

Abstract
BACKGROUND:
Gene regulatory networks control tissue homeostasis and disease progression in a cell-type specific manner. Ubiquitously expressed chromatin regulators modulate these networks, yet the mechanisms governing how tissue-specificity of their function is achieved are poorly understood. BRD4, a member of the BET (Bromo- and Extra-Terminal domain) family of ubiquitously expressed acetyl-lysine reader proteins, plays a pivotal role as a coactivator of enhancer signaling across diverse tissue types in both health and disease, and has been implicated as a pharmacologic target in heart failure. However, the cell-specific role of BRD4 in adult cardiomyocytes remains unknown.
METHODS:
We combined conditional mouse genetics, unbiased transcriptomic and epigenomic analyses, and classical molecular biology and biochemical approaches to understand the role of BRD4 in adult cardiomyocyte homeostasis.
RESULTS:
Here, we show that cardiomyocyte-specific deletion of Brd4 in adult mice leads to acute deterioration of cardiac contractile function with mutant animals demonstrating a transcriptomic signature enriched for decreased expression of genes critical for mitochondrial energy production. Genome-wide occupancy data show that BRD4 enriches at many downregulated genes (including the master co-activators Ppargc1a, Ppargc1b, and their downstream targets) and preferentially co-localizes with GATA4, a lineage determining cardiac transcription factor not previously implicated in regulation of adult cardiac metabolism. BRD4 and GATA4 form an endogenous complex in cardiomyocytes and interact in a bromodomain-independent manner, revealing a new functional interaction partner for BRD4 that can direct its locus and tissue specificity.
CONCLUSIONS:
These results highlight a novel role for a BRD4-GATA4 module in cooperative regulation of a cardiomyocyte specific gene program governing bioenergetic homeostasis in the adult heart.
Keywords: epigenetics, mitochondria, cardiomyocyte
INTRODUCTION
Heart failure (HF) is a clinical syndrome that occurs when a weakened heart is unable to maintain organ perfusion at a level adequate to meet tissue demand, resulting in shortness of breath, fatigue, and early death. This condition represents a challenge to the healthcare system accounting for almost 2% of all medical expenditures annually and, despite the current standard of care, carries a dismal prognosis with a 5-year mortality approaching 50%1,2. The mainstays of therapy for HF target neurohormonal signaling pathways with beta-adrenergic receptor antagonism, inhibition of the renin-angiotensin system, and augmentation of the natriuretic peptide system, all of which have improved survival in HF patients3. Despite these successes, the residual burden of morbidity and mortality in HF remains high, underscoring the need for novel treatment approaches1.
During HF pathogenesis, hemodynamic and neurohormonal stressors activate a network of signal transduction cascades that converge upon the nucleus, where specific transcription factors (TFs) drive maladaptive gene expression programs and modulate cell state4,5. In response, the heart undergoes pathologic remodeling characterized by cardiomyocyte (CM) hypertrophy, interstitial fibrosis, and altered substrate/energy utilization that culminates in organ-level contractile dysfunction. Studies in animal models of HF have implicated several nodal TFs (e.g., NFAT, GATA4, MEF2, and NF-κB) as drivers of disease progression through their induction of gene expression programs that may provide short-term adaptation to pathologic stress but whose sustained activation progressively weakens cardiac performance4,6. This stress-coupled activation of TFs in HF elicits global changes in chromatin structure and post-translational modifications on histone proteins, including lysine acetylation of histone tails, TFs, and other chromatin associated proteins. Given the central role of signal-coupled gene transcription in cardiac plasticity, manipulation of chromatin-dependent signaling as a therapeutic approach for HF is of intense interest7,8.
Previous work has established a crucial role for the BET (Bromo- and Extra-Terminal domain) family of acetyl-lysine reader proteins in the epigenetic control of adverse cardiac remodeling and HF pathogenesis9,10. There are four mammalian BET proteins (BRD2, BRD3, and BRD4 that are ubiquitously expressed and BRDT, which is testis-specific) that each contain two tandem bromodomains (BDs) that mediate acetyl-lysine binding11,12. BRD4 is the most studied member of this family and is a highly pursued target in cancer13,14. BRD4 associates with acetylated chromatin at active enhancers and promoters, where it interacts with the transcriptional machinery to activate transcription15. The ability to probe BET function in mammalian biology was accelerated by the creation of JQ1, a potent and specific small molecule tool compound that reversibly binds the BDs of all BET proteins with high affinity16,17. JQ1 competitively and reversibly displaces BET proteins from their acetyl-lysine interaction partners on enhancers (e.g., acetylated histones or acetylated TFs), thereby disrupting signaling between enhancers and promoters16,17. Prior studies have demonstrated that systemic delivery of JQ1 potently ameliorates HF pathogenesis in an array of rodent models9,10,18,19. However, the precise identity of cell-types and BET isoforms that mediate these therapeutic benefits remain a major unanswered question with important translational implications. As systemic delivery of pan-BET inhibitors such as JQ1 are unable to probe the gene-specific and cell-compartment-specific functions of BRD4 in vivo, we set out to discover the role of BRD4 in adult CMs using a conditional genetic approach in mice.
Here, we identify a critical role for BRD4 in maintaining murine cardiac homeostasis in vivo. As germline deletion of Brd4 results in zygotic implantation defects and haploinsufficiency results in multisystem developmental abnormalities20, we generated a Brd4 conditional allele and genetically deleted Brd4 in adult murine CMs. Tamoxifen-inducible genetic ablation of Brd4 in these cells resulted in a rapid and severe reduction in global left ventricular (LV) systolic function, LV cavity dilation, and uniform death. Integration of gene expression, genomic occupancy, and chromatin accessibility datasets demonstrated that BRD4 regulates mitochondrial gene expression. These analyses unveiled a new interaction between BRD4 and GATA4 in regulating mitochondrial gene expression and CM homeostasis. We confirmed a physical interaction between BRD4 and GATA4 that occurs in a BD-independent fashion, with co-occupancy at thousands of promoters and enhancers across the genome in mouse cardiomyocytes. Taken together, our data reveal that BRD4 is a critical regulator of basal CM homeostasis and identify an unappreciated role for GATA4 in regulating metabolic gene programs in the adult heart.
METHODS
The data, analytic methods, and study materials will be made available to other researchers for purposes of reproducing the results or replicating the procedure. All relevant reagents will be maintained within the Srivastava and Jain laboratories and will be supplied upon reasonable request.
RNA sequencing, mapping, and quantification
For adult CM-enriched samples, paired-end poly(A)-enriched RNA libraries were prepared with the ovation RNA-seq Universal kit (NuGEN; strand specific). High-throughput sequencing was done using a PE75 run on a NextSeq 500 instrument (Illumina). For embryonic samples, single-end poly(A)-enriched RNA libraries were prepared with the NEBNext Ultra II DNA Library Prep kit (NEB). High-throughput sequencing was done using a SE75 run on a NextSeq 500 instrument (Illumina). Reads were mapped to the mm10 reference mouse genome using STAR (v 2.7.3a) and assigned to Ensembl genes. After read quality control, we obtained quantifications for 38,293 genes in all 16 adult samples (2 Day2 Cre-control, 2 Day5 Cre-control, 3 Day2 Control, 3 Day5 Control, 3 Day2 Brd4-KO and 3 Day5 Brd4-KO) and 19,453 genes in all 6 embryonic samples (Tnnt2-Cre; Brd4flox/+ and Tnnt2-Cre; Brd4flox/flox).
Differential gene expression and pathway enrichment analysis
In order to identify genes differentially expressed in Cre-control, Control, and Brd4-KO samples, we quantified gene expression using raw counts and performed differential expression gene testing with DESeq221 (v.1.24.0 R package) using default settings. Statistical significance was set at a 5% false discovery rate (FDR; Benjamini-Hochberg) and significant genes were determined using an adj. p<0.05 and |Log2 FC|>1. Functional enrichment gene-set analysis for GO (Gene Ontology) terms was performed using Enrichr22 with adjusted p-values reported. Heat maps were generated using the Bioconductor package pheatmap (v.1.0.12) using rlog-transformed counts (values shown are rlog-transformed and row-normalized). Volcano plots were generated using the Bioconductor package EnhancedVolcano (v.1.2.0).
Comparison of RNA-seq datasets
To compare our RNA-seq results to the transcriptional changes associated with JQ1-mediated BET BD inhibition, we used published expression profiles of JQ1- and vehicle-treated sham-operated mouse hearts (GSE96561). The raw counts were analyzed using DEseq2 (v1.22.2 R package), and the correlation of the results were done by representing the Log2 FC values from each study using ggplot2 (v3.2.0 R package). Data was taken from three biological replicates in each condition.
Electron microscopy
Tissues for electron microscopic examination were fixed with 2.5% glutaraldehyde and 2.0% paraformaldehyde in 0.1M sodium cacodylate buffer (pH7.4) overnight at 4°C. After subsequent buffer washes, the samples were post-fixed in 2.0% osmium tetroxide with 1.5% K3Fe(CN)6 for 1 hour at room temperature, and rinsed in distilled water. After dehydration through a graded ethanol series, the tissues were infiltrated and embedded in EMbed-812 (Electron Microscopy Sciences, Fort Washington, PA). Thin sections were stained with uranyl acetate and SATO lead and examined with a JEOL 1010 electron microscope fitted with a Hamamatsu digital camera and AMT Advantage NanoSprint500 software.
Mice
All mouse manipulations were performed in accordance with protocols approved by the Institutional Animal Care and Use Committee (IACUC) following guidelines described in the US National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Myh6-MCM and Tnnt2-Cre mice have been described previously23,24. Brd4flox mice were produced by targeting C57BL/6 ES cells with a targeting vector designed to flank exon 3 (containing the canonical Brd4 ATG) with loxP sites. The selection strategy included a FRT flanked neomycin resistance cassette that, after excision, leaves a single FRT site within intron 2. Brd4flox mice were genotyped using the polymerase chain reaction (PCR) primers listed below which produce a 311 bp wild-type band and a 414 bp knock-in band (Supplemental Figure ID). Brd4flox mice used for embryonic studies have been described previously25.
Brd4flox_01F: 5′-GAAAGAGAAGAAGCTAACTGGC
Brd4flox_02R: 5′-GAGCAAGTATATTGGAGGGGAG.
Mouse echocardiography
Echocardiography was performed blindly with the Vevo 770 High-Resolution Micro-Imaging System (VisualSonics) with a 15-MHz linear-array ultrasound transducer. The left ventricle was assessed in both parasternal long-axis and short-axis views at a frame rate of 120 Hz. End-systole or end-diastole were defined as the phases in which the left ventricle appeared the smallest and largest, respectively, and used for ejection-fraction measurements. To calculate the shortening fraction, left-ventricular end-systolic and end-diastolic diameters were measured from the left-ventricular M-mode tracing with a sweep speed of 50 mm/s at the papillary muscle. B-mode was used for two-dimensional measurements of end-systolic and end-diastolic dimensions. Imaging and calculations were done by an individual who was blinded to the treatment applied to each animal and code was broken only after all data acquired.
Histology
Isolated hearts were fixed in 2% paraformaldehyde (4°C overnight), dehydrated through an ethanol series, embedded in paraffin, and sectioned. Antibodies used for immunohistochemistry were: Brd4 (rabbit, Bethyl 00396) and Cleaved caspase 3 (rabbit, Cell Signaling 9664). Hematoxylin and Eosin staining was performed using standard protocols. Sections were imaged on a Nikon Eclipse 80i fluorescence microscope or Leica DMi8 inverted fluorescence microscope.
ATAC-sequencing, library preparation, and analysis
CM samples were prepared for ATAC-seq as previously described26. Aliquots of 50,000 cells were lysed with 3 mL of chilled lysis buffer (3.75mM PIPES, 450mM KCl, 1% NP-40, 1% Tween-20, 1% Triton X-100 in water; pH 7.3) for 10 minutes. Nuclear pellets were transposed with 25 μL of Tagment DNA Buffer, 2.5 μL of Tagment DNA Enzyme (Nextera Sample Prep Kit from Illumina; #FC-121–1030), and 22.5 μL of nuclease-free water. Samples were incubated at 37°C for 60 min and stored at 20°C. Transposed samples were purified using the QIAGEN MinElute Reaction Cleanup Kit (28204) and amplified using 25 μL of NEBNext High Fidelity 2x PCR Master Mix, 1.25 mM Nextera custom primers with unique barcodes, and nuclease-free water. Samples were amplified using the following PCR conditions: 72°C for 5 min; 98°C for 30 s; and cycled at 98°C for 10 s, 63°C for 30 s and 72°C for 1 min. Half of each sample was amplified for 12 cycles, purified, and assessed by Bioanalyzer (Agilent) for library quality. Sample concentration was quantified by Qubit (Invitrogen) prior to pooling. Pooled samples were sequenced using a PE75 run on a NextSeq 500 instrument (Illumina). Alignment to the mm10 reference genome was performed using Bowtie 2.2.4. Peaks were called using macs2 callpeak with options: “-p 0.1–nomodel–shift 100–extsize 200 -B–SPMR–call-summits”. Peaks concordant between the two replicates were considered for further analysis. Motif enrichment analysis was performed using HOMER27.
Analysis of publicly available ChIP-seq data
Occupancy analysis for BRD4 and GATA4 was done using publicly available ChIP-seq datasets (GSE52123 and GSE124008, respectively). Deeptools v.3.3.1 was used to generate the aggregation and heatmap plots centered at the TSS of mm10 gene annotations (GRCm38.p6). Analysis of TFs representation in our list of DEGs from Day2 and Day5 was done using published data (GSE124008) after filtering for peaks within 5Kb of the closest annotated gene.
Immunoblotting
Adult CMs were isolated via Langendorff perfusion of Myh6-MCM; Brd4flox/flox treated with TAM (75 μg/g/day or 50 μg/g/day) or VEH (corn oil) for 5-days. Whole cell extracts were prepared by lysis in RIPA buffer supplemented with protease and phosphatase inhibitors (Roche). Protein concentration was quantified by BCA assay (Thermo Fisher; 23225). Lysates were diluted in 4X LDS sample buffer (Invitrogen), boiled at 95°C for 5 min, resolved on a 3–8% Tris-Acetate SDS PAGE gel (Invitrogen), and transferred onto a PVDF membrane. Membranes were blocked with 5% milk in TBST for 1 hour at room temperature and incubated in primary antibody at the indicated dilution overnight at 4°C. Appropriate secondary HRP-conjugated antibody was added for 1 hour at a dilution of 1:5000 followed by detection with ECL Prime Western Blotting Detection Reagent (GE Life Sciences; RPN2232) and exposure to autoradiography film at various time intervals or by digital imaging (LI-COR Odyssey). Antibodies used in this study: rabbit anti-Brd4 (Abcam ab128874; 1:1000) and mouse anti-vinculin (Sigma-Aldrich V9131; 1:1000).
Luciferase reporter assay
GATA4–BRD4 transcriptional synergy reporter assay was performed using the pANF638L vector28. Briefly, HeLa cells were cultured in 24-well plates at 10^5 cells per well and transfected within 24 hrs of seeding. Cells were co-transfected with 200 ng of pANF638L vector and 20ng of pRL-TK (a Renilla luciferase control vector; Promega) in 2.4 μL FuGENE HD (Promega) and 43 μL Opti-MEM (Thermofisher). The transfection mix was aliquoted in 4 tubes (1 per condition) and the following conditions were prepared: 1.) Control: 600 ng of empty vector (EV); 2.) GATA4: 200 ng of GFP-GATA4 vector plus 400 ng EV; 3.) BRD4: 400 ng FLAG-BRD4 vector plus 200 ng EV and 4.) GATA4+BRD4: 200 ng of GFP-GATA4 vector with 400 ng FLAG-BRD4 vector. Media was changed 24 hrs after transfection and cells collected 48 hrs following transfection. Samples were processed with the Dual Luciferase Assay System (E1960, Promega) following manufacturer’s instructions and measured with a luminometer (SpectraMax i3). For analyzing the GATA4–BRD4 transcriptional activity within the Ppargc1a promoter, this DNA regions was cloned with Cold Phusion by designing gBlocks (IDT) for the selected target region (Ppargc1a promoter: Mm10 chr5:51551646–51553274) with homology arms complementary to the pGL4.23 Luciferase reporter vector (Promega).
Gene knockdown experiments on Neonatal Rat Ventricular Myocytes
Neonatal Rat Ventricular Myocytes (NRVMs) were isolated from hearts of 2-day-old Sprague-Dawley rat pups (Charles River) under aseptic conditions and transfected with siRNAs as previously described18,29. In brief, NRVMs were pre-plated for 2 hrs on tissue culture plates followed by 48 hrs of exposure to BrdU in culture medium to remove contaminating nonmyocytes. NRVMs were plated in growth medium (DMEM, 5% FBS, 100 U/mL penicillin-streptomycin) for 48 hrs. NRVMs were then transfected with RNAiMax (Invitrogen) and 50 nM of siRNA in serum-free medium for 48 hrs. Two independent siRNA probes for each target were used in tandem at a 1:1 ratio. siRNAs were purchased from Sigma-Aldrich (scramble control siRNA, SIC001; Brd4 siRNAs, SASI_Rn02_00315745, SASI_Rn02_00315746; Gata4 siRNAs, SASI_Rn01_00070083, SASI_Rn01_00070084).
Co-Immunoprecipitation in HEK 293T cells
HEK293T cells were plated in 6-well plate and 1.25 μg of GFP-GATA4 and Flag-BRD4 vectors were transfected with lipofectamine 2000, following manufacturer’s instruction. A day after transfection, DMSO and JQ1 were treated. After 48 hrs, cells were lysed with 500 μL of buffer A [10mM HEPES (pH7.9), 1.5mM MgCl2, 10mM KCl, 340mM Sucrose, 10% Glycerol, 0.1% Triton X-100 and protease inhibitors], incubated for 5 min at 4°C and centrifuged at 1,300g for 4 min at 4°C to get nuclei pellet. Pellet was washed twice with buffer A and suspended with 500 μL of buffer B [3mM EDTA, 0.2mM EGTA and protease inhibitors]. After centrifugation at 1,700g for 5 min at 4°C, pellet was suspended with 400 μL of lysis buffer [10mM HEPES (pH7.9), 3mM MgCl2, 5mM KCl, 140 mM NaCl, 0.1mM EDTA, 0.5% NP-40, 0.5mM DTT, protease inhibitor, 62.5U benzonase], incubated for 45 min at 4°C and centrifuged at 13,200 rpm for 20 min at 4°C. Clear supernatant was transferred into a new tube. 30 μL of lysate was saved as an input. Prior to IP, 15 μL of magnetic Protein G Dynabeads were coated with 0.5 μg of M2 anti-FLAG antibody (F1804, Sigma) for 1 hr at room temperature. 300 μL of lysate was mixed with antibody-coated Dynabeads. The mixture of lysate and Dynabeads was incubated overnight at 4°C and washed four times with lysis buffer. Beads were boiled for 5 min at 95 °C in 30 μL of sample buffer.
Co-Immunoprecipitation in cardiac progenitors cells
Human induced-pluripotent stem cells were differentiated into cardiomyocytes using Wnt pathway modulation30 and 4 × 12-well plates were collected at cardiac progenitor stage (day 6 of differentiation), pooled together, and snap frozen in liquid nitrogen. Cardiac progenitor cell pellets were lysed with 1 ml of cell lysis buffer [20 mM Tris-HCl pH 8, 85 mM KCl, 0.5% NP-40, and protease inhibitors], incubated 10 min at 4°C, and centrifuged at 2,500 rpm for 5 min at 4°C to pellet nuclei. Supernatant was removed and pellets resuspended in 600 μL of nuclei extraction buffer (NE buffer) [20 mM HEPES pH 7.4; 0.5 M NaCl, 2 mM MgCl2, 1 mM CaCl2, 0.5% NP-40, 110 mM K-Acetate, 1 μM ZnCl2, benzonase, and protease/phosphatase inhibitors], and incubated for 30 min at 4°C. Nuclear enriched lysates were centrifuged at maximum speed for 10 min at 4°C and supernatants transferred to a new tube and diluted with 1200 μL (1:3) IP dilution buffer [20 mM HEPES pH 7.9, 1 mM EDTA, 0.02% NP-40, and protease/phosphatase inhibitors]. Prior to IP, 30 μL was saved as input. 50 μL of magnetic Protein G Dynabeads were coated with 4 μg of anti-GATA4 (G4) antibody (sc-25310X, Santa Cruz) for 1 hr at 4°C. For IP, 50 μL of GATA4-coated beads were added to 2 mg of nuclear-enriched total protein, incubated overnight at 4 °C with agitation, and washed three times in IP dilution buffer. Beads were boiled for 10 min at 95 °C in 20 μl of sample buffer. Extracts and immunoprecipitates were examined by SDS–PAGE and blotted with antibodies for the indicated targets.
Statistics and reproducibility
Standard statistical analyses were performed using GraphPad Prism 8. When more than two conditions were to be compared, a one-way ANOVA followed by a Tukey range test or two-way ANOVA was used to assess the significance amongst pairs of conditions. When only two conditions were compared, a Student’s t-test was used. For survival analysis, a Mantel–Cox test was used. In all the figures showing RNA-seq DE analysis (with DESeq2) the p-values attained by the Wald test are corrected for multiple testing using the Benjamini and Hochberg method and thus referred to as adjusted p-values (adj. p). We used an adj. p<0.05 and a |Log2 FC|> 1 to determine significant genes. For all quantifications related to cardiac function and gene expression by RT-qPCR, the means ± SD are reported in the figures. For all quantifications related to cardiac function, the number of replicates is indicated as data points in the graphs. The level of significance in all graphs is represented as follow: * represents p<0.05, ** represents p<0.01, *** represents p<0.001, **** represents p<0.0001.
RESULTS
Loss of BRD4 in adult CMs results in contractile dysfunction and lethality
We generated a Brd4 conditional allele by engineering a homologous recombination event in mouse embryonic stem (ES) cells (Supplemental Figure IA) in a pure C57BL/6 background. We designed a targeting strategy that inserted flanking loxP sites around the third exon of Brd4, which contains the canonical translational start site, and verified appropriate targeting in several ES cell clones by Southern blotting (Supplemental Figure IB,C). These were used to generate chimeric mice that were subsequently bred for germline transmission. Mice harboring this allele (designated Brd4flox) were normal, viable, and able to breed in both the heterozygous and homozygous state (Supplemental Figure ID). These were crossed to mice harboring the Myh6-Mer-Cre-Mer (Myh6-MCM) allele23, which permits for tamoxifen (TAM)-inducible CM-specific deletion of Brd4. Immunofluorescence of heart tissue (Supplemental Figure II) and immunoblotting of isolated CM lysates (Figure 1A) confirmed efficient loss of BRD4 protein after 5 days of TAM administration.
Figure 1: Adult cardiomyocyte-specific Brd4 deletion results in acute and persistent contractile dysfunction and lethality.
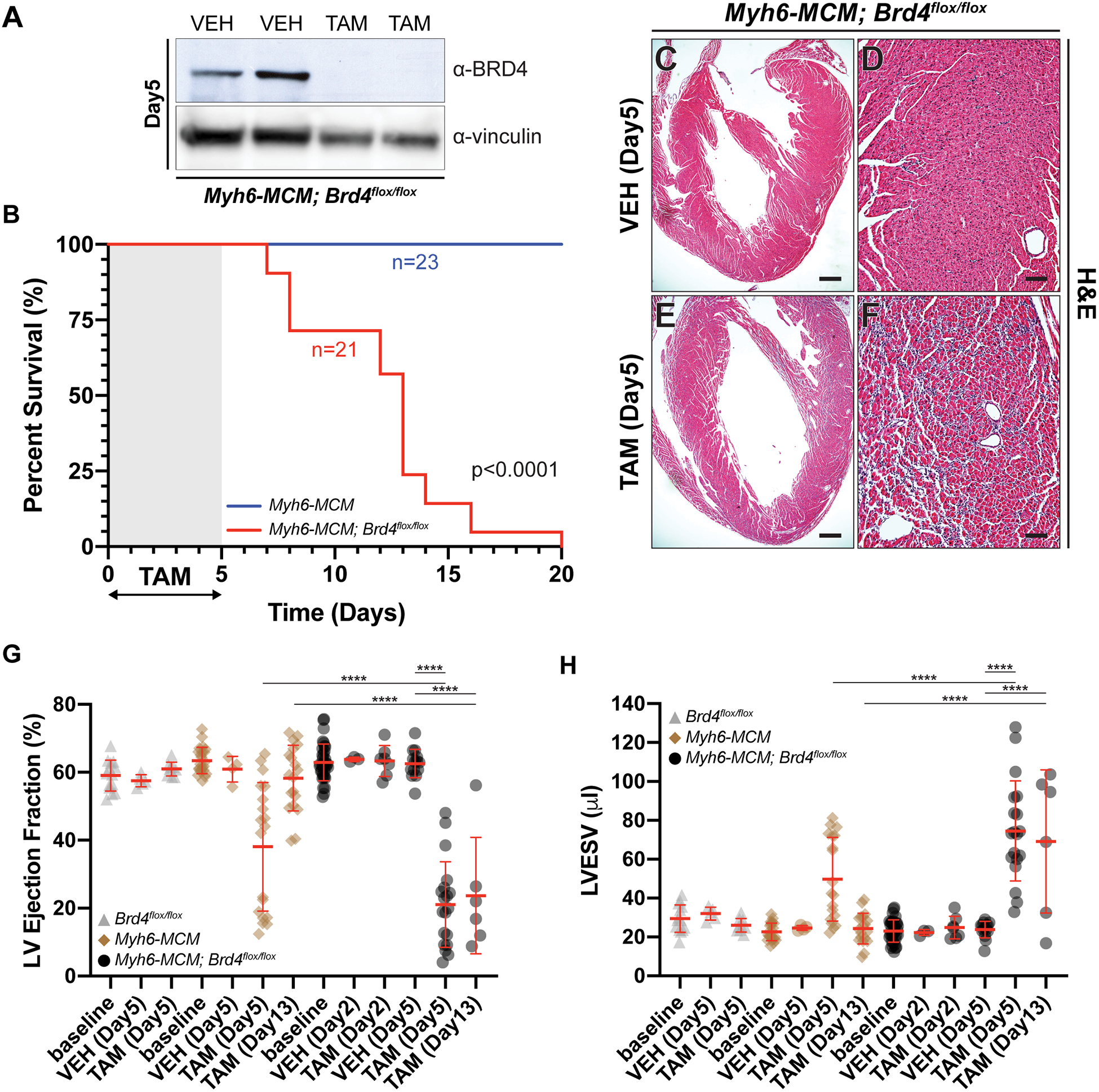
(A) Immunoblot of isolated Myh6-MCM; Brd4flox/flox cardiomyocyte lysates from mice treated with vehicle (VEH) or tamoxifen (TAM) for 5 days using BRD4 or vinculin (loading control) antibodies. (B) Kaplan-Meier curve demonstrating survival of indicated mice treated with tamoxifen (TAM; 75 μg/g/day). p-values were calculated using a Mantel–Cox test. (C-F) H&E images of Myh6-MCM; Brd4flox/flox mice treated with vehicle (VEH) or tamoxifen (TAM) at low and high magnification. (G) Ejection fraction and (H) left ventricular end systolic volume (LVESV) of indicated mice treated with tamoxifen (TAM) or vehicle (VEH) at indicated days after injection. Individual points and mean ± SD shown. One-way ANOVA analysis coupled with a Tukey test was used to assess significance.
For B, G, and H, **** represents p<0.0001 for indicated comparison.
Scale bars = 100 μm (D, F) and 500 μm (C, E)
Following administration of intraperitoneal TAM (75 μg/g/day) or vehicle (VEH; corn oil) for 5 consecutive days to adult mice, we noticed that Myh6-MCM; Brd4flox/flox animals became lethargic and ill-appearing, with several succumbing shortly thereafter. In a survival analysis of Myh6-MCM, Brd4flox/flox (n=21) animals treated with TAM, 83% died within 14 days of the last dose of TAM with 100% mortality by 21 days. In contrast, 100% of TAM-treated Myh6-MCM (n=23) animals survived (Figure 1B). Histological analyses revealed interstitial infiltrates in TAM treated Myh6-MCM; Brd4flox/flox animals when compared with VEH-treated controls of the same genotype (Figure 1C–F). We did not find any significant differences in apoptosis 5 days post-TAM initiation (Supplemental Figure III).
We next assessed cardiac function by transthoracic echocardiography. Cohorts of Brd4flox/flox (n=14), Myh6-MCM (n=23), or Myh6-MCM; Brd4flox/flox mice (n=41) at 8–12 weeks of age were administered intraperitoneal TAM (75 μg/g/day) or VEH (corn oil) for 5 consecutive days. Echocardiography was performed at baseline, and on day 2, 5, and 13 after commencing TAM. CM-specific deletion of Brd4 resulted in a significant reduction in LV ejection fraction (EF) when compared with VEH (21% vs. 63%, p<0.0001) and LV chamber dilation (LV end systolic volume [LVESV] 24 μl vs. 75 μl, p<0.0001) (Figures 1G,H) after 5 days of TAM administration. No appreciable changes in these indices were detected following 2-days of TAM treatment (EF 63% vs 63%, ns; LVESV 22 μl vs. 25 μl, ns). Treatment of Brd4flox/flox animals with TAM or VEH did not result in any significant changes in these parameters. Consistent with previous reports31, administration of TAM alone for 5 days in Myh6-MCM animals led to a transient decrease in LV systolic function (EF 38% vs. 61%, p<0.0001) and LV chamber dilation immediately following TAM exposure (LVESV 25 μl vs. 50 μl, p<0.0001). However, the degree of LV systolic dysfunction and chamber dilation was significantly greater in Myh6-MCM; Brd4flox/flox animals when compared to Myh6-MCM controls at the 5 day time point (EF 21% vs. 38%, p<0.0001; LVESV 75 μl vs. 50 μl, p<0.0001). Repeat echocardiographic analysis at day 13 revealed normalization of these indices in Myh6-MCM controls consistent with a transient and reversible Cre-mediated toxicity. In contrast, Myh6-MCM; Brd4flox/flox animals that survived to this time point failed to demonstrate any recovery (Figure 1G,H). These data are consistent with our finding that only TAM-treated Myh6-MCM; Brd4flox/flox mice, and not equivalently treated Myh6-MCM mice, had a striking propensity for early mortality after TAM treatment (Figure 1B). Notably, intraperitoneal administration of TAM at a lower dose (50 μg/g/day) for 5 consecutive days in Myh6-MCM (n=23) or Myh6-MCM; Brd4flox/flox mice (n=6) did not result in depletion of BRD4 protein (Supplemental Figure IVA). Accordingly, noninvasive analysis demonstrated LV systolic dysfunction at day 5 that resolved by day 13 in both groups (Supplemental Figure IVB,C). Taken together, these data demonstrate that postnatal deletion of Brd4 in CMs of adult mice leads to early mortality and severe systolic HF within 5 days that is sustained, suggesting an essential role for BRD4 in maintaining expression of key homeostatic gene programs in the adult CM.
BRD4 regulates mitochondrial bioenergetic gene pathways
To better understand the mechanism by which BRD4, a potent transcriptional coactivator, regulates homeostasis in adult CMs, we performed bulk RNA-sequencing (RNA-seq) on isolated CMs from Myh6-MCM; Brd4flox/flox treated with TAM (Brd4-KO) or VEH (control) and Myh6-MCM mice treated with TAM (Cre-control) (Supplemental Figure VA,B). We collected samples at two early time points that occurred prior to the onset of mortality (n≥2 biological replicates per condition): day 2 post-TAM (prior to the decrease in LVEF) and day 5 post-TAM (when acute HF was first detected).
While Cre-control mice did not display mortality, we noted a transient decrease in LV systolic function after 5 days of TAM treatment. Therefore, we sought to account for any gene dysregulation associated with tamoxifen-induced Cre activation in CMs, particularly as the transcriptomic effects of this manipulation have not been reported in the literature. Using a statistical threshold to capture the vast majority of Cre-related effects on gene expression (Log2 FC±1, adj. p<0.05), we found that Cre-control CMs demonstrated differential expression of 5,038 genes compared with control CMs at the 5-day time point (Supplemental Figure VC,D). Using these data, we defined a molecular signature following 2 and 5 days of tamoxifen treatment in Myh6-MCM CMs and removed it from all subsequent analyses comparing Brd4-KO and control CMs (Supplemental Tables I–IV).
Comparison of Brd4-KO and control samples at day 2 and 5 reveal a temporal increase in the number of dysregulated genes, consistent with the degree of cardiac dysfunction at each time point. We observed 101 dysregulated genes at day 2, prior to cardiac dysfunction (Figure 2A, Log2 FC±1, adj. p<0.05). By day 5, Brd4-KO CMs demonstrated differential expression of 2,094 genes when compared with controls (1326 upregulated, 768 downregulated, Log2 FC±1, adj. p<0.05) (Figure 2B), after removal of Cre-control dysregulated genes. GO analysis revealed a striking signature for mitochondrial bioenergetics amongst those genes preferentially downregulated in Brd4-KO CMs, a pathway wherein BRD4 has not been implicated. GO analysis of upregulated genes in Brd4-KO CMs were enriched broadly for general terms associated with cardiac stress, including fibrotic and inflammatory cellular processes, suggesting that many of these upregulated genes may be a secondary response to loss of BRD4 coactivator function in CMs (Figure 2B).
Figure 2: BRD4 regulates mitochondrial metabolic pathways in adult cardiomyocytes.
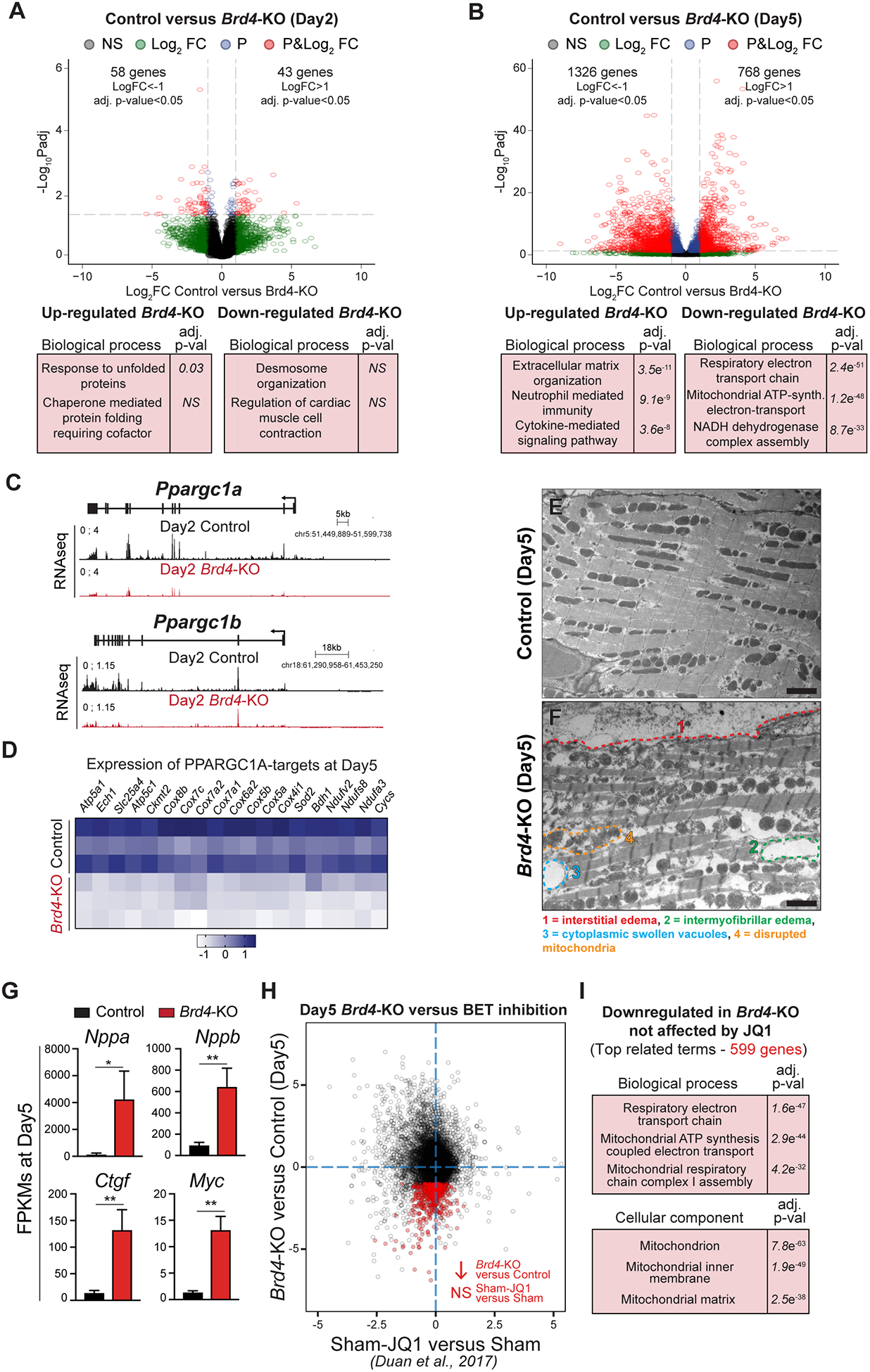
(A, B) Volcano plots showing Log2 fold change and adj. p-value of individual genes 2 days (A) or 5 days (B) after Brd4 deletion; genes differentially expressed between Cre-control and Control samples have been excluded. Selected categories identified from gene ontology analysis from up or down regulated genes are shown below the volcano plot. Genes are assigned with specific colors following DE analysis: grey (not significant), green (Log2 FC<−1 or >+1), blue (adj. p<0.05) or red (Log2 FC<−1 or >+1 and adj. p<0.05). (C) Track view of Ppargc1a and Ppargc1b genes showing sequencing reads mapping from RNA-seq signature at day 2 post-tamoxifen (TAM) treatment for Control and Brd4-KO samples. (D) Heatmap of expression of PPARGC1A known targets32 in Control and Brd4-KO samples at day 5 post-TAM treatment. (E, F) Electron micrographs of Brd4-KO and Control animals at day 5 highlighting the loss of normal mitochondrial morphology. (G) FPKMs of indicated genes related to cardiac stress and homeostasis in Control (n=3) or Brd4-KO (n=3) cardiomyocytes at day 5. Error bars represent standard deviation (SD). (H) Correlation analysis of difference in gene expression in day 5 Brd4-KO cardiomyocytes compared to sham-operated animals administered JQ1 (normalized to their respective controls). Genes highlighted in red are those which are downregulated upon BRD4 loss but not significantly changed by JQ1. (I) Gene ontology analysis of genes that were downregulated upon BRD4 loss but not affected by JQ1 treatment.
For G, * represents p<0.05; ** represents p<0.01 for indicated comparison.
Scale bars = 2 μm (E, F)
Given the marked transcriptional dysregulation of gene programs linked to mitochondrial homeostasis in Brd4-KO CMs at day 5, we turned our attention to the earliest time point in our analysis (day 2) to interrogate if established nodal regulators of mitochondrial metabolism were downregulated early after BRD4 loss. Indeed, we discovered that the expression of the transcriptional co-activators Ppargc1a (PPARGC1A; PGC-1α) and Ppargc1b (PPARGC1B; PGC-1β) were downregulated in Brd4-KO CMs when compared to control CMs at this early time point, prior to the appearance of any LV systolic dysfunction (Figure 2C). PGC-1α and PGC-1β are known to be master regulators of mitochondrial biogenesis and oxidative phosphorylation gene programs in cardiac and skeletal muscle cells32,33, suggesting the mitochondrial dysregulation may be a primary consequence of Brd4 deletion. At day 5, expression of known targets of PPARGC1A in the heart32 were downregulated in Brd4-KO CMs when compared with controls, including genes involved in oxidative phosphorylation, fatty acid oxidation, and ATP synthesis (Figure 2D). Given this signature of broad mitochondrial dysfunction identified at the transcriptional level, we performed electron microscopy to assess cardiac ultrastructure and mitochondrial morphology following BRD4 loss. While control samples showed an organized arrangement of myofibers with normal sarcomeres (Figure 2E), Brd4-KO hearts demonstrated interstitial edema between CMs (Figure 2F,1), intermyofibrillar edema within CMs (Figure 2F,2), swollen cytoplasmic vacuoles (Figure 2F,3), and disrupted mitochondria that showed mild swelling (Figure 2F,4; Supplemental Figure VI). These findings are often observed with severe disruption of cellular metabolism, underscoring a CM-intrinsic gene program controlling basal mitochondrial energetics in the adult heart that is acutely sensitive to BRD4 abundance.
Figure 3: Constitutive cardiomyocyte-specific Brd4 deletion is embryonic lethal and reveals BRD4 as a regulator of mitochondrial metabolic pathways in embryonic cardiomyocytes.
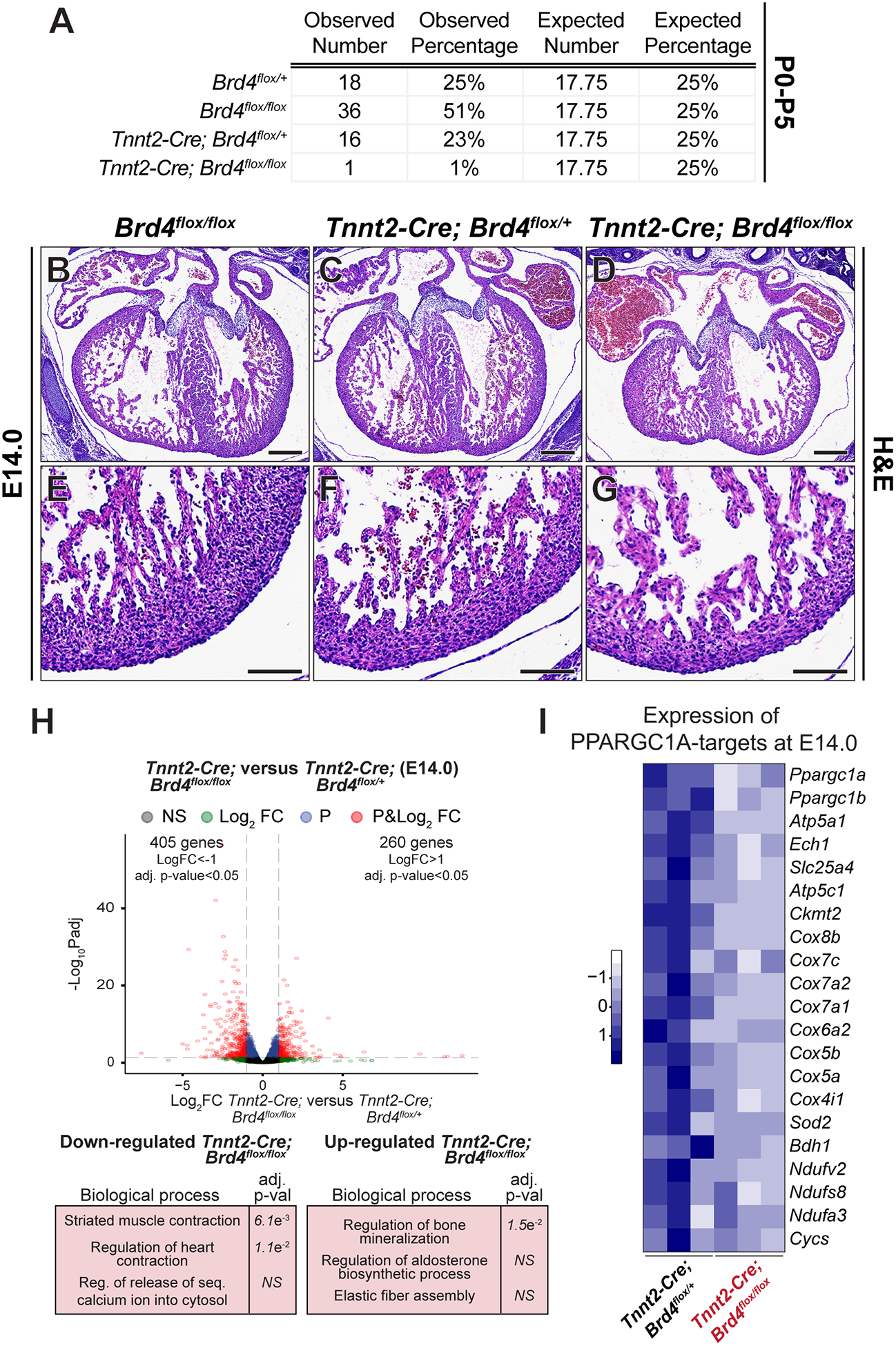
(A) Genotyping data from parental cross of Tnnt2-Cre; Brd4flox/+ and Brd4flox/flox animals demonstrating embryonic lethality in Tnnt2-Cre; Brd4flox/flox offspring at postnatal days 0 to 5 (P0–5). (B-G) H&E images of cross sections from Brd4flox/flox, Tnnt2-Cre; Brd4flox/+, and Tnnt2-Cre; Brd4flox/flox embryonic hearts at E14.0 at low and high magnification. (H) Volcano plots showing Log2 fold change and adj. p-value of individual genes at E14.0 between Tnnt2-Cre; Brd4flox/+ and Tnnt2-Cre; Brd4flox/flox microdissected embryonic hearts. Selected categories identified from gene ontology analysis from up or down regulated genes are shown below the volcano plot. Genes are assigned with specific colors following DE analysis: grey (not significant), green (Log2 FC<−1 or >+1), blue (adj. p<0.05) or red (Log2 FC<−1 or >+1 and adj. p<0.05). (I) Heatmap of expression of PPARGC1A known targets32 in Tnnt2-Cre; Brd4flox/+ and Tnnt2-Cre; Brd4flox/flox in embryonic hearts at E14.0.
Scale bars = 250 μm (B-D) and 100 μm (E-G)
Figure 4: BRD4 and GATA4 co-occupy and regulate genes controlling mitochondrial homeostasis.
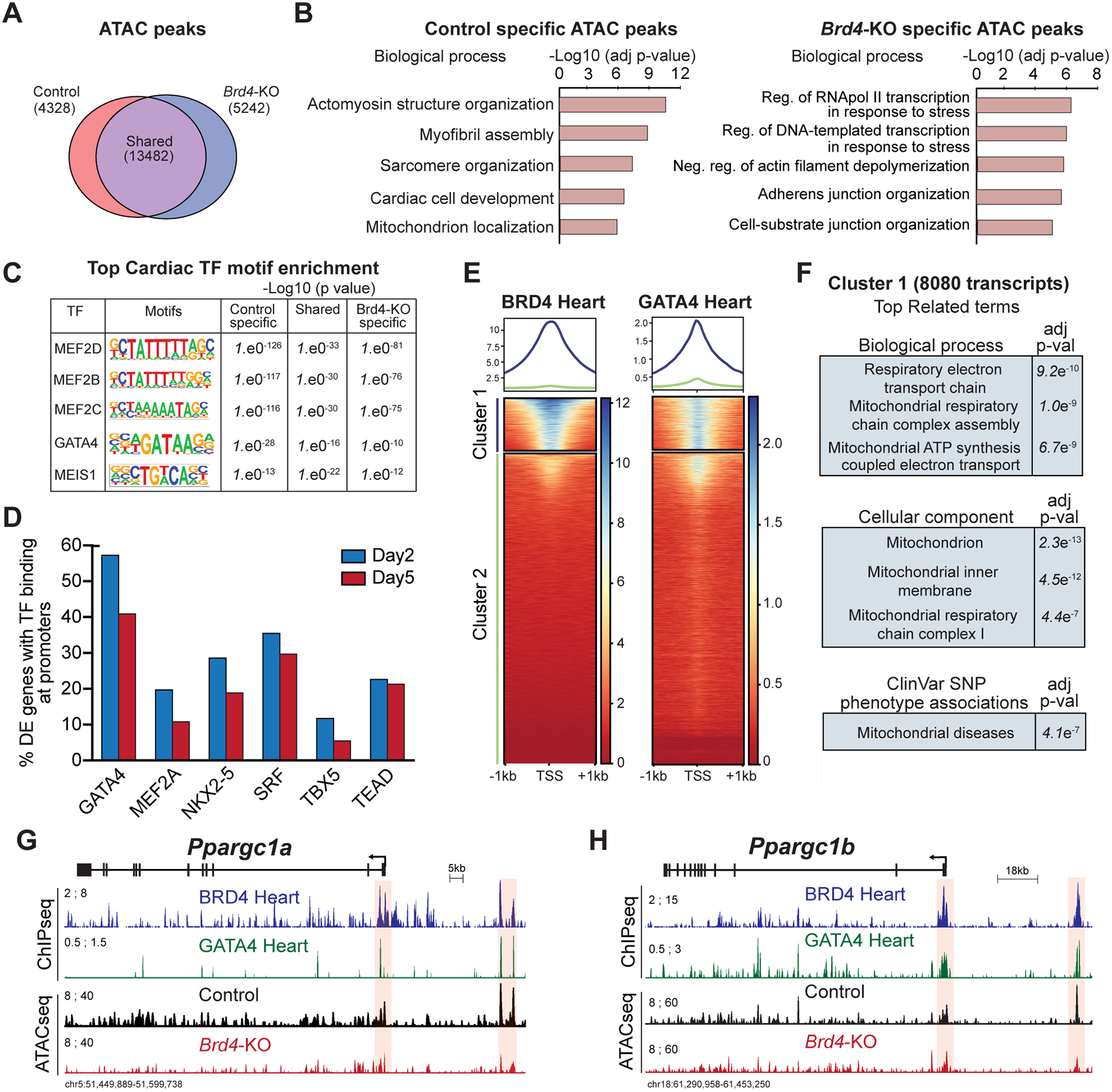
(A) Venn diagram showing number of unique and shared accessible chromatin regions between Control and Brd4-KO samples. (B) Top selected categories identified from gene ontology analysis from Control and Brd4-KO specific ATAC regions. (C) Motif enrichment analysis for cardiac TFs in unique and shared accessible chromatin regions between Control and Brd4-KO samples. (D) Number of differentially expressed genes between Control and Brd4-KO at day 2 and day 5 occupied by cardiac TFs40,41 at their promoters (±1 TSS). (E) Heatmaps showing enrichment of BRD4 and GATA4 ChIP signals from adult mouse hearts under basal homeostatic conditions at gene promoters (±1 TSS, 55,386 mm10 annotated transcripts) ordered by BRD4 intensity identifies a cluster of strongly bound transcripts (Cluster 1, n=8080). (F) Gene ontology analysis of Cluster 1 genes identified enriched terms for biological processes, cellular components, and SNP-phenotype associations. (G-H) Track view of Ppargc1a and Ppargc1b genes showing sequencing reads mapping from BRD4 and GATA4 ChIP-seq as well as Control and Brd4-KO ATAC-seq at day 5 post-tamoxifen treatment. The promoter region and a putative regulatory element for each gene is highlighted in red.
Pharmacologic BET BD inhibition with JQ1 ameliorates adverse cardiac remodeling and HF in several murine models9,10,18. However, JQ1-treatment does not affect exercise-induced physiological cardiac hypertrophy18, a form of plasticity that features upregulation of genes involved in mitochondrial biogenesis and oxidative metabolism. Transcriptional profiling of the sham-operated animals treated with JQ1 at these doses did not reveal dramatic changes, suggesting that BET BD inhibition in homeostatic conditions is not associated with a pronounced gene dysregulation signature in the heart18. This is in stark contrast to the broad transcriptional changes that follow Brd4 CM deletion that results in marked upregulation of canonical cardiac stress markers, including Nppa, Nppb, Ctgf, and Myc (Figure 2G) with concomitant downregulation of mitochondrial and metabolic genes. To assess this quantitatively, we compared gene expression changes seen with JQ1-treatment in sham-operated mice to those that follow CM-specific BRD4 loss in adult mice. We confirmed that global gene expression changes were poorly correlated between pharmacologic BET BD inhibition and BRD4 loss at day 5 post-TAM administration (Figure 2H). GO analysis of genes downregulated in Brd4-KO CMs whose expression was unaffected by JQ1 treatment were enriched specifically for mitochondrial-related terms (Figure 2I), highlighting that transient exposure to small molecule BET BD inhibitors and Brd4 CM-deletion are markedly different molecular perturbations7,8,34.
We next sought to validate the importance of BRD4 in basal CM homeostasis using a constitutively active cardiomyocyte-specific Cre transgene with onset of expression during cardiogenesis. We crossed Brd4flox mice with the cTnT-Cre (Tnnt2-Cre) allele24 in which the rat troponin T2 cardiac promoter drives Cre recombinase expression. We only identified 1 Tnnt2-Cre; Brd4flox/flox animal out of 71 pups collected between postnatal day 0 and 5 (P0–5) (Figure 3A, 1% observed vs. 17.75% expected; χ2=34.75, p<0.001). Immunofluorescence confirmed efficient cardiomyocyte-loss of BRD4 in Tnnt2-Cre; Brd4flox/flox mutants (Supplemental Figure VII) by mid-gestation. Histologic analysis revealed cardiac hypoplasia in Tnnt2-Cre; Brd4flox/flox mutants at embryonic day 14.0 (E14.0) when compared with littermate controls (Figure 3B–G). Bulk RNA-seq of microdissected whole hearts from Tnnt2-Cre; Brd4flox/flox mutants and Tnnt2-Cre; Brd4flox/+ littermate controls at E14.0 (n=3 biological replicates per condition) revealed 260 upregulated and 405 downregulated genes (Log2 FC±1, adj. p<0.05) (Figure 3H; Supplemental Excel Table V). Importantly, consistent with our findings in adult CMs following BRD4 loss, Ppargc1a, Ppargc1b, and known PPARGC1A-regulated genes were downregulated in Tnnt2-Cre; Brd4flox/flox hearts when compared with littermate controls (Figure 3I).
BRD4 colocalizes with GATA4 at genes controlling mitochondrial bioenergy production
Given the specific changes in gene expression caused by BRD4 deletion and the known role of BRD4 as a coactivator of transcription, we queried changes in chromatin accessibility following CM-specific BRD4 loss. We performed Assay for Transposase-Accessible Chromatin followed by sequencing (ATAC-seq)26 on control and Brd4-KO CMs isolated at day 5 (n=2 per condition). We found that 58% of ATAC-seq peaks identified in control and Brd4-KO samples were shared (13,428 of 23,052 total peaks) (Figure 4A). In addition, we found accessible regions unique to Brd4-KO samples (n=5,242 peaks) or control samples (n=4,328 peaks), defined as regions of the genome which became accessible (gained in Brd4-KOs) or lost accessibility (lost in Brd4-KOs) upon BRD4 loss, respectively (Figure 4A). Ontology analysis of the regions which lose accessibility in Brd4-KO CMs showed enrichment for regulatory elements of genes linked to CM identity (e.g., sarcomere organization and myofibril assembly) and mitochondrial function, suggesting that BRD4 is required for those regions to be accessible (Figure 4B). Interestingly, accessible regions gained in Brd4-KO CMs were related to elements associated with stress responses (Figure 4B).
As BRD4 has previously been demonstrated to interact with sequence-specific TFs in other contexts35–39, we hypothesized that tissue-restricted TFs may facilitate preferential enrichment of BRD4 at specific genomic loci and contribute to the sensitivity of certain gene programs to BRD4 depletion. Therefore, we performed motif analyses27 of all the elements identified in our ATAC-seq and found enrichment for several cardiac TF motifs, including those for members of the MEF2 family, GATA4, and MEIS1 (Figure 4C). GATA4 was the only cardiac TF motif whose enrichment demonstrated a graded decrease in significance from those peaks unique to control CMs (lost in Brd4-KO CMs), shared between both conditions, and those unique to Brd4-KO CMs (gained in Brd4-KO CMs), suggesting that GATA4 may have preferential function in regions of active chromatin that were most sensitive to the presence of BRD4. Importantly, publicly available datasets defining the occupancy of cardiac TFs in adult heart tissue40,41 under basal homeostatic conditions demonstrate that promoters of differentially expressed genes in Brd4-KO CMs are enriched in GATA4 occupancy as compared to other cardiac TFs (Figure 4D).
Given this enrichment for GATA4 occupancy in the promoters of differentially expressed genes of Brd4-KO CMs, we posited that BRD4 and GATA4 may co-occupy these regions. Thus, we analyzed our previously performed BRD4 ChIP-seq9 and publicly available GATA4 ChIP-seq datasets defining occupancy of these factors in adult mouse myocardium under basal homeostatic conditions40. We clustered the enrichment of BRD4 and GATA4 occupancy in regions proximal to the transcriptional start site (TSS, ±1 kb) of all annotated transcripts and found that these separated into two clusters, one with strong occupancy of BRD4 and GATA4 (cluster 1; n=8,080) and a second with little or absent co-occupancy (cluster 2; n=47,305) (Figure 4E). Like our RNA-seq data, GO analysis of cluster 1 revealed genes for mitochondrial bioenergetics. Similar ontology analyses identified the mitochondrion as the most enriched cellular component, and genome-wide association studies linked single nucleotide polymorphisms (SNPs) from these transcripts with mitochondrial disease (Figure 4F). These data indicate that BRD4 and GATA4 co-occupy promoter regions of transcripts involved in mitochondrial energy production in adult CMs.
Given the known role of BRD4 in enhancer-mediated transcriptional activity, we also clustered the enrichment of BRD4 and GATA4 occupancy in distal chromatin regions marked by acetylation of lysine 27 on histone H3 (H3K27Ac) in the heart40 (Supplemental Figure VIIIA). We defined 10,800 putative regulatory elements co-occupied by H3K27Ac, BRD4, and GATA4 (Supplemental Figure VIIIB–D) and found that 28% of the dysregulated genes following BRD4 loss lied within 1 kb of a H3K27Ac/BRD4/GATA4 positive DNA element, and 49% were within 25 kb (Supplemental Figure VIIIE).
Given the downregulation of Ppargc1a and Ppargc1b at day 2 after Brd4 deletion, we specifically examined these loci for evidence of cooperative regulation by BRD4 and GATA4. Both of these genes exhibited strong enrichment for BRD4 and GATA4 at their promoters and were characterized by a putative upstream regulatory element that was also co-occupied by BRD4 and GATA4 (Figure 4G,H). Notably, both of these upstream DNA elements showed decreased accessibility in Brd4-KO CMs when compared with controls, suggesting that accessibility of these regulatory regions are BRD4-dependent (Figure 4G,H). Although HF can be associated with a secondary downregulation of CM metabolic genes, our findings suggest a model in which a BRD4-GATA4 module controls expression of both nodal upstream transcriptional coactivators of mitochondrial metabolism as well as their downstream gene targets.
BRD4 forms a complex with GATA4 in an BD-independent fashion to regulate the transcriptional coactivator Ppargc1a
Given the striking colocalization of BRD4 and GATA4 at regulatory regions controlling mitochondrial bioenergetics, we hypothesized that BRD4 and GATA4 may functionally co-regulate these genes. We first tested the ability of BRD4 and GATA4 to transactivate the Nppa promoter-luciferase reporter, a well-established gene reporter construct and known GATA4 target42,43, in transient transfection assays. Transfection of BRD4 or GATA4 alone resulted in activation of this reporter, while co-transfection of both together led to additive transcriptional activation (Figure 5A). We then performed the same assay for the promoter element of Ppargc1a that showed strong co-occupancy of BRD4 and GATA4 (Figure 4G). A luciferase reporter construct containing the Ppargc1a promoter also demonstrated additive co-regulation by BRD4 and GATA4 (11-fold activation with BRD4, 22-fold activation with GATA4, and 37-fold activation with GATA4 + BRD4) (Figure 5A). Conversely, in neonatal rat ventricular myocytes, we observed a greater downregulation of Ppargc1a and Ppargc1b expression upon siRNA-mediated knockdown of both Brd4 and Gata4 when compared with either factor individually (Figure 5B). We do note that individual Brd4 knockdown did not phenocopy the decrease in Ppargc1a/Ppargc1b expression seen in vivo, which may reflect differences attributable to this immature cellular system and/or the partial knockdown of Brd4. Taken together, these data suggest a functional cooperativity between BRD4 and GATA4.
Figure 5. BRD4 and GATA4 interact in a bromodomain-independent manner to control the master regulator of mitochondrial homeostasis Ppargc1a.
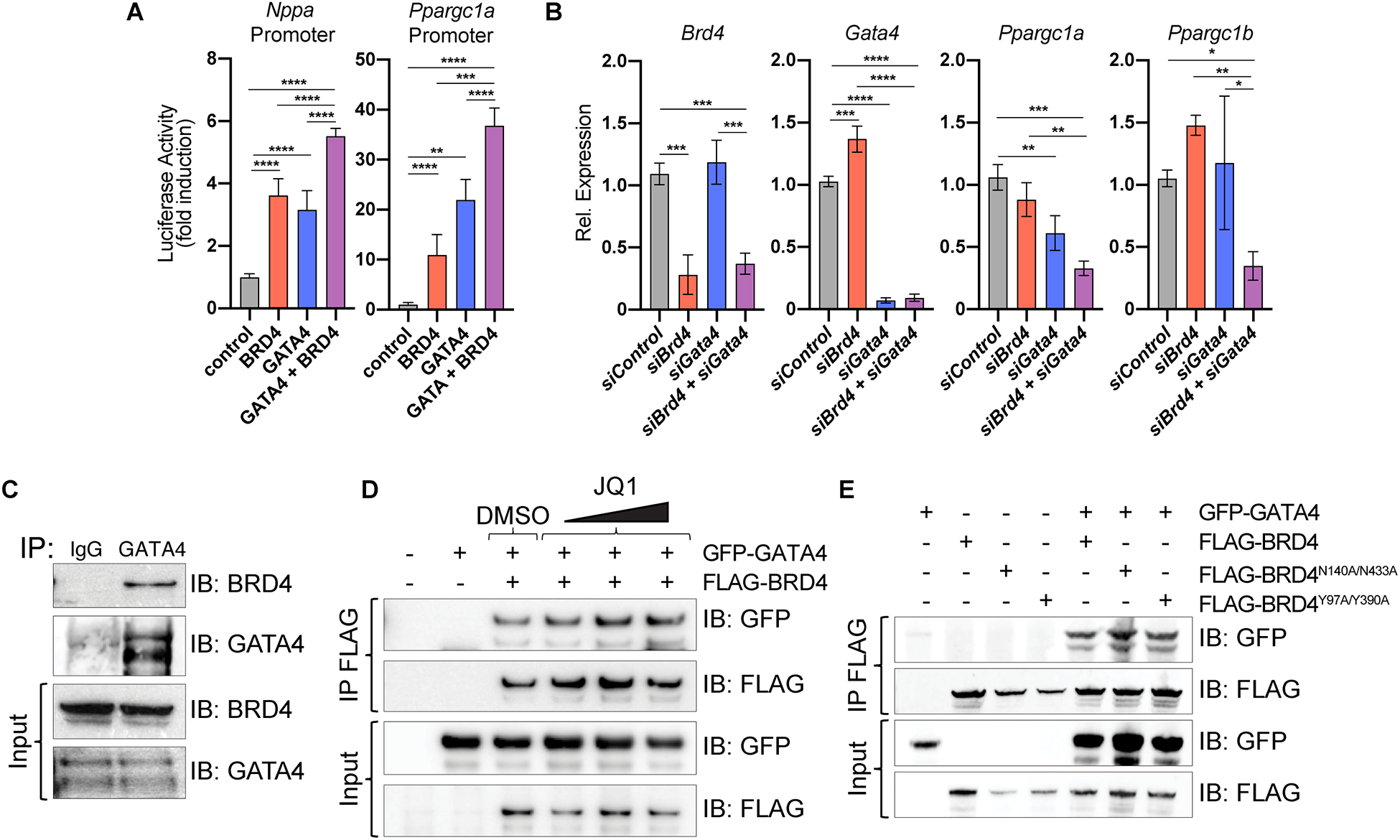
(A) Gene reporter assay showing activation of indicated luciferase reporters upon addition of plasmids encoding indicated proteins. Statistical significance is shown between control and GATA4 + BRD4 vs. all other individual conditions (n=4). (B) Relative expression by RT-qPCR in neonatal rat ventricular myocytes. Statistical significance is indicated between siControl and siGata4 + siBrd4 vs. all other individual conditions (n=3). One-way ANOVA analysis coupled with a Tukey test was used to assess significance. (C) Immunoprecipitation (IP) of endogenous protein from human cardiac progenitor cells using anti-GATA4 or anti-IgG antibody and immunoblotting (IB) with anti-BRD4 or anti-GATA4 antibody demonstrates endogenous BRD4 co-immunoprecipitates with GATA4 but not IgG. (D) Immunoprecipitation of FLAG-BRD4 overexpressed in HEK293 cells followed by immunoblotting with anti-GFP or anti-FLAG antibodies demonstrates GFP-GATA4 still co-IPs with BRD4 even in the presence of increasing doses of JQ1 or DMSO as control. (E) Immunoprecipitation of FLAG-BRD4, FLAG-BRD4N140A/N433A, and FLAG-BRD4Y97A/Y390A in HEK293 cells followed by immunoblotting with anti-GFP or anti-FLAG antibodies indicates co-immunoprecipitation of BRD4 mutants with GFP-GATA4.
For A and B, * represents p<0.05, ** represents p<0.01, *** represents p<0.001 and **** represents p<0.0001 for indicated comparison.
Immunoprecipitation of endogenous GATA4 protein from human induced pluripotent stem cell–derived cardiac progenitor cell lysates (which express high levels of GATA4) followed by immunoblotting for BRD4, demonstrated that endogenous human GATA4 and BRD4 interact in CM progenitors (Figure 5C). When co-transfected in 293T cells, GFP-tagged GATA4 and FLAG-tagged BRD4 also co-immunoprecipitated (Figure 5D). GATA4 contains four lysine residues that are targets of p300-mediated acetylation44. These residues are conserved in GATA1, which has previously been demonstrated to bind to BRD3 in an acetyl-lysine dependent fashion via BRD3’s BDs45,46. However, increasing concentrations of JQ1 had no appreciable effect on the ability of BRD4 and GATA4 to interact in 293T cells (Figure 5D). The tertiary structure of the BRD4 BD bound to acetyl-lysine11 and JQ117 has been extensively characterized, revealing 2 amino acids in each BD that are critical for mediating interactions with acetylated-lysine targets (N140 and Y97 of BD1; N433 and Y390 of BD2)47–50. BRD4 mutant constructs harboring N140A and N433A mutations or Y97A and Y390A retained the ability to interact with GATA4 (Figure 5E), consistent with this interaction occurring independent of the acetyl-lysine recognition activity of BRD4. This observation is also consistent with the disparity in cardiac phenotype of CM-specific Brd4 deletion and JQ1 treatment in mice.
DISCUSSION
In this report, we leveraged a newly developed mouse line harboring a conditional Brd4 allele and unbiased transcriptomic and epigenomic assays to dissect the role of this transcriptional coactivator in adult CM homeostasis in vivo. Acute depletion of BRD4 in adult CMs caused rapid onset of systolic HF within 5 days, leading to 100% lethality by 20 days. Transcriptomic profiling of CMs from Brd4-KO mice at day 5 revealed robust and relatively specific downregulation of gene programs important for mitochondrial energetics and homeostasis. Integrated analysis of BRD4 dependent gene expression and chromatin accessibility in adult CMs identified that GATA4 binding sites were preferentially enriched in the regulatory regions of BRD4-dependent genes. Our results reveal that BRD4 and GATA4 co-localize across the genome at loci relevant to mitochondrial bioenergy production and identify a novel BD-independent protein complex formation between BRD4 and this key cardiac TF. These findings highlight an unexpected role for both BRD4 and GATA4 in controlling a metabolic gene expression program in CMs, including direct regulation of Ppargc1a and Ppargc1b. While the interaction with GATA4 is important, our data do not preclude the possibility of BRD4 to complex with other cardiac TFs in activating cardiac gene expression programs. Important aspects of the BRD4 CM-deletion phenotype are likely mediated through interactions with GATA4 and other transcriptional regulators. Future work aimed at defining the specific regions of BRD4 and GATA4 that mediate their interaction and the consequences of selectively abrogating BRD4-GATA4 complex formation will be an important area of study.
BET proteins have emerged as pharmacologic targets in cancer and chronic diseases, including HF7,8,14,51–53. In animal models of HF, JQ1 administration improves cardiac function, decreases fibrosis, and attenuates activation of inflammatory and pro-fibrotic gene programs9,10,18. Clinical grade BET BD inhibitors are in trials for the treatment of a variety of malignancies14 and, although chemically diverse, share a common structural motif that reversibly binds the BDs of all BET family members and transiently displaces them from their endogenous binding partners. BRD2, BRD3, and BRD4 are all ubiquitously expressed and the relevant tissue compartments in which they function in vivo remains unknown. Small molecule proteolysis targeting chimeras (PROTACs) that degrade BET proteins represent a closer equivalent to genetic deletion54–56, but these again are neither cell- or BET-isoform specific. Therefore, these pharmacologic approaches offer limited insight into the cell-type and isoform specificities associated with beneficial responses of these compounds in disease models. Our data show that acute BRD4 depletion in CMs leads to rapid-onset systolic HF and mortality, highlighting a sharp contrast to the beneficial effects of small molecule BET BD inhibitors in treating adult mice with HF. These data are consistent with earlier studies and suggest that the dominant cellular targets of JQ1 in the context of HF are non-CMs, such as fibroblasts and immune cells18,57. Further dissection of these cell compartment- and gene-specific effects in the context of HF using conditional mice for each BET allele will be a fruitful area of study. While small molecule BET BD inhibitors and conditional gene deletion approaches manipulate BET proteins by very different mechanisms, our findings underscore the importance of understanding the specific cell-types that mediate the beneficial effects and potential liabilities of BET BD inhibitor therapy.
Our studies also suggest a previously unrecognized role for GATA4, one of the most studied TFs in cardiac biology, in CM metabolism and mitochondrial homeostasis. GATA4 is a well-established lineage determining TF for CMs whose role in cardiac development and human congenital heart disease has been extensively studied58,59. As ectopic Gata4 expression (along with Mef2c and Tbx5) can induce conversion of cardiac fibroblasts into induced CMs60–62, much effort has focused on dissecting the molecular mechanisms by which GATA4 regulates cellular identity63,64. In the adult heart, GATA4 plays an important role in stress responsiveness and regulation of pro-angiogenic genes65,66. Our data indicate that BRD4 also governs mitochondrial homeostasis in the adult heart and suggest that the preference of BRD4 to occupy certain loci may be mediated, in part, through an interaction with GATA4. They further demonstrate that a BRD4-GATA4 co-regulatory module orchestrates mitochondrial gene expression programs by regulating both nodal upstream regulators of these transcriptional programs (PGC-1α and PGC-1β) along with their downstream targets, suggesting transcriptional control via a tiered, feed-forward circuitry.
Cardiac contractile function is highly dependent on the ability of working CMs to sustain very high rates of mitochondrial oxidative phosphorylation and efficiently transfer energy67,68. Cardiomyopathies caused by loss-of-function variants in key mitochondrial genes underscore the direct link between mitochondrial dysfunction and contractile failure69,70. Consistent with these findings, deletion of key transcriptional regulators of mitochondrial homeostasis in murine CMs, including PPARGC1A and ERRα, cause cardiomyopathy and HF32,33,71. Electron microscopy of Brd4-KO hearts revealed myofiber degeneration and sarcomere disarray. This phenotype may be a downstream consequence of mitochondrial dysfunction, which can lead to pleiotropic defects in cellular homeostatic processes (e.g., protein quality control and calcium handling). In parallel, BRD4 may also play a primary role in coactivating key genes that are critical for sarcomere assembly and maintenance. GATA4 is required for cardiac morphogenesis and directly activates sarcomere genes during cardiac lineage commitment72, raising the possibility that the BRD4-GATA4 axis in adult CMs may regulate sarcomere genes in addition to mitochondrial programs. Given our finding that BRD4 is required for normal cardiogenesis and the previous discovery of GATA4 haploinsufficiency in congenital heart disease58, it will be interesting to determine if BRD4-GATA4 regulation is also critical for normal cardiac development. Dissecting the mechanisms by which BRD4 deficiency leads to both mitochondrial dysfunction and sarcomere disarray in CMs will be an important area of future investigation.
Our experiments have also revealed critical new knowledge about a commonly used reagent, the Myh6-MCM transgenic mouse23. This mouse strain has become a standard tool for inducible CM-targeted deletion of conditional alleles. This transgene expresses a Cre recombinase flanked by mutated estrogen receptor ligand-binding domains insensitive to endogenous estrogen (but responsive to exogenously administered tamoxifen) under the control of a ɑ-myosin heavy chain (Myh6) promoter. Prior reports have demonstrated a transient myopathy associated with Cre nuclear translocation following tamoxifen administration in these animals and warned of potential confounding in assessment of acute phenotypes that result from ablation of any gene of interest31. We define the first transcriptomic characterization of this transient myopathy (Supplemental Figure V) at days 2 and 5 after tamoxifen administration. Hence, accounting for gene expression changes upon induction of this transgene will be critical in the interpretation of results generated using these mice, providing a major resource for the field.
Apart from potential clinical application of BET BD inhibitors for therapeutic purposes, compounds like JQ1 have emerged as powerful tools for investigating enhancer biology73. Indeed, much of the field focuses on the BD-mediated functions of these proteins. In addition to acetylated histones, BET proteins bind acetylated TFs and modulate their transcriptional output. A BD-dependent interaction between BRD3 and GATA1 is important for erythroid maturation45. Likewise, BD-dependent interactions between BRD4 and p65/RelA-K310ac have been implicated in innate immunity35 and similar interactions with hematopoietic TFs (including PU.1, FLI1, and ERG) are essential for acute myeloid leukemia pathogenesis39. The advent of small molecules with a selectivity for BD1 vs. BD2 have stimulated further interest in dissecting the relative contribution of each of these domains in a variety of cellular contexts74,75. However, the BETs are large proteins that can scaffold complexes via multiple domains outside of their BDs. The BRD4 C-terminal domain interacts with the PTEFb complex76,77 and the BRD4 extra-terminal domain interacts with the histone methyltransferase NSD378,79. Unbiased interaction screens for BET family members have been performed in the presence and absence of JQ1, demonstrating a number of BD-independent interacting partners for each of the ubiquitously expressed BET family members80. Our data identify a novel, BD-independent interaction between BRD4 and GATA4, suggesting a mechanism by which a broadly expressed chromatin coactivator can be preferentially targeted to specific genomic loci by associating with a tissue-enriched DNA-binding TF. JQ1-mediated inhibition in vivo would not be expected to disrupt the BRD4-GATA4 interaction, consistent with the differences seen between the therapeutic effects of JQ1 and the deleterious consequences of Brd4 deletion in vivo. Our work highlights a critical need to better understand structure-function relationships of BRD4 outside of the BDs, findings which will provide general insight into the molecular underpinnings of cell-specific gene regulation and inform novel therapeutic approaches for HF and other cardiovascular diseases.
CONCLUSIONS
In this study, we discover a BRD4-GATA4 protein module that regulates mitochondrial gene expression and bioenergetic homeostasis in adult CMs.
Supplementary Material
CLINICAL PERSPECTIVE.
What is new:
Genetic loss of BRD4, an epigenetic reader protein, in adult cardiomyocytes results in cardiac dysfunction and death in mice.
BRD4 forms a transcriptional regulatory module with GATA4, a lineage-determining transcription factor in cardiomyocytes.
The BRD4-GATA4 module is a critical orchestrator mitochondrial bioenergetics in the adult heart.
What are the clinical implications:
Disruptions in substrate and energy metabolism, hallmark features of human heart failure, may be driven by dysregulation of BRD4 and GATA4 function in cardiomyocytes.
The beneficial effects of small molecule BET bromodomain inhibitors in mouse models of heart failure are unlikely to be mediated exclusively by inhibition of BRD4 in cardiomyocytes, suggesting roles for other cardiac cell types and BET family members as effectors of their pharmacology.
Identification of new BRD4 interaction partners such as GATA4 can provide new insights into developing epigenetic-based therapies for heart failure.
ACKNOWLEDGEMENTS
We thank the Srivastava and Jain laboratories for critical discussions and feedback; the Penn Electron Microscopy Core for assistance with sample preparation and imaging for electron microscopy; and the Gladstone Genomics Core for library preparation and sequencing for transcriptional analyses. We thank Keiko Ozato for experimental reagents. We thank Reuben Thomas from the Gladstone Bioinformatics Core for assistance with statistical analyses.
SOURCES OF FUNDING
A.P. is supported by the Tobacco‐Related Disease Research Program (578649), A.P. Giannini Foundation (P0527061), Michael Antonov Charitable Foundation Inc., and the Sarnoff Cardiovascular Research Foundation. M.A. is supported by the Swiss National Science Foundation (P400PM_186704). R.L.S. is supported by NIH F31 HL147416. S.M.H. was supported by NIH R01 HL127240. R.J. is supported by the Burroughs Wellcome Career Award for Medical Scientists, Gilead Research Scholars Award, American Heart Association, Allen Foundation, NSF CMMI-1548571, and NIH R01 HL139783. D.S. is supported by NIH P01 HL098707, P01 HL146366, R01 HL057181, R01 HL127240, and by the Roddenberry Foundation, the L.K. Whittier Foundation, and the Younger Family Fund. This work was also supported by NIH/NCRR grant C06 RR018928 to the Gladstone Institutes.
Nonstandard Abbreviations/Nonstandard Acronyms:
- HF
heart failure
- TF
transcription factor
- CM
cardiomyocyte
- BET
Bromo- and Extra-Terminal domain
- BD
bromodomain
- LV
left ventricle
- ES
embryonic stem
- TAM
tamoxifen
- VEH
vehicle
- EF
ejection fraction
- LVESV
LV end systolic volume
- RNA-seq
RNA sequencing
- KO
knockout
- GO
gene ontology
- ATAC-seq
assay for transposase-accessible chromatin sequencing
Footnotes
DISCLOSURES
D.S. is scientific co-founder, shareholder and director of Tenaya Therapeutics. S.M.H. is an executive, officer, and shareholder of Amgen, Inc. and is a scientific co-founder and shareholder of Tenaya Therapeutics.
REFERENCES
- 1.Virani SS, Alonso A, Benjamin EJ, Bittencourt MS, Callaway CW, Carson AP, Chamberlain AM, Chang AR, Cheng S, Delling FN, Djousse L, Elkind MSV, Ferguson JF, Fornage M, Khan SS, Kissela BM, Knutson KL, Kwan TW, Lackland DT, Lewis TT, Lichtman JH, Longenecker CT, Loop MS, Lutsey PL, Martin SS, Matsushita K, Moran AE, Mussolino ME, Perak AM, Rosamond WD, Roth GA, Sampson UKA, Satou GM, Schroeder EB, Shah SH, Shay CM, Spartano NL, Stokes A, Tirschwell DL, VanWagner LB, Tsao CW, American Heart Association Council on Epidemiology and Prevention Statistics Committee and Stroke Statistics Subcommittee. Heart Disease and Stroke Statistics-2020 Update: A Report From the American Heart Association. Circulation. 2020;141:e139–e596. [DOI] [PubMed] [Google Scholar]
- 2.Timmis A, Townsend N, Gale CP, Torbica A, Lettino M, Petersen SE, Mossialos EA, Maggioni AP, Kazakiewicz D, May HT, De Smedt D, Flather M, Zuhlke L, Beltrame JF, Huculeci R, Tavazzi L, Hindricks G, Bax J, Casadei B, Achenbach S, Wright L, Vardas P, European Society of Cardiology. European Society of Cardiology: Cardiovascular Disease Statistics 2019. Eur Heart J. 2020;41:12–85. [DOI] [PubMed] [Google Scholar]
- 3.Burnett H, Earley A, Voors AA, Senni M, McMurray JJV, Deschaseaux C, Cope S. Thirty Years of Evidence on the Efficacy of Drug Treatments for Chronic Heart Failure With Reduced Ejection Fraction: A Network Meta-Analysis. Circ Heart Fail. 2017;10:e003529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.van Berlo JH, Maillet M, Molkentin JD. Signaling effectors underlying pathologic growth and remodeling of the heart. J Clin Invest. 2013;123:37–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hill JA, Olson EN. Cardiac plasticity. N Engl J Med. 2008;358:1370–1380. [DOI] [PubMed] [Google Scholar]
- 6.Akazawa H, Komuro I. Roles of cardiac transcription factors in cardiac hypertrophy. Circ Res. 2003;92:1079–1088. [DOI] [PubMed] [Google Scholar]
- 7.Alexanian M, Padmanabhan A, McKinsey TA, Haldar SM. Epigenetic therapies in heart failure. J Mol Cell Cardiol. 2019;130:197–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Padmanabhan A, Haldar SM. Drugging transcription in heart failure. J Physiol. 2020;598:3005–3014. [DOI] [PubMed] [Google Scholar]
- 9.Anand P, Brown JD, Lin CY, Qi J, Zhang R, Artero PC, Alaiti MA, Bullard J, Alazem K, Margulies KB, Cappola TP, Lemieux M, Plutzky J, Bradner JE, Haldar SM. BET bromodomains mediate transcriptional pause release in heart failure. Cell. 2013;154:569–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Spiltoir JI, Stratton MS, Cavasin MA, Demos-Davies K, Reid BG, Qi J, Bradner JE, McKinsey TA. BET acetyl-lysine binding proteins control pathological cardiac hypertrophy. J Mol Cell Cardiol. 2013;63:175–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dhalluin C, Carlson JE, Zeng L, He C, Aggarwal AK, Zhou MM. Structure and ligand of a histone acetyltransferase bromodomain. Nature. 1999;399:491–496. [DOI] [PubMed] [Google Scholar]
- 12.Taniguchi Y. The Bromodomain and Extra-Terminal Domain (BET) Family: Functional Anatomy of BET Paralogous Proteins. Int J Mol Sci. 2016;17:1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xu Y, Vakoc CR. Targeting Cancer Cells with BET Bromodomain Inhibitors. Cold Spring Harb Perspect Med. 2017;7:a026674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stathis A, Bertoni F. BET Proteins as Targets for Anticancer Treatment. Cancer Discov. 2018;8:24–36. [DOI] [PubMed] [Google Scholar]
- 15.Lovén J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR, Bradner JE, Lee TI, Young RA. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell. 2013;153:320–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nicodeme E, Jeffrey KL, Schaefer U, Beinke S, Dewell S, Chung C-W, Chandwani R, Marazzi I, Wilson P, Coste H, White J, Kirilovsky J, Rice CM, Lora JM, Prinjha RK, Lee K, Tarakhovsky A. Suppression of inflammation by a synthetic histone mimic. Nature. 2010;468:1119–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, Morse EM, Keates T, Hickman TT, Felletar I, Philpott M, Munro S, McKeown MR, Wang Y, Christie AL, West N, Cameron MJ, Schwartz B, Heightman TD, La Thangue N, French CA, Wiest O, Kung AL, Knapp S, Bradner JE. Selective inhibition of BET bromodomains. Nature. 2010;468:1067–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Duan Q, McMahon S, Anand P, Shah H, Thomas S, Salunga HT, Huang Y, Zhang R, Sahadevan A, Lemieux ME, Brown JD, Srivastava D, Bradner JE, McKinsey TA, Haldar SM. BET bromodomain inhibition suppresses innate inflammatory and profibrotic transcriptional networks in heart failure. Sci Transl Med. 2017;9:eaah5084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Antolic A, Wakimoto H, Jiao Z, Gorham JM, DePalma SR, Lemieux ME, Conner DA, Lee DY, Qi J, Seidman JG, Bradner JE, Brown JD, Haldar SM, Seidman CE, Burke MA. BET bromodomain proteins regulate transcriptional reprogramming in genetic dilated cardiomyopathy. JCI Insight. 2020;5: e138687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Houzelstein D, Bullock SL, Lynch DE, Grigorieva EF, Wilson VA, Beddington RSP. Growth and early postimplantation defects in mice deficient for the bromodomain-containing protein Brd4. Mol Cell Biol. 2002;22:3794–3802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, Clark NR, Ma’ayan A. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics. 2013;14:128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sohal DS, Nghiem M, Crackower MA, Witt SA, Kimball TR, Tymitz KM, Penninger JM, Molkentin JD. Temporally regulated and tissue-specific gene manipulations in the adult and embryonic heart using a tamoxifen-inducible Cre protein. Circ Res. 2001;89:20–25. [DOI] [PubMed] [Google Scholar]
- 24.Jiao K, Kulessa H, Tompkins K, Zhou Y, Batts L, Baldwin HS, Hogan BLM. An essential role of Bmp4 in the atrioventricular septation of the mouse heart. Genes Dev. 2003;17:2362–2367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee J-E, Park Y-K, Park S, Jang Y, Waring N, Dey A, Ozato K, Lai B, Peng W, Ge K. Brd4 binds to active enhancers to control cell identity gene induction in adipogenesis and myogenesis. Nat Commun. 2017;8:2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods. 2013;10:1213–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell. 2010;38:576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Knowlton KU, Baracchini E, Ross RS, Harris AN, Henderson SA, Evans SM, Glembotski CC, Chien KR. Co-regulation of the atrial natriuretic factor and cardiac myosin light chain-2 genes during alpha-adrenergic stimulation of neonatal rat ventricular cells. Identification of cis sequences within an embryonic and a constitutive contractile protein gene which mediate inducible expression. J Biol Chem. 1991;266:7759–7768. [PubMed] [Google Scholar]
- 29.Hsu A, Duan Q, McMahon S, Huang Y, Wood SA, Gray NS, Wang B, Bruneau BG, Haldar SM. Salt-inducible kinase 1 maintains HDAC7 stability to promote pathologic cardiac remodeling. J Clin Invest. 2020;130:2966–2977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Burridge PW, Matsa E, Shukla P, Lin ZC, Churko JM, Ebert AD, Lan F, Diecke S, Huber B, Mordwinkin NM, Plews JR, Abilez OJ, Cui B, Gold JD, Wu JC. Chemically defined generation of human cardiomyocytes. Nat Methods. 2014;11:855–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Koitabashi N, Bedja D, Zaiman AL, Pinto YM, Zhang M, Gabrielson KL, Takimoto E, Kass DA. Avoidance of transient cardiomyopathy in cardiomyocyte-targeted tamoxifen-induced MerCreMer gene deletion models. Circ Res. 2009;105:12–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Arany Z, He H, Lin J, Hoyer K, Handschin C, Toka O, Ahmad F, Matsui T, Chin S, Wu P-H, Rybkin II, Shelton JM, Manieri M, Cinti S, Schoen FJ, Bassel-Duby R, Rosenzweig A, Ingwall JS, Spiegelman BM. Transcriptional coactivator PGC-1 alpha controls the energy state and contractile function of cardiac muscle. Cell Metab. 2005;1:259–271. [DOI] [PubMed] [Google Scholar]
- 33.Lehman JJ, Boudina S, Banke NH, Sambandam N, Han X, Young DM, Leone TC, Gross RW, Lewandowski ED, Abel ED, Kelly DP. The transcriptional coactivator PGC-1alpha is essential for maximal and efficient cardiac mitochondrial fatty acid oxidation and lipid homeostasis. Am J Physiol Heart Circ Physiol. 2008;295:H185–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Alexanian M, Haldar SM. BETs that cover the spread from acquired to heritable heart failure. J Clin Invest. 2020;130:4536–4539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang B, Yang X-D, Zhou M-M, Ozato K, Chen L-F. Brd4 coactivates transcriptional activation of NF-kappaB via specific binding to acetylated RelA. Mol Cell Biol. 2009;29:1375–1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shi J, Wang Y, Zeng L, Wu Y, Deng J, Zhang Q, Lin Y, Li J, Kang T, Tao M, Rusinova E, Zhang G, Wang C, Zhu H, Yao J, Zeng Y-X, Evers BM, Zhou M-M, Zhou BP. Disrupting the interaction of BRD4 with diacetylated Twist suppresses tumorigenesis in basal-like breast cancer. Cancer Cell. 2014;25:210–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wu S-Y, Lee A-Y, Lai H-T, Zhang H, Chiang C-M. Phospho switch triggers Brd4 chromatin binding and activator recruitment for gene-specific targeting. Mol Cell. 2013;49:843–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Asangani IA, Dommeti VL, Wang X, Malik R, Cieslik M, Yang R, Escara-Wilke J, Wilder-Romans K, Dhanireddy S, Engelke C, Iyer MK, Jing X, Wu Y-M, Cao X, Qin ZS, Wang S, Feng FY, Chinnaiyan AM. Therapeutic targeting of BET bromodomain proteins in castration-resistant prostate cancer. Nature. 2014;510:278–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Roe J-S, Mercan F, Rivera K, Pappin DJ, Vakoc CR. BET Bromodomain Inhibition Suppresses the Function of Hematopoietic Transcription Factors in Acute Myeloid Leukemia. Mol Cell. 2015;58:1028–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.He A, Gu F, Hu Y, Ma Q, Ye LY, Akiyama JA, Visel A, Pennacchio LA, Pu WT. Dynamic GATA4 enhancers shape the chromatin landscape central to heart development and disease. Nat Commun. 2014;5:4907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Akerberg BN, Gu F, VanDusen NJ, Zhang X, Dong R, Li K, Zhang B, Zhou B, Sethi I, Ma Q, Wasson L, Wen T, Liu J, Dong K, Conlon FL, Zhou J, Yuan G-C, Zhou P, Pu WT. A reference map of murine cardiac transcription factor chromatin occupancy identifies dynamic and conserved enhancers. Nat Commun. 2019;10:4907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Durocher D, Chen CY, Ardati A, Schwartz RJ, Nemer M. The atrial natriuretic factor promoter is a downstream target for Nkx-2.5 in the myocardium. Mol Cell Biol. 1996;16:4648–4655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Grépin C, Dagnino L, Robitaille L, Haberstroh L, Antakly T, Nemer M. A hormone-encoding gene identifies a pathway for cardiac but not skeletal muscle gene transcription. Mol Cell Biol. 1994;14:3115–3129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Takaya T, Kawamura T, Morimoto T, Ono K, Kita T, Shimatsu A, Hasegawa K. Identification of p300-targeted acetylated residues in GATA4 during hypertrophic responses in cardiac myocytes. J Biol Chem. 2008;283:9828–9835. [DOI] [PubMed] [Google Scholar]
- 45.Lamonica JM, Deng W, Kadauke S, Campbell AE, Gamsjaeger R, Wang H, Cheng Y, Billin AN, Hardison RC, Mackay JP, Blobel GA. Bromodomain protein Brd3 associates with acetylated GATA1 to promote its chromatin occupancy at erythroid target genes. Proc Natl Acad Sci U S A. 2011;108:E159–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gamsjaeger R, Webb SR, Lamonica JM, Billin A, Blobel GA, Mackay JP. Structural basis and specificity of acetylated transcription factor GATA1 recognition by BET family bromodomain protein Brd3. Mol Cell Biol. 2011;31:2632–2640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wu S-Y, Chiang C-M. The double bromodomain-containing chromatin adaptor Brd4 and transcriptional regulation. J Biol Chem. 2007;282:13141–13145. [DOI] [PubMed] [Google Scholar]
- 48.Filippakopoulos P, Picaud S, Mangos M, Keates T, Lambert J-P, Barsyte-Lovejoy D, Felletar I, Volkmer R, Müller S, Pawson T, Gingras A-C, Arrowsmith CH, Knapp S. Histone recognition and large-scale structural analysis of the human bromodomain family. Cell. 2012;149:214–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jung M, Philpott M, Müller S, Schulze J, Badock V, Eberspächer U, Moosmayer D, Bader B, Schmees N, Fernández-Montalván A, Haendler B. Affinity map of bromodomain protein 4 (BRD4) interactions with the histone H4 tail and the small molecule inhibitor JQ1. J Biol Chem. 2014;289:9304–9319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Romero FA, Taylor AM, Crawford TD, Tsui V, Côté A, Magnuson S. Disrupting Acetyl-Lysine Recognition: Progress in the Development of Bromodomain Inhibitors. J Med Chem. 2016;59:1271–1298. [DOI] [PubMed] [Google Scholar]
- 51.Dawson MA. The cancer epigenome: Concepts, challenges, and therapeutic opportunities. Science. 2017;355:1147–1152. [DOI] [PubMed] [Google Scholar]
- 52.Piha-Paul SA, Sachdev JC, Barve M, LoRusso P, Szmulewitz R, Patel SP, Lara PN Jr, Chen X, Hu B, Freise KJ, Modi D, Sood A, Hutti JE, Wolff J, O’Neil BH. First-in-Human Study of Mivebresib (ABBV-075), an Oral Pan-Inhibitor of Bromodomain and Extra Terminal Proteins, in Patients with Relapsed/Refractory Solid Tumors. Clin Cancer Res. 2019;25:6309–6319. [DOI] [PubMed] [Google Scholar]
- 53.Ray KK, Nicholls SJ, Buhr KA, Ginsberg HN, Johansson JO, Kalantar-Zadeh K, Kulikowski E, Toth PP, Wong N, Sweeney M, Schwartz GG, BETonMACE Investigators and Committees. Effect of Apabetalone Added to Standard Therapy on Major Adverse Cardiovascular Events in Patients With Recent Acute Coronary Syndrome and Type 2 Diabetes: A Randomized Clinical Trial. JAMA. 2020;323:1565–1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Winter GE, Buckley DL, Paulk J, Roberts JM, Souza A, Dhe-Paganon S, Bradner JE. DRUG DEVELOPMENT. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science. 2015;348:1376–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lu J, Qian Y, Altieri M, Dong H, Wang J, Raina K, Hines J, Winkler JD, Crew AP, Coleman K, Crews CM. Hijacking the E3 Ubiquitin Ligase Cereblon to Efficiently Target BRD4. Chem Biol. 2015;22:755–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhou B, Hu J, Xu F, Chen Z, Bai L, Fernandez-Salas E, Lin M, Liu L, Yang C-Y, Zhao Y, McEachern D, Przybranowski S, Wen B, Sun D, Wang S. Discovery of a Small-Molecule Degrader of Bromodomain and Extra-Terminal (BET) Proteins with Picomolar Cellular Potencies and Capable of Achieving Tumor Regression. J Med Chem. 2018;61:462–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Stratton MS, Bagchi RA, Felisbino MB, Hirsch RA, Smith HE, Riching AS, Enyart BY, Koch KA, Cavasin MA, Alexanian M, Song K, Qi J, Lemieux ME, Srivastava D, Lam MPY, Haldar SM, Lin CY, McKinsey TA. Dynamic Chromatin Targeting of BRD4 Stimulates Cardiac Fibroblast Activation. Circ Res. 2019;125:662–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Garg V, Kathiriya IS, Barnes R, Schluterman MK, King IN, Butler CA, Rothrock CR, Eapen RS, Hirayama-Yamada K, Joo K, Matsuoka R, Cohen JC, Srivastava D. GATA4 mutations cause human congenital heart defects and reveal an interaction with TBX5. Nature. 2003;424:443–447. [DOI] [PubMed] [Google Scholar]
- 59.Molkentin JD, Lin Q, Duncan SA, Olson EN. Requirement of the transcription factor GATA4 for heart tube formation and ventral morphogenesis. Genes Dev. 1997;11:1061–1072. [DOI] [PubMed] [Google Scholar]
- 60.Ieda M, Fu J-D, Delgado-Olguin P, Vedantham V, Hayashi Y, Bruneau BG, Srivastava D. Direct reprogramming of fibroblasts into functional cardiomyocytes by defined factors. Cell. 2010;142:375–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fu J-D, Stone NR, Liu L, Spencer CI, Qian L, Hayashi Y, Delgado-Olguin P, Ding S, Bruneau BG, Srivastava D. Direct reprogramming of human fibroblasts toward a cardiomyocyte-like state. Stem Cell Reports. 2013;1:235–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Qian L, Huang Y, Spencer CI, Foley A, Vedantham V, Liu L, Conway SJ, Fu J-D, Srivastava D. In vivo reprogramming of murine cardiac fibroblasts into induced cardiomyocytes. Nature. 2012;485:593–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Stone NR, Gifford CA, Thomas R, Pratt KJB, Samse-Knapp K, Mohamed TMA, Radzinsky EM, Schricker A, Ye L, Yu P, van Bemmel JG, Ivey KN, Pollard KS, Srivastava D. Context-Specific Transcription Factor Functions Regulate Epigenomic and Transcriptional Dynamics during Cardiac Reprogramming. Cell Stem Cell. 2019;25:87–102.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hashimoto H, Wang Z, Garry GA, Malladi VS, Botten GA, Ye W, Zhou H, Osterwalder M, Dickel DE, Visel A, Liu N, Bassel-Duby R, Olson EN. Cardiac Reprogramming Factors Synergistically Activate Genome-wide Cardiogenic Stage-Specific Enhancers. Cell Stem Cell. 2019;25:69–86.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Oka T, Maillet M, Watt AJ, Schwartz RJ, Aronow BJ, Duncan SA, Molkentin JD. Cardiac-specific deletion of Gata4 reveals its requirement for hypertrophy, compensation, and myocyte viability. Circ Res. 2006;98:837–845. [DOI] [PubMed] [Google Scholar]
- 66.Heineke J, Auger-Messier M, Xu J, Oka T, Sargent MA, York A, Klevitsky R, Vaikunth S, Duncan SA, Aronow BJ, Robbins J, Crombleholme TM, Molkentin JD. Cardiomyocyte GATA4 functions as a stress-responsive regulator of angiogenesis in the murine heart. J Clin Invest. 2007;117:3198–3210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Neubauer S. The failing heart—an engine out of fuel. N Engl J Med. 2007;356:1140–1151. [DOI] [PubMed] [Google Scholar]
- 68.Huss JM, Kelly DP. Mitochondrial energy metabolism in heart failure: a question of balance. J Clin Invest. 2005;115(3):547–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bates MGD, Bourke JP, Giordano C, d’Amati G, Turnbull DM, Taylor RW. Cardiac involvement in mitochondrial DNA disease: clinical spectrum, diagnosis, and management. Eur Heart J. 2012;33:3023–3033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.El-Hattab AW, Scaglia F. Mitochondrial Cardiomyopathies. Front Cardiovasc Med. 2016;3:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dufour CR, Wilson BJ, Huss JM, Kelly DP, Alaynick WA, Downes M, Evans RM, Blanchette M, Giguère V. Genome-wide orchestration of cardiac functions by the orphan nuclear receptors ERRalpha and gamma. Cell Metab. 2007;5:345–356. [DOI] [PubMed] [Google Scholar]
- 72.Stefanovic S, Christoffels VM. GATA-dependent transcriptional and epigenetic control of cardiac lineage specification and differentiation. Cell Mol Life Sci. 2015;72:3871–3881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Shi J, Vakoc CR. The mechanisms behind the therapeutic activity of BET bromodomain inhibition. Mol Cell. 2014;54:728–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Faivre EJ, McDaniel KF, Albert DH, Mantena SR, Plotnik JP, Wilcox D, Zhang L, Bui MH, Sheppard GS, Wang L, Sehgal V, Lin X, Huang X, Lu X, Uziel T, Hessler P, Lam LT, Bellin RJ, Mehta G, Fidanze S, Pratt JK, Liu D, Hasvold LA, Sun C, Panchal SC, Nicolette JJ, Fossey SL, Park CH, Longenecker K, Bigelow L, Torrent M, Rosenberg SH, Kati WM, Shen Y. Selective inhibition of the BD2 bromodomain of BET proteins in prostate cancer. Nature. 2020;578:306–310. [DOI] [PubMed] [Google Scholar]
- 75.Gilan O, Rioja I, Knezevic K, Bell MJ, Yeung MM, Harker NR, Lam EYN, Chung C-W, Bamborough P, Petretich M, Urh M, Atkinson SJ, Bassil AK, Roberts EJ, Vassiliadis D, Burr ML, Preston AGS, Wellaway C, Werner T, Gray JR, Michon A-M, Gobbetti T, Kumar V, Soden PE, Haynes A, Vappiani J, Tough DF, Taylor S, Dawson S-J, Bantscheff M, Lindon M, Drewes G, Demont EH, Daniels DL, Grandi P, Prinjha RK, Dawson MA. Selective targeting of BD1 and BD2 of the BET proteins in cancer and immunoinflammation. Science. 2020;368:387–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Jang MK, Mochizuki K, Zhou M, Jeong H-S, Brady JN, Ozato K. The Bromodomain Protein Brd4 Is a Positive Regulatory Component of P-TEFb and Stimulates RNA Polymerase II-Dependent Transcription. Mol Cell. 2005;19:523–534. [DOI] [PubMed] [Google Scholar]
- 77.Yang Z, Yik JHN, Chen R, He N, Jang MK, Ozato K, Zhou Q. Recruitment of P-TEFb for Stimulation of Transcriptional Elongation by the Bromodomain Protein Brd4. Mol Cell. 2005;19:535–545. [DOI] [PubMed] [Google Scholar]
- 78.Shen C, Ipsaro JJ, Shi J, Milazzo JP, Wang E, Roe J-S, Suzuki Y, Pappin DJ, Joshua-Tor L, Vakoc CR. NSD3-Short Is an Adaptor Protein that Couples BRD4 to the CHD8 Chromatin Remodeler. Mol Cell. 2015;60:847–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Rahman S, Sowa ME, Ottinger M, Smith JA, Shi Y, Harper JW, Howley PM. The Brd4 extraterminal domain confers transcription activation independent of pTEFb by recruiting multiple proteins, including NSD3. Mol Cell Biol. 2011;31:2641–2652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lambert J-P, Picaud S, Fujisawa T, Hou H, Savitsky P, Uusküla-Reimand L, Gupta GD, Abdouni H, Lin Z-Y, Tucholska M, Knight JDR, Gonzalez-Badillo B, St-Denis N, Newman JA, Stucki M, Pelletier L, Bandeira N, Wilson MD, Filippakopoulos P, Gingras A-C. Interactome Rewiring Following Pharmacological Targeting of BET Bromodomains. Mol Cell. 2019;73:621–638.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.