

Abstract
Methamphetamine is a derivative of amphetamines, a highly addictive central stimulant with multiple systemic toxicity including the brain, heart, liver, lung, and spleen. It has adverse effects such as apoptosis and breakdown of the blood-brain barrier. Methamphetamine is a fatal and toxic chemical substance, and its lethal mechanism has been widely studied in recent years. The possible mechanism is that methamphetamine can cause cardiotoxicity and neurotoxicity mainly by inducing oxidative stress so as to generate heat, eliminate people's hunger and thirst, and maintain a state of excitement so that people can continue to exercise. According to many research, there is no doubt that methamphetamine triggers neurotoxicity by inducing reactive oxygen species (ROS) production and redox imbalance. This review summarized the mechanisms of methamphetamine-induced neurotoxicity including apoptosis and blood-brain barrier breakdown through oxidative stress and analyzed several possible antioxidative mechanisms of tert-butylhydroquinone (TBHQ) which is a kind of food additive with antioxidative effects. As a nuclear factor E2-related factor 2 (Nrf2) agonist, TBHQ may inhibit neurotoxicity caused by oxidative stress through the following three mechanisms: the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase system, the astrocytes activation, and the glutathione pathway. The mechanism about methamphetamine's toxic effects and its antioxidative therapeutic drugs would become a research hotspot in this field and has very important research significance.
1. Introduction
According to data from 2018, the global seizures of methamphetamine reached 228 tons and increased by 23% compared with the previous years. Methamphetamine abuse has been in the first place among amphetamine-type stimulants. During the period 2014-2018, global seizures of methamphetamine accounted for 71% of the total amphetamine-type stimulants, followed by amphetamine with 21% [1]. Therefore, related studies on the prevention and treatment of methamphetamine addiction have become increasingly important. Brain hyperthermia is an important death cause of methamphetamine. Toxic doses of methamphetamine can cause hyperthermia, and high core temperature can cause a large amount of dopamine loss in the brain. Experiments showed that hyperthermia was one of the causes of methamphetamine neurotoxicity [2–4]. Methamphetamine induces dose-dependent brain hyperthermia. Compared with the core of the body, the brain warms up faster and to a higher degree [5, 6]. At an ambient temperature of 23°C, 9 mg/kg methamphetamine made brain temperature (nucleus accumbens) and muscle temperature (temporal muscle) increased to 38.92 ± 0.34°C and 37.92 ± 0.32°C, respectively [7]. Brain hyperthermia may play a role in destroying neural cells, causing brain edema, increasing the permeability of the blood-brain barrier, and eventually lead to apoptosis and the blood-brain barrier breakdown [8]. Fish were exposed to a suitable temperature (25 ± 1°C) and a high temperature (32 ± 1°C). The study found that under high temperature, the concentration of antisuperoxide anion free radicals and the activity of superoxide dismutase (SOD) were significantly reduced, proving that hyperthermia caused oxidative stress [9]. Another study found that the ROS and reactive nitrogen (RNS) levels in oysters exposed to a high temperature (32°C) were significantly increased, and the incidence of apoptosis was significantly increased [10]. The concrete results were that the contents of protein carbonyl (an indicator of ROS) were increased 11.7 times, the intensity of nitrotyrosine protein (an indicator of RNS) was increased 3.5 times, and the activity of the apoptosis indicator caspase-3/7 was increased 5 times [10]. A scholar randomly selected 96 methamphetamine abusers to measure the plasma iron-reducing ability and the serum malondialdehyde (MDA) content to assess the total antioxidant capacity and lipid peroxidation status in the body [11]. The statistical analysis showed that the total antioxidant capacity in the methamphetamine group (0.31 ± 0.04 μmol/L) was significantly lower than the control group (0.46 ± 0.05 μmol/L), and the level of MDA in the methamphetamine group (4.38 ± 5.05 μmol/L) was significantly higher than the control group (1.72 ± 2.04 μmol/L) [11]. The scholar's research showed that abuse of methamphetamine enhanced the level of oxidative stress and lipid peroxidation in the body. The neurotoxicity of methamphetamine is mainly produced by inducing oxidative stress, so antioxidation is of great significance in the prevention and treatment of methamphetamine toxicity. This review briefly explains the neurotoxicity of methamphetamine from apoptosis and blood-brain barrier breakdown, and summarizes the possible antioxidative mechanism of TBHQ from three perspectives. At present, there are only a few related literatures about the effects of TBHQ on methamphetamine, and this review is expected to open up new ideas for the prevention and treatment of methamphetamine neurotoxicity.
2. Methamphetamine and Neurotoxicity
Methamphetamine can induce hyperthermia, thereby increasing the level of oxidative stress at the dopamine terminal and ultimately causing neurotoxicity [12–14]. The hyperthermic reaction induced by methamphetamine can affect the oxidation process of dopamine from the following two aspects: (1) In the autooxidation process of dopamine, free iron and other transition metals play an inducing role [15]. Methamphetamine can promote the release of free iron by inducing a hyperthermic reaction [16] and ultimately increase the autooxidation level of dopamine. (2) During the enzymatic oxidative degradation of dopamine, hyperthermia may cause enzyme activation [12], thereby accelerating the enzyme reaction process and accelerating the degradation of dopamine. The oxidation products of dopamine include dopamine quinone and free radicals [12]. The production of free radicals is a key factor in inducing methamphetamine neurotoxicity. A large amount of ROS causes oxidative stress damage to various cells in the central nervous system so as to trigger neurotoxicity [17–19]. Excessive nitric oxide (NO) causes neurotoxicity by affecting the normal energy supply of mitochondria [20]. The formation of NO and superoxide (O2−) and the peroxynitrite (ONOO−) synthesized by both trigger long-term neurotoxicity by affecting the outflow of glutamate, increasing the concentration of extracellular glutamate, and excessively stimulating glutamate receptors [21]. The neurotoxicity induced by methamphetamine can be reflected in two aspects: apoptosis and blood-brain barrier breakdown.
2.1. Methamphetamine and Apoptosis
Methamphetamine causes damage to mitochondria in the brain, thereby leading to apoptosis and a series of other neurotoxicities. Oxidative stress is an important link between mitochondrial damage and methamphetamine-induced neurotoxicity. Studies showed that methamphetamine interfered with mitochondrial energy metabolism by inhibiting the Krebs cycle and electron transport chain to ultimately cause neurotoxicity [22–24]. After methamphetamine administration, the activity of electron transport chain complex IV (cytochrome oxidase) decreased [25]. Other studies showed that a large amount of methamphetamine in a short time significantly reduced the activity of electron transport chain complex II (succinate dehydrogenase) [26–28]. The mechanism of inhibiting the electron transport chain complex is mainly due to the large amount of ROS and RNS produced by intensive oxidative stress after methamphetamine exposure. ROS and RNS directly act on the electron transport chain complex, reducing their activity. At the same time, the inhibition on the electron transport chain complex increases the leakage of electrons and produces O2− to form a positive feedback loop [29]. The damaged mitochondria are subsequently degraded by autophagy [30]. Many studies showed that methamphetamine-induced mitochondrial damage enhanced susceptibility to proapoptosis [22, 31–33]. Methamphetamine exposure causes intensive oxidative stress, and the produced ROS and RNS can block the electron transport chain of mitochondria so that the energy metabolism of mitochondria is interfered to cause the damage of mitochondria. Mitochondria damage induces neurotoxicity such as apoptosis. Therefore, oxidative stress and ROS are important factors for methamphetamine-induced apoptosis. In addition, PUMA, PKCδ, miRNA, and lncRNA also seem to be involved in the process of methamphetamine-induced neurocyte apoptosis [29].
2.2. Methamphetamine and Blood-Brain Barrier Breakdown
The blood-brain barrier is a barrier between plasma and brain cells, and its role is to prevent certain substances from entering the brain tissue from the blood. The innermost layer of the blood-brain barrier is vascular endothelial cells. The two endothelial cells rely on tight junction proteins to connect and closely overlap each other. There are some pericytes outside the endothelial cells. The outer side of the endothelial cells and pericytes is covered by a basement membrane. The outermost layer of the blood-brain barrier consists of astrocytic end-feet. Among these structures, closely overlapping endothelial cells are the most important structure of the blood-brain barrier. Tight junction proteins include claudin-5, ZO-1, and occludin. Methamphetamine disrupts the tight overlap of the endothelium by downregulating or redistributing these tight junction proteins, thereby increasing endothelial permeability and making the blood-brain barrier collapse. Endothelial cells are very sensitive to the redox imbalance induced by methamphetamine, and endothelial cells produce ROS through oxidative stress [34]. There are many reasons for methamphetamine to cause oxidative stress. Methamphetamine downregulates glutamate-cysteine ligase (GCL) [35]. GCL is the rate-limiting enzyme for the synthesis of the antioxidant glutathione [36]. Methamphetamine can also affect the outflow of glutamate, increasing the concentration of extracellular glutamate [21]. Glutamate is one of the raw materials of glutathione, and it combines glycine and cysteine to form glutathione [37]. The outflow of glutamate reduces its intracellular concentration, thereby reducing the content of glutathione. Methamphetamine reduces the concentration of glutathione through the above two factors, which causes redox imbalance and ROS production. Methamphetamine can also induce oxidative stress through other two pathways, activating NADPH oxidase or making astrocytes overactivated. Oxidative stress and ROS can cause intracellular protein thiol oxidation [38], which can activate nonmuscle myosin light chain kinase (nmMLCK) [39]. The activation of nmMLCK causes the phosphorylation of claudin-5 and occludin, makes them lose their functions, weakens the connection between endothelial cells, and reduces the barrier function [40]. After methamphetamine administration, the blood-brain barrier is eventually destroyed by the series of effects mentioned above.
3. TBHQ and Antioxidative Stress
TBHQ is a food antioxidant. Many research showed that TBHQ reduced the level of oxidative stress in mammals [35, 41–43]. Antioxidant response elements are mainly mediated by Nrf2 and can participate in the transcriptional regulation process of phase II detoxification enzymes and antioxidant proteins, including quinone oxidoreductase, glutathione S-transferase (GST), and GCL. [44]. Nrf2 is one of the key factors to prevent excessive oxidative stress in brain cells. TBHQ, as an Nrf2 agonist [45], mainly plays a role in inhibiting oxidative stress in the brain through the following mechanisms: On one hand, Nrf2 is responsible for activating transcription in response to oxidative stress. In the presence of a large number of stimuli, Nrf2 moves from the cytoplasm to the nucleus, and in turn combines with antioxidant response elements [46], so TBHQ can play an antioxidative stress role by activating Nrf2. On the other hand, antioxidant response elements/electrophile response elements are activated transcription by TBHQ, and the increased gene expression caused by this process can also prevent excessive oxidative stress [47]. Research data showed that TBHQ increased the levels of glutathione and GCL in astrocytes and neurons, while in astrocytes, the increase in both was greater [48]. In astrocytes, after treatment with 20 μM TBHQ, glutathione levels were increased by 50% and GCL activity increased by 150%. However, in neurons, when treated with 20 μM TBHQ, glutathione levels only increased by 20%, and GCL activity only increased by 40% [48]. This phenomenon may be due to TBHQ activating Nrf2, and the Nrf2 pathway has been shown to regulate glutathione metabolism [49]. In addition, another study showed that TBHQ moderately increased the number of astrocytes in the brain, and in the central nervous system, astrocytes played an extremely important role in antioxidative stress [50]. NADPH, also known as quinone oxidoreductase 1 (NQO1), can accelerate quinone excretion by reducing quinone to hydroquinone; however, in the absence of this enzyme, quinone is reduced to hemihydroquinone, which generates ROS through redox. NADPH oxidase system is one of the sources of ROS [34]. Nrf2 may resist oxidative stress by downregulating or inhibiting the expression of NADPH oxidase. Therefore, TBHQ plays an antioxidative role by activating Nrf2.
3.1. TBHQ against Oxidative Stress through NADPH Oxidase System
TBHQ exerts an antioxidative stress effect through the NADPH oxidase system, and the mechanism is shown in Figure 1. NADPH oxidase family includes 7 isoforms of NOX1, NOX2, NOX3, NOX4, NOX5, DUOX1, and DUOX2. NADPH oxidase is a membrane protein, mainly responsible for the transmission of electrons, transferring electrons to molecular oxygen, generating ROS and O2−[51]. All NADPH oxidase isoforms act as catalysts, transferring two electrons from NADPH to molecular oxygen through its FAD domain and two heme repair groups [51, 52]. NOX1, NOX2, NOX3, and NOX5 produce O2−, and NOX4, DUOX1, and DUOX2 release hydrogen peroxide [51, 53, 54]. NADPH oxidase can be stimulated and activated by many factors and subsequently produces a large amount of ROS, including drug factors (e.g., methamphetamine), hormones, and environmental factors (e.g., noise stimulation). NOX2 is highly expressed in the cells of the central nervous system, including cerebrovascular endothelial cells. Endothelial NOX2 has low activity before being activated (i.e., under physiological conditions), and overexpressed NOX2 after activation can cause endothelial cells to produce a large amount of ROS, resulting in the occurrence of oxidative stress. It was found that ROS derived from NOX2 caused severe oxidative damage to neurocytes and cerebrovascular endothelium [55]. NOX2 deficiency prevents brain oxidative stress, reverses the production of cerebrovascular O2−, and reverses the functional damage of endothelial cells, making it tend to normalize [56]. NOX4 also plays a very important role in the regulation of oxidative stress [57]. The increased expression of NOX4 in the brain leads to increased production of ROS, which not only causes excessive consumption of endogenous antioxidase in the brain but also reduces its activity, thereby weakening the brain's ability to scavenge ROS. Excessive ROS may also cause oxidation of proteins and fats, cause damage to DNA, and affect energy metabolism, thereby eventually inducing neuronal death and apoptosis in the brain [58]. SOD scavenges oxygen free radicals and blocks the pathological chain reaction. It is an important defending enzyme, and its activity has become a key index for measuring the scavenging ability of oxygen free radicals [58, 59]. Oxygen free radicals attack cells and produce large amounts of lipid peroxides, including MDA, and MDA can be used as an important index to indirectly reflect changes in free radical content [58, 60]. A study found that upregulating NOX4 increased oxygen free radicals, decreased the SOD expression, and increased the MDA expression [58].
Figure 1.
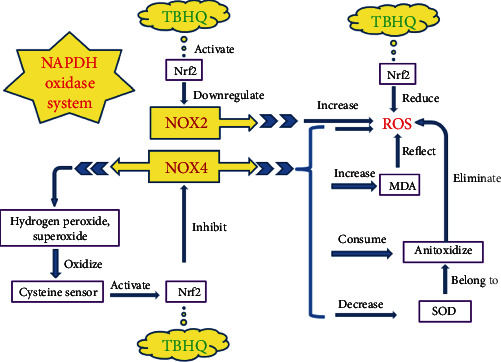
NADPH oxidase system and antioxidative stress. NADPH oxidase system contains NOX2 and NOX4. TBHQ activates Nrf2, then Nrf2 negatively regulates NOX2, and NOX2 can produce ROS, and Nrf2 can also directly reduce ROS. Under homeostasis, NOX4 produces hydrogen peroxide and superoxide which both oxidize cysteine sensor and cysteine sensor activate Nrf2; then, Nrf2 inhibits the transcription of NOX4, making NOX4 under homeostasis, forming a positive feedback loop. However, with upregulation of NOX4, the homeostasis and feedback loop mentioned above are destroyed, creating a prooxidative environment; then, ROS and MDA will increase, and MDA can indirectly reflect ROS. In addition, NOX4 consumes and inhibits antioxidases including SOD. NADPH: nicotinamide adenine dinucleotide phosphate; TBHQ: tert-butyl hydroquinone; Nrf2: nuclear factor E2 related factor 2; ROS: reactive oxygen species; SOD: superoxide dismutase; MDA: malondialdehyde; NOX: NADPH oxidase.
NADPH oxidase is present in cells capable of producing ROS (including neurons, glial cells, macrophages) in the brain and is considered to be an important factor for the generation of ROS and the maintenance of ROS homeostasis [61]. The two isoforms NOX2 and NOX4 of the NADPH oxidase system are involved in the process of brain oxidative damage. The knockout of Nrf2 upregulates ROS of mitochondrial origin, and this process involves oxidative phosphorylation. Nrf2 can also prevent the oxidation of mitochondrial fatty acids, and at the same time regulate the availability of substrates, thereby affecting the cellular bioenergetics [62, 63]. This process involves oxidative phosphorylation and ROS production. Nrf2 not only interferes with the production of ROS in the mitochondria but also affects the process of ROS production by NADPH oxidase. Experiments showed that the specific regulation mechanism of Nrf2 was related to NOX2 and NOX4 of the NADPH oxidase system [64–66]. The mechanism of Nrf2 acting on NADPH oxidase is complex, and it may be through direct and indirect regulation through promoter binding and chromatin remodeling [64]. The expression of NOX2 was upregulated in primary brain hippocampal glio-neuronal cultures of Nrf2-KO mice, indicating that Nrf2 negatively regulated NOX2 [64]. A positive feedback loop is formed between Nrf2 and homeostatic NOX4. Activated Nrf2 inhibits the NOX4 transcription [65], and then, homeostatic NOX4 produces hydrogen peroxide and O2−, which subsequently oxidize the cysteine sensor, and the cysteine sensor can activate Nrf2 [66] to form a positive feedback loop. However, in the absence of Nrf2, NOX4 homeostasis is disrupted, and there is no such feedback loop. Overexpressed NOX4 consumes antioxidase and increases ROS production [64]. Therefore, TBHQ, as an Nrf2 agonist, plays a role in reducing ROS and resisting oxidative stress.
3.2. TBHQ against Oxidative Stress by Regulating Astrocytes
TBHQ plays an antioxidative stress role by regulating astrocytes, and the mechanism is shown in Figure 2. Astrocytes are an important part of the blood-brain barrier. They secrete a variety of neuroactive molecules and respond to a variety of immune regulatory signals to counteract methamphetamine-induced oxidative stress [67]. The outermost layer of the blood-brain barrier is covered by astrocytic end-feet. A study found that in the blood-brain barrier coculture model, the removal of astrocytes destroyed the tight junctions of the blood-brain barrier and led to increased permeability [68]. Astrocytes express glutamatergic, GABAergic, adrenergic, purinergic, serotonergic, muscarinic, and peptidergic receptors [67, 69]. Activated astrocytes release glutamate, prostaglandins, ATP and NO, and other neuroactive molecules [67, 70]. Changes in astrocyte activity directly affect the central nervous system [71], and the regulatory mechanism of astrocytes is complex. Abnormal activity or function of astrocytes can promote nervous system damage. Sigma-1 receptor is closely related to methamphetamine-induced neurotoxicity. Recent studies have found that Sigma-1 receptor antagonists can reduce methamphetamine-induced oxidative stress, cerebral hyperthermia, and behavioral abnormalities [67, 72, 73]. Glial fibrillary acidic protein (GFAP) is an index of astrocyte activation. After knocking out the Sigma-1 receptor, it was found that GFAP expression was abrogated [74]. Both Sigma-1 receptor antagonists BD1047 and SN79 can block the overactivation of astrocytes and attenuate the expression of proinflammatory cytokines after methamphetamine exposure [74–76]. Methamphetamine can regulate the Sigma-1 receptor in astrocytes so as to change astrocyte activity [76]. A study showed that methamphetamine simultaneously increased the oxidative burden and antioxidative capacity of astrocytes. This effect may be exerted through abnormal regulation of mitochondria. Under physiological conditions, astrocytes protect the central nervous system from damage by maintaining redox homeostasis and a delicate balance. Stimulation of methamphetamine can lead to dichotomous dysregulation of redox. Attenuating methamphetamine-mediated secondary messenger signaling downstream of the Sigma-1 receptor can target dysregulation of mitochondrial regulatory proteins in astrocytes [77]. Heme oxygenase-1 (HO-1) is a phase II antioxidative enzyme, and its expression level can be regulated by TBHQ and Nrf2. Another study showed that inducing the activation of Nrf2 and the expression of HO-1 in astrocytes downregulated the expression of proinflammatory cytokines and upregulated the antioxidative mechanism [78]. Phosphatidylinositol 3-kinase (PI3K) is also involved in resisting methamphetamine-induced oxidative stress. PI3K works by activating protein kinase B (also known as AKT) [79]. PI3K/AKT has been reported as the upstream signaling pathway of Nrf2 in many papers [79–81].
Figure 2.
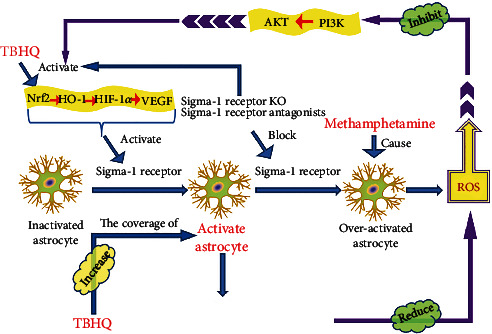
Double-edged sword effects of astrocyte activation. TBHQ activates Nrf2 and then regulates its downstream HO-1, HIF-1α, and VEGF. This process activates the Sigma-1 receptor, so that astrocytes become activated from inactivated, and activated astrocytes can play a role in resisting methamphetamine-induced oxidative stress. TBHQ can also increase activated astrocytes and their coverage to reduce ROS. ROS inhibits the PI3K/AKT signaling pathway. TBHQ, as an Nrf2 agonist, can also indirectly activate Nrf2 by positively regulating the PI3K/AKT signaling pathway. Sigma-1 receptor KO or Sigma-1 receptor antagonists can activate Nrf2 as well as block excessive Sigma-1 receptors, thereby preventing the overactivation of astrocytes. However, methamphetamine can induce overactivation of astrocytes. TBHQ: tert-butyl hydroquinone; Nrf2: nuclear factor E2 related factor 2; HO-1: heme oxygenase-1; HIF-1α: hypoxia-inducible factor-1α; VEGF: vascular endothelial growth factor; KO: knockout; ROS: reactive oxygen species; PI3K: phosphatidylinositol 3-kinase; AKT: protein kinase B.
A recent experiment showed that in astrocytes, the Sigma-1 receptor was activated by the Nrf2/HO-1 signaling pathway, thereby making astrocytes from inactivation to activation and then reducing the production of ROS [82]. The activation of Nrf2 and HO-1 partially mediates the activation of Sigma-1 receptor and its anti-inflammatory and antioxidative effects [82]. The Sigma-1 receptor is equivalent to a molecular chaperone and participates in various mental diseases by interacting with multiple protein or lipid molecules. Knockout or loss of Sigma-1 receptors may activate the Nrf2/HO-1 signaling pathway. Astrocytes and neurons contain a large number of Sigma-1 receptors, and their silencing will induce a decrease in mitochondrial membrane potential and aberrant formation of mitochondrial aggregates [83–85]. Astrocyte activation and overactivation show double-edged sword effects in multiple mental diseases [86]. After methamphetamine administration, Sigma-1 receptor activates astrocytes through a self-activation mechanism [76]. Methamphetamine exposure makes astrocytes overactivated. Sigma-1 receptor exerts neuroprotective effects by maintaining homeostasis of astrocytes and neurons and maintaining the proper and balanced degree of activation of astrocytes. Knock-out of Sigma-1 receptor leads to the imbalance of astrocyte populations and enhancement of the Nrf2 signaling pathway, which can attenuate excessive oxidative stress, promote neuronal survival, and reduce methamphetamine-induced neurotoxicity [83].
PI3K has both Ser/Thr kinase activity and phosphatidylinositol kinase activity [87]. The Ser/Thr kinase activity of PI3K can activate its downstream target AKT [79]. PI3K/AKT signaling pathway can phosphorylate Ser/Thr residues, which is a key for Nrf2 activation [79, 81]. Under hyperoxia conditions, PI3K is inhibited. In the presence of a large amount of ROS, Nrf2 and its downstream are inhibited through the PI3K/AKT signaling pathway [79]. Methamphetamine leads to the overactivation of astrocytes and generates ROS, thereby inhibiting the expression of Nrf2. This process can be reversed by TBHQ. TBHQ reverses methamphetamine-induced oxidative stress damage by directly activating the Nrf2/HO-1 signaling pathway and indirectly regulating Nrf2 through the PI3K-AKT signaling pathway [79]. Hypoxia-inducible factor-1α (HIF-1α) and vascular endothelial growth factor (VEGF) are downstream of the Nrf2/HO-1 signaling pathway. An experimental result showed that TBHQ activated astrocytes and increased astrocytic end-feet coverage by activating the Nrf2/HO-1/VEGF pathway [88]. The antioxidative stress and neuroprotective effect of astrocytes are closely related to the degree of activation. Both inactivated and overactivated astrocytes can induce dysfunction. TBHQ increases the nuclear accumulation of Nrf2 and at the same time enhances the expression of antioxidative genes downstream of Nrf2 (including HO-1 and NQO1), moderately activates astrocytes, and reduces the production of inflammatory cytokines, thereby reducing apoptosis and neuronal death [89]. Under physiological conditions, Nrf2 binds to Keap1 to form the Nrf2 Keap1 complex [90]. Under oxidative stress, Nrf2 is released from Keap1's antioxidant response element; in this way, Nrf2 is activated. Meanwhile, Nrf2 achieves its accumulation in the nucleus by transferring from the cytoplasm to the nucleus, thereby playing a series of neuroprotective effects such as antioxidative stress [90, 91]. HO-1 is induced after Nrf2 activation and is a target gene for antioxidative stress [92, 93]. The Nrf2/HO-1 signaling pathway is closely related to oxidative stress in the brain [94–96] and some antioxidants work by upregulating Nrf2 and HO-1 [97–99]. The HIF-1α/VEGF pathway may be related to oxidative stress in astrocytes. The Nrf2/HO-1 signaling pathway may inhibit oxidative stress in astrocytes by regulating HIF-1α and VEGF [90]. HO-1 is a phase II antioxidative enzyme, and its expression level can be regulated by TBHQ. TBHQ promotes the translocation of Nrf2 to the nucleus, and Nrf2 participates in the regulation of HO-1 expression. PI3K/AKT signaling pathway is the upstream of Nrf2, and the protective effect of TBHQ on methamphetamine-induced neurotoxicity is also closely related to the activation of PI3K/AKT [79]. TBHQ regulates Sigma-1 receptors through the Nrf2/HO-1 and PI3K/AKT signaling pathways, thereby ultimately regulating the activation state of astrocytes and playing a role in resisting oxidative stress. In astrocytes, TBHQ increases the mRNA and protein levels of HO-1 by inducing a coordinated interaction between Nrf2 and c-Jun [100].
3.3. TBHQ against Oxidative Stress via Glutathione Pathway
TBHQ can also inhibit the oxidative stress of cerebrovascular endothelial cells and neurocytes via the glutathione pathway, and the mechanism is shown in Figure 3. Glutathione is an important antioxidant in the body. It is a ROS scavenger, especially to clear the main form of ROS -O2−[101]. Glutathione is also the electron donor of glutathione peroxidase (GSH-Px) to reduce peroxides [102]. First, Nrf2 increases the expression of GCL [103], which is the rate-limiting enzyme for glutathione synthesis [36], so Nrf2 increases glutathione synthesis. Second, Nrf2 can also regulate GSH-Px and glutathione reductase (GR), and the two enzymes coordinate the regulation of glutathione regeneration. Third, Nrf2 can also positively regulate GST (ROS detoxification enzyme). Glutathione reduces ROS through antioxidative stress to resist neurocytes apoptosis and inhibit blood-brain barrier breakdown. In addition, glutathione can also reverse the process of thiol oxidation to disulfide in the presence of ROS (reversed by the reduction of disulfide through sulfhydryl/disulfide exchange) [38], thereby blocking the activation process of nmMLCK, blocking the middle link of blood-brain barrier breakdown, thereby protecting the blood-brain barrier from being destroyed. Therefore, the regulation of glutathione-related enzymes by Nrf2 has extremely important therapeutic significance for methamphetamine-induced oxidative stress. TBHQ regulates the above process by activating Nrf2, thereby reducing methamphetamine-induced neurotoxicity.
Figure 3.
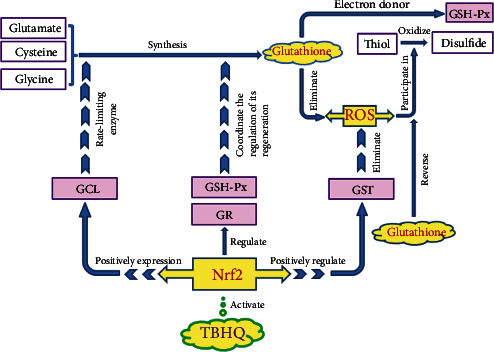
Glutathione pathway and antioxidative stress. Glutathione is synthesized by three substances glutamate, cysteine, and glycine. Glutathione can eliminate ROS and act as an electron donor for GSH-Px to reduce peroxides. TBHQ activates Nrf2, and Nrf2 positively regulates GST. GST is a ROS detoxification enzyme that eliminates ROS. At the same time, Nrf2 promotes the expression of GCL. GCL is the rate-limiting enzyme for glutathione synthesis. Nrf2 can also regulate GSH-Px and GR, which coordinate the regulation of glutathione regeneration. ROS is also involved in the oxidation of thiols to disulfides, which can be reversed by glutathione. TBHQ: tert-butyl hydroquinone; Nrf2: nuclear factor E2 related factor 2; GCL: glutamate-cysteine ligase; GSH-Px: glutathione peroxidase; GR: glutathione reductase; GST: glutathione S-transferase.
4. Concluding Remarks
Methamphetamine addiction and chronic poisoning have been widely concerned by society. The toxicity of methamphetamine mainly includes cardiotoxicity and neurotoxicity, and its toxicity is closely related to oxidative stress. There are also scholars who explored the lung toxicity of methamphetamine, and they found that its lung toxicity was also related to oxidative stress [104–106]. TBHQ is a food additive often used as an antioxidant or preservative. Many studies have confirmed that TBHQ can activate Nrf2 [43, 45, 46] and can play the role of antioxidative stress through Nrf2, but the specific mechanism is not very clear. This review synthesizes many scholars' research and systematically explains how TBHQ can resist methamphetamine-induced oxidative stress from three perspectives, including regulating the NADPH oxidase system, adjusting astrocytes activation, and regulating the glutathione pathway. Methamphetamine induces neurotoxicity including apoptosis and blood-brain barrier breakdown through oxidative stress. This review also briefly introduces the mechanisms of methamphetamine-induced neurotoxicity. Brain hyperthermia is a major cause of death from methamphetamine, and hyperthermia is one of the factors that produce excessive ROS and induce oxidative stress. All in all, the redox imbalance induced by methamphetamine is an important reason for its neurotoxicity, so the prevention and treatment of methamphetamine-induced neurotoxicity can be started from the aspect of antioxidation. As a food additive, TBHQ has almost no toxic and side effects. If the antioxidative effect of TBHQ can be used for the prevention and treatment of methamphetamine toxicity, it will provide a new and safer treatment for methamphetamine addiction. This review comprehensively and systematically introduces TBHQ, which is expected to provide new clues and develop new methods for the detoxification treatment of methamphetamine. As the number of methamphetamine users increases year by year, research and reviews in this area have increasingly important clinical significance.
Acknowledgments
This research was funded by the National Natural Science Foundation of China (No. 81973404, 81503058), Department of Education of Liaoning Province (No. JC2019034), Natural Science Foundation of Liaoning Province (No. 2014021065), and Department of Science and Technology of Liaoning Province (No. 20170520137).
Conflicts of Interest
The authors declare that there are no competing interests.
References
- 1.Nations U. World Drug Report. United Nations publication; 2020. Sales No. E.20.XI.6. [Google Scholar]
- 2.Albers D. S., Sonsalla P. K. Methamphetamine-induced hyperthermia and dopaminergic neurotoxicity in mice: pharmacological profile of protective and nonprotective agents. The Journal of Pharmacology and Experimental Therapeutics. 1995;275(3):1104–1114. [PubMed] [Google Scholar]
- 3.Kikuchi-Utsumi K., Ishizaka M., Matsumura N., et al. Involvement of the α(1D)-adrenergic receptor in methamphetamine-induced hyperthermia and neurotoxicity in rats. Neurotoxicity Research. 2013;24(2):130–138. doi: 10.1007/s12640-012-9369-9. [DOI] [PubMed] [Google Scholar]
- 4.Kiyatkin E. A., Sharma H. S. Leakage of the blood-brain barrier followed by vasogenic edema as the ultimate cause of death induced by acute methamphetamine overdose. International Review of Neurobiology. 2019;146:189–207. doi: 10.1016/bs.irn.2019.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brown P. L., Wise R. A., Kiyatkin E. A. Brain hyperthermia is induced by methamphetamine and exacerbated by social interaction. The Journal of Neuroscience. 2003;23(9):3924–3929. doi: 10.1523/JNEUROSCI.23-09-03924.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kiyatkin E. A., Sharma H. S. Expression of heat shock protein (HSP 72 kDa) during acute methamphetamine intoxication depends on brain hyperthermia: neurotoxicity or neuroprotection? Journal of Neural Transmission (Vienna) 2011;118(1):47–60. doi: 10.1007/s00702-010-0477-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kiyatkin E. A., Sharma H. S. Breakdown of blood-brain and blood-spinal cord barriers during acute methamphetamine intoxication: role of brain temperature. CNS & Neurological Disorders Drug Targets. 2016;15(9):1129–1138. doi: 10.2174/1871527315666160920112445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kiyatkin E. A., Brown P. L., Sharma H. S. Brain edema and breakdown of the blood-brain barrier during methamphetamine intoxication: critical role of brain hyperthermia. The European Journal of Neuroscience. 2007;26(5):1242–1253. doi: 10.1111/j.1460-9568.2007.05741.x. [DOI] [PubMed] [Google Scholar]
- 9.Liu B., Xu P., Brown P. B., et al. The effect of hyperthermia on liver histology, oxidative stress and disease resistance of the Wuchang bream, Megalobrama amblycephala. Fish & Shellfish Immunology. 2016;52:317–324. doi: 10.1016/j.fsi.2016.03.018. [DOI] [PubMed] [Google Scholar]
- 10.Nash S., Johnstone J., Rahman M. S. Elevated temperature attenuates ovarian functions and induces apoptosis and oxidative stress in the American oyster, Crassostrea virginica: potential mechanisms and signaling pathways. Cell Stress & Chaperones. 2019;24(5):957–967. doi: 10.1007/s12192-019-01023-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Solhi H., Malekirad A., Kazemifar A. M., Sharifi F. Oxidative stress and lipid peroxidation in prolonged users of methamphetamine. Drug Metabolism Letters. 2014;7(2):79–82. doi: 10.2174/187231280702140520191324. [DOI] [PubMed] [Google Scholar]
- 12.LaVoie M. J., Hastings T. G. Dopamine quinone formation and protein modification associated with the striatal neurotoxicity of methamphetamine: evidence against a role for extracellular dopamine. The Journal of Neuroscience. 1999;19(4):1484–1491. doi: 10.1523/JNEUROSCI.19-04-01484.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Barayuga S. M., Pang X., Andres M. A., Panee J., Bellinger F. P. Methamphetamine decreases levels of glutathione peroxidases 1 and 4 in SH-SY5Y neuronal cells: protective effects of selenium. Neurotoxicology. 2013;37:240–246. doi: 10.1016/j.neuro.2013.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Quinton M. S., Yamamoto B. K. Causes and consequences of methamphetamine and MDMA toxicity. The AAPS Journal. 2006;8(2):E337–E347. doi: 10.1208/aapsj080238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Graham D. G. Oxidative pathways for catecholamines in the genesis of neuromelanin and cytotoxic quinones. Molecular Pharmacology. 1978;14(4):633–643. [PubMed] [Google Scholar]
- 16.Skibba J. L., Gwartney E. A. Liver hyperthermia and oxidative stress: role of iron and aldehyde production. International Journal of Hyperthermia. 1997;13(2):215–226. doi: 10.3109/02656739709012384. [DOI] [PubMed] [Google Scholar]
- 17.Ferraro S. A., Domingo M. G., Etcheverrito A., Olmedo D. G., Tasat D. R. Neurotoxicity mediated by oxidative stress caused by titanium dioxide nanoparticles in human neuroblastoma (SH-SY5Y) cells. Journal of Trace Elements in Medicine and Biology. 2020;57:p. 126413. doi: 10.1016/j.jtemb.2019.126413. [DOI] [PubMed] [Google Scholar]
- 18.Dai C., Xiao X., Sun F., et al. T-2 toxin neurotoxicity: role of oxidative stress and mitochondrial dysfunction. Archives of Toxicology. 2019;93(11):3041–3056. doi: 10.1007/s00204-019-02577-5. [DOI] [PubMed] [Google Scholar]
- 19.Chandravanshi L. P., Gupta R., Shukla R. K. Developmental neurotoxicity of arsenic: involvement of oxidative stress and mitochondrial functions. Biological Trace Element Research. 2018;186(1):185–198. doi: 10.1007/s12011-018-1286-1. [DOI] [PubMed] [Google Scholar]
- 20.Nash J. F., Yamamoto B. K. Methamphetamine neurotoxicity and striatal glutamate release: comparison to 3,4-methylenedioxymethamphetamine. Brain Research. 1992;581(2):237–243. doi: 10.1016/0006-8993(92)90713-J. [DOI] [PubMed] [Google Scholar]
- 21.Bolanos J. P., Almeida A., Stewart V., et al. Nitric oxide-mediated mitochondrial damage in the brain: mechanisms and implications for neurodegenerative diseases. Journal of Neurochemistry. 1997;68(6):2227–2240. doi: 10.1046/j.1471-4159.1997.68062227.x. [DOI] [PubMed] [Google Scholar]
- 22.Shin E. J., Tran H. Q., Nguyen P. T., et al. Role of mitochondria in methamphetamine-induced dopaminergic neurotoxicity: involvement in oxidative stress, neuroinflammation, and pro-apoptosis-a review. Neurochemical Research. 2018;43(1):66–78. doi: 10.1007/s11064-017-2318-5. [DOI] [PubMed] [Google Scholar]
- 23.Barbosa D. J., Capela J. P., Feio-Azevedo R., Teixeira-Gomes A., Bastos M. L., Carvalho F. Mitochondria: key players in the neurotoxic effects of amphetamines. Archives of Toxicology. 2015;89(10):1695–1725. doi: 10.1007/s00204-015-1478-9. [DOI] [PubMed] [Google Scholar]
- 24.Feier G., Valvassori S. S., Varela R. B., et al. Lithium and valproate modulate energy metabolism in an animal model of mania induced by methamphetamine. Pharmacology, Biochemistry, and Behavior. 2013;103(3):589–596. doi: 10.1016/j.pbb.2012.09.010. [DOI] [PubMed] [Google Scholar]
- 25.Burrows K. B., Gudelsky G., Yamamoto B. K. Rapid and transient inhibition of mitochondrial function following methamphetamine or 3,4-methylenedioxymethamphetamine administration. European Journal of Pharmacology. 2000;398(1):11–18. doi: 10.1016/S0014-2999(00)00264-8. [DOI] [PubMed] [Google Scholar]
- 26.Brown J. M., Quinton M. S., Yamamoto B. K. Methamphetamine-induced inhibition of mitochondrial complex II: roles of glutamate and peroxynitrite. Journal of Neurochemistry. 2005;95(2):429–436. doi: 10.1111/j.1471-4159.2005.03379.x. [DOI] [PubMed] [Google Scholar]
- 27.Mashayekhi V., Eskandari M. R., Kobarfard F., Khajeamiri A., Hosseini M. J. Induction of mitochondrial permeability transition (MPT) pore opening and ROS formation as a mechanism for methamphetamine-induced mitochondrial toxicity. Naunyn-Schmiedeberg's Archives of Pharmacology. 2014;387(1):47–58. doi: 10.1007/s00210-013-0919-3. [DOI] [PubMed] [Google Scholar]
- 28.Feier G., Valvassori S. S., Lopes-Borges J., et al. Behavioral changes and brain energy metabolism dysfunction in rats treated with methamphetamine or dextroamphetamine. Neuroscience Letters. 2012;530(1):75–79. doi: 10.1016/j.neulet.2012.09.039. [DOI] [PubMed] [Google Scholar]
- 29.Yang X., Wang Y., Li Q., et al. The main molecular mechanisms underlying methamphetamine- induced neurotoxicity and implications for pharmacological treatment. Frontiers in Molecular Neuroscience. 2018;11:p. 186. doi: 10.3389/fnmol.2018.00186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Michel S., Wanet A., De Pauw A., Rommelaere G., Arnould T., Renard P. Crosstalk between mitochondrial (dys)function and mitochondrial abundance. Journal of Cellular Physiology. 2012;227(6):2297–2310. doi: 10.1002/jcp.23021. [DOI] [PubMed] [Google Scholar]
- 31.Park J. H., Seo Y. H., Jang J. H., Jeong C. H., Lee S., Park B. Asiatic acid attenuates methamphetamine-induced neuroinflammation and neurotoxicity through blocking of NF-kB/STAT3/ERK and mitochondria-mediated apoptosis pathway. Journal of Neuroinflammation. 2017;14(1):p. 240. doi: 10.1186/s12974-017-1009-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sharikova A. V., Quaye E., Park J. Y., et al. Methamphetamine induces apoptosis of microglia via the intrinsic mitochondrial-dependent pathway. Journal of Neuroimmune Pharmacology. 2018;13(3):396–411. doi: 10.1007/s11481-018-9787-4. [DOI] [PubMed] [Google Scholar]
- 33.Nam Y., Wie M. B., Shin E. J., et al. Ginsenoside Re protects methamphetamine-induced mitochondrial burdens and proapoptosis via genetic inhibition of protein kinase C δ in human neuroblastoma dopaminergic SH-SY5Y cell lines. Journal of Applied Toxicology. 2015;35(8):927–944. doi: 10.1002/jat.3093. [DOI] [PubMed] [Google Scholar]
- 34.Sajja R. K., Rahman S., Cucullo L. Drugs of abuse and blood-brain barrier endothelial dysfunction: a focus on the role of oxidative stress. Journal of Cerebral Blood Flow and Metabolism. 2016;36(3):539–554. doi: 10.1177/0271678x15616978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang Y., Gu Y. H., Liu M., Bai Y., Liang L. Y., Wang H. L. TBHQ alleviated endoplasmic reticulum stress-apoptosis and oxidative stress by PERK-Nrf2 crosstalk in methamphetamine-induced chronic pulmonary toxicity. Oxidative Medicine and Cellular Longevity. 2017;2017:12. doi: 10.1155/2017/4310475.4310475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lu S. C. Glutathione synthesis. Biochimica et Biophysica Acta. 2013;1830(5):3143–3153. doi: 10.1016/j.bbagen.2012.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Weinberg J. M., Davis J. A., Abarzua M., Rajan T. Cytoprotective effects of glycine and glutathione against hypoxic injury to renal tubules. The Journal of Clinical Investigation. 1987;80(5):1446–1454. doi: 10.1172/jci113224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Terrill J. R., Radley-Crabb H. G., Iwasaki T., Lemckert F. A., Arthur P. G., Grounds M. D. Oxidative stress and pathology in muscular dystrophies: focus on protein thiol oxidation and dysferlinopathies. The FEBS Journal. 2013;280(17):4149–4164. doi: 10.1111/febs.12142. [DOI] [PubMed] [Google Scholar]
- 39.Samak G., Gangwar R., Meena A. S., et al. Calcium channels and oxidative stress mediate a synergistic disruption of tight junctions by ethanol and acetaldehyde in Caco-2 cell monolayers. Scientific Reports. 2016;6(1, article 38899) doi: 10.1038/srep38899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Haorah J., Heilman D., Knipe B., et al. Ethanol-induced activation of myosin light chain kinase leads to dysfunction of tight junctions and blood-brain barrier compromise. Alcoholism, Clinical and Experimental Research. 2005;29(6):999–1009. doi: 10.1097/01.ALC.0000166944.79914.0A. [DOI] [PubMed] [Google Scholar]
- 41.Bai J., Yu X. J., Liu K. L., et al. Tert-butylhydroquinone attenuates oxidative stress and inflammation in hypothalamic paraventricular nucleus in high salt-induced hypertension. Toxicology Letters. 2017;281:1–9. doi: 10.1016/j.toxlet.2017.08.018. [DOI] [PubMed] [Google Scholar]
- 42.Gavia-García G., Rosas-Trejo M. Á., García-Mendoza E., et al. T-BHQ protects against oxidative damage and maintains the antioxidant response in malnourished rats. Dose-Response. 2018;16(3) doi: 10.1177/1559325818796304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Song H., Xu Y., Yang X., Rong X., Wang Y., Wei N. Tertiary butylhydroquinone alleviates gestational diabetes mellitus in C57BL/KsJ-Lep db/+ mice by suppression of oxidative stress. Journal of Cellular Biochemistry. 2019;120(9):15310–15319. doi: 10.1002/jcb.28798. [DOI] [PubMed] [Google Scholar]
- 44.Lee J. M., Calkins M. J., Chan K., Kan Y. W., Johnson J. A. Identification of the NF-E2-related factor-2-dependent genes conferring protection against oxidative stress in primary cortical astrocytes using oligonucleotide microarray analysis. The Journal of Biological Chemistry. 2003;278(14):12029–12038. doi: 10.1074/jbc.M211558200. [DOI] [PubMed] [Google Scholar]
- 45.Ye F., Li X., Li L., Yuan J., Chen J. t-BHQ provides protection against lead neurotoxicity via Nrf2/HO-1 pathway. Oxidative Medicine and Cellular Longevity. 2016;2016:15. doi: 10.1155/2016/2075915.2075915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang Z., Ji C., Wu L., et al. Tert-butylhydroquinone alleviates early brain injury and cognitive dysfunction after experimental subarachnoid hemorrhage: role of Keap1/Nrf2/ARE pathway. PLoS One. 2014;9(5, article e97685) doi: 10.1371/journal.pone.0097685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ahlgren-Beckendorf J. A., Reising A. M., Schander M. A., Herdler J. W., Johnson J. A. Coordinate regulation of NAD(P)H:quinone oxidoreductase and glutathione-S-transferases in primary cultures of rat neurons and glia: role of the antioxidant/electrophile responsive element. Glia. 1999;25(2):131–142. doi: 10.1002/(SICI)1098-1136(19990115)25:2<131::AID-GLIA4>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 48.Lavoie S., Chen Y., Dalton T. P., et al. Curcumin, quercetin, and tBHQ modulate glutathione levels in astrocytes and neurons: importance of the glutamate cysteine ligase modifier subunit. Journal of Neurochemistry. 2009;108(6):1410–1422. doi: 10.1111/j.1471-4159.2009.05908.x. [DOI] [PubMed] [Google Scholar]
- 49.Sun X., Erb H., Murphy T. H. Coordinate regulation of glutathione metabolism in astrocytes by Nrf2. Biochemical and Biophysical Research Communications. 2005;326(2):371–377. doi: 10.1016/j.bbrc.2004.11.031. [DOI] [PubMed] [Google Scholar]
- 50.Eftekharpour E., Holmgren A., Juurlink B. H. Thioredoxin reductase and glutathione synthesis is upregulated by t-butylhydroquinone in cortical astrocytes but not in cortical neurons. Glia. 2000;31(3):241–248. doi: 10.1002/1098-1136(200009)31:3<241::AID-GLIA50>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 51.Tarafdar A., Pula G. The role of NADPH oxidases and oxidative stress in neurodegenerative disorders. International Journal of Molecular Sciences. 2018;19(12, article 3824) doi: 10.3390/ijms19123824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Altenhöfer S., Radermacher K. A., Kleikers P. W., Wingler K., Schmidt H. H. Evolution of NADPH oxidase inhibitors: selectivity and mechanisms for target engagement. Antioxidants & Redox Signaling. 2015;23(5):406–427. doi: 10.1089/ars.2013.5814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.García-Redondo A. B., Aguado A., Briones A. M., Salaices M. NADPH oxidases and vascular remodeling in cardiovascular diseases. Pharmacological Research. 2016;114:110–120. doi: 10.1016/j.phrs.2016.10.015. [DOI] [PubMed] [Google Scholar]
- 54.Martyn K. D., Frederick L. M., von Loehneysen K., Dinauer M. C., Knaus U. G. Functional analysis of Nox4 reveals unique characteristics compared to other NADPH oxidases. Cellular Signalling. 2006;18(1):69–82. doi: 10.1016/j.cellsig.2005.03.023. [DOI] [PubMed] [Google Scholar]
- 55.Fan L. M., Geng L., Cahill-Smith S., et al. Nox2 contributes to age-related oxidative damage to neurons and the cerebral vasculature. The Journal of Clinical Investigation. 2019;129(8):3374–3386. doi: 10.1172/jci125173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kröller-Schön S., Daiber A., Steven S., et al. Crucial role for Nox2 and sleep deprivation in aircraft noise-induced vascular and cerebral oxidative stress, inflammation, and gene regulation. European Heart Journal. 2018;39(38):3528–3539. doi: 10.1093/eurheartj/ehy333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hong S., Kwon J., Kim D. W., Lee H. J., Lee D., Mar W. Mulberrofuran G protects ischemic injury-induced cell death via inhibition of NOX4-mediated ROS generation and ER stress. Phytotherapy Research. 2017;31(2):321–329. doi: 10.1002/ptr.5754. [DOI] [PubMed] [Google Scholar]
- 58.Yu F., Xue W., Dong L., Hu X., Huang D., Wang K. Tetrahydroxystilbene glucoside suppresses NAPDH oxidative stress to mitigate apoptosis and autophagy induced by cerebral ischemia/reperfusion injury in mice. Evidence-based Complementary and Alternative Medicine. 2019;2019:9. doi: 10.1155/2019/3913981.3913981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Adibhatla R. M., Hatcher J. F. Lipid oxidation and peroxidation in CNS health and disease: from molecular mechanisms to therapeutic opportunities. Antioxidants & Redox Signaling. 2010;12(1):125–169. doi: 10.1089/ars.2009.2668. [DOI] [PubMed] [Google Scholar]
- 60.Xia Z., Chen Y., Fan Q., Xue M. Oxidative stress-mediated reperfusion injury: mechanism and therapies. Oxidative Medicine and Cellular Longevity. 2014;2014:2. doi: 10.1155/2014/373081.373081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dröge W. Free radicals in the physiological control of cell function. Physiological Reviews. 2002;82(1):47–95. doi: 10.1152/physrev.00018.2001. [DOI] [PubMed] [Google Scholar]
- 62.Holmstrom K. M., Baird L., Zhang Y., et al. Nrf2 impacts cellular bioenergetics by controlling substrate availability for mitochondrial respiration. Biology Open. 2013;2(8):761–770. doi: 10.1242/bio.20134853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ludtmann M. H. R., Angelova P. R., Zhang Y., Abramov A. Y., Dinkova-Kostova A. T. Nrf2 affects the efficiency of mitochondrial fatty acid oxidation. The Biochemical Journal. 2014;457(3):415–424. doi: 10.1042/bj20130863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kovac S., Angelova P. R., Holmström K. M., Zhang Y., Dinkova-Kostova A. T., Abramov A. Y. Nrf2 regulates ROS production by mitochondria and NADPH oxidase. Biochimica et Biophysica Acta. 2015;1850(4):794–801. doi: 10.1016/j.bbagen.2014.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Goettsch C., Goettsch W., Brux M., et al. Arterial flow reduces oxidative stress via an antioxidant response element and Oct-1 binding site within the NADPH oxidase 4 promoter in endothelial cells. Basic Research in Cardiology. 2011;106(4):551–561. doi: 10.1007/s00395-011-0170-3. [DOI] [PubMed] [Google Scholar]
- 66.Brewer A. C., Murray T. V., Arno M., et al. Nox4 regulates Nrf2 and glutathione redox in cardiomyocytes in vivo. Free Radical Biology & Medicine. 2011;51(1):205–215. doi: 10.1016/j.freeradbiomed.2011.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Borgmann K., Ghorpade A. HIV-1, methamphetamine and astrocytes at neuroinflammatory crossroads. Frontiers in Microbiology. 2015;6, article 1143 doi: 10.3389/fmicb.2015.01143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hamm S., Dehouck B., Kraus J., et al. Astrocyte mediated modulation of blood-brain barrier permeability does not correlate with a loss of tight junction proteins from the cellular contacts. Cell and Tissue Research. 2004;315(2):157–166. doi: 10.1007/s00441-003-0825-y. [DOI] [PubMed] [Google Scholar]
- 69.Porter J. T., McCarthy K. D. Astrocytic neurotransmitter receptors in situ and in vivo. Progress in Neurobiology. 1997;51(4):439–455. doi: 10.1016/s0301-0082(96)00068-8. [DOI] [PubMed] [Google Scholar]
- 70.Haydon P. G. GLIA: listening and talking to the synapse. Nature Reviews. Neuroscience. 2001;2(3):185–193. doi: 10.1038/35058528. [DOI] [PubMed] [Google Scholar]
- 71.Abudara V., Roux L., Dallérac G., et al. Activated microglia impairs neuroglial interaction by opening Cx43 hemichannels in hippocampal astrocytes. Glia. 2015;63(5):795–811. doi: 10.1002/glia.22785. [DOI] [PubMed] [Google Scholar]
- 72.Kaushal N., Matsumoto R. Role of sigma receptors in methamphetamine-induced neurotoxicity. Current Neuropharmacology. 2011;9(1):54–57. doi: 10.2174/157015911795016930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Robson M. J., Seminerio M. J., McCurdy C. R., Coop A., Matsumoto R. R. σ Receptor antagonist attenuation of methamphetamine-induced neurotoxicity is correlated to body temperature modulation. Pharmacological Reports. 2013;65(2):343–349. doi: 10.1016/s1734-1140(13)71009-0. [DOI] [PubMed] [Google Scholar]
- 74.Robson M. J., Turner R. C., Naser Z. J., et al. SN79, a sigma receptor antagonist, attenuates methamphetamine-induced astrogliosis through a blockade of OSMR/gp130 signaling and STAT3 phosphorylation. Experimental Neurology. 2014;254:180–189. doi: 10.1016/j.expneurol.2014.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Robson M. J., Turner R. C., Naser Z. J., McCurdy C. R., Huber J. D., Matsumoto R. R. SN79, a sigma receptor ligand, blocks methamphetamine-induced microglial activation and cytokine upregulation. Experimental Neurology. 2013;247:134–142. doi: 10.1016/j.expneurol.2013.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhang Y., Lv X., Bai Y., et al. Involvement of sigma-1 receptor in astrocyte activation induced by methamphetamine via up-regulation of its own expression. Journal of Neuroinflammation. 2015;12(1):p. 29. doi: 10.1186/s12974-015-0250-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Borgmann K., Ghorpade A. Methamphetamine augments concurrent astrocyte mitochondrial stress, oxidative burden, and antioxidant capacity: tipping the balance in HIV-associated neurodegeneration. Neurotoxicity Research. 2018;33(2):433–447. doi: 10.1007/s12640-017-9812-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Youn G. S., Kwon D. J., Ju S. M., et al. Celastrol ameliorates HIV-1 Tat-induced inflammatory responses via NF-kappaB and AP-1 inhibition and heme oxygenase-1 induction in astrocytes. Toxicology and Applied Pharmacology. 2014;280(1):42–52. doi: 10.1016/j.taap.2014.07.010. [DOI] [PubMed] [Google Scholar]
- 79.Meng X., Zhang C., Guo Y., et al. TBHQ attenuates neurotoxicity induced by methamphetamine in the VTA through the Nrf2/HO-1 and PI3K/AKT Signaling pathways. Oxidative Medicine and Cellular Longevity. 2020;2020:13. doi: 10.1155/2020/8787156.8787156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Reddy N. M., Potteti H. R., Vegiraju S., Chen H. J., Tamatam C. M., Reddy S. P. PI3K-AKT signaling via Nrf2 protects against hyperoxia-induced acute lung injury, but promotes inflammation post-injury independent of Nrf2 in mice. PLoS One. 2015;10(6, article e0129676) doi: 10.1371/journal.pone.0129676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Baird L., Dinkova-Kostova A. T. The cytoprotective role of the Keap1-Nrf2 pathway. Archives of Toxicology. 2011;85(4):241–272. doi: 10.1007/s00204-011-0674-5. [DOI] [PubMed] [Google Scholar]
- 82.Wang Y., Zhao C. S. Sigma-1 receptor activation ameliorates LPS-induced NO production and ROS formation through the Nrf2/HO-1 signaling pathway in cultured astrocytes. Neuroscience Letters. 2019;711:p. 134387. doi: 10.1016/j.neulet.2019.134387. [DOI] [PubMed] [Google Scholar]
- 83.Weng T. Y., Hung D. T., Su T. P., Tsai S. A. Loss of sigma-1 receptor chaperone promotes astrocytosis and enhances the Nrf2 antioxidant defense. Oxidative Medicine and Cellular Longevity. 2017;2017:14. doi: 10.1155/2017/4582135.4582135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tsai S. Y., Hayashi T., Harvey B. K., et al. Sigma-1 receptors regulate hippocampal dendritic spine formation via a free radical-sensitive mechanism involving Rac1xGTP pathway. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(52):22468–22473. doi: 10.1073/pnas.0909089106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Tsai S. Y., Pokrass M. J., Klauer N. R., Nohara H., Su T. P. Sigma-1 receptor regulates Tau phosphorylation and axon extension by shaping p35 turnover via myristic acid. Proceedings of the National Academy of Sciences of the United States of America. 2015;112(21):6742–6747. doi: 10.1073/pnas.1422001112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Pekny M., Wilhelmsson U., Pekna M. The dual role of astrocyte activation and reactive gliosis. Neuroscience Letters. 2014;565:30–38. doi: 10.1016/j.neulet.2013.12.071. [DOI] [PubMed] [Google Scholar]
- 87.Voigt P., Brock C., Nürnberg B., Schaefer M. Assigning functional domains within the p101 regulatory subunit of phosphoinositide 3-kinase gamma. The Journal of Biological Chemistry. 2005;280(6):5121–5127. doi: 10.1074/jbc.M413104200. [DOI] [PubMed] [Google Scholar]
- 88.Chen Y., Zhang X., Yang Y., et al. Tert-butylhydroquinone enhanced angiogenesis and astrocyte activation by activating nuclear factor-E2-related factor 2/heme oxygenase-1 after focal cerebral ischemia in mice. Microvascular Research. 2019;126:p. 103891. doi: 10.1016/j.mvr.2019.103891. [DOI] [PubMed] [Google Scholar]
- 89.Zhang Z. W., Liang J., Yan J. X., et al. TBHQ improved neurological recovery after traumatic brain injury by inhibiting the overactivation of astrocytes. Brain Research. 2020;1739, article 146818 doi: 10.1016/j.brainres.2020.146818. [DOI] [PubMed] [Google Scholar]
- 90.Liu L., Zhao Z., Yin Q., Zhang X. TTB protects astrocytes against oxygen-glucose deprivation/reoxygenation-induced injury via activation of Nrf2/HO-1 signaling pathway. Frontiers in Pharmacology. 2019;10:p. 792. doi: 10.3389/fphar.2019.00792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Cao S., Chao D., Zhou H., Balboni G., Xia Y. A novel mechanism for cytoprotection against hypoxic injury: δ-opioid receptor-mediated increase in Nrf2 translocation. British Journal of Pharmacology. 2015;172(7):1869–1881. doi: 10.1111/bph.13031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kang J. S., Choi I. W., Han M. H., et al. The cytoprotective effects of 7,8-dihydroxyflavone against oxidative stress are mediated by the upregulation of Nrf2-dependent HO-1 expression through the activation of the PI3K/Akt and ERK pathways in C2C12 myoblasts. International Journal of Molecular Medicine. 2015;36(2):501–510. doi: 10.3892/ijmm.2015.2256. [DOI] [PubMed] [Google Scholar]
- 93.Wu L., Li H. H., Wu Q., et al. Lipoxin A4 activates Nrf2 pathway and ameliorates cell damage in cultured cortical astrocytes exposed to oxygen-glucose deprivation/reperfusion insults. Journal of Molecular Neuroscience. 2015;56(4):848–857. doi: 10.1007/s12031-015-0525-6. [DOI] [PubMed] [Google Scholar]
- 94.Bellaver B., Souza D. G., Souza D. O., Quincozes-Santos A. Hippocampal astrocyte cultures from adult and aged rats reproduce changes in glial functionality observed in the aging brain. Molecular Neurobiology. 2017;54(4):2969–2985. doi: 10.1007/s12035-016-9880-8. [DOI] [PubMed] [Google Scholar]
- 95.Meng X. E., Zhang Y., Li N., et al. Effects of hyperbaric oxygen on the Nrf2 signaling pathway in secondary injury following traumatic brain injury. Genetics and Molecular Research. 2016;15(1) doi: 10.4238/gmr.15016933. [DOI] [PubMed] [Google Scholar]
- 96.Zhao Y. J., Nai Y., Ma Q. S., et al. Dl-3-n-butylphthalide protects the blood brain barrier of cerebral infarction by activating the Nrf-2/HO-1 signaling pathway in mice. European Review for Medical and Pharmacological Sciences. 2018;22(7):2109–2118. doi: 10.26355/eurrev_201804_14744. [DOI] [PubMed] [Google Scholar]
- 97.Hsu Y. Y., Chen C. S., Wu S. N., Jong Y. J., Lo Y. C. Berberine activates Nrf2 nuclear translocation and protects against oxidative damage via a phosphatidylinositol 3-kinase/Akt-dependent mechanism in NSC34 motor neuron-like cells. European Journal of Pharmaceutical Sciences. 2012;46(5):415–425. doi: 10.1016/j.ejps.2012.03.004. [DOI] [PubMed] [Google Scholar]
- 98.Jung E., Kim J. H., Kim M. O., Lee S. Y., Lee J. Melanocyte-protective effect of afzelin is mediated by the Nrf2-ARE signalling pathway via GSK-3β inactivation. Experimental Dermatology. 2017;26(9):764–770. doi: 10.1111/exd.13277. [DOI] [PubMed] [Google Scholar]
- 99.Yao J., Zhang B., Ge C., Peng S., Fang J. Xanthohumol, a polyphenol chalcone present in hops, activating Nrf2 enzymes to confer protection against oxidative damage in PC12 cells. Journal of Agricultural and Food Chemistry. 2015;63(5):1521–1531. doi: 10.1021/jf505075n. [DOI] [PubMed] [Google Scholar]
- 100.Park J. S., Kim H. S. Regulation of hemeoxygenase-1 gene expression by Nrf2 and c-Jun in tertiary butylhydroquinone-stimulated rat primary astrocytes. Biochemical and Biophysical Research Communications. 2014;447(4):672–677. doi: 10.1016/j.bbrc.2014.04.073. [DOI] [PubMed] [Google Scholar]
- 101.Shin H. R., You B. R., Park W. H. PX-12-induced HeLa cell death is associated with oxidative stress and GSH depletion. Oncology Letters. 2013;6(6):1804–1810. doi: 10.3892/ol.2013.1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Deng Y., Xu Z. F., Liu W., Xu B., Yang H. B., Wei Y. G. Riluzole-triggered GSH synthesis via activation of glutamate transporters to antagonize methylmercury-induced oxidative stress in rat cerebral cortex. Oxidative Medicine and Cellular Longevity. 2012;2012:12. doi: 10.1155/2012/534705.534705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Cortese M. M., Suschek C. V., Wetzel W., Kroncke K. D., Kolb-Bachofen V. Zinc protects endothelial cells from hydrogen peroxide via Nrf2-dependent stimulation of glutathione biosynthesis. Free Radical Biology & Medicine. 2008;44(12):2002–2012. doi: 10.1016/j.freeradbiomed.2008.02.013. [DOI] [PubMed] [Google Scholar]
- 104.Liang L. Y., Wang M. M., Liu M., et al. Chronic toxicity of methamphetamine: oxidative remodeling of pulmonary arteries. Toxicology In Vitro. 2020;62:p. 104668. doi: 10.1016/j.tiv.2019.104668. [DOI] [PubMed] [Google Scholar]
- 105.Orcholski M. E., Khurshudyan A., Shamskhou E. A., et al. Reduced carboxylesterase 1 is associated with endothelial injury in methamphetamine-induced pulmonary arterial hypertension. American Journal of Physiology. Lung Cellular and Molecular Physiology. 2017;313(2):L252–l266. doi: 10.1152/ajplung.00453.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Wang X., Liu M., Zhu M. J., et al. Resveratrol protects the integrity of alveolar epithelial barrier via SIRT1/PTEN/p-Akt pathway in methamphetamine-induced chronic lung injury. Cell Proliferation. 2020;53(3, article e12773) doi: 10.1111/cpr.12773. [DOI] [PMC free article] [PubMed] [Google Scholar]