

Abstract
Withaferin A (WA) exhibits cancer chemopreventive efficacy in preclinical models representative of two different subtypes of breast cancer. However, the mechanism(s) underlying breast cancer chemoprevention by WA is not fully elucidated. We performed RNA-seq analyses using a non-tumorigenic mammary epithelial cell line (MCF-10A) and human breast cancer cells (BCC) belonging to the luminal-type (MCF-7), HER2-enriched (SK-BR-3) and basal-like subtype (MDA-MB-231) to identify novel cancer-selective mechanistic targets of WA. The WA-regulated transcriptome was strikingly different between MCF-10A versus BCC. The KEGG pathway analysis revealed downregulation of genes associated with cellular senescence in WA-treated BCC. Consequently, the number of senescence-associated β-galactosidase positive cells was decreased significantly in WA-treated BCC but not in the MCF-10A cells. WA treatment caused upregulation of senescence marker p21 more robustly in BCC than in MCF-10A. Breast cancer prevention by WA in rats was also associated with up-regulation of p21 protein expression. The Reactome pathway analyses indicated up-regulation of genes associated with cellular response to stress/external stimuli in WA-treated BCC but not in MCF-10A. Two proteins represented in these pathways (HSPA6 and NRF2) were further investigated. While HSPA6 was dispensable for WA-mediated apoptosis and autophagy or inhibition of cell migration, the NRF2 knockout cells were more resistant to apoptosis resulting from WA treatment than control cells. Finally, expression of some glycolysis-related proteins was decreased by WA treatment both in vitro and in vivo. In summary, this study provides novel insights into cancer-selective pathways affected by WA that may contribute to its chemopreventive efficacy in breast cancer.
Keywords: Withaferin A, Breast Cancer, Chemoprevention
1. INTRODUCTION
Chemoprevention, which broadly implies use of natural or synthetic small molecules, extracts from medicinal or dietary plants or biological agents like vaccines to impede carcinogenesis at its earliest stages, is desirable to reduce mortality and suffering from breast cancer that affects thousands of women each year in the United States.1,2 Clinical feasibility of breast cancer chemoprevention is demonstrated with selective estrogen receptor modulators and aromatase inhibitors.3–5 However, these interventions are clinically useful for chemoprevention of only estrogen-receptor positive (luminal-type) breast cancers.3–5 Accordingly, a broader chemopreventative intervention efficacious against other subtypes of breast cancers [human epidermal growth factor receptor 2 (HER2)-enriched and basal-like] is still lacking. We have demonstrated previously that withaferin A (WA), a small molecule steroidal lactone derived from a medicinal plant (Withania somnifera), prevents development of two different subtypes of breast cancer in preclinical rodent models including HER2-driven breast cancer in MMTV-neu transgenic mice and N-methyl-N-nitrosourea (MNU)-induced luminal-type mammary neoplasia in rats.6,7 In the MMTV-neu model, WA administration (0.1 mg/mouse, three times/week by intraperitoneal route) delayed progression of breast cancer as evidenced by smaller size of macroscopic tumors and reduced area of microscopic mammary tumor lesions when compared to solvent-treated control mice.6 On the other hand, WA administration (4 and 8 mg/kg body weight, five times/week by intraperitoneal route) significantly decreased the incidence as well as burden (wet weight) of mammary cancer in the MNU-rat model in comparison of control.7 The in vivo growth inhibitory effects of WA have also been reported in subcutaneous or orthotopic xenograft models of human breast cancer cells (MDA-MB-231) and 4T1 mouse mammary carcinoma cell line.8,9 WA administration also inhibited self-renewal of breast cancer stem-like cells in the MMTV-neu mice as well as in the MNU-rat model.7,10
Several pharmacological attributes are essential for clinical development of chemopreventative interventions. While safety (lack of any toxicity) is one of the most essential requirement for long-term administration of chemopreventative agents in high-risk but otherwise healthy subjects, favorable pharmacokinetic profile including oral bioavailability and target tissue exposure is also desirable. WA meets these criteria as no major toxicity was observed in any of the in vivo studies to date with this agent.6–10 We have shown previously that WA is detectable in the rat tumors following intraperitoneal administration.7 The oral bioavailability of WA has been demonstrated in rats.11 While pharmacokinetics and bioavailability of WA in human mammary tissue/tumor is yet to be determined, most of the cellular mechanistic studies have used pharmacologically-relevant plasma achievable concentrations of this phytochemical.12
Elucidation of the mechanism(s) underlying cancer chemoprevention by WA is an equally important consideration for its development as this knowledge could lead to identification of biomarkers potentially useful in future clinical studies. Interestingly, mechanistic studies have revealed that normal human mammary epithelial cells (MCF-10A and HMEC) are resistant to apoptosis induction and/or mitotic arrest following WA treatment in comparison with human breast cancer cells (BCC) belonging to different subtypes, including MCF-7 (luminal-type), SK-BR-3 (HER2-enriched), and MDA-MB-231 (basal-type).13,14 To gain insights into this level of cancer selectivity and to identify novel mechanistic targets, we compared WA-regulated transcriptome in MCF-10A, MCF-7, SK-BR-3, and MDA-MB-231 cells by RNA-seq analyses.
2. Materials and Methods
2.1. Ethics statement
The present study used the rat mammary tumor sections from our previously published study, which was approved by the Animal Care and Use Committee of the University of Pittsburgh,7 for immuoblotting and immunohistochemistry of different proteins.
2.2. Reagents
The WA (purity > 95%) was purchased from ChromaDex (Irvine, CA), dissolved in dimethyl sulfoxide (DMSO; 20 mM stock), and stored at −80°C. Cell culture media were from MediaTech (Manassas, VA). Fetal bovine serum was obtained from Atlanta Biologicals (Norcross, GA). Antibodies against ataxia telangiectasia and Rad3-related protein (ATR), phospho(S345)-checkpoint kinase 1 (CHK1), total CHK1, cleaved poly (ADP-ribose) polymerase (PARP), cleaved caspase-3, microtubule associated protein 1 light chain 3 beta (LC3B or MAP1LC3B), sequestosome 1 (SQSTM1), pyruvate kinase M2 (PKM2), and lactate dehydrogenase A (LDHA) (for immunoblotting) were from Cell Signaling Technology (Danvers, MA). Anti-phospho(T1989)-ATR antibody and anti-glyceraldehyde 3-phosphate dehydrogenase (GAPDH) antibody were from GeneTex (Irvine, CA). Anti-phospho(S10)-histone H3 antibody was from MilliporeSigma (Burlington, MA). Antibodies against glucose transporter (GLUT) 1, GLUT4, and nuclear factor erythroid 2-related factor 2 (NRF2) were from Proteintech Group (Rosemont, IL). Anti-p21 antibody and Annexin V/propidium iodide (PI) assay kit for apoptosis detection were purchased from BD Biosciences (San Jose, CA). Anti-heat shock protein family A member 6 (HSPA6) antibody was from Santa Cruz Biotechnology (Dallas, TX). Anti-LDHA antibody for confocal microscopy and immunohistochemistry was from Novus Biologicals (Centennial, CO). Anti-sirtuin 1 (SIRT1) and anti-hexokinase 2 (HK2) antibodies were from Abcam (Cambridge, MA). Anti-β-Tubulin anti β-Actin antibodies were from Sigma-Aldrich (St. Louis, MO). Nonspecific control small interfering RNA (siRNA) and HSPA6-targeted siRNA were purchased from Qiagen and Santa Cruz Biotechnology, respectively. Alexa Fluor 488- or 568-conjugated goat anti-rabbit antibody was from Life Technologies-Thermo Fisher Scientific (Waltham, MA).
2.3. Cell culture
The MCF-10A, MCF-7, SK-BR-3, and MDA-MB-231 cell lines were procured from the American Type Culture Collection (Manassas, VA) and each cell line was maintained as suggested by the supplier. The MCF-7, SK-BR-3, and MDA-MB-231 cells were last authenticated by us in March of 2017. Immortalized wild-type (WT) mouse embryonic fibroblasts (MEF) and NRF2 knockout (NRF2KO) MEF were a generous gift from Dr. Thomas W. Kensler while he was at the University of Pittsburgh. The MEF were cultured as described previously by his group.15
2.4. RNA-seq analyses
The MCF-10A, MCF-7, SK-BR-3, and MDA-MB-231 cells were plated in 6-cm dishes at a density of 5 × 105 cells/dish in triplicate and incubated overnight for attachment. The cells were treated with DMSO (final concentration: 0.01%) or 2 μM of WA for 16 hours. RNA isolation, RNA quality determination, and RNA-seq analyses were performed by Novogene (Sacramento, CA). Data analyses were done as described by us previously.16
The RNA-seq data presented in this study have been submitted to the Gene Expression Omnibus of NCBI (GSE158085).
2.5. Quantitative real-time reverse transcription polymerase chain reaction (qRT-PCR)
Cells were plated at a density of 5 × 105 cells/dish in 6-cm dishes in triplicate, allowed to attach by overnight incubation, and then treated with DMSO (control) or 2 μM of WA for 8 or 16 hours. RNA isolation, reverse transcription, and qRT-PCR were performed essentially as described by us previously.16 Primers for retinoblastoma-like 2 (RBL2), nibrin (NBN), nuclear factor of activated T cells 4 (NFATC4), protein phosphatase 1 catalytic subunit gamma (PPP1CC), and ZFP36 ring finger protein like 2 (ZFP36L2) were purchased from GeneCopoeia (Rockville, MD). Primers for GAPDH were as follows. GAPDH-forward: 5’-GGACCTGACCTGCCGTCTAGAA-3’ GAPDH-reverse: 5’-GGTGTCGCTGTTGAAGTCAGAG-3’. The PCR conditions were as follows: 95°C for 10 minutes followed by 40 cycles of 95°C for 15 seconds, 60°C for 1 minute and 72°C for 30 seconds. Relative gene expression levels were calculated using the method of Livak and Schmittgen.17
2.6. Senescence-associated β-galactosidase (SA-β-gal) assay
The SA-β-gal staining was performed using Senescence β-Galactosidase staining kit from Cell Signaling according to the protocol of the manufacturer. At least four nonoverlapping fields on each well were captured and scored under an inverted microscope.
2.7. Western blotting
Preparation of lysates from cultured cells and tumors have been detailed by us previously.6,18 Blots were stripped and re-probed with anti-β-Actin or anti-GAPDH antibody for normalization. Immunoreactive bands were visualized by the enhanced chemiluminescence method. Quantitation of protein expression was performed using UN-SCAN-IT gel analysis & graph digitization software (Version 7.1, Silk Scientific Corporation, Orem, UT).
2.8. RNA interference of HSPA6
SK-BR-3 cells were seeded in six-well plates and transfected at 50% confluency with a control (non-specific) siRNA or HSPA6 siRNA. Twenty-four hours after transfection, the cells were treated with DMSO (control) or 2 μM of WA for 24 hours. Cells were then collected and processed for immunoblotting and cell migration assay.
2.9. Cell migration assay
Cell migration assay was performed using a Corning Transwell Boy den chamber containing 8.0 μm pore size polycarbonate filter as described by us previously.19
2.10. Annexin V/PI assay
The Annexin V/PI assay kit was used to measure total (early and late) apoptosis. The MEF (WT and NRF2KO) were plated in triplicate in 6-well plates at a density of 2 × 105 cells/well and incubated overnight to allow attachment. The MEF were then treated with DMSO (control) or 2 μM WA for 24 hours. At the end of the treatment, the MEF were collected by trypsinization, resuspended in Annexin V/PI solution, incubated for 30 minutes at room temperature with gentle shaking in the dark, and analyzed using a BD Accuri™ C6 flow cytometer.
2.11. Confocal microscopy and immunohistochemistry
The SK-BR-3 and MDA-MB-231 cells were plated in triplicate on coverslips in 24-well plates. After overnight incubation, the cells were treated with DMSO (control) or the indicated doses of WA for 24 hours. The cells were then fixed and permeabilized with 2% paraformaldehyde and 0.5% Triton X-100, respectively. After blocking with phosphate-buffered saline (PBS) containing 0.5% bovine serum albumin and 0.15% glycine, cells were treated overnight at 4°C with the desired primary antibody (PKM2 or LDHA) followed by incubation with Alexa Fluor 488-conjugated or Alexa Fluor 568-conjugated goat anti-rabbit antibody for 1 hour, and then counterstained with DRAQ5 (nuclear stain) for 5 minutes at room temperature in the dark. Corrected total cell fluorescence (CTCF) was determined using ImageJ software.
Archived rat mammary tumor sections from our previous study7 were de-paraffinized, hydrated, and immersed in boiling citrate retrieval buffer solution (pH 6.0) for 30 minutes followed by treatment with 0.3% hydrogen peroxide in 100% methanol for 20 minutes at room temperature. Sections were exposed to PBS containing 5% normal goat serum and 0.1% Triton X-100 for 24 hours at 4°C followed by incubation with anti-HK2, anti-PKM2 or anti-LDHA antibody in humidified chambers at 4°C. Sections were washed with PBS, incubated with horseradish peroxidase-conjugated secondary antibody for 2 hours at room temperature, followed by incubation with 3,3’-diaminobenzi dine tetrahydrochloride for 10 minutes. The sections were counter stained with hematoxylin. Stained sections were examined under Leica microscope at 40× objective magnification. Non-overlapping and non-necrotic regions were captured from each section and analyzed by Aperio ImageScope software (positive pixel count V9 algorithm) to quantitate H-score as described previously.6
2.12. Statistical analysis
GraphPad Prism (version 8.0.0) was used to perform statistical analyses. Statistical significance of difference for two group comparisons was determined by unpaired Student’s t-test. One-way analysis of variance (ANOVA) followed by Dunnett’s test (for dose-response comparison) or Bonferroni’s test (for multiple comparisons) was used for other statistical analyses.
3. RESULTS
3.1. The WA-regulated transcriptome was different between MCF-10A and BCC
The mapping results are summarized in Table S1. The distribution of differentially expressed genes between control and WA-treated cells are depicted by Volcano plots in Figure 1A. The heatmaps of three replicates of each group are shown in Figure 1B highlighting data consistency and WA-mediated gene expression changes in each cell line. The Venn diagrams for control and WA-treated cells showing unique and overlapping genes are shown in Figure S1. The Venn diagram shown in Figure 1C shows unique and overlapping gene expression between control and WA-treated BCC. There were 99 genes that were upregulated by WA treatment in BCC but not in the MCF-10A cell line (Figure 1C). Downregulation of 59 genes was observed in WA-treated BCC but not in the MCF-10A cell line (Figure 1C).
FIGURE 1.
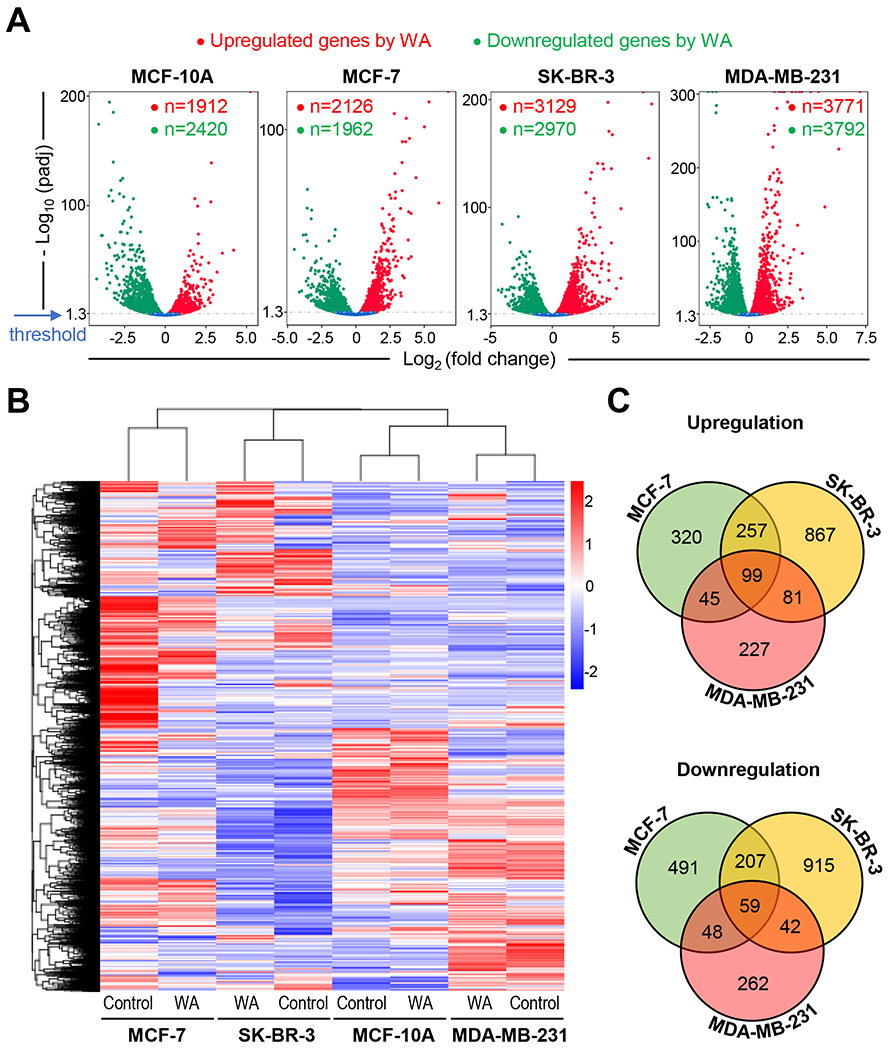
RNA-seq revealed differential effect on gene expression between human breast cancer cells and a normal mammary epithelial cell line following withaferin A (WA) treatment. A, Volcano plots displaying genes upregulated or downregulated by WA exposure in MCF-10A, MCF-7, SK-BR-3, and MDA-MB-231 cells. B, Heatmaps depicting differentially expressed genes by WA treatment in four cell lines. C, Venn diagram showing unique as well as common genes upregulated or downregulated by WA treatment (cut-off > ± 2-fold change in gene expression and Padj < 0.05). The genes that were commonly affected by WA treatment in MCF-10A as well as BCC are not included in the Venn diagram.
3.2. WA treatment inhibited senescence in BCC but not in MCF-10A cells
The Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analyses showed WA-mediated downregulation of genes associated with senescence in each BCC but not in the MCF-10A cells (Figure S2). In the RNA-seq data, many senescence-associated genes were downregulated by WA treatment in BCC (Table S2). Expression changes for some senescence-associated genes from the RNA-seq data are shown in Figure 2A. Strikingly, WA treatment either did not affect or increased expression of these genes in the MCF-10A cell line (Figure 2A). These RNA-seq results were confirmed by qRT-PCR (Figure 2B). Except for PPP1CC, expression of other genes was decreased following 8-hour and/or 16-hour treatment with WA in at least two or all three BCC (Figure 2B). Eight-hour exposure of SK-BR-3 cells resulted in induction of NFATC4 but its level was decreased after 16 hours of treatment (Figure 2B).
FIGURE 2.
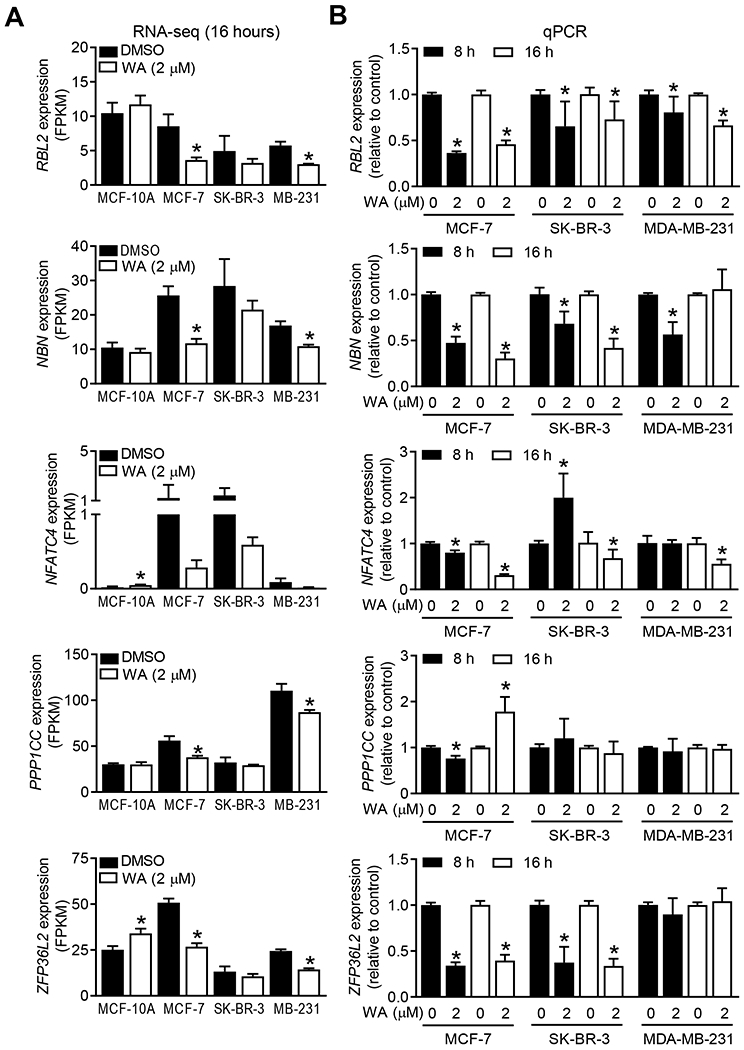
Withaferin A (WA) treatment modulated expression of genes associated with cellular senescence. A, Expression of retinoblastoma transcriptional corepressor like 2 (RBL2), nibrin (NBN), nuclear factor of activated T cells 4 (NFATC4), protein phosphatase 1 catalytic subunit gamma (PPP1CC), and ZFP36 ring finger protein like 2 (ZFP36L2) genes in four cell lines after 16-hour treatment with dimethyl sulfoxide (DMSO, control) or 2 μM WA. Results shown are mean ± SD (n=3) and statistical analysis was done by unpaired Student’s t-test (*, P < 0.05). B, Quantitative real-time reverse transcription polymerase chain reaction analysis of above-mentioned genes in breast cancer cell lines treated with DMSO (control) or 2 μM WA for 8 and 16 hours. Combined results from two independent experiments are shown as mean ± SD (n=6). Statistical analysis was done by unpaired Student’s t-test (*, P < 0.05).
SA-β-gal staining is a widely used technique for visualization and quantitation of senescent cells. Representative SA-β-gal staining in control and WA-treated MCF-7 cells is shown in Figure 3A. As shown in Figure 3B, WA treatment resulted in a statistically significant increase in the number of SA-β-gal positive cells in the MCF-10A cultures (Figure 3B). On the other hand, the number of SA-β-gal positive cells was significantly decreased by WA treatment in each BCC line (Figure 3B). Induction of p21 but suppression of SIRT1 are biomarkers of senescence20,21 As can be seen in Figure 3C, WA treatment resulted in marked induction of p21 protein in each BCC as well as in the MCF-10A cells. However, the extent of p21 induction by WA treatment was much higher in the BCC than in the MCF-10A cell line (Figure 3C). WA treatment resulted in suppression of SIRT1 in each cell line except SK-BR-3 (Figure 3C). Finally, WA administration to rats in which mammary tumor was induced chemically resulted in induction of p21 protein (Figure 3D). Four out of six tumors from the WA treatment group exhibited induced expression of p21 protein whose level was very low in each tumor of the control group (Figure 3D, E). Collectively, these results indicated for the first time, inhibition of senescence in BCC following WA treatment.
FIGURE 3.
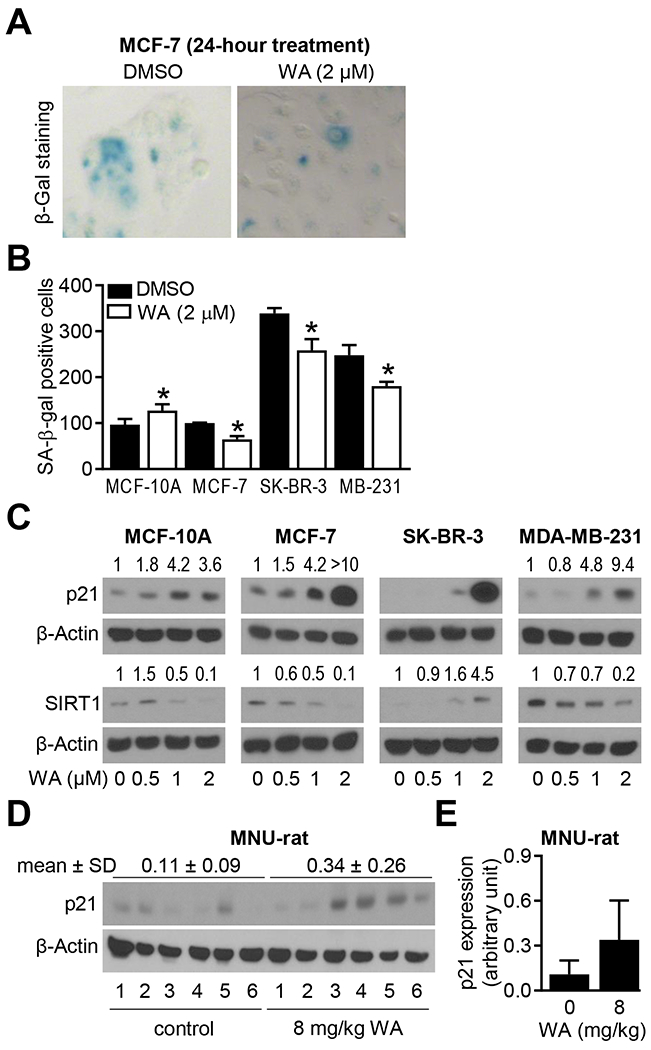
Withaferin A (WA) treatment inhibited senescence in breast cancer cells. A, Representative images for senescence-associated β-galactosidase (SA-β-Gal) staining (blue color) in MCF-7 cells treated with dimethyl sulfoxide (DMSO, control) or 2 μM WA for 24 hours. B, Quantification of number of SA β-gal positive cells in four cell lines after 24 hours of treatment with DMSO (control) or 2 μM WA. Data shown are mean ± SD (n=3). Statistical significance was determined by unpaired Student’s t-test (*, P < 0.05). Experiments were repeated with comparable results. C, Western blots for p21, sirtuin 1 (SIRT1), and β-Actin using lysates from four cell lines after 24 hours of treatment with DMSO (control) or the indicated doses of WA. The numbers above bands represent fold change in expression relative to corresponding DMSO-treated controls. Experiments were repeated with comparable results. D, Western blot for p21 using mammary tumor lysates from control and WA-treated rats. E, Quantification of p21 expression. Results are shown as mean ± SD (n=6 each).
3.3. HSPA6 was dispensable for antitumor effects of WA
The Reactome pathway analyses revealed WA-mediated upregulation of genes associated with cellular response to external stimuli in each BCC but not in the MCF-10A cells (Figure S3). Top ten upregulated genes associated with cellular response to external stimuli in WA-treated BCC are summarized in Table S3. Because HSPA6 was the most induced gene following WA treatment in each BCC, we focused on this molecule for further investigation of its role in antitumor effects of WA. Figure 4A shows that WA-mediated upregulation of HSPA6 gene was more pronounced in BCC than in the MCF-10A cell line. Western blotting confirmed WA-mediated induction of HSPA6 protein in BCC (Figure 4B). Apoptosis induction reflected by PARP cleavage, mitotic arrest characterized by increased S10 phosphorylation of histone H3 and mediated by downregulation of β-Tubulin protein, and autophagy induction reflected by increased levels of SQSTM1 and LC3B are known anticancer effects of WA in breast cancer.12–14,22 None of these pathways were affected by knockdown of HSPA6 protein in SK-BR-3 cell line (Figure 4C). Moreover, inhibition of SK-BR-3 cell migration resulting from WA treatment was also not impacted by HSPA6 knockdown (Figure 4D, E). Because of these negative data in SK-BR-3 cells, these experiments were not attempted in MCF-7 or MDA-MB-231 cells.
FIGURE 4.
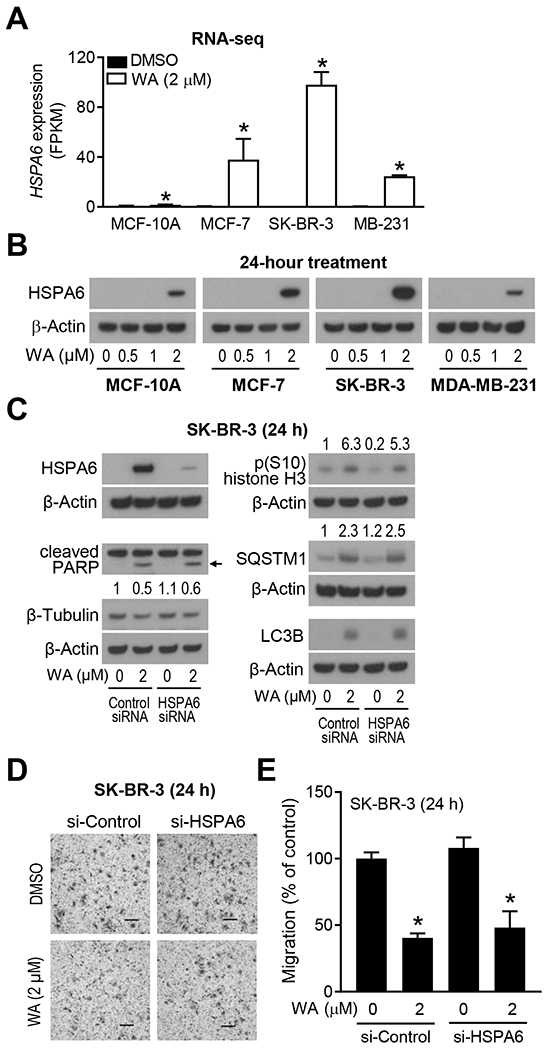
Heat shock protein family A member 6 (HSPA6) was dispensable for antitumor mechanisms of withaferin A (WA). A, Quantification of HSPA6 level from RNA-seq data. Results shown are mean ± SD (n=3). Statistical significance was determined by unpaired Student’s t-test (*, P < 0.05). B, Western blot for HSPA6 protein using lysates from four cell lines treated for 24 hours with dimethyl sulfoxide (DMSO, control) or the indicated doses of WA. Experiments were repeated twice with comparable results. C, Western blots for HSPA6, cleaved poly (ADP-ribose) polymerase (PARP), β-Tubulin, p(S10)-histone H3, sequestosome 1 (SQSTM1), microtubule associated protein 1 light chain 3 beta (LC3B), and β-Actin using lysates from transiently transfected SK-BR-3 cells with a control small interfering RNA (siRNA) or an HSPA6-targeted siRNA and treated for 24 hours with DMSO (control) or 2 μM WA. The numbers above bands represent fold change of each protein relative to DMSO-treated control siRNA-transfected cells. Experiments were repeated twice with comparable results. Arrow indicates correct band for cleaved PARP. D, Representative hematoxylin and eosin-stained images showing cell migration in SK-BR-3 cells transiently transfected with a control siRNA or an HSPA6-targeted siRNA and then treated for 24 hours with DMSO (control) or 2 μM WA. E, Quantification of cell migration. Results are shown as mean ± SD (n=3). Statistical analysis was done by unpaired Student’s t-test (*, P < 0.05). Experiments were repeated with comparable results.
3.4. Antitumor effects of WA were mediated by NRF2
NRF2 is a known target of WA.23 The expression of NRF2 gene was increased significantly by WA treatment in each BCC cell line and this gene was represented in the cellular response to stress/external categories of the Reactome pathway analyses (data not shown). We used immortalized MEF from WT and NRF2KO mice to study the function of NRF2 in antitumor effects of WA. The level of NRF2 protein was increased after WA treatment in WT MEF (Figure 5A). Level of β-Tubulin was higher in NRF2KO cells than in WT cells (Figure 5A). Decrease in β-Tubulin protein expression resulting from WA treatment was more pronounced in NRF2KO than in WT cells (Figure 5A). Phosphorylation of S10 histone H3 was increased by NRF2 deficiency that was partly suppressed by WA treatment in NRF2KO MEF (Figure 5A). We have shown previously that WA treatment inhibits ATR/CHK1 signaling to enhance sensitivity to cisplatin in BCC.24 Similar to the β-Tubulin protein, levels of ATR, total CHK1, and p(S345) CHK1 proteins were increased by knockdown of NRF2. WA-mediated induction of autophagy (SQSTM1 and LC3B) and apoptosis (cleaved caspase-3) markers were markedly attenuated in NRF2KO in comparison with WT cells (Figure 5A). Apoptosis results were confirmed by flow cytometry by Annexin V/PI method (Figure 5B, C). These results are summarized by a cartoon in Figure 5D.
FIGURE 5.
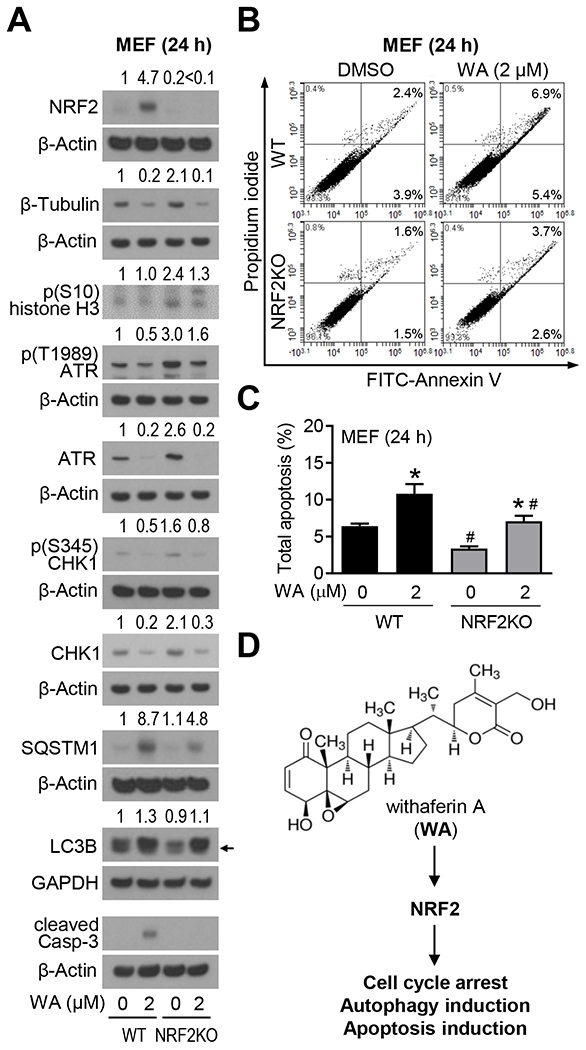
Withaferin A (WA)-mediated apoptosis was partially dependent on nuclear factor erythroid-2-related factor 2 (NRF2). A, Western blots for NRF2 and proteins associated with cell cycle, DNA damage response, autophagy, and apoptosis using lysates from wild-type (WT) or NRF2 knockout (NRF2KO) mouse embryonic fibroblasts (MEF). The MEF were treated for 24 hours with dimethyl sulfoxide (DMSO, control) or 2 μM WA prior to preparation of lysates. The numbers above bands indicate fold change of each protein relative to DMSO-treated WT MEF. Experiments were repeated at least twice with comparable results. Arrow indicates right band for LC3BII protein. B, Representative flow histograms showing apoptotic cells in WT and NRF2KO MEF following 24 hours of treatment with DMSO or 2 μM WA. C, Quantification of total apoptosis. Results shown are mean ± SD (n=3). Statistical analysis was done by one-way analysis of variance (ANOVA) followed by Bonferroni’s multiple comparisons test (*statistically significant compared with corresponding DMSO-treated control, #statistically significant compared with the same dose between WT and NRF2KO). D, A cartoon showing structure of WA and involvement of NRF2 in antitumor effects of WA.
3.5. WA treatment suppressed levels of HK2 and LDHA in vitro and in vivo
Through global metabolomics and proteomics, we have shown previously that breast cancer chemoprevention by WA in the MMTV-neu model is associated with a decrease in plasma and tumor levels of lactic acid,6 which is the end-product of glycolysis. The plasma level of lactate was also significantly lower in WA-treated rats compared with control in the MNU-rat study.7 However, the mechanism underlying glycolysis inhibition by WA is still not clear. As can be seen in Figure 6A, western blotting showed downregulation of GLUT1, HK2, PKM2, and/or LDHA in BCC but not in the MCF-10A cell line. Surprisingly, GLUT4 expression was downregulated in MCF-10A but not in BCC. The WA-mediated suppression of PKM2 and LDHA was confirmed by confocal microscopy using SK-BR-3 and MDA-MB-231 cell lines (Figure 6B, C). Representative immunohistochemistry images for HK2, PKM2, and LDHA in mammary tumor sections of control and WA-treated rats are shown in Figure 6D. The protein levels of HK2 and LDHA, but not PKM2, were decreased significantly in the mammary tumors of WA-treated rats when compared to that of control rats (Figure 6E). These results showed WA-mediated downregulation of HK2 and LDHA in vitro and in vivo.
FIGURE 6.
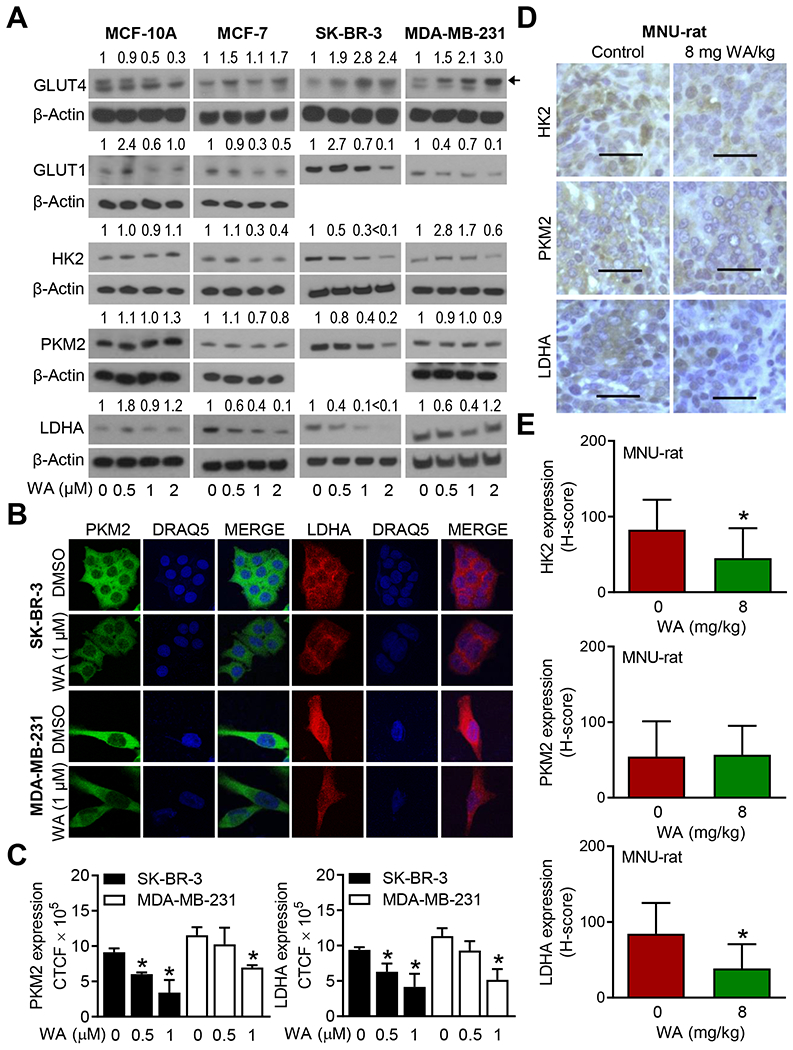
Withaferin A (WA) treatment inhibited expression of hexokinase 2 (HK2) and lactate dehydrogenase A (LDHA) in breast cancer cells in vitro and in vivo. A, Western blotting for glucose transporter 1 (GLUT1), glucose transporter 4 (GLUT4), HK2, pyruvate kinase M2 (PKM2), and LDHA using lysates from four cell lines treated for 24 hours with dimethyl sulfoxide (DMSO) or the indicated doses of WA. Results were consistent in replicate experiments. The numbers above bands represent fold change in level relative to corresponding DMSO-treated control. B, Confocal microscopy for PKM2 and LDHA in SK-BR-3 and MDA-MB-231 cells after 24 hours of treatment with DMSO or WA. Nucleus was stained with DRAQ5. C, Quantitation of corrected total cellular fluorescence (CTCF) for PKM2 and LDHA. Results shown are mean ± SD (n=3). Statistical significance was determined by one-way analysis of variance (ANOVA) followed by Dunnett’s test (*, P < 0.05). Results were consistent in independent replicate experiments. D, Immunohistochemistry for HK2, PKM2, and LDHA in a control rat mammary tumor and a mammary tumor of WA treatment group (objective magnification ×40, scale bar = 50 μm). E, H-score for HK2, PKM2, and LDHA expression. Results shown are mean ± SD (n=10). Statistical significance was determined by unpaired Student’s t-test (*, P < 0.05).
4. DISCUSSION
The RNA-seq analyses presented herein reveal that the WA-regulated transcriptome is strikingly different in a normal mammary epithelial cell line than in BCC. For example, the Gene Ontology (GO) pathway analysis showed upregulation of genes associated with endoplasmic reticulum stress, response to topologically incorrect protein, protein folding, response to unfolded protein, and regulation of ketone metabolic process in the WA-treated MCF-10A cells, but none of these pathways were top 5 enrichment in the BCC (Figure S4). While the cellular consequences of the upregulation of genes associated with these pathways in the MCF-10A cell line is unclear without detailed further investigation, it is logical to propose that differences in WA-regulated transcriptome between normal versus cancerous breast cells may be partly responsible for their differential response to WA with respect to cell viability inhibition and apoptosis induction. WA treatment results in cell viability inhibition and apoptosis induction in MCF-7 and MDA-MB-231 cells but not in a normal mammary epithelial cell line.13
It is satisfying to observe that many previously published findings were validated by the RNA-seq analyses performed in this study. For example, WA is known to induce autophagy in BCC.22 The GO pathway analyses revealed upregulation of genes associated with autophagy in each BCC, which was not the case for MCF-10A cells (Figure S4). Likewise, G2 phase and mitotic arrest is a known antitumor mechanism of WA in BCC.14,25,26 The GO pathway analyses showed most significant downregulation of genes associated with cell cycle in each BCC but not in MCF-10A cells (Figure S4). We have also shown previously that the G2 arrest in MCF-7 and MDA-MB-231 cells following WA exposure is associated with downregulation of cyclin-dependent kinase 1, β-Tubulin, and cell division cycle 25C.25 The RNA-seq data from the present study was consistent with these published findings with statistically significant downregulation observed in all 3 cells or 2 of the three cells (Figure S5).
The Reactome pathway analyses revealed upregulation of genes associated with cellular response to stress and external stimuli in WA-treated BCC, but these pathways were not affected in MCF-10A cells (Figure S3). From a list of top 10 induced genes by WA treatment that were common in MCF-7, SK-BR-3, and MDA-MB-231 included HSPA6, HSPA1A, HSPA1B, HSPB8, and GABARAPL1 (Table S3). While HSPA6 is dispensable to antitumor effects of WA, including apoptosis and autophagy induction and inhibition of cell migration, the possibility that other affected proteins may contribute to antitumor mechanisms of WA requires further investigation. In this context, we have shown recently that GABARAPL1 gene is also upregulated by WA treatment in prostate cancer cells.27 The WA treatment induced GABARAPL1 (ATG8L) protein expression in prostate cancer cells and its knockdown by RNA interference partly attenuated WA-mediated apoptosis.27 It is reasonable to postulate that GABARAPL1 may play a similar role in WA-mediated autophagy in BCC.
The present study shows suppression of genes associated with senescence in WA-treated BCC, whereas this pathway is not affected in MCF-10A cells. In agreement with these results, WA treatment significantly decreases the number of SA-β-gal positive cells in every tested BCC. Senescence is a state of irreversible growth arrest, and this cellular response can be initiated by multiple mechanisms such as shortening of the telomere, epigenetic silencing of INK4a/ARF, DNA damage, and so forth.28,29 However, the exact role of senescence in cancer is context dependent. It has been proposed that senescence can serve to inhibit cancer initiation but promote tumorigenesis at later stages of development.28,29 The possibility that inhibition of senescence also contributes to breast cancer chemoprevention by WA cannot be excluded.
The present study shows a role for NRF2, which is a transcription factor belonging to the cap n collar subfamily,30 in regulation of antitumor mechanisms of WA. We found robust induction of NRF2 in WA-treated WT MEF. The NRF2KO cells were significantly more resistant to apoptosis and autophagy induction as evidenced by western blotting for markers of apoptosis (cleaved caspase-3) and autophagy (SQSTM1 and LC3B). However, NRF2-independent mechanisms may also contribute to WA-induced apoptosis as this effect was not fully abolished in the NRF2KO cells.
Metabolic deregulation is one of the hallmarks of different cancers including breast cancer.31 Cancer cells preferentially utilize glucose even in an aerobic environment, a phenomenon first noted by Otto Warburg several decades ago.32 The prevailing hypothesis is that dependence of cancer cells on aerobic glycolysis, which is a much less efficient process for ATP production than oxidative phosphorylation, allows them to meet metabolic demand beyond ATP for the synthesis of bio-molecules such as lipids, amino acids, and nucleotides.33,34 We showed previously that breast cancer prevention by WA in the MMTV-neu mice and MNU-rat model was associated with a significant decrease in circulating and/or tumor levels of lactic acid.6,7 The present study indicates that decrease in lactic acid production by WA in the MNU-rat model is due to suppression of HK2, which catalyzes the first reaction in the glycolysis pathway and converts glucose to glucose 6-phosphate, and LDHA that is responsible for conversion of pyruvate to lactate. These results are corroborated by cellular studies showing downregulation of HK2 and LDHA in WA-treated BCC. Thus, circulating and tumor levels of lactic acid, HK2, and LDHA may serve as biomarkers of WA response in future clinical trials.
In conclusion, the present study shows differential gene expression changes in BCC versus a normal mammary epithelial cell line following WA treatment. The present study not only validates previously published findings (e.g., downregulation of proteins involved in G2/M progression)14,25 but also identifies novel mechanistic target of WA in BCC (e.g., inhibition of senescence).
Supplementary Material
Acknowledgments
Funding: This study was supported by the USPHS grant RO1 CA142604 awarded by the National Cancer Institute. This research used the Tissue and Research Pathology Facility supported in part by Cancer Center Support Grant from the National Cancer Institute (P30 CA047904).
Abbreviations
- ANOVA
analysis of variance
- ATR
ataxia telangiectasia and Rad3-related protein
- BCC
breast cancer cell(s)
- CHK1
checkpoint kinase 1
- CTCF
corrected total cell fluorescence
- DMSO
dimethyl sulfoxide
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- GLUT
glucose transporter
- GO
Gene Ontology
- HER2
human epidermal growth factor receptor 2
- HK2
hexokinase 2
- HSPA6
heat shock protein family A member 6
- KEGG
Kyoto Encyclopedia of Genes and Genomes
- LC3B (or MAP1LC3B)
microtubule associated protein 1 light chain 3 beta
- LDHA
lactate dehydrogenase A
- MEF
mouse embryonic fibroblasts
- MNU
N-methyl-N-nitrosourea
- NBN
nibrin
- NFATC4
nuclear factor of activated T cells 4
- NRF2
nuclear factor erythroid 2-related factor 2
- NRF2KO
immortalized mouse embryonic fibroblasts from NRF2 knockout mice
- PARP
poly (ADP-ribose) polymerase
- PBS
phosphate-buffered saline
- PI
propidium iodide
- PKM2
pyruvate kinase M2
- PPP1CC
protein phosphatase 1 catalytic subunit gamma
- qRT-PCR
quantitative real-time reverse transcription polymerase chain reaction
- RBL2
retinoblastoma transcriptional corepressor like 2
- SA-β-gal
senescence-associated β-galactosidase
- siRNA
small interfering RNA
- SIRT1
sirtuin 1
- SQSTM1
sequestosome 1
- WA
withaferin A
- WT
wild-type
- ZFP36L2
ZFP36 ring finger protein like 2
Footnotes
Conflict of interest: None of the authors has any conflict of interest.
DATA AVAILABILITY STATEMENT
The data and the reagents used for this publication are available upon formal request to the senior author.
REFERENCES
- 1.Hong WK, Sporn MB. Recent advances in chemoprevention of cancer. Science. 1997;278:1073–1077. [DOI] [PubMed] [Google Scholar]
- 2.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020;70:7–30. [DOI] [PubMed] [Google Scholar]
- 3.Fisher B, Costantino JP, Wickerham DL, et al. Tamoxifen for prevention of breast cancer: report of the National Surgical Adjuvant Breast and Bowel Project P-1 study. J Natl Cancer Inst. 1998;90:1371–1388. [DOI] [PubMed] [Google Scholar]
- 4.Cauley JA, Norton L, Lippman ME, et al. Continued breast cancer risk reduction in postmenopausal women treated with raloxifene: 4-year results from the MORE trial: Multiple outcomes of raloxifene evaluation. Breast Cancer Res Treat. 2001;65:125–134. [DOI] [PubMed] [Google Scholar]
- 5.Goss PE, Ingle JN, Ales-Martinez JE, et al. Exemestane for breast-cancer prevention in postmenopausal women. N Eng J Med. 2011;364:2381–91. [DOI] [PubMed] [Google Scholar]
- 6.Hahm ER, Lee J, Kim SH, Sehrawat A, et al. Metabolic alterations in mammary cancer prevention by withaferin A in a clinically relevant mouse model. J Natl Cancer Inst. 2013;105:1111–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Samanta SK, Sehrawat A, Kim SH, et al. Disease subtype-independent biomarkers of breast cancer chemoprevention by the ayurvedic medicine phytochemical withaferin A. J Natl Cancer Inst. 2016;109(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stan SD, Hahm ER, Warin R, Singh SV. Withaferin A causes FOXO3a- and Bim-dependent apoptosis and inhibits growth of human breast cancer cells in vivo. Cancer Res. 2008;68:7661–7669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thaiparambil JT, Bender L, Ganesh T, et al. Withaferin A inhibits breast cancer invasion and metastasis at sub-cytotoxic doses by inducing vimentin disassembly and serine 56 phosphorylation. Int J Cancer. 2011;129:2744–2755. [DOI] [PubMed] [Google Scholar]
- 10.Kim SH, Singh SV. Mammary cancer chemoprevention by withaferin A is accompanied by in vivo suppression of self-renewal of cancer stem cells. Cancer Prev Res (Phila). 2014;7:738–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang F, Zhao J, Bai J, et al. Liquid chromatography-tandem mass spectrometry to assess the pharmacokinetics and tissue distribution of withaferin A in rats. J Chromatogr B Analyt Technol Biomed Life Sci. 2019; 1122-1123:90–95. [DOI] [PubMed] [Google Scholar]
- 12.Hahm ER, Kim SH, Singh KB, Singh K, Singh SV. A comprehensive review and perspective on anti cancer mechanisms of withaferin A in breast cancer. Cancer Prev Res (Philla.). 2020;13:721–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hahm ER, Moura MB, Kelley EE, Van Houten B, Shiva S, Singh SV. Withaferin A-induced apoptosis in human breast cancer cells is mediated by reactive oxygen species. PLoS One. 2011;6:e23354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Antony ML, Lee J, Hahm ER, et al. Growth arrest by the antitumor steroidal lactone withaferin A in human breast cancer cells is associated with down-regulation and covalent binding at cysteine 303 of β-tubulin. J Biol Chem. 2014;289:1852–1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wakabayashi N, Dinkova-Kostova AT, Holtzclaw WD, et al. Protection against electrophile and oxidant stress by induction of the phase 2 response: fate of cysteines of the Keapl sensor modified by inducers. Proc Natl Acad Sci USA. 2004; 101:2040–2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim SH, Hahm ER, Singh KB, Shiva S, Stewart-Ornstein J, Singh SV. RNA-seq reveals novel mechanistic targets of withaferin A in prostate cancer cells. Carcinogenesis. 2020;41:778–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods. 2001;25:402–8. [DOI] [PubMed] [Google Scholar]
- 18.Xiao D, Srivastava SK, Lew KL, et al. Allyl isothiocyanate, a constituent of cruciferous vegetables, inhibits proliferation of human prostate cancer cells by causing G2/M arrest and inducing apoptosis. Carcinogenesis. 2003;24:891–897. [DOI] [PubMed] [Google Scholar]
- 19.Lee J, Hahm ER, Singh SV. Withaferin A inhibits activation of signal transducer and activator of transcription 3 in human breast cancer cells. Carcinogenesis. 2010;31:1991–1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Romanov VS, Pospelov VA, Pospelova TV. Cyclin-dependent kinase inhibitor p21(Wafl): contemporary view on its role in senescence and oncogenesis. Biochemistry (Mosc). 2012;77:575–584. [DOI] [PubMed] [Google Scholar]
- 21.Yi J, Luo J. SIRT1 and p53, effect on cancer, senescence and beyond. Biochim Biophys Acta. 2010;1804:1684–1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ghosh K, De S, Mukherjee S, Das S, Ghosh AN, Sengupta SB. Withaferin A induced impaired autophagy and unfolded protein response in human breast cancer cell-lines MCF-7 and MDA-MB-231. Toxicol In Vitro. 2017;44:330–338. [DOI] [PubMed] [Google Scholar]
- 23.Palliyaguru DL, Chartoumpekis DV, Wakabayashi N, et al. Withaferin A induces Nrf2-dependent protection against liver injury: Role of Keap1-independent mechanisms. Free Radic Biol Med. 2016;101:116–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hahm ER, Lee J, Abella T, Singh SV. Withaferin A inhibits expression of ataxia telangiectasia and Rad3-related kinase and enhances sensitivity of human breast cancer cells to cisplatin. Mol Carcinog. 2019;58:2139–2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stan SD, Zeng Y, Singh SV. Ayurvedic medicine constituent withaferin a causes G2 and M phase cell cycle arrest in human breast cancer cells. Nutr Cancer. 2008;60(suppl.1):51–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Royston KJ, Paul B, Nozell S, Rajbhandari R, Tollefsbol TO. Withaferin A and sulforaphane regulate breast cancer cell cycle progression through epigenetic mechanisms. Exp Cell Res. 2018;368:67–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hahm ER, Singh SV. Cytoprotective autophagy induction by withaferin A in prostate cancer cells involves GABARAPL1. Mol Carcinog. 2020;59:1105–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Collado M, Blasco MA, Serrano M. Cellular senescence in cancer and aging. Cell. 2007;130:223–233. [DOI] [PubMed] [Google Scholar]
- 29.Pare R, Yang T, Shin JS, Lee CS. The significance of the senescence pathway in breast cancer progression. J Clin Pathol. 2013;66:491–495. [DOI] [PubMed] [Google Scholar]
- 30.Jaramillo MC, Zhang DD. The emerging role of the Nrf2–Keap1 signaling pathway in cancer. Genes Dev. 2013;27:2179–2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011; 144:646–674. [DOI] [PubMed] [Google Scholar]
- 32.Warburg O On respiratory impairment in cancer cells. Science. 1956;124:269–270. [PubMed] [Google Scholar]
- 33.Ogrodzinski MP, Bernard JJ, Lunt SY. Deciphering metabolic rewiring in breast cancer subtypes. Transl Res. 2017;189:105–122. [DOI] [PubMed] [Google Scholar]
- 34.Wu Z, Wu J, Zhao Q, Fu S, Jin J. Emerging roles of aerobic glycolysis in breast cancer. Clin Transl Oncol. 2020;22:631–646. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.