

Abstract
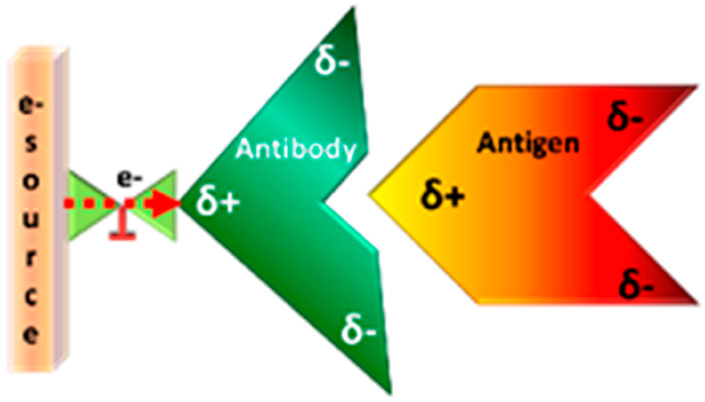
A new mechanism of allostery in proteins, based on charge rather than structure, is reported. We demonstrate that dynamic redistribution of charge within a protein can control its function and affect its interaction with a binding partner. In particular, the association of an antibody with its target protein antigen is studied. Dynamic charge shifting within the antibody during its interaction with the antigen is enabled by its binding to a metallic surface that serves as a source for electrons. The kinetics of antibody–antigen association are enhanced when charge redistribution is allowed, even though charge injection happens at a position far from the antigen binding site. This observation points to charge-reorganization allostery, which should be operative in addition or parallel to other mechanisms of allostery, and may explain some current observations on protein interactions.
Introduction
There is a well-established relation between structure and function in proteins, which is supported by multiple experimental and theoretical tools. An important tenet of the structure–function paradigm is the allosteric effect,1 i.e. the modulation of the function of a protein through the binding of a small molecule or of another protein at a location far away from the active site.2−4 Classical allosteric mechanisms involve protein conformational changes,5,6 and more recently it has been shown that changes in conformational dynamics may also lead to allosteric effects.7 Protein–protein interactions (PPA) are ubiquitous in living systems and are often subject to allosteric modulation. It has been known for a long time that PPA may involve electrostatic interactions between charged groups located at the interaction site, and may modify the diffusion-limited association rate in a multiplicative manner.8−11 Theoretical analysis of PPA often takes into account the static distribution of charges on the surfaces of the proteins, which is related to the location of charged amino acid residues.11 In this work, it is found that the electrostatic effect can be nonlocal and can be controlled by the ability of the protein to withdraw charge at sites remote to its reaction site and redistribute these charges throughout its structure. This leads to a new allosteric mechanism, charge-reorganization allostery, which must accompany any situation in which a protein is interacts with another species or is exposed to an electric field within the biological environment.
We tested the effect of charge reorganization on PPA kinetics by studying a model system, as shown in Figure 1. An antibody is attached through a linker to a metal surface that serves as a source for charge. The antibody recognizes a polyhistidine tag (His-tag) attached to another protein, the antigen (ClpB from Thermus thermophilus). By controlling the charge flow between the antibody and the surface, we can modulate the rate of association of the antigen (His-tagged ClpB), which takes place at a site far away from the antibody’s adsorption site. We tested that the effect observed here is indeed due to charge flow (and not purely structural) by controlling charge motion using a virtual “valve” (see Figure 1). This particular “valve” is based on the Chiral Induced Spin Selectivity (CISS) effect (see Supplementary Text),12 which relates to the ability of charge to flow through a chiral molecule (the protein) due to electronic spin. By changing the direction of the magnetic dipole of the substrate (or the chirality of a linker molecule, see below), we enable or disable charge injection. To prove that the CISS effect is operative with the anti-His antibody used in this study, we adsorbed the protein on a Hall device (see Supplementary Text for details, Figure S1). Upon applying an external electric field, we observed a Hall voltage that indicates spin-selective charge flow. This allows us to prove unequivocally that the modulation of the association kinetics arises from charge reorganization within the protein, which is a novel allosteric mechanism.
Figure 1.
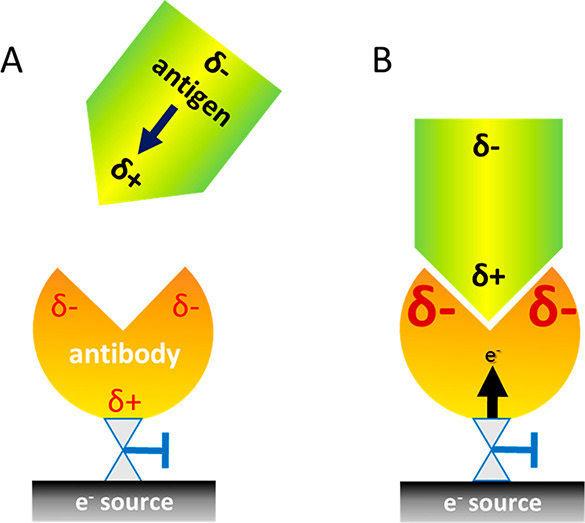
Scheme of the experimental system. (A) When an antigen with a dipole moment is approaching the antibody, it induces charge reorganization in the antibody. δ- and δ+ designate negative and positive charge accumulation, respectively. (B) The rate at which the antigen binds to the antibody is modulated by charge reorganization within the antibody. This novel allosteric effect is facilitated here by the motion of charge from the “electron source” into the antibody, but it can operate under any setup where a protein is inserted into an external electric field.
Results and Discussion
We measured the rate of binding of a His-tagged variant of ClpB to the anti-His antibody attached to a magnetized metal surface, which served as a source of electrons. In particular, the antibody was adsorbed on a gold-coated Ni surface (2 nm Au on top of 120 nm Ni) using dithiobis[succinimidyl]propionate (DSP) as a linker. The His-tagged ClpB could interact with the antibody in two configurations, through either its C terminus or its N terminus (Figure 2), depending on the location of the His-tag. Notably, the direction of the dipole moment of the antigen (which was calculated using the Protein Dipole Moments Server)13 with respect to the antibody was reversed when it was bound through the C terminus or the N terminus. The rate of binding of His-tagged ClpB molecules was measured with the substrate magnetized with the North magnetic pole pointing either UP (H+) or DOWN (H−), which was achieved by changing the polarity of a magnet. (Relevant control experiments are presented in Figure S2.)
Figure 2.

Effect of surface magnetization on the kinetics of antigen–antibody binding. (A) Fluorescence microscope images of individual complexes formed between adsorbed anti-His antibodies and ClpB molecules His-tagged either at the C terminus (C-ter) or at the N terminus (N-ter). The interaction time is 2 s, with the North magnetic pole of the magnetized substrate pointing either UP (H+) or DOWN (H−) and also without the magnet as a control. For the C-terminal His-tag the number of adsorbed antigen molecules is larger for H+ while for the N-terminal His-tag the number is larger for H–. The number of molecules is calculated as described in the Methods section (Figure S3). (B) Reaction kinetics of the antibody with C-terminal His-tagged ClpB under the two magnetic orientations. (C) Reaction kinetics of the antibody with N-terminal His-tagged ClpB under the two magnetic orientations. (D) Schematic representation of the mechanism of the effect of spin on the antigen–antibody interaction. When the spin on the ferromagnet is pointing opposite to the momentary spin of the charge at the interface of the antibody and the ferromagnetic, charge flows more efficiently between the antibody and the surface. The charge flow facilitates charge redistribution in the antibody, which in turn increases the antigen–antibody binding rate.
As revealed by Figure 2A and 2B, it was found that when ClpB binds through a C-terminal His-tag, the binding to the antibody is faster for the magnetic field pointing up versus down, while the opposite is true for the case that ClpB binds through an N-terminal His-tag (Figures 2A and 2C). No difference is observed between N- and C-terminal His-tagged proteins in the absence of a magnetic field (Figure 2A). Further, the difference is only seen in the kinetics of binding and vanishes when the system reaches equilibrium.
To understand the inversion of the relative binding kinetics with the magnet direction and the position of the His-tag, we note that ClpB carries a net dipole moment with a significant projection along the axis connecting its two termini. When the direction of interaction of ClpB with the antibody is inverted, the dipole moment direction is also inverted. Therefore, while in the case of binding through the C-terminus, which is negatively charged, electrons are displaced through the antibody toward the surface, in the case of N-terminal binding (positively charged), electrons are displaced away from the surface toward the protein; that is, the sign of the displacement current changes in the two cases.
Importantly, in each direction of the protein dipole, the rate of charge flow from the ferromagnetic surface into the chiral antibody depends on the direction of the magnetization of the ferromagnet (Figure 2).14 Based on the CISS effect15 (Figure 2D), in one magnetization direction charge flows more readily into the antibody when the antigen is approaching, changing the charge distribution within the antibody and thereby modulating the antibody–antigen association rate. Put more simply, for one spin orientation the connection of the antibody to the charge reservoir in the substrate is better than that for the other spin orientation, allowing for more facile charge flow.14 When more electrons flow into or away from the antibody, its internal charge distribution is dynamically modulated in a way that enhances its interaction with the antigen, thereby increasing the rate of binding between the two. This experiment therefore establishes modulation of charge distribution as a means to allosterically affect PPA kinetics, an effect we term charge-reorganization allostery.
It is important to appreciate that the spin associated with the ferromagnetic substrate cannot affect charge reorganization within the biomolecule without actually transferring spin-polarized charge. The only way a ferromagnet can affect spin in a nonmagnetic adsorbate without charge flow is by the proximity effect, which cannot be extended beyond the range of a few C–C bonds.16 Hence, the spin control discussed here must result from charge transfer between the magnetic surface and the protein.
To exhibit that the effect is indeed related to charge changes within the proteins rather than to some other involvement of the ferromagnetic surface, we investigated antigen–antibody association kinetics when the antibody is connected to a nonmagnetic gold substrate through a chiral amino acid, either l-cysteine or d-cysteine (Figure 3A). As shown in Figure 3B, the rate of PPA is larger when the antibody is bound to the gold through d-cysteine rather than l-cysteine. This finding indicates that the chiral linker replaces the direct interaction of the ferromagnetic surface and the protein. The scheme in Figure 3C shows that in the case of d-cysteine the preferred spin of the electrons transmitted through the linker to the antibody is such that it is antiparallel to the spin polarization in the positive pole of the antibody. As a result, and based on the CISS effect, electrons can penetrate into the antibody and strengthen its electrostatic interaction with the antigen. In the case of the l-cysteine, the electrons that have to pass through the molecule have a preferred spin that is parallel to the spin polarization on the positive pole of the antibody; therefore these electrons penetrate more slowly into the antibody. The chiral linker molecules and the ferromagnetic substrate, used in the experiment of in Figure 2, are functioning similarly as valves that regulate electron flow from the substrate into the adsorbed protein. The similarity in the effect between the two “valves” demonstrates that we are probing here the response of the proteins themselves to charge redistribution.
Figure 3.
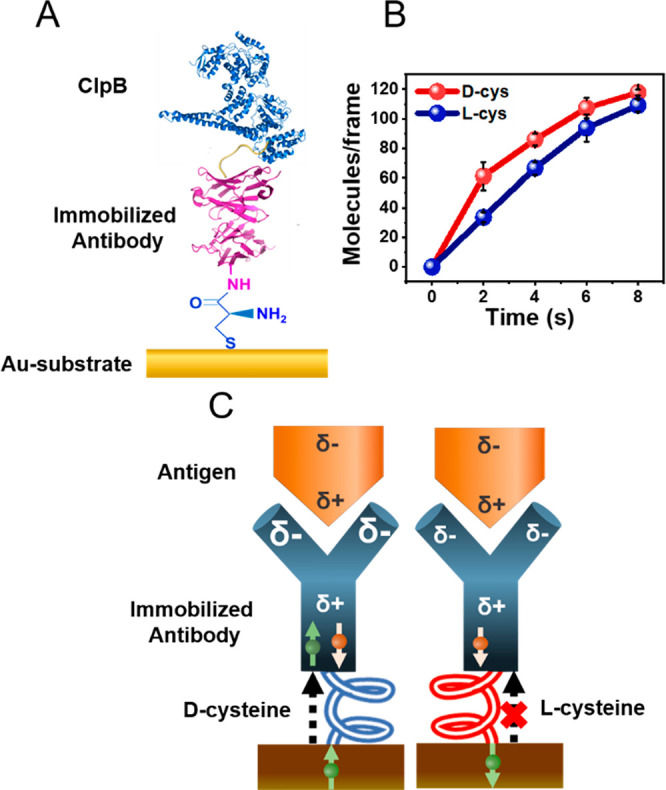
Kinetics of binding of the antigen to the antibody when bound to a gold substrate through a chiral molecule. (A) Schematic showing an anti-His antibody attached to a gold substrate via either a d- or an l-cysteine linker. The surface is exposed to a solution containing the His-tagged antigen protein. (B) Number of antibody–antigen complexes as a function of time when the antibody is linked to the surface via d-cysteine (red) or l-cysteine (blue). (C) A model for the effect of the handedness of the chiral linker on the association rate (see details in text).
Since the valves controlling electron flow into the adsorbed antibody molecules depend on spin, one may wonder whether electron spin may have a role in modulating PPA. To probe this question, short oligopeptides, HS-CH2CH2CO-(l-Ala-Aib)8-His and HS-CH2CH2CO-(d-Ala-Aib)8-His, referred to as l-PAL and d-PAL, respectively, were adsorbed on a ferromagnetic substrate (Ni/Au film, as above) through a thiol group on their N-termini, while their C-termini were His-tagged. The handedness of the oligopeptides determines the preferred spin direction for electrons that flow through them. Spin flows more readily through d-PAL when the magnet is pointing up, and through l-PAL when the magnet is pointing down. The oligopeptides serve as antigens for anti-His antibodies from the solution.
In this experiment, the surfaces coated with either l-PAL or d-PAL were exposed to the antibodies in the presence of a magnetic field that pointed either up or down. The results (Figure 4A and B) indicate that the binding between the oligopeptide and the antibody is faster when the electron transport through the oligopeptide is efficient. Namely, for d-PAL with the magnet pointing UP and for the l-PAL with the magnet pointing DOWN the charge injection is efficient and results in fast binding of the antibody. This is despite the spin being opposite in sign in the two cases. Hence, spin orientation does not control the rate of the association process occurring through electrostatic interaction. Rather, it is the amount of charge that reaches the binding region within the proteins that is dominant. Hence, we conclude that the effect of the binding to the antigen is purely electrostatic and does not depend on spin alignment.
Figure 4.

Association of the antibody to d- or l- His-tagged polyalanine (PAL) adsorbed on a ferromagnetic substrate. (A) Fluorescence microscopy images of individual complexes of His-tagged l-PAL or d-PAL molecules with anti-His antibodies. The reaction was carried out for 2 s with two substrate magnetization directions. (B) Histogram of the number of antigen–antibody complexes formed (averaged over 10 frames) with the magnetization pointing either Up or Down. (C) Schematic of the effect of the direction of magnetization of the substrate for right- or left-handed PAL. Note that the spin alignment is opposite to the direction of the magnet. Starting from the left, when the spins in the ferromagnet are aligned so that their direction coincides with the preferred spin transport through the chiral PAL, charge moves from the ferromagnetic substrate into the PAL monolayer, modulating the electrostatic interaction with the antibody and making complex formation faster. When the spins in the ferromagnet are oriented in the opposite direction, charge flows less efficiently, as this spin orientation is not the preferred one, and electron transfer through the l-PAL is slow. For d-PAL molecules, shown schematically on the right, the preferred spin is opposite to the spin that passes through l-PAL and therefore a faster association is observed for the opposite magnet orientation.
Conclusion
The present work points to a new mechanism for controlling protein activity, charge-redistribution allostery. We demonstrated here that the electrostatic interaction at the binding site of two proteins can be modulated by charge injection at a position remote to the binding site. We used the CISS effect to control the charge flow between metal substrates (ferromagnets) and adsorbed molecules that present recognition sites for His-tagged ClpB molecules. However, we showed that the effect does not depend on the spin of electrons flowing from the surface into the antibody, but rather on the charge redistribution within the protein. Our results indicate the importance of polarizability, namely the response of the charge in the molecule to external electric field, in controlling PPA in particular, and allosteric interactions in general. The observations highlight the fact that even parts of a protein that are remote to its active site may contribute to its activity by serving as a “reservoir” for charge and thereby regulating the overall protein polarizability. The new mechanism does not exclude conformational changes that are accompanied by the charge redistribution.
Electric field effects are ubiquitous in biology.17,18 They have been observed on multiple levels, from the organismal level, e.g. the development of morphological asymmetries, down to the molecular level, e.g. voltage-gated ion channels that open and close in response to changes in electric field.19 The powerful role of electric fields at the active site of an enzyme has been demonstrated recently.20 The new polarizability-related allosteric mechanism, introduced in this paper, is unique, as it involves dynamic rearrangement of charge throughout a protein. The new mechanism implies that an electric field acting on a protein may affect its activity due to its electric polarizability. This phenomenon may be quite abundant and should be found wherever electric field gradients are operative within the cell or the organism, as future experiments are likely to demonstrate.
Materials and Methods
Microscopy Setup and Data Analysis
The fluorescence imaging of the samples was carried out using a home-built total internal reflection fluorescence microscope (TIRFM). A detailed description of the TIRFM setup is given elsewhere.21 Only one of the lasers of this setup, a 647 nm laser source (Toptica iBEAM-SMART-640-S), was used for the current experiments. A polarizer (GT10-A; Thorlabs) was used to modulate laser polarization direction. Laser power was controlled using the computer. We used achromatic lenses to expand and collimate the laser beam to a diameter of 6 mm. The expanded laser beam was focused at the back focal plane of the microscope objective lens (UAPON 100×OTIRF; N.A., 1.49; Olympus) with an achromatic lens (f = 500 mm; LAO801; CVI Melles Griot). To attain total internal reflection at the sample, the position of the focused beam was shifted from the center of the objective to its edge to generate a beam with an angle of incidence of 66.8°. Emitted fluorescence was separated from the excitation by utilizing a quad-edge super-resolution laser dichroic beam splitter (Di03-R405/488/532/635-t1-25x36). It was then coupled out from the side port of the microscope. The residual scattered laser light was blocked by notch filters (NF01-405/488/532/635 StopLine Quad-notch filter and ZET635NF; Semrock). An EMCCD camera (iXonEM + 897 back-illuminated; Andor) was employed to collect images. The final magnification of the setup was 240×, along with a pixel size of 66.67 nm.
In each experiment, 10 different TIRFM movies were recorded on 10 different regions of the sample. On each region (with a size of 101 pixel × 101 pixel, i.e. 6.73 μm × 6.73 μm), we recorded 100 ms frames until all the molecules in the designated area were photobleached.
TIRFM movies were analyzed using custom-written Matlab (MathWorks) routines. Individual spots were identified in the first frame of a movie by steps of thresholding and center of mass (CM) analysis as described previously.22 Then the intensity of center of mass of each individual spot was plotted with respect to time, and change-point analysis was performed on the obtained trajectory to identify the number of change points and hence the number of emitters in each spot. Some examples are shown in Figure S3.
Expression, Purification, and Labeling of ClpB
Expression and purification of Thermus thermophilus ClpB used in these experiments were similar to the previously reported procedures.23 In brief, the ClpB gene, cloned into a pET28b vector with the addition of a six-histidine tag preceded by a tobacco etch virus protease (TEV) cleavage site at the N-terminus, was transformed into E. coli BL21 bacteria. Cysteine mutations (S359C and S771C) were then introduced into the protein using a standard site-directed mutagenesis procedure.24 Protein expression was initiated by growing the bacteria at 37 °C to reach 0.8 OD, and then protein expression was induced by adding 1 mM IPTG, followed by incubation at 25 °C overnight. Bacteria were then harvested, and the protein was purified on a Ni-NTA resin (GE Healthcare) with an elution step involving 250 mM imidazole. The protein was dialyzed overnight to remove imidazole from the solution. We further purified the protein using a HiPrep DEAE FF (Ge Healthcare) column equilibrated with 50 mM HEPES, 20 mM KCl, and 2 mM TCEP at pH 7.4 (DEAE buffer). The peak containing the purified protein was collected and stored at −80 °C.
C-terminal His-tagged ClpB was generated using a standard site-directed mutagenesis protocol. The C-terminal His-tagged ClpB gene was then inserted into a pET 41 plasmid. The expression and purification of this ClpB variant were the same as described above.
For the protein labeling reaction, 4 mg of the protein were thawed and then desalted on a Sephadex G25 column (GE Healthcare) against a labeling buffer that contained 25 mM HEPES, 25 mM KCl at pH 7, using a desalting column. The protein was then reacted with Alexa 647 C2 maleimide at a 1:1 molar ratio and incubated for 2–3 h. Reacted protein was separated from unreacted free dye using a desalting column as above with a buffer that contained 25 mM HEPES, 25 mM KCl, and 1 mM TCEP at pH 7.5. In the presence of nucleotide (ATP) ClpB forms a homohexameric structure; however, under the conditions of our measurements the protein is disassembled and a distribution of smaller assemblies is formed.23 The monomeric structure of the protein (PDB: 1QVR) is presented in Figure S4.
Ferromagnetic Surface Preparation
Gold-coated ferromagnetic substrate was prepared using e-beam evaporation on a p-doped silicon wafer ⟨100⟩, with 8 nm of titanium as the adhesion layer and a 120 nm Ni layer. Antigen–antibody reaction kinetics were studied on 2 nm gold coated Ni layer. The effect of thickness of the gold layer on the spin selectivity was checked by studying the kinetics of ClpB adsorption on gold using three thicknesses (2, 6, and 10 nm). Eventually, the 2 nm gold layer was used in the studies reported here. After deposition, substrates were cut into 0.5 cm × 0.5 cm squares and cleaned before the experiments by boiling them first in acetone and then in ethanol for 10 min.
Adsorption Kinetics of ClpB on Gold Coated Magnetic Surface
To verify that indeed the CISS effect influences ClpB molecules, we investigated the dependence of their rate of binding to a ferromagnetic substrate on the direction of spin orientation in the substrate. The adsorption kinetics of ClpB were studied on gold-coated ferromagnetic surfaces (120 nm Ni/8 nm Ti on Si ⟨100⟩). The gold layer thickness was varied (2, 6, and 10 nm). The surface was magnetized with a permanent 0.55 T magnet.
The adsorption of molecules was carried out using a protein concentration of 1 μM in a 25 mM HEPES buffer solution (pH = 7), with 25 mM KCl and 1 mM tris(2-carboxyethyl) phosphine (TCEP, purchased from Sigma-Aldrich, purity >98%), and probed at different time intervals, 2 s, 5 s, 10 s, 20 s, 40 s, with the surface magnetized either with the positive pole (H+) of the permanent magnet or with the negative pole (H−) of the permanent magnet. Control experiments were performed using similar magnetic surfaces but without magnetization. After adsorption, the gold surfaces with the attached protein molecules were rinsed with the same buffer solution as above to remove nonspecifically bound molecules and kept in buffer solution for fluorescence imaging, which was done immediately after preparing the sample as described above. The adsorption kinetics of the protein molecules denatured in 6 M guanidinium chloride were also studied following the same procedure. The results of this experiment are presented in Figure S5.
Panel A of Figure S5 shows the microscope images obtained at various time points after a 2 nm gold coated nickel (120 nm) substrate was exposed to a solution containing the protein, and Figure S5B shows a histogram of the average number of adsorbed molecules as a function of time for different conditions. Clearly, the adsorption rate is faster when the substrate is magnetized with the positive pole (H+) of the magnet, though at the longest adsorption time the number of adsorbed molecules does not depend anymore on the magnetization direction. This is consistent with the spin affecting the rate of adsorption but not the adsorption equilibrium. As the thickness of gold is increased, spin selectivity of the protein is decreased, which is clearly observed in Figure S5C. When the protein is denatured, the spin selectivity is significantly reduced and the rates of adsorption depend only weakly on the spin alignment in the substrate (Figure S5D).
Immobilization of Antibody on Gold-Coated Surface and Kinetics of Association with ClpB
Ultra-LEAF Purified anti-His Tag antibody was attached to a gold-coated surface using dithiobis[succinimidyl]propionate (DSP) as a linker. A DSP monolayer was formed on the gold surfaces by incubating them in the solution of DSP in DMSO (4 mg/mL) for 30 min. After the surfaces were rinsed with DMSO and water, they were immersed in the antibody solution in PBS (1 mg/mL) and incubated for 4 h. After immobilizing the antibody on the gold, the surfaces were then rinsed with PBS (pH = 7.1) and HEPES buffer solution (in the presence of 25 mM KCl). They were then immersed in the ClpB solution (0.1 μM) in a MAKTEK glass bottom Petri-dish kept on a permanent magnet for different time intervals (2 s, 4 s, 6 s, 8 s, 10 s), then immediately taken out, and rinsed with buffer. The reaction kinetics were studied with both orientations (either H+ or H−) of the magnet and also in the absence of the magnet as a control. Fluorescence imaging was carried out immediately following sample preparation. All samples were prepared twice to test reproducibility of the results.
Antigen–Antibody Reaction Kinetics with the Antibody Immobilized on a Gold Substrate through l-Cysteine or d-Cysteine
A cysteine monolayer was prepared on top of a 120 nm gold-coated Si surface using 1 mM cysteine in phosphate buffer solution in the presence of 10 mM TCEP. After rinsing the surface with cold water, it was incubated in a cold aqueous solution of 1-ethyl-3-(3-(dimethylamino)propyl) carbodiimide (EDC, purchased from Fluka, purity >98%) maintaining the concentration at 20 mg/mL. The incubation was performed in a refrigerator (2–8 °C) for 30 min. The surface was rinsed with cold water and a MES buffer solution (pH = 6) quickly and inserted into the antibody solution in MES buffer for 30 min at room temperature to couple antibody molecules to the cysteine. Afterward, the surface was rinsed with the MES buffer solution and then with a HEPES buffer solution. The antibody-coated surface was immersed into a ClpB solution (0.1 μM) for different time intervals 2 s, 4 s, 6 s, and 8 s, immediately taken out, rinsed with buffer, and studied under the microscope. Control experiments were carried out to check the background signals in the images, and the number of molecules detected was not significant with respect to the experimental data. Details are given in the Supplementary Text. Polarization Modulation Infrared Reflectance Absorption Spectroscopy (PMIRRAS) was used to characterize the cysteine monolayers on the gold surface, and the similar intensity of peaks for both cysteine enantiomers infers the similar coverage of the surface (Figure S6A). Details are given in the Supplementary Text.
Reaction Kinetics of Antibodies with Immobilized l-or d-Polyalanine Tagged with Histidine on a Gold-Coated Magnetic Substrate
Labeling of Antibody
To study the effect of chirality of the antigen molecule on the antigen–antibody reaction kinetics, l- or d-polyalanine (PAL) with the histidine tag [HS-CH2CH2CO-(l- or d-Ala-AiB)8-(l-His)6] purchased from Genemed Synthesis Inc. (purity >98%) was used as the antigen. For this study, the anti-His tag antibody was tagged with the dye Alex 647 as follows. Unlabeled antibody molecules in PBS buffer were reacted with the NHS ester of the dye in a 1:10 ratio in the presence of 0.1 M sodium bicarbonate buffer for 1 h at room temperature in the dark. The Micro Bio-Spin column with Bio-Gel P-30 (Bio-Rad) was used to remove the unlabeled dye molecules.
Preparation of PAL Monolayers and Reaction with Antibody
For this experiment, a mixed monolayer of His-tagged polyalanine and polyalanine without the His tag was generated on the top of a gold-coated magnetic surface using a 0.5 mM polyalanine solution in trifluoroethanol (TFE). The surface was rinsed with TFE and then with a PBS buffer solution, then immersed for 2 s in the antibody solution (5 nM) within a thin glass bottom Petri dish kept on a permanent magnet, immediately taken out, rinsed with buffer, and studied under the microscope. Oligopeptide monolayers on the gold coated magnetic surface were characterized using PMIRRAS. The similar intensity of the peaks of the l- and d- oligopeptide proves similar coverage of the surface (Figure S6B). Details are given in the Supplementary Text.
Acknowledgments
We acknowledge Dorit Levy for generating a C-terminal His tagged version of ClpB. R.N. acknowledges the partial support of the Israel Science Foundation, The MINERVA Foundation, and the John Templeton Foundation. G.H. acknowledges partial support of the Israel Science Foundation.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.0c10105.
Control experiments, methods and the materials used (PDF)
Author Contributions
† K.B.-G. and S.G. contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Changeux J. P. The feedback control mechanisms of biosynthetic Lthreonine deaminase by l-isoleucine. Cold Spring Harbor Symp. Quant. Biol. 1961, 26, 313–318. 10.1101/SQB.1961.026.01.037. [DOI] [PubMed] [Google Scholar]
- Gunasekaran K.; Ma B.; Nussinov R. Is allostery an intrinsic property of all dynamic proteins?. Proteins: Struct., Funct., Genet. 2004, 57, 433–443. 10.1002/prot.20232. [DOI] [PubMed] [Google Scholar]
- Motlagh H. N.; Wrabl J. O.; Hilser V. J. The ensemble nature of allostery. Nature 2014, 508, 331–339. 10.1038/nature13001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski R. A.; Gerick F.; Thornton J. M. The structural basis of allosteric regulation in proteins. FEBS Lett. 2009, 583, 1692–1698. 10.1016/j.febslet.2009.03.019. [DOI] [PubMed] [Google Scholar]
- Monod J.; Wyman J.; Changeux J. P. On the nature of allosteric transitions: a plausible model. J. Mol. Biol. 1965, 12, 88–118. 10.1016/S0022-2836(65)80285-6. [DOI] [PubMed] [Google Scholar]
- Koshland D. E. Jr; Némethy G.; Filmer D. Comparison of experimental binding data and theoretical models in proteins containing subunits. Biochemistry 1966, 5, 365–385. 10.1021/bi00865a047. [DOI] [PubMed] [Google Scholar]
- Cooper A.; Dryden D. T. Allostery without conformational change. A plausible model. Eur. Biophys. J. 1984, 11, 103–109. 10.1007/BF00276625. [DOI] [PubMed] [Google Scholar]
- Sheinerman F. B.; Norel R.; Honig B. Electrostatic aspects of protein-protein interactions. Curr. Opin. Struct. Biol. 2000, 10, 153–159. 10.1016/S0959-440X(00)00065-8. [DOI] [PubMed] [Google Scholar]
- Schreiber G.; Fersht A. R. Rapid, electrostatically assisted association of proteins. Nat. Struct. Biol. 1996, 3, 427–431. 10.1038/nsb0596-427. [DOI] [PubMed] [Google Scholar]
- Bauer M. R.; Mackey M. D. Electrostatic Complementarity as a Fast and Effective Tool to Optimize Binding and Selectivity of Protein–Ligand Complexes. J. Med. Chem. 2019, 62 (6), 3036–3050. 10.1021/acs.jmedchem.8b01925. [DOI] [PubMed] [Google Scholar]
- Schreiber G.; Haran G.; Zhou H. – X. Fundamental Aspects of Protein–Protein Association Kinetics. Chem. Rev. 2009, 109, 839–860. 10.1021/cr800373w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naaman R.; Paltiel Y.; Waldeck D. Chiral Molecules and the Electron’s Spin. Nature Reviews Chemistry 2019, 3, 250–260. 10.1038/s41570-019-0087-1. [DOI] [Google Scholar]
- Felder C. E.; Prilusky J.; Silman I.; Sussman J. L. A server and database for dipole moments of proteins. Nucleic Acids Res. 2007, 35, W512–W521. 10.1093/nar/gkm307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh S.; Mishra S.; Avigad E.; Bloom B. P.; Baczewski L. T.; Yochelis S.; Paltiel Y.; Naaman R.; Waldeck D. H. Effect of Chiral Molecules on the Electron’s Spin Wavefunction at Interfaces. J. Phys. Chem. Lett. 2020, 11, 1550–1557. 10.1021/acs.jpclett.9b03487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckshtain-Levi M.; Capua E.; Refaely-Abramson S.; Sarkar S.; Gavrilov Y.; Mathew S. P.; Paltiel Y.; Levy Y.; Kronik L.; Naaman R. Cold Denaturation Induces Inversion of Dipole and Spin Transfer in Chiral Peptide Monolayers. Nat. Commun. 2016, 7, 10744. 10.1038/ncomms10744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzger T. S.; Mishra S.; Bloom B. P.; Goren N.; Neubauer A.; Shmul G.; Wei J.; Yochelis S.; Tassinari F.; Fontanesi C.; Waldeck D. H.; Paltiel Y.; Naaman R. The Electron Spin as a Chiral Reagent. Angew. Chem., Int. Ed. 2020, 59, 1653–1658. 10.1002/anie.201911400. [DOI] [PubMed] [Google Scholar]
- McCaig C. D.; Rajnicek A. M.; Song B.; Zhao M. Controlling Cell Behavior Electrically: Current Views and Future Potential. Physiol. Rev. 2005, 85, 943–978. 10.1152/physrev.00020.2004. [DOI] [PubMed] [Google Scholar]
- Shaik S.; de Visser S. P.; Kumar D. External Electric Field Will Control the Selectivity of Enzymatic-Like Bond Activations. J. Am. Chem. Soc. 2004, 126, 11746–11749. 10.1021/ja047432k. [DOI] [PubMed] [Google Scholar]
- Catterall W. A. Ion Channel Voltage Sensors: Structure, Function, and Pathophysiology. Neuron 2010, 67, 915–928. 10.1016/j.neuron.2010.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fried S. D.; Bagchi S.; Boxer S. G. Extreme electric fields power catalysis in the active site of ketosteroid isomerase. Science 2014, 346, 1510–1514. 10.1126/science.1259802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung Y.; Riven I.; Feigelson S. W.; Kartvelishvily E.; Tohya K.; Miyasaka M.; Alon R.; Haran G. Three-dimensional localization of T-cell receptors in relation to microvilli using a combination of superresolution microscopies. Proc. Natl. Acad. Sci. U. S. A. 2016, 113 (40), E5916–E5924. 10.1073/pnas.1605399113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henriques R.; Lelek M.; Fornasiero E. F.; Valtorta F.; Zimmer C.; Mhlanga M. M. QuickPALM:3D real-time photoactivation nanoscopy image processing in Image J. Nat. Methods 2010, 7, 339–340. 10.1038/nmeth0510-339. [DOI] [PubMed] [Google Scholar]
- Mazal H.; Iljina M.; Barak Y.; Elad N.; Rosenzweig R.; Goloubinoff P.; Riven I.; Haran G. Tunable microsecond dynamics of an allosteric switch regulate the activity of a AAA+ disaggregation machine. Nat. Commun. 2019, 10, 1438. 10.1038/s41467-019-09474-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho S. N.; Hunt H. D.; Horton R. M.; Pullen J. K.; Pease L. R. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene 1989, 77, 51–59. 10.1016/0378-1119(89)90358-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.