

Abstract
Background.
We reported 3 novel non-synonymous single-nucleotide variants of Bcl2-associated athanogene 3 (BAG3) in African Americans with heart failure (HF) which are associated with 2-fold increase in cardiac events (HF hospitalization, heart transplant or death).
Methods.
We expressed BAG3 variants (P63A, P380S and A479V) via adenovirus-mediated gene transfer in adult left ventricular myocytes isolated from either wild-type (WT) or cardiac-specific BAG3 haplo-insufficient (cBAG3+/−) mice: the latter to simulate the clinical situation in which BAG3 variants are only found on one allele.
Results.
Compared to WT myocytes, cBAG3+/− myocytes expressed ~50% of endogenous BAG3 levels and exhibited decreased [Ca2+]i and contraction amplitudes after isoproterenol due to reduced L-type Ca2+ current. BAG3 repletion with WT BAG3 but not P380S, A479V or P63A/P380S variants restored contraction amplitudes in cBAG3+/− myocytes to those measured in WT myocytes, suggesting excitation-contraction (EC) abnormalities partly account for HF in patients harboring these mutants. Since P63A is near the WW domain (residues 21–55) and A479V is in the BAG domain (residues 420–499), we expressed BAG3 deletion mutants (Δ1–61 and Δ421–575) in WT myocytes and demonstrated that the BAG but not the WW domain was involved in enhancement of EC by isoproterenol.
Conclusion.
The BAG3 variants contribute to heart failure in African American patients partly by reducing myocyte excitation-contraction under stress, and that both the BAG and PXXP domains are involved in mediating β-adrenergic responsiveness in myocytes.
Keywords: BAG3, dilated cardiomyopathy, adenovirus-mediated gene transfer, excitation-contraction coupling, isolated adult cardiac myocytes
Introduction
B cell lymphoma 2 (Bcl2)-associated anthogene 3 (BAG3) is an evolutionarily conserved, stress-activated 575 amino acid protein predominantly expressed in the heart, skeletal muscle and many cancers1. BAG3 contains multiple protein-protein binding domains (Fig. 1) which confer pleotropic effects on the cell. In the heart, BAG3 regulates protein quality control (autophagy), apoptosis2, 3 and mitophagy4, maintains nuclear envelope integrity5 and sarcomere stability6, plays a key role in electrical impulse generation and propagation7, and facilitates adrenergic enhancement of excitation-contraction coupling8.
Figure 1. Consensus protein-protein binding domains in BAG3.

BAG3 serves as a co-chaperone for both the constituitively expressed and the inducible heat shock protein 70 (Hsc70/Hsp70): its BAG domain binding with the ATPase binding site of Hsc70/Hsp70. Hsp70-BAG3 interactions mediate chaperone assisted autophagy by shuttling client proteins along the cellular micro-tubular apparatus that are dedicated to eliminating mis-folded proteins33. The BAG domain also couples with the anti-apoptotic protein Bcl-2 resulting in inhibition of apoptosis32. Two highly conserved IPV (Ile-Pro-Val) motifs allow BAG3 to interact with the small heat shock proteins HspB8 and HspB6 to support macroautophagy, a process in which mis-folded or damaged proteins and organelles (e.g. mitochondria) are sequestered in autophagosomes and degraded39 whereas the second IPV domain binds to αβ-crystallin with the subsequent inhibition of protein aggregation40. The WW domain plays a pivotal role in chaperone-assisted selective autophagy or CASA, a tension-induced autophagy pathway that is essential for mechano-transduction in muscle and immune cells34, 35. The WW region also couples with the PXXP region to modify the three-dimensional structure of the protein. The proline-rich PXXP region also facilitates binding to the SH3 (Src homology 3)-containing protein phospholipase C-γ (PLC-γ) with subsequent stimulation of invasion, adhesion and migration of cancer cells36, 37 and the retrograde transport of mis-folded proteins to peri-nuclear aggresomes38.
BAG3 protein levels are reduced in swine hearts 12 weeks post-myocardial infarction (MI)1, in mouse hearts 18 weeks after transverse aortic constriction1 or after ischemia-reperfusion (I/R)3 or 11 weeks post-MI9, and in human hearts with severe left ventricular (LV) dysfunction secondary to idiopathic dilated cardiomyopathy (IDC) or ischemic heart disease10, 11. In addition, lowering BAG3 in wild-type (WT) mouse myocytes by shRNA-BAG3 (~54% reduction)8 or by engineering cardiac-specific BAG3 haplo-insufficient (cBAG3+/−) mouse (~51% reduction)2 resulted in a significant decrease in myocyte contraction amplitude (by 15.3% and 10.1%, respectively) in the presence of isoproterenol. When compared to WT littermates, cBAG3+/− mice maintained a normal ejection fraction (EF) at 8 weeks but suffered ~29% decline in EF by 10 weeks of age2. By contrast, repletion of the depleted BAG3 by virus-mediated gene transfer in mice post-MI9 or post-I/R3 dramatically improved the globally depressed myocardial function. These observations in animal models of heart failure (HF), human HF and molecular approaches to manipulate BAG3 levels are all consistent with the hypothesis that reduction in BAG3 levels plays an important pathogenetic role in contractile abnormalities in HF, and that restoring BAG3 levels in failing hearts improves contractile function. Whether the protein-protein binding domains of BAG3 are involved in the regulation of contractility is unknown.
Recently, by analyzing genomic DNA from 509 African American patients with dilated cardiomyopathy enrolled in 3 independent US studies [Genetic Risk Assessment of African Americans with Heart Failure (GRAHF), Intervention in Myocarditis and Acute Cardiomyopathy Trial-2 (IMAC-2), and Genetic Risk Assessment of Cardiac Events (GRACE)], we identified 3 novel non-synonymous single-nucleotide BAG3 variants (P380S, A479V or P63A) in African Americans (but not in patients of European ancestry) with HF11. The presence of any 1 of these BAG3 variants was associated with a worse outcome, as reflected by the combined outcome variable of HF hospitalization, heart transplant or death11. Two important observations are that the BAG3 variants were found on one allele and patients who harbored the P63A variant also carried the P380S variant. Since P63A is near the WW domain (residues 21–55), P380S is in the PXXP domain (residues 302–412) and A479V is in the BAG domain (residues 420–499)(Fig. 1), we took advantage of our cBAG3+/− mouse (to simulate WT BAG3 on one allele) and expressed either WT BAG3 or the BAG3 variants (P380S, P479V or P63A/P380S) to test if the BAG3 variants behaved as loss-of-function mutants: the readout being reduced β-adrenergic enhancement of EC coupling. We also expressed BAG3 mutants in which either the WW or the BAG domain was deleted to corroborate our findings with non-synonymous single-nucleotide BAG3 variants.
Methods
Generation of cBAG+/− mouse and animal care.
Cardiac-specific haplo-insufficient BAG3 mice were generated as described previously2. Adult cBAG3+/− mice used in this study were 8 weeks old at which time they had normal EF when compared to their WT littermates2. Mice were housed and fed on a 12:12h light-dark cycle at the Temple University Animal Facility supervised by full-time veterinarian staff members. Standard care was provided to all mice used for experiments.
Isolation, adenoviral infection, and culture of adult murine cardiac myocytes.
Cardiac myocytes were isolated from the septum and LV free wall of WT and cBAG3+/− mice (8 wks old) according to the protocol of Zhou et al.12, and plated on laminin-coated glass coverslips13. Myocytes were used either on the same day (within 2–6 hr after isolation) or after 24 hr of culture.
To express exogenous genes via adenovirus (Adv)-mediated gene transfer, two hours after isolation, myocytes were infected with replication-deficient Adv expressing either green fluorescent protein (GFP)(Adv-GFP, 6.0 × 106 pfu/ml), WT BAG3 (Adv-BAG3, 4.0 × 106 pfu/ml), P380S (Adv-380, 4.0 × 106 pfu/ml), A479V (Adv-479, 4.2 × 106 pfu/ml), P63A/P380S (Adv-63/380, 4.8 × 106 pfu/ml), WW domain deletion (residues 62–575; abbreviated as Adv-575, 3.75 × 106 pfu/ml) or BAG domain deletion (residues 1–420; abbreviated as Adv-420, 3.7 × 106 pfu/ml) mutants in 1 ml of fetal bovine serum-free Eagle minimal essential medium (MEM) containing 0.2% bovine serum albumin (BSA), creatine (5 mM), carnitine (2 mM), taurine (5 mM), NaHCO3 (4.2 mM), penicillin (30 mg/L), gentamicin (4 mg/L), insulin-transferrin-selenium supplement, and 2,3-butanedione monoxime (BDM, 10 mM) for 3 hours. An additional 1 ml of MEM (with same supplements) was then added, and myocytes were cultured for 24h before experiments14, 15. Before measurements of myocyte function, cells were bathed with MEM without BDM and returned to the incubator (37°C) for 30 min. We have previously demonstrated that under our culture conditions, adult mouse LV myocytes cultured for up to 48h maintained rod-shape morphology, transverse-tubule organization, and normal contractile function; and that infection with Adv-GFP had no effect on single myocyte contractility when compared to uninfected myocytes14.
Myocyte shortening measurements.
LV myocytes (freshly isolated or cultured) adherent to laminin-coated glass coverslips were bathed in 0.6 ml of air- and temperature equilibrated (37°C), HEPES-buffered (20 mM, pH 7.4) medium 199 containing 1.8 mM extracellular Ca2+ concentration ([Ca2+]o). Myocytes were electrically paced to contract at 2 Hz and myocyte motion was captured by charge-coupled device video camera (Ionoptix; Milton, MA). Isoproterenol (Iso), when indicated, was present at 1 μM. Myocyte contraction dynamics was analyzed off-line with edge-detection algorithm as previously described13–15.
Measurement of intracellular Ca2+ concentration ([Ca2+]i) in cardiac myocytes.
Fura-2 loaded (0.67 μM fura-2 AM, Molecular Probes, Eugene, OR; 15 min, 37°C) LV myocytes attached to laminin-coated coverslips were incubated in medium 199 containing 1.8 mM [Ca2+]o and field-stimulated to contract (2 Hz, 37°C). Myocytes were exposed to excitation light (360 and 380 nm) only during data acquisition. Epifluorescence (510 nm) was measured in steady-state twitches both before after addition of Iso (1 μM). Daily calibration of fura-2 signals and [Ca2+]i analyses were performed as previously described13, 14, 16–19.
L-type Ca2+ current (ICa) measurements.
Whole cell patch-clamp recordings were performed at 30°C as described previously13, 20. Pipette diameter was 4 – 6 μm and pipette resistance was 0.8 to 1.4 MΩ when filled with standard internal solution. For ICa measurements, pipette solution contained (in mM): CsCl 110, TEA.Cl 20, HEPES 10, MgATP 5, and EGTA 10; pH 7.2. Extracellular bathing solution contained (in mM): N-methyl-D-glucamine 137, CsCl 5.4, CaCl2 2, MgSO4 1.3, HEPES 20, 4-aminopyridine 4, and glucose 15; pH 7.4. Holding potential was at −90 mV21. To ensure steady-state sarcoplasmic reticulum (SR) Ca2+ loading, 6 conditioning pulses (from −70 to 0 mV, 100 ms, 2 Hz) were delivered to the myocyte before the arrival of each test pulse (from −90 to +50 mV, 10 mV increments, 60 ms)21. Leak-subtracted inward currents were used in analysis for ICa amplitudes. Inward currents obtained under these conditions were blocked by 1 μM verapamil8, 21. ICa was normalized to membrane capacitance (Cm) before comparison between WT and cBAG3+/− myocytes, and among Adv-GFP, Adv-BAG3, Adv-420 and Adv-575 myocytes.
Statistics.
All results are expressed as means ± SE. For analysis of contraction amplitudes as a function of mouse (WT vs. cBAG3+/−), single nucleotide BAG3 variant and Iso, three-way ANOVA was used. For analysis of [Ca2+]i transient, contraction and maximal ICa (at 0 mV) amplitudes as a function of BAG3 deletion mutant and Iso or as a function of mouse (WT vs. cBAG3+/−) and Iso, two-way ANOVA was used. A commercially available software package (JMP version 12; SAS Institute, Cary, NC) was used. In all primary analyses, p≤0.05 was taken to be statistically significant. In subgroup analyses, the Bonferroni adjustment (statistical significance at p≤0.05/n, where n=number of subgroup comparisons) was applied.
Results
[Ca2+]i transient, contraction and maximal ICa amplitudes are lower in cBAG3+/− myocytes after isoproterenol.
We have previously demonstrated that shRNA-mediated reduction in BAG3 (by ~54%) resulted in lower [Ca2+]i transient, contraction and ICa amplitudes after Iso stimulation8. Similarly, in cBAG3+/− myocytes in which BAG3 levels are reduced by ~51%2, baseline [Ca2+]i transient and contraction amplitudes were similar but were significantly lower after Iso (Fig. 2; for [Ca2+]i transient: p=0.0077, group × Iso interaction effect; for contraction: p=0.0003, group × Iso interaction effect). Reduced maximal [Ca2+]i transient amplitude in cBAG3+/− myocytes was due to lower systolic [Ca2+]i (p=0.0028, group × Iso interaction effect) since diastolic [Ca2+]i (p=0.9728, group × Iso interaction effect) were similar (Fig. 2G). The t1/2 of [Ca2+]i transient decline, a measure of SR Ca2+ uptake22, 23, was significantly (p=0.0425; group effect) prolonged in cBAG3+/− when compared to WT myocytes (Fig. 2H), indicating that SR Ca2+-ATPase activity was lower in cBAG3+/− myocytes. Lower systolic [Ca2+]i and [Ca2+]i transient amplitudes after Iso in cBAG3+/− myocytes were largely due to reduced ICa amplitudes (Fig. 3; p=0.0021, group × Iso interaction effect).
Figure 2. Abnormal Ca2+ homeostasis and reduced contraction in cardiac-specific haplo-insufficient BAG3 murine myocytes.
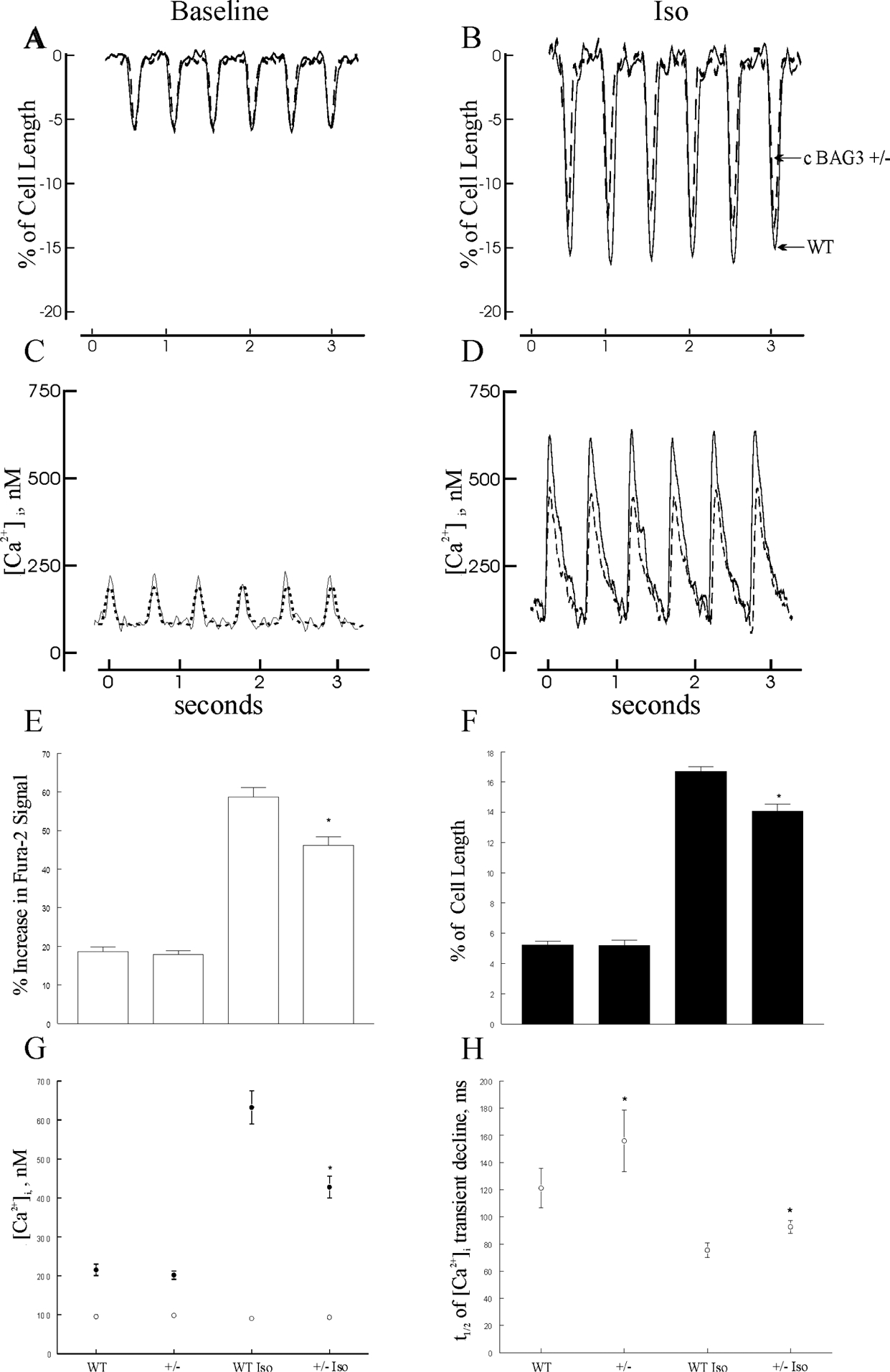
[Ca2+]i and contraction were measured in field-stimulated (2 Hz, 37°C) LV myocytes isolated from 8-wk old WT and cBAG3+/− mice (Methods). Representative traces of contraction at baseline (A) and after 1 μM isoproterenol (Iso)(B), and [Ca2+]i transients at baseline (C) and after Iso (D) of WT (solid lines) and cBAG3+/− (dashed lines) myocytes are shown. E, G and H: Means ± SE of [Ca2+]i transient amplitudes (E), systolic and diastolic [Ca2+]i (G) and t1/2 of [Ca2+]i transient decline (H) from 11 WT and 13 cBAG3+/− myocytes (2 mice each) at baseline and 15 WT and 19 cBAG3+/− myocytes (2 mice each) after 1 μM Iso. For systolic and diastolic [Ca2+]i, error bars are not shown if they fall within the boundaries of the symbols. F: Means ± SE of contraction amplitudes from 21 WT and 16 cBAG3+/− myocytes (3 WT and 2 cBAG3+/− mice) at baseline and 19 WT and 12 cBAG3+/− myocytes after Iso. For E, F, G: *p<0.008, group (WT vs. cBAG3+/−) × Iso interaction effect. For H: *p=0.0425, WT vs. cBAG3+/−.
Figure 3. Reduced L-type Ca2+ current (ICa) in cardiac-specific haplo-insufficient BAG3 myocytes.

ICa was measured in myocytes isolated from 8-wk old WT and cBAG3+/− myocytes (Methods). A and B: Representative traces of ICa (0 mV) from WT and cBAG3+/− myocytes before (A) and after (B) 1 μM Iso. C: IV relationship of ICa in WT (▄, baseline; □, after Iso; n=7 cells from 3 mice) and cBAG3+/− (●, baseline; ○, after Iso; n=5 cells from 3 mice) myocytes. 3-way ANOVA indicates significant group (WT vs. cBAG3+/−, p<0.0001), voltage (p<0.0001), Iso (p<0.0001), group × voltage (p=0.0425) and group × Iso (p=0.0021) interaction effects.
WT BAG3, but not P380S, A479V or P63A/P380S BAG3 mutant restores maximal contraction amplitude in cBAG3+/− myocytes.
To simulate the clinical observation that the 3 non-synonymous single-nucleotide BAG3 variants detected in African Americans with heart failure were heterozygous, we expressed GFP, WT BAG3 or its variants in either cBAG3+/− or WT myocytes to test if myocyte excitation-contraction in response to β-adrenergic stimulation was affected differently. In cBAG3+/− myocytes expressing GFP, contraction amplitudes were similar to those observed in WT-GFP myocytes under basal conditions (Fig. 4A). After Iso, contraction amplitude in cBAG3+/−-GFP myocytes was significantly lower than that measured in WT-GFP myocytes (Fig. 4B; p=0.0090, group × Iso interaction effect). The observation that contraction amplitudes were similar under basal conditions but lower in cBAG3+/−-GFP myocytes after Iso closely mimicked those measured in uninfected WT and cBAG3+/− myocytes (Fig. 2), indicating adenovirus infection followed by short-term culture did not affect the intrinsic differences between WT and cBAG3+/− myocytes.
Figure 4. Non-synonymous, single nucleotide BAG3 variants in African-American patients with heart failure do not enhance β-adrenergic responsiveness compared to WT BAG3.
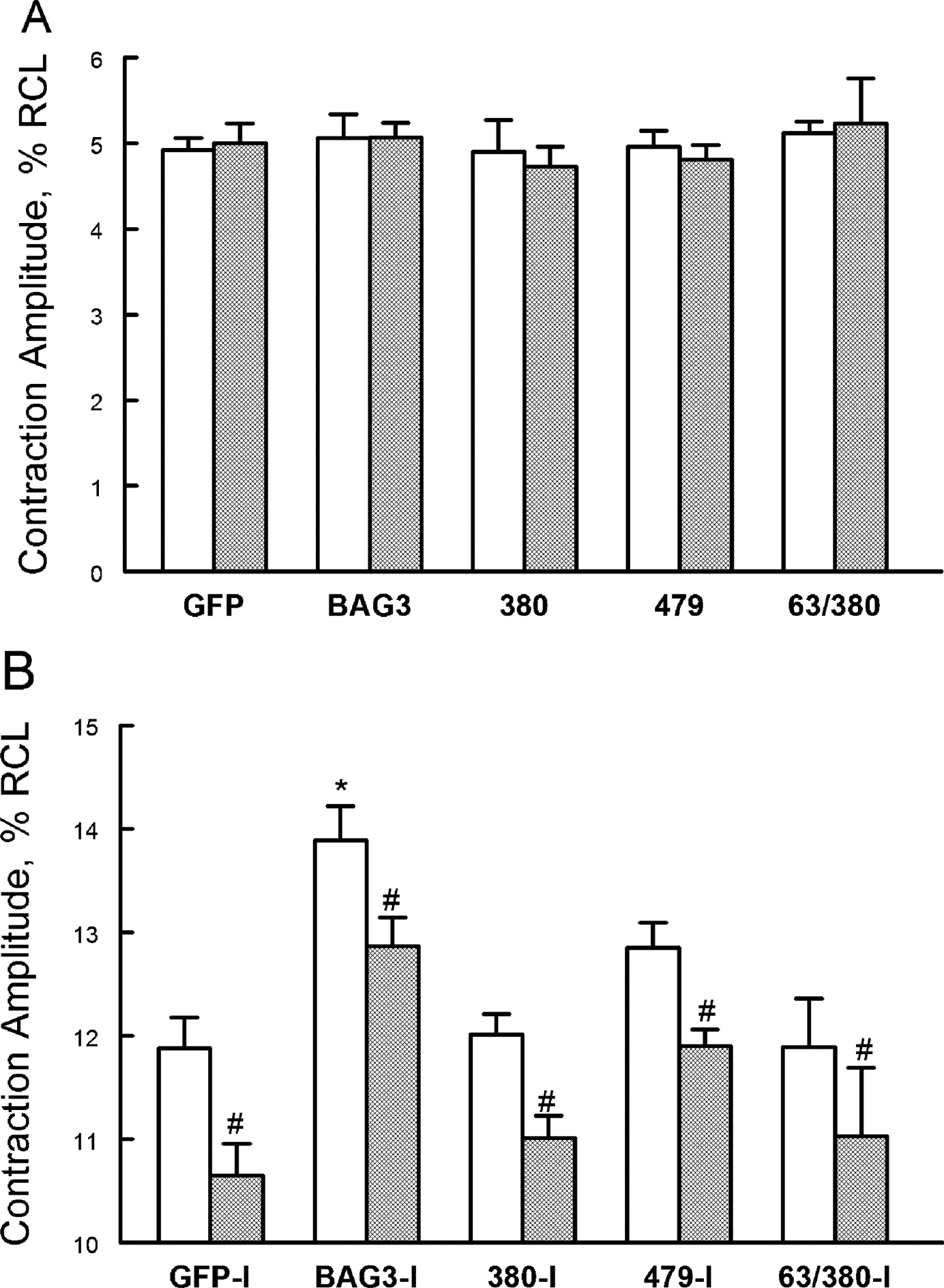
LV myocytes isolated from 8-wk old WT (open bars) and cBAG3+/− (hatched bars) mice were infected with adenovirus (Adv) expressing GFP, WT BAG3, P380S, A479V or P63A/P380S BAG3 variants and cultured for 24h before contraction (2 Hz, 37°C) measurements, both before and after 1 μM Iso (Methods). A. Means ± SE of baseline contraction amplitudes [% resting cell length (RCL)] measured in WT and cBAG3+/− myocytes. B. Means ± SE of contraction amplitudes after Iso. Note the difference in Y-axis scales. For WT-GFP, there are 20 myocytes at baseline and 15 myocytes after Iso (n=6 mice); for cBAG3+/−-GFP, there are 19 myocytes at baseline and 17 myocytes after Iso (n=6 mice); for WT-BAG3, there are 14 myocytes at baseline and 14 myocytes after Iso (n=5 mice); for cBAG3+/−-BAG3, there are 16 myocytes at baseline and 21 myocytes after Iso (n=5 mice); for WT-P380S, there are 7 myocytes at baseline and 8 myocytes after Iso (n=3 mice); for cBAG3+/−-P380S, there are 8 myocytes at baseline and 8 myocytes after Iso (n=2 mice); for WT-A479V, there are 17 myocytes at baseline and 22 myocytes after Iso (n=3 mice); for cBAG3+/−-A479V, there are 18 myocytes at baseline and 19 myocytes after Iso (n=3 mice); for WT-P63A/P380S, there are 7 myocytes at baseline and 7 myocytes after Iso (n= 1 mouse); and for cBAG3+/−-P63A/P380S, there are 5 myocytes at baseline and 6 myocytes after Iso (n=1 mouse). 3-way ANOVA indicate significant group (WT vs. cBAG3+/−; p=0.0002), Adv (p<0.0001), Iso (p<0.0001) and group × Iso interaction (p=0.0003) effects. *p<0.002, BAG3 vs. GFP, P380S, A479V or P63A/P380S by subgroup analysis. #p=0.0003, group (WT vs. cBAG3+/−) × Iso interaction effects.
To analyze contraction amplitude data (Fig. 4) while taking into account the effects of WT and cBAG+/− mice (group), adenovirus expressing GFP, BAG3 or its variants (Adv), and isoproterenol (Iso), we used 3-way ANOVA. The primary analysis on 20 sets of data (WT-GFP ± Iso, cBAG3+/−-GFP ± Iso, etc.) indicates significant group (p=0.0002), Adv (p<0.0001), Iso (p<0.0001) and group × Iso interaction (p=0.0003) effects; indicating contraction amplitudes were lower in cBAG3+/− myocytes in the presence of Iso, and that expressing GFP, BAG3 or its variants had different effects on contraction amplitudes. To further dissect whether the effects of expressing WT BAG3 on contraction amplitude were different from GFP or BAG3 variants, we performed subgroup analysis (BAG3 vs. GFP, BAG3 vs. P380S, BAG3 vs. P63A/P380S, and BAG3 vs. A479V) with p≤0.0125 taken to be statistically significant (Bonferroni adjustment). The analysis indicates that the effects of expressing WT BAG3 on contraction are significantly different than those of expressing GFP (p<0.0001), P380S (p<0.0001), P63A/P380S (p=0.0012) or A479V (p=0.0007). Stated differently, the 3 BAG3 variants tested behaved like loss-of-function mutants in terms of enhancing excitation-contraction in the presence of isoproterenol.
BAG but not WW domain is required to endow β-adrenergic responsiveness in adult mouse LV myocytes.
The results from non-synonymous single-nucleotide BAG3 variants implicate WW (P63A), PXXP (P380S) and BAG (A479V) domains as important in regulation of excitation-contraction by BAG3. To test this hypothesis, we engineered 2 BAG3 mutants in which either the WW (Adv-575) or BAG (Adv-420) domain was deleted and expressed them in WT myocytes. Since there are 5 subgroup comparisons (GFP vs. BAG3, GFP vs. 420, GFP vs. 575, BAG3 vs. 420 and BAG3 vs. 575), p≤0.01 was taken to be statistically significant. Compared to WT-GFP myocytes, WT-BAG3 myocytes contracted more vigorously in the presence of Iso (Fig. 5; p=0.0022, Adv × Iso interaction effect). Enhanced contraction observed in Iso-treated WT-BAG3 myocytes as compared to WT-GFP myocytes was due to increased systolic [Ca2+]i (Fig. 6; p=0.0052, Adv × Iso interaction effect), resulting in larger [Ca2+]i transient amplitudes (Fig. 6; p=0.0009, Adv × Iso interaction effect). Augmented [Ca2+]i transient amplitudes in Iso-stimulated WT-BAG3 myocytes were primarily due to larger ICa (Fig. 7; p=0.01, Adv × Iso interaction effect), resulting in increased Ca2+ influx during the action potential and higher systolic [Ca2+]i. Compared to WT-BAG3 myocytes, deletion of the BAG (Adv-420) domain abolished the enhanced β-adrenergic response in terms of contraction (Fig. 5; p=0.0008, Adv × Iso interaction effect), [Ca2+]i transient (Fig. 6; p=0.0071, Adv × Iso interaction effect) and ICa (Fig. 7; p=0.0013, Adv × Iso interaction effect) amplitudes; indicating the BAG domain is intimately involved in Ca2+ homeostasis in adult cardiac myocytes. By contrast, deletion of the WW domain (Adv-575) did not negatively impact enhanced β-adrenergic responsiveness in terms of contraction (Fig. 5; p=0.9022, Adv × Iso interaction effect), [Ca2+]i transient (Fig. 6; p=0.2907, Adv × Iso interaction effect) and ICa (Fig. 7; p=0.0321, Adv × Iso interaction effect) amplitudes.
Figure 5. BAG3 but not WW domain is required for enhanced contraction amplitudes after isoproterenol.
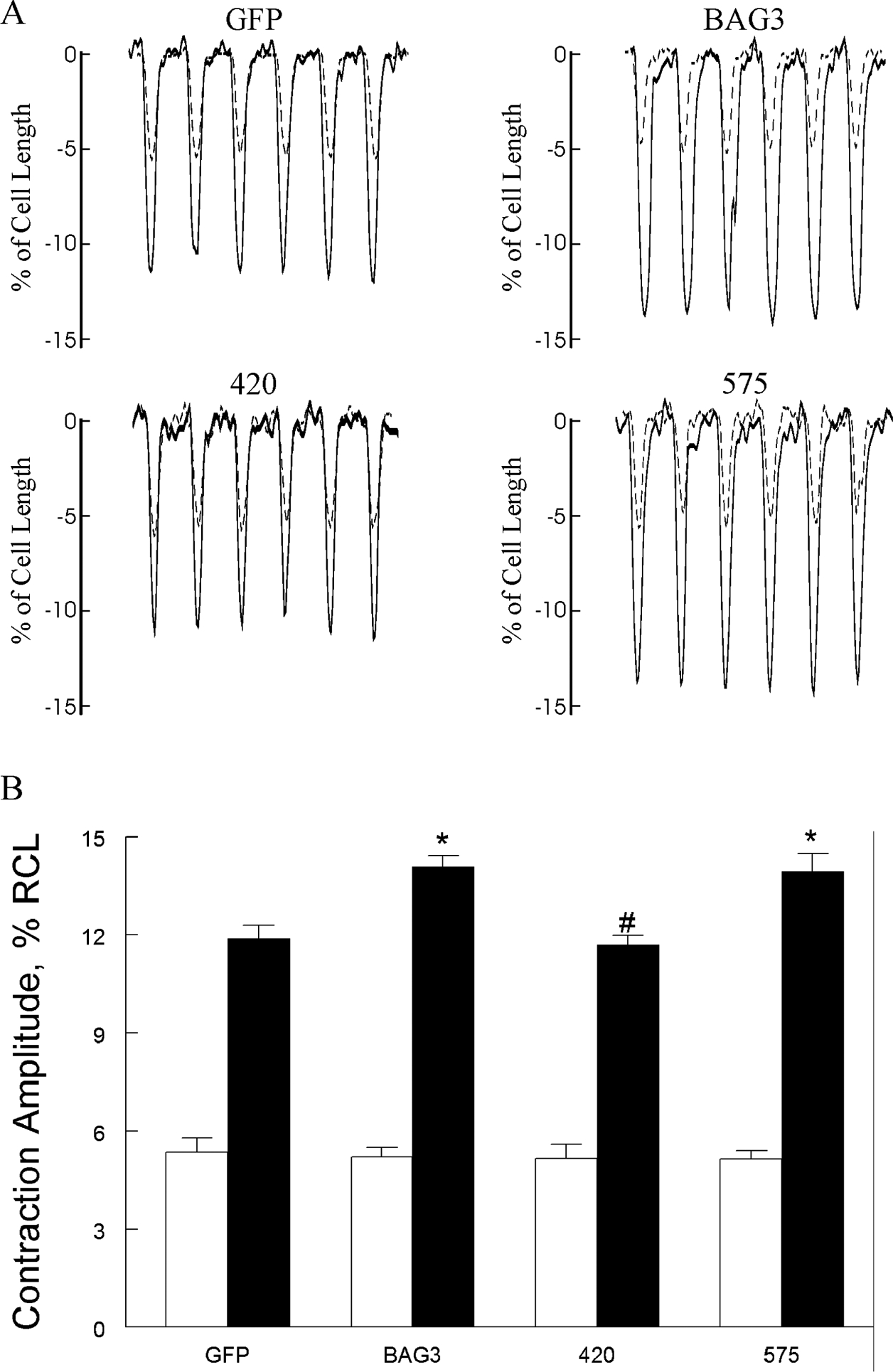
LV myocytes isolated from 8 wk-old WT mice were infected with adenovirus (Adv) expressing GFP, WT BAG3, WW domain (residues 62 to 575 and abbreviated as 575) or BAG domain (residues 1 to 420 and abbreviated as 420) deletion mutants and cultured for 24h before contraction (2 Hz, 37°C) measurements (Methods). A. Representative traces of contraction for GFP, BAG3, 420 and 575 myocytes, both before (dotted lines) and after (solid lines) 1 μM isoproterenol (Iso). B. Means ± SE of contraction amplitudes [% resting cell length (RCL)] before (open bars) and after Iso (solid bars). For GFP, there are 11 myocytes at baseline and 13 myocytes after Iso (n=4 mice); for BAG3, there are 14 myocytes at baseline and 12 myocytes after Iso (n=5 mice); for 420, there are 10 myocytes at baseline and 14 myocytes after Iso (n=2 mice); and for 575, there are 9 myocytes at baseline and 12 myocytes after Iso (n=3 mice). 2-way ANOVA of all 4 groups indicate significant Adv (p=0.0021), Iso (p<0.0001) and Adv × Iso (p=0.0011) interaction effects. *p<0.02, GFP vs. BAG3 or GFP vs. 575 by subgroup analysis (p≤0.01 is statistically significant after Bonferroni adjustment); #p<0.001, BAG3 vs. 420 by subgroup analysis.
Figure 6. BAG3 but not WW domain is required for higher systolic [Ca2+]i and larger [Ca2+]i transient amplitudes after isoproterenol.

LV myocytes isolated from 8 wk-old WT mice were infected with adenovirus (Adv) expressing GFP, WT BAG3, WW domain (575) or BAG domain (420) deletion mutants and cultured for 24h before [Ca2+]i (2 Hz, 37°C) measurements (Methods). A. Representative traces of [Ca2+]i transients for GFP, BAG3, 420 and 575 myocytes, both at baseline (dotted lines) and after 1 μM Iso (solid lines). B. Means ± SE of systolic (●) and diastolic (○) [Ca2+]i at baseline and after Iso (I). Error bars are not shown if they fall within the boundaries of the symbols. For GFP, there are 13 myocytes at baseline and 10 myocytes after Iso (n=2 mice); for BAG3, there are 9 myocytes at baseline and 14 myocytes after Iso (n=3 mice); for 420, there are 9 myocytes at baseline and 9 myocytes after Iso (n=2 mice); and for 575, there are 12 myocytes at baseline and 13 myocytes after Iso (n=3 mice). 2-way ANOVA of systolic [Ca2+]i from all 4 groups indicate significant Adv (p=0.0001), Iso (p<0.0001) and Adv × Iso (p=0.0006) interaction effects. *p≤0.005, GFP vs. BAG3 or GFP vs. 575 by subgroup analysis (p≤0.01 is statistically significant after Bonferroni adjustment). C. Means ± SE of [Ca2+]i transient amplitudes (% increase in Fura2 signal) at baseline (open bars) and after Iso (solid bars). 2-way ANOVA of all 4 groups indicate significant Adv (p=0.0002), Iso (p<0.0001) and Adv × Iso (p=0.0003) interaction effects. *p≤0.0009, GFP vs. BAG3 or GFP vs. 575 by subgroup analysis (p≤0.01 is statistically significant after Bonferroni adjustment).
Figure 7. BAG3 but not WW domain is required for enhanced L-type Ca2+ current (ICa) after isoproterenol.
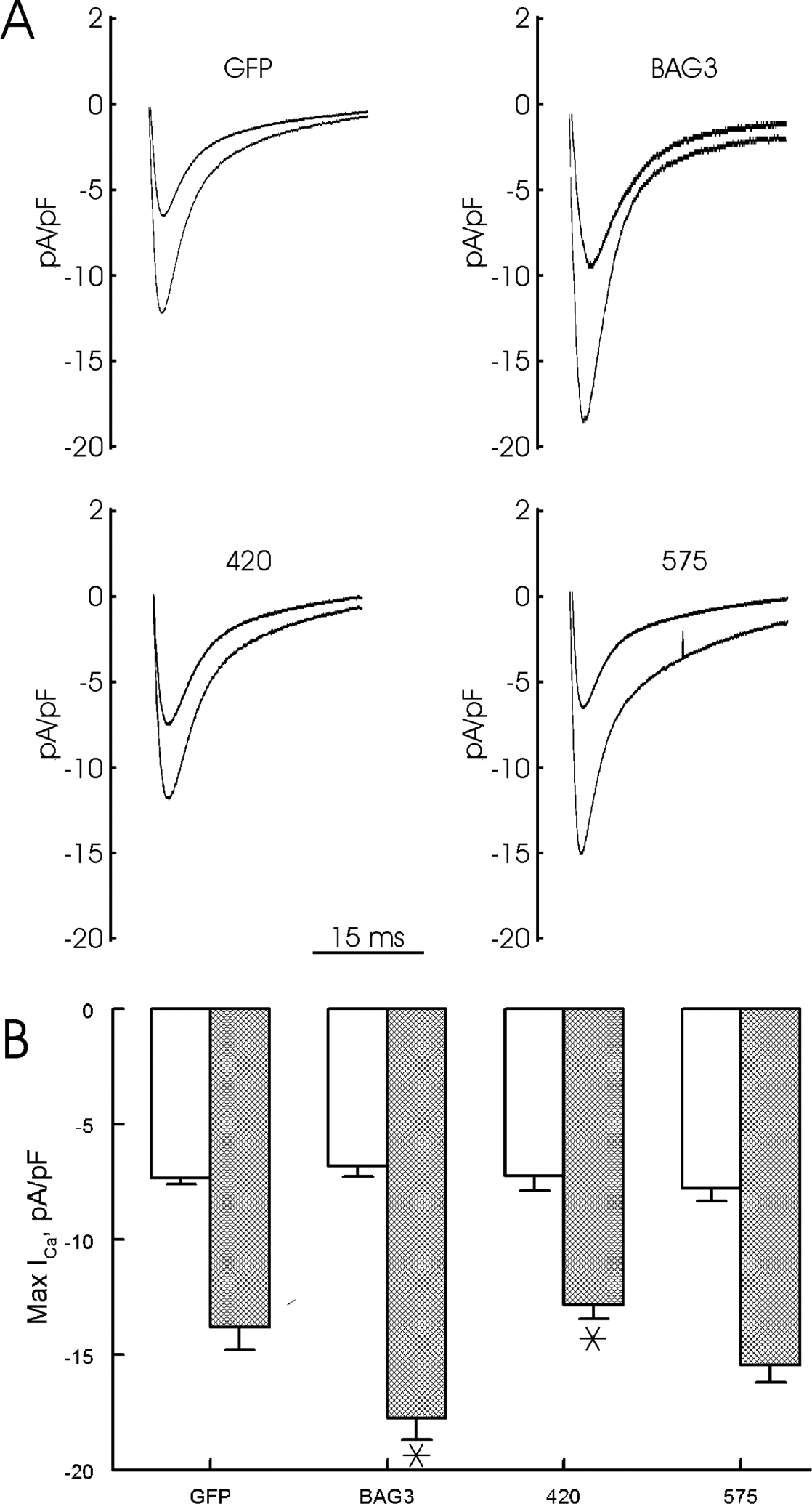
LV myocytes isolated from 8 wk-old WT mice were infected with adenovirus (Adv) expressing GFP, WT BAG3, WW domain (575) or BAG domain (420) deletion mutants and cultured for 24h before ICa measurements (Methods). A. Representative ICa traces at 0 mV for GFP, BAG3, 420 and 575 myocytes, both at baseline (black) and after 1 μM Iso (red). B. Means ± SE of maximal ICa amplitudes (0 mV) before (open bars) and after Iso (hatched bars). For GFP, there are 7 myocytes each at baseline and after Iso (n=5 mice); for BAG3, there are 4 myocytes each at baseline and after Iso (n=3 mice); for 420, there are 6 myocytes each at baseline and after Iso (n=3 mice); and for 575, there are 6 myocytes each at baseline and after Iso (n=3 mice). 2-way ANOVA of all 4 groups indicate significant Adv (p=0.0158), Iso (p<0.0001) and Adv × Iso (p=0.0078) interaction effects. *p<0.01, BAG3 vs. GFP or BAG3 vs. 420 by subgroup analysis (p≤0.01 is statistically significant after Bonferroni adjustment).
Discussion
Recent studies have demonstrated the increasing importance of BAG3 in maintaining cardiac health and function. Specifically, in the heart BAG3 regulates autophagy, apoptosis2, 3, mitophagy4 and [Ca2+]i homeostasis8, maintains sarcomere stability6 and nuclear membrane integrity5, and facilitates electrical signal propagation7. The pathophysiological significance of BAG3 in the heart is highlighted by the observation that patients with severe cardiac failure from dilated cardiomyopathy (both ischemic and non-ischemic causes) have marked BAG3 depletion 10, 11. Multiple animal models of heart failure corroborate the clinical observation that BAG3 levels were severely reduced1, 3, 9. Mechanistically, downregulation of BAG3 in cardiac myocytes either by shRNA3, 7, 8 or by heterozygous gene deletion2 resulted in decreased autophagy flux, increased apoptosis, reduced myocyte L-type Ca2+ current, [Ca2+]i transient and contraction amplitudes, prolonged action potential duration, decreased electrical impulse propagation, and global LV dysfunction. The clinical observations and experimental results support the exciting hypothesis that BAG3 is a novel and rational target for therapeutic intervention in patients with heart failure.
In 2009, Selcen was the first to describe 3 children harboring a non-synonymous single nucleotide BAG3 variant (P209L) with severe myofibrillar degeneration, skeletal muscle weakness, giant axon disease and a hypertrophic/restrictive cardiomyopathy24. Subsequently, cardiomyopathies due to BAG3 deletion, truncation and other non-synonymous single nucleotide polymorphism (SNP) mutants (which can lead to BAG3 haplo-insufficiency) have been reported10,11,25–30. Indeed, BAG3 has been proposed to be a major cardiomyopathy locus27 and may be efficacious in gene rescue in hereditary BAG3 cardiomyopathy.
The first major finding of our current study is that BAG3 haplo-insufficiency resulted in decreased β-adrenergic responsiveness in terms of lower systolic [Ca2+]i due to reduced ICa in isolated cardiac myocytes, leading to decreased myocyte contraction amplitudes. This observation mirrors the observations in myocytes in which BAG3 was reduced by shRNA8. We have previously demonstrated that in BAG3-deficient cardiac myocytes, addition of either forskolin (10 μM; directly activates adenylyl cyclase) or dibutyrl-cAMP (5 mM; directly activates protein kinase A) restores maximal ICa amplitudes to those measured in BAG3-replete myocytes8. These observations suggest that the defects in β-adrenergic signaling in BAG3-deficient myocytes occur proximal to adenylyl cyclase. In addition, we demonstrated that BAG3, β1-adrenergic receptor (β1AR), α1c-subunit of the L-type Ca2+ channel (Cav1.2) and phospholemman (PLM) co-immunoprecipitate together as a macromolecular complex.8 We speculated that there is a specific pool of β1AR that complexes with BAG3 and Cav1.2 and facilitates β1-adrenergic signaling to increase ICa. In this light, recent elegant studies by Marx’s group indicate that the protein-protein interaction between the β subunit of Cav1.2 and the α-interaction domain (AID) of the α1c-subunit of Cav1.2, while not affecting basal ICa, is absolutely required for the β-adrenergic modulation of ICa31. Therefore mechanistically, when BAG3 levels are low (either genetically or in heart failure), there is less β-subunit interaction with the α1c-subunit of Cav1.2, leading to normal baseline ICa but reduced β-adrenergic induced ICa increase. An alternative hypothesis is that since PLM regulates ICa in cardiac myocytes21, and since PLM, BAG3, β1AR and Cav1.2 form a macromolecular complex8, reduced BAG3 levels may alter interaction of PLM with Cav1.2, thereby affecting ICa amplitudes.
Our most recent contribution to the role of BAG3 in dilated cardiomyopathy is the identification of 3 novel non-synonymous BAG3 SNPs in African American patients with heart failure but not found in patients of European ancestry11. Thus the second major finding of our current study is that these novel BAG3 SNPs (P380S, A479V and P63A/P380S) behave like loss-of-function mutants in terms of enhancing myocyte contraction amplitudes after β-adrenergic stimulation. In HF patients in which circulating catecholamine levels are elevated, the inability to respond properly to β-adrenergic signals in African American HF patients harboring these BAG3 SNPs may partly account for the LV contractile dysfunction.. An important inference from these loss-of-function BAG3 SNPs is that the WW (P63A), PXXP (P380S) and BAG (A479V) domains may be involved in mediating β-adrenergic response in terms of augmenting ICa.
When viewed purely in the context of Ca2+ homeostasis, reduced β-adrenergic responsiveness in myocytes that are either BAG3-deficient or harboring one of these BAG3 variants may very well represent a protective mechanism to minimize Ca2+ overload under adrenergic stress. Indeed, this is one of the mechanisms by which β-adrenergic blockers exert their beneficial effects in HF patients. In addition, other than modulating excitation-contraction coupling, BAG3 also regulates autophagy, apoptosis2, 3, mitophagy4, maintains sarcomere stability6 and nuclear membrane integrity5 and facilitates electrical signal propagation7, the worse clinical outcomes observed in African American patients harboring these BAG3 variants may relate to a combination of these pleotropic effects of BAG311.
The third major finding is that the BAG but not WW domain is absolutely required for the β-adrenergic receptor induced increase in ICa, systolic [Ca2+]i and myocyte contraction amplitudes. The BAG domain is known to inhibit apoptosis by binding to Bcl-232 and mediate autophagy33(Fig. 1). The WW domain is involved in chaperone-assisted selective autophagy or CASA34, 35 and interacts with the PXXP region (Fig. 1). The PXXP domain binds to phospholipase C-γ and promotes metastasis of cancer cells36, 37 and the retrograde transport of mis-folded proteins to peri-nuclear aggresomes38. We speculate that the PXXP (P380S) and the BAG (A479V and deletion mutant) domains may be involved in the interaction of BAG3 to the L-type Ca2+ channel to implement β-adrenergic increases in ICa.
In summary, low BAG3 levels observed in acquired and some forms of hereditary heart failure results in reduced L-type Ca2+ current, lower systolic [Ca2+]i and decreased contraction amplitudes in cardiac myocytes under adrenergic stress, thereby further aggravating left ventricular dysfunction. In addition, the three novel non-synonymous single nucleotide BAG3 variants detected in African American patients with heart failure behave like loss-of-function mutants in terms of augmentation of myocyte contraction after β-adrenergic stimulation. Finally, the PXXP and BAG but not WW domains of BAG3 are required to implement β-adrenergic increases in L-type Ca2+ current, systolic [Ca2+]i and contraction amplitudes. We speculate that BAG3 may be an exciting new target for therapeutic intervention for heart failure.
Acknowledgments
Core facilities were provided by Comprehensive NeuroAIDS Center P30-MH92177 at Temple University. We wish to thank Manish Gupta, a former member of Dr. Khalili’s laboratory, for assistance with cloning of the Ad-BAG3 deletion mutant constructs.
This work was supported in part by the National Institutes of Health Grants RO1-HL123093, RO1-HL137426, UO1-NS097162, R21-NS098991, RO1-HL86699 and PO1-HL91799 (Project 2).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures
KK is a board member, scientific advisor, and holds equity in Excision Biotherapeutics, a biotech start-up who has licensed the viral gene editing technology from Temple University for commercial development, and clinical trials. AMF and KK have a pending US patent #611934,483 for BAG3 as a target for heart failure therapy. AMF and JYC have a pending US patent #621205,990 for BAG3 composition and methods. Exclusive rights to the patents have been optioned by Temple University to Renovacor, Inc. AMF and JYC hold equity in Renovacor, Inc.
References
- 1.Knezevic T, Myers VD, Gordon J, Tilley DG, Sharp TE 3rd, Wang J, Khalili K, Cheung JY and Feldman AM. BAG3: a new player in the heart failure paradigm. Heart Fail Rev. 2015;20:423–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Myers VD, Tomar D, Madesh M, Wang J, Song J, Zhang XQ, Gupta MK, Tahrir FG, Gordon J, McClung JM, Kontos CD, Khalili K, Cheung JY and Feldman AM. Haplo-insufficiency of Bcl2-associated athanogene 3 in mice results in progressive left ventricular dysfunction, β-adrenergic insensitivity, and increased apoptosis. Journal of cellular physiology. 2018;233:6319–6326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Su F, Myers VD, Knezevic T, Wang J, Gao E, Madesh M, Tahrir FG, Gupta MK, Gordon J, Rabinowitz J, Ramsey FV, Tilley DG, Khalili K, Cheung JY and Feldman AM. Bcl-2-associated athanogene 3 protects the heart from ischemia/reperfusion injury. JCI Insight. 2016;1:e90931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tahrir FG, Knezevic T, Gupta MK, Gordon J, Cheung JY, Feldman AM and Khalili K. Evidence for the role of BAG3 in mitochondrial quality control in cardiomyocytes. Journal of cellular physiology. 2017;232:797–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gupta MK, Gordon J, Glauser GM, Myers VD, Feldman AM, Cheung JY and Khalili K. Lamin B is a target for selective nuclear PQC by BAG3: implication for nuclear envelopathies. Cell death & disease. 2019;10:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pappas CT, Bhattacharya N, Cooper JA and Gregorio CC. Nebulin interacts with CapZ and regulates thin filament architecture within the Z-disc. Mol Biol Cell. 2008;19:1837–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tahrir FG, Gordon J, Feldman AM, Cheung J, Khalili K and Mohseni Ahooyi T. Evidence for the impact of BAG3 on electrophysiological activity of primary culture of neonatal cardiomyocytes. Journal of cellular physiology. 2019;234:18371–18381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Feldman AM, Gordon J, Wang J, Song J, Zhang XQ, Myers VD, Tilley DG, Gao E, Hoffman NE, Tomar D, Madesh M, Rabinowitz J, Koch WJ, Su F, Khalili K and Cheung JY. BAG3 regulates contractility and Ca2+ homeostasis in adult mouse ventricular myocytes. J Mol Cell Cardiol. 2016;92:10–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Knezevic T, Myers VD, Su F, Wang J, Song J, Zhang XQ, Gao E, Gao G, Muniswamy M, Gupta MK, Gordon J, Weiner KN, Rabinowitz J, Ramsey FV, Tilley DG, Khalili K, Cheung JY and Feldman AM. Adeno-associated virus serotype 9 - driven expression of BAG3 improves left ventricular function in murine hearts with left ventricular dysfunction secondary to a myocardial infarction. JACC Basic Transl Sci. 2016;1:647–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Feldman AM, Begay RL, Knezevic T, Myers VD, Slavov DB, Zhu W, Gowan K, Graw SL, Jones KL, Tilley DG, Coleman RC, Walinsky P, Cheung JY, Mestroni L, Khalili K and Taylor MR. Decreased levels of BAG3 in a family with a rare variant and in idiopathic dilated cardiomyopathy. Journal of cellular physiology. 2014;229:1697–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Myers VD, Gerhard GS, McNamara DM, Tomar D, Madesh M, Kaniper S, Ramsey FV, Fisher SG, Ingersoll RG, Kasch-Semenza L, Wang J, Hanley-Yanez K, Lemster B, Schwisow JA, Ambardekar AV, Degann SH, Bristow MR, Sheppard R, Alexis JD, Tilley DG, Kontos CD, McClung JM, Taylor AL, Yancy CW, Khalili K, Seidman JG, Seidman CE, McTiernan CF, Cheung JY and Feldman AM. Association of variants in BAG3 with cardiomyopathy outcomes in African American individuals. JAMA Cardiol. 2018;3:929–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhou YY, Wang SQ, Zhu WZ, Chruscinski A, Kobilka BK, Ziman B, Wang S, Lakatta EG, Cheng H and Xiao RP. Culture and adenoviral infection of adult mouse cardiac myocytes: methods for cellular genetic physiology. Am J Physiol Heart Circ Physiol. 2000;279:H429–36. [DOI] [PubMed] [Google Scholar]
- 13.Tucker AL, Song J, Zhang XQ, Wang J, Ahlers BA, Carl LL, Mounsey JP, Moorman JR, Rothblum LI and Cheung JY. Altered contractility and [Ca2+]i homeostasis in phospholemman-deficient murine myocytes: Role of Na+/Ca2+ exchange. Am J Physiol Heart Circ Physiol. 2006;291:H2199–H2209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Song J, Zhang XQ, Wang J, Cheskis E, Chan TO, Feldman AM, Tucker AL and Cheung JY. Regulation of cardiac myocyte contractility by phospholemman: Na+/Ca2+ exchange vs. Na+-K+-ATPase. Am J Physiol Heart Circ Physiol. 2008;295:H1615–H1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cheung JY, Gordon J, Wang J, Song J, Zhang XQ, Tilley DG, Gao E, Koch WJ, Rabinowitz J, Klotman PE, Khalili K and Feldman AM. Cardiac dysfunction in HIV-1 transgenic mouse: role of stress and BAG3. Clinical and translational science. 2015;8:305–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Song J, Gao E, Wang J, Zhang XQ, Chan TO, Koch WJ, Shang X, Joseph JI, Peterson BZ, Feldman AM and Cheung JY. Constitutive overexpression of phospholemman S68E mutant results in arrhythmias, early mortality and heart failure: Potenial involvement of Na+/Ca2+ exchanger. Am J Physiol Heart Circ Physiol. 2012;302:H770–H781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang J, Gao E, Song J, Zhang XQ, Li J, Koch WJ, Tucker AL, Philipson KD, Chan TO, Feldman AM and Cheung JY. Phospholemman and b-adrenergic stimulation in the heart. Am J Physiol Heart Circ Physiol. 2010;298:H807–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang J, Chan TO, Zhang XQ, Gao E, Song J, Koch WJ, Feldman AM and Cheung JY. Induced overexpression of Na+/Ca2+ exchanger transgene: Altered myocyte contractility, [Ca2+]i transients, SR Ca2+ contents and action potential duration. Am J Physiol Heart Circ Physiol. 2009;297:H590–H601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang J, Gao E, Rabinowitz J, Song J, Zhang XQ, Koch WJ, Tucker AL, Chan TO, Feldman AM and Cheung JY. Regulation of in vivo cardiac contractility by phospholemman: role of Na+/Ca2+ exchange. Am J Physiol Heart Circ Physiol. 2011;300:H859–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tadros GM, Zhang XQ, Song J, Carl LL, Rothblum LI, Tian Q, Dunn J, Lytton J and Cheung JY. Effects of Na+/Ca2+ exchanger downregulation on contractility and [Ca2+]i transients in adult rat myocytes. Am J Physiol Heart Circ Physiol. 2002;283:H1616–26. [DOI] [PubMed] [Google Scholar]
- 21.Zhang XQ, Wang J, Song J, Rabinowitz J, Chen X, Houser SR, Peterson BZ, Tucker AL, Feldman AM and Cheung JY. Regulation of L-type calcium channel by phospholemman in cardiac myocytes. J Mol Cell Cardiol. 2015;84:104–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bers DM and Berlin JR. Kinetics of [Ca]i decline in cardiac myocytes depend on peak [Ca]i. Am J Physiol Cell Physiol. 1995;268:C271–7. [DOI] [PubMed] [Google Scholar]
- 23.Zhang XQ, Ng YC, Moore RL, Musch TI and Cheung JY. In situ SR function in postinfarction myocytes. J Appl Physiol. 1999;87:2143–50. [DOI] [PubMed] [Google Scholar]
- 24.Selcen D, Muntoni F, Burton BK, Pegoraro E, Sewry C, Bite AV and Engel AG. Mutation in BAG3 causes severe dominant childhood muscular dystrophy. Annals of neurology. 2009;65:83–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Norton N, Li D, Rieder MJ, Siegfried JD, Rampersaud E, Zuchner S, Mangos S, Gonzalez-Quintana J, Wang L, McGee S, Reiser J, Martin E, Nickerson DA and Hershberger RE. Genome-wide studies of copy number variation and exome sequencing identify rare variants in BAG3 as a cause of dilated cardiomyopathy. American journal of human genetics. 2011;88:273–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Arimura T, Ishikawa T, Nunoda S, Kawai S and Kimura A. Dilated cardiomyopathy-associated BAG3 mutations impair Z-disc assembly and enhance sensitivity to apoptosis in cardiomyocytes. Human mutation. 2011;32:1481–91. [DOI] [PubMed] [Google Scholar]
- 27.Franaszczyk M, Bilinska ZT, Sobieszczanska-Malek M, Michalak E, Sleszycka J, Sioma A, Malek LA, Kaczmarska D, Walczak E, Wlodarski P, Hutnik L, Milanowska B, Dzielinska Z, Religa G, Grzybowski J, Zielinski T and Ploski R. The BAG3 gene variants in Polish patients with dilated cardiomyopathy: four novel mutations and a genotype-phenotype correlation. J Transl Med. 2014;12:192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Villard E, Perret C, Gary F, Proust C, Dilanian G, Hengstenberg C, Ruppert V, Arbustini E, Wichter T, Germain M, Dubourg O, Tavazzi L, Aumont MC, DeGroote P, Fauchier L, Trochu JN, Gibelin P, Aupetit JF, Stark K, Erdmann J, Hetzer R, Roberts AM, Barton PJ, Regitz-Zagrosek V, Cardiogenics C, Aslam U, Duboscq-Bidot L, Meyborg M, Maisch B, Madeira H, Waldenstrom A, Galve E, Cleland JG, Dorent R, Roizes G, Zeller T, Blankenberg S, Goodall AH, Cook S, Tregouet DA, Tiret L, Isnard R, Komajda M, Charron P and Cambien F. A genome-wide association study identifies two loci associated with heart failure due to dilated cardiomyopathy. European heart journal. 2011;32:1065–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Toro R, Perez-Serra A, Campuzano O, Moncayo-Arlandi J, Allegue C, Iglesias A, Mangas A and Brugada R. Familial dilated cardiomyopathy caused by a novel frameshift in the BAG3 gene. PloS one. 2016;11:e0158730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rafiq MA, Chaudhry A, Care M, Spears DA, Morel CF and Hamilton RM. Whole exome sequencing identified 1 base pair novel deletion in BCL2-associated athanogene 3 (BAG3) gene associated with severe dilated cardiomyopathy (DCM) requiring heart transplant in multiple family members. Am J Med Genet A. 2017;173:699–705. [DOI] [PubMed] [Google Scholar]
- 31.Yang L, Katchman A, Kushner J, Kushnir A, Zakharov SI, Chen BX, Shuja Z, Subramanyam P, Liu G, Papa A, Roybal D, Pitt GS, Colecraft HM and Marx SO. Cardiac CaV1.2 channels require b subunits for b-adrenergic-mediated modulation but not trafficking. J Clin Invest. 2019;129:647–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chao DT and Korsmeyer SJ. BCL-2 family: regulators of cell death. Annu Rev Immunol. 1998;16:395–419. [DOI] [PubMed] [Google Scholar]
- 33.Rodriguez AE, Lopez-Crisosto C, Pena-Oyarzun D, Salas D, Parra V, Quiroga C, Morawe T, Chiong M, Behl C and Lavandero S. BAG3 regulates total MAP1LC3B protein levels through a translational but not transcriptional mechanism. Autophagy. 2016;12:287–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Arndt V, Dick N, Tawo R, Dreiseidler M, Wenzel D, Hesse M, Furst DO, Saftig P, Saint R, Fleischmann BK, Hoch M and Hohfeld J. Chaperone-assisted selective autophagy is essential for muscle maintenance. Curr Biol. 2010;20:143–8. [DOI] [PubMed] [Google Scholar]
- 35.Ulbricht A, Eppler FJ, Tapia VE, van der Ven PF, Hampe N, Hersch N, Vakeel P, Stadel D, Haas A, Saftig P, Behrends C, Furst DO, Volkmer R, Hoffmann B, Kolanus W and Hohfeld J. Cellular mechanotransduction relies on tension-induced and chaperone-assisted autophagy. Curr Biol. 2013;23:430–5. [DOI] [PubMed] [Google Scholar]
- 36.Liao Q, Ozawa F, Friess H, Zimmermann A, Takayama S, Reed JC, Kleeff J and Buchler MW. The anti-apoptotic protein BAG-3 is overexpressed in pancreatic cancer and induced by heat stress in pancreatic cancer cell lines. FEBS letters. 2001;503:151–7. [DOI] [PubMed] [Google Scholar]
- 37.Xie Z, Peng J, Pennypacker SD and Chen Y. Critical role for the catalytic activity of phospholipase C-gamma1 in epidermal growth factor-induced cell migration. Biochemical and biophysical research communications. 2010;399:425–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Behl C Breaking BAG: The co-chaperone BAG3 in health and disease. Trends Pharmacol Sci. 2016;37:672–88. [DOI] [PubMed] [Google Scholar]
- 39.Behl C BAG3 and friends: co-chaperones in selective autophagy during aging and disease. Autophagy. 2011;7:795–8. [DOI] [PubMed] [Google Scholar]
- 40.Hishiya A, Salman MN, Carra S, Kampinga HH and Takayama S. BAG3 directly interacts with mutated alphaB-crystallin to suppress its aggregation and toxicity. PloS one. 2011;6:e16828. [DOI] [PMC free article] [PubMed] [Google Scholar]