

Abstract
The reaction of the bis(1,2-dithiolene) complex [Pd(Me2timdt)2] (1; Me2timdt•– = monoreduced 1,3-dimethyl-2,4,5-trithioxoimidazolidine) with Br2 yielded the complex [Pd(Me2timdt)Br2] (2), which was reacted with Na2mnt (mnt2– = 1,2-dicyano-1,2-ethylenedithiolate) to give the neutral mixed-ligand complex [Pd(Me2timdt)(mnt)] (3). Complex 3 shows an intense solvatochromic near-infrared (NIR) absorption band falling between 955 nm in DMF and 1060 nm in CHCl3 (ε = 10700 M–1 cm–1 in CHCl3). DFT calculations were used to elucidate the electronic structure of complex 3 and to compare it with those of the corresponding homoleptic complexes 1 and [Pd(mnt)2] (4). An in-depth comparison of calculated and experimental structural and vis–NIR spectroscopic properties, supported by IEF-PCM TD-DFT and NBO calculations, clearly points to a description of 3 as a dithione-dithiolato complex. For the first time, a broken-symmetry (BS) procedure for the evaluation of the singlet diradical character (DC) of heteroleptic bis(1,2-dithiolene) complexes has been developed and applied to complex 3. The DC, predominant for 1 (nDC = 55.4%), provides a remarkable contribution to the electronic structures of the ground states of both 3 and 4, showing a diradicaloid nature (nDC = 24.9% and 27.5%, respectively). The computational approach developed here clearly shows that a rational design of the DC of bis(1,2-ditiolene) metal complexes, and hence their linear and nonlinear optical properties, can be achieved by a proper choice of the 1,2-dithiolene ligands based on their electronic structure.
Short abstract
The reaction of [Pd(Me2timdt)2] (1; Me2timdt•− = monoreduced 1,3-dimethyl-2,4,5-trithioxoimidazolidine) with Br2 yielded [Pd(Me2timdt)Br2] (2), which was reacted with Na2mnt (mnt2− = 1,2-dicyano-1,2-ethylenedithiolate) to give [Pd(Me2timdt)(mnt)] (3), showing an intense solvatochromic near-infrared (NIR) absorption band. The singlet diradical character (DC), predominant for 1, provides a remarkable contribution to the ground states of 3 and 4. A rational design of the DC of bis(1,2-dithiolene) complexes can be achieved by a proper choice of the 1,2-dithiolene ligands.
Introduction
The interest of the scientific community toward bis(1,2-dithiolene) metal complexes has been continuously increasing during the past few decades,1−6 accompanied by a growing number of applications relying on the superconducting,7−12 photoconducting,13−17 magnetic, and linear and nonlinear optical properties18−22 of this class of compounds. Bis(1,2-dithiolene) complexes [M(R2C2S2)2]q− of d8 metal ions Mx+, such as NiII, PdII, PtII, and AuIII, feature peculiar properties,5,23 such as molecular planarity and the ability to exist in well-defined oxidation states q typically ranging between x – 4 and x – 2,24−26 also assuming fractional charges in nonintegral oxidation state (NIOS) salts.9,12 The redox noninnocence of the 1,2-dithiolene ligands (Scheme 1) renders it difficult to partition the charge of the complexes between the ligands L and the central metal ion Mx+.27,28
Scheme 1. Redox Noninnocence of 1,2-Dithiolene Ligands: Neutral 1,2-Dithiete (a) and 1,2-Dithione (b), Radical 1,2-Dithiolene Anion (c), and Dianionic Ene-1,2-dithiolate (d).

The typical redox steps accessible to bis(1,2-dithiolene) complexes of group 10 metals (M = Ni, Pd, Pt) are summarized in Scheme 2. Dianionic bis(1,2-dithiolene) complexes [ML2]2– are diamagnetic species, which can be isolated as stable anions in salts such as (Ph4P)2[Ni(mnt)2] (mnt2– = maleonitrile-1,2-dithiolate, 1,2-dicyano-1,2-ethylenedithiolate).29 These species are fully described as [MII(L2–)2]2– complexes, featuring the ene-1,2-dithiolato form L2– of the ligands (d in Scheme 1). Paramagnetic monoanionic 1,2-dithiolene complexes [ML2]− can be represented as [MIII(L2–)2]− compounds30,31 or by two resonance forms showing the dianionic ligand L2– (d in Scheme 1) and a monoanionic radical ligand L•– (c in Scheme 1): i.e., [MII(L•–)(L2–)]−.27 In diamagnetic neutral complexes [ML2], the central metal ion can carry formal charges varying between 0 and +4, while the ligands can assume a neutral, monoanionic, or dianionic charge (Scheme 2), indicating a large degree of π-electron delocalization involving the metal as well as the L ligands (metalloaromaticity).32 Spectroscopic and theoretical results suggest that the complexes are better described as formed by the metal dication MII, whatever the charge on the complex.27,33 Therefore, the oxidation/reduction steps leading from [ML2]2– to [ML2] are mainly located on the ligands,33−35 analogously to what has been reported for [AuIII(Ar-edt)2]0/– complexes (Ar-edt2– = arylethylene-1,2-dithiolate; Ar = phenyl, 2-naphthyl, 2-pyrenyl).36 Hence, the neutral MII complexes can be described as diamagnetic singlet species formed by two antiferromagnetically coupled monoanionic radical ligands, [MII(L•–)2].33,37 Indeed, neither the closed-shell (CS) restricted delocalized nor the localized singlet diradical description represents reliably the ground state (GS) of neutral bis(1,2-dithiolene) complexes, so that an index nDC of the diradical character (DC) can be calculated to evaluate the relative weight of the diradical singlet description.37−39 Notably, different optical properties in the visible–near-infrared (vis–NIR) region are associated with the differently charged forms of bis(1,2-dithiolene) complexes (electrochromism).16,40,56 Neutral complexes [ML2] show a peculiar intense absorption in the region above 800 nm.2,5,30 This band, attributed to a π–π* HOMO → LUMO (H → L) one-electron excitation,5,6 is shifted to lower energies and lowered in intensity in the corresponding monoreduced forms [ML2]−,41 while the dianions [ML2]2– do not show any vis–NIR absorption. In this context, for a few decades, some authors have been investigating the [M(R′2timdt)2]q– class of photoconducting42−44 complexes (R′2timdt•– = monoreduced 1,3-disubstituted imidazoline-2,4,5-trithione; M = Ni, Pd, Pt; q = 0, 1, 2; Chart S1).45−52 Neutral [M(R′2timdt)2] complexes show a strikingly intense absorption at about 1000 nm (molar extinction coefficient ε as large as 120000 M–1 cm–1 in toluene),48 whose energy can be fine-tuned by a proper choice of the metal M and the substituents R′.46,48 The corresponding reduced forms show a NIR absorption falling at about 1450 nm for M = Ni, Pt and at about 1700 nm for M = Pd.51
Scheme 2. Differently Charged Species and Resonance Forms of Bis(1,2-dithiolene) Metal Complexes (M = Ni, Pd, Pt; q = 0, 1, 2).

Mixed-ligand bis(1,2-dithiolene) complexes [M(L)(L′)] have been much less investigated than homoleptic complexes48,53 and are often prepared by metathesis reactions.54−56 The synthetic way of obtaining [M(L)(L′)] complexes by replacement of halides in MLX2 complexes has been previously reported in a few cases.48 In these complexes, most often containing a NiII ion,31,57−61 the most electron withdrawing “pull” ligand L tends to assume the ene-1,2-dithiolate form L2– (d in Scheme 1), with shorter C–C and longer C–S bond distances, while the other “push” ligand (L′) assumes a 1,2-dithione form (b in Scheme 1), with longer C–C and shorter C–S distances, so that the complex is generally described as the dithione-dithiolato species [MII(L2–)(L′)]. The electronic structure of these complexes in their neutral state, reminiscent of that of diimine-dichalcogenolato complexes,62−64 shows the HOMO featuring a larger contribution from the “pull” ligand L2– and the LUMO from the “push” ligand L′. The peculiar visible–near-IR (vis–NIR) electron transition of the neutral species assumes a partial charge-transfer (CT) character from the 1,2-dithiolato L2– ligand to the 1,2-dithione L′ (LL′CT), testified by a remarkable negative solvatochromism of the resulting absorption band.54 In comparison to homoleptic complexes, the DC of heteroleptic bis(1,2-dithiolene) complexes has not been investigated, implicitly accepting that the GS configuration of these complexes is fully defined by the dithione-dithiolato CS description.48 Nevertheless, it is conceivable that a continuous variation from ideally pure open-shell singlet diradicals [MII(L•–)(L′•–)] to CS dithione-dithiolato complexes [MII(L)(L′2–)] occurs as the difference in the donor properties of the L and L′ ligands increases. Therefore, we have considered as a case study the mixed-ligand 1,2-dithiolene PdII complex featuring the well-known mnt “pull” ligand coupled to the “push” ligand Me2timdt. Herein, we report an experimental and theoretical investigation on the resulting complex [Pd(Me2timdt)(mnt)], in comparison with the relevant parent complexes [Pd(Me2timdt)2] and [Pd(mnt)2], aimed at evaluating the role of the electronic structure of the ligands in tailoring the DC in homoleptic and heteroleptic bis(1,2-dithiolene) palladium complexes.
Experimental Section
Materials and Methods
Reagents were purchased from Honeywell, Alfa Aesar, and Sigma-Aldrich and used without further purification. Solvents (reagent grade) were purchased from Honeywell, VWR, and Merck and dried by using standard techniques when required. Manipulations were performed using standard Schlenk techniques under a dry dinitrogen atmosphere. Elemental analyses were performed with a CHNS/O PE 2400 series II CHNS/O elemental analyzer (T = 925 °C). FT-IR spectra were recorded with a Thermo-Nicolet 5700 spectrometer at room temperature: KBr pellets with a KBr beam splitter and KBr windows (4000–400 cm–1, resolution 4 cm–1) were used. Absorption spectra were recorded at 25 °C in a quartz cell of 10.00 mm optical path with either a Thermo Evolution 300 (190–1100 nm) spectrophotometer or an Agilent Cary 5000 UV–vis–NIR (190–2000 nm) dual-beam spectrophotometer. Absorption spectra were decomposed into their constituent Gaussian peaks using the Specpeak 2.065 and Fityk 1.3.166 programs. The Crystallographic Structural Database was accessed by using CCDC ConQuest 2020.1.67
X-ray Diffraction Measurements
Single-crystal X-ray diffraction data were collected with a Rigaku MM007/Mercury diffractometer with Mo Kα radiation. The structure was solved by direct methods with SHELXS-9768 and refined on F2 by using SHELXL-97.69
Synthesis
1,3-Dimethyl-2-thioxoimidazolidine-4,5-dione and complex 1 were prepared according to a previously reported procedure (yield 86%).45,46,70
Synthesis of [Pd(Me2timdt)Br2] (2)
[Pd(Me2timdt)2] (1; 35.4 mg; 7.27 × 10–2 mmol) was reacted with an excess of molecular dibromine in an Aldrich pressure tube using 30 mL of a CHCl3/CH3CN (2/1) solvent mixture. The glass vessel was heated to 130 °C for 15 min and slowly cooled to room temperature. The precipitate was filtered and washed with petroleum ether (brown solid; yield 32.1 mg, 97%). Mp: >230 °C. FT-IR (4000–400 cm–1): ν 1477 (s), 1399 (s), 1360 (m), 1345 (m), 1291 (s), 1080 (s), 1032 (mw), 548 cm–1 (w). Anal. Calcd for C5H6Br2N2PdS3: C, 14.10; H, 0.76; N, 8.11. Found: C, 14.86; H, 0.33; N 9.02.
Synthesis of [Pd(Me2timdt)(mnt)] (3)
Complex 2 (30.2 mg; 6.61 × 10–2 mmol) and a molar excess of sodium 1,2-dicyanoethylene-1,2-dithiolate (21.5 mg; 0.115 mmol) were suspended in CH3CN (30 mL) in an Aldrich pressure tube. The mixture was heated to 130 °C for 30 min and slowly cooled to room temperature. The product was isolated as black needles by filtration, washed with water, and dried under vacuum (yield 8.3 mg, 29%). Mp: >230 °C. FT-IR (4000–400 cm–1): ν 2925 (w), 2204 (m), 1459 (m), 1397 (ms), 1285 (s), 1150 (m), 1079 (ms), 864 (m), 551 (mw), 500 cm–1 (mw). Anal. Calcd for C9H6N4PdS5: C, 24.74; H, 1.38; N, 12.82. Found: C, 24.68; H, 0.45; N, 13.07.
Theoretical Calculations
Quantum-chemical calculations were carried out on [Pd(Me2timdt)2]q− (1q–; Chart S2 for q = 0), [Pd(Me2timdt)Br2] (2; Chart S2), [Pd(Me2timdt)(mnt)]q− (3q–; q = 0, 1, 2; Chart S2 for q = 0), [Pd(mnt)2]q− (4q–; q = 0, 1, 2; Chart S2 for q = 0), [Pt(phen)(tdt)] (phen = 1,10-phenanthroline; tdt2– = 3,4-toluenedithiolate; Chart S2),71 and [Ni(bdt)2] (bdt2– = benzene-1,2-dithiolate)33,37 and the compounds (Me2timdt)2 (Chart S2), Li(R′′2timdt)·2THF (R′′ = 2,6-diisopropylphenyl; 5; Chart S2),72,73 and Me2timdt(SPh)2 (7; Chart S2)74 at the density functional theory (DFT)75 level with the commercial suite Gaussian 16.76 The computational setup was validated as previously described for the strictly related [Ni(Me2timdt)2] complex and derivatives,52 and it took into account three hybrid functionals (B3LYP,77 mPW1PW,78 and PBE079) and six basis sets with relativistic effective core potentials (RECPs)80,81 for the central metal ion (LANL08(f),82 SBKJC,83 Stuttgart 1997 RC,84 CRENBL,85 LANL2DZ,86 and LANL2TZ82). DFT calculations were eventually carried out with the hybrid mPW1PW functional,78 including a modified Perdew and Wang (PW) exchange functional coupled with the PW correlation functional.87,88 Schäfer, Horn, and Ahlrichs double-ζ plus polarization (pVDZ)89 all-electron basis sets for light atomic species (C, H, N, S) and CRENBL basis sets85 with RECPs for heavier atomic species (Pd and Br) were used. Basis sets and RECPs were obtained from Basis Set Exchange and Basis Set EMSL Library.90 Dianionic bis(1,2-dithiolene) complex species were modeled according to a closed-shell (CS) restricted description (RDFT), monoanionic paramagnetic species within an unrestricted formalism (UDFT), while neutral species were investigated (a) in their triplet ground state (2S + 1 = 3, two unpaired electrons), (b) in the closed-shell singlet state (2S + 1 = 1), or (c) as antiferromagnetically coupled singlet diradicals in a broken-symmetry (DFT-BS) approach. The last description was obtained by starting from the triplet state, with the two antiparallel electrons attributed to the two 1,2-dithiolene ligands (Chart S3 in the Supporting Information). The BS electron density guess was obtained through a fragment approach (guess = fragment = 3, the three fragments being the PdII ion and the two monoanionic 1,2-dithiolene radical ligands), eventually allowing optimization (opt) of the geometry of the complex (BS1 solution). Finally, only in the case of 1, the guess of the electron density of the lowest BS singlet excited state (ES) was used to reoptimize the geometry of the GS electronic structure of the complex, giving an alternative minimum for the BS GS configuration (BS2), degenerate with respect to the BS1 description (|ΔεBS2–BS1| ≈ 8 × 10–4 hartree).
For all compounds, tight SCF convergence criteria and fine numerical integration grids (grid = ultrafine) were used. In order to evaluate the singlet diradical contribution to the GS in the BS approach, the differences between the total electronic energy of the singlet state (εS), the BS-singlet state (εBS), and the triplet (εT) states were considered:38,91,92
| 1 |
| 2 |
The effective electron exchange integrals Jab(93) were calculated as follows:
| 3 |
where ⟨S2⟩T and ⟨S2⟩BS represent the spin expectation values94,95 determined at the optimized geometry for the triplet and broken-symmetry GSs, respectively, after verification of the wave function stability (stable = opt). The singlet–triplet energy gap ΔεSTSC = εS – εT, accounting for the effect of spin contamination to the energy of the singlet GS,95 corresponding to a mixing of the singlet and triple state, was calculated as
Therefore, the εSSCvalue was obtained:
| 4 |
The diradical character index nDC can be directly calculated from ⟨S2⟩BS:17,37
| 5 |
A complete natural population analysis (NPA) was carried out with a natural bonding orbital (NBO)96 partitioning scheme (pop = nboread, with boao and bndidx keywords in the NBO section of the input file) in order to investigate the charge distributions and Wiberg bond indexes.97 Absorption vertical transition energies and oscillator strengths were calculated at the time-dependent (TD) DFT level.98,99 TD-DFT calculations were carried out at the optimized geometry in the gas phase and in a selection of solvents (CHCl3, CH2Cl2, DMF, THF, acetonitrile), implicitly taken into account by means of the polarizable continuum model in its integral equation formalism (IEF-PCM),100 describing the cavity of the complexes within the reaction field (SCRF) through a set of overlapping spheres. Oscillator strength values calculated at the TD-DFT level along with experimental full width at half maximum (FWHM) values of the NIR band were used to evaluate the molar extinction coefficients ε.101 Experimental FWHM values on an energy scale (eV) were evaluated from the corresponding values w determined in nm from the experimental NIR spectra:
The one-photon absorption oscillator strength f0n for each transition 0 → n is102
where me and e are the mass and the charge of the electron, ν0n is the frequency (s–1) of the transition between the states 0 and n, μ0n is the transition dipole moment, and h is Planck’s constant. f0n is related to the experimental intensity of each absorption band:
where ε is the molar extinction coefficient (M–1 cm–1) and ϖ is the frequency (cm–1). By adoption of Gaussian curve shapes for the absorption bands
where the width parameter θ is related to W by
Therefore, the equation
| 6 |
allows evaluating the molar extinction coefficients of the NIR transition at the TD-DFT level. Calculated molar extinction coefficients were scaled on experimental available data to give a corrected εcalccorr value.
The nature of the minima of each structure optimized at the DFT and DFT-BS levels was verified by harmonic frequency calculations (freq = raman), including the determination of thermochemistry parameters (zero-point energy (ZPE) corrections and thermal corrections to enthalpy and Gibbs free energy) and the calculation of FT-Raman frequencies. Gibbs free energies were used to calculate absolute reduction potentials at 298 K (EAbs298K) according to the following equation:103−106
| 7 |
where ΔGneutral298K and ΔGanion are the free energy values calculated at 298 K and ΔGe°/F represents the potential of the free electron (−0.03766 eV at 298 K; ΔGneutral is calculated on the most stable neutral form).107EAbs298K values were also referenced to the Fc+/Fc couple, taken into account at the same level of theory.
The total static (i.e., under zero frequency)108 second-order (quadratic) hyperpolarizability (the first hyperpolarizability)109 βtot was calculated as previously described.110
Throughout all this work, molecules in their optimized standard orientation were rotated in order to align the main symmetry axis (bisecting C–C 1,2-dithiolene bonds and passing through the central metal ion) with the z axis and lie on the yz plane. Molden 6.2111 and GaussView 6.0.16112 were used to analyze Kohn–Sham (KS) molecular orbital (MO) compositions and energies. GaussSum 3.0113 and Chemissian 4.54114 were used to evaluate the atomic orbital contributions to KS-MOs and to analyze TD-DFT data.
Results and Discussion
Synthesis
The synthesis of [Pd(R′2timdt)2] neutral complexes can be achieved by direct sulfuration with Lawesson’s reagent (2,4-bis(4-methoxyphenyl)-1,3,2,4-dithiadiphosphetane-2,4-disulfide)115 of the corresponding 1,3-disubstituted 2-thioxoimidazolidine-4,5-diones, followed by addition of PdCl2 (reactions i and ii in Scheme 3).45,46 The reaction of a suspension of [Pd(Me2timdt)2] (1) in a 2/1 CHCl3/CH3CN solvent mixture with a molar excess of molecular dibromine in a high-pressure tube at 130 °C yielded the neutral complex [Pd(Me2timdt)Br2] (2; reaction iii in Scheme 3), which was successively made to react with sodium 1,2-dicyanoethylene-1,2-dithiolate (Na2mnt) in CHCl3 at 130 °C to give the mixed-ligand neutral complex [Pd(Me2timdt)(mnt)] (3; reaction iv in Scheme 3).
Scheme 3. Reaction Pathways for the Synthesis of 3.

Legend: (i) Lawesson’s reagent, refluxing toluene, N2; (ii) PdCl2; (iii) Br2, CHCl3/CH3CN 2/1, 130 °C; (iv) Na2mnt, CH3CN, 130 °C.
X-ray Diffraction Studies
Tiny needle crystals of 3, suitable for a structural characterization by X-ray diffraction analysis (Figure 1, Tables S1–S6, and Figures S1 and S2), were isolated from the reaction mixture. The structural features of 3 (Figure 1) closely resemble those determined previously for [Pd(Et2timdt)(mnt)].48 The molecule is planar except for the methyl substituents, with the central Pd ion coordinated by the two 1,2-dithiolene ligands in a pseudo-square-planar fashion, with unbalanced Pd–S bond distances (Pd–S(1), 2.314(2); Pd–S(4), 2.258(2) Å). The C(1)–C(1)i bond length within the Me2timdt ligand (1.454(16) Å) is longer than that previously reported for [Pd(Et2timdt)2] (1.397(9) Å),45 while the C(1)–S(1)/C(1)i–S(1)i distances (1.660(8) Å) are shorter (corresponding average value in [Pd(Et2timdt)2] 1.689(8) Å), suggesting a larger character of ene-1,2-dithiolate of the Me2timdt ligand in 3 as compared to the Et2timdt ligands in [Pd(Et2timdt)2]. When the two C2S2Pd metallacycles in 3 are compared, the C(4)–C(4)i length in the mnt unit (1.364(16) Å) is shorter than that in the Me2timdt unit, while the C–S distances are longer in the mnt ligand (C(4)–S(4) 1.724(8) Å). The C–C distance in the Me2timdt ligand of 3 (1.454(16) Å) is intermediate between the corresponding distance in [Pd(Et2timdt)Br2] (1.474(10) Å; Chart S2),48 featuring an authentic neutral Et2timdt ligand, and that of Li(5, R″ = 2,6-diisopropylphenyl, Chart S2; 1.417(4) Å), featuring the R″2timdt•– radical monoanion.72,73 Analogously, the C(4)–C(4)i distance in 3 (1.364(16) Å) is shorter than that found in [Pd(mnt)2] (4; 1.39(2) Å; Chart S2)116 and the average value for 4– monoanions (1.377(24) Å)117 but slightly larger than the average value found in salts of the complex 42– (1.359(19) Å).118 This suggests that the Me2timdt ligand in 3 should be considered to carry a partial negative charge and that the GS of 3 should include a partial DC. The unit cell contains pairs (Z = 2) of symmetry-related complex molecules, each forming slipped stacks along the a vector (Figures S1 and S2 in the Supporting Information) with an interplanar distance of 3.619 Å, very close to that featured by the stacks found in the crystal structure of [Pd(Et2timdt)2] (3.6 Å).45 Along the stacks, the terminal thione groups of the Me2timdt ligands weakly interact with the π-system of the imidazoline ring (C(1)ii/iii–S(2) 3.420 Å; (ii) −1 + x, y, z; (iii) −1 + x, 1/2 – y, z; Figure S1). Weak contacts between the methyl substituents at the Me2timdt ligands and the terminal N atoms of the mnt ligands (H(3A)···N(5)iv 2.644 Å; (iv) −1 + x, y, 1 + z) are responsible for the interactions between adjacent stacks aligned along the c direction. Notably, the crystal packing is sensibly different from that found in [Pd(Et2timdt)(mnt)], where the complex molecules are stacked in an alternate head-to-tail disposition, allowing for shorter interplanar distances (3.570 Å).48
Figure 1.
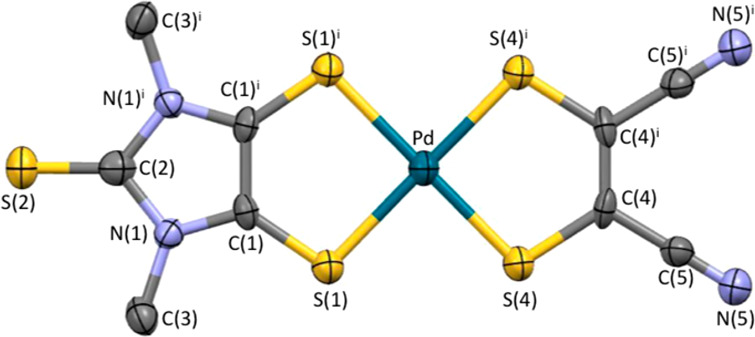
ORTEP view of complex 3 with the adopted labeling scheme. Selected bond distances (Å) and angles (deg): Pd–S(1) 2.314(2), C(1)–S(1) 1.660(8), C(1)–C(1)i 1.454(16), C(1)–N(1) 1.342(9), N(1)–C(3) 1.454(10), N(1)–C(2) 1.401(10), C(2)–S(2) 1.603(13), Pd–S(4) 2.258(2), C(4)–S(4) 1.724(8), C(4)–C(4)i 1.364(16), C(4)–C(5) 1.423(11), N(5)–C(5) 1.139(10) Å; S(1)–Pd–(S1)i 91.53(12), S(1)–Pd–(S1)i 90.61(12), S(1)–Pd–S(4) 88.92(8), C(1)–S(1)–Pd–S(4) 179.6(2), S(1)–Pd–S(4)–C(4) 177.3(2). Displacement ellipsoids are drawn at the 30% probability level. Hydrogen atoms are omitted for clarity. Symmetry operation: (i) x, 1/2 – y, z.
Absorption Spectroscopy
Neutral [Pd(R′2timdt)2] bis(1,2-dithiolene) complexes featuring alkyl R substituents show a peculiar, intense NIR absorption falling at about 1010 nm, with molar extinction coefficients ε as high as 70000 M–1 cm–1 in CH2Cl2.45,46 The UV–vis–NIR absorption spectrum of a CH2Cl2 solution of 1 shows a NIR absorption maximum falling at 1008 nm (full width at half-maximum (FWHM) w = 131 nm; Figure S3). Notably, the NIR peak shows at least three Gaussian components (λ1 = 1004.8 nm, w1 = 121.9 nm, integral ratio 74.5%; λ2 = 890.3 nm, w2 = 93.5 nm, 6.6%; λ3 = 1120.2 nm, w3 = 155.4 nm, 18.9%; Figure S3),65 in agreement with the spectral decomposition reported for [Pd(2,4-tBu2C6H2S2)2], for which a series of d–d transitions with different spin couplings to the open-shell ligands were envisaged.33 Complex 3 shows a well-defined intense NIR peak at 1060 nm in CHCl3 (ε = 10700 M–1 cm–1; Figure 2), in perfect agreement with the spectral features shown by [Pd(Et2timdt)(mnt)] in the same solvent (λmax = 1061 nm, ε = 12500 M–1 cm–1).48 The NIR band can be decomposed into two main peaks, each accounting for about half the area of the band (λ1 = 1066.2 nm, w1 = 140.6 nm, 51.6%; λ2 = 1025.1 nm, w2 = 249.5 nm, 48.4% in CHCl3; Figures S4 and S5). The NIR band displays a remarkable negative solvatochromism, with absorption maxima wavelengths ranging between 955 nm in DMF and 1060 nm in CHCl3 (Table 1). When the solvent polarity is increased, the change in the experimental spectral shapes (Figure S6) suggests that the relative weight and the energy difference between the red component and the main peak of the solvatochromic NIR band increases, so that a greater polar nature should be attributed to the higher energy peak as compared to the main peak.
Figure 2.

UV–vis–NIR absorption spectrum (350–1300 nm) of 3 in CHCl3 solution.
Table 1. Experimental NIR Absorption Maximum Wavelengths λ (nm), Energies E (eV), and FWHM Values w on a Wavelength Scale (nm) Recorded for 3, in Comparison with the Corresponding Values λcalc and Ecalc Calculated at the TD-DFT IEF-PCM Level (CS GS) and Calculated Oscillator Strengths f, Extinction Coefficients εcalcdcorr (M–1 cm–1), and HOMO–LUMO Energy Gaps ΔEH–L (eV) in Selected Solvents.
| λ | λcalc | E | Ecalc | w | f | εcalccorr | ΔEH–L | |
|---|---|---|---|---|---|---|---|---|
| CHCl3 | 1060 | 876.0 | 1.167 | 1.416 | 180 | 0.385 | 10700 | 1.68 |
| CH2Cl2 | 1020 | 863.8 | 1.216 | 1.436 | 151 | 0.368 | 11895 | 1.73 |
| THF | 1011 | 864.4 | 1.226 | 1.435 | 168 | 0.368 | 10675 | 1.71 |
| CH3CN | 966 | 843.9 | 1.284 | 1.469 | 236 | 0.340 | 6630 | 1.77 |
| DMF | 955 | 851.8 | 1.298 | 1.456 | 217 | 0.356 | 7712 | 1.77 |
Theoretical Calculations
During the past few decades, DFT calculations have been used successfully to investigate the structural features and the redox and spectroscopic properties of homoleptic and heteroleptic complexes containing 1,2-dithiolene ligands.36−39,48,50,52,62,64,72 DFT calculations were applied here on Me2timdtq–, mntq– (q = 0, 1, 2), and related compounds and the relevant neutral, monoanionic, and dianionic homoleptic and heteroleptic Pd complexes.
Ligands
The relative stability of the variously charged forms of 1,2-dithiolene ligands (Scheme 1) depends on the nature of the R substituents. The mnt ligand is generally encountered in its 1,2-dithiolate form, and the corresponding neutral species is unreported. In fact, neutral 1,2-dithiolene species are generally unstable,6 but depending on the R substituents they can be found as either 1,2-dithiones (Scheme 1, b), for instance embedded in 1,2-dithioxamides,119 or stabilized as 1,2-dithietes (Scheme 1, a).120−122 Since vicinal dithioxamides in five-membered rings are reportedly unstable,123 R′2timdt ligands cannot be isolated as neutral 2,4,5-trithiones and the sulfuration of disubstituted 2-thioxomidazolidine-4,5-diones leads to tetrasubstituted 4,5,6,7-tetrathiocino[1,2-b:3,4-b′]diimidazolyl-2,9-dithione or 4,5,9,10-tetrathiocino[1,2-b:5,6-b′]-2,7-dithione (a and b in Chart S4 in the Supporting Information, respectively), the latter type of compounds being the final product of the Br2 oxidation of [Pd(R′2timdt)2] complexes (R′ = Et).124 The only example of an authentic radical monoanion R″2timdt•– has been isolated in compound 5.72 Neutral 1,2-dithiolene ligands stabilized in the form of 3,4-disubstituted 1,2-dithietes have been characterized in few cases (R = CF3,120 COOCH3,121 1-adamantyl122). An examination of the ZPE-corrected total electronic energies εZPE0 of the neutral 1,2-dithiolene ligands in the dithione and dithiete forms (Table 2) shows that the dithiete form is more favored for the mnt ligand in comparison to the Me2timdt ligand (εZPE = 16.28 and −42.58 kcal/mol, respectively). A comparison of the C–C bond distances calculated for Me2timdtq– (dC–C = 1.500, 1.434, and 1.394 Å for q = 0, 1, and 2, respectively; Table 2) can be made with the corresponding bond distances determined structurally in [Pd(Et2timdt)Br2] (average dC–C value 1.47(1) Å; q = 0; Chart S2), 5 (dC–C = 1.417(4) Å, q = 1; Chart S2),48 [6·2Br]2+(Br2)2(Br2)3 (6 = 4,5,9,10-tetrathiocino[1,2-b:5,6-b′]-1,3,6,8-tetraethyldiimidazolyl-2,7-dithione; dC–C = 1.37(2) Å, q = 2; Chart S2), and 1,3-dimethyl-4,5-bis(phenylsulfanyl)-1,3-dihydro-2H-imidazole-2-thione (7, dC–C = 1.361(6) Å, q = 2; Chart S2).74 As expected, on passing from dianions to the corresponding neutral species the C–C bond lengths increase and the C–S distances decrease. It is worth noting that mntq– and Me2timdtq– (q = 0, 1, 2) share closely related frontier MO compositions (Figures S7 and S8). For both ligands, the KS-HOMO-1 and KS-HOMO of neutral species L are nonbonding molecular orbitals (NBMOs) built up from the in-phase and out-of-phase combinations, respectively, of the sulfur 3p AOs lying on the ligand plane. The LUMO is a π-in-nature MO derived from the combination of C 2pz and S 3pz AOs of the ene-1,2-dithiolate system, bonding with respect to the C–C bond and antibonding with respect to the C–S bonds. The LUMO of neutral L ligands becomes the SOMO and the HOMO in monoanionic L•– and dianionic L2– species, respectively (Figures S7 and S8). The absolute reduction potential105,125,126EAbs298K (eq 7) of the redox step L/L•– increases on passing from Me2timdt to mnt (2.245 and 3.014 V, respectively), indicating that the mnt ligand displays the largest tendency to reduction. The difference in the EAbs values for the L/L•– and L’/L′•– couples, which only depends on the choice of the substituents R at the ene-1,2-dithiolate, can be considered a useful parameter to evaluate the “push” and “pull” nature48,54 of 1,2-dithiolene ligands in heteroleptic mixed-ligand metal complexes [M(L)(L′)].
Table 2. Optimized C–C and C–S Bond Distances (dC–C and dC–S, Å) within the 1,2-Dithiolene Moiety and Corresponding Wiberg Bond Indices (WBIC–C and WBIC–S), Variations in ZPE Corrected Total Electronic Energies (εZPE0 = ε0 + ZPE, kcal mol–1), Sum of Electronic Energies and Thermal Enthalpies (Hcorr, kcal mol–1), and Free Energies (Gcorr, kcal mol–1) Calculated for the 1,2-Dithiolene Ligands mntq– and Me2timdtq– (q = 0, 1, 2).
| q | dC–C | dC–S | WBIC–C | WBIC–S | ΔεZPE0 | Δ(ε0 +Hcorr)c | Δ(ε0+Gcorr)d | |
|---|---|---|---|---|---|---|---|---|
| mnta | 0 | 1.501 | 1.625 | 1.018 | 1.787 | |||
| mntb | 0 | 1.371 | 1.756 | 1.501 | 1.100 | –16.28e | –16.58e | –15.46e |
| Me2timdta | 0 | 1.500 | 1.629 | 1.007 | 1.712 | |||
| Me2timdtb | 0 | 1.343 | 1.765 | 1.538 | 1.061 | 42.58e | 41.28e | 45.27e |
| mnt– | 1 | 1.431 | 1.681 | 1.397 | 1.235 | –84.47f | –84.58f | –84.09f |
| Me2timdt– | 1 | 1.434 | 1.675 | 1.217 | 1.423 | –51.33f | –51.35f | –50.90f |
| mnt2– | 2 | 1.406 | 1.736 | 1.365 | 1.178 | –34.75f | –34.87f | –33.87f |
| Me2timdt2– | 2 | 1.394 | 1.738 | 1.453 | 1.201 | 13.09f | 13.07f | 14.23f |
1,2-Dithione form.
1,2-Dithiete form.
Sum of electronic and thermal enthalpies.
Sum of electronic and thermal free energies.
Relative energy
difference between
the 1,2-dithiete and the 1,2-dithione form: 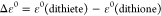 .
.
Relative energy difference
between
reduced and neutral species: 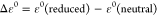
Diradical Character (DC) in Bis(1,2-dithiolene) Complexes
The ground state (GS) of neutral bis(1,2-dithiolene) complexes is characterized by a significant degree of DC,33−37 due to the very narrow HOMO–LUMO energy gap ΔEH–L that renders the singlet and triplet GSs very close in energy. In a pure diradical the nonbonding molecular orbitals (NBMOs) ψa and ψb hosting the two electrons at the highest energy are degenerate, being localized on the two different ligands in bis(1,2-dithiolene) metal complexes,127 and they have consequently a negligible overlap integral Sab = ⟨ψa|ψb⟩. Under these conditions, the two possible spin states, i.e. singlet (2S + 1 = 1) and triplet (2S + 1 = 3), are degenerate, their energy difference ΔεST = εS – εT being related to the exchange interaction J = 1/2(εS – εT) = 1/2ΔεST.91 When Sab is not negligible and the two NBMOs are quasi-degenerate, the triplet configuration is the most stable (εS > εT) and J assumes positive values in the so-called diradicaloids or diradical-like compounds. Wirz proposed discriminating between diradicals and diradicaloids depending on the singlet–triplet energy gaps (ΔεST ≤ 10 and 100 kJ mol–1 for diradicals and diradicaloids, respectively).128 When the energy difference between the two involved MOs is larger and a significant gap exists, the energy stabilization competes with the electron–electron exchange interaction, and the CS singlet GS becomes progressively more stable. The theoretical evaluation of the GS in diradical and diradicaloid species is a challenging task, which requires the evaluation of the stability of the triplet and singlet GSs of the investigated compound. The triplet GS can generally be calculated by theoretical methods with unrestricted wave functions, such an unrestricted HF (UHF) or density functional theory (UDFT). The modeling of open-shell singlet diradicals requires multireference approaches: for instance, multireference coupled-cluster calculations, such as Mk-CCSD(T),129 complete active-space self-consistent field (CASSCF),94 or the complete-active-space second-order perturbation theory (CASPT2).130 In fact, although they are computationally very efficient, DFT calculations cannot accurately describe open-shell singlet states of diradicals, so that ΔεST and hence J values are largely uncertain. The broken-symmetry (BS) DFT (DFT-BS) spin-unrestricted reference configuration with antiparallel spins has been proposed as a compromise to extend UDFT calculations to diradical species,91,131−135 by correcting J and ΔεST for spin contamination94,133,134 that affects the expectation value of the total spin ⟨S2⟩ with respect to ⟨S(S + 1)⟩.94,95,136 In different studies, the DC index nDC of homoleptic bis(1,2-dithiolene) complexes was evaluated. A comparison of nDC calculated at different levels of theory suggests that the DFT-BS approach underestimates the DC of bis(1,2-dithiolene) metal complexes. In fact, the DC of the neutral complex [Ni(bdt)2] (bdt2– = benzene-1,2-dithiolate) was calculated to be as large as 69.1% at the CASSCF level,37 32% at the ZORA-SORCI level (ZORA = zeroth-order regular approximation; SORCI = spectroscopy oriented configuration interaction),33 and 17.2% at the DFT-BS level. The DC depends not only on the 1,2-dithiolene ligand but also on the nature of the central metal ion. In the series of complexes of MII ions derived from the 3,5-di-tert-butyl-1,2-benzene-dithiolato ligand, the nDC values were calculated to be 32%, 50%, and 30% for M = Ni, Pd, Pt, respectively,33 suggesting that PdII species may show particularly stable singlet diradicals in comparison to the corresponding NiII/PtII analogues.
Homoleptic Bis(1,2-dithiolene) Complexes
Members of the class of complexes [M(R′2timdt)2]q− (M = Ni, Pd, Pt; q = 0, 1, 2) are mostly stable as neutral species, and quite severe conditions are needed to achieve a reversible chemical or electrochemical reduction to the corresponding monoanionic radical species.16,46,48,52 With regard to M = Pd, while the crystal structures of [Pd(Et2timdt)2] and the CT adduct [Pd(Et2timdt)2]·I2·CHCl3 have long since been published,45 no anionic complexes [Pd(R′2timdt)2]−/2– have been structurally characterized so far. Conversely, neutral [M(mnt)2] complexes (M = Ni, Pd, Pt) are extremely rare, and (perylene)2(4) is the only compound characterized structurally incorporating the neutral species 4,116 while to date 37 examples of compounds incorporating the anions 4–/2– have been deposited with the Cambridge Crystallographic Database.137 Accordingly, 1 is calculated to be sensibly more stable to reduction in the gas phase and in CH2Cl2 than 4 (EAbs298K = 3.057 and 4.349 eV for complex 1 and 4.786 and 5.752 eV for complex 4, in the gas phase and in CH2Cl2, respectively). In Table 3, the metric parameters optimized for the bis(1,2-dithiolene) complexes 1q– and 4q– (q = 0, 1, 2) are summarized. In the case of neutral complexes (q = 0), the geometry was optimized (i) for the singlet CS (RDFT), (ii) for the triplet open-shell (UDFT), and (iii) for the singlet diradical (DFT-BS) GS configurations. The total electronic energy εT of the 3B1u triplet state is calculated to be lower by about 2.4 kcal mol–1 (10 kJ mol–1) in comparison to that (εS) of the uncorrected singlet 1Ag GS (ε2 < 0, eq 2), thus classifying 1 as a diradical species.128 In fact, the DFT-BS GS of 1 shows a large ⟨S2⟩BS value (0.80, Table 4), indicating a considerable spin contamination from the triplet state.94 An evaluation of the total electronic energy of the BS GS shows that it is the most stable configuration in comparison to both the triplet and CS-singlet configurations (eq 1), reflected by the diradical character nDC = 55.4% (Table 4, eq 5). The singlet GS calculated for 4 is sensibly lower in energy in comparison to the relevant triplet state (Table 4), indicating a diradicaloid character. Accordingly, the singlet diradical configuration is only slightly more stable than the uncorrected CS singlet state and has an ⟨S2⟩BS value smaller than 0.5. In fact, the spin-contamination corrected state (eq 4) was found to be the most stable state with only a partial diradical character (nDC = 27.5%, Table 4). A comparison between structural and DFT-optimized bond distances for complexes 1 and 4 shows that the Pd–S and C–C distances are slightly overestimated, while C–S bond lengths are very close. Pd–S bond lengths are very sensitive to the GS configuration and follow the trend Pd–S (triplet) > Pd–S (CS singlet) ≥ Pd–S (singlet diradical) > Pd–S (structure). The C–S bond distances optimized for the singlet diradical GSs of 1 and 4 (1.699 Å; Table 3) are very close to those calculated for the hypothetical free Me2timdt•– and mnt•– radical anions (1.681 and 1.675 Å, respectively) but remarkably different from those calculated for the relevant 1,2-dithiones and 1,2-dithiolates (Table 2). This supports the description of neutral homoleptic complexes as [Pd(L•–)2] for both classes of complexes. Although the agreement between structural and optimized C–C distances is less accurate in comparison to C–S bond lengths, the former values are affected very greatly by the charge on the ligands. In Figure 3, the optimized C–C distances and the corresponding Wiberg bond indices (WBIs)97 are compared for a variety of R′2timdt derivatives showing a C=C double bond, as in 4,5,9,10-tetrathiocino[1,2-b:5,6-b′]diimidazolyl-1,3,6,8-tetramethyl-2,7-dithione (Me2timdt)2 and compound 7,74 or a single bond, as in the neutral complex 2 and in compound 5 (Chart S2).72,73 For these compounds, a clear correlation (R2 = 0.99) holds between the optimized C–C bond distance dC–C within the R2timdt ring and the corresponding WBIC–C values. This clearly shows that WBIs calculated at the optimized distances represent a reliable parameter for evaluating the charge distribution and hence the oxidation state of noninnocent 1,2-dithiolene ligands. When 1,2-dithiolene complexes 1q– (q = 0, 1, 2) are considered, the dC–C and WBIC–C data, while not exactly fitting the correlation, point out that complex 12– falls in the area of C=C double bonds, very close to (Me2timdt)2 and compound 7, therefore confirming the 2-thioxoimidazoline-4,5-dithiolate nature of the ligands in the dianionic complex. Complexes 1 (whatever the approach adopted for describing its GS) and 1– fall in the central area of the graph, indicating an intermediate character of the C–C bond between a single and a double bond.
Table 3. Optimized Pd–S, C–C, and C–S Bond Lengths (d, Å) and Wiberg Bond Indices (WBI) within the 1,2-Dithiolene Ligands L and L′ of [Pd(L)(L′)]2q– Complexes (L = L′ = Me2timdt for 1q–; L = Me2timdt, L′ = mnt for 3q–; L = L′ = mnt for 4q–; q = 0, 1, 2)a.
|
L |
L′ |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| GS | symmb | dPd–S | dC–C | dC–S | WBIC–C | WBIC–S | dPd–S | dC–C | dC–S | ΔdC–Cc | WBIC–C | WBIC–S | |
| 1 | triplet | 3B1u | 2.351 | 1.410 | 1.698 | 1.177 | 1.378 | ||||||
| singlet | 1Ag | 2.314 | 1.404 | 1.697 | 1.269 | 1.276 | |||||||
| BS | 2.307 | 1.401 | 1.699 | 1.251 | 1.274 | ||||||||
| [Pd(Et2timdt)2] (structural data)d,e | 2.295(2) | 1.397(9) | 1.689(8) | ||||||||||
| 1– | doublet | 2B2g | 2.337 | 1.381 | 1.717 | 1.385 | 1.192 | ||||||
| 12– | singlet | 1Ag | 2.376 | 1.365 | 1.739 | 1.482 | 1.126 | ||||||
| 3 | triplet | 3B1 | 2.359 | 1.408 | 1.700 | 1.226 | 1.267 | 2.305 | 1.404 | 1.701 | 0.004 | 1.307 | 1.098 |
| singlet | 1B1 | 2.342 | 1.428 | 1.674 | 1.168 | 1.397 | 2.263 | 1.393 | 1.725 | 0.035 | 1.417 | 1.098 | |
| BS | – | 2.344 | 1.422 | 1.682 | 1.186 | 1.354 | 2.273 | 1.389 | 1.717 | 0.033 | 1.384 | 1.098 | |
| 3 (structural data)f | 2.314(2) | 1.454(16) | 1.660(8) | 2.258(2) | 1.364(16) | 1.724(8) | 0.090 | ||||||
| 3– | doublet | 2A1 | 2.369 | 1.397 | 1.697 | 1.290 | 1.276 | 2.286 | 1.376 | 1.740 | 0.021 | 1.451 | 1.138 |
| 32– | singlet | 1B1 | 2.384 | 1.364 | 1.737 | 1.477 | 1.131 | 2.323 | 1.381 | 1.744 | –0.017 | 1.424 | 1.130 |
| 4 | triplet | 3B1u | 2.325 | 1.410 | 1.696 | 1.277 | 1.329 | ||||||
| singlet | 1Ag | 2.275 | 1.403 | 1.701 | 1.325 | 1.305 | |||||||
| BS | 2.289 | 1.451 | 1.699 | 1.311 | 1.312 | ||||||||
| 4– | doublet | 2B2g | 2.295 | 1.386 | 1.725 | 1.405 | 1.195 | ||||||
| 42– | singlet | 1Ag | 2.332 | 1.380 | 1.743 | 1.428 | 1.131 | ||||||
| 4 (structural data)e,g | 2.263(7) | 1.39(2) | 1.71(1) | ||||||||||
For neutral species (q = 0), the distances in the open-shell paramagnetic triplet, CS diamagnetic singlet, and singlet diradical (BS) configurations are reported.
GS-symmetry representations are referenced to the D2h (1, 4) and C2v (3) point groups, with the complex molecule laying on the yz plane and the main axis being coincident with z.
ΔdC–C = difference in dC–C values calculated within the two ligands Me2timdt–q/2–1 and mnt–q/2–1 in 3q–.
Reference (45).
Average value.
This work.
Isolated in (perylene)2(4). Reference (116).
Table 4. Energy Differences (kcal mol−1) between the Singlet and the Broken–Symmetry Configurations (ε1), the Singlet and the Triplet Configurations (ε2), the Spin Contamination Corrected Singlet and the Triplet Configurations (ΔεSTSC), Expectation Value of the Spin Contaminant ⟨S2⟩BS for the Singlet Diradical Configuration, Effective Electron Exchange Integrals Jab and Diradical Characters nDC (%) Calculated for Complexes 1, 3, and 4.
| 1 | 3 | 4 | |
|---|---|---|---|
| ε1 | –4.325 | –0.789 | –1.015 |
| ε2 | –2.404 | 5.817 | 6.197 |
| ΔεSTSC | 3.986 | –7.420 | –8.096 |
| ⟨S2⟩BS | 0.801 | 0.436 | 0.474 |
| |Jab| | 1.579 | 4.175 | 4.664 |
| nDC (%) | 55.4 | 24.9 | 27.5 |
Figure 3.

Correlation between optimized C–C bond distances (dC–C) and Wiberg bond indices (WBIC–C) within the 1,2-dithiolene ligand calculated for selected systems (circle, singlet; star, doublet; triangle, triplet; square, singlet diradical).
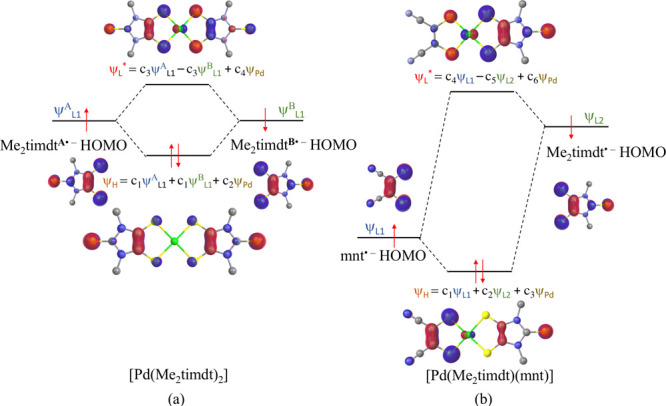
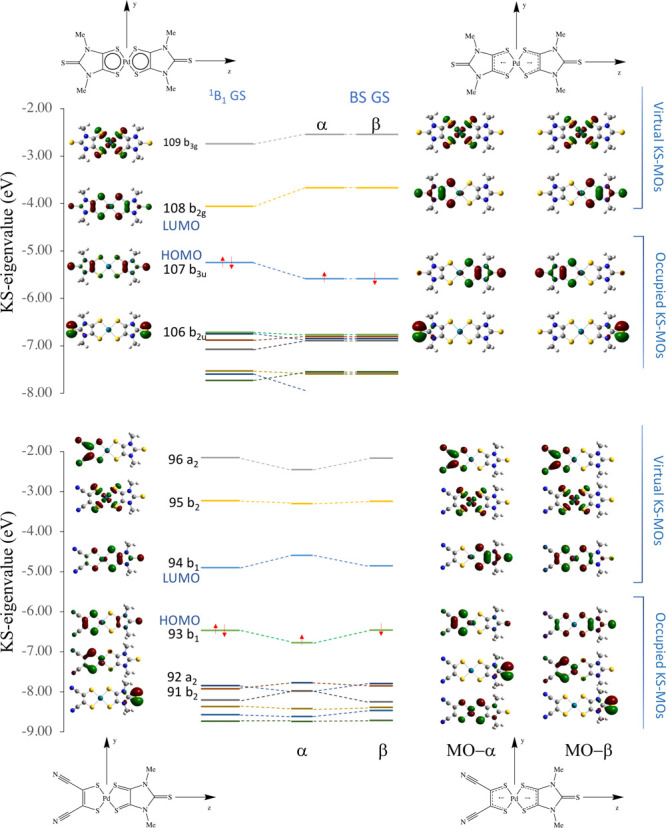
An examination of the frontier KS-MOs composition shows that, according to the CS description of the GS of 1, the KS-HOMO and the KS-LUMO are π-MOs represented by the b3u in-phase and b2g out-of-phase combinations of the singly occupied molecular orbitals (SOMOs) of the two Me2timdt•– ligands (Figure 4a and Figure S8). In fact, the b3u KS-HOMO (MO 107 according to a progressive labeling based on an energy scale) is mainly made up of the four 3px AOs of the four donor S atoms, perpendicular to the molecular yz plane, and the four C 2px AOs taken with opposite phases. The terminal S atoms also participate in this MO, while the contribution from the central Pd ion is very poor (4%). The 108 b2g KS-LUMO involves the same atomic species as the HOMO with a larger contribution from the bonding sulfur atoms, but the contributions from the two ligands are opposite in phase. In the KS-LUMO, the metal ion is only marginally involved (5%) as well through its 3dyz AOs. In the singlet diradical DFT-BS GS configuration, the α- and β-HOMOs show the same composition as the HOMOs of the constituent 1,2-dithiolene Me2timdt•– ligands, analogously to what was previously reported for different Ni and Pt bis(1,2-dithiolene) metal complexes,52 the central Pd ion participating to both α and β MOs (3%). Notably, the DFT-BS approach results in a stabilization of the KS-HOMO and destabilization of the KS-LUMO with respect to the restricted CS solution, thus increasing the ΔEH–L gap (Figure 5, top). The CS description of complex 1 features a single allowed NIR one-electron excitation calculated at the TD-DFT level. This corresponds to the 1Ag → 1B1u transition, involving almost exclusively (97%) the one-electron HOMO–LUMO (H → L) excitation. This is calculated to fall at 963.0 nm (oscillator strength f = 0.436) in the gas phase and 1068.3 nm (f = 0.581) in CH2Cl2. The oscillator strength calculated at the TD-DFT level along with the experimental FWHM value of the NIR band were used to evaluate the molar extinction coefficient ε for 1 (eq 6).101 The symmetric and antisymmetric combinations of the α-107 → α-108 and β-107 → β-108 excitations (H,H → L,L double exciton states) are calculated as BS-GS → ES 1 and BS-GS → ES 2 transitions. Double exciton states have been reported for conjugated chromophores with open-shell diradical character,127 such as polyenes138,139 and quinoidal oligothiophenes.140 The double exciton state is one-photon forbidden, and it has been observed as a weak band at lower energies in comparison to the main absorption band due to the one-photon allowed single exciton state.127 The symmetry-allowed transition BS-GS → ES 2 falls at wavelength values lower (E = 1.487 eV, λmax = 833.6 nm, f = 0.310) than those predicted for the singlet GS (see above). The complex envelope of the NIR absorption band of neutral [M(R,R′timdt)2] bis(1,2-dithiolene) complexes can be attributed to the contribution of doubly excited states to the main single exciton states, thus possibly accounting for the unusually high molar extinction coefficients observed for the NIR absorption in this class of bis(1,2-dithiolene) complexes.46,49 The forbidden BS-GS → ES 1 transition (1.142 eV, λmax = 1085.6 nm) may provide a low-energy weak contribution138 to the NIR absorption due to the vibronic coupling with the B1u antisymmetric combination of the stretching Pd–S vibrations, calculated at 294.1 and 293.2 cm–1 at the RDFT and DFT-BS levels, respectively.
Figure 4.
Qualitative MO diagram showing the contributions of the HOMOs of the 1,2-dithiolene ligands to the HOMO and LUMO of complex 1 (a; c1 48%, c2 4%; c3 47.5%, c4 5%) and complex 3 (b; c1 61%, c2 31%, c3 8%; c4 24%, c5 70%, c6 6%) in the CS GS description. In the KS-MO drawings hydrogens have been omitted for clarity. Cutoff value: 0.05 |e|.
Figure 5.
Frontier KS-MO energy diagram (−2 to −8 eV) showing the energy and MO drawing in the restricted (CS GS, left) and singlet diradical (BS GS, right) descriptions for 1 (top) and 3 (bottom). Cutoff value: 0.05 |e|.
Electronic Structure of [Pd(Me2timdt)(mnt)] (3)
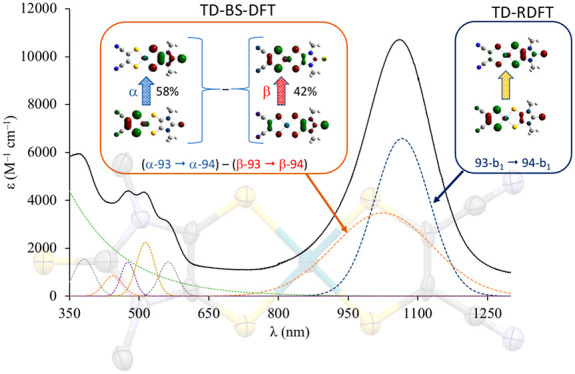
The GS geometry for 3 (EAbs298K = 3.784 and 4.872 eV in the gas phase and CH2Cl2, respectively) was optimized in its singlet CS (1B1), triplet open shell (3B1), and singlet diradical (BS) configurations. The triplet–singlet energy gap classifies 3 as a diradicaloid (Table 4).91,128 While the 3B1 state is sensibly less stable than the singlet state, the singlet diradical BS (⟨S2⟩BS = 0.436) and the 1B1 CS singlet configurations differ by less than 1 kcal mol–1 (Table 4). The singlet configurations calculated at the RDFT and DFT-BS levels show very close optimized Pd–S, C–C, and C–S bond distances, only very slightly overestimated (by less than 0.03 Å) in comparison to the relevant structural distances (Table 3). The difference ΔdC–C between the C–C bond distances dC–C of the two 1,2-dithiolene ligands (corresponding to the C(1)–C(1)i and C(4)–C(4)i structural bond lengths in Figure 1 for the Me2timdt and mnt ligands, respectively) is evaluated correctly (C(1)–C(1)i > C(4)–C(4)i), but it is slightly underestimated, so that the calculated dithione-dithiolato character is less pronounced than expected on the basis of the structural data (Table 3). The DFT-BS description of the GS provides a lower ΔdC–C value in comparison to the CS description. Accordingly, while the charge QPd on the central Pd ion in the CS and in the singlet diradical GSs is essentially unchanged (ΔQPd = 0.024 |e|), the difference in the charges calculated on the two 1,2-dithiolene ligands at the NBO level is sensibly larger in the former (0.514 and 0.390 |e|, respectively). In the RDFT approach, the optimized values for the C–C distances of the Me2timdt and mnt ligands correspond to noticeably different WBIs (1.168 and 1.417 in the 1B1 CS GS) and clearly fall in different regions of the dC–C vs WBIC–C correlation (Figure 3). Therefore, a comparison between structural and DFT-optimized data indicates that the CS description is more suitable than the singlet diradical description in modeling the GS of the mixed-ligand complex 3. Notably, the value of the C–C bond lengths within the mnt ligand in 3 (1.393 Å, Table 3) is even shorter than the value calculated for the free mnt2– ligand (1.406 Å, Table 2). On passing from 3 to the radical anion 3•– and the dianion 32–, only minor differences (lower than 0.02 Å) are observed on the mnt ligand, while the Me2timdt ligand is more greatly affected, the dC–C distance being progressively shortened (dC–C = 1.428, 1.397, and 1.364 Å within the Me2timdt ligand for 3, 3–, and 32–, respectively; Table 3). In summary, a comparison between calculated and experimental data supports the hypothesis that the GS of 3 can be better described as a dithione-dithiolato [PdII(Me2timdt)(mnt2–)] complex with a minor contribution from the singlet diradical [PdII(Me2timdt•–)(mnt•–)] description. Accordingly, the diradical character nDC = 24.9% (eq 5) is calculated for complex 3 (Table 4).141 The KS-HOMO and KS-LUMO in the CS description (MOs 93 and 94, respectively) are π-in-nature MOs mainly located on the mnt (61%) and Me2timdt (70%) ligands, respectively, with only minor contributions from the 4dxz AO of the Pd central atom (6% and 8%, respectively; Table S7). Hence, KS-HOMO and KS-LUMO in the heteroleptic complex can be considered as being derived from the in-phase and out-of-phase combinations of the SOMOs of the constituent ligands mnt•– and Me2timdt•– (Figure 4b and Figures S7 and S8). The former (“pull” electron-withdrawing ligand, HOMO at lower energy) contributes mostly to the KS-HOMO of 3 and assumes a larger character of 1,2-dithiolate, while the latter (“push” ligand, HOMO at higher energy) contributes mostly to the KS-LUMO of 3 and assumes a larger character of 1,2-dithione, in agreement with the structural data discussed above. In the DFT-BS description, the eigenvalues of the corresponding α- and β-MOs are remarkably unequal, reflecting their different compositions (Figure 5 (bottom) and Figure S9). In particular, the α-MO 93 is located on the mnt ligand (76%; Table S8 and (a) in Figure S9), while the β-MO 93 is less stable and is located largely on the Me2timdt ligand (63%; Table S8 and (c) in Figure S9). Conversely, the α-MO 94 is located almost entirely on the Me2timdt ligand (91%; Table S8 and (b) in Figure S9), while the β-MO 94 shows contributions from both ligands (mnt 54%, Me2timdt 36%; Table S8 and (d) in Figure S9).
Therefore, both the RDFT and DFT-BS approaches agree in attributing an LL′CT character to the lowest energy transition, from the mnt “pull” ligand to the Me2timdt “push” ligand. TD-RDFT calculations show, in excellent agreement with experimental data (Figure 2 and Figure S3 in the Supporting Information), three main spectral regions, namely (i) an overlap of intense transitions in the UV region (λ < 280 nm), (ii) a band in the visible region (300 ≤ λ ≤ 500 nm), and (iii) a single very intense NIR transition (λ > 800 nm). In Figure 6, the UV–vis–NIR spectrum of 3 in CHCl3 solution, simulated on the basis of singlet IEF-PCM TD-RDFT calculations (Table 5), is depicted. The absorption bands in the UV region are due to the overlap of several peaks involving the frontier orbitals, with intraligand (GS → ES 9), interligand (GS → ES 13), or ligand-to-metal (GS → ES 15 and GS → ES 17) character. The absorption in the visible region is mainly due to the GS → ES 7 intraligand transition, involving mainly the KS-HOMO-3 and KS-LUMO, centered on the Me2timdt ligand and the PdII ion. A single intense transition is calculated in the NIR region, falling at 1.489 eV (Table 5). This transition involves almost exclusively (88.4%) the one-electron excitation from the KS-HOMO (MO 93) to the KS-LUMO (MO 94). Notably, the oscillator strength f calculated for the NIR transition (0.315) is sensibly lower than that calculated for 1 (f = 0.436, see above), in agreement with the experimental values determined for the corresponding molar extinction coefficients. TD-RDFT calculations were carried out at the IEF-PCM level of theory in the same solvent systems experimentally adopted to record UV–vis–NIR spectra. The calculated NIR transition energies are generally overestimated but are linearly correlated to the experimental energies (R2 = 0.88; Ecalc (eV) = 0.374; Eexp + 0.979; Table 1 and Figure S10). Both in the gas phase and in the solvents considered at the IEF-PCM level, the NIR transition is attributed exclusively to the H → L one-electron excitation. Accordingly, a linear correlation holds between the calculated transition energies in the NIR region and the ΔEH–L energy gap evaluated in each of the examined solvents (Table 1; R2 = 0.92). Along the series CHCl3, CH2Cl2, THF, CH3CN, and DMF the contribution of the mnt fragment to KS-HOMO and KS-LUMO slightly increases (68% to 70%) and decreases (16% to 13%), respectively (Table S7). The contribution of the Me2timdt fragment to the KS-HOMO and to the KS-LUMO decreases (23% to 20%) and increases (78% to 82%), respectively. Therefore, on passing to CHCl3 to DMF, the NIR transition assumes a larger LL′CT character, and the ΔEH–L energy gap increases from 1.68 eV in CHCl3 to 1.77 eV in DMF and CH3CN, in agreement with the experimental trend of NIR absorption energies (Table 1). Calculated oscillator strengths f fall between 0.356 and 0.385 in DMF and CHCl3, respectively (Table S9). Oscillator strength values calculated at the TD-DFT level have been used along with experimental full widths at half-maximum (FWHM, w) to evaluate the ratio between the extinction coefficient in each solvent and that in CHCl3 solution. The resulting scaled calculated extinction coefficients εcalccorr (Table 1) progressively decrease with an increase in the transition energies. TD-DFT calculations were carried out on 3 in the singlet diradical electron configuration. Since the two α-93 → α-94 and β-93 → β-94 (H → L) excitations are not degenerate (Figure 5, bottom), the α-excitation contributes mainly (58.1%) to the symmetry-allowed transition at higher energy (0.853(α-93 → α-94) – 0.464(β-93 → β-94); E = 1.629 eV, λ = 761 nm, f = 0.267 in the gas phase), while the β-excitation contributes to the transition at lower energy (0.486(α-93 → α-94) + 0.881(β-93 → β-94); E = 0.746 eV, λ = 1663 nm, f = 0.033), forbidden in the C2v point group. Although the contribution of the singlet diradical description to the GS of 3 is limited, it is conceivable that the former transition, corresponding to a double exciton state, can provide a high-energy component to the NIR transition. Notably, due to its nature, the double exciton transition is predicted to show remarkable solvatochromic effects, thus accounting for the different spectral shapes observed on varying the solvent (Table 1 and Figure S6).
Figure 6.
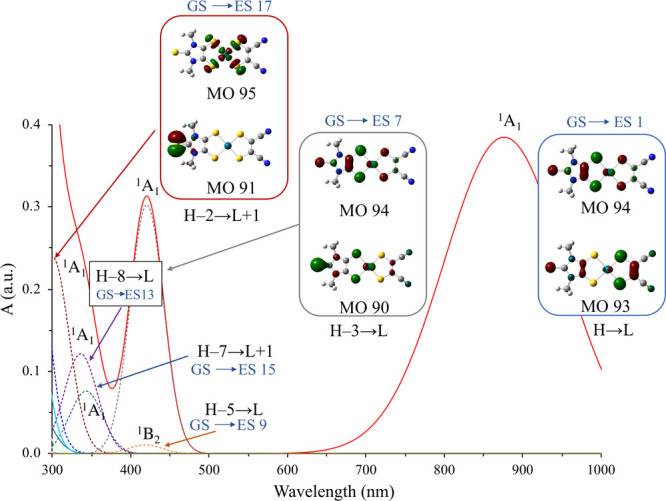
UV–vis–NIR spectrum of 3 simulated on the basis of IEF-PCM TD-RDFT calculations in CHCl3. FWHM w = 90 nm for the 1A1 (H → L) transition, and w = 25 nm for the UV–vis bands. Cutoff value: 0.05 |e|.
Table 5. Energies E (eV), Wavelengths λ (nm), and Oscillator Strengths f of the Main (f ≥ 0.005) UV–vis–NIR Electronic Transitions Calculated for 3 in the Gas Phase and in CH2Cl2 and CHCl3 at the IEF-PCM TD-RDFT Levela.
| gas phase |
CHCl3 |
CH2Cl2 |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| ESb | symm | E | λ | f | E | λ | f | E | λ | f | main contribution |
| 1 | 1A1 | 1.489 | 832.6 | 0.315 | 1.415 | 876.0 | 0.385 | 1.436 | 863.8 | 0.368 | H(93) → L(94) (100%) |
| 7 | 1A1 | 2.931 | 423.0 | 0.153 | 2.950 | 420.3 | 0.303 | 2.974 | 416.9 | 0.324 | H-3(90) → L(94) (91%) |
| 9 | 1B2 | 2.983 | 415.7 | 0.009 | 2.962 | 418.6 | 0.011 | 2.966 | 418.0 | 0.008 | H-5(88) → L(94) (92%) |
| 13 | 1A1 | 3.586 | 345.8 | 0.096 | 3.691 | 335.9 | 0.121 | 3.719 | 333.4 | 0.122 | H-8(85) → L(94) (91%) |
| 15 | 1B2 | 3.787 | 327.4 | 0.046 | 3.616 | 342.9 | 0.076 | 3.583 | 346.1 | 0.076 | H-7(86) → L+1(95 (15%), H(93) → L+2(96) (75%) |
| 17 | 1A1 | 4.133 | 300.1 | 0.121 | 4.135 | 299.9 | 0.240 | 4.132 | 300.1 | 0.250 | H-2(91) → L+1(95) (96%) |
| 20 | 1B2 | 4.364 | 284.2 | 0.005 | 4.294 | 288.8 | 0.007 | 4.274 | 290.1 | 0.006 | H-13(80) → L+1(95) (19%), H-9(84) → L+1(95) (65%) |
KS-HOMO (H) = MO 93; KS-LUMO (L) = MO 94.
Excited state (ES) numbering taken from gas-phase calculations.
Finally, the lack of an inversion center in the title complexes suggests a possible application of heteroleptic bis(1,2-dithiolene) complexes as second-order nonlinear optical (SONLO) materials. Prompted by the results obtained at TD-DFT level, since small geometrical differences can determine large differences in NLO properties,38,39 we calculated static dipole moments (μ) and static first (quadratic) hyperpolarizabilities (βtot) for 3 in the gas phase and in CH2Cl2 and CHCl3 solutions (Table S10).109 Calculations were also carried out at the DFT-BS level (Table S10) in the gas phase. For the sake of comparison, the same calculations were also undertaken, at the same level of theory, on [Pt(phen)(tdt)] (phen = 1,10-phenanthroline; tdt2– = 3,4-toluenedithiolate; Chart S2), a neutral diimine-dithiolate Pt complex showing a very large hyperpolarizability value among those investigated experimentally by means of EFISH measurements (λmax = 583 nm; βμ = −28 × 10–30 esu with ω = 1.569 × 1011 GHz; zero-frequency β0 = −16 × 10–30 esu).71 In agreement with the charge distribution within complex 3, the μ vector lies along the molecular z axis and β shows only tensor z components. As previously observed for different heteroleptic metal complexes containing 1,2-dithiolato ligands,62 a dramatic increase in βtot was calculated when solvation is taken into account (|βtot| = 37.6 × 10–30, 475.6 × 10–30, and 330.5 × 10–30 esu in the gas phase, CH2Cl2, and CHCl3, respectively). In addition, when the diradical character of 3 is evaluated at the DFT-BS level, the βtot value dramatically increases (177.4 × 10–30 esu in the gas phase), reaching the same order of magnitude computed for [Pt(phen)(tdt)].
Conclusions
DFT calculations have been exploited to investigate the structural and spectroscopic features of the heteroleptic mixed-ligand neutral complex PdII bis(1,2-dithiolene) 3, to highlight the differences between the homoleptic related complexes 1 and 4 and to develop sound structure–property relationships. The closed-shell (CS) description is only partially suitable to describe the electronic structure of bis(1,2-dithiolene) complexes, and—whatever the nature of the ligands—the singlet diradical character (DC) must be taken into account. The broken-symmetry (BS) approach within DFT, although itself a dramatic approximation underestimating the DC of bis(1,2-dithiolene) metal complexes, is a useful tool in supplementing the description of the ground state (GS). A few general conclusions can be drawn
-
(1)
The nature of the 1,2-dithiolene ligand is responsible for the relevance of diradical character (DC) in the GS of 1,2-dithiolene complexes. In homoleptic neutral bis(1,2-dithiolene) complexes, on passing from complex 4 to complex 1, the nDC index is roughly doubled. This can be related to the capability of the ligands mnt•– and Me2timdt•–, respectively, to stabilize the unpaired electron. In heteroleptic mixed-ligand complexes, the absolute one-electron-reduction potentials EAbs298K calculated for the L/L•– and L′/L′•– couples can be used to evaluate the nature of the [PdII(L)(L′)] complex. The 1,2-dithiolene ligand displaying the largest reduction potential (“pull” ligand) features its π-NBMO at lower energy and contributes largely to the KS-HOMO of the heteroleptic complex, while that with the lowest potential (“pull” ligand) contributes to the KS-LUMO. As a consequence, it is conceivable that the difference ΔEAbs in the absolute reduction potentials of the ligands L and L′ can be adopted as a useful parameter to estimate the push–pull nature of the resulting heteroleptic neutral complexes [PdII(L)(L′)] and the different localizations of the KS-HOMO and KS-LUMO. A larger push–pull character points to a larger dithione-dithiolato nature and a lower DC of the complex. This implies that the DC is the largest in homoleptic bis(1,2-dithiolene) complexes [PdII(L)2] with ligands L featuring low values of EAbs298K, such as Me2timdt, and decreases in heteroleptic complexes [PdII(L)(L′)] in dependence on ΔEAbs.
-
(2)
Several authors have observed that metal–sulfur bond lengths optimized at the DFT level are slightly overestimated in comparison to structural bond distances. This can be attributed to the use of RDFT calculations in complexes featuring a significant DC. The DFT-BS approach leads to bond distances closer to the structural distances. It can be deduced that, in the case of complexes with a large DC, such as complex 1, the difference between CS-optimized distances and the relevant experimental metric parameters increases with the DC of the complex.
-
(3)
The spectral shape of the NIR band of neutral bis(1,2-dithiolene) metal complexes has been indicated to be a complex envelope resulting from a series of d–d transitions with different spin couplings to the open-shell ligands. The intensity of this band, peculiar to metal bis(1,2-dithiolene) complexes, may be attributed not only to the very large oscillator strength f calculated for the HOMO–LUMO one-electron excitation within a CS description but also to the contribution of double exciton states typical of diradical species. To a lower extent, double exciton states are possible also in heteroleptic bis(1,2-dithiolene) complexes and can be related to the spectral shapes observed for the NIR band in different solvents.
-
(4)
The intrinsic optical nonlinearity of heteroleptic bis(1,2-dithiolene) complexes is enhanced by their DC, providing a further criterion, in addition to the lack of an inversion center and large electric dipole moment values, for the rational design of NLO materials active in the vis–NIR region.
Summarily, this investigation shows that the DC of bis(1,2-dithiolene) metal complexes can be extensively modulated by means of the choice of the substituents R at the 1,2-dithiolene core, allowing for the rational design of the linear and nonlinear optical properties of the resulting complexes and hence the possibility of applying them in fields as varied as nonlinear optics, photoconductivity, and electrochromism.
Further studies are ongoing in our laboratory to investigate in detail the role of the central metal ion and to generalize the limited findings described here for the PdII complexes with the mnt and Me2timdt ligands to other homoleptic and heteroleptic bis(1,2-dithiolene) complexes differing in the nature of the central metal ions and 1,2-dithiolene ligands.
Acknowledgments
M.A., M.C.A., C.C., and V.L. thank the Fondazione di Sardegna (FdS) and Regione Autonoma della Sardegna (RAS) (Progetti Biennali di Ateneo FdS/RAS annualità 2018) for financial support. A.P. acknowledges the RAS for funding in the context of the POR FSE 2014–2020 (CUP F24J17000190009).
Glossary
Abbreviations
- BS
broken symmetry
- CS
closed shell
- DC
diradical character
- GS
ground state
- MO
molecular orbital
- HOMO
highest occupied molecular orbital
- KS
Kohn–Sham
- LUMO
lowest unoccupied molecular orbital
- NBMO
nonbonding molecular orbital
- NLO
nonlinear optics
- SONLO
second-order nonlinear optics.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.inorgchem.0c02696.
Details on theoretical calculations, molecular schemes for the compound discussed in the paper, crystallographic data and packing details for complex 3, experimental UV–vis–NIR spectra decomposed into their component Gaussian peaks, NIR spectra in MeCN, DMF, THF, CH2Cl2, and CHCl3, KS frontier MO drawings calculated for Me2timdtq–, mntq– (q = 0, 1, 2), and complex 3, RDFT, DFT-BS, and (IEF-PCM) TD-DFT data for 1 and 3, and calculated second-order hyperpolarizabilities β and dipole moments μ (PDF)
Accession Codes
CCDC 2027023 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.
Author Contributions
The manuscript was written through contributions of all authors.
The authors declare no competing financial interest.
Supplementary Material
References
- McCleverty J. A. Metal 1, 2-dithiolene and related complexes. Prog. Inorg. Chem. 2007, 10, 49–221. 10.1002/9780470166116.ch2. [DOI] [Google Scholar]
- Mueller-Westerhoff U. T.; Vance B.; Yoon D. I. The synthesis of Dithiolene Dyes with Strong Near-IR Absorption. Tetrahedron 1991, 47, 909–932. 10.1016/S0040-4020(01)80932-7. [DOI] [Google Scholar]
- Mueller-Westerhoff U. T.; Vance B.. Dithiolenes and Related Species Comprehensive Coordination Chemistry 2; Pergamon Press: 1987; Chapter 16.5, pp 595–631. [Google Scholar]
- Dithiolene Chemistry: Synthesis, Properties, and Applications; Stiefel E. I., Ed.; Wiley: Hoboken, 2004. [Google Scholar]
- Garreau-de Bonneval B.; Moineau-Chane Ching K. I.; Alary F.; Bui T.-T.; Valade L. Neutral d8 metal bis-dithiolene complexes: synthesis, electronic properties and applications. Coord. Chem. Rev. 2010, 254, 1457–1467. 10.1016/j.ccr.2010.02.019. [DOI] [Google Scholar]
- Arca M.; Aragoni M. C.; Pintus A. In Handbook of chalcogen chemistry ;Devillanova F. A., du Mont W.-W., Eds.; RSC Publishing: Cambridge, 2013; pp 127–179. [Google Scholar]
- Robertson N.; Cronin L. Metal bis-1,2-dithiolene complexes in conducting or magnetic crystalline assemblies. Coord. Chem. Rev. 2002, 227, 93–127. 10.1016/S0010-8545(01)00457-X. [DOI] [Google Scholar]
- Kobayashi A.; Fujiwara E.; Kobayashi H. Single-Component Molecular Metals with Extended-TTF Dithiolate Ligands. Chem. Rev. 2004, 104, 5243–5264. 10.1021/cr030656l. [DOI] [PubMed] [Google Scholar]
- Kato R. Conducting metal dithiolene complexes: Structural and electronic properties. Chem. Rev. 2004, 104, 5319–5346. 10.1021/cr030655t. [DOI] [PubMed] [Google Scholar]
- Faulmann C.; Jacob K.; Dorbes S.; Lampert S.; Malfant I.; Doublet M.-L.; Valade L.; Real J. A. Electrical Conductivity and Spin Crossover: A New Achievement with a Metal Bis Dithiolene Complex. Inorg. Chem. 2007, 46 (21), 8548–8559. 10.1021/ic062461c. [DOI] [PubMed] [Google Scholar]
- Nunes J.; Figuera M.; Belo D.; Santos I.; Ribeiro B.; Lopes E.; Heniriques R.; Vidal-Gancedo J.; Veciana J.; Rovira C.; Almeida M. Transition Metal Bisdithiolene Complexes Based on Extended Ligands with Fused Tetrathiafulvalene and Thiophene Moieties: New Single-Component Molecular Metals. Chem. - Eur. J. 2007, 13, 9841–9849. 10.1002/chem.200701050. [DOI] [PubMed] [Google Scholar]
- Faulmann C.; Cassoux P. Solid-State Properties (Electronic, Magnetic, Optical) of Dithiolene Complex-Based Compounds. Prog. Inorg. Chem. 2004, 52, 399–489. 10.1002/0471471933.ch8. [DOI] [Google Scholar]
- Shen W.-C.; Huo P.; Huang Y.-D.; Yin J.-X.; Zhu Q.-Y.; Dai J. Photocurrent responsive films prepared from a nickel-dithiolate compound with directly bonded pyridyl groups. RSC Adv. 2014, 4, 60221–60226. 10.1039/C4RA10991K. [DOI] [Google Scholar]
- Naito T.; Karasudani T.; Nagayama N.; Ohara K.; Konishi K.; Mori S.; Takano T.; Takahashi Y.; Inabe T.; Kinose S.; Nishihara S.; Inoue K. Giant Photoconductivity in NMQ[Ni(dmit)2]. Eur. J. Inorg. Chem. 2014, 2014, 4000–4009. 10.1002/ejic.201402035. [DOI] [Google Scholar]
- Dalgleish S.; Matsushita M. M.; Hu L.; Li B.; Yoshikawa H.; Awaga K. Utilizing photocurrent transients for dithiolene-based photodetection: stepwise improvements at communications relevant wavelengths. J. Am. Chem. Soc. 2012, 134, 12742–12750. 10.1021/ja304228c. [DOI] [PubMed] [Google Scholar]
- Aragoni M. C.; Arca M.; Caironi M.; Denotti C.; Devillanova F. A.; Grigiotti E.; Isaia F.; Laschi F.; Lippolis V.; Natali D.; Pala L.; Sampietro M.; Zanello P. Monoreduced [M(R,R′timdt)2]− dithiolenes (M = Ni, Pd, Pt; R,R′timdt = disubstituted imidazolidine-2,4,5-trithione): solid state photoconducting properties in the third optical fiber window. Chem. Commun. 2004, 1882–1883. 10.1039/B406406B. [DOI] [PubMed] [Google Scholar]
- Pintus A.; Ambrosio L.; Aragoni M. C.; Binda M.; Coles S. J.; Hursthouse M. B.; Isaia F.; Lippolis V.; Meloni G.; Natali D.; Orton J. B.; Podda E.; Sampietro M.; Arca M. Photoconducting Devices with Response in the Visible–Near-Infrared Region Based on Neutral Ni Complexes of Aryl-1,2-dithiolene Ligands. Inorg. Chem. 2020, 59, 6410–6421. 10.1021/acs.inorgchem.0c00491. [DOI] [PubMed] [Google Scholar]
- Mueller-Westerhoff U. T.; Vance B.; Yoon D. I. The synthesis of dithiolene dyes with strong near-IR absorption. Tetrahedron 1991, 47, 909–932. 10.1016/S0040-4020(01)80932-7. [DOI] [Google Scholar]
- Liu Y.; Zhang Z.; Chen X.; Xu S.; Cao S. Near-infrared absorbing dyes at 1064 nm: Soluble dithiolene nickel complexes with alkylated electron-donating groups as Peripheral substituents. Dyes Pigm. 2016, 128, 179–189. 10.1016/j.dyepig.2016.01.026. [DOI] [Google Scholar]
- Chatzikyriakos G.; Papagiannouli I.; Couris S.; Anyfantis G.C.; Papavassiliou G.C. Nonlinear optical response of a symmetrical Au dithiolene complex under ps and ns laser excitation in the infrared and in the visible. Chem. Phys. Lett. 2011, 513, 229–235. 10.1016/j.cplett.2011.07.091. [DOI] [Google Scholar]
- Guo W. F.; Sun X. B.; Sun J.; Wang X. Q.; Zhang G. H.; Ren Q.; Xu D. Nonlinear optical absorption of a metal dithiolene complex irradiated by different laser pulses at near-infrared wavelengths. Chem. Phys. Lett. 2007, 435, 65–68. 10.1016/j.cplett.2006.12.057. [DOI] [Google Scholar]
- Cassano T.; Tommasi R.; Nitti L.; Aragoni M. C.; Arca M.; Denotti C.; Devillanova F. A.; Isaia F.; Lippolis V.; Lelj F.; Romaniello P. Picosecond absorption saturation dynamics in neutral [M(R,R′timdt)2] metal-dithiolenes. J. Chem. Phys. 2003, 118, 5995–6002. 10.1063/1.1556612. [DOI] [Google Scholar]
- Arca M.; Aragoni M. C.; Pintus A. In Handbook of chalcogen chemistry ;Devillanova F. A., du Mont W.-W., Eds.; RSC Publishing: Cambridge, 2013; pp 127–179. [Google Scholar]
- Barriere F.; Camire N.; Geiger W. E.; Mueller-Westerhoof U. T.; Sanders R. Use of Medium Effects to Tune the ΔE1/2 Values of Bimetallic and Oligometallic Compounds. J. Am. Chem. Soc. 2002, 124, 7262–7263. 10.1021/ja020309d. [DOI] [PubMed] [Google Scholar]
- Chirik P. J. Preface: forum on redox-active ligands. Inorg. Chem. 2011, 50, 9737–9740. 10.1021/ic201881k. [DOI] [PubMed] [Google Scholar]
- Allwright E.; Silber G.; Crain J.; Matsushita M. M.; Awaga K.; Robertson N. Electrochemical deposition of highly-conducting metal dithiolene films. Dalton Trans. 2016, 45, 9363–9368. [DOI] [PubMed] [Google Scholar]
- Eisenberg R.; Gray H. B. Noninnocence in metal complexes: A dithiolene dawn. Inorg. Chem. 2011, 50, 9741–9751. 10.1021/ic2011748. [DOI] [PubMed] [Google Scholar]
- Periyasamy G.; Burton N. A.; Hillier I. H.; Vincent M. A.; Disley H.; McMaster J.; Garner C. D. The dithiolene ligand--’innocent’ or ’non-innocent’? A theoretical and experimental study of some cobalt-dithiolene complexes. Faraday Discuss. 2007, 135, 469–488. 10.1039/B607144A. [DOI] [PubMed] [Google Scholar]
- Lewis G. R.; Dance I. Crystal supramolecular motifs for [Ph4P]+ salts of [M(mnt)2]2–, [M(mnt)2]−, [{M(mnt)2}2]2–, [M(mnt)3]3– and [M(mnt)3]2– (mnt2– = maleonitriledithiolate). J. Chem. Soc., Dalton Trans. 2000, 3176–3185. 10.1039/b000093k. [DOI] [Google Scholar]
- Lim B. S.; Fomitchev D. V.; Holm R. H. Nickel Dithiolenes Revisited: Structures and Electron Distribution from Density Functional Theory for the Three-Member Electron-Transfer Series [Ni(S2C2Me2)2]0,1–,2–. Inorg. Chem. 2001, 40, 4257–4262. 10.1021/ic010138y. [DOI] [PubMed] [Google Scholar]
- Madhu V.; Das S. K. New Series of Asymmetrically Substituted Bis(1,2-dithiolato)-Nickel (III) Complexes Exhibiting Near IR Absorption and Structural Diversity. Inorg. Chem. 2008, 47, 5055–5070. 10.1021/ic701408m. [DOI] [PubMed] [Google Scholar]
- Masui H. Metalloaromaticity. Coord. Chem. Rev. 2001, 219–221, 957–992. 10.1016/S0010-8545(01)00389-7. [DOI] [Google Scholar]
- Ray K.; Weyhermüller T.; Neese F.; Wieghardt K. Electronic Structure of Square Planar Bis(benzene-1,2-dithiolato)metal Complexes [M(L)2]z (z = 2–, 1–, 0; M = Ni, Pd, Pt, Cu, Au): An Experimental, Density Functional, and Correlated ab Initio Study. Inorg. Chem. 2005, 44, 5345–5360. 10.1021/ic0507565. [DOI] [PubMed] [Google Scholar]
- Szilagyi R. K.; Lim B. S.; Glaser T.; Holm R. H.; Hedman B.; Hodgson K. O.; Solomon E. I. Description of the Ground State Wave Functions of Ni Dithiolenes Using Sulfur K-edge X-ray Absorption Spectroscopy. J. Am. Chem. Soc. 2003, 125, 9158–9169. 10.1021/ja029806k. [DOI] [PubMed] [Google Scholar]
- Li X. Y.; Sun Y. G.; Huo P.; Shao M. Y.; Ji S. F.; Zhu Q. Y.; Dai J. Metal centered oxidation or ligand centered oxidation of metal dithiolene? Spectral, electrochemical and structural studies on a nickel-4-pyridine-1,2-dithiolate system. Phys. Chem. Chem. Phys. 2013, 15, 4016–4023. 10.1039/c3cp44054k. [DOI] [PubMed] [Google Scholar]
- Ambrosio L.; Aragoni M. C.; Arca M.; Devillanova F. A.; Hursthouse M. B.; Huth S. L.; Isaia F.; Lippolis V.; Mancini A.; Pintus A. Synthesis and Characterization of Novel Gold (III) Complexes of Asymmetrically Aryl-Substituted 1,2-Dithiolene Ligands Featuring Potential-Controlled Spectroscopic Properties. Chem. - Asian J. 2010, 5, 1395–1406. 10.1002/asia.200900690. [DOI] [PubMed] [Google Scholar]
- Bachler V.; Olbrich G.; Neese F.; Wieghardt K. Theoretical evidence for the singlet diradical character of square planar nickel complexes containing two o-semiquinonato type ligands. Inorg. Chem. 2002, 41, 4179–4193. 10.1021/ic0113101. [DOI] [PubMed] [Google Scholar]
- Avramopoulos A.; Reis H.; Mousdis G.; Papadopoulos M. G. Ni Dithiolenes – A Theoretical Study on Structure–Property Relationships. Eur. J. Inorg. Chem. 2013, 2013, 4839–4850. 10.1002/ejic.201300534. [DOI] [Google Scholar]
- Avramopoulos A.; Otero N.; Reis H.; Karamanis P.; Papadopoulos M. G. A computational study of photonic materials based on Ni bis(dithiolene) fused with benzene, possessing gigantic second hyperpolarizabilities. J. Mater. Chem. C 2018, 6, 91–110. 10.1039/C7TC05047J. [DOI] [Google Scholar]
- Dalgleish S.; Robertson N. A stable near IR switchable electrochromic polymer based on an indole-substituted nickel dithiolene. Chem. Commun. 2009, 5826–5828. 10.1039/b913174d. [DOI] [PubMed] [Google Scholar]
- Basu P.; Nigam A.; Mogesa B.; Denti S.; Nemykin V. Synthesis, characterization, spectroscopy, electronic and redox properties of a new nickel dithiolene system. Inorg. Chim. Acta 2010, 363, 2857–2864. 10.1016/j.ica.2010.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aragoni M. C.; Arca M.; Cassano T.; Denotti C.; Devillanova F. A.; Isaia F.; Lippolis V.; Natali D.; Nitti L.; Sampietro M.; Tommasi R. Photoinduced conductivity and nonlinear optical properties of [M(R,R’timdt)2] dithiolenes (M = Ni, Pd, Pt; R,R’timdt = monoreduced imidazolidine-2,4,5-trithione) as materials for optically driven switches and photodetectors. Inorg. Chem. Commun. 2002, 5, 869–872. 10.1016/S1387-7003(02)00582-8. [DOI] [Google Scholar]
- Natali D.; Sampietro M.; Arca M.; Denotti C.; Devillanova F. A. Wavelenght selective photodetectors for Near-Infrared applications based on novel neutral dithiolenes. Synth. Met. 2003, 137, 1489–1490. 10.1016/S0379-6779(02)01212-2. [DOI] [Google Scholar]
- Caironi M.; Natali D.; Sampietro M.; Ward M.; Meacham A.; Devillanova F. A.; Arca M.; Denotti C.; Pala L. Near-infrared detection by means of coordination complexes. Synth. Met. 2005, 153, 273–276. 10.1016/j.synthmet.2005.07.107. [DOI] [Google Scholar]
- Arca M.; Demartin F.; Devillanova F. A.; Garau A.; Isaia F.; Lelj F.; Lippolis V.; Pedraglio S.; Verani G. Synthesis, X-ray crystal structure and spectroscopic characterization of the new dithiolene [Pd(Et2timdt)2] and of its adduct with molecular diiodine [Pd(Et2timdt)2]·I2·CHCl3 (Et2timdt = monoanion of 1,3-diethylimidazolidine-2,4,5-trithione). J. Chem. Soc., Dalton Trans. 1998, 3731–3736. 10.1039/a805494k. [DOI] [Google Scholar]
- Aragoni M.; Arca M.; Demartin F.; Devillanova F. A.; Garau A.; Isaia F.; Lelj F.; Lippolis V.; Verani G. New [M(R,R’timdt)2] metal-dithiolenes and related compounds (M = Ni, Pd, Pt; R,R’timdt = monoanion of di-substituted imidazolidine-2,4,5-trithiones): an experimental and theoretical investigation. J. Am. Chem. Soc. 1999, 121, 7098–7107. 10.1021/ja990827x. [DOI] [Google Scholar]
- Cassano T.; Tommasi R.; Nitti L.; Aragoni M. C.; Arca M.; Denotti C.; Devillanova F. A.; Isaia F.; Lippolis V.; Lelj F.; Romaniello P. Picosecond absorption saturation dynamics in neutral [M(R,R’timdt)2] metal-dithiolenes. J. Chem. Phys. 2003, 118, 5995–6002. 10.1063/1.1556612. [DOI] [Google Scholar]
- Aragoni M. C.; Arca M.; Denotti C.; Devillanova F. A.; Grigiotti E.; Isaia F.; Laschi F.; Lippolis V.; Pala L.; Slawin A. M. Z.; Zanello P.; Woollins J. D. A facile synthesis via [Pd(Et2timdt)Br2] of a push-pull mixed-ligand Pd-dithiolene containing the Et2timdt ligand (Et2timdt = diethylimidazolidine-2,4,5-trithione). Eur. J. Inorg. Chem. 2003, 2003, 1291–1295. 10.1002/ejic.200390165. [DOI] [Google Scholar]
- Aragoni M. C.; Arca M.; Cassano T.; Denotti C.; Devillanova F. A.; Frau R.; Isaia F.; Lelj F.; Lippolis V.; Nitti L.; Romaniello P.; Tommasi R.; Verani G. NIR Dyes Based on [M(R,R’timdt)2] Metal-Dithiolenes: Additivity of M, R, and R’ Contributions To Tune the NIR Absorption (M = Ni, Pd, Pt; R,R’timdt = Monoreduced Form of Disubstituted Imidazolidine-2,4,5-trithione). Eur. J. Inorg. Chem. 2003, 2003, 1939–1947. 10.1002/ejic.200200602. [DOI] [Google Scholar]
- Aragoni M. C.; Arca M.; Demartin F.; Devillanova F. A.; Lelj F.; Isaia F.; Lippolis V.; Mancini A.; Pala L.; Verani G. A theoretical investigation of the donor ability of [M(R,R’timdt)2] dithiolenes towards molecular diiodine (M = Ni, Pd, Pt; R,R’timdt = formally monoreduced disubstituted imidazolidine-2,4,5-trithione). Eur. J. Inorg. Chem. 2004, 2004, 3099–3109. 10.1002/ejic.200300953. [DOI] [Google Scholar]
- Aragoni M.; Arca M.; Caironi M.; Denotti C.; Devillanova F. A.; Grigiotti E.; Isaia F.; Laschi F.; Lippolis V.; Natali D.; Pala L.; Sampietro M.; Zanello P. Monoreduced [M(R,R’timdt)2]− dithiolenes: solid state photoconducting properties in the third optical fiber window. Chem. Commun. 2004, 1882–1883. 10.1039/B406406B. [DOI] [PubMed] [Google Scholar]
- Deiana C.; Aragoni M. C.; Isaia F.; Lippolis V.; Pintus A.; Slawin A. M. Z.; Woollins J. D.; Arca M. Structural tailoring of the NIR-absorption of bis(1,2-dichalcogenolene) Ni/Pt electrochromophores deriving from 1,3-dimethyl-2-chalcogenoxo-imidazoline-4,5-dichalcogenolato. New J. Chem. 2016, 40, 8206–8210. 10.1039/C6NJ01854H. [DOI] [Google Scholar]
- Ahmadi M.; Fischer C.; Ghosh A. C.; Schultze C. An Asymmetrically Substituted Aliphatic Bis-Dithiolene Mono-Oxido Molybdenum (IV) Complex With Ester and Alcohol Functions as Structural and Functional Active Site Model of Molybdoenzymes. Front. Chem. 2019, 7, 486. 10.3389/fchem.2019.00486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C. – T.; Liao S. – Y.; Lin K. – J.; Lai L. – L. Syntheses, Charge Distribution, and Molecular Second-Order Nonlinear Optical Properties of Push–Pull Bisdithiolene Nickel Complexes. Adv. Mater. 1998, 10, 334–338. . [DOI] [Google Scholar]
- Chen X.-R.; Xue C.; Liu S.-X.; Cai B.; Wang J.; Tao J.-Q.; Xue Y.-S.; Huang X.-C.; Ren X.-M.; Liu J.-L. Investigation on crystal structure, magnetic and near-infrared absorption properties of a novel heteroleptic nickel-bis-1,2-dithiolene compound. Polyhedron 2017, 132, 12–19. 10.1016/j.poly.2017.04.025. [DOI] [Google Scholar]
- Obanda A.; Martinez K.; Schmehl R. H.; Mague J. T.; Rubtson I. V.; MacMillan S. N.; Lancaster K. M.; Sproules S.; Donahue J. P. Expanding the Scope of Ligand Substitution from [M(S2C2Ph2] (M = Ni2+, Pd2+, Pt2+) To Afford New Heteroleptic Dithiolene Complexes. Inorg. Chem. 2017, 56, 10257–10267. 10.1021/acs.inorgchem.7b00971. [DOI] [PubMed] [Google Scholar]
- A search of the Cambridge structural database for heteroleptic bis(1,2-dithiolene) metal complexes [M(L)(L′)] shows 33 examples for M = Ni and only 7 with M = Pd. Among these 40 complexes, 9 have the mnt ligand.
- Jeannin O.; Delaunay J.; Barrière F.; Fourmigué M. Between Ni(mnt)2 and Ni(tfd)2 Dithiolene Complexes: the Unsymmetrical 2-(Trifluoromethyl)acrylonitrile-1,2-dithiolate and Its Nickel Complexes. Inorg. Chem. 2005, 44, 9763–9770. 10.1021/ic051226b. [DOI] [PubMed] [Google Scholar]
- Papavassiliou G. C.; Anyfantis G. C.; Mousdis G. A. Neutral Metal 1,2-Dithiolenes: Preparations, Properties and Possible Applications of Unsymmetrical in Comparison to the Symmetrical. Crystals 2012, 2, 762–811. 10.3390/cryst2030762. [DOI] [Google Scholar]
- Anyfantisa G. C.; Papavassiliou G. C.; Aloukos P.; Couris S.; Weng Y. F.; Yoshino H.; Murata K. Unsymmetrical Single-Component Nickel 1,2-Dithiolene Complexes with Extended Tetrachalcogenafulvalenedithiolato Ligands. Z. Naturforsch., B: J. Chem. Sci. 2007, 62, 200–204. 10.1515/znb-2007-0209. [DOI] [Google Scholar]
- Papavassiliou G. C.; Anyfantis G. C.; Steele B. R.; Terzis A.; Raptopoulou C. P.; Tatakis G.; Chaidogiannos G.; Glezos N.; Weng Y.; Yoshino H.; Murata K. Some New Nickel 1,2-Dichalcogenolene Complexes as Single-component Semiconductors. Z. Naturforsch., B: J. Chem. Sci. 2007, 62, 679–684. 10.1515/znb-2007-0509. [DOI] [Google Scholar]
- Pintus A.; Aragoni M. C.; Bellec N.; Devillanova F. A.; Lorcy D.; Isaia F.; Lippolis V.; Randall R. A. M.; Roisnel T.; Slawin A. M. Z.; Woollins J. D.; Arca M. Structure–Property Relationships in PtII Diimine-Dithiolate Nonlinear Optical Chromophores Based on Arylethylene-1,2-dithiolate and 2-Thioxothiazoline-4,5-dithiolate. Eur. J. Inorg. Chem. 2012, 2012, 3577–3594. 10.1002/ejic.201200346. [DOI] [Google Scholar]
- Pintus A.; Aragoni M. C.; Coles S. J.; Coles S. L.; Isaia F.; Lippolis V.; Musteti A.-D.; Teixidor F.; Viñas C.; Arca M. New PtII diimine-dithiolate complexes containing a 1,2-dithiolate-1,2-closo-dicarbadodecarborane: an experimental and theoretical investigation. Dalton Trans. 2014, 43, 13649–13660. 10.1039/C4DT01929F. [DOI] [PubMed] [Google Scholar]
- Pintus A.; Aragoni M. C.; Isaia F.; Lippolis V.; Lorcy D.; Slawin A. M. Z.; Woollins J. D.; Arca M. On the Role of Chalcogen Donor Atoms in Diimine-Dichalcogenolate PtII SONLO Chromophores: Is It Worth Replacing Sulfur with Selenium?. Eur. J. Inorg. Chem. 2015, 2015, 5163–5170. 10.1002/ejic.201500777. [DOI] [Google Scholar]
- Aragoni M. C.; Arca M.; Crisponi G.; Nurchi V. M. Simultaneous decomposition of several spectra into the constituent Gaussian peaks. Anal. Chim. Acta 1995, 316, 195–204. 10.1016/0003-2670(95)00354-3. [DOI] [Google Scholar]
- Wojdyr M. Fityk: a general-purpose peak fitting program. J. Appl. Crystallogr. 2010, 43, 1126–1128. 10.1107/S0021889810030499. [DOI] [Google Scholar]
- Bruno I. J.; Cole J. C.; Edgington P. R.; Kessler M.; Macrae C. F.; McCabe P.; Pearson J.; Taylor R. New software for searching the Cambridge Structural Database and visualizing crystal structures. Acta Crystallogr., Sect. B: Struct. Sci. 2002, 58, 389–397. 10.1107/S0108768102003324. [DOI] [PubMed] [Google Scholar]
- Sheldrick G. M.SHELXS-97, Program for Crystal Structure Solution; University of Göttingen: Göttingen, Germany, 1997.
- Sheldrick G. M.; SHELXL-97, Program for X-ray Crystal Structure Refinement; University of Göttingen: Göttingen, Germany, 1997.
- Arca M.; Demartin F.; Devillanova F. A.; Isaia F.; Lelj F.; Lippolis V.; Verani G. An experimental and theoretical approach to the study of the properties of parabanic acid and related compounds: synthesis and crystal structure of diethylimidazolidine-2-selone-4,5- dione. Can. J. Chem. 2000, 78, 1147–1157. 10.1139/v00-117. [DOI] [Google Scholar]
- Cummings S. D.; Cheng L.-T.; Eisenberg R. Metalloorganic Compounds for Nonlinear Optics: Molecular Hyperpolarizabilities of M(diimine)(dithiolate) Complexes (M = Pt, Pd, Ni). Chem. Mater. 1997, 9, 440–450. 10.1021/cm960222p. [DOI] [Google Scholar]
- Wang Y.; Hickox H. P.; Xie Y.; Wei P.; Blair S. A.; Johnson M. K.; Schaefer H. F. III; Robinson G. H. A Stable Anionic Dithiolene Radical. J. Am. Chem. Soc. 2017, 139, 6859–6862. 10.1021/jacs.7b03794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Xie Y.; Wei P.; Blair S. A.; Cui D.; Johnson M. K.; Schaefer H. F. III; Robinson G. H. Stable Boron Dithiolene Radicals. Angew. Chem., Int. Ed. 2018, 57, 7865–7868. 10.1002/anie.201804298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karthik V.; Bhat I. A.; Anantharaman G. Backbone Thio-Functionalized Imidazol-2-ylidene–Metal Complexes: Synthesis, Structure, Electronic Properties, and Catalytic Activity. Organometallics 2013, 32, 7006–7013. 10.1021/om400585b. [DOI] [Google Scholar]
- Koch W.; Holthausen M. C.. A Chemist’s Guide to Density Functional Theory, 2nd ed.; Wiley-VCH: Weinheim, 2002. [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Petersson G. A.; Nakatsuji H.; Li X.; Caricato M.; Marenich A. V.; Bloino J.; Janesko B. G.; Gomperts R.; Mennucci B.; Hratchian H. P.; Ortiz J. V.; Izmaylov A. F.; Sonnenberg J. L.; Williams-Young D.; Ding F.; Lipparini F.; Egidi F.; Goings J.; Peng B.; Petrone A.; Henderson T.; Ranasinghe D.; Zakrzewski V. G.; Gao J.; Rega N.; Zheng G.; Liang W.; Hada M.; Ehara M.; Toyota K.; Fukuda R.; Hasegawa J.; Ishida M.; Nakajima T.; Honda Y.; Kitao O.; Nakai H.; Vreven T.; Throssell K.; Montgomery J. A. Jr.; Peralta J. E.; Ogliaro F.; Bearpark M. J.; Heyd J. J.; Brothers E. N.; Kudin K. N.; Staroverov V. N.; Keith T. A.; Kobayashi R.; Normand J.; Raghavachari K.; Rendell A. P.; Burant J. C.; Iyengar S. S.; Tomasi J.; Cossi M.; Millam J. M.; Klene M.; Adamo C.; Cammi R.; Ochterski J. W.; Martin R. L.; Morokuma K.; Farkas O.; Foresman J. B.; Fox D. J.. Gaussian 16, Rev. B.01; Gaussian, Inc.: Wallingford, CT, 2016.
- Becke A. D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. 10.1063/1.464913. [DOI] [Google Scholar]
- Adamo C.; Barone V. Exchange functionals with improved long-range behavior and adiabatic connection methods without adjustable parameters: The mPW and mPW1PW models. J. Chem. Phys. 1998, 108, 664–675. 10.1063/1.475428. [DOI] [Google Scholar]
- Adamo C.; Barone V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. 10.1063/1.478522. [DOI] [Google Scholar]
- Dunning T. H. Jr.; Hay P. J. In Methods of Electronic Structure, Theory; Schaefer H. F. III, Ed.; Plenum Press: 1977; Vol. 2. [Google Scholar]
- Ortiz J. V.; Hay P. J.; Martin R. L. Role of d and f orbitals in the geometries of low-valent actinide compounds. J. Am. Chem. Soc. 1992, 114, 2736–2737. 10.1021/ja00033a068. [DOI] [Google Scholar]
- Roy L. E.; Hay P. J.; Martin R. L. Revised Basis Sets for the LANL Effective Core Potentials. J. Chem. Theory Comput. 2008, 4, 1029–1031. 10.1021/ct8000409. [DOI] [PubMed] [Google Scholar]
- Stevens W. J.; Krauss M.; Basch H.; Jasien P. G. Relativistic compact effective potentials and efficient, shared- exponent basis sets for the third-, fourth-, and fifth row atoms. Can. J. Chem. 1992, 70, 612–630. 10.1139/v92-085. [DOI] [Google Scholar]
- Kaupp M.; Schleyer P. V. R.; Stoll H.; Preuss H. Pseudopotential approaches to Ca, Sr, and Ba hydrides. Why are some alkaline earth MX2 compounds bent?. J. Chem. Phys. 1991, 94, 1360–1366. 10.1063/1.459993. [DOI] [Google Scholar]
- Ermler W. C.; Ross R. B.; Christiansen P. A. Ab initio relativistic effective potentials with spin-orbit operators. VI. Fr through Pu. Int. J. Quantum Chem. 1991, 40, 829–846. 10.1002/qua.560400611. [DOI] [Google Scholar]
- Wadt W. R.; Hay P. J. Ab initio effective core potentials for molecular calculations. Potentials for the transition metal atoms Sc to Hg. J. Chem. Phys. 1985, 82, 270–283. 10.1063/1.448799. [DOI] [Google Scholar]; 284 - 298.10.1063/1.448800; 299 - 310.10.1063/1.448975
- Perdew J. P.; Chevary J. A.; Vosko S. H.; Jackson K. A.; Pederson M. R.; Singh D. J.; Fiolhais C. Atoms, molecules, solids, and surfaces: Applications of the generalized gradient approximation for exchange and correlation. Phys. Rev. B: Condens. Matter Mater. Phys. 1992, 46, 6671–6687. 10.1103/PhysRevB.46.6671. [DOI] [PubMed] [Google Scholar]
- Perdew J. P.; Chevary J. A.; Vosko S. H.; Jackson K. A.; Pederson M. R.; Singh D. J.; Fiolhais C. Atoms, molecules, solids, and surfaces: Applications of the generalized gradient approximation for exchange and correlation. Phys. Rev. B: Condens. Matter Mater. Phys. 1993, 48, 4978–4978. 10.1103/PhysRevB.48.4978.2. [DOI] [PubMed] [Google Scholar]
- Schäfer A.; Horn H.; Ahlrichs R. Fully optimized contracted Gaussian basis sets for atoms Li to Kr. J. Chem. Phys. 1992, 97, 2571–2577. 10.1063/1.463096. [DOI] [Google Scholar]
- Pritchard B. P.; Altarawy D.; Didier B.; Gibson T. D.; Windus T. L. A New Basis Set Exchange: An Open, Up-to-date Resource for the Molecular Sciences Community. J. Chem. Inf. Model. 2019, 59, 4814–4820. 10.1021/acs.jcim.9b00725. [DOI] [PubMed] [Google Scholar]
- Abe M. Diradicals. Chem. Rev. 2013, 113, 7011–7088. 10.1021/cr400056a. [DOI] [PubMed] [Google Scholar]
- Srinivas K.; Prabhakar Ch.; Lavanya Devi C.; Yesudas K.; Bhanuprakash K.; Jayathirtha Rao V. Enhanced Diradical Nature in Oxyallyl Derivatives Leads to Near Infra Red Absorption: A Comparative Study of the Squaraine and Croconate Dyes Using Computational Techniques. J. Phys. Chem. A 2007, 111, 3378–3386. 10.1021/jp067410f. [DOI] [PubMed] [Google Scholar]
- Mostafanejad M. Basics of the Spin Hamiltonian Formalism. Int. J. Quantum Chem. 2014, 114, 1495–1512. 10.1002/qua.24721. [DOI] [Google Scholar]
- Bendikov M.; Duong H. M.; Starkey K.; Houk K. N.; Carter E. A.; Wudl F. Oligoacenes: Theoretical Prediction of Open-Shell Singlet Diradical Ground States. J. Am. Chem. Soc. 2004, 126, 7416–7417. 10.1021/ja048919w. [DOI] [PubMed] [Google Scholar]
- Schmidt J. R.; Shenvi N.; Tully J. C. Controlling spin contamination using constrained density functional theory. J. Chem. Phys. 2008, 129, 114110. 10.1063/1.2978168. [DOI] [PubMed] [Google Scholar]
- Reed A. E.; Weinstock R. B.; Weinhold F. Natural population analysis. J. Chem. Phys. 1985, 83, 735–746. 10.1063/1.449486. [DOI] [Google Scholar]
- Wiberg K. B. Application of the pople-santry-segal CNDO method to the cyclopropylcarbinyl and cyclobutyl cation and to bicyclobutane. Tetrahedron 1968, 24, 1083–1096. 10.1016/0040-4020(68)88057-3. [DOI] [Google Scholar]
- Adamo C.; Jacquemin D. The calculations of excited-state properties with Time-Dependent Density Functional Theory. Chem. Soc. Rev. 2013, 42, 845–856. 10.1039/C2CS35394F. [DOI] [PubMed] [Google Scholar]
- Laurent A. D.; Jacquemin D. TD-DFT benchmarks: A review. Int. J. Quantum Chem. 2013, 113, 2019–2039. 10.1002/qua.24438. [DOI] [Google Scholar]
- Tomasi J.; Mennucci B.; Cammi R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3094. 10.1021/cr9904009. [DOI] [PubMed] [Google Scholar]
- Jodaian V.; Mirzaei M.; Arca M.; Aragoni M. C.; Lippolis V.; Tavakoli E.; Langeroodi N. S. First example of a 1:1 vanadium(IV)–citrate complex featuring the 2,2′-bipyridine co-ligand: Synthesis, X-ray crystal structure and DFT calculations. Inorg. Chim. Acta 2013, 400, 107–114. 10.1016/j.ica.2013.02.012. [DOI] [Google Scholar]
- Day P. N.; Nguyen K. A. Pachter, TDDFT Study of One- and Two-Photon Absorption Properties: Donor−π–Acceptor Chromophores R. J. Phys. Chem. B 2005, 109, 1803–1814. 10.1021/jp047511i. [DOI] [PubMed] [Google Scholar]
- Huong V. T. T.; Tai T. B.; Nguien M. T. A theoretical study on charge transport of dithiolene nickel complexes. Phys. Chem. Chem. Phys. 2016, 18, 6259–6267. 10.1039/C5CP07277H. [DOI] [PubMed] [Google Scholar]
- Davis A. P.; Fry A. J. Experimental and computed absolute redox potentials of polycyclic aromatic hydrocarbons are highly linearly correlated over a wide range of structures and potentials. J. Phys. Chem. A 2010, 114, 12299–12304. 10.1021/jp106088n. [DOI] [PubMed] [Google Scholar]
- Yan L.; Lu Y.; Li X. A density functional theory protocol for the calculation of redox potentials of copper complexes. Phys. Chem. Chem. Phys. 2016, 18, 5529–5536. 10.1039/C5CP06638G. [DOI] [PubMed] [Google Scholar]
- Baik M. – H.; Friesnel R. A. Computing Redox Potentials in Solution: Density Functional Theory as A Tool for Rational Design of Redox Agents. J. Phys. Chem. A 2002, 106, 7407–7412. 10.1021/jp025853n. [DOI] [Google Scholar]
- Bartmess J. E. Thermodynamics of the Electron and the Proton. J. Phys. Chem. 1994, 98, 6420–6424. 10.1021/j100076a029. [DOI] [Google Scholar]
- Jiang S.; Xu M. Hyperpolarizabilities for the one-dimensional infinite single-electron periodic systems. I. Analytical solutions under dipole-dipole correlations. J. Chem. Phys. 2005, 123, 064901. 10.1063/1.1989307. [DOI] [PubMed] [Google Scholar]
- Cifuentes M. P.; Humphrey M. G. Alkynyl compounds and nonlinear optics. J. Organomet. Chem. 2004, 689, 3968–3981. 10.1016/j.jorganchem.2004.06.027. [DOI] [Google Scholar]
- Mendes P. J.; Carvalho A. J. P.; Ramalho J. P. P. Role played by the organometallic fragment on the first hyperpolarizability of iron–acetylide complexes: A TD-DFT study. J. Mol. Struct.: THEOCHEM 2009, 900, 110–117. 10.1016/j.theochem.2008.12.037. [DOI] [Google Scholar]
- Schaftenaar G.; Noordik J. H. Molden: a pre-and post-processing program for molecular and electronic structures. J. Comput.-Aided Mol. Des. 2000, 14, 123–134. 10.1023/A:1008193805436. [DOI] [PubMed] [Google Scholar]
- Dennington R.; Keith T. A.; Millam J. M.; GaussView, Ver. 6; Semichem Inc.: Shawnee Mission, KS, 2016.
- O’Boyle N. M.; Tenderholt A. L.; Langner K. M. Clib: a library for package-independent computational chemistry algorithms. J. Comput. Chem. 2008, 29, 839–845. 10.1002/jcc.20823. [DOI] [PubMed] [Google Scholar]
- Chemissian, a computer program to analyze and visualize quantum-chemical calculations written by L. Skripnikov. For the current version, see http://www.chemissian.com.
- Scheibye S.; Pedersen B. S.; Lawesson S.-O. Studies on organophosphorus compounds XXI. The dimer of p-methoxyphenylthionophosphine sulfide as thiation reagent. A new route to thiocarboxamides. Bull. Soc. Chim. Belg. 1978, 87, 229–238. 10.1002/bscb.19780870311. [DOI] [Google Scholar]
- Domingos A.; Henriques R. T.; Gama V.; Almeida M.; Lopes Vieira A.; Alcacer L. Crystalline structure/transport properties relationship in the (perylene)2M(mnt)2 family M = Au, Pd, Pt, Ni). Synth. Met. 1988, 27, 411–416. 10.1016/0379-6779(88)90177-4. [DOI] [Google Scholar]
- Average value calculated on 13 structures deposited at the CCDC and featuring a [Pd(mnt)2]− anion (JUJGOK, JUJGOK, JUJGOK01, JUJGOK01, KOJTUY, MOSXID, SAHFEM, SAMCIT, SAMCIT, SATVAK, SICFOB, UCUHIK, WAPBAS).
- Average value calculated on 21 structures deposited at the CCDC and featuring a [Pd(mnt)2]2– anion (LEHFOU, PAZSIT, BAHMOO, BUTSUF, CAJQEM, EBOZEB, HAVYOT, ICUBIS, JUJGIE, JUJGIE01, MOQCIF, NALTEZ, OPAMAU, VUQWOU, WABDIN, WASKUW, YICKUR, YOBMEH, KANZOQ, SAHFOW, XOJXAX).
- Damas A.; Chamereau L. – M.; Cooksy A. L.; Jutand A.; Amouri H. π-Bonded Dithiolene Complexes: Synthesis, Molecular Structures, Electrochemical Behavior, and Density Functional Theory Calculations. Inorg. Chem. 2013, 52, 1409–1417. 10.1021/ic302128q. [DOI] [PubMed] [Google Scholar]
- Hencher J. L.; Shen Q.; Tuck D. G. Molecular structure of 1,2-bis(trifluoromethyl)dithiete by vapor phase electron diffraction. J. Am. Chem. Soc. 1976, 98, 899–902. 10.1021/ja00420a006. [DOI] [Google Scholar]
- Shimizu T.; Murakami H.; Kobayashi Y.; Iwata K.; Kamigata N. Synthesis, Structure, and Ring Conversion of 1,2-Dithiete and Related Compounds. J. Org. Chem. 1998, 63, 8192–8199. 10.1021/jo9806714. [DOI] [Google Scholar]
- Donahue J. P.; Holm R. H. 3,4-Bis(1-adamantyl)-1,2-dithiete: the First Structurally Characterized Dithiete Unsupported by a Ring or Benzenoid Frame. Acta Crystallogr., Sect. C: Cryst. Struct. Commun. 1998, 54, 1175–1178. 10.1107/S0108270198002935. [DOI] [PubMed] [Google Scholar]
- Roesky H. W.; Hofmann H.; Clegg W.; Noltemeyer M.; Sheldrick G. M. Preparation and crystal structure of cyclic dithiooxamides. Inorg. Chem. 1982, 21, 3798–3800. 10.1021/ic00140a039. [DOI] [Google Scholar]
- Aragoni M. C.; Arca M.; Devillanova F. A.; Isaia F.; Lippolis V.; Mancini A.; Pala L.; Slawin A. M. Z.; Woollins J. D. First example of infinite polybromide 2D network. Chem. Commun. 2003, 2226–2227. 10.1039/B306841B. [DOI] [PubMed] [Google Scholar]
- Davis A. P.; Fry A. J. Experimental and computed absolute redox potentials of polycyclic aromatic hydrocarbons are highly linearly correlated over a wide range of structures and potentials. J. Phys. Chem. A 2010, 114, 12299–12304. 10.1021/jp106088n. [DOI] [PubMed] [Google Scholar]
- Baik M. – H.; Friesnel R. A. Computing Redox Potentials in Solution: Density Functional Theory as A Tool for Rational Design of Redox Agents. J. Phys. Chem. A 2002, 106, 7407–7412. 10.1021/jp025853n. [DOI] [Google Scholar]
- Canola S.; Casado J.; Negri F. The double exciton state of conjugated chromophores with strong diradical character: insights from TDDFT calculations. Phys. Chem. Chem. Phys. 2018, 20, 24227–24238. 10.1039/C8CP04008G. [DOI] [PubMed] [Google Scholar]
- Wirz J. Spectroscopic and kinetic investigations of conjugated biradical intermediates. Pure Appl. Chem. 1984, 56 (9), 1289–1300. 10.1351/pac198456091289. [DOI] [Google Scholar]
- Evangelista F. A.; Allen W. D.; Schaefer H. F. III Coupling term derivation and general implementation of state-specific multireference coupled cluster theories. J. Chem. Phys. 2007, 127, 024102. 10.1063/1.2743014. [DOI] [PubMed] [Google Scholar]
- Matsumoto M.; Antol I.; Abe M. Curve Effect on Singlet Diradical Contribution in Kekulé-type Diradicals: A Sensitive Probe for Quinoidal Structure in Curved π-Conjugated Molecules. Molecules 2019, 24, 209. 10.3390/molecules24010209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noodleman L. J. Valence bond description of antiferromagnetic coupling in transition metal dimers. J. Chem. Phys. 1981, 74, 5737–5743. 10.1063/1.440939. [DOI] [Google Scholar]
- Noodleman L.; Baerends E. J. Electronic structure, magnetic properties, ESR, and optical spectra for 2-iron ferredoxin models by LCAO-Xα valence bond theory. J. Am. Chem. Soc. 1984, 106, 2316–2327. 10.1021/ja00320a017. [DOI] [Google Scholar]
- Yamaguchi K.; Takahara Y.; Fueno T.; Nasu K. Ab Initio MO Calculations of Effective Exchange Integrals between Transition-Metal Ions via Oxygen Dianions: Nature of the Copper-Oxygen Bonds and Superconductivity. Jpn. J. Appl. Phys. 1987, 26, L1362–L1364. 10.1143/JJAP.26.L1362. [DOI] [Google Scholar]
- Yamaguchi K.; Jensen F.; Dorigo A.; Houk K. N. A spin correction procedure for unrestricted Hartree-Fock and Møller-Plesset wavefunctions for singlet diradicals and polyradicals. Chem. Phys. Lett. 1988, 149, 537–542. 10.1016/0009-2614(88)80378-6. [DOI] [Google Scholar]
- Malrieu J. – P.; Trinquier G. Communication: Proper use of broken-symmetry calculations in antiferromagnetic polyradicals. J. Chem. Phys. 2016, 144, 211101. 10.1063/1.4953040. [DOI] [PubMed] [Google Scholar]
- Young D. C., In Computational Chemistry: A Practical Guide for Applying Techniques to Real-World Problems; Wiley: 2001, Chapter 27. [Google Scholar]
- CSD codes: BAHMOO, BUTSUF, CAJQEM, EBOZEB, HAVYOT, ICUBIS, JUJGIE, JUJGIE01, JUJGOK, JUJGOK01, KANZOQ, MOQCIF, MOSXID, NALTEZ, OPAMAU, SAHFEM, SAHFOW, SAMCIT, SATVAK, SICFOB, TENVIU, TENVOA, TENVUG, TEPCUP, TUKYAZ, UCUHIK, VUQWOU, WABDIN, WAPBAS, WASKUW, XOJXAX, YICKUR, YOBMEH, YOFCEB.
- Hudson B.S.; Kohler B.E. A low-lying weak transition in the polyene α,ω-diphenyloctatetraene. Chem. Phys. Lett. 1972, 14, 299–304. 10.1016/0009-2614(72)80119-2. [DOI] [Google Scholar]
- Schulten K.; Karplus M. On the origin of low-lying forbidden transition in polyenes and related molecules. Chem. Phys. Lett. 1972, 14, 305–309. 10.1016/0009-2614(72)80120-9. [DOI] [Google Scholar]
- Di Motta S.; Negri F.; Fazzi D.; Castiglioni C.; Canesi E. V. Biradicaloid and Polyenic Character of Quinoidal Oligothiophenes Revealed by the Presence of a Low-Lying Double-Exciton State. J. Phys. Chem. Lett. 2010, 1, 3334–3339. 10.1021/jz101400d. [DOI] [Google Scholar]
- For the sake of comparison, calculations were repeated by adopting the B3LYP hybrid functional, including a 20% HF exchange, in place of mPW1PW (25% HF exchange). The corresponding DC index was nDC = 17.4%.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.