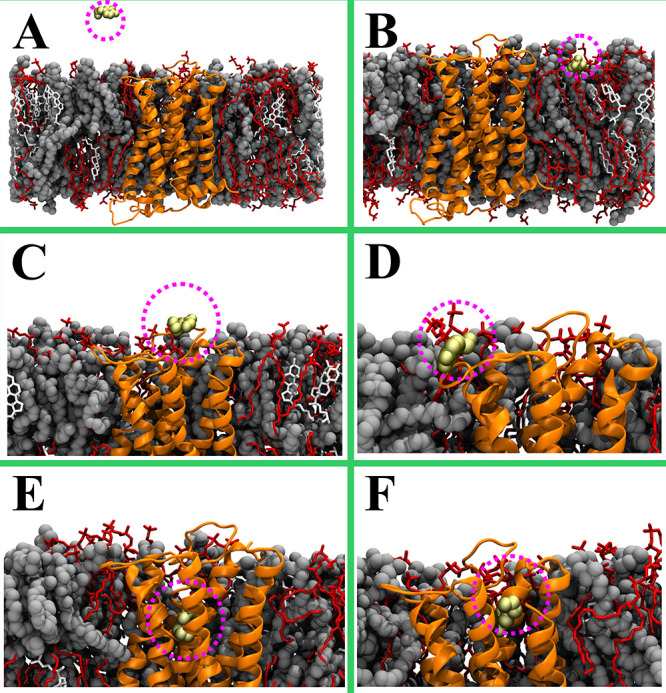
Selected snapshots taken from 20 ns atomistic MD simulations. (A) An example of the initial (0 ns) system. (B–F) Representative snapshots highlighting the position of dopamine molecules (yellow space-filling representation) after 20 ns. (B) Dopamine positions itself to the water–membrane interface; (C, D) Dopamine binds to the protein–lipid interface; (E, F) Dopamine enters directly the receptor’s ligand-binding cavity. The dopamine D3 receptor is shown in an orange cartoon representation, dioleoylphosphatidylcholine (DOPC) in a gray van der Waals representation, sphingomyelin (SSM) in a red stick, cholesterol (CHOL) in a white stick, and the dopamine in a yellow van der Waals representation. Water and ions are not shown for clarity. The figure was prepared with VMD.27
Table 2. Probability of Dopamine Molecule Location at the End of an MD Simulation and Distribution of the Membrane Surface Area between the Protein and Lipidsa.
| dopamine in contact with | % | surface area | % |
|---|---|---|---|
| the binding pocket | 2.2 | protein | 14 |
| protein surface | 6.2 | neighboring lipids | 10 |
| protein–lipid interface | 18 | protein and neighboring lipids | 24 |
| lipids only | 73.6 | lipids not in contact with the protein | 74 |
The maximum value of standard error for the given probabilities is smaller than 1.5% (estimated based on a binomial distribution with 1000 trials).
To assess whether the observed propensities can simply be attributed to the relative surfaces of the protein and lipids exposed to the water phase, we also calculated the surface area occupied by the protein and lipids using the APL@Voro tool (Table 2).25 The protein was found to occupy 14% of the membrane surface and be surrounded by ∼25 lipids, which occupied 10% of the membrane surface. This ratio is lower than what is present in biological membranes in general, ∼25%.26 However, the area where dopamine is in contact with the protein or with both the protein and lipids covered ∼24% of the entire surface area. Normalizing for the relative exposed surface area, dopamine did not appear to have a preference for binding either to the membrane or the membrane-embedded protein. However, because under physiological conditions the lipid head groups cover a considerably larger surface area than proteins, our results indicate that a vast majority of amphipathic NTs released to the synaptic cleft interact initially with the membrane surface.
Dopamine May Enter/Exit the Receptor Directly from the Water Phase and Directly from the Membrane
To explore the exit pathways for a bound dopamine, we performed 10 independent RAMD simulations (Figure 5B). In RAMD simulations, a force is applied to the ligand in a random direction as long as the ligand moves away from its initial position. Once the force cannot sustain the motion of the ligand, a new direction is chosen randomly for the applied force to drive the molecule in a different direction. This approach allows sampling of reasonable pathways for ligand exit and has been successfully employed in this context.28−31
Figure 5.
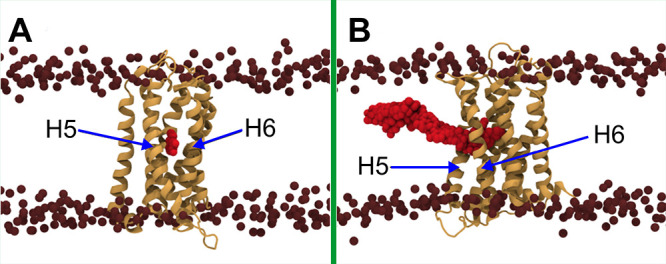
Exit pathway of dopamine through the membrane. (A) Membrane-exposed pocket between helix 5 (H5) and helix 6 (H6) sampled in the RAMD simulation. (B) Overlaid positions of the dopamine molecule highlighting the exit pathway as reconstructed from the RAMD and SMD simulations. The protein is shown as an orange cartoon, and dopamine in red van der Waals spheres. The nitrogen atoms of DOPC and SSM head groups are shown as brown spheres to indicate the location of the membrane.
RAMD simulations revealed two distinct ligand exit pathways: one that vertically reaches to the aqueous phase through the protein medium and a second one that drives dopamine laterally into the hydrophobic core of the membrane. In nine of the ten RAMD simulations, dopamine exited from the binding pocket directly into the water phase through the vertical channel. We note that a similar spontaneous binding pathway was captured in a few of the aforementioned unbiased MD simulations (Figure 4E, F). In one case, however, the dopamine molecule moved into a pocket between transmembrane alpha-helices 5 and 6 (Figure 5A). This pocket is accessible to the membrane and suggests a new exit pathway to the membrane phase, which we further explored using SMD simulations (Figure 5B).
The Lateral Membrane-Mediated Route for Dopamine Binding Is Energetically More Favorable
We next estimated the free energy profiles for the aforementioned lateral and vertical routes using separate sets of umbrella sampling simulations: (A) Dopamine translocation from the membrane surface to the water phase along the bilayer normal (black arrow in Figure 6); (B) dopamine translocation from the receptor’s ligand-binding pocket into the water phase through the main opening at the top of the receptor along the bilayer normal (blue arrow in Figure 6); and (C) Dopamine translocation from the ligand-binding pocket into the membrane (red arrow in Figure 6). In the case (C), the ligand was restrained along a vector that extended from the dopamine binding site to the membrane surface, while in the cases (A) and (B), the restraints were only active along the membrane normal direction. The umbrella sampling simulations for (B) and (C) were initiated from the combined RAMD-SMD simulations, while the simulations for (A) were reported earlier.1,32
Figure 6.
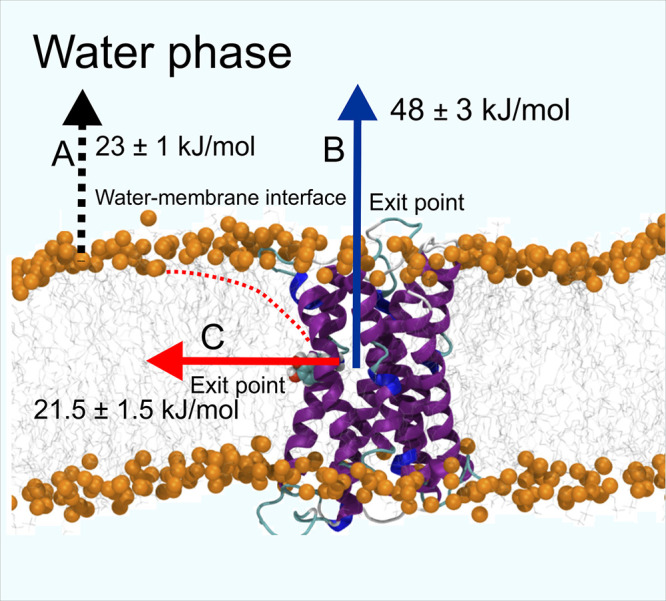
Pulling directions used in free energy calculations. Three directions or reaction coordinates were used in pulling the dopamine molecule: (A) From the membrane surface into the bulk water phase (black-dotted vertical arrow as a process; (B) from the ligand-binding site of the dopamine receptor to the water phase (blue vertical arrow); (C) from the receptor’s binding site to the membrane surface (red arrow). A red-dotted line shows the approximate location of dopamine in the membrane.
We had previously estimated the free energy profile of dopamine translocation from the membrane interior to the water phase (A; black arrow in Figure 6, Figure 7A) in our previous studies (receptor not being present in the membrane).1,32 The free energy minimum corresponds to the membrane–water interface, where it is −23 kJ/mol with respect to the water phase. Further penetration of dopamine into the lipid bilayer is associated with a steep increase in the free energy: the transfer of dopamine from the headgroup region to the center of the bilayer requires an additional 94 kJ/mol. These results show that dopamine strongly partitions into the lipid headgroup region but a membrane is not readily permeable to dopamine.
Figure 7.
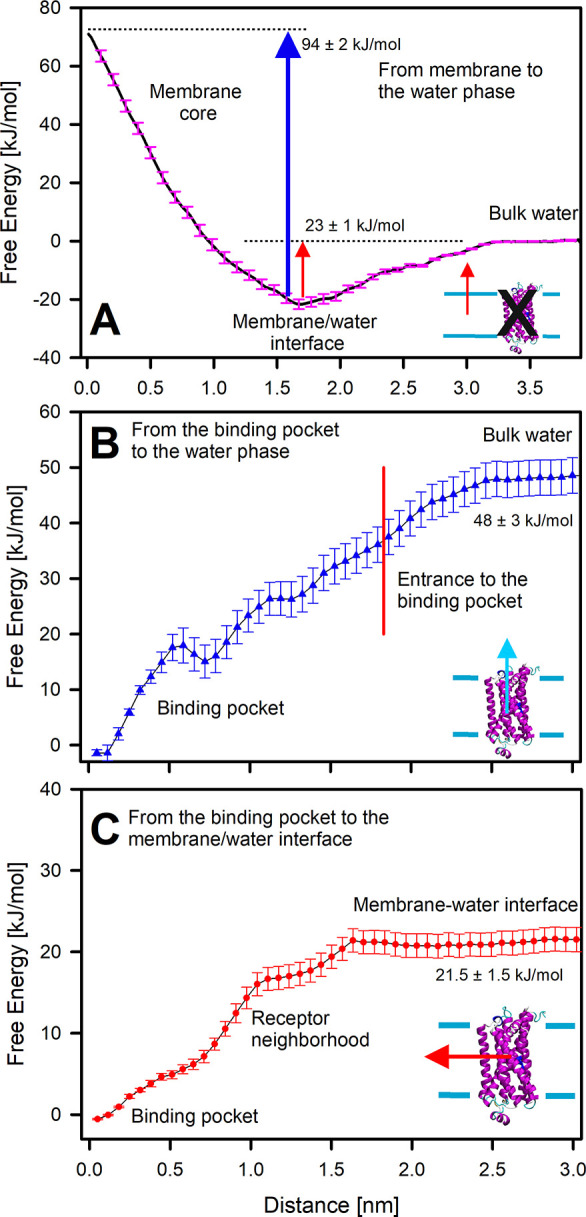
Free energy profiles of the studied exit/entry pathways from the dopamine receptor’s ligand-binding cavity. (A) Pathway in which dopamine translocates from the water phase to the bilayer center (black in Figure 6). (B) Pathway in which dopamine exits the receptor’s ligand-binding site directly into the bulk water phase (blue in Figure 6). (C) Pathway in which dopamine exits the receptor’s ligand-binding site via the membrane (red dotted line in Figure 6).
We also estimated the free energy profile for dopamine traversing the vertical pathway and exiting directly into the water phase from the cavity opening located at the top of the receptor (B; blue arrow in Figure 6). The profile for (B) shown in Figure 7B features a sharp increase in free energy within the vicinity of the binding pocket. This indicates strong resistance for the ligand movement along this pathway. While the profile demonstrates an increasing trend until the ligand reaches the water phase, it features no substantial barrier at the gate of the binding pocket. A small minimum at the distance around 0.7 nm results from formation of a transient hydrogen bond between dopamine and residues at this position.
Comparison of the free energies associated with the (B) and (C) (Figure 7C) pathways reveals that the free energy profile for dopamine exit along (C) requires a lower energy and feature smaller barriers. The free energy difference between the membrane–water interface and the binding pocket is 21.5 ± 1.5 kJ/mol as compared to 48.0 ± 1.5 kJ/mol between the water phase and the binding pocket. There is no energy barrier associated with the translocation.
Notably, in case (C), the average position of the ligand follows a curved path (indicated by the red dotted line in Figure 6) deviating from the initial positions shown in Figure 5. This reflects the preferential partitioning of dopamine to the lipid headgroup region, as discussed above. In this way, these simulations form a closed thermodynamic cycle, where the sum of end-state free energies for (A) and (C) is equal to that of (B). Indeed, our free energy calculations confirm this, as expected upon convergence: the difference between the two different routes is within statistical uncertainty (see the free energy values reported in Figure 6: 44.5 ± 2.5 kJ/mol for (A) plus (C) vs 48.0 ± 3.0 kJ/mol for (B)).
Overall, these results show that dopamine preferably partitions to the membrane headgroup region. Moreover, we revealed a new lateral pathway from the membrane core region to the binding pocket. Taken together with the enhanced local concentration of dopamine in the membrane headgroup region, this lateral pathway will likely dominate over the vertical pathway under physiological conditions.
Summary and Conclusions
The diffusion rates reported for molecules in pure water33 suggest that the translocation of neurotransmitters (NTs) from the presynaptic compartment into the receptor embedded in the postsynaptic membrane takes only a few microseconds. Synapses, however, are complex structures with numerous macromolecules protruding into or spanning the synaptic cleft.34,35 Thus, the translocation of NTs is more complicated than simple diffusion in water. Moreover, amphipathic NTs have a strong affinity for lipid bilayers’ headgroup region, especially those containing anionic lipids.1,4,5 Therefore, many research groups have previously postulated that the membrane actively participates in the entry of amphipathic NTs (e.g., dopamine) and drugs into the binding pocket of their respective G protein-coupled receptors (Figure 1B).1,36−38
The results of the unbiased MD simulations performed in this study are in line with this membrane-dependent entry model (Figure 4). Only a small fraction (2.2%) of the unbiased dopamine binding simulations showed dopamine to enter the receptor’s ligand-binding site directly from the water phase (Figure 4E, F). Meanwhile, in 91.6% of the cases, dopamine first associated with the water–membrane interface (Figure 4B) or the water–membrane–receptor interface (Figure 4D). In the remaining fraction of simulations, dopamine first directly interacted with the receptor surface (Figure 4C). Even if the relative surface areas of the receptors and the lipids exposed to the synaptic cleft are taken into consideration, the fraction of dopamine molecules that directly enter the binding cavity from the water phase is unlikely to be sufficient for effective signal transduction.
In the membrane-dependent model of neurotransmission (Figure 1B),38 the 3D diffusion of NTs is transformed into 2D diffusion at the membrane plane. This reduction in dimensionality can, in principle, enhance the rate of dopamine entry into the binding pocket of its receptor. However, our free energy calculations revealed an obstacle in the process. Dopamine needs to detach from the membrane headgroup region to enter the binding cavity through the central opening at the top of the receptor. Meanwhile, our calorimetric data (Table 1) suggest that dopamine molecules bind membranes strongly enough to significantly slow down neurotransmission. This observation is in agreement with prior studies, highlighting strong interactions between dopamine and lipids.3−6,39
RAMD simulations (Figure 5) uncovered a tangible explanation for this puzzling issue. Unlike conventional MD simulations (Figure 4), which directly explore the NT entry/binding, the biased RAMD simulations provide information on NT unbinding from the receptor’s ligand-binding site. By combining the RAMD, SMD, and free energy calculations (Figures 5 and 6), we discovered an unexpected new dopamine entry/exit pathway (red dotted line in Figure 6). In this pathway, dopamine enters the cavity directly through a membrane patch near the protein. This pathway does not require dopamine detaching from the membrane back to the water phase against a free energy barrier. Although this entry path fulfills the conditions of the membrane-dependent mechanism (Figure 1B), both the unbiased and biased calculations also suggest that the dopamine entry could take place directly from the water phase (Figure 1A), too, but with weaker odds.
The potential exit/entry pathway, taking place between two transmembrane alpha-helices (red dotted line in Figure 5A), does not have unfavorable free energy barriers (red line in Figure 7). This means that the entry could depend only on the diffusion rate of dopamine in the membrane. In prior studies of GPCRs, including β-adrenergic receptor,40 cannabinoid CB2 receptor,41 opsin,42 and sphingosine 1-phosphate receptor,43 unconventional exit pathways of drug molecules have also been reported. These exit points were located at the interfaces of helices 4–5, 5–6, 6–7, and 7–1 in the representative GPCRs. There are two separate openings in opsin and five in the β-adrenergic receptor that could facilitate the entry/exit. Interestingly, direct entrance from the water phase is blocked by the N-terminus and the extracellular loops in the sphingosine 1-phosphate receptor.
The importance of membranes for the ligand entry into membrane proteins has been recognized in pharmacology, although the number of studies on the topic is limited. In a pioneering study of 1,4-dihydropyridines, compounds that block calcium channels, the authors provided kinetic data favoring membrane-mediated entry into the channels.37 The significance of the membrane–drug association in drug design and drug kinetics has been discussed.36 Recently, extensive studies of β-adrenergic receptor ligands indicated that lipophilic drugs accumulate on the lipid bilayer, increasing the rate of drug–target protein association without affecting the dissociation rate.44 Thus, the binding affinities of drugs toward their target proteins should be calculated based on the local environment without assuming a uniform distribution within the system. This was highlighted by an example of two drugs with identical pharmacophores but differing by 3 orders of magnitude in affinity on the target protein. A drug with a higher affinity also had a 5000-fold higher membrane partition coefficient.44
The free energy difference associated with dopamine translocation from the water phase to the membrane environment depends on the membrane lipid composition and is sensitive to Ca2+ cations.1,32,45 The association constant KA that quantitatively describes the process of dopamine association on the liposomes show that dopamine connects with a zwitterionic membrane almost 6 times less compared to an anionic membrane (Table 1). Nevertheless, studies reported so far were performed with a limited number of different lipid species. Thus, one may expect a broader range of differences in more complex membranes containing, e.g., negatively charged lipids such as complex gangliosides and galactosulfatides, which appear in the central nervous system in higher concentrations.46−48 The mechanisms that could explain how the difference in lipid composition affects dopamine entry processes are likely intricate and require detailed studies. The decrease of the dopamine affinity on the membrane reduces the local concentration of dopamine, which in turn can slow down signal transduction. Accordingly, changes in membrane lipid composition can hinder or enhance NT receptor binding by the membrane-dependent and independent mechanisms, even leading to diseases such as depression.
Materials and Methods
Materials
Dopamine hydrochloride (dopamine, pharmaceutical secondary standard) was purchased from Supelco. 1-Palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC, ≥99.0%), cholesterol (Chol), phosphate buffered saline (PBS) tablets, and chloroform (HPLC grade, ≥99.9%) were obtained from Sigma-Aldrich. 1-Palmitoyl-2-oleoyl-sn-glycero-3-phospho-l-serine (sodium salt) (POPS, ≥99.0%) and sphingomyelin (SM, ≥98.0%) were purchased from Avanti Polar Lipids, Inc. The ultrapure Milli-Q water used in the experiments had a surface tension of 72.6 mN/m (at 20 °C) and a resistivity of 18 MΩ cm.
Preparation of Liposomes
POPS and POPC/SM/Chol 1:1:1 liposomes were prepared by sonication using the modified procedure described previously.24 Briefly, lipid stock solutions were prepared in chloroform. Appropriate volumes of the stock solutions were combined in a flask, and a lipid film was formed by evaporation of chloroform. PBS (1 mM) was added to reach a desired lipid concentration (approximately 40 mg/mL), and the sample was vortex mixed for several minutes. The lipid dispersion was subjected to five freeze–thaw cycles from liquid nitrogen temperature to 60 °C and sonication at ice temperature for 10 min using a SONICS VC 130 sonicator.
Isothermal Titration Calorimetry (ITC) Measurements
A Microcal PEAQ-ITC calorimeter (Malvern Instruments Limited, Worcestershire, UK) equipped with two 200 μL cells was used in the experiments. Data were analyzed using a MicroCal PEAQ-ITC analysis software. The “one set of sites” mathematic model was applied to analyze the thermodynamic parameters. Three types of experiments were performed at 25 °C: (i) 19 injections of 2 μL each of the dopamine solution (4.2 mM) in 1 mM PBS to the liposome dispersion (for the POPS liposomes the lipid concentration was 51 mM; for the POPC/SM/Chol liposomes, the total concentration of POPC and SM was 54 mM) in 1 mM PBS, duration of injection = 4 s; (ii) 39 injections of 1 μL each (the first injection of 0.4 μL) of the DA solution (11 mM) to the POPS liposomes (the lipid concentration was 3.2 mM), injection duration = 2 s; (iii) 13 injections of 3 μL each (the first injection of 0.4 μL) of the dopamine solution (16 mM) to the POPC/SM/Chol liposomes (the total concentration of POPC and SM was 6.8 mM), injection duration = 6 s. In all experiments, the interval between injections was 150 s and the stirring speed was 500 rpm. The heats were corrected for the dilution effects determined in separate experiments. The experiments were repeated at least three times.
Molecular Dynamics: System Setup
The 3D structure of the human dopamine D3 receptor was retrieved from the Protein Data Bank (PDB: 3PBL).49 Eticlopride was first removed from the complex. The 10 missing residues that connect helices V and VI (residues VAL213 to ARGR222) were modeled using MODELER 9.17.50 The receptor was then embedded into a lipid bilayer composed of 159 dioleoylphosphatidylcholine (DOPC), 141 sphingomyelin (SSM), and 110 cholesterol (CHOL) molecules. The system was solvated with 29 594 water molecules, and 8 Cl– ions were added to neutralize the system. The system was subjected to steepest descent minimization followed by consecutive NVT and NpT equilibration steps with the protein heavy atoms restrained. This relaxed membrane-embedded apo D3 receptor system served as the initial model for the rest of the simulations.
Molecular Dynamics: Unbiased Molecular Dynamics Simulations
All unbiased simulations reported in this work were performed using GROMACS 4.6.3,51 together with the OPLS all-atom (OPLS-AA) force field.52
Dopamine–D3 Receptor Complex Simulation
The dopamine–D3 receptor complex was constructed by docking dopamine into the binding pocket of the D3 receptor. The binding mode of dopamine was computationally determined using the docking software GOLD.53 The highest-ranking pose was selected and the receptor-dopamine complex was energy minimized to relieve steric and geometric strains. An unbiased simulation was performed in the NpT ensemble for 100 ns.
Spontaneous Dopamine Binding Simulations
To explore the spontaneous binding of dopamine to the membrane or the receptor from the water phase, a pool of 1000 simulations was prepared. In each simulation, a single dopamine molecule was introduced in a random orientation and position to the water phase (∼2 nm away from the membrane surface), starting from the relaxed simulation system of the apo D3 receptor embedded into the membrane. Each system was first energy minimized and, then, simulated for 20 ns.
Molecular Dynamics: Biased Molecular Dynamics Simulations
Random Acceleration Molecular Dynamics (RAMD) simulations
To identify the low energy exit pathways for dopamine from the receptor, the random acceleration molecular dynamics (RAMD)28,29 method implemented in NAMD 2.1054 was employed. Ten independent RAMD simulations were performed, with each initiated from the final configuration of 100 ns long unbiased simulation of the dopamine–D3 receptor complex. Seven acceleration values (accel parameter) were tested in the range of 10.5—0.4 kJ/(mol·nm). The smallest acceleration value (1.3 kJ/(mol·nm)) that allowed dopamine to exit the binding cavity within the first 5 ns of the simulations without distorting the receptor structure was chosen for the production RAMD simulations. For other RAMD parameters, the NAMD defaults were used: the number of steps in one RAMD block (ramdSteps) was set to 50, the threshold for the minimum distance traveled by the ligand in one RAMD block (rMinRamd) to 0.1 nm, and the distance between the centers of mass of the ligand and the protein for stopping the simulation (maxDist) to 5 nm.
Steered Molecular Dynamics (SMD) Simulations
Two sets of steered molecular dynamics (SMD) simulations were performed to probe (i) the vertical exit pathway from the binding pocket to the water phase and (ii) the lateral exit pathway from the binding pocket to the membrane. All SMD simulations were performed using GROMACS 4.6.3.
The vertical-route SMD simulations were initiated from the final configuration of the 100 ns long unbiased simulation of the dopamine–D3 receptor complex. The z-component of the distance between the centers of mass of the receptor and the dopamine molecule (receptor–dopamine distance) was used as the collective variable. The force constant and the velocity of the moving harmonic restraint was set to 1000 kJ/(mol·nm2) and 0.2 nm/ns, respectively.
The lateral-route SMD simulations were initiated from the final configuration of the single RAMD simulation that captured dopamine relocation into a membrane facing pocket between helices 5 and 6. The pulling was performed along the receptor–dopamine distance until the dopamine reached the membrane surface. The force constant and the velocity of the moving harmonic restraint was set to 1000 kJ/(mol·nm2) and 1 nm/ns, respectively.
Umbrella Sampling Simulations
The free energy profiles were calculated using the umbrella sampling method for both the lateral and the vertical routes described above. The umbrella sampling simulations were performed using GROMACS 4.6.3.51 The potential of mean force (PMF) profiles were reconstructed using the GROMACS tool g_wham.(55,56)
The Free Energy Profile for the Vertical Route
The z-component of the receptor–dopamine distance was used as the reaction coordinate. 32 umbrella windows were prepared with a spacing of 0.1 nm along this reaction coordinate covering a range from the binding pocket to the water phase. The initial configuration for each window was extracted from the vertical-route SMD simulation. For all umbrella windows, the force constant of the harmonic potential was set to 1000 kJ/(mol·nm2). The windows in which dopamine was inside the receptor were simulated for 100 ns, whereas those in which dopamine was in the water phase were simulated for 30 ns. The first 10 ns was discarded from each window before the analyses.
The Free Energy Profile for the Lateral Route
The receptor–dopamine distance was used as the reaction coordinate. 36 umbrella windows were prepared with a spacing of 0.1 nm spanning a range from the binding pocket to the membrane surface. The initial configurations were sampled from the lateral route RAMD and the SMD simulations. Each window was simulated for 100 ns with the force constant of the harmonic potential set to 1000 kJ/(mol·nm2). The first 50 ns from each umbrella window was discarded before the analyses.
Molecular Dynamics: Simulation Protocols
Unbiased, SMD, and umbrella sampling simulations were performed using GROMACS 4.6.3.51 The parametrization of all molecules was performed using the OPLS all-atom (OPLS-AA) force field.52,57,58 Recently developed models, including OPLS parameters specifically derived for lipids, were used.59−61 Partial charges of dopamine were adopted from a previous study.1 The TIP3P model that is compatible with the OPLS force field was employed for water.62
The equations of motion were integrated using a leapfrog algorithm with a 2 fs time step. All bonds were constrained using the LINCS algorithm.63 Periodic boundary conditions were used in all three dimensions. Electrostatic interactions were evaluated using the particle-mesh Ewald (PME) summation with a fourth-order (cubic) interpolation and direct sum tolerance of 10–5.64 A cutoff of 1 nm was applied for the real space and Lennard-Jones interactions. The list of nonbonded pairs was updated every 10 steps.
All production simulations were carried out in the isobaric–isothermal (NpT) ensemble; the reference temperature and pressure were set to 310 K and 1 bar, respectively. The temperature was controlled with the V-rescale thermostat,65 using a time constant of 0.1 ps. The pressure was controlled with a semi-isotropic scheme using the Parrinello–Rahman barostat with a time constant of 5 ps.66
RAMD simulations were performed using NAMD 2.10.54 The CHARMM36 force field was used for the lipids67 and the protein68 (with the φ/ψ cross term map (CMAP) corrections).69 For dopamine, CHARMM general force field parameters were generated with the CGneFF server (https://cgenff.umaryland.edu/commonFiles/aboutUs.php).70 A CHARMM compatible TIP3P model was used for water.62 In these calculations, we used different force fields due to a lack of OPLS-AA parameters for lipids in NAMD. Nevertheless, RAMD calculations were used only for qualitative observations, and free energy calculations performed with the OPLS-AA force field confirmed RAMD results.
The simulations were performed in the NpT ensemble using periodic boundary conditions in all three dimensions. An integration time step of 2 fs and bonds involving hydrogen atoms were constrained using the SHAKE algorithm.71 A cutoff of 12 Å was applied with a switching distance of 10 Å for nonbonded interactions. Long-range electrostatic interactions were calculated with the PME method with a grid spacing of 1 Å.72 The temperature was maintained at 310 K using Langevin dynamics with a damping coefficient of 1 ps–1 for non-hydrogen atoms. The pressure was maintained semi-isotopically at 1 atm with the Langevin piston Nosé–Hoover method73,74 using an oscillation period and a damping time scale of 200 fs.
Molecular Dynamics: Analysis
The statistical errors for the reconstructed free energy profiles were estimated with the Bayesian bootstrap method implemented in g_wham,75 using 200 bootstraps. The statistical error for the occupancy values reported in Table 1 was estimated analytically assuming a binomial distribution (1000 trials). The maximum error for any category falls below 1.5%.
Acknowledgments
This work was supported by the Academy of Finland (Center of Excellence program). I.V. also thanks Sigrid Juselius Foundation and the Helsinki Institute of Life Science (HiLIFE) Fellow program. For computational resources, we thank CSC – IT Centre for Science Ltd (Espoo, Finland). Experimental research was carried out with equipment purchased thanks to the financial support of the National Science Centre, Poland (Grant No. 2016/21/B/ST5/00250).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acschemneuro.9b00656.
Thermodynamic parameters of interactions between dopamine and lipids obtained in individual ITC measurements (PDF)
Author Contributions
◆ F.L., H.J., and A.Z. contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Postila P. A.; Vattulainen I.; Róg T. (2016) Selective Effect of Cell Membrane on Synaptic Neurotransmission. Sci. Rep. 6, 19345. 10.1038/srep19345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postila P. A.; Róg T. (2020) A Perspective: Active Role of Lipids in Neurotransmitter Dynamics. Mol. Neurobiol. 57, 910–925. 10.1007/s12035-019-01775-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orłowski A.; Grzybek M.; Bunker A.; Pasenkiewicz-Gierula M.; Vattulainen I.; Männistö P. T.; Róg T. (2012) Strong Preferences of Dopamine and l -Dopa towards Lipid Head Group: Importance of Lipid Composition and Implication for Neurotransmitter Metabolism. J. Neurochem. 122 (4), 681–690. 10.1111/j.1471-4159.2012.07813.x. [DOI] [PubMed] [Google Scholar]
- Jodko-Piorecka K.; Litwinienko G. (2013) First Experimental Evidence of Dopamine Interactions with Negatively Charged Model Biomembranes. ACS Chem. Neurosci. 4 (7), 1114–1122. 10.1021/cn4000633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matam Y.; Ray B. D.; Petrache H. I. (2016) Direct Affinity of Dopamine to Lipid Membranes Investigated by Nuclear Magnetic Resonance Spectroscopy. Neurosci. Lett. 618, 104–109. 10.1016/j.neulet.2016.02.052. [DOI] [PubMed] [Google Scholar]
- Das S.; Purkayastha P. (2017) A Fluorescence Lifetime Imaging Microscopy Supported Investigation on Temperature-Dependent Penetration of Dopamine in a 1,2-Ditetradecanoyl- Sn -Glycero-3-Phospho-(1′- Rac -Glycerol) Lipid Bilayer. Langmuir 33 (29), 7281–7287. 10.1021/acs.langmuir.7b01173. [DOI] [PubMed] [Google Scholar]
- Peters G. H.; Wang C.; Cruys-Bagger N.; Velardez G. F.; Madsen J. J.; Westh P. (2013) Binding of Serotonin to Lipid Membranes. J. Am. Chem. Soc. 135 (6), 2164–2171. 10.1021/ja306681d. [DOI] [PubMed] [Google Scholar]
- Drolle E.; Kučerka N.; Hoopes M. I.; Choi Y.; Katsaras J.; Karttunen M.; Leonenko Z. (2013) Effect of Melatonin and Cholesterol on the Structure of DOPC and DPPC Membranes. Biochim. Biophys. Acta, Biomembr. 1828 (9), 2247–2254. 10.1016/j.bbamem.2013.05.015. [DOI] [PubMed] [Google Scholar]
- Choi Y.; Attwood S. J.; Hoopes M. I.; Drolle E.; Karttunen M.; Leonenko Z. (2014) Melatonin Directly Interacts with Cholesterol and Alleviates Cholesterol Effects in Dipalmitoylphosphatidylcholine Monolayers. Soft Matter 10 (1), 206–213. 10.1039/C3SM52064A. [DOI] [PubMed] [Google Scholar]
- Dies H.; Cheung B.; Tang J.; Rheinstädter M. C. (2015) The Organization of Melatonin in Lipid Membranes. Biochim. Biophys. Biochim. Biophys. Acta, Biomembr. 1848 (4), 1032–1040. 10.1016/j.bbamem.2015.01.006. [DOI] [PubMed] [Google Scholar]
- Wang C.; Ye F.; Velardez G. F.; Peters G. H.; Westh P. (2011) Affinity of Four Polar Neurotransmitters for Lipid Bilayer Membranes. J. Phys. Chem. B 115 (1), 196–203. 10.1021/jp108368w. [DOI] [PubMed] [Google Scholar]
- Pérez-Isidoro R.; Ruiz-Suárez J. C. (2016) Calcium and Protons Affect the Interaction of Neurotransmitters and Anesthetics with Anionic Lipid Membranes. Biochim. Biophys. Biochim. Biophys. Acta, Biomembr. 1858 (9), 2215–2222. 10.1016/j.bbamem.2016.06.017. [DOI] [PubMed] [Google Scholar]
- Rangel-barajas C.; Coronel I.; Floran B. (2015) Dopamine Receptors and Neurodegeneration. Aging Dis. 6 (5), 349–368. 10.14336/AD.2015.0330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levant B. (1997) The D 3 Dopamine Receptor: Neurobiology and Potential Clinical Relevance. Pharmacol. Rev. 49, 231–252. [PubMed] [Google Scholar]
- Groman S. M.; Smith N. J.; Petrullli J. R.; Massi B.; Chen L.; Ropchan J.; Huang Y.; Lee X. D.; Morris E. D.; Taylor J. R. (2016) Dopamine D 3 Receptor Availability Is Associated with Inflexible Decision Making. J. Neurosci. 36 (25), 6732–6741. 10.1523/JNEUROSCI.3253-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fresnoza S.; Stiksrud E.; Klinker X. F.; Liebetanz D.; Paulus W.; Kuo M.; Nitsche M. A. (2014) Dosage-Dependent Effect of Dopamine D 2 Receptor Activation on Motor Cortex Plasticity in Humans. J. Neurosci. 34 (32), 10701–10709. 10.1523/JNEUROSCI.0832-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arias-Carrion O.; Stamelou M.; Murillo-Rodríguez E.; Menéndez-Gonzlez M.; Pöppel E. (2010) Dopaminergic Reward System: A Short Integrative Review. Int. Arch. Med. 3 (1), 24. 10.1186/1755-7682-3-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wise R. A. (2004) Dopamine, Learning and Motivation. Nat. Rev. Neurosci. 5 (6), 483–494. 10.1038/nrn1406. [DOI] [PubMed] [Google Scholar]
- Van Meer G.; Voelker D. R.; Feigenson G. W. (2008) Membrane Lipids: Where They Are and How They Behave. Nat. Rev. Mol. Cell Biol. 9 (2), 112–124. 10.1038/nrm2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lingwood D.; Simons K. (2012) Lipid Rafts As a Membrane-Organizing Principle. Science 327 (5961), 46–50. 10.1126/science.1174621. [DOI] [PubMed] [Google Scholar]
- Heerklotz H. (2004) The Microcalorimetry of Lipid Membranes. J. Phys.: Condens. Matter 16, R441. 10.1088/0953-8984/16/15/R01. [DOI] [Google Scholar]
- Heerklotz H.; Seelig J. (2000) Titration Calorimetry of Surfactant-Membrane Partitioning and Membrane Solubilization. Biochim. Biophys. Acta, Biomembr. 1508, 69–85. 10.1016/S0304-4157(00)00009-5. [DOI] [PubMed] [Google Scholar]
- Indyk L.; Fisher H. F. (1998) Theoretical Aspects of Isothermal Titration Calorimetry. Methods Enzymol. 295, 350–364. 10.1016/S0076-6879(98)95048-0. [DOI] [PubMed] [Google Scholar]
- Kepczynski M.; Kumorek M.; Stepniewski M.; Róg T.; Kozik B.; Jamróz D.; Bednar J.; Nowakowska M. (2010) Behavior of 2,6-Bis(Decyloxy)Naphthalene inside Lipid Bilayer. J. Phys. Chem. B 114 (47), 15483–15494. 10.1021/jp103753f. [DOI] [PubMed] [Google Scholar]
- Lukat G.; Krüger J.; Sommer B. (2013) APL@Voro: A Voronoi-Based Membrane Analysis Tool for GROMACS Trajectories. J. Chem. Inf. Model. 53 (11), 2908–2925. 10.1021/ci400172g. [DOI] [PubMed] [Google Scholar]
- Enkavi G.; Javanainen M.; Kulig W.; Róg T.; Vattulainen I. (2019) Multiscale Simulations of Biological Membranes: The Challenge to Understand Biological Phenomena in a Living Substance. Chem. Rev. 119, 5607–5774. 10.1021/acs.chemrev.8b00538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey W.; Dalke A.; Schulten K. (1996) VMD-Visual Molecular Dynamics. J. Mol. Graphics 14, 33–38. 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- Lüdemann S. K.; Lounnas V.; Wade R. C. (2000) How Do Substrates Enter and Products Exit the Buried Active Site of Cytochrome P450cam? 1. Random Expulsion Molecular Dynamics Investigation of Ligand Access Channels and Mechanisms. J. Mol. Biol. 303 (5), 797–811. 10.1006/jmbi.2000.4154. [DOI] [PubMed] [Google Scholar]
- Carlsson P.; Burendahl S.; Nilsson L. (2006) Unbinding of Retinoic Acid from the Retinoic Acid Receptor by Random Expulsion Molecular Dynamics. Biophys. J. 91 (9), 3151–3161. 10.1529/biophysj.106.082917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klvana M.; Pavlova M.; Koudelakova T.; Chaloupkova R.; Dvorak P.; Prokop Z.; Stsiapanava A.; Kuty M.; Kuta-Smatanova I.; Dohnalek J.; Kulhanek P.; Wade R. C.; Damborsky J. (2009) Pathways and Mechanisms for Product Release in the Engineered Haloalkane Dehalogenases Explored Using Classical and Random Acceleration Molecular Dynamics Simulations. J. Mol. Biol. 392 (5), 1339–1356. 10.1016/j.jmb.2009.06.076. [DOI] [PubMed] [Google Scholar]
- Kalyaanamoorthy S.; Chen Y. P. P. (2012) Exploring Inhibitor Release Pathways in Histone Deacetylases Using Random Acceleration Molecular Dynamics Simulations. J. Chem. Inf. Model. 52 (2), 589–603. 10.1021/ci200584f. [DOI] [PubMed] [Google Scholar]
- Mokkila S.; Postila P. A.; Rissanen S.; Juhola H.; Vattulainen I.; Róg T. (2017) Calcium Assists Dopamine Release by Preventing Aggregation on the Inner Leaflet of Presynaptic Vesicles. ACS Chem. Neurosci. 8 (6), 1242–1250. 10.1021/acschemneuro.6b00395. [DOI] [PubMed] [Google Scholar]
- Sabatini B. L.; Regehr W. G. (1999) Timing of Synaptic Transmission. Annu. Rev. Physiol. 61 (1), 521–542. 10.1146/annurev.physiol.61.1.521. [DOI] [PubMed] [Google Scholar]
- Koopmans F.; Pandya N. J.; Franke S. K.; Phillippens I. H.C.M.H.; Paliukhovich I.; Li K. W.; Smit A. B. (2018) Comparative Hippocampal Synaptic Proteomes of Rodents and Primates: Differences in Neuroplasticity-Related Proteins. Front. Mol. Neurosci. 11, 364. 10.3389/fnmol.2018.00364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hondius D. C.; van Nierop P.; Li K. W.; Hoozemans J. J.M.; van der Schors R. C.; van Haastert E. S.; van der Vies S. M.; Rozemuller A. J.M.; Smit A. B. (2016) Profiling the Human Hippocampal Proteome at All Pathologic Stages of Alzheimer ’ s Disease. Alzheimer's Dementia 12 (6), 654–668. 10.1016/j.jalz.2015.11.002. [DOI] [PubMed] [Google Scholar]
- Mason R. P.; Rhodes D. G.; Herbette L. G. (1991) Reevaluating Equilibrium and Kinetic Binding Parameters for Lipophilic Drugs Based on a Structural Model for Drug Interaction with Biological Membranes. J. Med. Chem. 34 (3), 869–877. 10.1021/jm00107a001. [DOI] [PubMed] [Google Scholar]
- Rhodes D. G.; Sarmiento J. G.; Herbette L. G. (1985) Kinetics of Binding of Membrane-Active Drugs to Receptor Sites. Diffusion-limited Rates for a Membrane Bilayer Approach of 1,4-dihydropyridine Calcium Channel Antagonists to Their Active Site. Mol. Pharmacol. 27, 612–623. [PubMed] [Google Scholar]
- Vauquelin G.; Packeu A. (2009) Ligands, Their Receptors and··· Plasma Membranes. Mol. Cell. Endocrinol. 311 (1–2), 1–10. 10.1016/j.mce.2009.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes D.; Jakobtorweihen S.; Nunes C.; Sarmento B.; Reis S. (2017) Shedding Light on the Puzzle of Drug-Membrane Interactions: Experimental Techniques and Molecular Dynamics Simulations. Prog. Lipid Res. 65, 24–44. 10.1016/j.plipres.2016.12.001. [DOI] [PubMed] [Google Scholar]
- Wang T.; Duan Y. (2009) Ligand Entry and Exit Pathways in the Β2-Adrenergic Receptor. J. Mol. Biol. 392 (4), 1102–1115. 10.1016/j.jmb.2009.07.093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurst D. P.; Grossfield A.; Lynch D. L.; Feller S.; Romo T. D.; Gawrisch K.; Pitman M. C.; Reggio P. H. (2010) A Lipid Pathway for Ligand Binding Is Necessary for a Cannabinoid G Protein-Coupled Receptor. J. Biol. Chem. 285 (23), 17954–17964. 10.1074/jbc.M109.041590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildebrand P. W.; Scheerer P.; Park J. H.; Choe H. W.; Piechnick R.; Ernst O. P.; Hofmann K. P.; Heck M. (2009) A Ligand Channel through the G Protein Coupled Receptor Opsin. PLoS One 4 (2), e4382 10.1371/journal.pone.0004382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanna M. G.; Cahalan S. M.; Han G. W.; Schuerer S. C.; Kuhn P.; Stevens R. C.; Desale H.; Scott F. L.; Clemons B.; Griffith M. T.; Jo E.; Reinhart G.; Rosen H.; Roth C. B.; Hanson M. A. (2012) Crystal Structure of a Lipid G Protein-Coupled Receptor. Science 335 (6070), 851–855. 10.1126/science.1215904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sykes D. A.; Parry C.; Reilly J.; Wright P.; Fairhurst R. A.; Charlton S. J. (2014) Observed Drug-Receptor Association Rates Are Governed by Membrane Affinity: The Importance of Establishing “Micro-Pharmacokinetic/Pharmacodynamic Relationships” at the Β2 -Adrenoceptors. Mol. Pharmacol. 85, 608–617. 10.1124/mol.113.090209. [DOI] [PubMed] [Google Scholar]
- Juhola H.; Postila P. A.; Rissanen S.; Lolicato F.; Vattulainen I.; Róg T. (2018) Negatively Charged Gangliosides Promote Membrane Association of Amphipathic Neurotransmitters. Neuroscience 384, 214–223. 10.1016/j.neuroscience.2018.05.035. [DOI] [PubMed] [Google Scholar]
- Eckhardt M. (2008) The Role and Metabolism of Sulfatide in the Nervous System. Mol. Neurobiol. 37 (2–3), 93–103. 10.1007/s12035-008-8022-3. [DOI] [PubMed] [Google Scholar]
- Takahashi T.; Suzuki T. (2012) Role of Sulfatide in Normal and Pathological Cells and Tissues. J. Lipid Res. 53 (8), 1437–1450. 10.1194/jlr.R026682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lingwood C. A. (2011) Glycosphingolipid Functions. Cold Spring Harbor Perspect. Biol. 3 (7), a004788. 10.1101/cshperspect.a004788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien E. Y. T.; Liu W.; Zhao Q.; Katritch V.; Won Han G.; Hanson M. A.; Shi L.; Hauck Newman A.; Javitch J. A.; Cherezov V.; Stevens S. C. (2010) Structure of the Human Dopamine D3 Receptor. Science 330, 1091–1095. 10.1126/science.1197410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiser A.; Do R. K. G.; Šali A. (2000) Modeling of Loops in Protein Structures. Protein Sci. 9 (9), 1753–1773. 10.1110/ps.9.9.1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pronk S.; Páll S.; Schulz R.; Larsson P.; Bjelkmar P.; Apostolov R.; Shirts M. R.; Smith J. C.; Kasson P. M.; Van Der Spoel D.; Hess B.; Lindahl E. (2013) GROMACS 4.5: A High-Throughput and Highly Parallel Open Source Molecular Simulation Toolkit. Bioinformatics 29 (7), 845–854. 10.1093/bioinformatics/btt055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaminski G. A.; Friesner R. A.; Tirado-Rives J.; Jorgensen W. L. (2001) Evaluation and Reparametrization of the OPLS-AA Force Field for Proteins via Comparison with Accurate Quantum Chemical Calculations on Peptides. J. Phys. Chem. B 105 (28), 6474–6487. 10.1021/jp003919d. [DOI] [Google Scholar]
- Verdonk M.; Cole J.; Hartshorn M.; Murray C.; Taylor R. (2003) Improved Protein-Ligand Docking Using GOLD. Proteins: Struct., Funct., Genet. 52, 609–623. 10.1002/prot.10465. [DOI] [PubMed] [Google Scholar]
- Phillips J. C.; Braun R.; Wang W.; Gumbart J.; Tajkhorshid E.; Villa E.; Chipot C.; Skeel R. D.; Kalé L.; Schulten K. (2005) Scalable Molecular Dynamics with NAMD. J. Comput. Chem. 26 (16), 1781–1802. 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitschke N.; Atkovska K.; Hub J. S. (2016) Accelerating Potential of Mean Force Calculations for Lipid Membrane Permeation: System Size, Reaction Coordinate, Solute-Solute Distance, and Cutoffs. J. Chem. Phys. 145 (12), 125101. 10.1063/1.4963192. [DOI] [PubMed] [Google Scholar]
- Hub J. S.; Winkler F. K.; Merrick M.; de Groot B. L. (2010) Potentials of Mean Force and Permeabilities for Carbon Dioxide, Ammonia, and Water Flux across a Rhesus Protein Channel and Lipid Membranes. J. Am. Chem. Soc. 132 (38), 13251–13263. 10.1021/ja102133x. [DOI] [PubMed] [Google Scholar]
- Jorgensen W. L.; Tirado-Rives J. (1988) The OPLS Potential Functions for Proteins. Energy Minimizations for Crystals of Cyclic Peptides and Crambin. J. Am. J. Am. Chem. Soc. 110 (6), 1657–1666. 10.1021/ja00214a001. [DOI] [PubMed] [Google Scholar]
- Jorgensen W. L.; Maxwell D. S.; Tirado-Rives J. (1996) Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. J. Am. Chem. Soc. 118 (45), 11225–11236. 10.1021/ja9621760. [DOI] [Google Scholar]
- Maciejewski A.; Pasenkiewicz-Gierula M.; Cramariuc O.; Vattulainen I.; Róg T. (2014) Refined OPLS-AA Force Field for Saturated Phosphatidylcholine Bilayers at Full Hydration. J. Phys. Chem. B 118 (17), 4571–4581. 10.1021/jp5016627. [DOI] [PubMed] [Google Scholar]
- Kulig W.; Pasenkiewicz-Gierula M.; Róg T. (2015) Topologies, Structures and Parameter Files for Lipid Simulations in GROMACS with the OPLS-Aa Force Field: DPPC, POPC, DOPC, PEPC, and Cholesterol. Data Br. 5, 333–336. 10.1016/j.dib.2015.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulig W.; Pasenkiewicz-Gierula M.; Róg T. (2016) Cis and Trans Unsaturated Phosphatidylcholine Bilayers: A Molecular Dynamics Simulation Study. Chem. Phys. Lipids 195, 12–20. 10.1016/j.chemphyslip.2015.07.002. [DOI] [PubMed] [Google Scholar]
- Jorgensen W. L.; Chandrasekhar J.; Madura J. D.; Impey R. W.; Klein M. L. (1983) Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 79 (2), 926–935. 10.1063/1.445869. [DOI] [Google Scholar]
- Hess B.; Bekker H.; Berendsen H. J. C.; Fraaije J. G. E. M. (1997) LINCS: A Linear Constraint Solver for Molecular Simulations. J. Comput. Chem. 18 (12), 1463–1472. 10.1002/(SICI)1096-987X(199709)18:12<1463::AID-JCC4>3.0.CO;2-H. [DOI] [Google Scholar]
- Essmann U.; Perera L.; Berkowitz M. L.; Darden T.; Lee H.; Pedersen L. G. (1995) A Smooth Particle Mesh Ewald Method. J. Chem. Phys. 103 (19), 8577–8593. 10.1063/1.470117. [DOI] [Google Scholar]
- Bussi G.; Donadio D.; Parrinello M. (2007) Canonical Sampling through Velocity Rescaling. J. Chem. Phys. 126, 014101 10.1063/1.2408420. [DOI] [PubMed] [Google Scholar]
- Parrinello M.; Rahman A. (1981) Polymorphic Transitions in Single Crystals: A New Molecular Dynamics Method. J. Appl. Phys. 52 (12), 7182–7190. 10.1063/1.328693. [DOI] [Google Scholar]
- Klauda J. B.; Venable R. M.; Freites J. A.; O’Connor J. W.; Tobias D. J.; Mondragon-Ramirez C.; Vorobyov I.; MacKerell A. D.; Pastor R. W. (2010) Update of the CHARMM All-Atom Additive Force Field for Lipids: Validation on Six Lipid Types. J. Phys. Chem. B 114 (23), 7830–7843. 10.1021/jp101759q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J.; Jr MacKerell A. D. (2013) CHARMM36 All-atom Additive Protein Force Field: Validation Based on Comparison to NMR Data. J. Comput. Chem. 34, 2135–2145. 10.1002/jcc.23354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackerell A. D.; Feig M.; Brooks C. L. (2004) Extending the Treatment of Backbone Energetics in Protein Force Fields: Limitations of Gas-Phase Quantum Mechanics in Reproducing Protein Conformational Distributions in Molecular Dynamics Simulation. J. Comput. Chem. 25 (11), 1400–1415. 10.1002/jcc.20065. [DOI] [PubMed] [Google Scholar]
- Vanommeslaeghe K.; Hatcher E.; Acharya C.; Kundu S.; Zhong S.; Shim J.; Darian E.; Guvench O.; Lopes P.; Vorobyov I.; MacKerell A. D. Jr. (2011) CHARMM General Force Field (CGenFF): A Force Field for Drug-like Molecules Compatible with the CHARMM All-Atom Additive Biological Force Fields. J. Comput. Chem. 31, 671–690. 10.1002/jcc.21367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryckaert J.-P.; Ciccotti G.; Berendsen H. J. C. (1977) Numerical Integration of the Cartesian Equations of Motion of a System with Constraints: Molecular Dynamics of n-Alkanes. J. Comput. Phys. 23, 327–341. 10.1016/0021-9991(77)90098-5. [DOI] [Google Scholar]
- Darden T.; York D.; Pedersen L. (1993) Particle Mesh Ewald: An N ·log(N) Method for Ewald Sums in Large Systems. J. Chem. Phys. 98 (12), 10089–10092. 10.1063/1.464397. [DOI] [Google Scholar]
- Feller S. E.; Zhang Y.; Pastor R. W.; Brooks B. R. (1995) Constant Pressure Molecular Dynamics Simulation: The Langevin Piston Method. J. Chem. Phys. 103, 4613–4621. 10.1063/1.470648. [DOI] [Google Scholar]
- Martyna G. J.; Tobias D. J.; Klein M. L. (1994) Constant Pressure Molecular Dynamics Algorithms. J. Chem. Phys. 101, 4177–4189. 10.1063/1.467468. [DOI] [Google Scholar]
- Hub J. S.; De Groot B. L.; Van Der Spoel D. (2010) G_wham A Free Weighted Histogram Analysis Implementation Including Robust Error and Autocorrelation Estimates. J. Chem. Theory Comput. 6, 3713–372. 10.1021/ct100494z. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.