

Abstract
When nanoparticles enter biological environments, proteins adsorb to form the “protein corona” which causes significant changes in biodistribution and toxicity. Herein, we measure protein corona formation on DNA-functionalized single-walled carbon nanotubes (ssDNA-SWCNTs), a nanoparticle ideal for sensing and delivery, in blood plasma and cerebrospinal fluid. We characterize corona composition by mass spectrometry, revealing high-abundance corona proteins involved in lipid binding, complement activation, and coagulation. We investigate roles of electrostatic and entropic interactions driving selective corona formation. Lastly, we study real-time protein binding on ssDNA-SWCNTs, obtaining agreement between enriched proteins binding strongly and depleted proteins binding marginally, while highlighting cooperative adsorption mechanisms. Knowledge of protein corona composition, formation mechanisms, and dynamics informs nanoparticle translation from in vitro design to in vivo application.
Keywords: Biophysics, Colloids, Nanoparticles, Nanotechnology, Proteins
Graphical Abstract
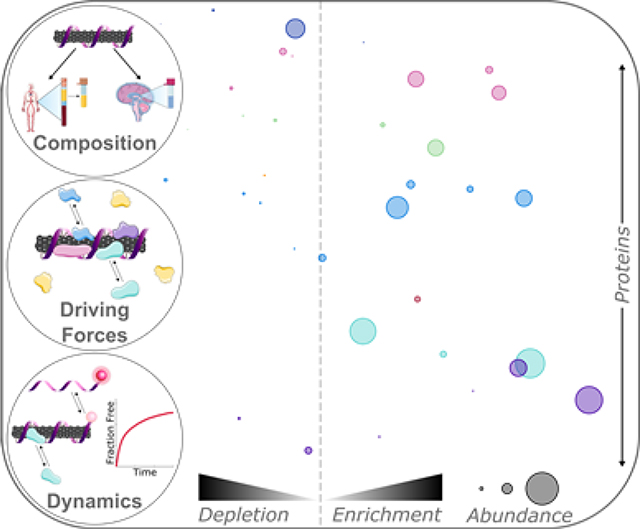
Introducing engineered nanoparticles into biosystems leads to formation of the interfacial protein corona, modifying nanoparticle identity and function. Herein, protein corona composition, interactions, and dynamics are analyzed on carbon nanotube-based sensors. This generic framework will enhance our fundamental understanding of protein corona formation and enable effective translation of nanotechnologies from in vitro design to in vivo use.
Introduction
Engineered nanoparticles are prominent in biological sensing, imaging, and delivery applications due to their distinctive optical and physical properties.[1,2] The critical − yet often overlooked − challenge with these nanoscale tools is understanding the mechanisms of interaction between the nanoprobe and the biological system they are designed to query.[3,4] Nanotechnologies are generally developed and validated in vitro, absent from the complexity of biological fluids.[5] However, when nanoparticles are introduced into biological systems, proteins spontaneously adsorb to the nanoparticle surfaces, leading to the formation of the “protein corona”.[6] Binding of proteins to pristine nanoparticles can adversely affect the structure and function of the bound proteins,[7,8] and carries the additional consequence of masking and re-defining the nanoparticle identity.[3,9] Accordingly, in vivo trafficking, biodistribution, clearance, and biocompatibility of the nanoparticle-corona complex become unpredictable.[3,9–11] These corona-mediated alterations manifest as decreased nanoparticle efficacy or loss of in vitro-validated results, whereby the nanoparticle no longer carries out its designated function.[12,13] Moreover, the protein corona is dynamic in nature.[3,14] Rapid protein binding events on the nanoparticle surface, in conjunction with differential protein affinities in the corona, give rise to further complications in understanding the timescales over which nanoparticles retain their corona-free attributes within biological environments.
Corona formation as a function of nanoparticle type, biological environment, and time remains poorly understood.[15] Prior work investigates the roles of nanoparticle surface charge and chemistry, among other factors, in governing protein corona formation, [16–19] revealing the variation and complexity of protein corona formation across nanoparticle types and bioenvironments.[4,20] Consequent work has sought to mitigate corona formation by nanoparticle surface passivation with polymers such as polyethylene glycol (PEG) to abrogate protein adsorption and sterically stabilize the nanoparticle. Of note, PEGylation of liposomes, a model nanoparticle for delivery, has been demonstrated to decrease protein adsorption and maintain some functionality of surface-exposed targeting moieties.[21–23] Yet, such strategies can display variable efficacy depending on the underlying nanoparticle and bioenvironmental factors.[24,25]
Although many studies classify protein corona composition around specific nanoparticle systems, significant debate persists as to which protein and nanoparticle characteristics are most important in determining corona composition, and how different biological environments contribute to compositional and temporal corona heterogeneity.[4,26] While prior studies clarify different aspects of bio-corona formation, system constraints such as surface-immobilization or treating the protein corona as existing at thermodynamic equilibrium make it difficult to reliably translate results to real biofluid systems.[3,27,28] Additionally, many nanosensor technologies are tested for biofouling and biocompatibility in blood serum, a blood-based fluid rich in albumin, the most abundant blood plasma protein, and devoid of blood clotting proteins. The assumptions that serum is a representative biofluid for confirming in vivo function and that protein abundance in a native biofluid determines its relative abundance in a nanoparticle corona both stand to be refined.
Understanding protein corona formation is essential to design nanoparticles that are robust and stable in biological environments. Our work focuses on single-walled carbon nanotubes (SWCNTs), a nanoparticle class that possesses unique optical and physical properties ideal for biological imaging, molecular sensing, and delivery applications.[1,29–31] To apply hydrophobic SWCNTs in aqueous biological systems, noncovalent functionalization with amphiphilic polymers imparts water solubility to the SWCNT, while retaining the near-infrared-emissive electronic structure.[29] Select polymers confer molecular recognition functionality when adsorbed to the SWCNT surface, such as single-stranded DNA (ssDNA). Specifically, ssDNA sequences (GT)6 or (GT)15 adsorbed to SWCNTs are implemented to image the neurotransmitter dopamine in the brain at spatiotemporal scales of relevance to endogenous neuromodulation.[30,32,33] ssDNA-functionalized SWCNTs have further been applied in intravenous in vivo scenarios, to monitor endolysosomal lipid accumulation[34] and nitric oxide production.[35] To design and apply these and other SWCNT-based nanotechnologies in biological systems, it is crucial to understand the composition, dynamics, and dominant mechanisms of protein corona formation.
Herein, we explore protein corona formation probed with a selective adsorption assay generalizable to different types of nanoparticles and biofluids. We focus on two nanoparticles: a model system of commonly studied polystyrene nanoparticles (PNPs)[5,14,16,28,36] and a newer system of noncovalently functionalized SWCNTs. Protein adsorption on these nanoparticles is assessed in two biofluids: blood plasma, a standard biofluid relevant for blood circulation applications, and cerebrospinal fluid (CSF), an understudied biofluid relevant for central nervous system studies. An understanding of the corona formed on SWCNTs in CSF has not been investigated and is imperative for developing SWCNT-based applications in the brain, including mapping of the brain extracellular space[37] and vasculature,[38] neurotransmitter imaging,[30,39] and delivery to the brain.[31] Corona composition characterized by quantitative, label-free mass spectrometry analysis reveals key protein corona contributors and isolation of protein factors governing corona formation. We identify interactions driving protein adsorption, where hydrophobic interactions dominate formation of the inner corona, while electrostatic interactions govern formation of the outer corona. To quantify the time-dependent protein corona formation process, we assess binding thermodynamics and kinetics by measuring adsorption of key proteins to (GT)15-SWCNTs via isothermal titration calorimetry (ITC) and a corona exchange assay.[40] Finally, the protein-SWCNT complex structure is ascertained by small-angle x-ray scattering (SAXS), demonstrating changing mass fractal morphology of ssDNA-SWCNTs in the presence of a high-binding protein (fibrinogen) otherwise absent with the low-binding protein (albumin). Overall, we present a holistic experimental approach and analysis methodology to understand the complexities of protein corona formation, and apply this framework to examine an understudied system of interest: SWCNT-based probes in brain.
Results and Discussion
1. Protein Corona Composition
Protein corona composition was studied on (GT)15-functionalized SWCNTs (see synthesis in SI; average 1 nm diameter, 500 nm length) and PNPs (100 nm diameter) in blood plasma (normal human, pooled donors; Innovative Research Inc.) and cerebrospinal fluid (CSF; normal human, pooled donors; Lee Biosolutions). Selective adsorption of proteins onto nanoparticles was evaluated by (i) incubating nanoparticles with biofluid for 1 h, (ii) isolating protein-nanoparticle complexes by centrifugation, (iii) removing unbound proteins by washing, (iv) eluting bound proteins from nanoparticles with surfactant and reducing agent, and (v) characterizing proteins by two-dimensional polyacrylamide gel electrophoretic separation (2D PAGE) or liquid chromatography-tandem mass spectrometry (LC-MS/MS) (Figure S2; see methods in SI).[16] Following workflow validation (Figure S2 and Figure S3), protein coronas were studied on these two distinct nanoparticle surfaces (PNPs and (GT)15-SWCNTs) in two biofluids (plasma and CSF). PAGE analysis confirmed that proteins showed selective enrichment or depletion fingerprints on nanoparticles (Figure S4). More in-depth protein corona composition studies were subsequently undertaken by performing in-solution trypsin digestion of proteins eluted from nanoparticles, followed by protein characterization with label-free, quantitative LC-MS/MS. Analysis by LC-MS/MS provides (i) molar corona protein abundances via comparison to an internal standard and (ii) enrichment or depletion in each nanoparticle corona, relative to protein concentrations in the native biofluid (see SI). Protein abundances, fold changes, and functional classes for protein coronas formed on PNPs and (GT)15-SWCNTs are represented graphically in Figure 1 (plasma) and Figure 2 (CSF) (full protein lists in Figure S5, Figure S6, and attached datasheet). The twenty most abundant proteins in the nanoparticle coronas are summarized in Table 1 (plasma) and Table 2 (CSF).
Figure 1. Blood plasma protein corona compositional map determined by proteomic mass spectrometry.

Protein corona formed from blood plasma on (a) PNPs and (b) (GT)15-SWCNTs. Full protein lists are available in SI (Figure S5 and attached datasheet). Circle size corresponds to protein abundance (femtomolar). Proteins are grouped by functional class according to color (PANTHER).[41] Log2 fold change is in comparison to the biofluid alone, e.g. log2 fold change of zero indicates the same relative amount of protein exists in the corona as in bulk solution of the native biofluid, while < 0 is depletion and > 0 is enrichment. Names are included for proteins of interest or used for subsequent experiments. Colored boxes at x-axis limits indicate no protein detected in either corona (x < 2−6 or 2−8) or biofluid (x > 28). Data represent experimental triplicate for plasma with nanoparticles, technical triplicate for plasma alone.
Figure 2. Cerebrospinal fluid (CSF) protein corona compositional map determined by proteomic mass spectrometry.

Protein corona formed from CSF on (a) PNPs and (b) (GT)15-SWCNTs. Full protein lists are available in SI (Figure S6 and attached datasheet). Circle size corresponds to protein abundance (femtomolar). Proteins are grouped by functional class according to color (PANTHER).[41] Log2 fold change is in comparison to the biofluid alone, e.g. fold change of zero indicates the same relative amount of protein exists in the corona as in bulk solution of the native biofluid, while < 0 is depletion and > 0 is enrichment. Names are included for proteins of interest or used for subsequent experiments. Colored boxes at x-axis limits indicate no protein detected in either corona (x < 2−6 or 2−8) or biofluid (x > 28). Data represent technical triplicate for CSF with and without nanoparticles.
Table 1.
Top 20 most abundant proteins identified by proteomic mass spectrometry in plasma nanoparticle coronas.
| Plasma | PNPs in plasma | (GT)15-SWCNTs in plasma | |
|---|---|---|---|
| 1 | Serum albumin | Alpha-2-HS-glycoprotein | Clusterin |
| 2 | Haptoglobin | Ig kappa constant | Histidine-rich glycoprotein |
| 3 | Ig kappa constant | Haptoglobin | Apolipoprotein A-I |
| 4 | Ig heavy constant gamma | Complement C3 | Complement C3 |
| 5 | Serotransferrin | Kininogen-1 | Haptoglobin |
| 6 | Apolipoprotein A-I | Ig heavy constant gamma 1 | A disintegrin and metalloproteinase with thrombospondin motifs 12 |
| 7 | Complement C4 | Apolipoprotein A-II | Complement C1r subcomponent |
| 8 | Telomeric repeat-binding factor 2-interacting protein | tRNA-dihydrouridine(47) synthase [NAD(P)(+)]-like | Vitronectin |
| 9 | Alpha-1-antitrypsin | Beta-2-glycoprotein 1 | Kininogen-1 |
| 10 | Alpha-2-HS-glycoprotein | Vitronectin | Prothrombin |
| 11 | Apolipoprotein A-II | Serum albumin | C4b-binding protein alpha chain |
| 12 | Ig heavy constant alpha 1 | Vitamin D-binding protein | Complement factor H |
| 13 | Integrin alpha-7 | A disintegrin and metalloproteinase with thrombospondin motifs 12 | Fibrinogen alpha chain |
| 14 | Alpha-2-macroglobulin | Hemopexin | Protein AMBP |
| 15 | Complement C3 | Apolipoprotein A-I | Beta-2-glycoprotein 1 |
| 16 | Complement C5 | Ig lambda-like polypeptide 5 | Apolipoprotein E |
| 17 | Hemopexin | Histidine-rich glycoprotein | Complement C1q subcomponent subunit B |
| 18 | Alpha-1-acid glycoprotein 1 | Clusterin | Ig heavy constant gamma 1 |
| 19 | Ig heavy constant mu | Alpha-1-antitrypsin | Ig J chain |
| 20 | Beta-2-glycoprotein 1 | Serum paraoxonase/arylesterase 1 | Galectin-3-binding protein |
Table 2.
Top 20 most abundant proteins identified by proteomic mass spectrometry in CSF nanoparticle coronas.
| CSF | PNPs in CSF | (GT)15-SWCNTs in CSF | |
|---|---|---|---|
| 1 | Serum albumin | Cystatin-C | Complement C3 |
| 2 | Transthyretin | Complement C3 | Clusterin |
| 3 | Alpha-1-antitrypsin | Clusterin | Histidine-rich glycoprotein |
| 4 | Prostaglandin-H2 D-isomerase | Prostaglandin-H2 D-isomerase | Galectin-3-binding protein |
| 5 | Serotransferrin | Alpha-2-HS-glycoprotein | Apolipoprotein E |
| 6 | Cystatin-C | Collagen alpha-2(XI) chain | Prostaglandin-H2 D-isomerase |
| 7 | Alpha-1-acid glycoprotein 1 | Beta-2-glycoprotein 1 | Kininogen-1 |
| 8 | Hemoglobin subunit alpha | Gelsolin | Apolipoprotein A-I |
| 9 | Ig heavy constant gamma 1 | Serotransferrin | Vitronectin |
| 10 | Vitamin D-binding protein | Vitronectin | Transthyretin |
| 11 | Ceruloplasmin | Ig heavy constant gamma 1 | Gelsolin |
| 12 | Hemopexin | Apolipoprotein E | Ig heavy constant gamma 1 |
| 13 | Apolipoprotein E | Fibulin-1 | Serotransferrin |
| 14 | Ig kappa constant | Major prion protein | Complement C1s subcomponent |
| 15 | Apolipoprotein A-I | Kininogen-1 | Complement C1q subcomponent subunit B |
| 16 | Hemoglobin subunit beta | EGF-containing fibulin-like extracellular matrix protein 1 | Fibulin-1 |
| 17 | Haptoglobin | Complement factor H | Complement factor H |
| 18 | Clusterin | Histidine-rich glycoprotein | Major prion protein |
| 19 | Suppression of tumorigenicity 18 protein | Fibrinogen beta chain | Fibrinogen alpha chain |
| 20 | Gelsolin | ProSAAS | Cystatin-C |
1.1. Blood plasma protein corona composition
Our LC-MS/MS analysis highlights the significant enrichment vs. depletion of specific plasma proteins in the nanoparticle coronas (Figure 1). First, plasma proteins identified in the PNP corona are corroborated by previous literature.[14,16] Of note for (GT)15-SWCNTs, serum albumin is the most abundant protein in plasma (55% w/v in plasma), yet does not appreciably adsorb: albumin is in low abundance and significantly depleted in the corona, with a 1300-fold lower bound concentration relative to native plasma. Broadly, corona proteins on PNPs are more diverse in functional classes than corona proteins on (GT)15-SWCNTs, representing a range of endogenous functions including adaptive immune response and transport, for which proteins are largely absent on (GT)15-SWCNTs. We quantify these protein functional roles implicated in corona formation by regressing ln-fold change against protein class using effect-coding while controlling for sample-to-sample variability (Figure S7 and Figure S8). The dissimilarities from this analysis highlight the less selective association of proteins to PNPs in comparison to (GT)15-SWCNTs, where this ability of (GT)15-SWCNTs to resist nonspecific protein biofouling is promising towards SWCNT-based biotechnology applications.
In particular, plasma proteins enhanced on (GT)15-SWCNTs are involved in (i) lipid binding/transport (150%-fold change over the average of all protein classes) and (ii) complement activation (140%-fold change). These LC-MS/MS results suggest that, due to corona formation, (GT)15-SWCNTs will have reduced nonspecific cellular uptake (high clusterin adsorption[36]), prolonged circulation in blood (overall high apolipoprotein adsorption[5]), and minimal activation of the adaptive immune response (low immunoglobulin representation) compared to other nanoparticles (i.e. PNPs). Detrimental responses, however, may include activating the innate immune response (high complement C3 and other complement protein adsorption[42,43]) and eliciting inflammatory responses (high fibrinogen adsorption[5,8,27]) (see SI).
We further compared the plasma corona formed on (GT)15-SWCNTs to that on (GT)6-SWCNTs, where the adsorbed ssDNA differs in length (30 vs. 12 nucleotides) and morphology (helical vs. ring wrapping).[32] Plasma proteins identified in the (GT)6-SWCNT corona approximately match those in the (GT)15-SWCNT corona (Figure S5 and Table S2), and the analogous regression of protein functional classes suggests that the SWCNT surface, rather than the initial ssDNA corona, determines protein adsorption selectivity (Figure S7 and Figure S9). Surface passivation with ssDNA does, however, lead to distinct results compared to prior protein corona characterization on pristine or carboxylated SWCNTs,[44,45] where adsorption of albumin and immunoglobulins is seemingly prohibited by an initial ssDNA corona.
1.2. Cerebrospinal fluid protein corona composition
We repeated our assay and analysis to study protein corona formation from CSF on PNPs and (GT)15-SWCNTs (Figure 2). Highly abundant proteins in the CSF corona formed on (GT)15-SWCNTs include complement C3 as the most abundant (26-fold enriched relative to native CSF), clusterin as the 2nd most abundant (15-fold enriched), and histidine-rich glycoprotein as the 3rd most abundant (41-fold enriched). Again, serum albumin is remarkably absent, with over 2.5 million-fold lower bound concentration relative to native CSF. Notably, the reproducible outlier of galectin-3-binding protein (G3BP) emerges, which is the 4th most abundant and most strongly enriched (80-fold) protein on the (GT)15-SWCNT surface. Identification of highly adsorbing and potentially interfering proteins could enable a priori design of future nanosensors to either promote selective or mitigate unfavorable protein adsorption.
LC-MS/MS analysis of the CSF-based protein corona revealed proteins across a range of functional classes for both nanoparticles (Figure S7). Protein classes that have higher than average fold change on (GT)15-SWCNTs are coagulation proteins (441% higher fold change than average), complement proteins (213% higher), and cell adhesion/signal transduction proteins (486% higher).
Many proteins have the same corona representation on (GT)15-SWCNTs across plasma and CSF, including clusterin, histidine-rich glycoprotein, and complement C3. Conversely, certain proteins show distinctive behaviors in the (GT)15-SWCNT corona formed from different biofluids, such as serotransferrin missing from the plasma corona and present in the CSF corona, despite serotransferrin’s higher native concentration in plasma. These discrepancies point to mechanisms such as adsorption cooperativity and the Vroman effect,[27] whereby surface adsorption is dictated by relative affinities and abundances of all protein constituents in the bulk to determine the end-state corona. We also find that while plasma protein content in the corona vs. native biofluid is positively correlated for plasma proteins on PNPs (R2 = 0.461), this scaling does not hold for either (GT)15-SWCNTs (R2 = 0.101) or (GT)6-SWCNTs (R2 = 0.072) (Figure S11). This again suggests complex mechanisms driving selective corona adsorption on SWCNTs and, depending on the biofluid, also PNPs.
2. Protein Corona Formation Mechanisms
2.1. Molecular phenomena involved in protein corona formation
To evaluate the nanoscale mechanisms involved in corona formation, we linearly regressed the ln-fold change against protein physicochemical properties including mass, post-translational modification frequency, binding site frequency, and amino acid percent compositions (Figure 3 and Figure S10; see SI).[46,47] Statistically, the calculated regression coefficients quantify the fractional difference of the fold change for a protein with a unit increase of the independent variable, holding all other independent variables constant. Thermodynamically, the regression coefficients quantify the free energy change of a protein adsorbing into the corona per unit of the independent variable in units of kbT (see derivation in SI). Proteins often denature upon surface adsorption, exposing otherwise solvent-inaccessible residues.[27,45,48–50] Accordingly, we include all amino acid groups in the regression analysis rather than only solvent exposed groups. Future work should isolate the roles residue accessibility and/or protein structure play on corona formation.
Figure 3. Molecular attributes of proteins that govern protein corona formation for each nanoparticle-biofluid pairing.

Ln-fold change regression coefficients for molecular attributes of proteins (rows) for each nanoparticle-biofluid pairing (columns). Cells are colored from dark purple (negative effect on fold change) to white (no effect) to dark blue (positive effect). Standard errors of the coefficients are given in parenthesis. Results that have false-discovery-rate-corrected p-values below 0.1 are bolded and noted with asterisks. Amino acid groupings include: non-aromatic hydrophobic (sum of alanine, valine, isoleucine, leucine, and methionine content), hydrophilic (sum of serine, threonine, asparagine, and glutamine content), and acidic (sum of aspartic acid and glutamic acid content).
This regression analysis reveals that PNPs are generally agnostic to physicochemical properties of proteins entering the corona, except for a slight favorable interaction associated with the aromatic phenylalanine residue and unfavorable interaction with tyrosine. In contrast, the plasma corona formed on (GT)15-SWCNTs is selective, showing unfavorable interactions with non-aromatic, hydrophobic amino acids (−0.53 kbT/%hydrophobic), despite the extremely hydrophobic SWCNT surface. However, we find aromatic residues are enhanced in the (GT)15-SWCNT plasma corona, namely phenylalanine (0.57 kbT/%F), tyrosine (0.22 kbT/%Y), and tryptophan (0.39 kbT/%W). Regarding basic residues, lysine (and to a lesser extent, arginine) content is also associated with unfavorable interactions with (GT)15-SWCNTs (−0.63 kbT/%K), which is surprising in that positively charged proteins are expected to have favorable electrostatic interactions with the negatively charged, solvent-exposed phosphate backbone of ssDNA on the SWCNT. Moreover, more negatively charged, acidic residue content led to enhancement in the protein corona (0.31 kbT/%acidic). This result indicates that protein charges are effectively screened by salt in solution or that proteins interact directly with the solvent-accessible SWCNT surface. The latter hypothesis is supported by the low initial ssDNA coverage on the SWCNT (~1–25%)[40,51,52] and the small fraction of ssDNA removed from the SWCNT during protein adsorption (~1–5%).[40] Direct protein interaction with the SWCNT surface could also explain the enhancement of aromatic residues, as they would favorably interact via π-π stacking directly with the graphitic SWCNT surface.
Apart from the favorable aromatic residue contribution, (GT)15-SWCNTs have strikingly different interactions with proteins in CSF. In CSF, unlike in plasma, acidic residues are associated with unfavorable interactions with SWCNTs (−0.52 kbT/%acidic), whereas positively charged arginine (and to a lesser extent, lysine) have favorable interactions (1.53 kbT/%R). The tendency of ssDNA-SWCNTs in CSF to interact favorably with positively charged residues and unfavorably with negatively charged residues suggests that the negatively charged ssDNA wrapping is less screened electrostatically by other adsorbed proteins (seeing as net ionic strength in both biofluids is comparable), or that more ssDNA remains immobilized on the SWCNT surface following protein exposure in CSF than in plasma (Figure S12). In sum, these regression results emphasize the non-intuitive nature of corona formation, in that seemingly important properties for corona formation such as in-solution protein stability are not predictive of presence in a nanoparticle corona.
2.2. Driving forces of protein corona formation
To gain further insight on interactions driving protein adsorption to nanoparticles, incubation conditions of (GT)15-SWCNTs exposed to plasma were varied and corona proteins were characterized by 2D PAGE. Specifically, conditions varied include dynamics (to probe corona stability), ionic strength (to probe electrostatic interactions), and temperature (to probe entropic contributions) (Figure 4). Under dynamic protein-nanoparticle incubation, proteins in the outer adsorbed plane undergo shear and are removed. Remaining proteins are postulated to represent the inner, more tightly bound “hard” corona proteins that interact more strongly with the nanoparticle surface, while removed proteins represent the outer, more loosely bound “soft” corona proteins that interact with other adsorbed proteins. We note that in both cases, the proteins are co-precipitated with the nanoparticles for characterization. For (GT)15-SWCNTs, apolipoproteins A-I, clusterin, complement C3, fibrinogen, and alpha-1-antitrypsin compose the hard corona, while some soft corona proteins of interest include albumin and haptoglobin. Elimination of salt during incubation increases the role of repulsive electrostatic forces by removing ionic charge screening. This absence of charge screening means that proteins and nanoparticles do not approach as closely in solution and that lateral electrostatic interactions of surface-adsorbed proteins increase, both of which result in less protein adsorption.[53] For (GT)15-SWCNTs, the three hard-corona proteins (apolipoprotein A-I, complement C3, and fibrinogen) still enter the corona despite the lack of charge shielding. This result points to the role of non-electrostatic forces facilitating formation of the hard corona, likely hydrophobic interactions, that drive protein-SWCNT adsorption even under electrostatically adverse conditions of electric double-layer repulsion (as supported by zeta potential measurements of nanoparticles and plasma proteins, separately, Figure S3). In addition, most soft corona proteins are missing in the no salt incubation, underscoring the need for charge-screening for soft corona formation. Finally, higher temperature incubation increases weighting of the entropic contribution to the overall free energy change of protein binding. Entropic contributions originate from the solvent (positive, as hydration shells of the protein and surface are released to bulk) and protein (negative, from the adsorbate losing degrees of freedom and potentially positive if proteins unfold upon adsorption). At physiological temperature, hard-corona proteins are still able to adsorb to (GT)15-SWCNTs, indicating that adsorption of these proteins is entropically favorable and/or enthalpically driven. Enthalpic contributions arise from noncovalent interactions between proteins and the SWCNT surface, and hydrogen bond formation within the bulk solvent as proteins leave solution to enter the adsorbed state. Thus, hard-corona proteins undergo high affinity binding to (GT)15-SWCNTs despite dynamic perturbation, low ionic strength, and increased temperature incubation conditions.
Figure 4. Effect of varying incubation parameters to probe corona stability, electrostatic interactions, and entropic contributions to corona formation of plasma proteins on (GT)15-SWCNTs.

(a) Schematics depicting incubation conditions affecting corona adsorption, with reference conditions (top) vs. varied conditions (bottom). (b) Proteins present in native plasma (left-most column) as compared to the plasma corona formed on (GT)15-SWCNTs under reference conditions (static, 0.1 M phosphate-buffered saline, and 25°C incubation) and varying incubation conditions, including: dynamic (on orbital shaker; to probe corona stability), no salt (water; to probe ionic effects), and temperature (37°C; to probe entropic contributions). Color indicates normalized appearance frequency of protein in corona characterized by 2D PAGE (N ≥ 3 experimental replicates; see details in SI).
3. Protein Corona Dynamics
Beyond probing corona composition at the end point of adsorption, we investigated corona formation dynamics to understand the time-dependent process and overall system energetics driving corona formation. Towards this end, isothermal titration calorimetry (ITC) was applied to probe the thermodynamics of protein adsorption to SWCNT surfaces.[6,25,54] We studied binding of (GT)15-SWCNTs with two proteins identified by LC-MS/MS with opposite binding affinities: albumin, selected as a model low-binding protein, and fibrinogen, a model high-binding protein. ITC results confirm that fibrinogen preferentially adsorbs to (GT)15-SWCNTs and albumin does not, as evidenced by the binding curve in the former and absence of changing heats upon injection in the latter (Figure S13). From the ITC binding curve of fibrinogen with (GT)15-SWCNTs, the change in enthalpy is −565.2 kJ/mol and the change in entropy is −1.756 kJ/K-mol. This favorable enthalpic term outweighs the net unfavorable entropic terms to ultimately drive formation as a spontaneous, energetically favorable process: the net change in free energy is −41.91 kJ/mol. However, these ITC results must be interpreted with the consideration that the equilibrium requirement for this thermodynamic analysis is not rigorously held (see SI).[3,27,28] This binding profile shape for protein-surface adsorption processes often emerges as a result of adsorption-induced protein spreading/denaturation, reorientation, and aggregation as functions of bulk protein concentration, in contrast to originating from the dynamic equilibrium between the fluid and surface-adsorbed phases required for Langmuirian adsorption.[48–50,55] Thus, although these binding curves confirm compositional findings of the relative binding affinities, it should be noted that ITC is not a suitable methodology to study all nanoparticle-protein systems and these limitations must be reflected in interpreting these energetics as overall changes in system energies, rather than a true deconvolution of protein-nanoparticle binding interactions.
We next implemented a real-time kinetic binding assay to study dynamic protein interactions with SWCNTs.[40] Briefly, multiplexed fluorescence enables tracking each entity involved in the corona formation process, with cyanine 5 (Cy5)-tagged ssDNA originally on the SWCNT surface exchanging with protein added to solution. We implemented this platform to track the binding of key plasma corona proteins to (GT)15-SWCNTs and (GT)6-SWCNTs (Figure 5a–b), with desorption of Cy5-tagged ssDNA originally on the SWCNT measured as an increase in Cy5 fluorescence and used as a proxy for protein adsorption to SWCNT. Specifically, we assayed the protein panel: clusterin, apolipoprotein A-I, fibrinogen, and complement C3, which are predicted to adsorb in high abundance to (GT)15-SWCNTs, and alpha-2-HS glycoprotein, immunoglobulin G, and albumin, which are predicted to adsorb less to (GT)15-SWCNTs based on LC-MS/MS compositional analysis (see expected ordering in Table 1). Interestingly, the order of protein adsorption was: fibrinogen > apolipoprotein A-I > alpha-2-HS glycoprotein > immunoglobulin G ≈ clusterin > complement C3 > albumin (Figure 5a). While this result affirms the high affinity of fibrinogen and apolipoprotein A-I vs. low affinity of albumin to (GT)15-SWCNTs, some of the single-protein end states do not match the relative ordering of protein abundances from the full-biofluid LC-MS/MS experiments. Accordingly, higher order interactions such as the Vroman effect are further supported in affecting protein adsorption in the full-biofluid experiments, absent in the single-protein experiments. Moreover, these time-dependent dynamics reveal that the rates of protein binding are distinct among proteins, even though some converge to the same final value (such as alpha-2-HS glycoprotein and clusterin). A comparison of this same protein panel binding to (GT)6-SWCNTs is provided because the shorter ssDNA strand is displaced more readily, providing a greater spread between protein species (Figure 5b; see expected ordering in Table S2). The dynamics of protein adsorption recapitulate similar high- vs. low-binding propensities, yet, complement C3 and clusterin again display significantly less adsorption than expected based on LC-MS/MS results, signifying that these proteins enter the corona with cooperative binding mechanisms (e.g. C3 binding to other surface-adsorbed proteins[56]) rather than by high binding affinity to the SWCNT surface on their own. To more quantitatively build a physical picture of protein-SWCNT association, we next expand to structural studies of these protein-nanoparticle complexes.
Figure 5. Protein corona dynamics and structure assessed for binding of key proteins to ssDNA-SWCNTs.

A corona exchange assay is employed to determine binding kinetics of a protein panel (each at 80 mg L−1 final concentration) to (a) (GT)15-SWCNTs and (b) (GT)6-SWCNTs (each at 5 mg L−1 final concentration). Shaded error bars indicate standard error between experimental replicates (N = 3). Small-angle x-ray scattering (SAXS) is applied to gain in-solution structural information of albumin vs. fibrinogen adsorption on (GT)15-SWCNTs. (c) Experimental SAXS profiles for 0.5 g L−1 (GT)15-SWCNTs with and without albumin or fibrinogen, each at 0.5 g L−1 final concentrations. Mass fractal model fits are included in purple together with fit residuals on the right. The accompanying illustration depicts the mass fractal dimension Dm increasing from approximately 1 (rod-like) to 2 (disk-like), with the fit Dm values for (GT)15-SWCNTs in the presence and absence of proteins.
4. Protein Corona Morphology
To evaluate in-solution structural changes of the (GT)15-SWCNTs due to protein corona formation, small-angle X-ray scattering (SAXS) was performed with two proteins, albumin and fibrinogen, as low-binding and high-binding proteins, respectively. SAXS results confirm formation of unique form factors and thus complexation for (GT)15-SWCNTs with fibrinogen, absent for the case of albumin (Figure 5c), therefore recapitulating corona compositional findings.
The intrinsically disordered experimental SAXS profiles were fit using mass fractal geometries, complemented by power-law dependencies from the Porod region, as detailed in SI (Figure 5c, Figure S14, and Table S5).[57–59] The mass fractal radii (all R~1 nm), traditionally defined as the radius of the uniform sphere used to cover the fractal, suggest that the overall topology of the (GT)15-SWCNTs remains constant with and without protein. The fractal dimension Dm and analogous power-law exponent p, found to be in close agreement, estimate the bulk geometries of the mass fractals, where the integer value represents the three dimensions in Euclidean space such that values of 1, 2, and 3 represent rod, disk, and sphere geometries, respectively. The decrease in fractal dimension from Dm~1.90 for (GT)15-SWCNTs with or without albumin to Dm=1.77 for (GT)15-SWCNTs with fibrinogen reveals an initial disk-like mass fractal geometry, then elongation to gain rod-like character in the presence of fibrinogen (Figure 5c). This is consistent with previous literature in which fibrinogen binds to SWCNTs in a lengthwise manner.[45,60] Furthermore, the decrease of (GT)15-SWCNT Dm in the presence of fibrinogen signifies increasing attractive forces between the molecular entities and consequent colloidal instability.[61] Finally, the cutoff length ζ, or the maximum distance between any two points of the mass fractal, undergoes a ten-fold increase for (GT)15-SWCNTs with fibrinogen, denoting a drastic increase in the aggregate size. Thus, SAXS confirms fibrinogen complexation with (GT)15-SWCNTs, suggests a side-on orientation (as reiterated by TEM, Figure S15), and enables quantification of the changing fractal structure, pointing to the role of multilayer adsorption mechanisms and aggregate formation.
Conclusion
As engineered nanoparticles are increasingly implemented as tools to study and alter biosystems, it is crucial to develop an understanding of how these nanoparticles interact with their biological surroundings. Accordingly, we have conducted a multimodal study to characterize protein corona formation in a biologically representative in-solution state. We focus on applying (GT)15-SWCNTs in the brain microenvironment, although the framework itself is generic to study protein corona composition on other nanoparticles and in other biofluids.
We find that while PNPs are largely agnostic to protein adsorption, (GT)15-SWCNTs show strong preferential binding of proteins involved in lipid transport, complement activation, and blood coagulation. Importantly, enrichment of complement proteins (especially C3) on ssDNA-SWCNTs is concerning due to the potential of nanoparticle opsonization and complement pathway activation. The selectivity of proteins binding to (GT)15-SWCNTs motivates either the rational design of sensors harnessing innate affinity for the SWCNT surface, or the development of SWCNT-based nanosensors passivated against detrimental biofouling. Additionally, (GT)15-SWCNTs show high binding of fibrinogen and low binding of albumin, despite the prevalence of albumin binding on other nanoparticles across a body of previous literature.[62] This raises cogent concern for the need to test nanotechnologies in blood plasma (with all protein constituents present) rather than blood serum (absent of fibrinogen), where fibrinogen may be a more important contributor to diminished in vivo efficacy than albumin.
We connect protein attributes that dictate protein-nanoparticle interactions to the thermodynamics and transient kinetics of protein-nanoparticle binding. Outer corona formation can be mitigated by tuning electrostatic interactions through nanoparticle design and by applying dynamic flow conditions (such as in circulating environments), whereas entropic considerations must be considered for the inner corona. Moreover, protein properties mediate adsorption differently in each biofluid, underscoring the complexity of corona formation. This phenomenon emphasizes that protein corona formation is a function of collective interactions at the nano-bio interface, rather than a property of isolated protein-nanomaterial interactions.
This work clarifies fundamental interactions for nanoscale systems in which development and optimization is done in vitro, with a desired application in vivo. Difficulties persist in the effective application of ssDNA-SWCNTs in brain imaging and delivery, including biofouling and the tendency of ssDNA-SWCNTs to aggregate in the presence of proteins. A more in-depth understanding of the protein corona could allow a priori prediction of biodistribution profiles and/or enable us to better understand these results in organisms. Elucidating protein corona composition, dynamics, structure, and driving forces that mediate nanoparticle-protein interactions will establish design considerations for nanosensor development and provide a framework for understanding how and why our engineered nanoparticles are affecting, and being affected by, complex bioenvironments.
Supplementary Material
Acknowledgements
Proteomic mass spectrometry data collection at the California Institute for Quantitative Biosciences (QB3)/College of Chemistry Mass Spectrometry Facility for LC-MS/MS was funded through grant 1S10OD020062-01. SAXS data collection at the SIBYLS beamline (bl12.3.1) was funded through DOE BER Integrated Diffraction Analysis Technologies (IDAT) program and NIGMS grant P30 GM124169-01, ALS-ENABLE. This work benefited from the use of the SasView application, originally developed under NSF award DMR-0520547. SasView contains code developed with funding from the European Union’s Horizon 2020 research and innovation programme under the SINE2020 project, grant agreement No 654000. We thank Behzad Rad, Blakely Tresca, and Ron Zuckerman for assistance with ITC at the Molecular Foundry, supported by the Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231. We thank Younghun Sung for TEM images acquired at the National Center for Electron Microscopy through proposal #5501, working with Dr. Karen Bustillo. We thank Matt Francis for Zetasizer Nano use. We acknowledge support of an NIH NIDA CEBRA award # R21DA044010 (to M.P.L.), a Burroughs Wellcome Fund Career Award at the Scientific Interface (CASI) (to M.P.L.), the Simons Foundation (to M.P.L.), a Stanley Fahn PDF Junior Faculty Grant with Award # PF-JFA-1760 (to M.P.L.), a Beckman Foundation Young Investigator Award (to M.P.L.), and a DARPA Young Investigator Award (to M.P.L.). M.P.L. is a Chan Zuckerberg Biohub investigator. R.L.P. and D.Y. acknowledge the support of NSF Graduate Research Fellowships (NSF DGE 1752814).
Footnotes
Supporting information for this article is given via a link at the end of the document
References
- [1].Liu Z, Tabakman S, Welsher K, Dai H, Nano Res. 2009, 2, 85–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Al-Jamal WT, Kostarelos K, Acc. Chem. Res 2011, 44, 1094–1104. [DOI] [PubMed] [Google Scholar]
- [3].Monopoli MP, Åberg C, Salvati A, Dawson KA, Nature Nanotechnology 2012, 7, 779–786. [DOI] [PubMed] [Google Scholar]
- [4].Nel AE, Mädler L, Velegol D, Xia T, Hoek EMV, Somasundaran P, Klaessig F, Castranova V, Thompson M, Nature Materials 2009, 8, 543–557. [DOI] [PubMed] [Google Scholar]
- [5].Monopoli MP, Walczyk D, Campbell A, Elia G, Lynch I, Baldelli Bombelli F, Dawson KA, J. Am. Chem. Soc 2011, 133, 2525–2534. [DOI] [PubMed] [Google Scholar]
- [6].Cedervall T, Lynch I, Lindman S, Bergg\a ard T, Thulin E, Nilsson H, Dawson KA, Linse S, Proceedings of the National Academy of Sciences 2007, 104, 2050–2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Karajanagi SS, Vertegel AA, Kane RS, Dordick JS, Langmuir 2004, 20, 11594–11599. [DOI] [PubMed] [Google Scholar]
- [8].Deng ZJ, Liang M, Monteiro M, Toth I, Minchin RF, Nature Nanotechnology 2011, 6, 39–44. [DOI] [PubMed] [Google Scholar]
- [9].Aggarwal P, Hall JB, McLeland CB, Dobrovolskaia MA, McNeil SE, Advanced Drug Delivery Reviews 2009, 61, 428–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Owens DE, Peppas NA, International Journal of Pharmaceutics 2006, 307, 93–102. [DOI] [PubMed] [Google Scholar]
- [11].Dutta D, Sundaram SK, Teeguarden JG, Riley BJ, Fifield LS, Jacobs JM, Addleman SR, Kaysen GA, Moudgil BM, Weber TJ, Toxicol Sci 2007, 100, 303–315. [DOI] [PubMed] [Google Scholar]
- [12].Gravely M, Safaee MM, Roxbury D, Nano Lett. 2019, 19, 6203–6212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Salvati A, Pitek AS, Monopoli MP, Prapainop K, Bombelli FB, Hristov DR, Kelly PM, Åberg C, Mahon E, Dawson KA, Nature Nanotechnology 2013, 8, 137–143. [DOI] [PubMed] [Google Scholar]
- [14].Tenzer S, Docter D, Kuharev J, Musyanovych A, Fetz V, Hecht R, Schlenk F, Fischer D, Kiouptsi K, Reinhardt C, Landfester K, Schild H, Maskos M, Knauer SK, Stauber RH, Nat Nanotechnol 2013, 8, 772–781. [DOI] [PubMed] [Google Scholar]
- [15].Röcker C, Pötzl M, Zhang F, Parak WJ, Nienhaus GU, Nature Nanotechnology 2009, 4, 577–580. [DOI] [PubMed] [Google Scholar]
- [16].Lundqvist M, Stigler J, Elia G, Lynch I, Cedervall T, Dawson KA, Proceedings of the National Academy of Sciences 2008, 105, 14265–14270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Walkey CD, Olsen JB, Song F, Liu R, Guo H, Olsen DWH, Cohen Y, Emili A, Chan WCW, ACS Nano 2014, 8, 2439–2455. [DOI] [PubMed] [Google Scholar]
- [18].Walkey CD, Olsen JB, Guo H, Emili A, Chan WCW, J. Am. Chem. Soc. 2012, 134, 2139–2147. [DOI] [PubMed] [Google Scholar]
- [19].Cai R, Ren J, Ji Y, Wang Y, Liu Y, Chen Z, Farhadi Sabet Z, Wu X, Lynch I, Chen C, ACS Appl. Mater. Interfaces 2020, 12, 1997–2008. [DOI] [PubMed] [Google Scholar]
- [20].Cox A, Andreozzi P, Dal Magro R, Fiordaliso F, Corbelli A, Talamini L, Chinello C, Raimondo F, Magni F, Tringali M, Krol S, Jacob Silva P, Stellacci F, Masserini M, Re F, ACS Nano 2018, DOI 10.1021/acsnano.8b03500. [DOI] [PubMed] [Google Scholar]
- [21].Hadjidemetriou M, Al-Ahmady Z, Mazza M, Collins RF, Dawson K, Kostarelos K, ACS Nano 2015, 9, 8142–8156. [DOI] [PubMed] [Google Scholar]
- [22].Kristensen K, Engel TB, Stensballe A, Simonsen JB, Andresen TL, Journal of Controlled Release 2019, 307, 1–15. [DOI] [PubMed] [Google Scholar]
- [23].Klibanov AL, Maruyama K, Torchilin VP, Huang L, FEBS Letters 1990, 268, 235–237. [DOI] [PubMed] [Google Scholar]
- [24].Sacchetti C, Motamedchaboki K, Magrini A, Palmieri G, Mattei M, Bernardini S, Rosato N, Bottini N, Bottini M, ACS Nano 2013, 7, 1974–1989. [DOI] [PubMed] [Google Scholar]
- [25].Gal N, Schroffenegger M, Reimhult E, J. Phys. Chem. B 2018, 122, 5820–5834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Cedervall T, Lynch I, Foy M, Berggård T, Donnelly SC, Cagney G, Linse S, Dawson KA, Angewandte Chemie International Edition 2007, 46, 5754–5756. [DOI] [PubMed] [Google Scholar]
- [27].Horbett TA, Journal of Biomedical Materials Research Part A 2018, 0, DOI 10.1002/jbm.a.36460. [DOI] [Google Scholar]
- [28].Milani S, Baldelli Bombelli F, Pitek AS, Dawson KA, Rädler J, ACS Nano 2012, 6, 2532–2541. [DOI] [PubMed] [Google Scholar]
- [29].Zhang J, Landry MP, Barone PW, Kim J-H, Lin S, Ulissi ZW, Lin D, Mu B, Boghossian AA, Hilmer AJ, Rwei A, Hinckley AC, Kruss S, Shandell MA, Nair N, Blake S, Şen F, Şen S, Croy RG, Li D, Yum K, Ahn J-H, Jin H, Heller DA, Essigmann JM, Blankschtein D, Strano MS, Nature Nanotechnology 2013, 8, 959–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Beyene AG, Delevich K, Bonis-O’Donnell JTD, Piekarski DJ, Lin WC, Thomas AW, Yang SJ, Kosillo P, Yang D, Prounis GS, Wilbrecht L, Landry MP, Science Advances 2019, 5, eaaw3108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Costa PM, Bourgognon M, Wang JT-W, Al-Jamal KT, Journal of Controlled Release 2016, 241, 200–219. [DOI] [PubMed] [Google Scholar]
- [32].Beyene AG, Alizadehmojarad AA, Dorlhiac G, Goh N, Streets AM, Král P, Vuković L, Landry MP, Nano Lett. 2018, 18, 6995–7003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Kruss S, Landry MP, Vander Ende E, Lima BMA, Reuel NF, Zhang J, Nelson J, Mu B, Hilmer A, Strano M, Journal of the American Chemical Society 2014, 136, 713–724. [DOI] [PubMed] [Google Scholar]
- [34].Galassi TV, Jena PV, Shah J, Ao G, Molitor E, Bram Y, Frankel A, Park J, Jessurun J, Ory DS, Haimovitz-Friedman A, Roxbury D, Mittal J, Zheng M, Schwartz RE, Heller DA, Science Translational Medicine 2018, 10, DOI 10.1126/scitranslmed.aar2680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Iverson NM, Barone PW, Shandell M, Trudel LJ, Sen S, Sen F, Ivanov V, Atolia E, Farias E, McNicholas TP, Reuel N, Parry NMA, Wogan GN, Strano MS, Nature Nanotechnology 2013, 8, 873–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Schöttler S, Becker G, Winzen S, Steinbach T, Mohr K, Landfester K, Mailänder V, Wurm FR, Nature Nanotechnology 2016, 11, 372–377. [DOI] [PubMed] [Google Scholar]
- [37].Godin AG, Varela JA, Gao Z, Danné N, Dupuis JP, Lounis B, Groc L, Cognet L, Nature Nanotechnology 2016, 12, 238–243. [DOI] [PubMed] [Google Scholar]
- [38].Hong G, Diao S, Chang J, Antaris AL, Chen C, Zhang B, Zhao S, Atochin DN, Huang PL, Andreasson KI, Kuo CJ, Dai H, Nature Photonics 2014, 8, 723–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Dinarvand M, Neubert E, Meyer D, Selvaggio G, Mann FA, Erpenbeck L, Kruss S, Nano Lett. 2019, DOI 10.1021/acs.nanolett.9b02865. [DOI] [PubMed] [Google Scholar]
- [40].Pinals RL, Yang D, Lui A, Cao W, Landry MP, J. Am. Chem. Soc. 2020, 142, 1254–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Thomas PD, Campbell MJ, Kejariwal A, Mi H, Karlak B, Daverman R, Diemer K, Muruganujan A, Narechania A, Genome Res. 2003, 13, 2129–2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Salvador-Morales C, Flahaut E, Sim E, Sloan J, Green MLH, Sim RB, Molecular Immunology 2006, 43, 193–201. [DOI] [PubMed] [Google Scholar]
- [43].Pondman KM, Sobik M, Nayak A, Tsolaki AG, Jäkel A, Flahaut E, Hampel S, Ten Haken B, Sim RB, Kishore U, Nanomedicine 2014, 10, 1287–1299. [DOI] [PubMed] [Google Scholar]
- [44].Lu N, Sui Y, Tian R, Peng Y-Y, Chem. Res. Toxicol. 2018, 31, 1061–1068. [DOI] [PubMed] [Google Scholar]
- [45].Ge C, Du J, Zhao L, Wang L, Liu Y, Li D, Yang Y, Zhou R, Zhao Y, Chai Z, others, Proceedings of the National Academy of Sciences 2011, 108, 16968–16973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Nucleic Acids Res 2019, 47, D506–D515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Findlay MR, Freitas DN, Mobed-Miremadi M, Wheeler KE, Environmental Science: Nano 2018, 5, 64–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Latour RA, J. Biomed. Mater. Res 2015, 103, 949–958. [DOI] [PubMed] [Google Scholar]
- [49].Wertz CF, Santore MM, Langmuir 2001, 17, 3006–3016. [Google Scholar]
- [50].Wertz CF, Santore MM, Langmuir 2002, 18, 706–715. [Google Scholar]
- [51].Jeng ES, Moll AE, Roy AC, Gastala JB, Strano MS, Nano Lett. 2006, 6, 371–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Schöppler F, Mann C, Hain TC, Neubauer FM, Privitera G, Bonaccorso F, Chu D, Ferrari AC, Hertel T, J. Phys. Chem. C 2011, 115, 14682–14686. [Google Scholar]
- [53].Tsapikouni TS, Missirlis YF, Colloids and Surfaces B: Biointerfaces 2007, 57, 89–96. [DOI] [PubMed] [Google Scholar]
- [54].Dutta AK, Rösgen J, Rajarathnam K, Methods Mol Biol 2015, 1229, 315–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Prozeller D, Morsbach S, Landfester K, Nanoscale 2019, 11, 19265–19273. [DOI] [PubMed] [Google Scholar]
- [56].Chen F, Wang G, Griffin JI, Brenneman B, Banda NK, Holers VM, Backos DS, Wu L, Moghimi SM, Simberg D, Nature Nanotechnology 2017, 12, 387–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Mildner DFR, Hall PL, J. Phys. D: Appl. Phys 1986, 19, 1535–1545. [Google Scholar]
- [58].Schaefer DW, Science 1989, 243, 1023–1027. [DOI] [PubMed] [Google Scholar]
- [59].Schmidt PW, J. Appl. Cryst 1991, 24, 414–435. [Google Scholar]
- [60].Bisker G, Dong J, Park HD, Iverson NM, Ahn J, Nelson JT, Landry MP, Kruss S, Strano MS, Nature Communications 2016, 7, 10241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Lazzari S, Nicoud L, Jaquet B, Lattuada M, Morbidelli M, Advances in Colloid and Interface Science 2016, 235, 1–13. [DOI] [PubMed] [Google Scholar]
- [62].Walkey CD, Chan WCW, Chem. Soc. Rev 2012, 41, 2780–2799. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.