

Abstract
The design of near-infrared (NIR)-active photosensitizers (PSs) for light-based cancer treatments such as photodynamic therapy (PDT) has been a challenge. While several NIR-Ru(II) scaffolds have been reported, this approach has not been proven in cells. This is the first report of NIR-Ru(II) PSs that are phototoxic to cancer cells, including highly pigmented B16F10 melanoma cells. The PS family incorporated a bis(1,8-naphthyridine)-based ligand (tpbn), a bidentate thiophene-based ligand (nT; n=0–4), and a monodentate 4-picoline ligand (4-pic). All compounds absorbed light >800 nm with maxima near 730 nm. Transient absorption (TA) measurements indicated that n≥4 thiophene rings (4T) positioned the PDT-active triplet intraligand charge transfer (3ILCT) excited state in energetic proximity to the lowest lying triplet metal-to-ligand charge transfer (3MLCT). 4T had low micromolar phototoxicity with PIvis and PI733nm values as large as 90 and 12, respectively. Spectroscopic studies suggested that the long-lived (TTA=3–6 μs) 3ILCT was accessible from the 3MLCT state, but energetically uphill in the overall photophysics. The study highlights that phototoxic effects can be achieved with NIR-absorbing Ru(II) PSs as long as the reactive 3ILCT states are energetically accessible from the low-energy 3MLCT states. It also demonstrates that tissue-penetrating NIR light can be used to activate the PSs in highly pigmented cells where melanin attenuates shorter wavelengths of light.
Keywords: near-infrared (NIR) photosensitizers, ruthenium anticancer agents, photodynamic therapy, melanoma, intraligand charge-transfer
Graphical Abstract
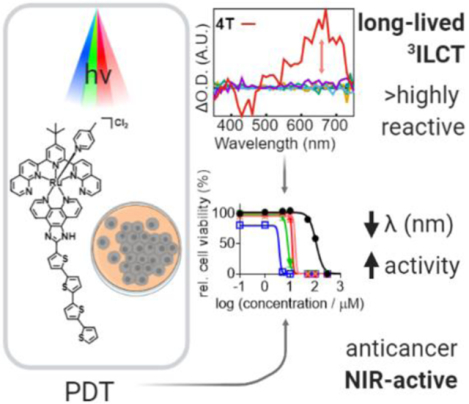
Four thiophenes are required to produce the requisite 3ILCT energy for eliciting PDT effects with this new class of NIR-absorbing photosensitizers.
1. INTRODUCTION
Photodynamic therapy (PDT) is a light-triggered therapy that has been used to treat cancer and infection.[1–9] PDT utilizes a photosensitizer (PS), light, and oxygen to create cytotoxic reactive oxygen species (ROS) with spatiotemporal selectivity. This selectivity allows for healthy tissue to be spared during treatment, which presents a sought-after advantage in cancer therapy. PDT-induced ROS generation is initiated from the triplet state of the PS and can be classified mechanistically as Type I or Type II.[10] Type I photoreactions involve electron transfer and the formation of radical species, whereas Type II photosensitization involves energy transfer to form singlet oxygen (1O2). Singlet oxygen produced via the Type II mechanism is thought to be the most important mediator of the PDT response. The antitumor effects of PDT result from a combination of direct action on the primary tumor, disruption of tumor vasculature, and the induction of an immune response.[11–22]
Photofrin® was the first approved PS for PDT[23–27] and continues to be the standard agent used in many clinical settings. It consists of a mixture of oligomeric tetrapyrroles that is administered systemically and then activated with 630 nm laser light.[27] The success of Photofrin inspired efforts to design subsequent generations of tetrapyrrole-based macrocycles[28] (porphyrins, chlorins, bacteriochlorins, phthalocyanins) that are single compounds with improved water solubility and reduced skin photosensitivity. Some of these and related systems have been altered further by introducing a central metal ion into the tetrapyrrolic scaffold to enhance the chemical, photophysical, and biological characteristics of the parent PSs.[29,30]
Design of the next generation of PSs has been focused on extending the absorption window to include NIR wavelengths (700–900 nm)[31–34] and to alter the excited state properties of the molecules to exploit oxygen-independent mechanistic pathways to achieve phototoxic effects in hypoxia.[35–40] Ru(II) polypyridyl complexes are particularly attractive scaffolds for PS design because the photophysics of these complexes is well-established[41–47] and polypyridyl ligand combinations can be chosen to systematically manipulate the excited state dynamics in a rational and consistent manner. Following this strategy, our group developed the photosensitizer TLD1433[47–49] and demonstrated that extremely efficient singlet oxygen sensitization can be obtained with Ru(II) complexes containing oligothienyl-based ligands with low-energy triplet intraligand charge transfer (3ILCT) excited states that are characterized by microsecond lifetimes. Some of the light-potentiated cytotoxicity of compounds with dominant 3ILCT states can even be maintained in hypoxia (1% O2, unpublished) due to their high reactivity. Our TLD1433 is the first Ru(II) PS to enter human clinical trials and is currently in a Phase II human clinical trial (NCT03945162) for treating nonmuscle invasive bladder cancer (NMIBC) with PDT.
While TLD1433 is preferentially activated in the clinic with green light (532 nm) and can also be activated with red light (630 nm), wavelengths near 700 nm do not produce photocytotoxic effects without special formulations.[49] We hypothesized that combining the PDT-active ligand in TLD1433 with a Ru(II) NIR-absorbing scaffold would generate NIR PDT effects and may be active in hypoxia. Various Ru(II)-based NIR scaffolds are known, but their phototoxic effects on cells were not reported.[31–34,50] Generally, the 1O2 quantum yields are low (and PDT effects thus poor) for metal complexes with low-energy triplet metal to ligand charge transfer (3MLCT) excited states due to efficient intersystem crossing (ISC) that competes effectively with 1O2 sensitization.[46] To circumvent this issue, we have designed Ru(II)-based NIR complexes with 3ILCT states that are lower in energy than the 3MLCT states and longer lived, having mostly organic 3ππ* character. In the present study, the goal was to examine whether such Ru(II)-based NIR-absorbing complexes could be useful for PDT.
A tris-heteroleptic Ru(II) NIR scaffold was chosen, having the general formula [Ru(NNN)(NN)(N)]Cl2 that coordinates three different types of ligands to the Ru(II) center in a pseudo-octahedral geometry: a tridentate chromophoric ligand (NNN), a bidentate and (potentially) PDT-active ligand (NN), and a monodentate axial ligand (N). The purpose of the chromophoric NNN ligand was to extend the absorption window into the NIR by introducing absorptive low-energy MLCT states, which allows for greater tissue penetration. The purpose of the most π-extended NN ligand(s) was to provide PDT-active 3ILCT states lower in energy and longer-lived than the 3MLCT state and thus well-suited for generating cytotoxic singlet oxygen. The axial ligand N completed the Ru(II) coordination sphere and modulated the solubility, the stability, and the absorption window of the complex.
Herein, we prepared a series of related tris-heteroleptic Ru(II) complexes with 2,2′-(4-(tertbutyl)pyridine-2,6-diyl)bis(1,8-naphthyridine) (tpbn), also referred to as 4-t-butyl-2,6-di-1′,8′-(naphthyrid-2′-yl)-pyridine in literature,[51] as the chromophoric ligand and 4-picoline (4-pic) as the axial monodentate ligand. The compounds varied by the number of thiophene rings in the bidentate IP-nT ligand (Chart 1), where some critical value of n was expected to yield PDT effects. The related Ru(II) complex (phen) incorporating 1,10-phenanthroline instead of the IP-nT ligand was used as a reference compound that lacks a low-energy 3ILCT state.[52] From a photophysical standpoint, the criterion to achieve photocytotoxic effects with Ru(II) complexes appears to be that the 3ILCT state must be lower in energy than the 3MLCT state for the 3ILCT state to remain accessible and thus exert its effects. It was anticipated that in our scaffold the lower energy 3MLCT state afforded by the chromophoric ligand might require a more extended thiophene chain in order to ensure that the 3ILCT state is lower in energy than the 3MLCT state. While n=3 is sufficient to achieve photocytotoxic activity with TLD1433, it was understood that the NIR absorbing Ru(II) complexes in this study could require n≥4 in order to achieve desired PDT effects.
Chart 1.

Molecular structures of tris-heteroleptic Ru(II) complexes 0T–4T and reference compound phen
2. EXPERIMENTAL DETAILS
Full description of methods, instrumentation, and additional synthetic details are included in the Supporting Information.
2.1. Synthesis and Characterization.
All solvents and reagents were purchased from commercial sources and used without further purification. Methanol was purchased from Fisher Scientific (ACS grade for synthesis and Optima™ grade for HPLC analysis) and ethanol (200 proof) was purchased from Decon Laboratories. Deuterated solvents for NMR were purchased from Cambridge Isotope Laboratories. Ruthenium(III) trichloride trihydrate was purchased from Ark Pharm and Acros Organics. 4-Picoline (4-pic) was purchased from Alfa Aesar. Triethylamine (TEA) was purchased from Fisher Scientific. Ligand 2,2′-(4-(tert-butyl)pyridine-2,6-diyl)bis(1,8-naphthyridine) (tpbn), also referred to as 4-t-butyl-2,6-di-1′,8′-(naphthyrid-2′-yl)-pyridine in literature,[51] was synthesized following reported literature procedures.[53,54] Ligand 1H-imidazo[4,5-f][1,10]-phenanthroline (IP) and ligands IP-nT (2-(thiophen-2-yl)-1H-imidazo[4,5-f][1,10]phenanthroline (IP-1T), 2-([2,2′-bithiophen]-5-yl)-1H-imidazo[4,5-f][1,10]phenanthroline (IP-2T), 2-([2,2′:5′,2″-terthiophen]-5-yl)-1H-imidazo[4,5-f][1,10]phenanthroline (IP-3T), and 2-([2,2′:5′,2″:5″,2‴-quaterthiophen]-5-yl)-1H-imidazo[4,5-f][1,10]phenanthroline (IP-4T)) were synthesized following Radziszewski protocol.[55,56] The synthesis of IP-based ligands follows the synthesis of IP-4T that is described below. [2,2′:5′,2″:5″,2‴-quaterthiophene]-5-carbaldehyde (4T-CHO) was prepared as previously described[57] via the coupling of 5-bromo-5″-formyl-2,2′:5′,2″-terthiophene with 2-(tributylstannyl)thiophene, which were purchased from Alfa Aesar and Fisher Scientific, respectively.
A CEM Discover microwave reactor was used to perform chemical reactions. Manual column chromatography was carried out using neutral aluminum oxide, activated, Brockmann Grade I (58Å, −60 Mesh Powder, S.A. 150 m2/g) as a stationary phase. Flash column chromatography was carried out on the Teledyne ISCO CombiFlash EZ Prep UV Prep using SILICYCLE SiliaSep™ Neutral Alumina cartridges. Size-exclusion chromatography was performed using a manual column packed with Sephadex® LH-20. The NMR spectra were collected on a JEOL ECA 500 spectrometer, operating at 500 MHz for 1H experiments (University of North Carolina at Greensboro) and on an Agilent 700 MHz Magnet spectrometer (The Joint School of Nanoscience and Nanoengineering at Greensboro) operating at 700 MHz for 1H experiments. The chemical shifts are reported in parts per million (ppm) and were referenced to the residual solvent peaks. ESI mass spectra were obtained using a Thermo Fisher Scientific LTQ Orbitrap XL instrument (Triad Mass Spectrometry Laboratory at University of North Carolina at Greensboro). HPLC analyses were carried out on an Agilent/Hewlett Packard 1100 series instrument in 100 μM solutions in methanol using a Hypersil GOLD C18 reversed-phase column with an A-B gradient (98% → 5% A; A = 0.1% formic acid in H2O, B = 0.1% formic acid in MeCN). Reported retention times are correct to within ±0.1 min.
Intermediate complexes 0T-Cl–4T-Cl and target complexes 0T–4T were synthesized using the microwave reactor, and purification of the complexes was performed by manual or flash column chromatography on alumina followed by size-exclusion chromatography on Sephadex® LH-20. Detailed synthesis of 0T is described below. 1T–4T were synthesized following the same protocol, and a detailed description of their syntheses is included in the Supporting Information. High resolution ESI+ mass spectra of 0T–4T showed two sets of peaks, with one set corresponding to the calculated [M-2Cl]2+ molecular ion and another (significantly lower abundance) matching that calculated for [M-2Cl-H]+. Observed molecular ions displayed isotopic patterns characteristic for mononuclear Ru(II)-containing molecular ions, with a separation of 0.5 m/z between the peaks for M2+ ions and 1 m/z between the peaks for M+ ions.
IP-4T. 1,10-phenanthroline-5,6-dione (175 mg, 0.83 mmol), 4T-CHO (200 mg, 0.56 mmol), and ammonium acetate (1.38 g, 18 mmol) were added to a 250 mL round-bottom flask with glacial acetic acid (100 mL). The orange mixture was heated at 100 °C for 96 hours. Once cooled, the reddish-brown mixture was neutralized with NH4OH. The precipitate was vacuum filtered using a Büchner funnel and washed with cold deionized water (50 mL) and cold ether (100 mL) to obtain the desired product as a brown solid (279 mg, 91%). 1H NMR (DMSO-d6, 500 MHz): δ 9.05 (dd, J = 4.2, 1.7 Hz, 2H), 8.85 (d, J = 7.9 Hz, 2H), 7.88–7.81 (m, 3H), 7.57 (d, J = 4.7 Hz, 1H), 7.52 (d, J = 3.8 Hz, 1H), 7.48 (d, J = 3.9 Hz, 1H), 7.41–7.36 (m, 3H), 7.32 (d, J = 3.7 Hz, 1H), 7.13 (dd, J = 5.1, 3.5 Hz, 1H).
[Ru(tpbn)(IP)(4-pic)]Cl2 (Complex 0T). Synthesis of [Ru(tpbn)(IP)(Cl)]Cl (intermediate for complex 0T): A mixture of EtOH (2.00 mL) and H2O (0.48 mL) was added to a microwave vial pre-filled with argon. The solution was degassed with argon for 10 minutes, and after that tpbn (0.033 g, 0.084 mmol) and RuCl3•3H2O (0.023 g, 0.087 mmol) were added to the reaction vial. The vial was capped, and the reaction mixture was exposed to microwave irradiation at 145°C for 15 min. Then, IP (0.021 g, 0.095 mmol) and TEA (20 drops) were added to the reaction mixture. The reaction mixture was exposed to microwave irradiation at 145°C for 30 min then concentrated under reduced pressure and the residue was treated with a few mL of H2O. The precipitate was isolated by filtration, washed with H2O (a few mL, dropwise, due to partial solubility of material in H2O) and Et2O, and dry-loaded on a manual column for purification. Manual column chromatography was performed on neutral alumina, using gradient elution with the following eluents: 1) CH2Cl2 2) 50% CH2Cl2 : 50% acetone 3) acetone 4) 99% acetone : 1% MeOH 5) 97% acetone : 3% MeOH. The product fraction (purple-violet band) eluted last with acetone 97% : MeOH 3%. The product fraction was concentrated under reduced pressure and the residue was treated with a few mL of Et2O. The precipitate was isolated by filtration, washed with Et2O and dried. Complex [Ru(tpbn)(IP)(Cl)]Cl 0T-Cl was obtained as a dark-purple powder (0.014 g, 21% yield). Rf = 0.54 (alumina; 8% H2O in acetonitrile). 1H NMR (MeOD-d3, 500 MHz): δ 10.65 (dd, J1 = 5.5 Hz, J2 = 1.0 Hz, 1H), a signal around 9.3 ppm is expected but is too broadened to be seen (1H), 9.08 (s, 2H), 8.90 (d, J = 9.0 Hz, 2H), 8.55 (s, 1H), 8.50 (d, J = 9.0 Hz, 2H), a signal around 8.5 ppm is expected but is too broadened to be seen (1H), 8.33 (dd, J1 = 8.5 Hz, J2 = 5.5 Hz, 1H), 8.23 (dd, J1 = 8.0 Hz, J2 = 2.0 Hz, 2H), 7.78 (dd, J1 = 4.0 Hz, J2 = 2.0 Hz, 2H), 7.69 (dd, J1 = 5.0 Hz, J2 = 1.0 Hz, 1H), 7.29 (dd, J1 = 8.0 Hz, J2 = 4.0 Hz, 2H), 7.21 (dd, J1 = 8.0 Hz, J2 = 5.5 Hz, 1H), 1.79 (s, 9H). HRMS (ESI+) m/z: [M-Cl]+ Calcd for C38H29ClN9Ru 748.1272; Found 748.1257. [M-Cl+H]2+ Calcd for C38H30ClN9Ru 374.5672; Found 374.5665. Synthesis of 0T from 0T-Cl: The reaction was performed in 2 batches, then combined for workup and purification. Procedure for batch 1: A microwave vial was charged with [Ru(tpbn)(IP)(Cl)]Cl 0T-Cl (0.025 g, 0.032 mmol), 4-picoline (0.30 mL, 3.09 mmol) and MeOH (2.8 mL). The vial was capped, and the reaction mixture was exposed to microwave irradiation at 150°C for 15 min. After that, H2O (0.8 mL) was added and the reaction mixture was exposed to microwave irradiation at 160°C for 20 min. Batch 2: procedure for batch 1 was reproduced. Reaction mixtures from batch 1 and batch 2 were combined and concentrated under reduced pressure. The residue was dried under high vacuum and purified with flash chromatography on alumina, using gradient elution from 100% acetonitrile to 93% acetonitrile : 7% H2O. The product fraction (maroon band) eluted with 93% acetonitrile : 7% H2O. The product fraction was concentrated under reduced pressure and further purified using size-exclusion chromatography on Sephadex® LH-20 (eluent: MeOH). The main band (maroon) was collected and concentrated under reduced pressure. The residue was treated with a few mL of Et2O, sonicated, and isolated by filtration. The precipitate was thoroughly washed with Et2O and dried. [Ru(tpbn)(IP)(4-pic)]Cl2 0T was obtained as a maroon powder (0.025 g, 45% yield). Rf = 0.41 (alumina; 8% H2O in acetonitrile). 1H NMR (MeOD-d3, 500 MHz): δ 9.39 (a′, dd, J1 = 5.5 Hz, J2 = 1.5 Hz, 1H), 9.26 (c′, broad d, J = 8.0 Hz, 1H), 9.15 (3, s, 2H), 9.01 (3′, d, J = 8.5 Hz, 2H), 8.68 (4′, d, J = 8.5 Hz, 2H), 8.55 (d, s, 1H), 8.51 (c, broad d, J = 8.0 Hz, 1H), 8.33 (5′, dd, J1 = 8.5 Hz, J2 = 2.0 Hz, 2H), 8.33 (b′, m, 1H), 7.95 (7′, dd, J1 = 4.0 Hz, J2 = 2.0 Hz, 2H), 7.65 (2″, d, J = 6.5 Hz, 2H), 7.61 (a, dd, J1 = 5.5 Hz, J2 = 1.5 Hz, 1H), 7.40 (6′, dd, J1 = 8.5 Hz, J2 = 4.0 Hz, 2H), 7.34 (b, dd, J1 = 8.0 Hz, J2 = 5.5 Hz, 1H), 6.97 (3″, d, J = 6.0 Hz, 2H), 2.24 (4″-Me, s, 3H), 1.76 (4-tBu, s, 9H) (for hydrogen labels, see Figure 1 and Figure S1). HRMS (ESI+) m/z: [M-2Cl-H]+ Calcd for C44H35N10Ru 805.2084; Found 805.2080. [M-2Cl]2+ Calcd for C44H36N10Ru 403.1079; Found 403.1074. HPLC retention time: 9.44 min (96.4% purity by peak area).
Figure 1.
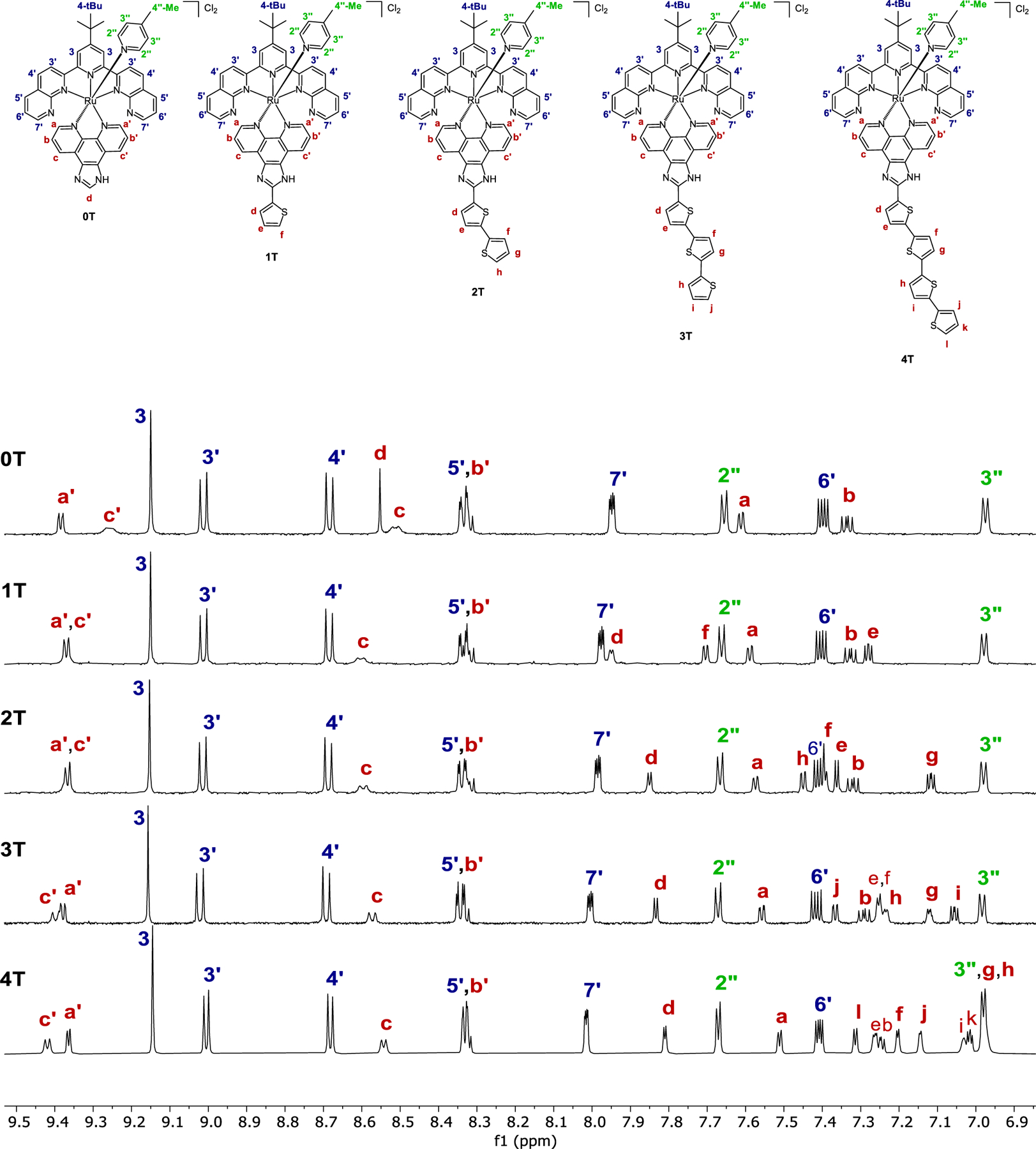
500 MHz (0T–3T) and 700 MHz (4T) 1H NMR spectra of Ru(II) complexes 0T–4T in MeOD-d3 at 298 K, aromatic region.
[Ru(tpbn)(IP-1T)(4-pic)]Cl2 (Complex 1T) was synthesized following a similar procedure as was used for the synthesis of 0T. Intermediate [Ru(tpbn)(IP-1T)(Cl)]Cl 1T-Cl was obtained as a dark-purple powder (0.057 g, 40% yield). Rf = 0.56 (alumina; 8% H2O in acetonitrile). 1H NMR (MeOD-d3, 500 MHz): δ 10.64 (d, J = 5.5 Hz, 1H), 9.31 (broad signal, 1H), 9.08 (s, 2H), 8.90 (d, J = 9.0 Hz, 2H), 8.50 (d, J = 8.5 Hz, 2H), 8.50 (m, 1H), 8.33 (dd, J1 = 9.0 Hz, J2 = 5.5 Hz), 8.22 (d, J = 8.0 Hz, 2H), 7.95 (d, J = 3.5 Hz, 1H), 7.81 (dd, J1 = 4.0 Hz, J2 = 2.0 Hz, 2H), 7.71 (d, J = 5.0 Hz, 1H), 7.67 (d, J = 5.5 Hz, 1H), 7.29 (dd, J1 = 6.5 Hz, J2 = 4.0 Hz, 2H), 7.29 (m, 1H), 7.20 (dd, J1 = 8.5 Hz, J2 = 5.5 Hz, 1H), 1.79 (s, 9H). HRMS (ESI+) m/z: [M-Cl]+ Calcd for C42H31ClN9RuS 830.1150; Found 830.1138. [M-Cl+H]2+ Calcd for C42H32ClN9RuS 415.5611; Found 415.5602. HPLC retention time: 23.09 min. [Ru(tpbn)(IP-1T)(4-pic)]Cl2 1T was obtained as a maroon powder (0.030 g, 52% yield). Rf = 0.44 (alumina; 8% H2O in acetonitrile). 1H NMR (MeOD-d3, 500 MHz): δ 9.37 (a′, d, J = 5.0 Hz, 1H), 9.37 (c′, m, 1H), 9.15 (3, s, 2H), 9.01 (3′, d, J = 9.0 Hz, 2H), 8.68 (4′, d, J = 8.5 Hz, 2H), 8.60 (c, broad d, J = 8.0 Hz, 1H), 8.34 (5′, dd, J1 = 8.5 Hz, J2 = 2.0 Hz, 2H), 8.33 (b′, m, 1H), 7.97 (7′, dd, J1 = 4.0 Hz, J2 = 2.0 Hz, 2H), 7.95 (d, dd, J1 = 4.0 Hz, J2 = 1.0 Hz, 1H), 7.70 (f, dd, J1 = 5.0 Hz, J2 = 1.0 Hz, 1H), 7.66 (2″, d, J = 6.5 Hz, 2H), 7.59 (a, dd, J1 = 5.5 Hz, J2 = 1.5 Hz, 1H), 7.40 (6′, dd, J1 = 8.5 Hz, J2 = 4.0 Hz, 2H), 7.33 (b, dd, J1 = 8.5 Hz, J2 = 5.5 Hz, 1H), 7.28 (e, dd, J1 = 5.0 Hz, J2 = 4.0 Hz, 1H), 6.98 (3″, d, J = 6.5 Hz, 2H), 2.24 (4″-Me, s, 3H), 1.76 (4-tBu, s, 9H) (for hydrogen labels, see Figure 1 and Figure S3). HRMS (ESI+) m/z: [M-2Cl-H]+ Calcd for C48H37N10RuS 887.1961; Found 887.1965. [M-2Cl]2+ Calcd for C48H38N10RuS, 444.1017; Found 444.1012. HPLC retention time: 21.36 min (97.0% purity by peak area).
[Ru(tpbn)(IP-2T)(4-pic)]Cl2 (Complex 2T) was synthesized following a similar procedure as was used for the synthesis of 0T. Intermediate [Ru(tpbn)(IP-2T)(Cl)]Cl 2T-Cl was obtained as a dark-purple powder (0.058 g, 36% yield). Rf = 0.58 (alumina; 8% H2O in acetonitrile). 1H NMR (DMSO-d6, 500 MHz): δ 10.47 (dd, J1 = 5.5 Hz, J2 = 1.0 Hz, 1H), 9.37 (broad signal, 1H), 9.18 (s, 2H), 9.16 (d, J = 8.0 Hz, 2H), 8.64 (d, J = 8.5 Hz, 2H), 8.55 (broad signal, 1H), 8.34 (dd, J1 = 8.0 Hz, J2 = 2.0 Hz, 2H), 8.34 (m, 1H), 7.74 (m, 2H), 7.65 (dd, J1 = 5.5 Hz, J2 = 1.0 Hz, 1H), 7.62 (m, 1H), 7.49 (m, 3H), 7.36 (dd, J1 = 8.0 Hz, J2 = 4.5 Hz, 2H), 7.24 (m, 1H), 7.16 (m, 1H), 1.74 (s, 9H). HRMS (ESI+) m/z: [M-Cl]+ Calcd for C46H33ClN9RuS2 912.1027; Found 912.1007. [M-Cl+H]2+ Calcd for C46H34ClN9RuS2 456.5550; Found 456.5534. HPLC retention time: 24.76 min. [Ru(tpbn)(IP-2T)(4-pic)]Cl2 2T was obtained as a maroon powder (0.030 g, 55% yield). Rf = 0.54 (alumina; 8% H2O in acetonitrile). 1H NMR (MeOD-d3, 500 MHz): δ 9.37 (a′, d, J = 6.0 Hz, 1H), 9.37 (c′, m, 1H), 9.15 (3, s, 2H), 9.01 (3′, d, J = 8.5 Hz, 2H), 8.68 (4′, d, J = 9.0 Hz, 2H), 8.59 (c, broad d, J = 8.5 Hz, 1H), 8.34 (5′, dd, J1 = 8.0 Hz, J2 = 2.0 Hz, 2H), 8.32 (b′, m, 1H), 7.98 (7′, dd, J1 = 4.5 Hz, J2 = 2.0 Hz, 2H), 7.85 (d, d, J = 4.0 Hz, 1H), 7.66 (2″, d, J = 7.0 Hz, 2H), 7.57 (a, dd, J1 = 5.5 Hz, J2 = 1.5 Hz, 1H), 7.45 (h, dd, J1 = 5.0 Hz, J2 = 1.0 Hz, 1H), 7.41 (6′, dd, J1 = 8.0 Hz, J2 = 4.0 Hz, 2H), 7.39 (f, dd, J1 = 3.0 Hz, J2 = 1.0 Hz, 1H), 7.36 (e, d, J = 4.0 Hz, 1H), 7.32 (b, dd, J1 = 8.0 Hz, J2 = 5.5 Hz, 1H), 7.11 (g, dd, J1 = 5.0 Hz, J2 = 3.5 Hz, 1H), 6.98 (3″, d, J = 6.0 Hz, 2H), 2.24 (4″-Me, s, 3H), 1.76 (4-tBu, s, 9H) (for hydrogen labels, see Figure 1 and Figure S5). HRMS (ESI+) m/z: [M-2Cl-H]+ Calcd for C52H39N10RuS2 969.1839; Found 969.1822. [M-2Cl]2+ Calcd for C52H40N10RuS2 485.0956; Found 485.0939. HPLC retention time: 22.75 min (95.1% purity by peak area).
[Ru(tpbn)(IP-3T)(4-pic)]Cl2 (Complex 3T) was synthesized following a similar procedure as was used for the synthesis of 0T. Intermediate [Ru(tpbn)(IP-3T)(Cl)]Cl 3T-Cl was obtained as a dark-brown powder (0.038 g, 22% yield). Rf = 0.55 (alumina; 8% H2O in acetonitrile). 1H NMR (DMSO-d6, 500 MHz): δ 10.47 (dd, J1 = 5.5 Hz, J2 = 1.0 Hz, 1H), 9.43 (broad signal, 1H), 9.18 (s, 2H), 9.16 (d, J = 9.0 Hz, 2H), 8.64 (d, J = 9.0 Hz, 2H), 8.56 (broad signal, 1H), 8.35 (m, 1H), 8.34 (dd, J1 = 8.0 Hz, J2 = 2.0 Hz, 2H), 7.75 (m, 2H), 7.65 (d, J = 5.5 Hz, 1H), 7.58 (d, J = 5.0 Hz, 1H), 7.52 (m, 1H), 7.49 (m, 1H), 7.46 (dd, J1 = 8.0 Hz, J2 = 4.0 Hz, 1H), 7.40 (m, 1H), 7.36 (dd, J1 = 8.0 Hz, J2 = 4.5 Hz, 2H), 7.35 (m, 1H), 7.24 (m, 1H), 7.14 (dd, J1 = 5.0 Hz, J2 = 3.5 Hz, 1H), 1.74 (s, 9H). HRMS (ESI+) m/z: [M-Cl]+ Calcd for C50H35ClN9RuS3 994.0904; Found 994.0895. [M-Cl+H]2+ Calcd for C50H36ClN9RuS3 497.5489; Found 497.5473. HPLC retention time: 26.83 min. [Ru(tpbn)(IP-3T)(4-pic)]Cl2 3T was obtained as a dark-brown powder (0.019 g, 42% yield). Rf = 0.46 (alumina; 8% H2O in acetonitrile). 1H NMR (MeOD-d3, 500 MHz): δ 9.39 (c′, d, J = 8.5 Hz, 1H), 9.38 (a′, dd, J1 = 5.5 Hz, J2 = 1.5 Hz, 1H), 9.16 (3, s, 2H), 9.02 (3′, d, J = 9.0 Hz, 2H), 8.69 (4′, d, J = 8.5 Hz, 2H), 8.57 (c, d, J = 8.5 Hz, 1H), 8.34 (5′, dd, J1 = 8.5 Hz, J2 = 2.0 Hz, 2H), 8.33 (b′, m, 1H), 8.00 (7′, dd, J1 = 4.5 Hz, J2 = 2.0 Hz, 2H), 7.83 (d, d, J = 3.5 Hz, 1H), 7.67 (2″, d, J = 7.0 Hz, 2H), 7.56 (a, dd, J1 = 5.5 Hz, J2 = 1.5 Hz, 1H), 7.42 (6′, dd, J1 = 8.5 Hz, J2 = 4.5 Hz, 2H), 7.37 (j, dd, J1 = 5.0 Hz, J2 = 1.0 Hz, 1H), 7.29 (b, dd, J1 = 8.5 Hz, J2 = 5.5 Hz, 1H), 7.25 (e, d, J = 3.5 Hz, 1H), 7.25 (f, d, J = 3.5 Hz, 1H), 7.23 (h, d, J = 3.5 Hz, 1H), 7.12 (g, d, J = 3.5 Hz, 1H), 7.05 (i, dd, J1 = 5.0 Hz, J2 = 3.5 Hz, 1H), 6.98 (3″, d, J = 6.5 Hz, 2H), 2.24 (4″-Me, s, 3H), 1.77 (4-tBu, s, 9H) (for hydrogen labels, see Figure 1 and Figure S7). HRMS (ESI+) m/z: [M-2Cl-H]+ Calcd for C56H41N10RuS3 1051.1716; Found 1051.1726. [M-2Cl]2+ Calcd for C56H42N10RuS3 526.0894; Found 526.0887. HPLC retention time: 24.09 min (100% purity by peak area).
[Ru(tpbn)(IP-4T)(4-pic)]Cl2 (Complex 4T) was synthesized following a similar procedure as was used for the synthesis of 0T. Intermediate [Ru(tpbn)(IP-4T)(Cl)]Cl 4T-Cl was obtained as a dark-brown powder (0.097 g, 35% yield). Rf = 0.57 (alumina; 8% H2O in acetonitrile). 1H NMR (DMSO-d6, 500 MHz): δ 10.48 (dd, J1 = 4.5 Hz, J2 = 1.0 Hz, 1H), 9.37 (broad signal, 1H), 9.18 (s, 2H), 9.16 (d, J = 8.5 Hz, 2H), 8.64 (d, J = 9.0 Hz, 2H), 8.45 (broad signal, 1H), 8.35 (m, 1H), 8.34 (dd, J1 = 8.5 Hz, J2 = 2.0 Hz, 2H), 7.75 (m, 2H), 7.65 (d, J = 5.5 Hz, 1H), 7.57 (d, J = 5.0 Hz, 1H), 7.53 (m, 1H), 7.52–7.34 (m, 5H), 7.36 (dd, J1 = 8.5 Hz, J2 = 4.0 Hz, 2H), 7.32 (d, J = 3.0 Hz, 1H), 7.24 (m, 1H), 7.13 (dd, J1 = 4.5 Hz, J2 = 4.0 Hz, 1H), 1.74 (s, 9H). HRMS (ESI+) m/z: [M-Cl]+ Calcd for C54H37ClN9RuS4 1076.0781; Found 1076.0771. [M-Cl+H]2+ Calcd for C54H38ClN9RuS4 538.5427; Found 538.5409. HPLC retention time: 28.81 min. [Ru(tpbn)(IP-4T)(4-pic)]Cl2 4T was obtained as a brown powder (0.031 g, 36% yield). Rf = 0.47 (alumina; 8% H2O in acetonitrile). 1H NMR (MeOD-d3, 700 MHz): δ 9.42 (c′, d, J = 8.4 Hz, 1H), 9.36 (a′, dd, J1 = 4.9 Hz, J1 = 1.4 Hz, 1H), 9.15 (3, s, 2H), 9.00 (3′, d, J = 8.4 Hz, 2H), 8.68 (4′, d, J = 8.4 Hz, 2H), 8.54 (c, d, J = 7.7 Hz, 1H), 8.33 (5′, dd, J1 = 8.4 Hz, J2 = 2.1 Hz, 2H), 8.33 (b′, m, 1H), 8.01 (7′, dd, J1 = 4.2 Hz, J2 = 2.1 Hz, 2H), 7.81 (d, d, J = 4.2 Hz, 1H), 7.67 (2″, d, J = 7.0 Hz, 2H), 7.51 (a, dd, J1 = 5.6 Hz, J2 = 1.4 Hz, 1H), 7.41 (6′, dd, J1 = 8.4 Hz, J2 = 4.2 Hz, 2H), 7.31 (l, d, J = 4.9 Hz, 1H), 7.26 (e, d, J = 3.5 Hz, 1H), 7.25 (b, dd, J1 = 7.7 Hz, J2 = 5.6 Hz, 1H), 7.20 (f, d, J = 3.5 Hz, 1H), 7.14 (j, d, J = 3.5 Hz, 1H), 7.03 (i, broad d, J1 = 3.5 Hz, 1H), 7.01 (k, dd, J1 = 5.6 Hz, J2 = 3.5 Hz, 1H), 6.98 (g, m, 1H), 6.98 (h, m, 1H), 6.98 (3″, d, J = 6.3 Hz, 2H), 2.24 (4″-Me, s, 3H), 1.76 (4-tBu, s, 9H) (for hydrogen labels, see Figure 1 and Figure S9–S10). HRMS (ESI+) m/z: [M-2Cl-H]+ Calcd for C60H43N10RuS4 1133.1593; Found 1133.1586. [M-2Cl]2+ Calcd for C60H44N10RuS4 567.0833; Found 567.0819. HPLC retention time: 25.28 min (98.8% purity by peak area).
3. RESULTS AND DISCUSSION
3.1. Synthesis and Characterization
The target complexes [Ru(tpbn)(IP-nT)(4-pic)]Cl2 0T–4T were synthesized following a procedure similar to that reported by Thummel and coworkers[53] but adapted for microwave irradiation to significantly shorten the reaction times (Scheme 1). Briefly, RuCl3•3H2O was combined with tpbn to form [Ru(tpbn)](Cl)3 in situ, which was then treated with the IP-nT (n=0–4) ligands to yield the intermediate [Ru(tpbn)(IP-nT)(Cl)]Cl (0-Cl–4T-Cl) complexes with chloro as the axial ligand. 0-Cl–4T-Cl were purified by column chromatography on neutral alumina to afford the products in 21–40% yields, which were characterized by thin-layer chromatography, 1H NMR (Figures S12–S16), HPLC (Figures S32–S35), and high resolution ESI+ mass spectrometry (Figures S17, S19, S21, S23, S25).
Scheme 1.
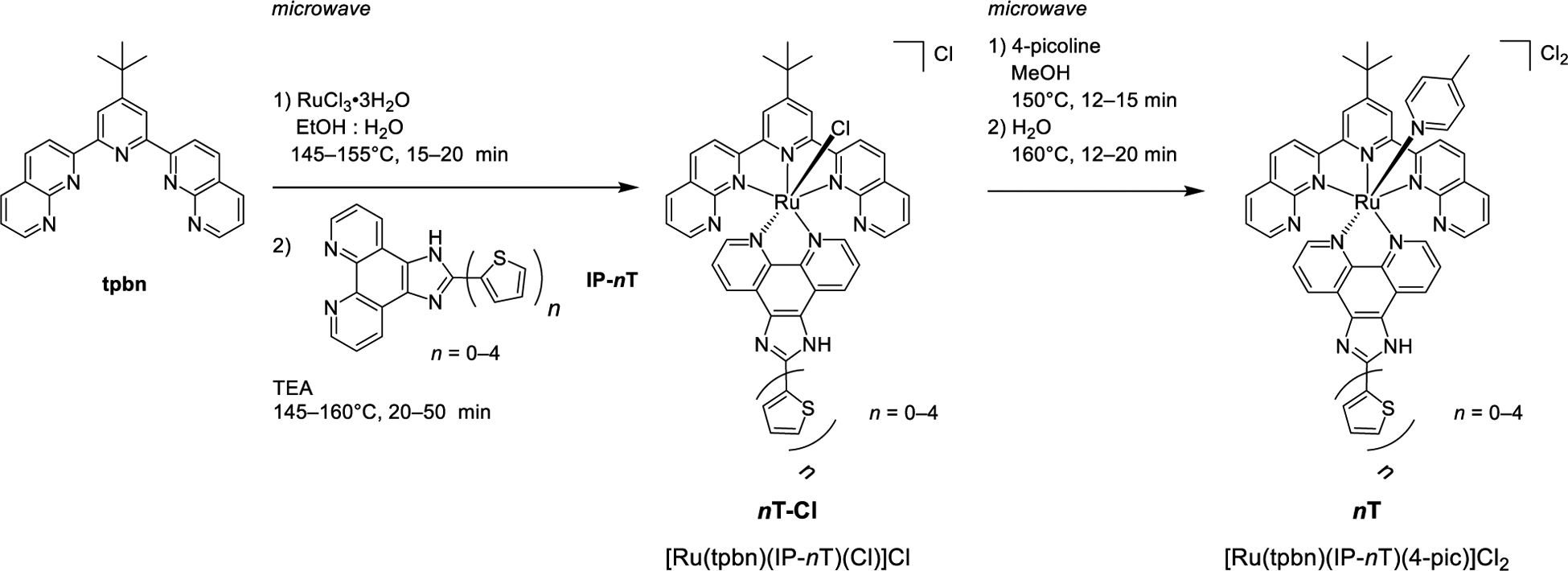
Synthetic scheme for Ru(II) complexes 0T–4T.
0-Cl–4T-Cl were converted to their corresponding 4-pic complexes (0T–4T) by microwave irradiation in the presence of excess 4-pic. 0T–4T were purified with flash column chromatography on neutral alumina followed by size-exclusion chromatography on Sephadex® LH-20 to afford the final products in 36–55% yield and >95% purity, which were characterized by thin-layer chromatography, 1D 1H NMR and 2D 1H–1H COSY NMR spectroscopy (Figure 1, Figures S1–S11), high resolution ESI+ mass spectrometry (Figures S18, S20, S22, S24, S26), and HPLC (Figures S27–S31).
3.2. NMR Assignments
The signals in the 1D 1H NMR and 2D 1H–1H COSY NMR spectra of complexes 0T–4T were analyzed and assigned to confirm the structures of the complexes (Figure 1). Assignments were made based on the connectivity observed in 2D 1H–1H COSY NMR spectra, coupling constants observed in 1D 1H NMR spectra, and assignments previously reported for similar systems.[52,54,58,59]
Complexes 0T–4T are tris-heteroleptic, and due to the pseudo-octahedral geometry of the complexes and relative positioning of the three ligands (tpbn, 4-pic, and IP-nT), the signals from ligands tpbn and 4-pic integrate as two hydrogens each, while signals from IP-nT ligand are non-symmetrical and integrate as one hydrogen each. Spin systems that consist of signals that integrate as two hydrogens each were identified and assigned to the signals from tpbn and 4-pic ligands. First, hydrogen 3 was assigned as a characteristic singlet that integrates as two hydrogens (9.15–9.16 ppm across the 0T–4T series). Spin system 5′−6′−7′ was identified as a three-hydrogen spin system, and 6′ was assigned as the most upfield signal of the three (7.40–7.42 ppm across the 0T–4T series) that appears as a clear doublet of doublets with a well-pronounced J2. Hydrogens 5′ (8.33–8.34 ppm) and 7′ (7.95–8.01) were assigned based on their 1H–1H COSY correlation with 6′ and with each other and based on observed J-values of the couplings. Hydrogen 7′ was assigned as a doublet that has a smaller J-value of coupling with 6′ (J = 4 Hz), and 5′ was assigned as a d that has a larger J-value of coupling with 6′ (J = 8 Hz). The remaining two two-hydrogen spin systems correspond to spin systems 3′−4′ from tpbn and 2″−3″ from 4-pic. It is typical for hydrogen 3″, which is in the meta-position relative to the nitrogen of 4-pic, to appear upfield in the aromatic region, and 3″ was assigned as the signal at 6.97–6.98 ppm, the most upfield hydrogen among 3′, 4′, 2″ and 3″, and the most upfield hydrogen in the structures of 0T–4T complexes overall. Hydrogen 2″ was assigned as a doublet at 7.65–7.67 ppm based on its 1H–1H COSY correlation with 3″. By the process of elimination, spin system 3′−4′ was assigned as a pair of doublets at 9.00–9.02 ppm and 8.68–8.69 ppm. Hydrogen 3′ was assigned as a signal at 9.00–9.02 ppm based on the assumption that it has a similar environment to that of 3 (9.15–9.16 ppm), while the environment of 4′ is different from 3 and appears more upfield at 8.68–8.69 ppm.[54] Overall, the resulting order of chemical-shift assignments for the tpbn signals (3 > 3′ > 4′ > 5′ > 7′ > 6′) is in good correlation with the assignments reported for a 2,2′:6′,2′′-terpyridine ligand (tpy) in [Ru(tpy)2]2+ coordination complexes.[59] Last, the methyl signals from tpbn (singlet, 9H) and 4-pic (singlet, 3H) are clearly observed in the aliphatic region and are easily assigned as the 4-tBu (1.76–1.77 ppm) and 2″-Me (2.24 ppm) groups. The assignments were further confirmed by a 2D 1H–1H ROESY experiment carried out for 3T, which revealed characteristic through-space couplings for the following hydrogens: 3 with 3′ ; 4’ with 5’ ; 3 with 4-tBu ; 3″ with 4″-Me ; and a′ with 2″ (Figures S8-b, S8-c).
All signals from IP-nT ligands are non-symmetrical and integrate as one hydrogen each. The IP-nT ligand consists of three parts: the phenanthroline portion (hydrogens a, b, c, a′, b′, c′ in all complexes), the phenathroline-fused imidazo group (hydrogen d in 0T complex), and a thiophene tail (hydrogens d–f in 1T complex, d–h in 2T complex, d–j in 3T complex, and d–l in 4T complex). While isolated phenanthroline is symmetrical, the hydrogens in the phenanthroline in the IP-nT ligand (a + a′, b + b′, and c + c′) are non-equivalent due to their asymmetric spatial positioning relative to the axial 4-pic ligand. The IP-nT ligand is bound to the Ru(II) center orthogonal to the tpbn ligand and in such way that one side of the phenanthroline is facing toward the axial ligand 4-pic (hydrogens a′, b′, c′), and the other side is pointing away from 4-pic (hydrogens a, b, c). Ligand 4-pic causes a deshielding effect, which is pronounced in the chemical shifts of a′, b′, and c′. The Ru(II) center causes a shielding effect on hydrogens nearby (a + a′, ortho-position),[58] while hydrogens c + c′ (para-position) are too far to be affected by this effect. An additional factor influencing the chemical shifts of c and c′ is their spatial proximity to the non-coordinated nitrogens from the imidazo group of IP, which causes a very pronounced deshielding effect. In the absence of the axial ligand’s influence, the combination of these effects would result in the following trends for these spin systems: c > a > b and c′ > a′ > b′ (where “>” stands for more downfield). However, while a′ and a are shielded similarly by Ru(II), the spatial proximity of a′ to the nitrogen of 4-pic affects its chemical shift such that it is very similar to that of c′ and the observed sequence is still c > a > b for the a-b-c spin system but is a′ ~ c′ > b′ for the a′-b′-c′ spin system. It should also be noted that while all other hydrogens appear on the spectrum as sharp signals, two hydrogens appear broadened. Those hydrogens were assigned as c and c′. This observed broadening is attributed to the proximity of the nitrogens of the -N=-NH- imidazo group of the IP-nT ligand.
Spin systems a-b-c and a′-b′-c′ were first assigned in complex 0T. Complex 0T lacks any thiophene groups, which makes the assignment of these spin systems more straightforward. All tpbn and 4-pic hydrogens were assigned following the strategies already discussed. Based on the arguments presented above, hydrogen a′ was assigned as a sharp doublet that appears the most downfield at 9.39 ppm (dd, J1 = 5.5 Hz, J2 = 1.5 Hz, 1H), with J1 corresponding to Ja′b′. Hydrogen b′ was assigned as a signal at 8.33 pm through its coupling to a′, observed by 1H–1H COSY NMR (b′ would appear as a doublet of doublets (dd) but it is overlapping with 5′ from tpbn). Hydrogen b, which does not overlap any other signal, is observed at 7.34 ppm as a clear dd with relatively large J-values (J1 = 8.0 Hz, J2 = 5.5 Hz), with J2 corresponding to Jab (identical to Ja′b′ mentioned above) and J1 corresponding to Jcb. Hydrogen a was assigned as a dd at 7.61 pm (J1 = 5.5 Hz, J2 = 1.5 Hz) through its coupling to b, observed by 1H–1H COSY NMR, and Jab derived from this assignment (5.5 Hz) is identical to Ja′b′. The assignment of signals from the a-b-c and a′-b′-c′ spin systems was completed by assigning hydrogens c and c′. Hydrogen c was assigned as a broad d at 8.51 ppm with J = 8.0 Hz, which matches the Jcb value derived from the assignment of hydrogen b. Hydrogen c′ was assigned as a broad d at 9.26 ppm with J = 8.0 Hz, which is identical to Jcb and corresponds to Jc′b′. Overall, while a′, b′, and c′ appear more downfield than a, b, and c, respectively, due to the influence of 4-pic on a′-b′-c′ spin system, observed multiplicities and J-values are identical for a and a′, b and b′, and c and c′.
Spin systems a-b-c and a′-b′-c′ were then assigned in 1T–4T complexes, following the same strategy as described above for 0T. The chemical shifts of these hydrogens occurred within a narrow range across the 0T–4T series: a (7.51–7.61), b (7.25–7.34), c (8.51–8.60), a′ (9.36–9.39), b′ (8.32–8.33), c′ (9.26–9.42). Overall, the following trend in chemical shifts is consistently observed across the series 0T–4T (excluding hydrogens from the thiophene tail): a′, c′ > 3 > 3′ > 4′ > c > 5′ = b′ > 7′ > 2″ > a > 6′ > b > 3″.
The hydrogens from the thiophene tail were assigned last (d–f in IP-1T, d–h in IP-2T, d–j in IP-3T, and d–l in IP-4T). Hydrogens d, e and f all appear as dd, but e was distinguished from d and f as a dd with a noticeably larger second J-value (7.28 ppm, J1 = 5.0 Hz, J2 = 4.0 Hz). With two hydrogens left to assign, hydrogen d was assigned as more downfield than f, due to its spatial proximity to the −N=−NH- portion of the imidazo group that causes a deshielding effect. Hydrogen f is also deshielded due to its position next to the sulfur from the thiophene unit, but d is more deshielded by -N=-NH- than f by sulfur. As a result, d appears at 7.95 (dd, J1 = 4.0 Hz, J2 = 1.0 Hz) and f appears at 7.70 (dd, J1 = 5.0 Hz, J2 = 1.0 Hz).
In complex 2T, spin systems d-e and f-g-h from the thiophene unit were identified as the last five hydrogens to be assigned. Spin system d-e was identified as a two-hydrogen spin system (signals at 7.85 ppm and 7.36 ppm) and spin system f-g-h as a three-hydrogen spin system (signals at 7.45 ppm, 7.39 ppm, and 7.11 ppm). Hydrogens d and e both appear as dd. Hydrogen d was assigned as being the more downfield of the two, due to its spatial proximity to the −N=−NH- portion of the imidazo group from the IP-2T ligand that causes a strong deshielding effect. As a result, d appears at 7.85 ppm (d, J = 4.0 Hz) and e appears at 7.36 ppm (d, J = 4.0 Hz). Hydrogens f, g and h all appear as dd, but g was distinguished from f and h as a dd with a noticeably larger second J-value (7.11 ppm, J1 = 5.0 Hz, J2 = 3.5 Hz). Hydrogen h was assigned as being more downfield than f, due to the deshielding effect of the sulfur positioned next to h. As a result, h appears at 7.45 ppm (dd, J1 = 5.0 Hz, J2 = 1.0 Hz) and f appears at 7.39 ppm (dd, J1 = 3.0 Hz, J2 = 1.0 Hz).
In complex 3T, three spin systems from the thiophene unit (d-e, f-g and h-i-j) were identified as the last seven hydrogens to be assigned. Spin system h-i-j appeared as a three-hydrogen spin system (signals at 7.37 ppm, 7.23 ppm, and 7.05 ppm). Hydrogen i was distinguished from h and j as a dd with a noticeably larger second J-value (7.05 ppm, J1 = 5.0 Hz, J2 = 3.5 Hz). Hydrogen j was assigned as being more downfield than h, due to the deshielding effect caused by sulfur positioned next to j. As a result, j appears at 7.37 ppm (dd, J1 = 5.0 Hz, J2 = 1.0 Hz) and h appears at 7.23 ppm (d, J = 3.5 Hz). Spin systems d-e and f-g were identified as two-hydrogen spin systems (one of the systems consists of the signals at 7.83 ppm and 7.25 ppm, and the other consists of the signals at 7.25 ppm and 7.12 ppm). All four signals d, e, f, g appeared as a d with J = 3.5 Hz. Hydrogen d was assigned as the most downfield of the four, due to its spatial proximity to the −N=−NH- portion of the imidazo group from the IP-3T ligand that causes a strong deshielding effect. As a result, d appears at 7.83 ppm. Hydrogen e was assigned as a signal at 7.25 pm through the observed coupling to d. By the process of elimination, the spin system consisting of signals at 7.25 ppm and 7.12 ppm was assigned to spin system f-g. Hydrogen f was assigned as the more downfield of the two (7.25 ppm), due to its spatial proximity to the −N=−NH- portion of the imidazo group from the IP-3T ligand that causes a strong deshielding effect. Even though f is positioned quite far from that group, it is possible that it still feels its influence, while g is positioned even farther away from that group. An additional argument for assigning the signal at 7.25 ppm as f is that this signal appears with the same chemical shift as e, and e would be expected to have an environment more similar to f than to g.
In complex 4T, four spin systems from the thiophene unit (d-e, f-g, h-i and j-k-l) were identified as the last nine hydrogens to be assigned. Spin system j-k-l was identified as a three-hydrogen spin system (signals at 7.01 ppm, 7.14 ppm, and 7.31 ppm). Hydrogen k was distinguished from j and l as a dd with a much larger second J-value (7.01 ppm, J1 = 5.6 Hz, J2 = 3.5 Hz). Hydrogen l was assigned as more downfield than j, due to the deshielding effect of sulfur positioned next to l. As a result, l appears at 7.31 ppm (d, J = 4.9 Hz) and j appears at 7.14 ppm (d, J = 3.5 Hz). Spin systems d-e, f-g, and h-i were identified as two-hydrogen spin systems (one of the systems consists of the signals at 7.81 ppm and 7.26 ppm, and the other consists of the signals at 7.20 ppm and 6.98 ppm, and another consists of the signals at 7.03 ppm and 6.98 ppm). Hydrogen d was assigned as the most downfield of the four, due to its spatial proximity to the −N=−NH- portion of the imidazo group of the IP-4T ligand that causes a strong deshielding effect. As a result, d appears at 7.81 ppm. Hydrogen e was assigned as a signal at 7.26 pm through the observed coupling to d. By the process of elimination, the spin systems consisting of signals at 7.20 ppm and 6.98 ppm, and 7.03 ppm and 6.98 ppm correspond to spin system f-g and h-i. Hydrogen f was assigned as the more downfield of the four (7.20 ppm), due to its spatial proximity to the −N=−NH portion of the imidazo group of the IP-4T ligand that causes a strong deshielding effect. Even though f is positioned quite far from that group, it is possible that it still feels its influence, while g, h and i are positioned even further away from that group. An additional argument for assigning the signal at 7.20 ppm as f is that this signal appears with a chemical shift that is close to that of e (7.26 ppm), and e would be expected to have an environment more similar to f than to g, h or i. Hydrogen g was assigned as a signal at 6.98 pm through the observed coupling to f. By the process of elimination, the spin system consisting of signals at 7.03 ppm and 6.98 ppm corresponds to spin system h-i. Hydrogen h is in an environment similar to that of g (6.98 ppm), and was therefore assigned as the signal at 6.98 ppm. By process of elimination and also through the observed correlation to h, i was assigned as a signal at 7.03 ppm.
Hydrogens from the thiophene tails in the 1T–4T complexes appear between 6.98 ppm and 7.95 ppm, which falls between the chemical shifts of 7′ and 3″. Hydrogens closest to the −N=−NH- portion of the imidazo group of the IP-nT ligand and hydrogens from the terminal thiophene unit are the most diagnostic, while hydrogens from internal thiophene units are less distinct. The most downfield hydrogen is d (7.81–7.95 across the 1T–4T series), which is significantly deshielded by its spatial proximity to the −N=−NH- portion of the imidazo group. Another deshielded hydrogen is the hydrogen on the terminal thiophene unit that is closest to the sulfur (f in 1T, h in 2T, j in 3T, l in 4T), which appears at 7.31–7.70 ppm across the series. Another characteristic hydrogen is the hydrogen in the middle of the three-hydrogen spin system of the terminal thiophene unit (e in 1T, g in 2T, i in 3T, k in 4T), which appears upfield relative to its neighboring hydrogens and as one of the most upfield hydrogens of the thiophene tail overall (7.01–7.28 ppm).
A detailed step-by-step description of the 1H NMR signal assignments in the NMR spectra of complexes 0T–4T is included in the Supporting Information.
3.3. Spectroscopy and Photophysics
3.3.1. UV-visible absorption
The UV-visible spectra of the complexes investigated in this series are shown in Figure 2 and local absorption maxima with extinction coefficients are tabulated in Table 1. Generally, the absorption spectra are defined by three regions. First, a series of sharp and intense IL (intraligand) ππ* bands dominate below 400 nm, involving transitions that are localized on the ligands. Diagnostic peaks occur for tpbn between 305 and 350 nm, while the those for phen (proximal phen of IP-nT) and 4-pic were evident near 260–270 and 210 nm, respectively. These positions are consistent with the degree of π-conjugation in the free ligands.
Figure 2.

Ground state electronic absorption spectra of the series as Cl− salts in MeCN at room temperature.
Table 1.
Peak wavelengths and log extinction coefficients
| Cmpd | Peak wavelength/nm (log ε) |
|---|---|
| phen | 265 (4.75), 291 (4.46), 306 (4.49), 318 (4.65), 350 (4.44), 371 (4.40), 500 (3.82), 548 (3.76), 724 (3.09) |
| 0T | 243 (4.61), 281 (4.42), 318 (4.40), 351 (4.23), 368 (4.13), 423 (3.53), 506 (3.57), 551 (3.50), 726 (2.82) |
| 1T | 241 (4.80), 293 (4.80), 302 (4.73), 318 (4.76), 348 (4.64), 429 (3.88), 505 (3.87), 549 (3.77), 726 (3.02) |
| 2T | 241 (4.68), 305 (4.50), 317 (4.58), 356 (4.60), 371 (4.63), 508 (3.80), 557 (3.67), 731 (2.92) |
| 3T | 242 (4.91), 302 (4.72), 317 (4.76), 353 (4.71), 372 (4.77), 408 (4.77), 501 (4.08), 553 (3.91), 726 (3.13) |
| 4T | 241 (4.92), 294 (4.77), 317 (4.78), 351 (4.71), 371 (4.71), 437 (4.87), 555 (3.94), 726 (3.15) |
The second region involves 1ππ* IL or ILCT transitions localized on the IP-appended thiophene rings, with bands that systematically increase in intensity and decrease in energy with increasing number of thienyl rings. For 1T and 2T these bands, typically occurring near 330 and 380 nm,[57,60] respectively, are obscured by the 1ππ* transitions on the chelating ligands. In 3T and 4T, the 1ILCT transitions are easily visible and centered near 408 and 437 nm, extending as far as 500 nm in 4T. Whether the transition is formally IL or ILCT depends on the number of thienyl rings, where n=3 or 4 likely have some charge transfer character and better described as ILCT.[61,62]
Third, lower intensity absorption peaks near 500, 550, and 725 nm corresponding to 1MLCT transitions in all the complexes. The local absorption maxima near 725 nm were also assigned as spin-allowed transitions because the estimated energies of the 3MLCT states from emission were significantly lower (vide infra). With the energy of this longest wavelength transition being largely unaffected by the different IP-nT ligands across the series, it is reasonable to ascribe this absorption to the Ru(dπ)→tpbn(π*) transition. This is an unusually long wavelength absorption for a Ru(II)-based 1MLCT state in a system with neutral diimine ligands and reflects the contributions of the chromophoric tpbn ligand. The analogous transition for the archetype [Ru(bpy)3]2+ occurs at around 450 nm, and attempts to drive the transition to longer wavelengths with π-expansive ligands usually do not extend much beyond the upper 500s.[63]
The key to achieving long-wavelength absorption in Ru(II) complexes with π-expansive ligands is to extend the conjugation orthogonal to the direction of the M–N bond such that the entire length of conjugation is in close proximity to the metal (rather than having the π-conjugation length extend away from the metal as in the case of the IP-nT ligand). This is why the NNN chromophoric ligand lowers the energies of the MLCT transitions, but the NN PDT-active ligand does not, despite both being π-extended. Additionally, the non-coordinating nitrogen atoms in the tpbn ligand serve to lower the energy of the tpbn(π*) orbital, which further lowers the energy of the MLCT transitions. This effect has been reported in a comparison of Ru(II) complexes derived from 2,2′-biquinoline versus 2,2′-binaphthyridyl ligands.[64,65] Unlike the tris-bidentate Ru(II) systems that are susceptible to ligand loss due to steric crowding of the coordination sphere, the combination of NNN, NN, and N ligands in the present series produced complexes that were relatively stable in the presence or absence of light under routine characterization and spectroscopy.
3.3.2. Emission spectroscopy
The compounds exhibited broad and featureless steady-state emission spectra, all having maxima at 805±5 nm (Table 2). This indicates that the lowest-lying emissive 3MLCT state is the same in all the complexes, and it is not influenced by the IP-nT or phen ligand. This conclusion is further supported by the essentially identical lifetimes (Tem=62±4 ns) of all the complexes. The energy of this emission (≈1.5 eV) indicates a very low-lying 3MLCT state, consistent with the inferences of the low energy 1MLCT absorption in the UV-vis spectrum, and suggests a tpbn-localized emissive 3MLCT state in all the complexes.
Table 2.
Room temperature emission and singlet oxygen data. Singlet oxygen quantum yields were measured at the wavelength indicated in parentheses, at the lowest energy peak in the excitation spectrum and at a representative NIR wavelength.
| Cmpd | λem/nrn | Tem/ns | ΦΔ |
|---|---|---|---|
| phen | 802 | 65 | 0.10 (555) 0.10 (630) |
| 0T | 810 | 58 | 0.06 (510) 0.05 (630) |
| 1T | 807 | 61 | 0.02 (506) 0.02 (630) |
| 2T | 803 | 61 | <0.01 (512) 0.02 (630) |
| 3T | 800 | 60 | <0.01 (422) 0.02 (630) |
| 4T | 800 | 66 | 0.01 (445) 0.05 (630) |
3.3.3. Transient absorption
Transient absorption lifetimes and differential excited state absorption (ESA) spectra were measured on deaerated MeCN solutions following excitation by a 355 nm laser pulse (pulse width around 5 ns). The lifetimes are tabulated in Table 3, and the short- and long-interval ESA spectra are shown in Figure 3.
Table 3.
Transient absorption lifetimes
| Cmpd | λta /nm : TTA/ns |
|---|---|
| phen | 400: 62 |
| 560: 62 | |
| 0T | 390: 56 |
| 560: 55 | |
| 1T | 400: 57 |
| 560: 56 | |
| 2T | 400: 59 |
| 550: 58 | |
| 3T | 390: 60 |
| 560: 54 | |
| 4T | 390: 69 |
| 500: 59 | |
| 600: 53–97, 2.9–5.6×103 | |
| (weak) |
Figure 3.
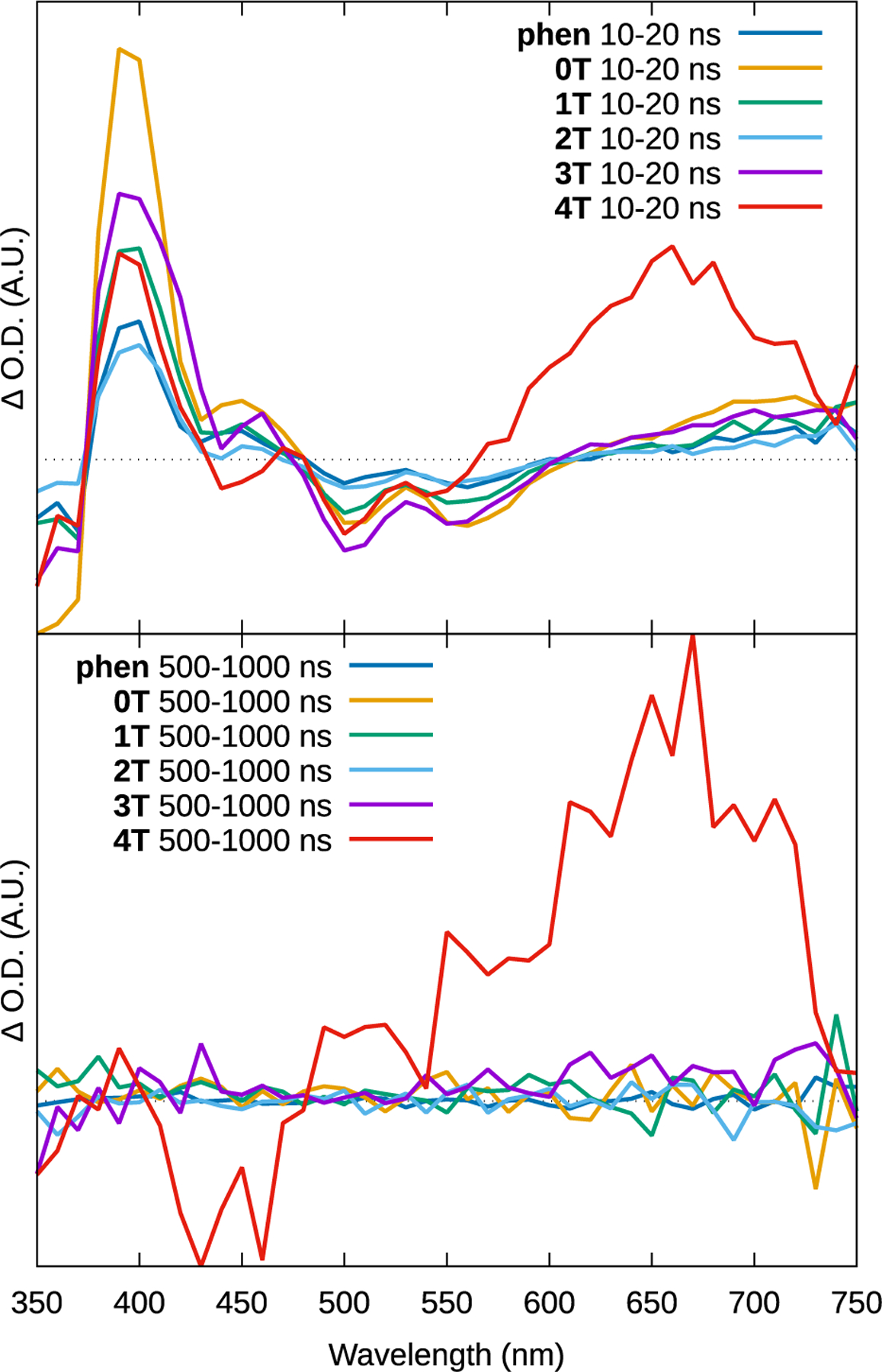
Excited state absorption profiles for the series, measured at room temperature in degassed MeCN.
Compounds phen and 0T–3T exhibited qualitatively similar ESA profiles. At short (≈10–20 ns) intervals after the excitation pulse, the spectra were dominated by the bleaching of the MLCT absorptions around 500 and 550 nm (the longer wavelength MLCT bleaches were not evident, likely owing to the instrument’s limited sensitivity in this range) plus the appearance of a new absorption near 400 nm associated with a new excited state transition. The TA lifetimes of the bleach and new absorption were the same for all complexes, and close for 4T, 58±4 ns. These lifetimes also corresponded well with the emission lifetimes and were assigned to the emissive 3MLCT state (cf. Table 2).
At early times following the excitation pulse, compound 4T showed a similar MLCT bleach and new absorption. There was also a broad absorption in the 550–750 nm range. This signal was longer lived, with an accompanying bleach centered around 440 nm, which corresponds to the IP-4T 1ππ* transition in the UV-vis spectrum. The shape of this longer-lived ESA spectrum is the characteristic signature of the 3ILCT state that we have observed in other complexes,[47,52,57,60,66] and compares well with the spectrum (but not the lifetime) of the free IP-4T ligand (T =28 μs)[62]. The intensity of this long-lived profile was comparitively weak, indicating that in 4T the population of the 3ILCT state is inefficient, and its absence from 3T suggests that the state is energetically inaccessible in complexes with the less π-extended NN ligands.
3.3.4. Excited state pathways
The Jablonski diagram in Figure 4 depicts the proposed excited state model in this series. The excitation by a 355 nm laser pulse to any upper 1MLCT or 1IL state is followed by rapid internal conversion to the low-lying phosphorescent tbpn-based 3MLCT state. Based on its emission energy, around 1.54 eV, this state is common across the series, and it is assigned as 3MLCT based on its <100 ns lifetime.[41] The lowest-lying 3ILCT should be accessible in the most conjugated 4T complex, the energy of which can be approximated from the quaterthiophene triplet at 1.8 eV.[67] Uphill population of the long-lived 3ILCT state from the 3MLCT is possible but very unfavorable in 4T, and not possible at all in 0T–3T. Our previous work[40,47,57,60,66] has shown that efficient population of the 3ILCT state coincides with potent 1O2 sensitization, and the present results are consistent.
Figure 4.

Jablonski diagram depicting the proposed excited state pathways in the series. The energies are not to scale.
The photophysics lead to a useful conclusion. The strategy of choosing ligands to drive the MLCT absorption to longer wavelengths comes at a cost: lowering the 3MLCT state too far prevents efficient population of the long-lived 3ILCT states, and this in turn negatively impacts the ability of the PS to effect photochemically useful reactions via 1O2.
3.3.5. Singlet oxygen sensitization
Table 2 reports the quantum yield for singlet oxygen sensitization (ΦΔ) for the series. As the photophysical model predicts, the performance was poor, no greater than 2% at the excitation maximum in any thiophene-containing complex. Sensitization was observed with NIR wavelengths, but the low signal-to-noise ratio for absorption led to high error so the data using 630 nm-excitation is reported in the table. The intensity of the 1O2 signal at the longer wavelengths was lower, but the efficiency was higher. This points to the tpbn 3MLCT state being involved in 1O2 sensitization, and that population of this state from the long-wavelength tpbn 1MLCT excitation is more efficient than by relaxation of an excited state localized elsewhere in the molecule, i.e., some significant portion of the excited state energy returns to ground state by fluorescence or thermal mechanisms, especially in the complexes with more π-expanded NN ligands. The values for ΦΔ measured for 4T, the only complex with a detectable 3ILCT state, were slightly higher than the other thienyl-containing complexes at the longer wavelengths but were no better than 0T and even lower than phen. These observations underscore the importance of having a sufficiently low-energy 3ILCT state (relative to the 3MLCT state) for 1O2 generation.
3.4. In Vitro (Photo)biological Activity
3.4.1. Cellular assays
The dark and light-induced cytotoxicities for phen and 0T–4T complexes were determined in three melanoma lines growing as 2D adherent monolayers. These cell lines were selected to represent variability in terms of gender and aggressiveness (male human SKMEL28 vs. female human A375)[68] in addition to a highly pigmented murine line (B16F10) for correlation to in vivo work. Briefly, log phase cells were seeded, dosed with compound in triplicate (0.001–300 μM), and incubated for 48 h before cell viability was measured using the resazurin-reduction viability assay. Photocytotoxicity was determined in parallel by irradiating the well plates after overnight incubation with compound (13–19 h DLI) and measured for cell viability a day later. Logistic fits of the dose-response curves for both dark and light treatments provided EC50 values, the effective concentration to reduce relative viability by 50%. The margin of toxicity amplified by a given light treatment is defined as the phototherapeutic index (PI) and calculated as the ratio of dark to light EC50 values. The synthesized family exhibited excellent solubility in aqueous solution, even with the high ionic strength DPBS used for compound dilutions, making comparisons in terms of activities between compounds straightforward by EC50 and PI.
3.4.2. Dark cytotoxicity
The magnitudes of the cytotoxicities depended on the cell line for each compound while the order of cytotoxicity among the compounds remained constant across all cell lines (Figure 5a–c, Table S1–S3). Dark EC50 values ranged between 49–187 μM in A375, 69.2–115 μM in B16F10, and 73–143 μM in SKMEL28. Those compounds lacking thiophenes, phen and 0T, were generally the least toxic in the family. Using SKMEL28 for example in Figure 5c, the highest dark EC50 values were 128 and 143 μM for compounds phen and 0T, respectively. Upon addition of a single thiophene as in 1T, toxicity increased by 2-fold. The compound 1T stood out in the series as the most (but still only mildly) toxic across all cell lines with EC50 values ranging from 49.0 to 73.0 μM. Additional thiophenes generally improved dark toxicity in an additive manner towards ~100 μM. For the complex with the longest thiophene chain (4T), toxicity was low and remained constant across all lines (103–108 μM).
Figure 5.

In vitro cytotoxicity and photocytotoxicity of the compounds toward A375 (a, d), B16F10 (b, e), and SKMEL28 (c, f) melanoma cell lines. Treatments include dark (0 J cm−2; black circles) and 100 J cm−2 doses of 733 nm (purple cross), 633 nm (red triangle), 523 nm (green inverted triangle), and visible (peak maxima ~450 nm; open blue square). Plots a–c show Log (EC50 ± SEM) and d–f show PI, where PI is the ratio of dark to light EC50 values. All linear-scale values are listed in the SI. Light source spectral outputs are shown in Figure S36.
3.4.3. Photocytotoxicity
Following activation with various wavelengths of light, compound cytotoxicity was amplified across all cell lines (Figure 5a–c, Table S1–S3). The photocytotoxicities of complexes phen and 0T–4T were determined using a fluence of 100 J cm−2 delivered from cool white visible (400–700, 19 mW cm−2), green (523 nm, 18.5 mW cm−2), red (633 nm, 20 mW cm−2), or NIR (733 nm, 9 mW cm−2) light sources. The spectral output of these light devices is shown in Figure S36. The light treatments in the absence of compound were generally nontoxic to cells, except that the visible light treatment resulted in 10–20% cell kill with light alone, which was attributed to the shorter wavelength blue components and likely due to heat and/or activation of endogenous photosensitizers in the cells. These effects can be minimized with smaller fluences.[69] Photocytotoxicity followed the order of wavelength energy, where visible treatments were the most potent followed by green (523 nm), red (633 nm), then NIR (733 nm).
Regardless of wavelength or cell line, photocytotoxicity increased with the number of thiophene rings in the IP-nT ligand. The compounds lacking any thiophene rings (phen, 0T) were the least photocytotoxic with EC50 values >30 μM and in many cases ≥100 μM. Compared to complex 0T, lacking any thiophenes, visible EC50 values improved by 5–10 fold upon inclusion of a single thiophene (1T). This improvement continued for each additional thiophene up to maximal effect in 4T with 10-fold (B16F10), 34-fold (SKMEL28), and 61-fold (A375) improvements over 0T. Compound 4T with the most π-extended ligand, was the most potent complex in the series and had single-digit micromolar EC50 values across the three cells lines with most of the light conditions. In the SKMEL28 cell line, 4T produced EC50 values of 1.18, 2.28, 5.85 and 8.79 μM for vis, green, red, and NIR, respectively. In the A375 cell lines these values ranged from 1.14 to 12.4 μM, and in the B16F10 cell line they ranged from 3.72 to 16.1 μM.
3.4.4. Phototherapeutic indices (PIs)
Because the light EC50 values include contributions from the inherent dark toxicities of the photosensitizers, the ratio of the dark and light EC50 values, or phototherapeutic index (PI), is a better approximation for comparing light-induced potencies. Across all cell lines, PIs increased systematically with number of thiophenes, with 4T being the most photoactive. PI values were largest for the amelanotic (no pigmentation)[70,71] cell lines A375 and SKMEL28 and strongly attenuated in the melanotic (pigmented)[70] B16F10 cell line.
For all compounds in all cell lines, the largest PIs were obtained with the highest photon energy treatments (visible). Values for PIvis ranged from 2–90 in A375, 2–29 in B16F10, and 4–88 in SKMEL28 cells. By comparison, the NIR treatments were less effective but promising nonetheless, with PI733nm ranging from 1–8 in A375, 1–7 in B16F10, and 1–12 in SKMEL28 cells. Compound 4T was most potent under all light conditions.
The PSs in this series clustered into four groups based on PI: inactive, low, moderate, and high. The reference complexes phen and 0T were either inactive or exhibited low activity (PIs <5) across the different cell lines and light conditions explored. Compound 1T had low activity, with almost all conditions yielding PIs less than 10. Complexes 2T and 3T were moderate in activity, having PIs near 10 and 20, respectively, for the visible light condition in A375 and B16F10 and about 5 to 9-fold higher in SKMEL28. These PI values were attenuated with the longer wavelengths of light, with lower photon energies yielding smaller PIs.
4T was the only compound in the present series that was considered to be highly active, with PIvis values of 90, 88, and 29 in A375, SKMEL28, and B16F10 cells, respectively. These values were reduced to 8, 12, and 7 on going from broadband visible light to 733-nm NIR light. As observed for the other compounds, the pigmented B16F10 cell line generally proved to be more resistant to PDT effects by 4T compared to the amelanotic lines and partially explains the lower PI values obtained in this cell line. These differences were less pronounced with longer wavelength NIR light as demonstrated for 4T where there was no significant difference in PI between the amelanotic A375 and pigmented B16F10 cell lines. This response agrees with what would be expected in that the more tissue-penetrating wavelengths of light should lead to less attenuation by melanin.
The much larger PIs for 4T could not be attributed to higher 1O2 quantum yields given that all of the thiophene-containing compounds exhibited similarly low efficiencies for 1O2 production at their excitation maxima under ambient oxygen (ΦΔ ≤2%). However, 4T was the only compound that produced the characteristic signature of the 3ILCT state in its TA spectrum and exhibited an excited state lifetime as long as 3–6 μs versus about 60 ns for 1T-3T. Accessible 3ILCT states are usually associated with much higher 1O2 quantum yields and PIs compared to 4T,[47,57,66] but these systems have been limited to Ru(II) polypyridyl complexes with 1MLCT/3MLCT states that are much higher in energy (i.e., systems that do not absorb NIR light) where the 3ILCT state is lower in energy than the 3MLCT state and thus dominates excited state relaxation. While the TA experiments show that the 3ILCT state is accessible in 4T, the energy of the 3MLCT state estimated from emission measurements (1.54 eV) is lower than that of the 3ILCT state (1.8 eV).[67] The uphill 3MLCT→3ILCT path is further corroborated by the very weak positive ESA signal observed in the TA measurements for the 3ILCT transient. Most of the excited state decay in 4T occurs through the 3MLCT state, which is similar in energy, lifetime, and 1O2 quantum yield in all of the complexes. Regardless, the small fraction of 3ILCT states that do contribute to excited state decay in 4T have a marked impact on the photobiological activity.
4. CONCLUSIONS
We hypothesized that by combining the [Ru(NNN)(NN)(N)]2+ NIR scaffold with potentially PDT-active IP-nT ligands that we could create photocytotoxic NIR PSs at some critical value of n. It was found that thiophenes had little influence on NIR absorption, but produced their diagnostic signatures in the UV-Vis region defined by the number of thiophenes. The chromophoric ligand tpbn was key to NIR absorption due to the installation of low lying Ru(dπ)→tpbn(π*) states for decreasing the 1MLCT/3MLCT energies. All compounds absorbed past 800 nm, with longest wavelength 1MLCT abs. maxima centered around 730 nm. Emission studies confirmed the complexes had a common 3MLCT state around 1.54 eV with a lifetime of approx. 60 ns. The only compound showing evidence of an additional triplet excited state was 4T, whereby TA experiments indicated a very weak transient with the characteristic 3ILCT signature and a 3–6 μs lifetime. All other compounds, having higher-lying 3ILCT states, had inaccessible ILCT states and their photophysics instead dominated by the MLCT state.
4T also had the largest PI values across three different cell lines and four different light treatments, and was the only compound considered to have high activity (single-digit EC50 values and PIvis up to 90) for a NIR PS. Photocytotoxicity from 3ILCT states generally arises from very efficient singlet oxygen sensitization. However, the singlet oxygen quantum yield for 4T was around 5% or less depending on the excitation wavelength and did not differ from the other thiophene-containing family members. Estimated excited state energies for the complex’s 3MLCT state and the quaterthiophene’s 3ILCT state placed the 3ILCT in 4T energetically uphill from the 3MLCT state. As a result, the 3ILCT did not dominate the observed photophysics as evidenced by both the weak ESA and the low singlet oxygen quantum yields but likely contributed to the more potent photocytotoxic effects. From this, it appears that n=4 is the minimum number of thiophenes required for photobiological potency with this NIR class of Ru(II) PSs and that n>4 may in fact prove even better as long as aqueous solubility is maintained and the reduced 3ILCT energies do not facilitate competing radiationless decay according to the energy gap law.
The study highlights a fundamental limit in creating Ru(II)-based NIR PSs for phototherapy: the 3ILCT energy must be kept substantially lower than the 3MLCT energy to achieve more potent photocytotoxic effects. In the IP-nT ligand class this means that IP-nT ligands work well in blue-green absorbing Ru(II) polypyridyl complexes (higher energy MLCT) but require larger n in the NIR class. Nevertheless, the ability to employ NIR light, as exemplified by the 733-nm effects with the highly pigmented B16F10 cell line, underscores the potential that such systems hold despite lower potencies relative to their Ru(II) trisdiimine counterparts. Efforts are underway to test our predictions regarding n and the role of 3ILCT states by preparing complexes of n>4.
Supplementary Material
5.2. Acknowledgements
S.A.M., C.G.C., and R.P.T. thank the National Cancer Institute (NCI) of the National Institutes of Health (NIH) (Award R01CA222227) for support. The content in this review is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. S.A.M. also thanks Dr. Daniel Todd as UNCG’s Triad Mass Spectrometry Facility manager and his assistants Jennifer Simpson and Diane Wallace. We thank Dr. Franklin Moy and Dr. Brian Edwards for their experimental support and instrument maintenance as UNCG’s NMR facility manager and UTA’s NMR facility manager, respectively. We also thank Dr. Brian Edwards for his help with ROESY NMR experiment. The table of contents graphic was created using Biorender.com.
Footnotes
Synthetic characterization (1D and 2D NMR, HPLC, HRMS) and (photo)biological data are included in the Supporting Information.
S.A.M. has a potential research conflict of interest due to a financial interest with Theralase Technologies, Inc. and PhotoDynamic, Inc. A management plan has been created to preserve objectivity in research in accordance with UTA policy.
6. REFERENCES
- [1].Henderson BW, Dougherty TJ, Eds. , Photodynamic Therapy: Basic Principles and Clinical Applications, CRC Press, 1992. [Google Scholar]
- [2].Dougherty TJ, Gomer CJ, Henderson BW, Jori G, Kessel D, Korbelik M, Moan J, Peng Q, JNCI J Natl. Cancer Inst 1998, 90, 889–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Bonnett R, Chemical Aspects of Photodynamic Therapy, Gordon And Breach Science Publishers, 2000. [Google Scholar]
- [4].Hamblin MR, Mroz P, Advances in Photodynamic Therapy: Basic, Translational, and Clinical, Artech House, Norwood, MA, 2008. [Google Scholar]
- [5].Agostinis P, Berg K, Cengel KA, Foster TH, Girotti AW, Gollnick SO, Hahn SM, Hamblin MR, Juzeniene A, Kessel D, Korbelik M, Moan J, Mroz P, Nowis D, Piette J, Wilson BC, Golab J, CA. Cancer J. Clin 2011, 61, 250–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Hamblin MR, Huang Y, Eds. , Handbook of Photomedicine, CRC Press, Boca Raton, FL, 2013. [Google Scholar]
- [7].Allison RR, Future Oncol 2014, 10, 123–124. [DOI] [PubMed] [Google Scholar]
- [8].Benov L, Med. Princ. Pract 2015, 24, 14–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Kostron H, Hasan T, Eds. , Photodynamic Medicine: From Bench to Clinic, Royal Society Of Chemistry, Cambridge, 2016. [Google Scholar]
- [10].Baptista MS, Cadet J, Di Mascio P, Ghogare AA, Greer A, Hamblin MR, Lorente C, Nunez SC, Ribeiro MS, Thomas AH, Vignoni M, Yoshimura TM, Photochem. Photobiol 2017, 93, 912–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Gollnick SO, Vaughan L, Henderson BW, Cancer Res. 2002, 62, 1604–1608. [PubMed] [Google Scholar]
- [12].Castano AP, Mroz P, Hamblin MR, Nat. Rev. Cancer 2006, 6, 535–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Mroz P, Hashmi JT, Huang Y-Y, Lange N, Hamblin MR, Expert Rev. Clin. Immunol 2011, 7, 75–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Gollnick SO, J. Natl. Compr. Cancer Netw. JNCCN 2012, 10 Suppl 2, S40–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Anzengruber F, Avci P, de Freitas LF, Hamblin MR, Photochem. Photobiol. Sci 2015, 14, 1492–1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Shams M, Owczarczak B, Manderscheid-Kern P, Bellnier DA, Gollnick SO, Cancer Immunol. Immunother 2015, 64, 287–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Vatansever F, Hamblin MR, in Cancer Immunol. (Ed.: Rezaei N), Springer Berlin Heidelberg, Berlin, Heidelberg, 2015, pp. 383–399. [Google Scholar]
- [18].Yang Y, Hu Y, Wang H, Oxid. Med. Cell. Longev 2016, 2016, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Hwang HS, Shin H, Han J, Na K, J. Pharm. Investig 2018, 48, 143–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Korbelik M, Lasers Surg. Med 2018, 50, 491–498. [DOI] [PubMed] [Google Scholar]
- [21].Nath S, Obaid G, Hasan T, Photochem. Photobiol 2019, 95, 1288–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Falk-Mahapatra R, Gollnick SO, Photochem. Photobiol 2020, php.13253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Dougherty TJ, Kaufman JE, Goldfarb A, Weishaupt KR, Boyle D, Mittleman A, Cancer Res. 1978, 38, 2628–2635. [PubMed] [Google Scholar]
- [24].Dougherty TJ, in Reg. Cancer Ther. Cancer Drug Discov. Dev (Eds.: Schlag PM, Stein U, Eggermont AMM), Humana Press, Totowa, NJ, 2007, pp. 117–132. [Google Scholar]
- [25].Dougherty TJ, in Photodyn. Ther. Dis. Head Neck (Ed.: Biel MA), Plural Publishing, San Diego, CA, 2008, pp. 1–18. [Google Scholar]
- [26].Levy J, Levy E, in Handb. Photodyn. Ther. Updat. Recent Appl. Porphyr.-Based Compd (Eds.: Pandey RK, Dougherty TJ, Kessel D), World Scientific Publishing, Singapore, 2016, pp. 531–548. [Google Scholar]
- [27].Hamblin MR, Photochem. Photobiol 2020, php.13190. [Google Scholar]
- [28].De Pinillos Bayona AM, Mroz P, Thunshelle C, Hamblin MR, Chem. Biol. Drug Des 2017, 89, 192–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].McFarland SA, Mandel A, Dumoulin-White R, Gasser G, Curr. Opin. Chem. Biol 2020, 56, 23–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Stranadko EF, Photodynamic Therapy and Photodyagnosis 2015, 4, 3–10. [Google Scholar]
- [31].Bergman SD, Gut D, Kol M, Sabatini C, Barbieri A, Barigelletti F, Inorg. Chem 2005, 44, 7943–7950. [DOI] [PubMed] [Google Scholar]
- [32].Al-Afyouni MH, Rohrabaugh TN, Al-Afyouni KF, Turro C, Chem. Sci 2018, 9, 6711–6720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Loftus LM, Al-Afyouni KF, Rohrabaugh TN, Gallucci JC, Moore CE, Rack JJ, Turro C, J. Phys. Chem . C 2019, 123, 10291–10299. [Google Scholar]
- [34].Notaro A, Jakubaszek M, Rotthowe N, Maschietto F, Vinck R, Felder PS, Goud B, Tharaud M, Ciofini I, Bedioui F, Winter RF, Gasser G, J. Am. Chem. Soc 2020, 142, 6066–6084. [DOI] [PubMed] [Google Scholar]
- [35].Lameijer LN, Ernst D, Hopkins SL, Meijer MS, Askes SHC, Le Dévédec SE, Bonnet S, Angew. Chem. Int. Ed 2017, 56, 11549–11553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Lv Z, Wei H, Li Q, Su X, Liu S, Zhang KY, Lv W, Zhao Q, Li X, Huang W, Chem. Sci 2018, 9, 502–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].van Rixel VHS, Ramu V, Auyeung AB, Beztsinna N, Leger DY, Lameijer LN, Hilt ST, Le Dévédec SE, Yildiz T, Betancourt T, Gildner MB, Hudnall TW, Sol V, Liagre B, Kornienko A, Bonnet S, J. Am. Chem. Soc 2019, 141, 18444–18454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Yu Q, Huang T, Liu C, Zhao M, Xie M, Li G, Liu S, Huang W, Zhao Q, Chem. Sci 2019, 10, 9091–9098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Bevernaegie R, Doix B, Bastien E, Diman A, Decottignies A, Feron O, Elias B, J. Am. Chem. Soc 2019, 141, 18486–18491. [DOI] [PubMed] [Google Scholar]
- [40].Roque J, Havrylyuk D, Barrett PC, Sainuddin T, McCain J, Colón K, Sparks WT, Bradner E, Monro S, Heidary D, Cameron CG, Glazer EC, McFarland SA, Photochem. Photobiol 2020, 96, 327–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Juris A, Balzani V, Barigelletti F, Campagna S, Belser P, von Zelewsky A, Coord. Chem. Rev 1988, 84, 85–277. [Google Scholar]
- [42].Kalyanasundaram K, Photochemistry of Polypyridine and Pophyrin Complexes, Acad. Pr, London, 1992. [Google Scholar]
- [43].Balzani V, Juris A, Venturi M, Campagna S, Serroni S, Chem. Rev 1996, 96, 759–834. [DOI] [PubMed] [Google Scholar]
- [44].Balzani V, Credi A, Venturi M, Coord. Chem. Rev 1998, 171, 3–16. [Google Scholar]
- [45].Balzani V, Juris A, Coord. Chem. Rev 2001, 211, 97–115. [Google Scholar]
- [46].Campagna S, Puntoriero F, Nastasi F, Bergamini G, Balzani V, in Photochem. Photophysics Coord. Compd. I (Eds.: Balzani V, Campagna S), Springer Berlin Heidelberg, Berlin, Heidelberg, 2007, pp. 117–214. [Google Scholar]
- [47].Monro S, Colón KL, Yin H, Roque J, Konda P, Gujar S, Thummel RP, Lilge L, Cameron CG, McFarland SA, Chem. Rev 2019, 119, 797–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Shi G, Monro S, Hennigar R, Colpitts J, Fong J, Kasimova K, Yin H, DeCoste R, Spencer C, Chamberlain L, Mandel A, Lilge L, McFarland SA, Coord. Chem. Rev 2015, 282–283, 127– 138. [Google Scholar]
- [49].Fong J, Kasimova K, Arenas Y, Kaspler P, Lazic S, Mandel A, Lilge L, Photochem. Photobiol. Sci 2015, 14, 2014–2023. [DOI] [PubMed] [Google Scholar]
- [50].Meijer MS, Talens VS, Hilbers MF, Kieltyka RE, Brouwer AM, Natile MM, Bonnet S, Langmuir 2019, 35, 12079–12090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Badiei YM, Polyansky DE, Muckerman JT, Szalda DJ, Haberdar R, Zong R, Thummel RP, Fujita E, Inorg. Chem 2013, 52, 8845–8850. [DOI] [PubMed] [Google Scholar]
- [52].Lifshits L, Roque III J, Konda P, Monro S, Cole HD, von Dohlen D, Kim S, Gagan D, Thummel RP, Cameron CG, Gujar S, McFarland SA, Chem Sci 2020, (In review). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Zong R, Thummel RP, J. Am. Chem. Soc 2005, 127, 12802–12803. [DOI] [PubMed] [Google Scholar]
- [54].Haberdar R, The Synthesis and Study of 4-Aza-Acridine Type Ligands and Their Ru(II) Complexes, University of Houston, 2012. [Google Scholar]
- [55].Wang Z, in Compr. Org. Name React. Reag, John Wiley & Sons, Inc., Hoboken, NJ., 2010. [Google Scholar]
- [56].Arenas Y, Monro S, Shi G, Mandel A, McFarland S, Lilge L, Photodiag Photodyn Ther 2013, 10, 615–625. [DOI] [PubMed] [Google Scholar]
- [57].Ghosh G, Colón KL, Fuller A, Sainuddin T, Bradner E, McCain J, Monro SMA, Yin H, Hetu MW, Cameron CG, McFarland SA, Inorg. Chem 2018, 57, 7694–7712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Pazderski L, Pawlak T, Sitkowski J, Kozerski L, Szłyk E, Magn. Reson. Chem 2010, 48, 450–457. [DOI] [PubMed] [Google Scholar]
- [59].Pazderski L, Pawlak T, Sitkowski J, Kozerski L, Szlyk E, Magn. Reson. Chem 2011, 49, 237–241. [DOI] [PubMed] [Google Scholar]
- [60].McCain J, Colón KL, Barrett PC, Monro SMA, Sainuddin T, Roque III J, Pinto M, Yin H, Cameron CG, McFarland SA, Inorg. Chem 2019, 58, 10778–10790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Wan J, Ferreira A, Xia W, Chow CH, Takechi K, Kamat PV, Jones G, Vullev VI, J. Photochem. Photobiol. Chem 2008, 197, 364–374. [Google Scholar]
- [62].Roque J, Barrett PC, Cole HD, Lifshits L, Shi G, Monro S, von Dohlen D, Kim S, Russo N, Deep G, Cameron CG, marta erminia alberto SA McFarland, Chem. Sci 2020, 10.1039.D0SC03008B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Anderson PA, Richard Keene F, Meyer TJ, Moss JA, Strouse GF, Treadway JA, J. Chem. Soc. Dalton Trans 2002, 3820. [Google Scholar]
- [64].Thummel RP, Lefoulon F, Inorg. Chem 1987, 26, 675–680. [Google Scholar]
- [65].Thummel RP, Hegde V, Jahng Y, Inorg. Chem 1989, 28, 3264–3267. [Google Scholar]
- [66].Monro S, Cameron CG, Zhu X, Colón KL, Yin H, Sainuddin T, Hetu M, Pinto M, Fuller A, Bennett L, Roque J, Sun W, McFarland SA, Photochem. Photobiol 2019, 95, 267–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].de Melo JS, Silva LM, Arnaut LG, Becker RS, J. Chem. Phys 1999, 111, 5427–5433. [Google Scholar]
- [68].Rossi S, Cordella M, Tabolacci C, Nassa G, D’Arcangelo D, Senatore C, Pagnotto P, Magliozzi R, Salvati A, Weisz A, Facchiano A, Facchiano F, J. Exp. Clin. Cancer Res 2018, 37, 326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Hopkins SL, Siewert B, Askes SHC, Veldhuizen P, Zwier R, Heger M, Bonnet S, Photochem. Photobiol. Sci 2016, 15, 644–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Chen KG, Leapman RD, Zhang G, Lai B, Valencia JC, Cardarelli CO, Vieira WD, Hearing VJ, Gottesman MM, JNCI J Natl. Cancer Inst 2009, 101, 1259–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Bracalente C, Ibañez IL, Berenstein A, Notcovich C, Cerda MB, Klamt F, Chernomoretz A, Durán H, Oncotarget 2016, 7, 41154–41171. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.