

Abstract
Mitochondria are the main organelles that produce adenosine 5′-triphosphate (ATP) and reactive oxygen species (ROS) in eukaryotic cells and meanwhile susceptible to oxidative damage. The irreversible oxidative damage in mitochondria has been implicated in various human diseases. Increasing evidence indicates the therapeutic potential of mitochondria-targeted antioxidants (MTAs) for oxidative damage-associated diseases. In this article, we introduce the advantageous properties of MTAs compared with the conventional (nontargeted) ones, review different mitochondria-targeted delivery systems and antioxidants, and summarize their experimental results for various disease treatments in different animal models and clinical trials. The combined evidence demonstrates that mitochondrial redox homeostasis is a potential target for disease treatment. Meanwhile, the limitations and prospects for exploiting MTAs are discussed, which might pave ways for further trial design and drug development.
1. Introduction
Mitochondria, subcellular organelles found in most eukaryotic cells, are responsible for numerous metabolic network processes, including the tricarboxylic acid cycle (TCA cycle), glycolysis, oxidative phosphorylation (OXPHOS), amino acid metabolism, and fatty acid oxidation. Among them, the most important physiological function of mitochondria is to generate ATP by oxidizing nutrients. To participate in adenosine 5′-triphosphate (ATP) production, mitochondria use a complex system interacting with these metabolic network processes, during which free radicals are produced. Generally, mitochondrial ROS production mainly occurs at the site of the electron transport chain located on the mitochondrial inner membrane, and the leakage of electrons from complex I and complex III leads to oxygen consumption and superoxide formation [1]. The mitochondrial redox homeostasis refers to an equilibrium between ROS production and scavenging, which is the basis for mitochondrial function and cell fate determination [2].
At present, it is recognized that many pathological changes are associated with impaired mitochondrial function [3], such as increased accumulation of ROS and decreased OXPHOS and ATP production. Although the production of intracellular ROS is itself an inevitable process, cells have an adaptive defense system to scavenge ROS [4]. However, under most oxidative stress conditions, the endogenous antioxidant system in the cells is not enough to scavenge excess ROS. In that case, the accumulation of ROS will cause oxidative damage to intracellular lipids, DNA, and proteins, thereby accelerating the development of related diseases [5]. In the past decade, research has focused on maintaining redox homeostasis and normal function of mitochondria via antioxidants [6]. Current medical projects are aimed at exploiting drugs that restore mitochondrial function and regulate mitochondrial ROS production [7]. To modulate mitochondrial redox homeostasis, the drug should selectively accumulate in the mitochondria and interact with mitochondrial targets, ultimately maintaining normal cellular functions [8]. Although this mitochondrial targeting strategy is attractive, the clinical applications are hampered by some challenges, such as the poor biological availability and the lack of evidence in animal models and clinical research studies [9]. Several drugs have been applied for clinical trials; however, no drug has been approved by the US Food and Drug Administration (FDA) for mitochondria-targeted treatment.
The present review article is aimed at summarizing experimental data on mitochondria-targeted antioxidants (MTAs) for various disease treatments in different models and clinical trials to present the evidence supporting the therapeutic potential of these MTAs. We specifically focused on brain neurological diseases [10, 11], cardiovascular diseases [12–14], and cancer development [15, 16], all of which are closely associated with oxidative damage and signal activation caused by the excess accumulation of ROS in mitochondria. Meanwhile, the potential MTA applications in disease treatment, their limitations, and prospects for exploiting MTAs are discussed.
2. Moving Forward from Nontargeted Antioxidants to MTAs
An increasing number of studies are aimed at developing conventional (nontargeted) antioxidants for restoring physiology conditions during oxidative stress. Although preliminary studies on many cell or animal models showed promising results, the results from clinical trials were sometimes contradictory. A recent review article [17] has summarized the adverse effects of nontargeted antioxidants (NAs) including vitamin A, vitamin C, vitamin E, and β-carotene. These adverse effects of NAs were mainly observed in the treatments of lung cancer and cardiovascular diseases [18]. Redox signaling is an important part of many physiological processes. Excessive or inappropriate use of antioxidants may abolish ROS production and result in compensatory upregulation of mitogen-activated protein kinase (MAPK) pathways [19], which in turn negatively affect the endogenous antioxidant system and normal cell growth [20]. Another concern is whether conventional (nontargeted) antioxidants can be absorbed properly and how they are metabolized in different organs. These uncertainties make it difficult to determine the dose of traditional antioxidants used for disease treatment. The most effective way for an antioxidant stepping forward to disease treatment is to conjugate with a carrier, such as lipophilic cations, liposomes, or peptides, to enable its bioactive ingredient to be targeted for transport into the mitochondria. This targeted delivery enables antioxidants to achieve high concentration accumulation in cells and mitochondria, thereby protecting cells and tissues from oxidative damage through different mechanisms. Ideal antioxidants should be bioavailable and can quickly enter the blood circulation via intestinal absorption or intravenous injection. The MTAs could accumulate in the mitochondria and protect the targeted tissues (brain, liver, kidney, muscle, ear, and heart) from oxidative damage (Figure 1). In the past decade, many studies focusing on the development of mitochondria-targeted antioxidants gave promising results, which we will discuss in detail.
Figure 1.
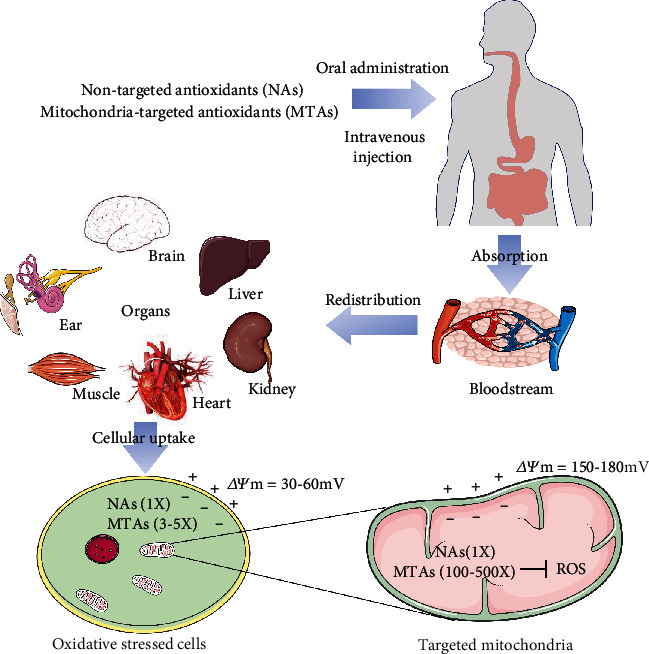
Administration and transport of nontargeted antioxidants (NAs) and mitochondria-targeted antioxidants (MTAs). Ideal antioxidants are bioavailable and can be quickly transported into the blood circulation via intestinal absorption or intravenous injection. The NA can hardly be efficiently delivered to the targeted tissues and mitochondria. The MTAs accumulate 100-500 times in the mitochondria and protect the tissues (brain, liver, kidney, muscle, ear, or heart) from oxidative damage.
3. Lipophilic Cation-Linked MTAs
The mitochondrial transmembrane potential theory was first proposed by Skulachev et al. in 1969 [21]. The lipophilic cation could easily penetrate cells and mitochondria with the help of ΔΨm, which is positive outside and negative inside. The targeted transport of antioxidants to mitochondria can be achieved by using a lipophilic cation as a transport vehicle. This strategy can be applied for a variety of bioactive substances, especially these hydrophobic ones that are not easily absorbed by cells and mitochondria. In the past decades, triphenylphosphonium (TPP) has been commonly used for the development of MTAs. Presentative studies on the TPP-linked MTAs are summarized in Table 1, and the chemical structures of MitoQ, SkQ1, MitoE, and Mito-TEMPO are shown in Figure 2. Among them, MitoQ and SkQ1 have been extensively studied in various animal models and several human clinical trials [22]. In a clinical trial on twenty healthy older adults (60-79 years) with impaired endothelial function (NCT02597023), oral MitoQ (20 mg/day) supplementation improved brachial artery flow-mediated dilation, decreased aortic stiffness, and lowered the plasma low-density lipoprotein [23]. In a clinical trial on hepatitis C virus- (HCV-) infected patients (NCT00433108), oral MitoQ (40 or 80 mg/day) supplementation decreased serum alanine transaminase (ALT), indicating a decreased necroinflammation in the liver [24]. SkQ1 was documented to relieve the dry eye symptoms in a phase 2 study (NCT02121301) [25]. Meanwhile, a vehicle-controlled study of SkQ1 as a treatment for dry eye syndrome is recruiting (NCT04206020).
Table 1.
TPP-linked MTAs.
| Mitochondria-targeted antioxidants | Bioactive component | Linker | Effects | Reference |
|---|---|---|---|---|
| MitoE | Vitamin E | 2-Carbon aliphatic linker | (1) Minimized lipid peroxidation and protected cells from oxidative damage (2) Eliminated H2O2-induced oxidative stress and caspase activation in cells (3) Accumulated in tissues (heart, brain, muscle, liver, and kidney) and protected tissues from oxidative damage |
[28, 30] |
| Mito-vitamin E derivation | Vitamin E | 11-Alkyl linker | (1) Inhibited energy metabolism and promote cell death (2) Antitumor properties |
[31, 32] |
| SkQ1 SkQR1 |
Plastoquinone | 10-Alkyl linker | (1) Minimized lipid peroxidation and ROS-induced apoptosis (2) Beneficial roles in many diseases including aging, stroke, myocardial infarction, sarcopenia, dry eye syndrome, vascular inflammation |
[33, 34] |
| MitoQ | Coenzyme Q | 10-Alkyl linker | (1) Penetrated the mitochondrial membrane and inhibited lipid peroxidation (2) Beneficial roles in animal models of alcoholic fatty liver, neurodegenerative diseases, ischemia-reperfusion, hypertension, sepsis, and kidney damage in type I diabetes |
[35, 36] |
| MitoC MitoVitC11 | Vitamin C | Thioalkyl linker | (1) Prevented mitochondrial lipid peroxidation and protected mitochondrial aconitase (2) Scavenged O2–, peroxyl radicals, and Fe3+ and could be rapidly recycled to the active ascorbate moiety |
[37] |
| MitoSOD | M40403 | Thioalkyl linker | (1) Regulated the mitochondrial redox system to convert ROS (2) Reversed the rapid and progressive inhibition of aconitase through redox cycling (3) Retained Mn2+ under nonacidic conditions |
[38, 39] |
Notes: ΔΨm: mitochondrial membrane potential; M40403: a macrocyclic Mn SOD mimetic system; ROS: reactive oxygen species; TPP: triphenylphosphonium.
Figure 2.
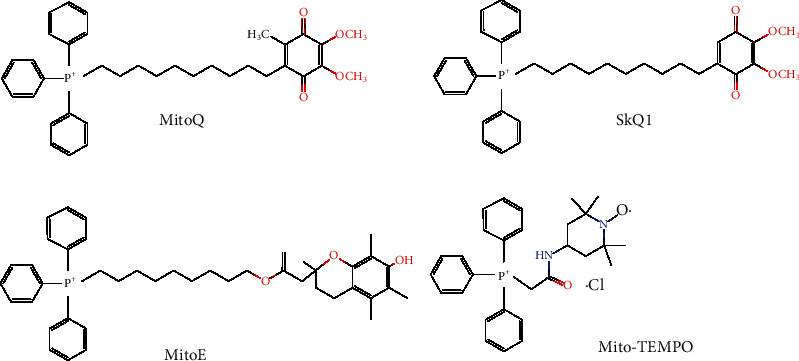
Chemical structures of representative TPP-linked mitochondria-targeted antioxidants (MitoQ, SkQ1, MitoE, and Mito-TEMPO are shown).
Toxicity to mitochondria is a major limiting factor for the application of TPP-linked antioxidants in disease treatment [26]. During the transport of the TPP-linked antioxidants, TPPs increasingly adhere to the surface of the mitochondrial inner membrane. This accumulation of TPPs could destroy the integrity of the mitochondrial membrane and limit aerobic respiration and ATP synthesis [27]. In the toxicity assessment of in vivo experiments [28] using a mouse model, the maximum tolerated doses of methyl TPP and MitoE2 are 3.8 and 6.0 mg/(kg∗bodyweight), respectively. Evident toxic effects of TPP and MitoE2 were observed at 6.4 and 10.2 mg/(kg∗bodyweight), respectively. Intravenous injection of MitoQ was not toxic to the mice at 20 mg/(kg∗bodyweight) but significantly toxic at 27.0 mg/(kg∗bodyweight). It is noteworthy that long-term and low-dose MitoQ administration did not exhibit any toxic effect to the mouse models [29], which indicates that the toxic effect is caused by the disruption of normal function of mitochondria in response to a concentrated accumulation of TPP, and an oral administration with low-dose TPP compound is feasible. Therefore, in clinical trials testing TPP-linked antioxidants, it is necessary to strictly control the dosage and to ensure the effective concentration of the MTAs is lower than the threshold that destroys the normal function of mitochondria. Although some of these TPP-linked antioxidants such as MitoQ and SkQ1 have been evaluated in a wide range of clinical trials (NCT03166800, NCT02597023, NCT00329056, NCT03764735, and NCT02121301), more studies are required to assess their optimal dosages for different disease phases, their long-term effects on redox signal activation, and their potential side effects.
4. Liposome-Encapsulated Antioxidants
Liposomes are lipid bilayer membrane vesicles first discovered in 1964 and have been commonly used as nanocarriers for pharmaceuticals and bioactive substances [40]. One advantage of the liposomal encapsulation strategy over lipophilic cations is that bioactive molecules can be encapsulated and delivered without altering their molecular structure and bioactivity. The liposome-encapsulated antioxidants are composed of phosphatidylcholine, phosphatidylglycerol, cholesterol, and antioxidant component. The encapsulated antioxidants such as quercetin, N-acetyl-L-cysteine (NAC), and vitamin E exhibited better therapeutic effects on the models of liver injuries [41] and MCF-7 carcinoma cells [42] when compared with those in nonencapsulated form. For example, only liposomal encapsulated NAC can long-lastingly prevent the cytokine-induced neutrophil chemoattractant expression in the lung, thereby protecting the rats against lipopolysaccharide-induced acute respiratory distress syndrome [43]. It has been reviewed that liposomal encapsulated analogs of vitamin E (α-tocopheryl succinate and α-tocopheryl ether-linked acetic acid) exerted better anticancer effects on various cancer models due to their higher solubility in aqueous solvents [44]. In a clinical study on fatty liver patients, the phospholipid-encapsulated silybin was revealed to protect the liver from oxidative damage via enhancing mitochondrial function and insulin sensitization [45]. Liposome-encapsulated curcumin administration with 100 mg/(kg∗bodyweight) increased the parameters of plasma antioxidant activity in the Sprague-Dawley rat [46]. Likewise, astaxanthin encapsulated within liposomes showed a better bioavailability than the nonencapsulated astaxanthin and ameliorated oxidative parameters in the Sprague-Dawley rat model of lipopolysaccharide- (LPS-) induced acute hepatotoxicity [47].
Liposome-based delivery systems can carry conventional antioxidants into the mitochondria of the living cells. The transport mechanism of liposome-encapsulated MTAs is shown in Figure 3. Liposome-encapsulated antioxidants enter the cells via micropinocytosis; after macropinosome disruption, the liposomal components fuse with the mitochondrial membrane, during which the antioxidant components are delivered into the matrix of targeted mitochondria. The main disadvantage of the liposome system for MTA delivery is the escape of endosome degradation, which limits the endosomes spontaneously degrading in the cytoplasm and mitochondria. To overcome this limitation, the MITO-Porter that consists of a condensed plasmid DNA and a lipid envelope was developed to deliver bioactive components to mitochondria [48]. The inventors have introduced the characteristics and potential development of MITO-Porter in a specific chapter [49]. Generally, the MITO-Porter-decorated liposomes consist of 1,2-dioleoyl-sn-glycero-3-phosphatidylethanolamine, sphingomyelin, and stearylated octaarginine peptide (R8). During a mitochondria-targeted delivery process, the MITO-Porter-decorated liposomes bind to the mitochondria via electrostatic interactions between R8 and negatively charged mitochondria and then fuse with the mitochondrial membrane. This delivery system can achieve efficient cytoplasmic and mitochondria-targeted delivery, which provides a new way for the treatment of mitochondrial disease. Besides, delivery experiments using fluorescent probes have verified MITO-Porter as an effective tool for macromolecule-targeted delivery [50].
Figure 3.
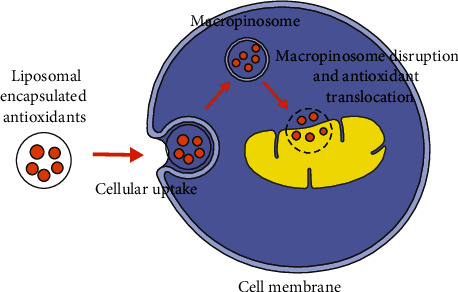
The mitochondrial transport of liposome-encapsulated antioxidants. Liposome-encapsulated antioxidants enter cell membranes via micropinocytosis; after macropinosome disruption, the liposomal components fuse with the mitochondrial membrane, during which the antioxidant components are delivered into the matrix of targeted mitochondria.
In a mouse model of liver ischemia/reperfusion injury, systemic injection of MITO-Porter-encapsulated CoQ10 (CoQ10-MITO-Porter) decreased serum alanine transaminase (ALT) and prevented kidney injury [51]. Recently, the mitochondrial delivery of methylated β-cyclodextrin-threaded polyrotaxanes using a MITO-Porter was revealed to mediate mitochondrial autophagy, which might be useful for mitochondria-associated disease treatment [52]. Moreover, the dual-function MITO-Porter (DF-MITO-Porter) that integrates both R8-modified liposomes and MITO-Porter was developed to effectively deliver exogenous macrobiomolecules into the mitochondria, providing an excellent delivery system for mitochondrial disease treatment [53]. More research studies on the MITO-Porter delivery system are expected to be conducted to shed more light on the mitochondrial therapeutic strategy and targeted antioxidant development.
5. Peptide-Based Mitochondrial Antioxidants
The Szeto-Schiller peptide (SS-peptide) and the mitochondria-penetrating peptide (MPP) are peptide chain-based antioxidant delivery systems. SS-peptides contain different small-molecule lipophilic antioxidant compounds and three positive charges and can be targeted-delivered to the mitochondria with the help of ΔΨm of the cellular membrane and mitochondrial membrane [54]. The advantageous properties of SS-peptides include the following: (1) alternating the MPP sequence between the basic and aromatic residues which favor their efficient absorption by cells; (2) unsaturated transport independently from the energy state or a dedicated peptide transporter [55]; (3) small and easily soluble in water, easy to synthesize, and the presence of D-amino acids at specific positions which prevents them from being degraded by aminopeptidases and allows them to be effectively transported into the mitochondria [56]; and (4) 1000-5000 times accumulation in the mitochondria.
Various experiments have confirmed that SS-peptides can be rapidly absorbed by different cell types, such as neurons [57], kidneys [58], epithelial cells, and endothelial cells [59]. It is noteworthy that the mitochondrial uptake speed of SS-peptides is ΔΨm-independent. The absorption of the SS-peptides does not affect the polarization of the mitochondrial membrane, which makes them ideal antioxidants for disease treatment [60]. For example, SS-02 was revealed to easily penetrate a single layer of intestinal epithelial cells from the basal and apical direction [61]. SS-02 has also been reported to penetrate the blood-brain barrier and thus serve as a neuroprotective agent [62]. The SS-peptides are effective in alleviating oxidative stress both in cell models and isolated mitochondria [63], among them SS-31 was widely validated to be effective. The therapeutic potential of SS-31 has been documented for many conditions including brain microvascular endothelial cell damage [64], lateral line hair cell damage [65], mitochondrial morphogenesis [66], atherosclerosis [67], Friedreich ataxia [68], renal fibrosis [69], limb ischemia-reperfusion injury [70], exercise tolerance [71], type 2 diabetes [72], hearing loss [73], neurovascular coupling responses [74], cardiac arrest [75], traumatic brain injury [76, 77], heart failure [78–81], and acute kidney injury [82]. Of importance, the phase 2a clinical trial of SS-31 (unique identifier: NCT01755858) on the atherosclerotic renal artery stenosis patients (ARASP) showed that supplementing with SS-31 during percutaneous transluminal renal angioplasty alleviated the pathological symptoms and improved kidney function, indicating a positive prospect of SS-31 in clinical application for ARASP [83].
A recent study on aged mice revealed that the disruption of mitochondrial redox homeostasis in muscle resulted in energy defect and exercise intolerance, and SS-31 administration restored redox homeostasis of the aged muscle, thereby increasing the exercise tolerance [71]. Five hours of SS-31 treatment significantly decreased mortality of cardiac arrest rats, during which the blood lactate level in the SS-31-treated rats was significantly decreased, suggesting improved mitochondrial aerobic respiration by SS-31 treatment [75]. The antioxidative roles of SS-31 have been also documented in kidney glomerular mitochondria [84]. SS-31 administration was revealed to prevent negative changes in pathological parameters in chronic kidney disease models [69]. More recently, an acute kidney injury- (AKI-) targeted nanopolyplex was designed for SS-31 delivery, which demonstrates a positive effect of combining the use of nanopolyplexes and SS-31 in the oxidative stressed and inflamed kidney [82]. Similarly, treatment with SS-31 was found to decrease cytoplasmic and mitochondrial O2- production by regulating the expression of NADPH oxidase subunit NOX4 in a model of traumatic brain injury [76].
Mitochondria-penetrating peptides (MPPs) consist of 4 to 8 alternating positively charged hydrophobically modified amino acids. They have been widely used for the targeted delivery of mitochondrial small molecules with the help of ΔΨm [85]. A series of XJB peptide-based antioxidants (XJB-5-131, XJB-5-125, and XJB-5-197) have been developed (Figure 4). XJB-5-131, a scavenger for mitochondrial ROS, is the most studied among all the XJB peptide-based antioxidants and has been reported to promote weight gain, prevent neuronal death, and reduce oxidative damage in a mouse model of neurodegeneration [86]. Besides, XJB-5-131 was demonstrated to alleviate oxidative damage of DNA and improve physiology behavior in a Huntington's disease model [87, 88]. Likewise, the compounds of XJB-5-131 and JP4-039 were reported to inhibit ferroptosis via scavenging ROS and altering the subcellular localization of the ferroptosis suppressors [89]. These findings encourage more therapeutic evaluation of XJB peptide-based antioxidants in clinical trials.
Figure 4.
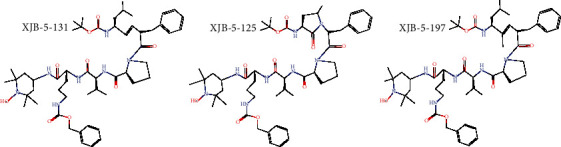
Chemical structures of XJB peptide-based mitochondria-targeted antioxidants (XJB-5-131, XJB-5-125, and XJB-5-197 are shown).
6. Potential Applications of MTAs in Disease Treatment
It is noteworthy that MTAs may exert multiple effects, such as alterations in redox status, ETC activity, and ATP synthesis during disease treatments, which could be affected by variations of disease types and phases. Proper dosage of the MTAs used in the trials lowers the ROS production in mitochondria and benefits the disease treatments; however, in some cases, a high dosage of MTAs may inhibit ETC activity and promote oxidative damage. Below, we reviewed the experimental and clinical results in Parkinson's disease (PD), traumatic brain injury (TBI), cardiovascular disorders/cardiovascular diseases (CVDs), or cancers, emphasizing the MTA dosage and potential target mechanisms (Tables 2–5).
Table 2.
MTAs in PD models and clinical trials.
| Mitochondria-targeted antioxidants | Models/clinical trials | Dosage | Effects/mechanism | Reference |
|---|---|---|---|---|
| MitoQ | Cellular MPP+ model | 50 nmol/L in culture medium | (1) Inhibited MPP+-induced decrease in dopamine levels | [93] |
| Mouse MPTP model | 4 mg/kg∗bodyweight; oral gavage | (1) Protected the nigrostriatal axis against MPTP toxicity (2) Improved locomotor activities in MPTP-treated mice (3) Inhibited mitochondrial aconitase inactivation |
[93, 96] | |
| Cellular 6-OHDA model | 10-200 nmol/L in culture medium | (1) Blocked 6-OHDA-induced mitochondrial fragmentation | [97] | |
| Mouse 6-OHDA model | 5 mg/kg∗bodyweight; intragastric administration | (1) Rescued dopamine neurons loss in SNc (2) Protected dopamine neurons via activating PGC-1α and enhance Mfn2-dependent mitochondrial fusion |
[97] | |
| Clinical trial | Daily 40/80 mg; oral administration | (1) Slowed the progression of Parkinson's disease as measured by the UPDRS (2) No difference in the measured parameters between the treatment and the placebo |
NCT00329056 | |
|
| ||||
| SS-20/Phe-D-Arg-Phe-Lys-NH2 | Cellular MPP+ model | 1-10 nmol/L in culture medium | (1) Rescued mitochondrial oxygen consumption and ATP production damaged by MPP+ (2) SS-20 (4 mg/kg∗bodyweight) protected against the loss of dopaminergic neurons in the substantia nigra pars compacta |
[94] |
| Mouse MPTP model | 0.5-5 mg/kg∗bodyweight; intraperitoneal injection | |||
|
| ||||
| SS-31/D-Arg-(2′6′-dimethyltyrosine)-Lys-Phe-NH2 | Cellular MPP+ model | 1-10 nmol/L in culture medium | (1) Improved cell survival and motor performance (2) Decreased cell loss and oxidative stress in the lumbar spinal cord (3) SS-31 (10 mg/kg∗bodyweight) protected against the loss of dopamine and its metabolites |
[94] |
| Mouse MPTP model | 0.5-10 mg/kg∗bodyweight; intraperitoneal injection | |||
|
| ||||
| P68+DQA nanocarriers NAC |
Cellular rotenone PD model | 1000 μmol/L in culture medium | (1) P68+DQA nanocarrier delivery system enhanced the stability, bioavailability, and brain penetrance of NAC (2) Formulation of NAC into P68+DQA nanocarriers rescued cell viability and alleviated oxidative stress |
[95] |
Notes: 6-OHDA: 6-hydroxydopamine; DQA: dequalinium; Mfn2: mitochondrial GTPase mitofusin-2; MPP+: 1-methyl-4-phenylpyridinium; MPTP: 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine; NAC: N-acetylcysteine; P68: Pluronic F68; PD: Parkinson's disease; PGC-1α: peroxisome proliferator-activated receptor gamma coactivator 1 alpha; SNc: substantia nigra pars compacta; UPDRS: Unified Parkinson's Disease Rating Scale.
Table 3.
MTAs in TBI models.
| Mitochondria-targeted antioxidants/bioactive component | Models/clinical trials | Dosage | Effects/mechanism | Reference |
|---|---|---|---|---|
| SkQR1 | Rat model by brain surgery | 100 nmol/kg; intraperitoneal injection | (1) Decreased the neurological deficit (2) Lowered the volume of the lesion in the brain cortex (3) Decreased mitochondrial ROS and GSK-3β activity |
[106] |
| Rat model of focal one-sided TBI | 250 nmol/kg; intraperitoneal injection | (1) Rescued the disruptions of limb functions (2) Increased survivability of neurons (3) Decreased astroglial expression and infiltration with segmented neutrophils (4) Beneficial effects are dependent on the reduction of mitochondrial reactive oxygen species |
[107] | |
|
| ||||
| XJB-5-131 | Rat CCI model after TBI | 10 mg/kg bodyweight; intravenous injection | (1) Protected brain thiols, GSH and PSH, oxidized by TBI (2) Decreased caspase 3/7 activity and attenuated apoptotic neuronal death (3) Scavenged the electrons leaking from electron carriers |
[108] |
|
| ||||
| Mito-TEMPO | Isolated MCAs from rats with traumatic injury | 30 nmol in the vessel chamber | (1) Alleviated myogenic constriction (2) Scavenged H2O2 (PEG-catalase) by blocking both BKCa channels and TRPV4 channels |
[109] |
|
| ||||
| SS-31 | Marmarou's weight drop model of TBI | 5 mg/kg; intraperitoneal administration | (1) Rescued mitochondrial dysfunction, and alleviated secondary brain injury (2) Decreased ROS, malondialdehyde, and cytochrome c release and prevented the decline of SOD activity (3) Attenuated neurological deficits, brain water content, DNA damage, and neural apoptosis |
[77] |
|
| ||||
| MitoQ | Marmarou's weight drop model | 4 mg/kg; intraperitoneal administration | (1) Alleviated neurological deficits and brain edema and inhibited cortical neuronal apoptosis (2) Increased the activity of SOD and GPx and decreased MDA level (3) Reduced Bax translocation to mitochondria and cytochrome c release into the cytosol (4) Accelerated the Nrf2 nuclear translocation and upregulated the Nrf2 downstream proteins, including HO-1 and Nqo1 |
[110] |
Notes: Bax: (Bcl-2)-associated X; BKCa: big conductance Ca2+-activated K+; CCI: chronic constriction injury; GPx: glutathione peroxidase; GSH: glutathione; GSK-3β: glycogen synthase kinase-3β; HO-1: heme oxygenase-1; MCAs: middle cerebral arteries; MDA: malondialdehyde; Nqo1: quinone oxidoreductase 1; Nrf2: nuclear factor erythroid 2; PEG-catalase: polyethylene glycol; PSH: protein thiols; SOD: superoxide dismutase; TBI: traumatic brain injury.
Table 4.
MTAs in cardiovascular disease (CVD) models.
| Mitochondria-targeted antioxidants/bioactive component | Models/clinical trials | Dosage | Effects/mechanism | Reference |
|---|---|---|---|---|
| Mito-TEMPO | THP-1 cell model induced by ox-LDL; high-fat dietary-fed rats | 20 μmol/L; 0.7 mg/kg∗bodyweight; intraperitoneal administration | (1) Attenuated foam cell formation via promoting autophagic flux (2) Increased cholesterol efflux via autophagy-dependent ABCA1 and ABCG1 upregulation (3) Reversed the accumulation of TC and LDL-c |
[125] |
|
| ||||
| MitoSNO | Open chest mouse model | 100 ng/kg∗bodyweight; intravenous injection | (1) Reduced infarct size and troponin release (2) Ineffectiveness on hemodynamics in the heart, dP/dtmax or heart rate (3) Alleviated infarction and myocardial fibrosis |
[126] |
|
| ||||
| SkQ1 | Lifelong treatment of mice | 1 or 30 nmol/kg∗bodyweight | (1) Prevented spontaneous cardiomyopathy (2) Decreased age-related heart hypertrophy and diffuse fibrosis (3) Affected cell adhesion-related gene expressions, one of which had mitochondrial localization |
[127] |
|
| ||||
| MitoQ | Pressure overload-induced heart failure in rats | 100 μmol/L in drinking water | (1) Reduced ventricular hypertrophy and lung congestion (2) Restored membrane potential in IFM (3) Improved retention capacity of mitochondrial calcium in the SSM and IFM |
[128] |
| Pressure overload-induced cardiac fibrosis in rats | 2 μmol; oral gavage | (1) Attenuated apoptosis, hypertrophic remodeling, fibrosis, and left ventricular dysfunction (2) Blunted TGF-β1 and NOX4 upregulation (3) Prevented Nrf2 downregulation and rescued TGF-β1 activation (4) Ameliorated the cardiac remodeling dysregulation in phenylephrine and TGF-β1-induced models |
[129] | |
| Rat model of prenatal hypoxia | 125 μmol; intravenous injection | (1) Improved vasorelaxation (2) Alleviated oxidative stress in placental cells (3) Prevented the decrease in vascular sensitivity to phenylephrine of their offspring |
[130] | |
| Mouse model of aortic stiffening | 250 μmol/L in drinking water | (1) Decreased pulse wave velocity in old mice (2) Rescued the decrease of elastin region elastic modulus and elastin expression (3) Reversed in vivo aortic stiffness |
[131] | |
|
| ||||
| MitoE | Bovine aortic endothelial cells induced by hydrogen peroxide and glucose oxidase | 1 μmol/L in culture medium | (1) Abrogated H2O2- and lipid peroxide-induced oxidative protein (2) Inhibited cytochrome c release, caspase 3 activation, and DNA fragmentation (3) Inhibited transferrin receptor-dependent iron uptake and apoptosis |
[132] |
|
| ||||
| SS-20, SS-31 | Rat model of myocardial infarction | 3 mg/kg∗bodyweight; intraperitoneal injection | (1) Reduced lipid peroxidation (2) Decreased the occurrence frequency and severity of arrhythmia |
[133] |
|
| ||||
| SS-31/elamipretide/MTP-131 | Clinical trials on heart failure patients | 20 mg subcutaneous injection 4 and 40 mg intravenous injection |
(1) High-dose SS-31 improved left ventricular volumes (2) Improved super complex-associated oxygen flux, complex (C) I activity |
NCT02388464
[78] |
| Clinical trials on reperfusion injury patients | Intravenous at 0.05 mg/kg/h | (1) Conjunction SS-31 with standard therapy is superior to placebo for reducing myocardial infarction |
NCT01572909
[124] |
|
Notes: CF: cardiac fibroblasts; IFM: interfibrillar mitochondria; LDL-c: high-density lipoprotein cholesterol; lncRNAs: long noncoding RNAs; NOX4: NADPH oxidase subunit 4; Nrf2: nuclear factor erythroid 2; ox-LDL: oxidized high-density lipoprotein; SSM: subsarcolemmal mitochondria; TC: total cholesterol; TEMPO: 4-hydroxy-2,2,6,6-tetramethylpiperidin-N-oxide; TGF-β1: transforming growth factor β 1.
Table 5.
MTAs in cancer models.
| Mitochondria-targeted antioxidants/bioactive component | Models/clinical trials | Dosage | Effects/mechanism | Reference |
|---|---|---|---|---|
| SkQ1 | HT1080 cells | 40 nmol/L in culture medium | (1) Suppressed cell growth and prolonged cell mitosis (2) Induced distribution and activation of Aurora family kinases |
[132] |
| Tumor cells in culture or mouse models | 40 nmol/L in culture medium; 250 nmol/kg∗bodyweight | (1) Decreased cell growth and the weight of subcutaneous tumors (2) Prolonged cell mitosis and apoptosis |
[132] | |
| p53(-/-) mice | 5 nmol/kg∗bodyweight per day | (1) Delayed appearance of tumors (2) Inhibited the growth of xenografts tumors and angiogenesis |
[133] | |
| BALB/c mice in SPF environment | 1 and 30 nmol/kg∗bodyweight per day | (1) Decreased the incidence of spontaneous cancers at the dosage of 30 nmol/kg∗bodyweight (2) Suppressed the cancer dissemination at 1 nmol/kg∗bodyweight dosage |
[134] | |
| Benzopyrene-induced carcinogenesis in SHR mice | 5 and 50 nmol/kg∗bodyweight per day | (1) Inhibited tumor growth (2) Dose-dependent effects were observed |
[135] | |
|
| ||||
| KRSH | HeLa and MCF-7 cells | 50 nmol/L in culture medium | (1) Inhibited greater tumor cell growth than the normal cells (2) Increased apoptosis of HeLa and MCF-7 cells, but not of MCF10A cells (3) Accumulated in mitochondria and increased mitochondrial depolarization |
[136] |
|
| ||||
| Mito-TEMPO | N-Nitrosodiethylamine-induced hepatocarcinogenesis in BALB/c mice | 0.1 mg/kg∗bodyweight weekly | (1) Increased animal survival ratio and decreased tumor incidence and tumor multiplicity (2) Rescued the gap junctions and gap junctional intercellular communication of tumor cells |
[137] |
Notes: HeLa cells: cervical cancer cell line taken from Henrietta Lacks; HT1080: human sarcoma cell line; MCF-7: breast cancer cell line that consisted of the acronym of Michigan Cancer Foundation-7; p53: tumor protein p53; SPF; specific pathogen free.
6.1. Parkinson's Disease
Parkinson's disease (PD) is a progressive neurodegenerative disease that mainly occurs in the elderly, without any acknowledged therapies. Evidence from in vitro cell models, animal models of PD, and genetic analysis has indicated the involvement of oxidative stress and mitochondrial dysfunction during PD development [90]. Thus, the antioxidative strategy shows great potential for PD therapy. In the past two decades, amounts of studies have been conducted and revealed the beneficial roles of antioxidants in the different cellular and animal models; however, the clinical trials using antioxidants (e.g., NAC (oral 1800, 3600 mg daily; 900 mg effervescent tablet daily), glutathione (100-200 mg daily), and vitamin E (1200 IU/day)+coenzyme Q10 (1200, 2400 mg daily)) to treat PD are mostly disappointing. The experimental factors including inefficient oral administration (NCT01470027, direct oral without any coating or carrier), inadequate patients' replicates (NCT01427517, totally 9 participants were involved; NCT02212678, 8 participants were enrolled), and inappropriate outcome measures (NCT00329056, only UPDRS results were provided; NCT02212678, only GSH levels were provided) may explain these frustrating outcomes from clinical studies. Another possible explanation for this ineffectiveness is that the phase for the antioxidant's treatment is too late for the neurons' rescue. Clinical trial showing high doses of CoQ10 administration benefits the PD patients (NCT01892176), which implies that the traditional antioxidants lack bioavailability, with small scales that can be absorbed into the mitochondria. A systematic review and meta-analysis concluded that CoQ10 cannot provide any symptomatic benefit for PD patients [91]. Consequently, approaches delivering antioxidants to mitochondria for PD treatment have been explored. MitoQ was firstly approved for the clinical trials of PD (NCT00329056) in 2006. A study showed MitoQ (40, 80 mg daily) could slow the progression of PD as measured by the Unified Parkinson's Disease Rating Scale (UPDRS); however, no significant difference between MitoQ and placebo on any measure of PD progression was observed [92]. In regard to dosage, although the experiments in vitro (50 nM, 10 μM) and on mouse models (daily 4 mg/kg∗bodyweight) have shown the beneficial effects of MitoQ against mitochondrial dysfunction via preserving striatal dopamine and improving motor functions [93], more study might be conducted to optimize the oral or injection dosage and for the preclinical trials. Besides, the peptide-based mitochondrial antioxidants, such as SS-31 and SS-20 (0.5-5.0 mg/kg∗bodyweight), have shown similar neuroprotective effects on cellular and mouse PD models induced by MPTP [94]; however, the clinical trials using peptide-based mitochondrial antioxidants to treat PD have not been approved until now, which might be hampered by the undesirable results of MitoQ in the clinical trials.
The nanocarrier delivery system that consisted of FDA-approved Pluronic F68 and dequalinium has been revealed to enhance the bioavailability of NAC protecting against the reduced cell viability and oxidative stress in the cellular model of PD, which raises a significant prospect of nanocarrier-based NAC to be transitioned for clinical trials [95]. As indicated by the results from previous clinical trials, the available clinical therapies using antioxidants for PD can only alleviate the symptoms; but none can prevent neuronal degeneration via regulating the dopaminergic system; thus, the combination of MTAs with the traditional drugs (dopamine receptor activator, such as pramipexole) could be a considerable strategy for the further experiments and clinical trials.
6.2. Traumatic Brain Injury (TBI)
TBI is a significant cause of death and disability, with an estimated 60-80 million cases per year worldwide, and has been considered an important medical and social problem. TBI can easily damage the cerebral circulatory, which in turn leads to cerebral artery contraction, glutamate poisoning, mitochondrial dysfunction, inflammatory response, and cell death, thereby increasing the severity of the primary damage and causing secondary brain damage [98]. Mechanistically, selective peroxidation of cardiolipin, impaired electron transport, decreased ATP production, and increased formation of ROS during TBI development lead to the final neurodegeneration and brain atrophy [99, 100]. In the past two decades, the beneficial effect of vitamins C and E, progesterone, and NAC to be used for adjuvant therapy in TBI has been evaluated [101]. Nontargeted antioxidants, commonly in very high concentrations, are used to achieve therapeutic effects. It should be noted that mitochondria are the main source of ROS and determine cell fate [102]; antioxidant delivery to mitochondria is an important target for TBI intervention therapy.
N-Acetylcysteine (NCT00822263), docosahexaenoic acid (NCT01903525), and melatonin (NCT04034771) had been approved for the clinical TBI trials. These clinical results indicated that the NAC (4 grams daily) administration could reduce the sequela of mild TBI [103]. Although the measurements of temperature, mean arterial pressure, intracranial pressure (ICP), use of ICP-directed therapies, surveillance serum brain injury biomarkers, and Glasgow Outcome Scale (GOS) at 3 months were not different between the NAC group and the placebo group [104], the metabolomic results support the antioxidative therapeutic target by the probenecid and N-acetylcysteine treatment [105]. Compared with nontargeted antioxidants, relatively low concentrations of MTAs show higher antioxidant activity. The antioxidant activity of SkQR1, XJB-5-131, Mito-TEMPO, SS-31, and MitoQ was revealed in TBI models (Table 3). Although the MTA dosages and mechanisms involved in the therapeutic efficiency are different, data from these different experimental models suggest that MTAs may be a more effective means for mitigating the negative effects of TBI. In terms of mechanism, in addition to antioxidant efficiency, the activation of anti-inflammatory and Nrf2-ARE signaling may also be key indicators for the effectiveness evaluation of MTAs. Until now, the clinical trials using MTAs to treat TBI have not been approved. In future studies, while focusing on the action mechanism and effective dose of various MTAs, the possible toxicological properties of these MTAs also need to be clarified.
6.3. Cardiovascular Diseases (CVDs)
Cell redox homeostasis maintains a healthy physiological state of cardiomyocytes and vascular endothelial cells. The content of superoxide anions in the failing human myocardium was found to be twice more than that in the healthy myocardium [111]. There are also similar observations in diabetes [112] and hypertensive cardiomyopathy [113]. Besides, the damage parameters of lipid oxidative, nucleic acid, and protein have been observed in the circulation or myocardial tissue of patients with myocardial infarction or heart failure in the animal models of these conditions [113–116]. Moreover, the oxidative damage of the mitochondria and ROS production by endothelial cells were significantly higher compared with the myocardium of young mice, indicating oxidative stress is also involved in age-related CVDs. The depletion of Sod2 (encoding mitochondrial superoxide dismutase) can aggravate the atherosclerotic process in mice, which confirms the detrimental role of ROS overproduction [117]. Interestingly, the absence of a specific cardiomyocyte Txnrd2 (encoding thioredoxin reductase 2) results in fatal dilated cardiomyopathy in the mouse embryo [118]. These observations indicate that overloaded oxidative stress is associated with a variety of cardiovascular diseases, such as atherosclerosis, cardiac hypertrophy, cardiomyopathy, and heart failure [119].
In the cases of overloaded oxidative stress, a timely supplement of antioxidants to maintain the normal function of the cardiovascular system is crucial for the prevention and treatment of cardiovascular diseases. Some conventional antioxidants such as CoQ10 [120], polyphenols [121], vitamin C [122], and vitamin E [123] have been shown to prevent and treat cardiovascular disease in models, with expanded ongoing clinical trials (NCT03133793, NCT01925937, NCT02779634, NCT02847585, NCT02934555, and NCT02218476). However, the majority of these clinical studies are in unknown status which might be attributed to the poor biopharmaceutical properties and the pharmacokinetics of the nontargeted antioxidants. Mitochondria are the main sites for the ROS production and oxidative energy metabolism, implying the MTAs might show better efficacy for the treatment of cardiovascular disease. In recent studies, promising results have been obtained with the MTAs, including Mito-TEMPO, MitoSNO, SkQ1, MitoQ, MitoE, SS-20, and SS-31, in the cellular and animal models (Table 4), fostering the initiation of clinical trials for CVD treatment. These promising results from preclinical experiments of MitoQ have fostered ongoing clinical trials for diastolic dysfunction (NCT03586414, suspended due to COVID-19 outbreak) and peripheral artery disease (NCT03506633, under recruiting). Clinical trials of SS-31 (formerly named as Bendavia or MTP-131) on the treatment of reperfusion injury (NCT01572909) and heart failure (NCT02388464, NCT02788747, NCT02245620, and NCT02814097) have been completed. The clinical results on reperfusion injury patients indicated that SS-31 combination therapy is superior to placebo [124]. These clinical trials on heart failure patients revealed that SS-31 brings favorable changes in left ventricular volumes in a dose-dependent manner. Recently, one study using an explanted human heart tissue model extended these clinical trials and revealed the beneficial effects of SS-31 on mitochondrial function in heart failure [78]. For further application, the clinical dosage and therapeutic effects of MTAs on the treatment of CVDs need to be further explored.
6.4. Cancer
The mitochondria-derived ROS are crucial in cancer development, which makes mitochondria-targeted antioxidants promising anticancer agents. The relationship between mitochondrial ROS and cancer development has been reviewed in recent publications [134, 135]. Generally, changes in the mitochondrial function of tumor cells (“aerobic glycolysis,” also known as “Warburg effect”) lead to further mitochondrial ROS production and nuclear DNA mutations that impair OXPHOS. As a consequence, oncogenic ROS promote the occurrence and development of tumors via inducing DNA damage and regulating various signaling pathways. For example, the production of H2O2 mediated by endogenous oncogenes can improve the proliferation rate of tumor cells via regulating MAPK signals and stimulating extracellular ERK pathway kinase. Mitochondrial production of O2- stimulates the growth of KRAS lung cancer cells through MAPK/ERK signaling [136]. The upregulation of ROS also activates transcription factors such as nuclear factor-κB (NF-κB), which increases the proliferation of cancer cells [137].
In the past two decades, several studies focused on targeting mitochondria for anticancer therapy. The anticancer mechanism of these agents includes the following: (1) increasing the conductivity of mitochondrial transition pore complex (PTPC), thereby promoting the rupture of mitochondrial membrane and the release of mitochondrial apoptotic factors [138]; (2) targeting proapoptotic Bcl-2 homology domain 3 (BH3) protein mimetics, during which the apoptotic factors are released [139]; (3) sensitizing the cancer cells to conventional treatments via inhibiting glycolysis of cancer cells [140]; and (4) interrupting glutamine catabolism, pyruvate dehydrogenase, and lactate dehydrogenase [138]. Comparatively, the MTAs with the properties of scavenging the mitochondrial ROS or inhibiting the ROS production could only inhibit the proliferation of cancer cells instead of triggering apoptosis. Several preclinical studies have shown the antigrowth effects of MTAs on cancer models (Table 5). Among the MTAs used for the preclinical experiments, the SkQ1 was the most promising and extensively studied one showing anticancer effects at a nanomole dosage in various models. For example, 40 nmol/L SkQ1 treatment suppressed the proliferation of HT1080 and RD tumor cells in culture via inhibiting mitosis [141]. Daily 5, 30, or 50 nmol/kg∗bodyweight SkQ1-supplemented diet decreased the incidence of spontaneous cancers in p53 knockout mice, BALB/c mice, and benzopyrene-induced mice, respectively [142–144]. Recently, a mitochondria-targeted peptide KRSH that consisted of lysine, arginine, tyrosine, and cysteine (Figure 5) was revealed to inhibit cell proliferation and increase apoptosis of HeLa and MCF-7 cell lines [145]. Mechanically, the positively charged lysine and arginine help the KRSH mitochondria-targeted delivery, meanwhile, the tyrosine and cysteine play antioxidative roles for scavenging mitochondrial ROS. The possible proapoptotic effects of KRSH on HeLa and MCF-7 cells might be attributed to the increased mitochondrial depolarization, but not the antioxidative effects, although the definite mechanism is unknown. The Mito-TEMPO, a TPP-linked MTA whose chemical structure is shown in Figure 2, was revealed to increase the survival ratio and decrease the tumor incidence and tumor multiplicity in the N-nitrosodiethylamine-induced hepatocarcinogenesis mice [146]; however, no in vitro evidence attributes its anticancer effect to the ROS scavenging.
Figure 5.
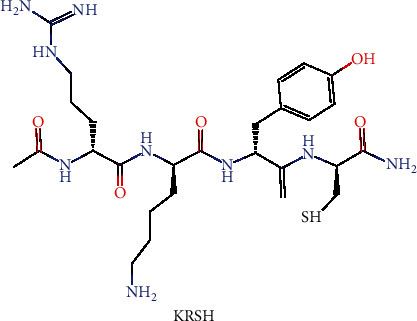
Chemical structure of KRSH.
MTAs have yielded promising results in several in vitro and animal models for cancer studies; however, it is noteworthy that some subsets of cancer cells, such as melanoma tumor cells, exhibit metabolic reprogramming heterogeneity, showing different bioenergy and ROS detoxification capabilities [147]. Besides, a study comparing the effects of MTAs and NAs on the hepatocarcinogenesis indicates contradictory results; that is, nontargeted antioxidants (NAC and vitamin E analog Trolox) prevented tumorigenesis, whereas MTAs (SS-31 and Mito-Q) aggravated tumorigenesis [148]. Therefore, it is critical to clarify the metabolic patterns of different cancer cells in a specific stage and to carefully adopt appropriate therapeutic strategies before clinical intervention with MTAs.
7. Conclusion
Mitochondria produce most of the energy and ROS in cells. Mitochondrial ROS are important signaling molecules involved in many cellular adaptative oxidative defense systems. However, excessive ROS accumulation or insufficient clearance results in damaged mitochondrial DNA and protein, both of which are pathophysiological features of a variety of diseases. In the past decades, many studies focused on developing NAs to restore the normal physiological function of oxidative stressed mitochondria. Research studies on various models were promising, but clinical trials sometimes showed contradictory results. Redox signaling is an important part of many physiological processes. Excessive or inappropriate use of antioxidants may abolish ROS production and result in compensatory upregulation of MAPK pathways, which in turn break down the endogenous antioxidant system. Thus, applying the appropriate dosage and delivery method of these antioxidants to balance ROS production and antioxidation is crucial for the clinical trials. Recently, a variety of mitochondria-targeted delivery systems and antioxidants have been exploited to recover mitochondrial function from the pathological conditions in different mechanisms. The outstanding advantages of MTAs over the nontargeted ones include (1) efficient pharmacokinetics and absorption and (2) specific accumulation at cells and mitochondria, avoiding nonspecific high concentration-induced side effects.
This article reviews the characteristics and applications of different mitochondria-targeting tools, including lipophilic cations, liposome vectors, peptide-based targeting, and their recent research reports. Overall, most of these tools have shown beneficial roles for mitochondria-targeting delivery. Based on these delivery tools, an increasing number of MTAs are currently being evaluated, some of which have been validated as effective agents in stage 2 clinical trials, providing unlimited possibilities for mitochondria-targeted therapies. Although the results from current studies are very promising, the human clinical trials on different disease stages should be firstly standardized to effectively translate these research results into usable medicines. Besides, these noteworthy questions should be preferentially considered to exploit more MTAs for disease treatment in the future (refer to noteworthy questions).
Acknowledgments
The authors apologize to scientists in this field whose papers are not cited due to space limitations. The authors are grateful to Emily Ammeter, who is one of the summer undergraduate students in the Department of Animal Science at the University of Manitoba, for her help in the manuscript preparation. This research was funded by the Young Elite Scientists Sponsorship Program by CAST (2019QNRC001), the Hunan Science Foundation for Outstanding Young Scholars (2020JJ3023), and the Open Project Program of Key Laboratory of Feed Biotechnology, the Ministry of Agriculture and Rural Affairs of the People's Republic of China.
Contributor Information
Jie Yin, Email: yinjie2014@126.com.
Bie Tan, Email: bietan@hunau.edu.cn.
Data Availability
No data were used to support this review article.
Additional Points
Noteworthy Questions. (1) What is the decisive mechanism in the development of mitochondria-targeted disease? (2) What is the most effective antioxidant for regulating the decisive mechanism (according to the experiment on separated mitochondria)? (3) Which delivery system will be the best choice for the mitochondria-targeted delivery of antioxidants? (4) How to optimize the effective MTA dosage in different administration approaches (e.g., oral, intravenous, or subcutaneous) for disease treatment? (5) How to standardize a clinical trial (e.g., how many patients to get involved, how long for patient tracking, and what parameters to assess side effects) for the evaluation of MTAs in disease treatment?
Conflicts of Interest
The authors declare no competing financial interest.
Authors' Contributions
Q. J., J. Y., JS. C., XK. M., MM. W., G. L., K. Y., BE. T., and YL. Y. cowrote the manuscript.
References
- 1.Cadenas E., Davies K. J. Mitochondrial free radical generation, oxidative stress, and aging. Free Radical Biology & Medicine. 2000;29(3-4):222–230. doi: 10.1016/s0891-5849(00)00317-8. [DOI] [PubMed] [Google Scholar]
- 2.Green D. R., Galluzzi L., Kroemer G. Metabolic control of cell death. Science. 2014;345(6203, article e1250256) doi: 10.1126/science.1250256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cai Q., Tammineni P. Alterations in mitochondrial quality control in Alzheimer's disease. Frontiers in Cellular Neuroscience. 2016;10 doi: 10.3389/fncel.2016.00024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Miura Y. Oxidative stress, radiation-adaptive responses, and aging. Journal of Radiation Research. 2004;45(3):357–372. doi: 10.1269/jrr.45.357. [DOI] [PubMed] [Google Scholar]
- 5.Kamat J. P., Ghosh A., Devasagayam T. P. A. Vanillin as an antioxidant in rat liver mitochondria: inhibition of protein oxidation and lipid peroxidation induced by photosensitization. Molecular and Cellular Biochemistry. 2000;209(1/2):47–53. doi: 10.1023/A:1007048313556. [DOI] [PubMed] [Google Scholar]
- 6.Picca A., Mankowski R. T., Burman J. L., et al. Mitochondrial quality control mechanisms as molecular targets in cardiac ageing. Nature Reviews Cardiology. 2018;15(9):543–554. doi: 10.1038/s41569-018-0059-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Picard M., Wallace D. C., Burelle Y. The rise of mitochondria in medicine. Mitochondrion. 2016;30:105–116. doi: 10.1016/j.mito.2016.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Szeto H. H. Cell-permeable, mitochondrial-targeted, peptide antioxidants. The AAPS Journal. 2006;8(2):E277–E283. doi: 10.1007/bf02854898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang W. J., Hu X. L., Shen Q., Xing D. Mitochondria-specific drug release and reactive oxygen species burst induced by polyprodrug nanoreactors can enhance chemotherapy. Nature Communications. 2019;10(1):p. 1704. doi: 10.1038/s41467-019-09566-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Takahashi M., Takahashi K. Water-soluble CoQ10 as a promising anti-aging agent for neurological dysfunction in brain mitochondria. Antioxidants. 2019;8(3):p. 61. doi: 10.3390/antiox8030061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Trist B. G., Hare D. J., Double K. L. Oxidative stress in the aging substantia nigra and the etiology of Parkinson's disease. Aging Cell. 2019;18(6) doi: 10.1111/acel.13031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ravarotto V., Simioni F., Carraro G., Bertoldi G., Pagnin E., Calò L. Oxidative stress and cardiovascular-renal damage in Fabry disease: is there room for a pathophysiological involvement? Journal of Clinical Medicine. 2018;7(11) 11:p. 409. doi: 10.3390/jcm7110409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang P. Y., Xu X., Li X. C. Cardiovascular diseases: oxidative damage and antioxidant protection. European Review for Medical and Pharmacological Sciences. 2014;18(20):3091–3096. [PubMed] [Google Scholar]
- 14.Hadi H., Vettor R., Rossato M. Vitamin E as a treatment for nonalcoholic fatty liver disease: reality or myth? Antioxidants. 2018;7(1):p. 12. doi: 10.3390/antiox7010012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ho U., Luff J., James A., et al. SMG1 heterozygosity exacerbates haematopoietic cancer development in Atm null mice by increasing persistent DNA damage and oxidative stress. Journal of Cellular and Molecular Medicine. 2019;23(12):8151–8160. doi: 10.1111/jcmm.14685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Reberio P., James A., Ling S., et al. ATM and SMG1 are tumour suppressors which co- regulate blood cancer development and cellular responses to DNA damage and oxidative stress. Leukemia & Lymphoma. 2015;56:78–78. doi: 10.3109/10428194.2015.108089306. [DOI] [Google Scholar]
- 17.Salehi B., Martorell M., Arbiser J. L., et al. Antioxidants: positive or negative actors? Biomolecules. 2018;8(4):p. 124. doi: 10.3390/biom8040124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Park Y., Spiegelman D., Hunter D. J., et al. Intakes of vitamins A, C, and E and use of multiple vitamin supplements and risk of colon cancer: a pooled analysis of prospective cohort studies. Cancer Causes & Control. 2010;21(11):1745–1757. doi: 10.1007/s10552-010-9549-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cerimele F., Battle T., Lynch R., et al. Reactive oxygen signaling and MAPK activation distinguish Epstein-Barr virus (EBV)-positive versus EBV-negative Burkitt's lymphoma. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(1):175–179. doi: 10.1073/pnas.0408381102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Burgoyne J. R., Mongue-Din H., Eaton P., Shah A. M. Redox signaling in cardiac physiology and pathology. Circulation Research. 2012;111(8):1091–1106. doi: 10.1161/CIRCRESAHA.111.255216. [DOI] [PubMed] [Google Scholar]
- 21.Liberman E. A., Topaly V. P. Permeability of bimolecular phospholipid membranes for fat-soluble ions. Biofizika. 1969;14(3):452–461. [PubMed] [Google Scholar]
- 22.Zinovkin R. A., Zamyatnin A. A. Mitochondria-targeted drugs. Current Molecular Pharmacology. 2019;12(3):202–214. doi: 10.2174/1874467212666181127151059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rossman M. J., Santos-Parker J. R., Steward C. A. C., et al. Chronic supplementation with a mitochondrial antioxidant (MitoQ) improves vascular function in healthy older adults. Hypertension. 2018;71(6):1056–1063. doi: 10.1161/HYPERTENSIONAHA.117.10787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gane E. J., Weilert F., Orr D. W., et al. The mitochondria-targeted anti-oxidant mitoquinone decreases liver damage in a phase II study of hepatitis C patients. Liver International. 2010;30(7):1019–1026. doi: 10.1111/j.1478-3231.2010.02250.x. [DOI] [PubMed] [Google Scholar]
- 25.Petrov A., Perekhvatova N., Skulachev M., Stein L., Ousler G. SkQ1 ophthalmic solution for dry eye treatment: results of a phase 2 safety and efficacy clinical study in the environment and during challenge in the controlled adverse environment model. Advances in Therapy. 2016;33(1):96–115. doi: 10.1007/s12325-015-0274-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Apostolova N., Victor V. M. Molecular strategies for targeting antioxidants to mitochondria: therapeutic implications. Antioxidants & Redox Signaling. 2015;22(8):686–729. doi: 10.1089/ars.2014.5952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ross M. F., Kelso G. F., Blaikie F. H., et al. Lipophilic triphenylphosphonium cations as tools in mitochondrial bioenergetics and free radical biology. Biochemistry-Moscow. 2005;70(2):222–230. doi: 10.1007/s10541-005-0104-5. [DOI] [PubMed] [Google Scholar]
- 28.Smith R. A., Porteous C. M., Gane A. M., Murphy M. P. Delivery of bioactive molecules to mitochondria in vivo. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(9):5407–5412. doi: 10.1073/pnas.0931245100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Adlam V. J., Harrison J. C., Porteous C. M., et al. Targeting an antioxidant to mitochondria decreases cardiac ischemia-reperfusion injury. FASEB Journal. 2005;19(9):1088–1095. doi: 10.1096/fj.05-3718com. [DOI] [PubMed] [Google Scholar]
- 30.Zielonka J., Joseph J., Sikora A., et al. Mitochondria-targeted triphenylphosphonium-based compounds: syntheses, mechanisms of action, and therapeutic and diagnostic applications. Chemical Reviews. 2017;117(15):10043–10120. doi: 10.1021/acs.chemrev.7b00042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dong L. F., Jameson V. J., Tilly D., et al. Mitochondrial targeting of vitamin E succinate enhances its pro-apoptotic and anti-cancer activity via mitochondrial complex II. Journal of Biological Chemistry. 2011;286(5):3717–3728. doi: 10.1074/jbc.M110.186643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jameson V. J. A., Cochemé H. M., Logan A., Hanton L. R., Smith R. A. J., Murphy M. P. Synthesis of triphenylphosphonium vitamin E derivatives as mitochondria-targeted antioxidants. Tetrahedron. 2015;71(44):8444–8453. doi: 10.1016/j.tet.2015.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bakeeva L. E., Barskov I. V., Egorov M. V., et al. Mitochondria-targeted plastoquinone derivatives as tools to interrupt execution of the aging program. 2. Treatment of some ROS- and age-related diseases (heart arrhythmia, heart infarctions, kidney ischemia, and stroke) Biochemistry-Moscow. 2008;73(12):1288–1299. doi: 10.1134/S000629790812002X. [DOI] [PubMed] [Google Scholar]
- 34.Skulachev V. P., Antonenko Y. N., Cherepanov D. A., et al. Prevention of cardiolipin oxidation and fatty acid cycling as two antioxidant mechanisms of cationic derivatives of plastoquinone (SkQs) Biochimica et Biophysica Acta-Bioenergetics. 2010;1797(6-7):878–889. doi: 10.1016/j.bbabio.2010.03.015. [DOI] [PubMed] [Google Scholar]
- 35.Murphy M. P. Understanding and preventing mitochondrial oxidative damage. Biochemical Society Transactions. 2016;44(5):1219–1226. doi: 10.1042/BST20160108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tauskela J. S. MitoQ--a mitochondria-targeted antioxidant. IDrugs: the investigational drugs journal. 2007;10(6):399–412. [PubMed] [Google Scholar]
- 37.Finichiu P. G., Larsen D. S., Evans C., et al. A mitochondria-targeted derivative of ascorbate: MitoC. Free Radical Biology and Medicine. 2015;89:668–678. doi: 10.1016/j.freeradbiomed.2015.07.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kelso G. F., Maroz A., Cochemé H. M., et al. A mitochondria-targeted macrocyclic Mn(II) superoxide dismutase mimetic. Chemistry & Biology. 2012;19(10):1237–1246. doi: 10.1016/j.chembiol.2012.08.005. [DOI] [PubMed] [Google Scholar]
- 39.Maroz A., Kelso G. F., Smith R. A. J., Ware D. C., Anderson R. F. Pulse radiolysis investigation on the mechanism of the catalytic action of Mn(II) - Pentaazamacrocycle compounds as superoxide dismutase mimetics. Journal of Physical Chemistry A. 2008;112(22):4929–4935. doi: 10.1021/jp800690u. [DOI] [PubMed] [Google Scholar]
- 40.Bangham A. D., Horne R. W. Negative staining of phospholipids and their structural modification by surface-active agents as observed in the electron microscope. Journal of Molecular Biology. 1964;8(5):660–IN10. doi: 10.1016/S0022-2836(64)80115-7. [DOI] [PubMed] [Google Scholar]
- 41.Alipour M., Omri A., Smith M. G., Suntres Z. E. Prophylactic effect of liposomal N-acetylcysteine against LPS-induced liver injuries. Journal of Endotoxin Research. 2016;13(5):297–304. doi: 10.1177/0968051907085062. [DOI] [PubMed] [Google Scholar]
- 42.Rezaei-Sadabady R., Eidi A., Zarghami N., Barzegar A. Intracellular ROS protection efficiency and free radical-scavenging activity of quercetin and quercetin-encapsulated liposomes. Artificial Cells Nanomedicine and Biotechnology. 2014;44(1):128–134. doi: 10.3109/21691401.2014.926456. [DOI] [PubMed] [Google Scholar]
- 43.Fan J., Shek P. N., Suntres Z. E., Li Y. H., Oreopoulos G. D., Rotstein O. D. Liposomal antioxidants provide prolonged protection against acute respiratory distress syndrome. Surgery. 2000;128(2):332–338. doi: 10.1067/msy.2000.108060. [DOI] [PubMed] [Google Scholar]
- 44.Koudelka S., Turanek Knotigova P., Masek J., et al. Liposomal delivery systems for anti-cancer analogues of vitamin E. Journal of Controlled Release. 2015;207:59–69. doi: 10.1016/j.jconrel.2015.04.003. [DOI] [PubMed] [Google Scholar]
- 45.Loguercio C., Federico A., Trappoliere M., et al. The effect of a silybin-vitamin E-phospholipid complex on nonalcoholic fatty liver disease: a pilot study. Digestive Diseases and Sciences. 2007;52(9):2387–2395. doi: 10.1007/s10620-006-9703-2. [DOI] [PubMed] [Google Scholar]
- 46.Takahashi M., Uechi S., Takara K., Asikin Y., Wada K. Evaluation of an oral carrier system in rats: bioavailability and antioxidant properties of liposome-encapsulated curcumin. Journal of Agricultural and Food Chemistry. 2009;57(19):9141–9146. doi: 10.1021/jf9013923. [DOI] [PubMed] [Google Scholar]
- 47.Chiu C. H., Chang C. C., Lin S. T., Chyau C. C., Peng R. Y. Improved hepatoprotective effect of liposome-encapsulated astaxanthin in lipopolysaccharide-induced acute hepatotoxicity. International Journal of Molecular Sciences. 2016;17(7):p. 1128. doi: 10.3390/ijms17071128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yasuzaki Y., Yamada Y., Harashima H. Mitochondrial matrix delivery using MITO-Porter, a liposome-based carrier that specifies fusion with mitochondrial membranes. Biochemical and Biophysical Research Communications. 2010;397(2):181–186. doi: 10.1016/j.bbrc.2010.05.070. [DOI] [PubMed] [Google Scholar]
- 49.Yamada Y., Harashima H. MITO-Porter for mitochondrial delivery and mitochondrial functional analysis. In: Singh H., Sheu S.-S., editors. Pharmacology of Mitochondria. Cham: Springer International Publishing; 2017. pp. 457–472. [DOI] [PubMed] [Google Scholar]
- 50.Yamada Y., Akita H., Kamiya H., et al. MITO-Porter: a liposome-based carrier system for delivery of macromolecules into mitochondria via membrane fusion. Biochimica et Biophysica Acta-Biomembranes. 2008;1778(2):423–432. doi: 10.1016/j.bbamem.2007.11.002. [DOI] [PubMed] [Google Scholar]
- 51.Yamada Y., Nakamura K., Abe J., et al. Mitochondrial delivery of coenzyme Q10 via systemic administration using a MITO-Porter prevents ischemia/reperfusion injury in the mouse liver. Journal of Controlled Release. 2015;213:86–95. doi: 10.1016/j.jconrel.2015.06.037. [DOI] [PubMed] [Google Scholar]
- 52.Yamada Y., Daikuhara S., Tamura A., Nishida K., Yui N., Harashima H. Enhanced autophagy inductionviathe mitochondrial delivery of methylated β-cyclodextrin-threaded polyrotaxanes using a MITO-Porter. Chemical Communications. 2019;55(50):7203–7206. doi: 10.1039/C9CC03272J. [DOI] [PubMed] [Google Scholar]
- 53.Yamada Y., Furukawa R., Yasuzaki Y., Harashima H. Dual function MITO-Porter, a nano carrier integrating both efficient cytoplasmic delivery and mitochondrial macromolecule delivery. Molecular Therapy. 2011;19(8):1449–1456. doi: 10.1038/mt.2011.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cerrato C. P., Pirisinu M., Vlachos E. N., Langel Ü. Novel cell-penetrating peptide targeting mitochondria. FASEB Journal. 2015;29(11):4589–4599. doi: 10.1096/fj.14-269225. [DOI] [PubMed] [Google Scholar]
- 55.Toyama S., Shimoyama N., Szeto H. H., Schiller P. W., Shimoyama M. Protective effect of a mitochondria-targeted peptide against the development of chemotherapy-induced peripheral neuropathy in mice. ACS Chemical Neuroscience. 2018;9(7):1566–1571. doi: 10.1021/acschemneuro.8b00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Qvit N., Rubin S. J. S., Urban T. J., Mochly-Rosen D., Gross E. R. Peptidomimetic therapeutics: scientific approaches and opportunities. Drug Discovery Today. 2017;22(2):454–462. doi: 10.1016/j.drudis.2016.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Oliver D. M. A., Reddy P. H. Small molecules as therapeutic drugs for Alzheimer's disease. Molecular and Cellular Neuroscience. 2019;96:47–62. doi: 10.1016/j.mcn.2019.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kezic A., Spasojevic I., Lezaic V., Bajcetic M. Mitochondria-targeted antioxidants: future perspectives in kidney ischemia reperfusion injury. Oxidative Medicine and Cellular Longevity. 2016;2016:12. doi: 10.1155/2016/2950503.2950503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.BÖHMOVÁ E., MACHOVÁ D., PECHAR M., et al. Cell-penetrating peptides: a useful tool for the delivery of various cargoes into cells. Physiological Research. 2018;67(Suppl 2):S267–S279. doi: 10.33549/physiolres.933975. [DOI] [PubMed] [Google Scholar]
- 60.Alder N. N., Mitchell W., Ng E., et al. Biophysical approaches toward understanding the molecular mechanism of action of the mitochondrial therapeutic SS-31 (elamipretide) Biophysical Journal. 2019;116(3):511a–512a. doi: 10.1016/j.bpj.2018.11.2759. [DOI] [Google Scholar]
- 61.Galdiero S., Gomes P. Peptide-based drugs and drug delivery systems. Molecules. 2017;22(12):p. 2185. doi: 10.3390/molecules22122185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Szeto H. H. Stealth peptides target cellular powerhouses to fight rare and common age-related diseases. Protein & Peptide Letters. 2018;25(12):1108–1123. doi: 10.2174/0929866525666181101105209. [DOI] [PubMed] [Google Scholar]
- 63.Serviddio G., Bellanti F., Sastre J., Vendemiale G., Altomare E. Targeting mitochondria: a new promising approach for the treatment of liver diseases. Current Medicinal Chemistry. 2010;17(22):2325–2337. doi: 10.2174/092986710791698530. [DOI] [PubMed] [Google Scholar]
- 64.Imai T., Mishiro K., Takagi T., et al. Protective effect of Bendavia (SS-31) against oxygen/glucose-deprivation stress-induced mitochondrial damage in human brain microvascular endothelial cells. Current Neurovascular Research. 2017;14(1):53–59. doi: 10.2174/1567202614666161117110609. [DOI] [PubMed] [Google Scholar]
- 65.Kuang X., Zhou S., Guo W., Wang Z., Sun Y., Liu H. SS-31 peptide enables mitochondrial targeting drug delivery: a promising therapeutic alteration to prevent hair cell damage from aminoglycosides. Drug Delivery. 2017;24(1):1750–1761. doi: 10.1080/10717544.2017.1402220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wu J., Hao S., Sun X. R., et al. Elamipretide (SS-31) ameliorates isoflurane-induced long-term impairments of mitochondrial morphogenesis and cognition in developing rats. Frontiers in Cellular Neuroscience. 2017;11:p. 119. doi: 10.3389/fncel.2017.00119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhang M., Zhao H., Cai J., et al. Chronic administration of mitochondrion-targeted peptide SS-31 prevents atherosclerotic development in ApoE knockout mice fed Western diet. PLoS One. 2017;12(9, article e0185688) doi: 10.1371/journal.pone.0185688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhao H., Li H., Hao S., et al. Peptide SS-31 upregulates frataxin expression and improves the quality of mitochondria: implications in the treatment of Friedreich ataxia. Scientific Reports. 2017;7(1):p. 9840. doi: 10.1038/s41598-017-10320-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhao H., Liu Y. J., Liu Z. R., et al. Role of mitochondrial dysfunction in renal fibrosis promoted by hypochlorite-modified albumin in a remnant kidney model and protective effects of antioxidant peptide SS-31. European Journal of Pharmacology. 2017;804:57–67. doi: 10.1016/j.ejphar.2017.03.037. [DOI] [PubMed] [Google Scholar]
- 70.Cai J., Jiang Y., Zhang M., et al. Protective effects of mitochondrion-targeted peptide SS-31 against hind limb ischemia-reperfusion injury. Journal of Physiology and Biochemistry. 2018;74(2):335–343. doi: 10.1007/s13105-018-0617-1. [DOI] [PubMed] [Google Scholar]
- 71.Campbell M. D., Duan J., Samuelson A. T., et al. Improving mitochondrial function with SS-31 reverses age-related redox stress and improves exercise tolerance in aged mice. Free Radical Biology and Medicine. 2019;134:268–281. doi: 10.1016/j.freeradbiomed.2018.12.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Escribano-Lopez I., Diaz-Morales N., Iannantuoni F., et al. The mitochondrial antioxidant SS-31 increases SIRT1 levels and ameliorates inflammation, oxidative stress and leukocyte-endothelium interactions in type 2 diabetes. Scientific Reports. 2018;8(1):p. 15862. doi: 10.1038/s41598-018-34251-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hou S., Yang Y., Zhou S., et al. Novel SS-31 modified liposomes for improved protective efficacy of minocycline against drug-induced hearing loss. Biomaterials Science. 2018;6(6):1627–1635. doi: 10.1039/C7BM01181D. [DOI] [PubMed] [Google Scholar]
- 74.Tarantini S., Valcarcel-Ares N. M., Yabluchanskiy A., et al. Treatment with the mitochondrial-targeted antioxidant peptide SS-31 rescues neurovascular coupling responses and cerebrovascular endothelial function and improves cognition in aged mice. Aging Cell. 2018;17(2, article e12731) doi: 10.1111/acel.12731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhang W., Tam J., Shinozaki K., et al. Increased survival time with SS-31 after prolonged cardiac arrest in rats. Heart Lung and Circulation. 2019;28(3):505–508. doi: 10.1016/j.hlc.2018.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Czigler A., Toth L., Szarka N., et al. Hypertension exacerbates cerebrovascular oxidative stress induced by mild traumatic brain injury: protective effects of the mitochondria-targeted antioxidative peptide SS-31. Journal of Neurotrauma. 2019;36(23):3309–3315. doi: 10.1089/neu.2019.6439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zhu Y., Wang H., Fang J., et al. SS-31 provides neuroprotection by reversing mitochondrial dysfunction after traumatic brain injury. Oxidative Medicine and Cellular Longevity. 2018;2018:12. doi: 10.1155/2018/4783602.4783602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chatfield K. C., Sparagna G. C., Chau S., et al. Elamipretide improves mitochondrial function in the failing human heart. JACC: Basic to Translational Science. 2019;4(2):147–157. doi: 10.1016/j.jacbts.2018.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Daubert M. A., Yow E., Dunn G., et al. Novel mitochondria-targeting peptide in heart failure Treatment. Circulation-Heart Failure. 2017;10(12, article e004389) doi: 10.1161/CIRCHEARTFAILURE.117.004389. [DOI] [PubMed] [Google Scholar]
- 80.Sabbah H. N., Gupta R. C., Singh-Gupta V., Zhang K. Effects of elamipretide on skeletal muscle in dogs with experimentally induced heart failure. ESC Heart Failure. 2019;6(2):328–335. doi: 10.1002/ehf2.12408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sabbah H. N., Gupta R. C., Singh-Gupta V., Zhang K., Lanfear D. E. Abnormalities of mitochondrial dynamics in the failing heart: normalization following long-term therapy with elamipretide. Cardiovascular Drugs and Therapy. 2018;32(4):319–328. doi: 10.1007/s10557-018-6805-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Liu D., Jin F., Shu G., et al. Enhanced efficiency of mitochondria-targeted peptide SS-31 for acute kidney injury by pH-responsive and AKI-kidney targeted nanopolyplexes. Biomaterials. 2019;211:57–67. doi: 10.1016/j.biomaterials.2019.04.034. [DOI] [PubMed] [Google Scholar]
- 83.Saad A., Herrmann S. M. S., Eirin A., et al. Phase 2a clinical trial of mitochondrial protection (elamipretide) during stent revascularization in patients with atherosclerotic renal artery stenosis. Circulation-Cardiovascular Interventions. 2017;10(9, article e005487) doi: 10.1161/CIRCINTERVENTIONS.117.005487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sweetwyne M. T., Pippin J. W., Eng D. G., et al. The mitochondrial-targeted peptide, SS-31, improves glomerular architecture in mice of advanced age. Kidney International. 2017;91(5):1126–1145. doi: 10.1016/j.kint.2016.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Yousif L. F., Stewart K. M., Horton K. L., Kelley S. O. Mitochondria-penetrating peptides: sequence effects and model cargo transport. Chembiochem. 2009;10(12):2081–2088. doi: 10.1002/cbic.200900017. [DOI] [PubMed] [Google Scholar]
- 86.Polyzos A., Holt A., Brown C., et al. Mitochondrial targeting of XJB-5-131 attenuates or improves pathophysiology in HdhQ150 animals with well-developed disease phenotypes. Human Molecular Genetics. 2016;25(9):1792–1802. doi: 10.1093/hmg/ddw051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Polyzos A. A., Wood N. I., Williams P., Wipf P., Morton A. J., McMurray C. T. XJB-5-131-mediated improvement in physiology and behaviour of the R6/2 mouse model of Huntington's disease is age- and sex- dependent. PLoS One. 2018;13(4, article e0194580) doi: 10.1371/journal.pone.0194580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Xun Z., Rivera-Sánchez S., Ayala-Peña S., et al. Targeting of XJB-5-131 to mitochondria suppresses oxidative DNA damage and motor decline in a mouse model of Huntington's disease. Cell Reports. 2012;2(5):1137–1142. doi: 10.1016/j.celrep.2012.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Krainz T., Gaschler M. M., Lim C., Sacher J. R., Stockwell B. R., Wipf P. A mitochondrial-targeted nitroxide is a potent inhibitor of ferroptosis. ACS Central Science. 2016;2(9):653–659. doi: 10.1021/acscentsci.6b00199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Subramaniam S. R., Chesselet M. F. Mitochondrial dysfunction and oxidative stress in Parkinson's disease. Progress in Neurobiology. 2013;106-107:17–32. doi: 10.1016/j.pneurobio.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Negida A., Menshawy A., el Ashal G., et al. Coenzyme Q10 for patients with Parkinson's disease: a systematic review and meta-analysis. CNS & Neurological Disorders-Drug Targets. 2016;15(1):45–53. doi: 10.2174/1871527314666150821103306. [DOI] [PubMed] [Google Scholar]
- 92.Snow B. J., Rolfe F. L., Lockhart M. M., et al. A double-blind, placebo-controlled study to assess the mitochondria-targeted antioxidant MitoQ as a disease-modifying therapy in Parkinson's disease. Movement Disorders. 2010;25(11):1670–1674. doi: 10.1002/mds.23148. [DOI] [PubMed] [Google Scholar]
- 93.Ghosh A., Chandran K., Kalivendi S. V., et al. Neuroprotection by a mitochondria-targeted drug in a Parkinson's disease model. Free Radical Biology and Medicine. 2010;49(11):1674–1684. doi: 10.1016/j.freeradbiomed.2010.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Yang L., Zhao K., Calingasan N. Y., Luo G., Szeto H. H., Beal M. F. Mitochondria targeted peptides protect against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine neurotoxicity. Antioxidants & Redox Signaling. 2009;11(9):2095–2104. doi: 10.1089/ars.2009.2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Mursaleen L., Noble B., Chan S. H. Y., Somavarapu S., Zariwala M. G. N-Acetylcysteine nanocarriers protect against oxidative stress in a cellular model of Parkinson's disease. Antioxidants. 2020;9(7):p. 600. doi: 10.3390/antiox9070600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Jin H., Kanthasamy A., Ghosh A., Anantharam V., Kalyanaraman B., Kanthasamy A. G. Mitochondria-targeted antioxidants for treatment of Parkinson's disease: preclinical and clinical outcomes. Biochimica et Biophysica Acta. 2014;1842(8):1282–1294. doi: 10.1016/j.bbadis.2013.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Xi Y., Feng D., Tao K., et al. MitoQ protects dopaminergic neurons in a 6-OHDA induced PD model by enhancing Mfn2-dependent mitochondrial fusion via activation of PGC-1α. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease. 2018;1864(9):2859–2870. doi: 10.1016/j.bbadis.2018.05.018. [DOI] [PubMed] [Google Scholar]
- 98.Fernández-Gajardo R., Matamala J. M., Carrasco R., Gutiérrez R., Melo R., Rodrigo R. Novel therapeutic strategies for traumatic brain injury: acute antioxidant reinforcement. CNS Drugs. 2014;28(3):229–248. doi: 10.1007/s40263-013-0138-y. [DOI] [PubMed] [Google Scholar]
- 99.Calabrese V., Lodi R., Tonon C., et al. Oxidative stress, mitochondrial dysfunction and cellular stress response in Friedreich's ataxia. Journal of Neurological Sciences. 2005;233(1-2):145–162. doi: 10.1016/j.jns.2005.03.012. [DOI] [PubMed] [Google Scholar]
- 100.Pointer C. B., Klegeris A. Cardiolipin in central nervous system physiology and pathology. Cellular and Molecular Neurobiology. 2017;37(7):1161–1172. doi: 10.1007/s10571-016-0458-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Shen Q., Hiebert J. B., Hartwell J., Thimmesch A. R., Pierce J. D. Systematic review of traumatic brain injury and the impact of antioxidant therapy on clinical outcomes. Worldviews on Evidence-Based Nursing. 2016;13(5):380–389. doi: 10.1111/wvn.12167. [DOI] [PubMed] [Google Scholar]
- 102.Singh A., Kukreti R., Saso L., Kukreti S. Oxidative stress: a key modulator in neurodegenerative diseases. Molecules. 2019;24(8):p. 1583. doi: 10.3390/molecules24081583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hoffer M. E., Balaban C., Slade M. D., Tsao J. W., Hoffer B. Amelioration of acute sequelae of blast induced mild traumatic brain injury by N-acetyl cysteine: a double-blind, placebo controlled study. PLoS One. 2013;8(1, article e54163) doi: 10.1371/journal.pone.0054163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Clark R. S. B., Empey P. E., Bayır H., et al. Phase I randomized clinical trial of N-acetylcysteine in combination with an adjuvant probenecid for treatment of severe traumatic brain injury in children. PLoS One. 2017;12(7, article e0180280) doi: 10.1371/journal.pone.0180280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Hagos F. T., Empey P. E., Wang P., et al. Exploratory application of neuropharmacometabolomics in severe childhood traumatic brain injury. Critical Care Medicine. 2018;46(9):1471–1479. doi: 10.1097/CCM.0000000000003203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Isaev N. K., Novikova S. V., Stelmashook E. V., et al. Mitochondria-targeted plastoquinone antioxidant SkQR1 decreases trauma-induced neurological deficit in rat. Biochemistry (Mosc) 2012;77(9):996–999. doi: 10.1134/S0006297912090052. [DOI] [PubMed] [Google Scholar]
- 107.Genrikhs E. E., Stelmashook E. V., Alexandrova O. P., et al. The single intravenous administration of mitochondria-targeted antioxidant SkQR1 after traumatic brain injury attenuates neurological deficit in rats. Brain Research Bulletin. 2019;148:100–108. doi: 10.1016/j.brainresbull.2019.03.011. [DOI] [PubMed] [Google Scholar]
- 108.Ji J., Kline A. E., Amoscato A., et al. Lipidomics identifies cardiolipin oxidation as a mitochondrial target for redox therapy of brain injury. Nature Neuroscience. 2012;15(10):1407–1413. doi: 10.1038/nn.3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Szarka N., Pabbidi M. R., Amrein K., et al. Traumatic brain injury impairs myogenic constriction of cerebral arteries: role of mitochondria-derived H2O2and TRPV4-dependent activation of BKcaChannels. Journal of Neurotrauma. 2018;35(7):930–939. doi: 10.1089/neu.2017.5056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Zhou J., Wang H., Shen R., et al. Mitochondrial-targeted antioxidant MitoQ provides neuroprotection and reduces neuronal apoptosis in experimental traumatic brain injury possibly via the Nrf2-ARE pathway. American Journal of Translational Research. 2018;10(6):1887–1899. [PMC free article] [PubMed] [Google Scholar]
- 111.Sam F., Kerstetter D. L., Pimental D. R., et al. Increased reactive oxygen species production and functional alterations in antioxidant enzymes in human failing myocardium. Journal of Cardiac Failure. 2005;11(6):473–480. doi: 10.1016/j.cardfail.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 112.Montaigne D., Marechal X., Coisne A., et al. Myocardial contractile dysfunction is associated with impaired mitochondrial function and dynamics in type 2 diabetic but not in obese patients. Circulation. 2014;130(7):554–564. doi: 10.1161/CIRCULATIONAHA.113.008476. [DOI] [PubMed] [Google Scholar]
- 113.Dai D. F., Chen T., Szeto H., et al. Mitochondrial targeted antioxidant peptide ameliorates hypertensive cardiomyopathy. Journal of the American College of Cardiology. 2011;58(1):73–82. doi: 10.1016/j.jacc.2010.12.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Canton M., Menazza S., Sheeran F. L., Polverino de Laureto P., di Lisa F., Pepe S. Oxidation of myofibrillar proteins in human heart failure. Journal of the American College of Cardiology. 2011;57(3):300–309. doi: 10.1016/j.jacc.2010.06.058. [DOI] [PubMed] [Google Scholar]
- 115.Kono Y., Nakamura K., Kimura H., et al. Elevated levels of oxidative DNA damage in serum and myocardium of patients with heart failure. Circulation Journal. 2006;70(8):1001–1005. doi: 10.1253/circj.70.1001. [DOI] [PubMed] [Google Scholar]
- 116.Mallat Z., Philip I., Lebret M., Chatel D., Maclouf J., Tedgui A. Elevated levels of 8-iso-prostaglandin F2αin pericardial fluid of patients with heart failure. Circulation. 1998;97(16):1536–1539. doi: 10.1161/01.CIR.97.16.1536. [DOI] [PubMed] [Google Scholar]
- 117.Vendrov A. E., Stevenson M. D., Alahari S., et al. Attenuated superoxide dismutase 2 activity induces atherosclerotic plaque instability during aging in hyperlipidemic mice. Journal of the American Heart Association. 2017;6(11) doi: 10.1161/JAHA.117.006775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Conrad M., Jakupoglu C., Moreno S. G., et al. Essential role for mitochondrial thioredoxin reductase in hematopoiesis, heart development, and heart function. Molecular and Cell Biology. 2004;24(21):9414–9423. doi: 10.1128/MCB.24.21.9414-9423.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Ooi B. K., Goh B. H., Yap W. H. Oxidative stress in cardiovascular diseases: involvement of Nrf2 antioxidant redox signaling in macrophage foam cells formation. International Journal of Molecular Sciences. 2017;18(11):p. 2336. doi: 10.3390/ijms18112336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Zozina V. I., Covantev S., Goroshko O. A., Krasnykh L. M., Kukes V. G. Coenzyme Q10 in cardiovascular and metabolic diseases: current state of the problem. Current Cardiology Reviews. 2018;14(3):164–174. doi: 10.2174/1573403X14666180416115428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Jiang Q., Zhang H., Yang R., et al. Red-osier dogwood extracts prevent inflammatory responses in Caco-2 cells and a Caco-2 BBe1/EA.hy926 cell co-culture model. Antioxidants. 2019;8(10):p. 428. doi: 10.3390/antiox8100428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Ashor A. W., Brown R., Keenan P. D., Willis N. D., Siervo M., Mathers J. C. Limited evidence for a beneficial effect of vitamin C supplementation on biomarkers of cardiovascular diseases: an umbrella review of systematic reviews and meta-analyses. Nutrition Research. 2019;61:1–12. doi: 10.1016/j.nutres.2018.08.005. [DOI] [PubMed] [Google Scholar]
- 123.Sozen E., Demirel T., Ozer N. K. Vitamin E: regulatory role in the cardiovascular system. IUBMB Life. 2019;71(4):507–515. doi: 10.1002/iub.2020. [DOI] [PubMed] [Google Scholar]
- 124.Chakrabarti A. K., Feeney K., Abueg C., et al. Rationale and design of the EMBRACE STEMI study: a phase 2a, randomized, double-blind, placebo-controlled trial to evaluate the safety, tolerability and efficacy of intravenous Bendavia on reperfusion injury in patients treated with standard therapy including primary percutaneous coronary intervention and stenting for ST-segment elevation myocardial infarction. American Heart Journal. 2013;165(4):509–514.e7. doi: 10.1016/j.ahj.2012.12.008. [DOI] [PubMed] [Google Scholar]
- 125.Ma Y., Huang Z. Y., Zhou Z. L., et al. A novel antioxidant Mito-Tempol inhibits ox-LDL-induced foam cell formation through restoration of autophagy flux. Free Radical Biology and Medicine. 2018;129:463–472. doi: 10.1016/j.freeradbiomed.2018.10.412. [DOI] [PubMed] [Google Scholar]
- 126.Methner C., Chouchani E. T., Buonincontri G., et al. Mitochondria selective S-nitrosation by mitochondria-targeted S-nitrosothiol protects against post-infarct heart failure in mouse hearts. European Journal of Heart Failure. 2014;16(7):712–717. doi: 10.1002/ejhf.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Manskikh V. N., Gancharova O. S., Nikiforova A. I., et al. Age-associated murine cardiac lesions are attenuated by the mitochondria-targeted antioxidant SkQ1. Histology and Histopathology. 2015;30(3):353–360. doi: 10.14670/HH-30.353. [DOI] [PubMed] [Google Scholar]
- 128.Ribeiro Junior R. F., Dabkowski E. R., Shekar K. C., O´Connell K. A., Hecker P. A., Murphy M. P. MitoQ improves mitochondrial dysfunction in heart failure induced by pressure overload. Free Radical Biology and Medicine. 2018;117:18–29. doi: 10.1016/j.freeradbiomed.2018.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Goh K. Y., He L., Song J., et al. Mitoquinone ameliorates pressure overload-induced cardiac fibrosis and left ventricular dysfunction in mice. Redox Biology. 2019;21, article 101100 doi: 10.1016/j.redox.2019.101100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Aljunaidy M. M., Morton J. S., Kirschenman R., et al. Maternal treatment with a placental-targeted antioxidant (MitoQ) impacts offspring cardiovascular function in a rat model of prenatal hypoxia. Pharmaceutical Research. 2018;134:332–342. doi: 10.1016/j.phrs.2018.05.006. [DOI] [PubMed] [Google Scholar]
- 131.Gioscia-Ryan R. A., Battson M. L., Cuevas L. M., Eng J. S., Murphy M. P., Seals D. R. Mitochondria-targeted antioxidant therapy with MitoQ ameliorates aortic stiffening in old mice. Journal of applied physiology : respiratory, environmental and exercise physiology. 2018;124(5):1194–1202. doi: 10.1152/japplphysiol.00670.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Dhanasekaran A., Kotamraju S., Kalivendi S. V., et al. Supplementation of endothelial cells with mitochondria-targeted antioxidants inhibit peroxide-induced mitochondrial iron uptake, oxidative damage, and apoptosis. The Journal of Biological Chemistry. 2004;279(36):37575–37587. doi: 10.1074/jbc.M404003200. [DOI] [PubMed] [Google Scholar]
- 133.Cho J., Won K., Wu D., et al. Potent mitochondria-targeted peptides reduce myocardial infarction in rats. Coronary Artery Disease. 2007;18(3):215–220. doi: 10.1097/01.mca.0000236285.71683.b6. [DOI] [PubMed] [Google Scholar]
- 134.Kalyanaraman B., Cheng G., Hardy M., Ouari O., Bennett B., Zielonka J. Teaching the basics of reactive oxygen species and their relevance to cancer biology: mitochondrial reactive oxygen species detection, redox signaling, and targeted therapies. Redox Biology. 2018;15:347–362. doi: 10.1016/j.redox.2017.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Yang Y. H., Karakhanova S., Hartwig W., et al. Mitochondria and mitochondrial ROS in cancer: novel targets for anticancer therapy. Journal of Cellular Physiology. 2016;231(12):2570–2581. doi: 10.1002/jcp.25349. [DOI] [PubMed] [Google Scholar]
- 136.Weinberg F., Hamanaka R., Wheaton W. W., et al. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(19):8788–8793. doi: 10.1073/pnas.1003428107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Morgan M. J., Liu Z. G. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Research. 2011;21(1):103–115. doi: 10.1038/cr.2010.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Fulda S., Galluzzi L., Kroemer G. Targeting mitochondria for cancer therapy. Nature Reviews Drug Discovery. 2010;9(6):447–464. doi: 10.1038/nrd3137. [DOI] [PubMed] [Google Scholar]
- 139.Zhang E. L., Zhang C., Su Y. P., Cheng T., Shi C. Newly developed strategies for multifunctional mitochondria-targeted agents in cancer therapy. Drug Discovery Today. 2011;16(3-4):140–146. doi: 10.1016/j.drudis.2010.12.006. [DOI] [PubMed] [Google Scholar]
- 140.Cao X. H., Fang L. Y., Gibbs S., et al. Glucose uptake inhibitor sensitizes cancer cells to daunorubicin and overcomes drug resistance in hypoxia. Cancer Chemotherapy and Pharmacology. 2007;59(4):495–505. doi: 10.1007/s00280-006-0291-9. [DOI] [PubMed] [Google Scholar]
- 141.Titova E., Shagieva G., Ivanova O., et al. Mitochondria-targeted antioxidant SkQ1 suppresses fibrosarcoma and rhabdomyosarcoma tumour cell growth. Cell Cycle. 2018;17(14):1797–1811. doi: 10.1080/15384101.2018.1496748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Agapova L. S., Chernyak B. V., Domnina L. V., et al. Mitochondria-targeted plastoquinone derivatives as tools to interrupt execution of the aging program. 3. Inhibitory effect of SkQ1 on tumor development from p53-deficient cells. Biochemistry-Moscow. 2008;73(12):1300–1316. doi: 10.1134/S0006297908120031. [DOI] [PubMed] [Google Scholar]
- 143.Fetisova E. K., Muntyan M. S., Lyamzaev K. G., Chernyak B. V. Therapeutic effect of the mitochondria-targeted antioxidant SkQ1 on the culture model of multiple sclerosis. Oxidative Medicine and Cellular Longevity. 2019;2019:10. doi: 10.1155/2019/2082561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Anikin I. V., Popovich I. G., Tyndyk M. L., et al. Effect of plastoquinone derivative SkQ1 on benzo(a)pyrene-induced soft tissue carcinogenesis. Voprosy Onkologii. 2013;59(1):89–93. [PubMed] [Google Scholar]
- 145.Zhan W., Liao X., Li L. H., et al. In vitro mitochondrial-targeted antioxidant peptide induces apoptosis in cancer cells. Oncotargets and Therapy. 2019;Volume 12:7297–7306. doi: 10.2147/OTT.S207640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Shetty S., Kumar R., Bharati S. Mito-TEMPO, a mitochondria-targeted antioxidant, prevents N-nitrosodiethylamine-induced hepatocarcinogenesis in mice. Free Radical Biology and Medicine. 2019;136:76–86. doi: 10.1016/j.freeradbiomed.2019.03.037. [DOI] [PubMed] [Google Scholar]
- 147.Vazquez F., Lim J. H., Chim H., et al. PGC1α expression defines a subset of human melanoma tumors with increased mitochondrial capacity and resistance to oxidative stress. Cancer Cell. 2013;23(3):287–301. doi: 10.1016/j.ccr.2012.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Wang B. B., Fu J., Yu T., et al. Contradictory effects of mitochondria- and non-mitochondria-targeted antioxidants on hepatocarcinogenesis by altering DNA repair in mice. Hepatology. 2017;67(2):623–635. doi: 10.1002/hep.29518. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
No data were used to support this review article.