

Abstract
Though immunotherapy has revolutionized the treatment of cancer to improve disease outcomes, an array of challenges remain that limit wider clinical success, including low rate of response and immune-related adverse events. Targeting immunomodulatory drugs to therapeutically relevant tissues offers a way to overcome these challenges by potentially enabling enhanced therapeutic efficacy and decreased incidence of side effects. Research highlighting the importance of lymphatic tissues in the response to immunotherapy has increased interest in the application of engineered drug delivery systems (DDSs) to enable specific targeting of immunomodulators to lymphatic tissues and cells that they house. To this end, a variety of DDS platforms have been developed that enable more efficient uptake into lymphatic vessels and lymph nodes to provide targeted modulation of the immune response to cancer. This can occur either by delivery of immunotherapeutics to lymphatics tissues or by direct modulation of the lymphatic vasculature itself due to their direct involvement in tumor immune processes. This review will highlight DDS platforms that, by enabling the activities of cancer vaccines, chemotherapeutics, immune checkpoint blockade (ICB) antibodies, and anti- or pro-lymphangiogenic factors to lymphatic tissues through directed delivery and controlled release, augment cancer immunotherapy.
Keywords: drug delivery system, controlled release, tumor immunotherapy, immunoengineering, lymph node, lymphatics
1. Introduction
Immunotherapy has revolutionized cancer treatment and represents a powerful treatment modality for patients with a variety of advanced cancers. This treatment class seeks to educate the patient’s own immune system in order to create an army of tumor-specific immune cells that can seek out and destroy malignant cells, preventing tumor progression and protecting against recurrence [1–3]. The approval of a variety of immunotherapies, including antibodies against immune checkpoint receptors [4, 5], cancer vaccines [6], oncolytic viral therapies [7], adoptive cell therapies [8, 9], and cytokine therapies [10–12] among others [13–17] has led in some indications to remarkable clinical success in patients with late-stage disease, benefits attributed to a potent and durable anti-tumor immune response. Nevertheless, these treatments still present an array of challenges that impede its success and hamper quality of life for patients [18]. While some cancer patients are seemingly cured, a large subset of patients treated with immunotherapies are left without significant clinical benefit [19]. In addition, most cancer immunotherapies have risk of substantial side effects including immune-related adverse events and dose-limiting kidney and liver toxicity [20–22].
Engineered drug delivery systems (DDSs) offer unique opportunities to address these obstacles [23]. For example, directing drug accumulation to within target cells and tissues where relevant tumor-immune mechanisms occur by leveraging the well-established benefits of controlled delivery and release afforded by DDSs [24, 25] both enhances therapeutic efficacy and simultaneously reduces the required dose to further enhance safety and tolerability drug profiles [26, 27]. To this end, lymphatic tissues are of high interest for targeted delivery of immune-modulating therapeutics due to their complex role in shaping the anti-cancer immune response [28–30]. These include lymph nodes (LNs) that house a substantial fraction of the body’s total immune cells, as well as lymphatic vessels that transport both immune signaling molecules and cells to draining LNs (dLNs) to enable the mounting of an adaptive anti-tumor response. Furthermore, the role of lymphatic tissues has additionally been demonstrated as critical in the production of lasting anti-tumor responses in the context of cancer immunotherapy [31–34]. Accordingly, lymphatic immunomodulation for cancer therapy can be achieved either by delivery of immunotherapeutics to lymphatic vessels and/or LNs, or direct modulation of the lymphatic tissue itself via immunotherapy or immunomodulatory interventions in order to support or suppress its direct immunological function(s). Application of such systems through systemic versus locoregional routes of administration can also enable delivery to lymphatic tissues that are systemically or locoregionally distributed, each of which have different advantages based on therapeutic application (e.g. treatment of liquid tumors versus metastases in sentinel LNs, as examples).
The unique challenges to achieving appreciable accumulation and retention of therapeutics in lymphatic tissues and relevant immune cell populations (reviewed extensively elsewhere [35–38]) motivates the application of engineered DDSs to enable lymphatic drug targeting. Engineered DDSs are versatile and dynamic technologies that have been widely investigated for enabling drug cargo to interface with tissues, cells, and pathways that they would otherwise be physiologically restricted from, as is the case with most immunomodulatory therapeutics and lymphatic tissues [37, 38]. In addition to permitting selective lymphatic tissue and immune cell targeting by tuning of physicochemical properties, engineered DDSs offer a multitude of benefits such as improving the bioavailability, pharmacokinetic profiles, and tolerability of immunotherapies [22, 39]. This review will highlight the unique potential for DDSs to enable therapeutic delivery to lymphatic tissues in the context of immune modulation for cancer, including directing immunomodulatory therapeutics to LNs and/or modulating lymphatic tissues themselves to enhance or mitigate their direct effect on the anti-tumor immune response, and discuss current strategies and future opportunities (Figure 1).
Figure 1.
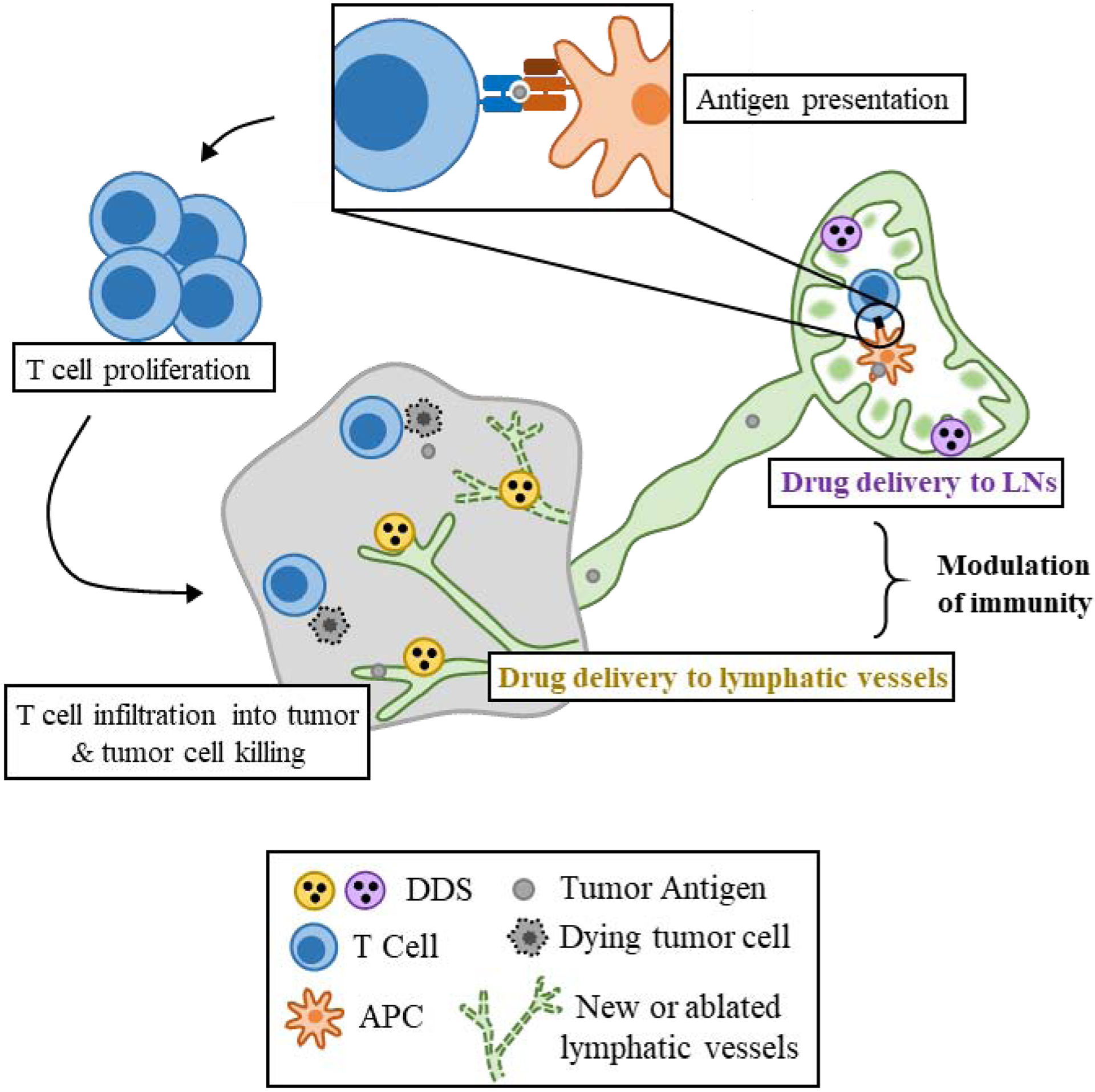
Potential for lymphatic drug delivery to enable modulation of the anti-tumor immune response. Tumor antigen drains to LNs via lymphatic vessels, where it is processed and presented via APCs to T cells. LN-targeted DDSs carrying immunomodulatory therapeutics can enhance this process by creating a more pro-inflammatory LN environment, more efficiently delivering tumor antigen to APCs, or stimulating specific immune cell subtypes, leading to increased antigen-specific T cell priming, proliferation, and subsequent infiltration into the tumor, potentially allowing increased tumor killing. Lymphatic vessel-targeted DDSs can modulate lymphatics by either promoting lymphangiogenesis via growth factory delivery, or preventing lymphangiogenesis by growth factor depletion or lymphatic ablation in order to support anti-tumor or suppress pro-tumor function to further reduce tumor burden.
2. Lymphatic system structure and function
As reviewed in depth elsewhere [30, 40–42], lymphatic tissues by virtue of their unique structure and function play an integral role in both mediating immunity and maintaining fluid balance in healthy and cancerous tissue [40]. In peripheral tissues, protein- and cell-containing interstitial fluid that does not re-enter circulation after capillary exchange is taken up into blunt-ended lymphatic capillaries, which are thin-walled and contain gaps between lymphatic endothelial cells (LECs), to facilitate fluid, cell, and solute uptake [43–45]. This solute- and cell-laden fluid, termed lymph, flows unidirectionally toward larger lymphatic collecting vessels and is filtered through LNs, which house cells of the innate and adaptive immune systems, eventually re-entering venous circulation [42, 46]. In this regard, lymphatic tissues function as a system that not only maintains tissue fluid balance, but also facilitates the immunological monitoring of peripheral tissues for presence of invading pathogens or other disease. In the context of cancer, lymph that flows from tumors towards the draining LN may contain tumor-associated factors or antigen [30, 47], which can be detected by specialized immune cells in the LN that mount a specific anti-tumor response with the potential to result in tumor cell killing [30, 47, 48]. The importance of lymphatic tissues in the context of cancer thus make lymphatic vessels and LNs valuable targets for immunotherapy, which can be enabled using DDSs.
3. Designing lymphatic targeting DDSs
A variety of physiological barriers exist in restricting the access of immunotherapeutics, including small molecules, peptides, or oligonucleotides, to lymphatic tissues [35–37]. Strategies employing biomaterial DDSs have been engineered to leverage these barriers to enable selective targeting of immune-modulating therapeutic molecules to lymphatic tissues. As with DDSs generally, the use of DDSs for lymphatic- and LN-directed drug delivery is motivated by enabling drug accumulation in target tissues and cells, extending the availability of the therapeutic within those tissues or to those target cell populations, and decreasing accumulation in off-target tissues in a controlled manner that is often difficult to achieve with administration of free drug [3, 36].
DDSs can be engineered to direct drug delivery to LNs and lymphatic vessels by a variety of means, including through the tuning of their various physical and chemical properties (Figure 2). Size is a critically important design feature that influences the lymphatic targeting capability of nanocarrier-based DDSs. Nanocarriers sized from 10–100 nm in hydrodynamic diameter [42, 49, 50] are utilized to deliver therapeutics into LNs from an injection directly into peripheral tissue [subcutaneous (s.c.), intramuscular (i.m.), or intradermal (i.d)] owing to the presence of interstitial lymphatic capillaries, which allow passive uptake and drainage in lymph toward LNs [30, 51], in contrast to systemic administration typically resulting in low lymphatic accumulation [35, 36, 52]. Nanocarriers above 100nm may still be transported to LNs via passive lymphatic drainage, albeit with drastically less efficiency [53]. Larger drug carriers (>200nm) that cannot move as freely through pores in the gel-like extracellular matrix (ECM) of tissues to directly access lymphatic capillaries are instead trafficked via lymphatics by antigen presenting cells (APCs) that migrate to LNs to facilitate antigen sensing [35, 54]. This strategy additionally allows DDSs to enable selective delivery into specific target cell populations based on carrier size as smaller (sub-100nm) nanocarriers passively drain to LNs and are taken up by LN-resident APCs, while larger (several hundred nm) carriers alternatively target peripheral tissue APCs that can migrate to LNs [53, 54]. Nanoscale DDSs in circulation can target cancerous LNs or metastatic lesions within LNs due the enhanced vascular permeability of these tissues that results from disease [55], though reports have suggested that elevated interstitial pressure in diseased LNs may prevent passive nanocarrier accumulation [56, 57]. Even larger, micron-scale injectable or implantable macromolecular delivery strategies, commonly hydrogel [58, 59], scaffold [60, 61], or microneedle [62, 63] DDSs also allow the formation of a depot in peripheral tissue [64]. Depot-forming materials protect therapeutics from a variety of clearance mechanisms to allow for sustained release and have been used to either directly accumulate in lymph or promote recruitment of APCs with immune modulatory functions that subsequently traffic to dLNs [64, 65]. Similar principles have been utilized to leverage DDS accumulation in mucosal-associated lymphatic tissues following locoregional administration to the mucosa via nasal or oral administration of DDSs (reviewed elsewhere [66–70]), though this is generally less common in preclinical cancer therapy applications.
Figure 2.
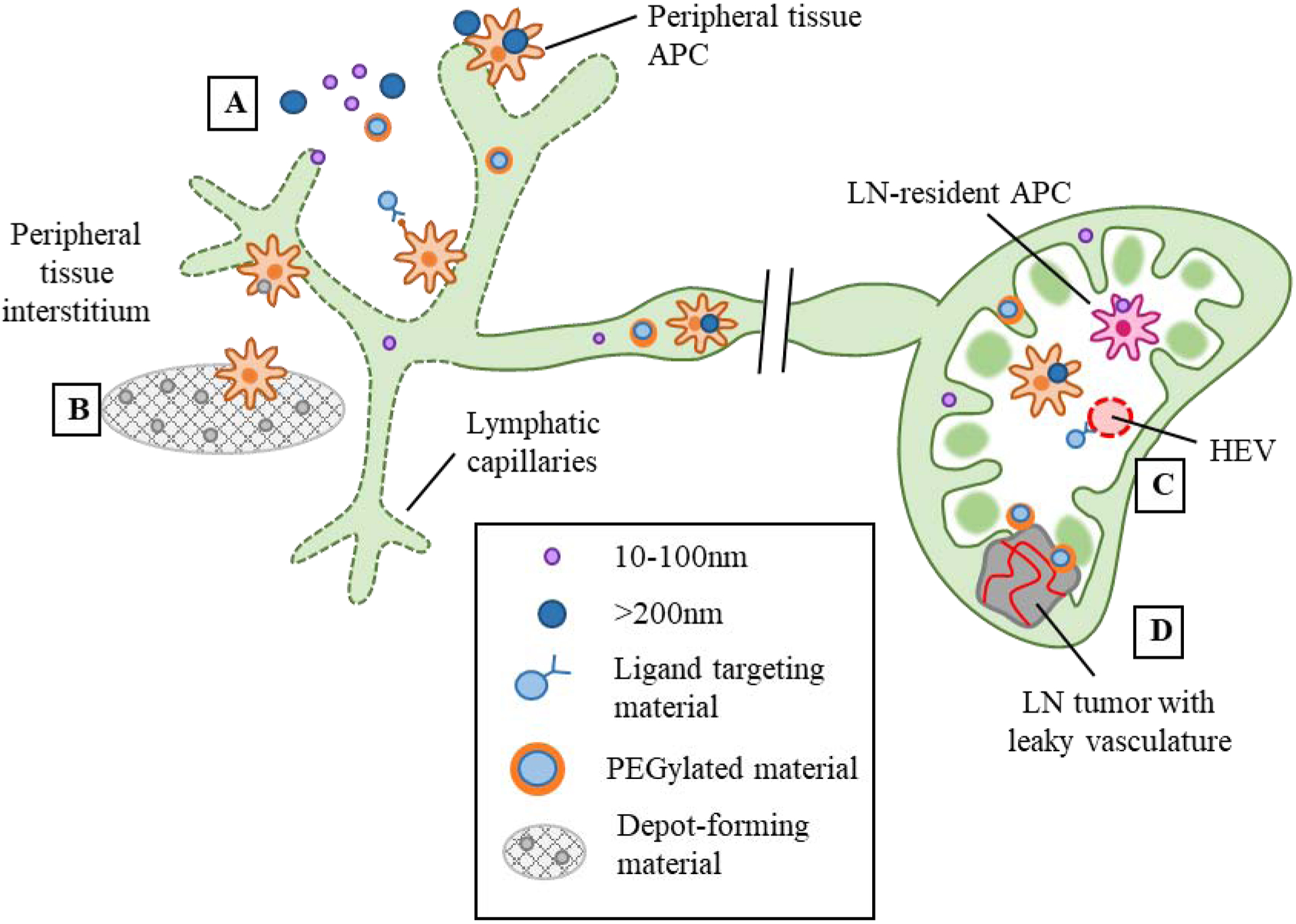
Design strategies for lymphatic-targeting drug delivery systems. A) From the peripheral tissue interstitium, particles sized 10–100nm drain passively via lymphatic capillaries and are taken up via LN-resident DCs, while larger >200nm particles are transported to the LN via peripheral tissue-resident APCs. In addition to size, PEGylation and incorporation of targeting ligands can also enhance lymphatic uptake. B) Depot-forming materials promote peripheral tissue APC infiltration followed by migration to the LN. C) Particles incorporated with high endothelial (HEV)-targeting ligands can enter via the bloodstream. D) Leaky vasculature of LN tumors or metastases may allow particles to enter from the bloodstream.
In addition to hydrodynamic size that is well established to be an important feature in controlling access of DDSs to lymphatic tissues, material composition plays an important role. One prominent strategy to impart biocompatibility and enhance lymphatic targeting of DDSs is coating the surface with poly(ethylene glycol) (PEG) chains, called PEGylation [71]. PEGylation is thought to reduce interactions between the DDS surface and the interstitial ECM, allowing easier passage toward and uptake into lymphatic vessels after injection or accumulation in peripheral tissues [71–74]. PEGylation additionally prevents the adhesion of proteins to the DDS surface [73] which is hypothesized to reduce recognition by phagocytic cells, allowing the DDS to circulate longer and increase dose available for lymphatic accumulation [71–73], a feature that is utilized to avoid clearance of DDSs in systemic circulation by phagocytic liver-resident cells [75, 76]. Lipophilicity also increases lymphatic absorption due to the role of the lymphatic system in lipid transport [37, 77], a property that has been exploited to improve drug delivery into lymphatic tissues [78–80], including oral drug delivery to intestinal lymphatics [81–84]. Lastly, materials can be designed to degrade in a spatiotemporally advantageous manner to deliver cargo into lymphatics and/or resident immune cells. DDSs designed using pH- or reactive oxygen species (ROS)-sensing materials can degrade inside phagolysosomes after uptake into APCs, allowing escape and intracellular delivery of therapeutic cargo [85], including in the application of antigen delivery to APCs for enhanced cross-presentation [86, 87]. Careful tuning of matrix macropores [88], hydrogel mesh size [89], or polymer molecular weight and crosslinking density [90] exerts a high level of spatiotemporal control over drug release or infiltration of local immune cells, allowing enhanced accumulation within lymphatic tissues [91–94]. Similarly, other material properties such as surface charge [35, 95] or shape and flexibility [35, 50, 96, 97] can be tuned to allow enhanced lymphatic uptake from different administration routes.
Another design approach that enables DDS-mediated lymphatic delivery is incorporating targeting ligands or antibodies to impart additional specificity. DDSs bearing ligands corresponding phagocytic APC populations can facilitate enhanced cellular uptake of the therapeutic cargo, and can additionally increase retention within targeted LNs [98, 99]. Similarly, DDSs bearing targeting moieties for LECs can specifically interact with LECs in order to retain the therapeutic drug payload within lymphatic vessels [100, 101]. Incorporation of targeting ligands for molecules expressed on specialized vessels within LNs, termed high endothelial venules, allow for LN targeting of therapeutic cargo by DDSs in systemic circulation [102].
In all, the large degree of tunability involved in DDS design has enabled the engineering of a multitude of platforms for different immunotherapeutic applications. The remainder of this Review will highlight examples of promising recent drug delivery strategies that allow unique and precise control over lymphatic immunomodulation for cancer therapy, as well as potential future applications.
4. Cancer vaccine DDSs for LN delivery
Due to the important role LNs play in mounting and regulating adaptive immune responses to cancer, it is unsurprising that cancer vaccines are a widely explored application for DDSs directed to LNs. Cancer vaccine formulations most often involve delivery of a tumor antigen and an immune adjuvant with the goal of increasing presentation of tumor antigen and enhancing the cytotoxic CD8+ T cell response. For this application, DDSs are uniquely capable of enabling co-delivery of antigen and adjuvant into specific APC populations in order to modulate their phenotype and enhance their function (Figure 3a). The studies highlighted below represent a variety of recent DDS design approaches for lymphatic delivery of cancer vaccines and underscore the potential for these systems to engineer a desirable anti-tumor immune response in a manner that cannot be achieved with traditionally formulated and/or administered therapeutics.
Figure 3.
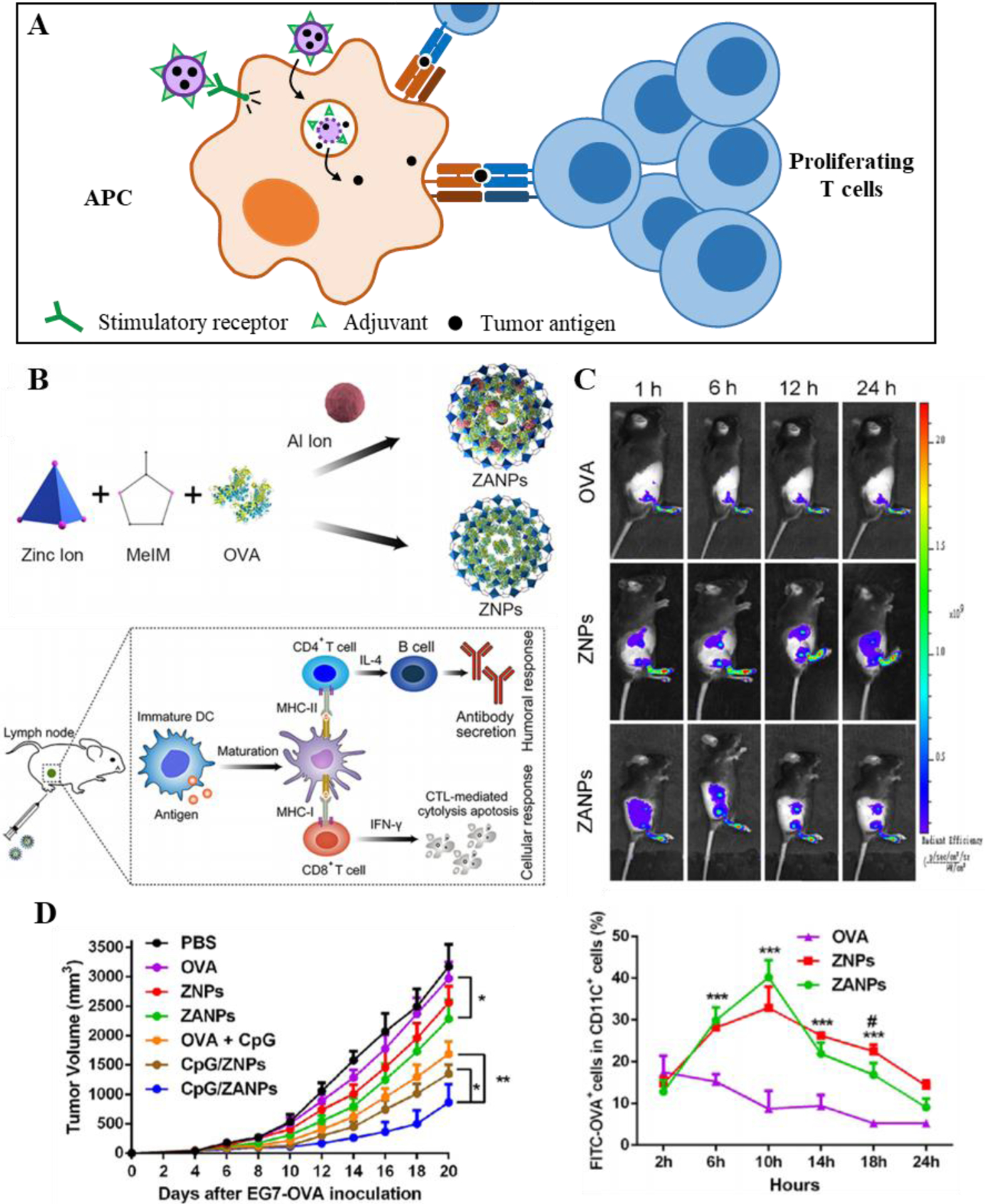
Lymphatic-targeting DDSs for cancer vaccine delivery and immune modulation. A) DDSs enable delivery of adjuvant and/or antigen to APCs, leading to enhanced maturation and expression of co-stimulatory factors, and efficient T cell priming and proliferation. B) Example of LN-targeting cancer vaccine: Aluminum metal organic framework (MOF) design encapsulating model antigen (ZANP), and its mechanism of action. C) Extended release and delivery of ZANPs to DCs in the draining LN; D) Enhanced anti-tumor efficacy of ZANP cancer vaccine DDS over free drug formulations. B-D Reproduced with permission from Ref [114] © 2019 Elsevier1.
4.1: Depot-forming DDSs
As discussed above, one explored category of DDSs for cancer vaccination involves the formation of a depot post-injection, commonly achieved using injectable hydrogels, large macromolecular scaffolds, or dissolving microneedle formulations [64, 65]. Though such formulations do not necessarily involve lymphatic targeting or immunomodulation, they offer opportunities to provide direct modulation of local lymphatic vessels and/or continuous drainage of the cancer vaccine components to the dLN. Therapeutic modulation of lymphatic tissues may therefore be accomplished using depot-forming DDSs by a variety of means, including modifying their physical or chemical properties to tune the recruitment of APC subtypes and subsequent activation and migration to the draining LN (dLN), as discussed below.
A recent approach to depot-forming DDSs employs an 88 μm mesoporous silica rod (MSR) system that spontaneously assembles into a three-dimensional scaffold in vivo following the diffusion of saline buffer away from the injection site [103, 104]. The MSRs feature a high aspect ratio that leads to large interparticle spaces on the scale of tens of microns, which was hypothesized to allow efficient cell recruitment into the MSR scaffold once assembled. Blank MSR scaffolds made with high aspect ratio MSRs allowed efficient in vivo recruitment of an APC subtype, CD11c+ dendritic cells (DCs), that are particularly potent in their T cell priming capabilities, after assembly compared to low aspect ratio MSR scaffolds featuring a smaller interparticle pore size and control MSR scaffolds lacking interparticle space, suggesting that recruited cells were able to migrate through the scaffold [103]. For use as a therapeutic, MSRs were loaded with granulocyte-macrophage-colony stimulating factor (GM-CSF) and Toll-like receptor (TLR) 9 ligand CpG oligonucleotide, which enhance DC recruitment and maturation, as well as a model tumor antigen. Superior in vivo retention of model antigen at the site of injection was achieved when incorporated into MSR scaffolds versus a bolus of free antigen as well as extended release of incorporated GM-CSF and CpG in vitro. Using this formulation, more DCs were recruited to s.c. implanted MSR scaffolds upon incorporation of all three vaccine components, which led to enhanced engulfment of the incorporated antigen, APC maturation and migration to the dLN, and T cell priming. When used as a prophylactic vaccine, this strategy led to marked reduction in tumor volume using a murine thymoma model and improved animal survival compared to bolus administration of therapeutics, demonstrating that effective immune-modulation in the LN leads to systemically functional immunity [103]. In a follow-up study using the same MSR scaffold system [104], the authors demonstrated even further tuning of the vaccine-induced immune response using a variety of covalent conjugation schemes to attach antigenic peptide to MSRs, showcasing the overall flexibility of this system to enable delivery of cancer antigen into LNs.
Another material approach with this goal includes a sponge-like macroporous alginate “cryogel” [105] for a whole tumor cell vaccine application. Methacrylated alginate covalently linked to an arginine-glycine-aspartate (RGD)-containing peptide was polymerized at low temperatures to allow formation of continuous macropores within the cryogel. Presentation of the RGD motif better allows this system to immobilize irradiated tumor cells as an antigen source within the gel, as RGD promotes integrin-mediated cell adhesion [106]. Formation of macropores within the scaffold was again crucial to DC recruitment and infiltration in vivo as cell density was significantly higher in explanted cryogel scaffolds compared to a hydrogel control that only features nanoscale pores and thus does not allow sufficient cell migration within the gel [105]. Combined with GM-CSF and CpG, which slowly release from cryogel scaffolds over the course of days, irradiated murine melanoma cells (B16-F10) incorporated in RGD-presenting cryogels increased the recruitment of DCs to the site of the scaffold as well as migration to the dLN, leading to an enhanced number of CD8+ T cells in the dLN, and conferring enhanced survival in vivo in a therapeutic setting versus free vaccine components [105].
In a similar study, a macroporous matrix of poly[lactide-co-glycolide] (PLG) [107], a commonly used material with excellent in vivo safety and biodegradability, was investigated for its ability to attract peripheral DCs and promote their maturation and migration to the dLN. A feature of this system is the ability to carefully tune the release of GM-CSF in a spatiotemporally defined manner. While GM-CSF promoted the recruitment of DCs in vitro in a dose-dependent manner as expected, it also decreased DC expression of CCR7 and reduced their ability to migrate towards CCL19, which helps direct the migration of activated DCs towards secondary lymphoid tissues [108]. The PLG scaffold initially allowed persistence of a high local concentration of GM-CSF long enough to attract DCs and allow antigen uptake, followed by a gradual decrease in local concentration over time that “released” DCs and allowed migration to LNs [107]. Incorporation of CpG into the scaffold additionally provided maturation of DCs, and further propagated their migration to the dLN. Combined with tumor lysate as an antigen source, GM-CSF and CpG-loaded PLG scaffold as a prophylactic vaccination led to higher overall survival than the combination of free adjuvant and antigen [107], owing to the high level of precise spatiotemporal control over delivery of therapeutic molecules enabled by the PLG scaffold.
While many depot-forming DDS designs focus on infiltration and recruitment of DCs to the local site of injection followed by migration to the dLN, an alternative strategy involves the self-assembly of 30–40 nm antigen and adjuvant-loaded nanomicelles (NMCs) from a depot formed by dissolving microneedles [109]. NMCs are formed in situ after dissolution of microneedles from a mixture of the triblock copolymer Pluronic F127 (PEG-block-poly(propylene glycol)-block-PEG) with additional PEG, which encapsulates the hydrophobic TLR7/8 agonist resiquimod (R848) and facilitates delivery of a model antigen to dLN-resident immune cells. NMCs co-localized with APCs including DCs and macrophages in the dLN. Microneedle-delivered model antigen and R848-containing NMCs conferred substantial reduction in tumor volume versus controls in both a prophylactic and therapeutic setting in a murine thymoma model. This demonstrates how combining the benefits of a depot-forming drug delivery platform with principles of passive dLN targeting (sub-100 nm sized particles) in order to elicit immune-modulation at the dLN can confer augmented anti-tumor immunity in vivo [109].
Overall, these studies demonstrate the potential for depot-forming DDSs to exert a high level of specific, spatiotemporal control over the delivery of therapeutics to the dLN, either by extended release of passively draining species or by enhanced cell trafficking to LNs, in order to engineer a desired immune outcome.
4.2: Nanocarrier DDSs
Given the efficiency of passive and cell-mediated lymphatic accumulation from local administration [42, 49, 50], long established use in drug delivery applications [110, 111], and versatility in chemical and physical properties [112], nanocarriers remain a popular strategy for the design of lymphatic-targeting DDSs. LN-targeting nanocarriers have been widely investigated in the context of cancer vaccines in a variety of material formulations, including polymeric, inorganic, peptide-based, and lipid-based nanoparticles, each of which can impart unique features and functionality to the DDS.
4.2.1. Inorganic nanocarriers
Inorganic materials have been studied for the application of LN-targeting DDSs due in part to the large degree of control over their synthesis. As a result, inorganic materials have the capacity to have highly specified and tailorable molecular and structural properties such as size, porosity, and mechanism of degradation [113], all of which affect drug release and subsequent lymphatic and immune cell accumulation [36].
One promising inorganic NP formulation involves the use of a zeolitic imidazole metal-organic framework (ZIF-8 MOF) that was investigated for its high biomolecule loading capability, tunable pore size and thus drug release rate, and pH sensitive degradation [114] (Figure 3b). ZIF-8 is used to form a 70–80 nm NP incorporating both a model tumor antigen and aluminum as adjuvant (ZANPs) that were further adsorbed with CpG. In vitro, ZANPs were taken up by DCs and induced activation and maturation versus non-aluminum containing ZNPs. Additionally, ZANPs led to an increase in model tumor antigen cross-presentation, likely owing to their pH sensitive degradation inside the phagolysosome. ZANPs accumulated efficiently in dLNs and delivered tumor antigen peptide and adsorbed CpG into DCs (Figure 3c), leading to a significant effector CD8+ T cell response and subsequent reduction in tumor burden in a model antigen [ovalbumin (OVA)] expressing murine thymoma model (EG.7-OVA) (Figure 3d) [114], demonstrating the potential for tunable inorganic nanocarriers to elicit lymphatic immunomodulation.
Another unique benefit of the use of inorganic materials for the design of dLN-targeting DDSs is the ability to incorporate imaging agents that enable in vivo tracking of nanocarrier or cellular biodistribution and LN mapping, creating a DDS that is both a therapeutic and a diagnostic [115]. In one application, iron oxide NPs (IONPs) 40 nm in diameter were microdosed with the radiotracer 67Ga, enabling ultra-specific in vivo imaging of IONP biodistribution via PET/SPECT, then were either covalently linked to model tumor antigen or adsorbed with CpG via a lipid coating [116]. The IONP size facilitated efficient drainage into LNs from several s.c. injection schemes in vivo, which was visualized using PET/SPECT imaging of 67Ga labeled NP. DCs in dLNs were efficiently activated and matured as a result of CpG-IONP treatment, leading to the presence of systemic tumor antigen-specific CD8+ T cells in the context of CpG-IONP and antigen-IONP co-delivery [116]. In another application, inorganic nanocarriers were used both as a DDS and as a tool to image dLN trafficking of a live DC vaccine [117]. In this approach, “upconversion” NPs were formulated using Yb and Er-doped NaY/GdF4 and then coated with PEG and polyethylenimine (PEI) in order facilitate adsorption of model tumor antigen, resulting in ~200 nm “UPP@OVA NPs” that enabled in vivo tracking of DCs via upconversion luminescence imaging. UPP@OVA NPs were efficiently taken up by isolated immature DCs ex vivo and caused enhanced intracellular delivery of tumor antigen resulting in DC maturation. Following s.c. injection of UPP@OVA-stimulated DCs, upconversion luminescence imaging revealed migration of DCs to dLNs, leading to increased T cell proliferation and effector function [117].
Together, these studies highlight inorganic nanomaterials as promising carriers for delivery of immune-modulating therapeutics to dLNs to enhance the anti-cancer immune response and lead to inhibition of tumor growth, and in some DDS applications to additionally function as a sensitive imaging tool to capture nanocarrier interactions with the LN microenvironment.
4.2.2. Polymeric nanocarriers
Synthetic polymer-based formulations have been long utilized in drug delivery applications due to their excellent in vivo biocompatibility [27, 111], and are particularly popular for application in lymphatic-targeting DDSs due to their wide range of bulk and molecular properties [118] that allow tuning of lymphatic and cellular accumulation. Properties of polymeric nanoparticles such as molecular weight, extent of chemical or physical crosslinking, and hydrophobicity can be altered in order to carefully tune drug encapsulation and release, whereas surface functionalization can facilitate conjugation or adsorption of therapeutics [119, 120], resulting in polymeric nanocarrier DDSs that can adopt a range of unique functionalities with the same goal of lymphatic immunomodulation.
As one example, poly(propylene sulfide) (PPS) nanoparticles (NPs) have been widely explored as versatile cancer vaccine nanocarriers [53, 121–125]. These block co-polymer (Pluronic F127)-stabilized NPs are formed by emulsion polymerization, yielding a hydrophobic, crosslinked PPS core that can be made amenable to derivatization post synthesis [125, 126]. The PPS-NP core also allows facile encapsulation of hydrophobic small molecule therapeutics [124], while the Pluronic NP corona is amenable to covalent attachment of molecules following end group derivatization [126]. PPS-NP synthesis can be tuned to yield nanoparticles from 20 to 100 nm in hydrodynamic diameter [53, 121], though they have been most frequently explored in the context of a 25–30 nm size range that allows efficient passive drainage to LNs from i.d. injection by way of dermal lymphatics [51, 121–124]. Additionally, PPS-NPs are found to co-localize with and be taken up by LN-resident DCs [53]. Owing to the presence of thioether groups in the PPS core, these NPs are also susceptible to release of encapsulated drug cargo via the solubilizing of NPs in the presence of concentrated reactive oxygen species (ROS), or release of disulfide-conjugated cargo in reducing conditions, such as that found in phagolysosomes [126]. Reducible conjugation of antigenic peptide via a disulfide bond, but not a non-reducible vinyl sulfone, was found to facilitate endosomal escape and cross-presentation of NP-conjugated antigenic peptide via major histocompatibility complex I in vivo, leading to proliferation and interferon-γ production of antigen specific T cells [127]. The combination of CpG-PPS-NP and tumor antigen-PPS-NP was effective as a therapeutic vaccine at reducing tumor burden and increasing survival in vivo using a variety of tumor models [123]. Notably, a separate study investigated the therapeutic potential of CpG-PPS-NP alone, using the TdLN as a source for antigen and thereby creating an “in situ” vaccine [124]. This strategy also led to a reduction in murine melanoma (B16-F10) tumor burden, but only from an injection resulting in drainage of adjuvanted NPs to the TdLN and not from an injection resulting in drainage to LNs not co-draining the tumor. Immune profiling in the TdLN of treated mice in several of the aforementioned studies indicates increased presence and maturation of LN-resident DCs [124], as well as enhanced tumor infiltration of antigen-specific effector CD8+ T cells [123, 124]. This platform has also been investigated for enabling nasal vaccine delivery of NP-conjugated model antigen to APCs in mucosal lymphatics [128], which yielded superior efficacy versus free antigen in mediating cross-presentation and eliciting antigen-specific cellular immunity locally in the lung and in systemic tissues and humoral immunity in the mucosa, further indicating its versatility.
Another application of the lymphatic-draining PPS-NP platform is in multistage delivery to LNs [129]. Through the incorporation of an oxanorbornadiene (OND) linker, which degrades in a temporally-defined manner by retro-Diels-Alder fragmentation independent of other factors [130, 131], cargo can be controllably attached and released from the NP with different half-lives depending on the molecular structure of the OND. Using this approach, cargo formulated into NP-OND and administered i.d. is able to accumulate in LNs by virtue of the 30 nm size of PPS-NPs (stage 1), then diffuse into the deeper portions of the LN following linker degradation (stage 2) [129] (Figure 4a). As such, delivery of fluorescent cargo via NP-OND to LN cortex- and paracortex-resident cells, including T cells, B cells, cDCs, and pDCs was dramatically improved versus free fluorophore, whereas NPs themselves were excluded to the LN subcapsular sinus (Figure 4b–c). Use of CpG as therapeutic drug cargo attached to NPs via an OND linker (NP-OND-CpG) led to an enhanced number of T cells, B cells (Figure 4d) and matured DCs in the dLN versus CpG covalently linked to NPs (non-degradable). NP-OND-CpG treatment also led to decreased primary tumor burden and incidence of LN metastases in a murine lymphoma model over CpG covalently linked to NPs, demonstrating the therapeutic potential of multistage LN drug delivery (Figure 4e). The principle of multi-stage delivery benefiting lymphatic and LN drug delivery was further proven using a scheme whereby hydrophobic dye was encapsulated into the NP core and passively released via first order diffusion. Similar to NP-OND, NPs demonstrated higher uptake into LN barrier cells whereas encapsulated and released dye partitioned into cortex- and paracortex-resident cells. Furthermore, the principle of time-dependent cargo release by OND linker degradation was demonstrated using a virus-like particle platform that was sized similarly to PPS-NP [129], demonstrating the versatility of this overall strategy to elicit highly controlled delivery of immunomodulatory therapeutics within LNs.
Figure 4.
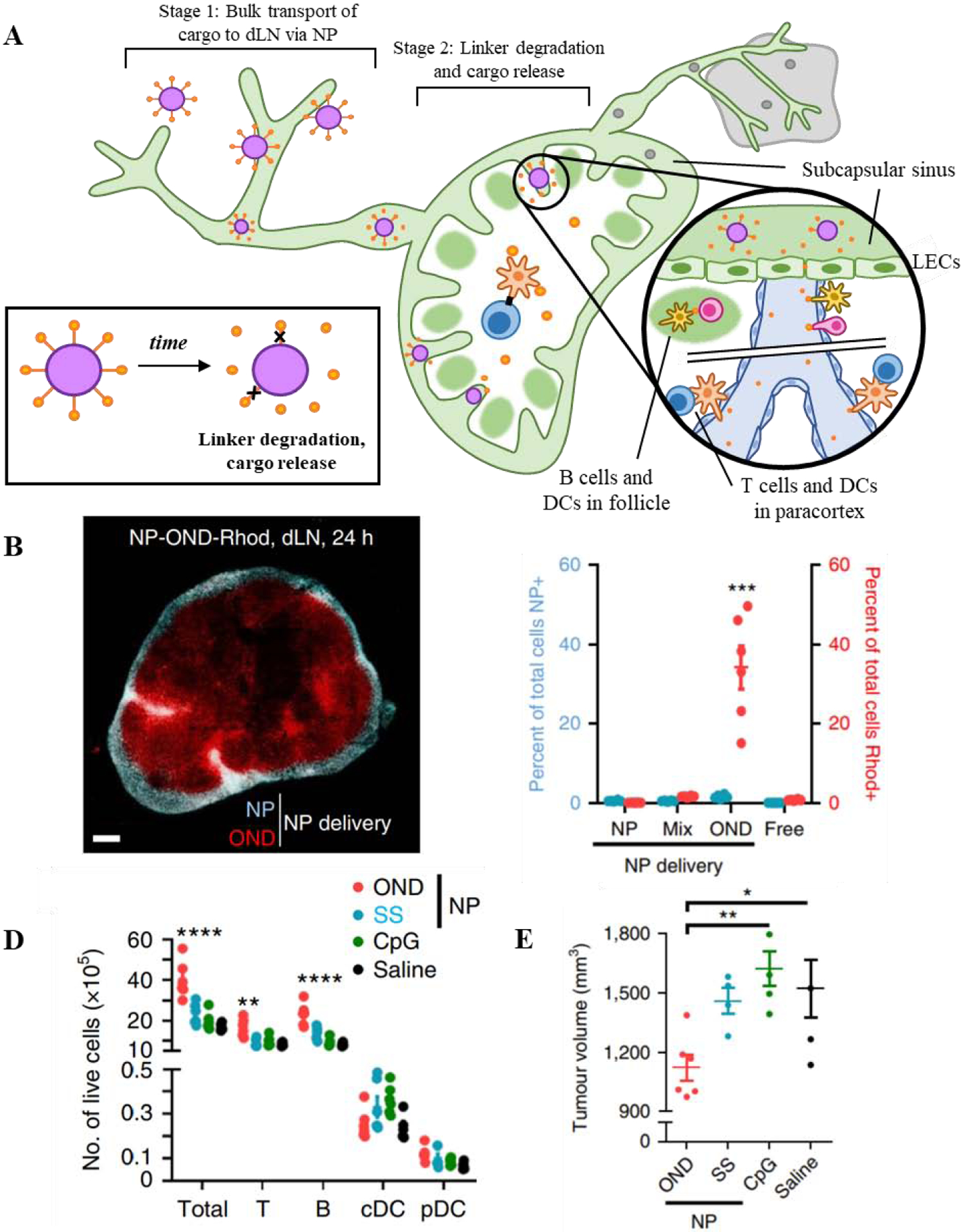
Concept of multistage drug delivery to TdLNs from a lymphatic-draining nanoparticle platform. A) Drug cargo is conjugated to 30nm poly(propylene sulfide) NPs via OND linkers which degrade in a temporally-defined manner. This allows cargo to transport to LNs via passive drainage from the peripheral interstitium (stage 1), then release from NPs via linker degradation (stage 2). B) Smaller molecular weight drug cargo linked to NPs via multistage OND chemistry (NP-OND-Rhod; red) freely diffuses throughout deeper portions of the LN, including B cell follicles and the paracortex, whereas NPs (blue) are restricted to the subcapsular sinus, C) increasing the total access of released drug cargo to dLN-resident cells. D) Delivery of immune adjuvant CpG linked to NPs via multistage OND chemistry (red) improves immune activation within the dLN over CpG linked to NP via non-degradable linker (blue) and free CpG (green) controls evidenced by increased dLN cellularity, E) leading to a reduction in tumor volume in a murine lymphoma model. Reproduced with permission from Ref [129] © 2020 Nature Publishing Group2.
Another example of a polymeric nanocarrier with diverse functionality is a 30 nm poly(2-(hexamethyleneimino)ethyl methacrylate) (PC7a) nanoparticle that serves as an innate immune adjuvant itself without the need to encapsulate traditional agonists, and additionally features a PEGylated surface capable of encapsulating peptide tumor antigen [132]. This allowed for highly efficient delivery of model tumor antigen to LN-resident DC and macrophage populations via passive drainage, and enhancement of APC maturation in vivo as well as efficient antigen cross-presentation in vitro owing to the adjuvant and pH-sensitive properties of the PC7a polymer. Furthermore, PC7a-NP delivery of tumor-associated antigen peptides in three distinct murine tumor models demonstrated superior efficacy in a therapeutic vaccination setting compared to controls [132].
Polymeric nanoparticles can similarly be designed to both encapsulate therapeutics and target them to specific dLN or migratory cell types via surface-conjugated ligands. In one application, poly(lactide-co-glycolide) (PLG or PLGA) NPs encapsulating the TLR7 ligand imiquimod (R837) were coated in cancer cell surface proteins as an antigen source, and additionally grafted with mannose moieties on the NP surface [133] in order to impart DC-targeting capability due to the interaction between mannose and DC lectin receptors [134]. Mannose and tumor cell surface protein functionalized PLGA-NPs with encapsulated R837 (NP-R@M-M) demonstrated increased DC uptake and maturation in vitro, as well as superior dLN targeting in vivo over non-mannose functionalized NP, demonstrating the value of this strategy for DC and dLN delivery applications [133]. A similar PLGA-containing, mannose-coated NP (Man-NP) ~170 nm in diameter was reported that encapsulated CpG, TLR4 agonist monophosphoryl lipid A, and D-α-tocopherol (vitamin E) as immune adjuvants along with tumor antigen peptide [135]. Significant Man-NP uptake occurred within migratory DC populations compared to dLN-resident DCs, which led to enhanced DC maturation compared to non-mannosylated-NP. Man-NP also demonstrated selective bulk accumulation and retention within dLNs and the site of injection [135], suggesting that mannose-lectin receptor targeting is an effective strategy for selective delivery of therapeutics into DCs and dLNs.
Together, these studies demonstrate that polymeric nanocarriers are functionally versatile and can be adapted with unique features that enhance lymphatic delivery and immunomodulation, such as conjugation schemes that allow controlled or staged drug release, material formulations that dually function to adjuvant immune cells, or ligand-targeting moieties that impart specificity to drug targeting.
4.2.3. Peptide/Protein nanocarriers
Protein-based nanocarrier formulations are often made of self-assembling subunits and have been investigated for lymphatic drug delivery due to their relative monodispersity, predictable morphology, and tunability afforded by protein engineering in that the presence, density, and location of functional groups within the nanocarrier structure can be precisely defined [136, 137]. These features impart the ability to control drug conjugation, loading, and release profiles [138] to maximize delivery of immunotherapeutics to lymphatic tissues. Several types of protein-based nanoparticles have been investigated for dLN delivery of cancer vaccines, including virus-like particles (VLPs) [138, 139] and non-virus derived caged protein nanoparticles [136, 137].
A well-investigated class of protein nanocarrier is VLPs, which are self-assembling protein-based particles that mimic the structure and conformation of a pathogenic virus, a feature often utilized in vaccine delivery [136, 139], but lack viral genetic material and thus do not have capability to replicate or harm healthy tissue [140]. VLPs are typically 20–200 nm in diameter and thus are optimal for LN-targeting, and can additionally be genetically fused or covalently coupled with tumor antigen or adjuvant for use in a variety of cancer vaccine applications [140]. One VLP system derived from the bacteriophage Qβ were recently investigated for its ability to target APCs in dLNs [141]. Qβ-VLPs were internally packaged with CpG as an adjuvant or chemically coupled with a model peptide tumor antigen. After s.c. injection, Qβ-VLPs efficiently drained to LNs, and the combination of Qβ(CpGs) and Qβ-antigen was co-delivered into a variety of APC subtypes, leading to maturation. This combination additionally substantially increased the frequency and effector function of systemic antigen-specific CD8+ T cells, indicating the induction of a successful adaptive anti-cancer immune response [141]. In a follow-up study, the authors further investigated the therapeutic efficacy of the Qβ-VLP system in the context of a personalized multi-target cancer vaccine incorporating CpG [142]. After identifying germline and neoantigen cytotoxic T lymphocyte epitopes in a murine melanoma cell line (B16-F10), the peptide sequences were synthesized and easily coupled to the Qβ-VLP platform via copper-free click chemistry. The resulting multi-target Qβ-VLP vaccine substantially inhibited the growth of B16-F10 murine melanoma tumors in a therapeutic setting [142], demonstrating versatility of this platform.
Other caged protein-based nanocarriers have been developed for cancer vaccine applications aside from VLPs that are generally similar in structure, properties, and drug loading profiles but are not derived from virus-associated proteins [137, 138]. In one study, protein NPs stemming from a variety of origins with sizes ranging from 10–30 nm were surface modified with model antigen and screened for their ability to accumulate in dLNs from an s.c. injection, identifying self-assembling human ferritin heavy chain (hFTN) protein NPs as the best candidate due their superior accumulation and retention in dLNs [143]. hFTN NPs were further engineered to densely display model antigen protein on the NP surface (hFTN-antigen), and prophylactic vaccination with hFTN-antigen led to increased populations of interferon-producing, antigen-specific cytotoxic T cells in the dLN and enhanced efficacy in a model antigen-expressing murine melanoma, likely afforded by the enhanced retention of hFTN-antigen in dLNs [143].
In all, protein-based materials represent another promising platform for dLN-targeted cancer vaccine delivery given their optimizable properties that can enhance lymphatic accumulation, and in the case of VLPs, their ability to mimic pathogenic viruses to enhance immune recognition of tumor antigen.
4.2.4. Lipid-based nanocarriers
Lipid-based materials have been extensively investigated in a wide variety of formulations with different structures including but not limited to liposomes [144, 145], nanoemulsions [146, 147], or hybrid lipid nanocarriers [148]. Given this versatility in structure and thus in drug loading capabilities [149, 150], as well as the propensity for lipophilic and fatty acid-containing molecules to traffic into lymph via albumin [151], a multitude of diverse lipid-containing DDSs have been have been developed for lymphatic targeting applications [152–157].
Liposomes are vesicles that contain an outer bilayer membrane and typically encapsulates an aqueous environment, lending to encapsulation of therapeutics or modification of the liposome surface [144, 145]. In one application, a liposomal DDS was engineered to deliver two immunostimulatory agents via conjugation to a PEG-modified surface, an antibody agonist for CD137 (anti-CD137) and a modified IL-2 construct fused with an antibody Fc domain (IL-2Fc), both of which serve to stimulate T cells in the TdLN to enhance the anti-tumor immune response [158]. This resulted in “Lip-aCD137 + Lip-IL-2Fc” which enhanced delivery of both therapeutics to the immune-privileged TdLN following i.t. administration and to dLN-resident T cells. This resulted in a significant increase in interferon-producing cytotoxic T cells in the dLN, as well as inhibition of tumor growth in both a primary, treated murine melanoma and a contralateral lesion and significant presence of infiltrating cytotoxic T cells in the primary tumor [158], indicating that dLN-targeted liposomes are a promising strategy for inducing systemic anti-tumor immunity.
Lipid-based materials can alternatively form versatile disc-shaped bilayer nanocarriers amenable to both covalent conjugation of tumor antigen and incorporation of cholesterol-modified CpG onto the nanocarrier surface [159]. In this application, 10nm synthetic high-density lipoprotein mimicking nanodiscs (sHDL-nanodiscs) decorated with CpG and peptide tumor antigen (sHDL-Ag/CpG) were investigated for their therapeutic efficacy as a personalized neoantigen tumor vaccine [159]. Using a model antigen, sHDL-Ag/CpG was efficiently taken up into DCs and enhanced antigen presentation in vitro and antigen-specific T cell proliferation in vivo versus free drug controls after accumulation in dLNs via the cutaneous injection route. A neoantigen for a murine colon adenocarcinoma model (MC-38) was additionally identified and loaded onto sHDL. In a therapeutic vaccination setting, sHDL-Ag/CpG significantly inhibited MC-38 tumor growth and induced markedly enhanced systemic antigen-specific CD8+ T cell proliferation and effector function versus therapeutics in their free form or delivered using the FDA-approved oil-in-water vaccine adjuvant Montanide. Furthermore, the combination of sHDL-Ag/CpG and checkpoint inhibitor therapy targeting programmed cell death 1 (anti-PD-1) led to a nearly complete response, further indicating that dLN-directed sHDL-Ag/CpG induced a large-scale T cell response that was exhausted and able to be rescued with checkpoint inhibition [159].
Further demonstrating the structural diversity of lipid-based nanocarriers for lymphatic drug delivery, a different HDL-mimicking lipid nanocarrier was developed that features a hydrophobic core stabilized with a monolayer shell amenable to both incorporation of a hydrophobic NIR dye in the core and surface decoration with peptide [160]. This resulted in a 30 nm core-shell lipid NP presenting tumor antigen and the DC-targeting peptide scavenger receptor B1 (α-Ap-NP) in order to target antigen to mature LN-resident DCs. From an s.c. injection, treatment with α-Ap-NP resulted in efficient accumulation within dLN and resident mature DCs and significant reduction of tumor burden in a murine thymoma model expressing model antigen [160]. In another study, the same nanocarrier α-Ap-NP was used to deliver inflammation-inducing melittin (α-Melittin-NP) intratumorally (i.t.) with the goal of increasing antigen spread to dLNs [161]. Following i.t. injection, α-Melittin-NPs accumulated in dLNs along with released tumor antigen, thereby creating an in situ cancer vaccine that resulted in local (dLN) and systemic activation of the anti-tumor immune response and significant reduction in both primary (treated) and distant (untreated) tumor burden [161], indicating the versatility of this core-shell lipid NP approach.
In all, lipid-based materials represent another option for the design of nanocarrier DDSs with unique functionalities given their diversity in shape, size, and method of incorporating therapeutic molecules or targeting ligands. Together, these properties enable lymphatic or cell-specific accumulation of immunotherapeutics in order to augment the anti-tumor immune response.
4.3: Strategies to exploit albumin transport to LNs
Another method of achieving drug delivery to LNs takes advantage of the natural transport of albumin from the interstitium to LNs after capillary exchange. Albumin-binding dyes have been long utilized for their ability to traffic to and identify sentinel LNs from an i.t. injection for proper determination of cancer stage or surgical resection [162]. Dye molecules that bind albumin successfully “hitchhike” into lymphatics and LNs and are taken up by LN-resident APCs [163]. Thus, another strategy to enable LN therapeutic exposure is the use of similar molecular albumin-binding domains as these dyes to engineer albumin hitchhiking derivatives of small molecule, peptide, or oligonucleotide therapeutics that otherwise exhibit low lymphatic accumulation.
An approach to LN targeting via albumin binding involves the creation of an amphiphilic albumin-binding derivative of CpG (“amph-CpG”) by conjugating a diacyl lipid albumin binding domain onto the 5’ end [164]. This engineered construct allowed efficient binding to albumin in vitro, bulk accumulation in dLNs from s.c. injection, and extended release into dLNs for >150h post-injection versus free CpG or other albumin binding domain candidates. Additionally, amph-CpG conjugates allowed accumulation within APC compartments in dLNs. To further apply this technique to a therapeutic cancer vaccine, peptide antigen was also conjugated to the amphiphilic diacyl lipid albumin binding domain via a PEG spacer incorporated to solubilize the construct (“amph-peptide”). Varying the diacyl lipid tail length and molecular weight of the PEG spacer allowed careful tuning of the accumulation of amph-peptide construct in dLNs. Treating mice with the combination of amph-CpG and endogenous tumor antigens E7 and Trp2 as amph-peptide (amph-vaccine) in a therapeutic setting led to significant reduction of tumor volume in murine TC-1 and B16-F10 tumor models, in comparison to free therapeutic controls, and additionally led to induction of systemic immunity, as there were more tumor antigen-specific T cells with higher effector function in circulation [164]. In follow-up studies, the potential for modified albumin-binding amph-vaccines to synergize with systemically-administered tumor antigen-specific antibody (A), cytokine interleukin-2 (IL-2) fused with mouse serum albumin (I), which was previously demonstrated to extend the immune-stimulatory therapeutic benefits of IL-2 by increasing the elimination half-life [165], and anti-PD-1 monoclonal antibody (P) was investigated [166]. While the combination of AIP allowed a significant increase in overall survival in a therapeutic setting compared to untreated mice, the addition of amph-CpG and amph-peptide (V) delivered s.c. to target LNs substantially increased survival benefit compared to AIP, leading to an ~80% cure rate in three murine tumor models with the quadruple combination therapy (AIPV) [166].
In another application, this technology is used in conjunction with chimeric antigen receptor (CAR) T cell therapy in order to stimulate LN-resident CAR-T cells for enhanced anti-tumor immunity [167]. Here, CARs were designed to bind a model (fluorescein isothiocyanate, FITC) or tumor-specific ligand. CAR-T cells encountering their cognate ligand, whether administered freely or expressed by tumor cells, stimulates the CAR-T cell in a non-major histocompatibility complex restricted manner and enhances effector function. To stimulate CAR-T cells residing in LNs, the cognate CAR ligand (FITC or tumor-specific peptide antigen) was modified as described above with an albumin-binding domain (“amph-ligand”) in order to target the stimulatory CAR ligand to the LN. Administration s.c. resulted in accumulation of amph-ligand in dLNs and subsequent transfer of amph-ligand the surface of APCs. CAR-T cell interaction with amph-ligand-decorated APCs within the LN allowed stimulation through the CAR as well as through other co-stimulatory pathways. In a tumor antigen-specific CAR T-cell model, vaccination with the relevant amph-ligand led to greatly increased CAR-T cell expansion in the LN and subsequent trafficking and infiltration into the tumor, which resulted in reduced tumor burden and enhanced survival [167]. Taken all together, delivery of cancer vaccine components to dLN-resident APCs via albumin-hitchhiking is an exciting strategy that enables the induction of a potent anti-tumor immune response, whether by endogenous or transferred cells.
5. Chemotherapy DDSs for lymphatic and LN delivery
Small molecule chemotherapies exert their therapeutic efficacy by direct cytotoxicity-mediated tumor cell killing. However, a mounting body of evidence suggests that traditionally tumoricidal chemotherapeutics can also have both a direct and indirect immune-stimulatory effect on the overall anti-cancer immune response [2, 168]. Some chemotherapies are able to indirectly adjuvant the immune system via induction of immunogenic cell death, a process involving the release of danger signals and tumor antigen in a manner that can be easily recognized by phagocytic APCs and activate the adaptive immune pathway [169, 170]. Alternatively, a variety of chemotherapies are able to directly stimulate immune cells, including DCs, macrophages, T cells, myeloid derived suppressor cells (MDSCs), and natural killer (NK) cells [2, 168, 171]. Because of their immune cell-rich environment and potential to host lymph-borne metastases, LNs are an interesting therapeutic target for the delivery of chemotherapeutics with immunomodulatory effects.
TdLN-targeting 30nm poly(propylene sulfide) (PPS) nanoparticles (Figure 5a) have been investigated for their ability to deliver the hydrophobic chemotherapeutic paclitaxel (PXL or PTX), which efficiently encapsulates into the PPS-NP core [124]. PXL has been widely used in the clinic for decades as an anti-mitotic agent for a variety of cancers but has more recently become implicated in a variety of immune-modulating effects. PXL has been shown to induce maturation and enhance antigen presentation in DCs without a significant decrease in viability [172, 173], and impair and decrease presence of suppressive regulatory T cells [174] while differentially favoring the activation and proliferation of effector CD8+ T cells [175, 176]. Upon encapsulation into PPS-NPs, PXL that is slowly released from the NP retains its bioactivity, as evidenced by efficient DC activation and maturation in vitro [124]. I.d. injection of low-dose PXL-PPS-NPs targeting the TdLN re-shaped the local immune response as expected with an increase in DCs (Figure 5b) and decrease in regulatory T cells. This allowed an increased infiltration of antigen-specific T cells into the tumor (Figure 5c), effects associated with substantial impairments in melanoma tumor growth [124] (Figure 5d).
Figure 5.
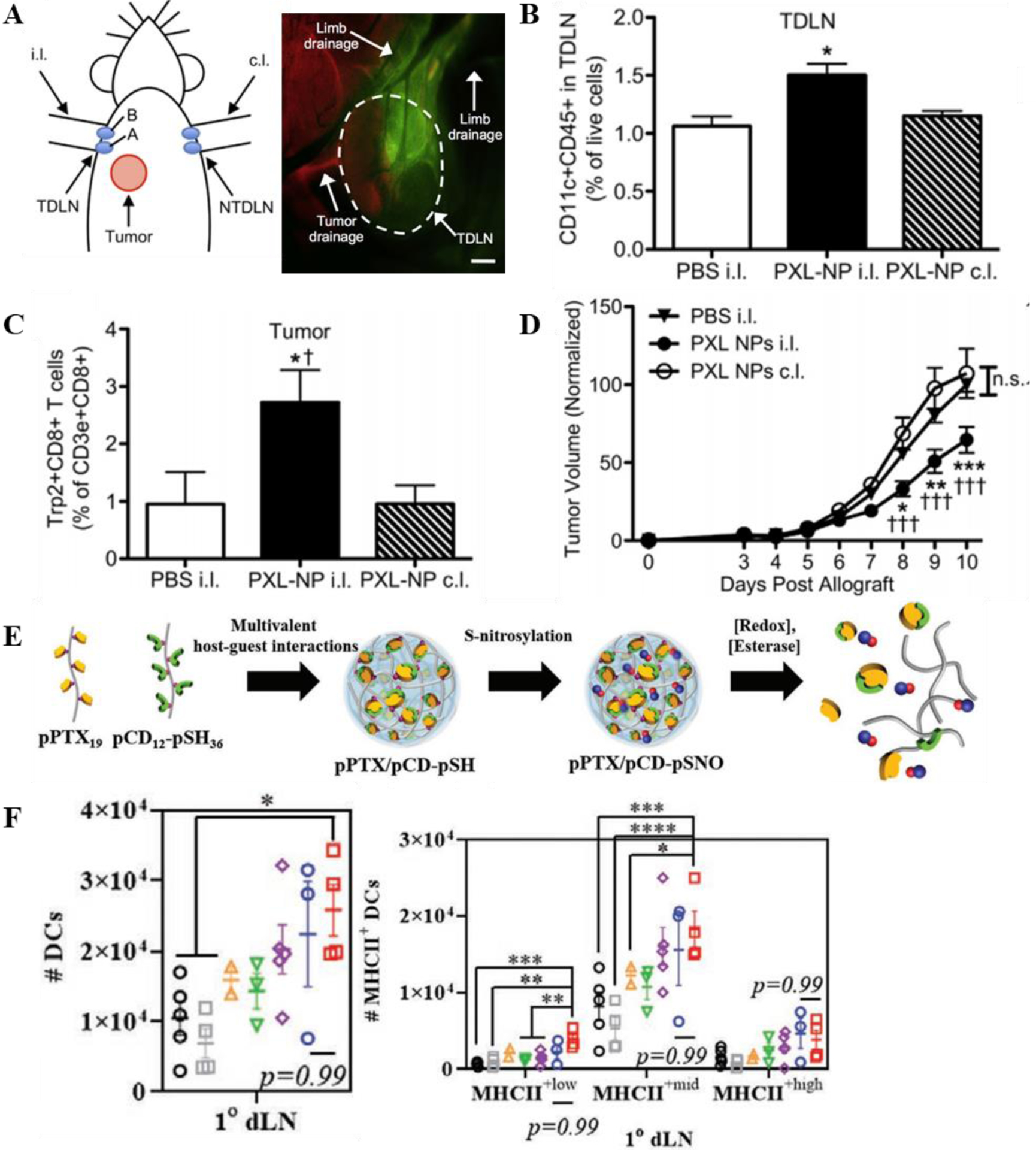
Chemotherapy delivery to LN re-shapes the anti-tumor immune response. A) Thomas et al.; 30 nm Poly(propylene sulfide) NPs target the TdLN from a cutaneous injection in the mouse forearm ipsilateral (i.l.) to the tumor. B) Treatment with PXL-NPs targeted to the TdLN, but not non-dLN, enhanced DC populations in the TdLN and C) tumor antigen-specific T cell infiltration into the tumor, D) resulting in a reduction in tumor burden. Reproduced with permission from Ref [124] © 2014 Elsevier3. E) Kim et al.; Self-assembled NPs made from poly(paclitaxel) and poly(cyclodextrin) and incorporated with NO. F) Treatment with pPTX/pCD-pSNO results in increased populations of matured, MHC-II+ DCs in the TdLN. Reproduced with permission from Ref [176] © 2020 John Wiley & Sons, Inc4.
In another application, a nanocomplex was formed by self-assembly of nitric oxide-incorporated poly(cyclodextrin) and a polymerized derivative of the chemotherapeutic paclitaxel (pPTX/pCD-SNO) [176] (Figure 5e). Nitric oxide (NO) participates in a variety of anti-tumor immune processes [177] and was investigated here for its ability to support the immunological effects of PXL. In a melanoma tumor model, local injection of the 60–70 nm pPTX/pCD-SNO nanocomplexes i.t. allowed significant expansion of DCs within the dLN and enhancement of maturation and antigen presentation markers over free agents (Figure 5f) [176]. Nanocomplexes additionally induced immunogenic cell death after i.t. injection, which presumably allows local release of tumor antigen that co-drains along with nanocomplexes to the TdLN. Additionally, pPTX/pCD-SNO nanocomplexes further synergized with cytotoxic T lymphocyte antigen 4 (CTLA-4) blocking therapy in vivo to reduce tumor burden in a secondary, untreated tumor, indicating the presence of a systemically functional cytotoxic T cell response [176]. Taken together, these results highlight the potential for pPTX/pCD-SNO nanocomplexes as a platform to enable lymphatic immunomodulation.
Overall, the application of traditional chemotherapeutics as immune modulators targeted to lymphoid tissues is in a nascent stage of development. There have however been a multitude of DDS platforms engineered for targeting chemotherapeutics to lymphatics in order to eliminate lymphatic metastases or LN cancers [55, 178–182]. Because of the dual function of chemotherapeutics, it is possible that these systems lead to direct LN immune cell stimulation and may in part account for their therapeutic efficacy, though this has not been directly explored. Future work in this area would certainly benefit from studies that decouple the direct tumor cell killing, immunogenic cell death-inducing, and direct immune adjuvanting functions of chemotherapies in order to design lymphatic targeting approaches that leverage these different mechanisms for enhanced therapeutic efficacy.
Another potential future direction of this work stems from recently investigated effects of traditional systemically administered chemotherapy on lymphatic vessels and lymphangiogenesis. Both PXL [183] and docetaxel [184] have been shown to increase VEGF-C/VEGFR3 signaling within tumors, leading to enlargement of tumor-draining lymphatic vessels and increased lymphatic vessel density. Additional evidence implicates PXL in causing LEC autophagy, hypothesized here to be a cytoprotective mechanism that allows the maintenance of lymphatic vessels, though they are considerably more permeable to metastasizing cancer cells [185]. Intratumoral VEGF-C/VEGFR3 signaling has been widely implicated in pro-tumor immune processes, such as the presence of a suppressive tumor immune microenvironment [186–188] and lymphatic metastasis [189]. That chemotherapeutic molecules can have deleterious effects resulting from tumor accumulation may provide additional motivation for LN-targeted chemotherapy enabled by DDSs to stimulate resident immune cells and prevent sentinel LN metastases. This also motivates investigation into the ability of anti-lymphangiogenic therapy to potentiate the effects of LN-targeted chemotherapy.
6. LN delivery of immune checkpoint blockade (ICB) antibodies
Monoclonal antibodies (mAbs) blocking the immune checkpoints PD-1 and CTLA-4 have demonstrated significant clinical benefit in a variety of cancers, but success has been stymied by ICB’s dose-efficacy relationship [3] being dose-limited by immune-related adverse events [20]. Following systemic administration, mAbs tend to accumulate in blood rich systemic organs but to a much lesser extent in tissues of interest including the tumor and LNs [3, 190–192], increasing the likelihood of off-target side effects. These therapies function by blocking the interaction between PD-1 on T cells and PD ligand (PD-L) 1 on tumor cells or PD-L1/2 on APCs (anti-PD-1), and between CTLA-4 on T cells and B-7 ligands (CD80/86) on APCs (anti-CTLA-4), which induce T cell exhaustion and anergy, respectively [3–5]. APC interactions with lymphocytes expressing PD-1 and CTLA-4 occur not only in site of viral infection and tumors but also within lymphoid tissues, namely the spleen and LNs [3]. As noted, LECs have substantial immune modulatory roles and have been widely reported to express PD-L1 in an inducible manner that modulates lymphocyte priming [193–195]. Additionally, significant evidence suggests a critically important role for TdLNs in the therapeutic success of ICB immunotherapy [31, 196]. This literature, in combination with the evidence that locoregional ICB mAb delivery (e.g. i.t., i.d., and s.c. routes of administration) reduces systemic toxicity and lowers required dose for therapeutic efficacy [197, 198] underscores the potential for targeting immune checkpoints active in lymphatic tissues.
The effect of modulating immune checkpoint pathways in lymphoid tissues was recently investigated by using routes of administration that result in differential accumulation of ICB mAbs in various combinations of tissues of interest, including the blood, spleen, LNs, and tumor [199]. Biodistribution analysis revealed efficient mAb accumulation within TdLNs using locoregional routes of administation (injection i.t. or i.d. administration into the i.l. lymphatic drainage basin, in line with previous reports [200]) not demonstrated using systemic i.p. administration. Furthermore, i.t. administration resulted in appreciable levels of mAb accumulating within the tumor and TdLNs, in contrast to i.d. administration that resulted in high levels of mAb accumulation only within draining LNs. Administration of mAb i.p. resulted in low to negligible levels of mAb accumulation within the tumor and TdLNs. Levels of circulating mAb, revealed by analysis of mAb levels within the blood as well as levels accumulating within the spleen, were equivalent between i.t., i.d. and i.p. administration routes. Thus, by comparing the theraeputic effects of dose-matched ICB administered via different routes of administration, direct comparisons of the importance of ICB mAb accumulation within the TdLN, tumor, blood and spleen could be made. In this manner, it was found that increased delivery of aPD-1 and/or aCTLA-4 mAb to TdLN achieved by i.d. injection within forelimbs co-draining a tumor (i.l.) elicited similar therapeutic efficacy as i.t. administration and far superior efficacy compared to i.p. administration, which was consistent in three murine tumor models (melanoma and breast). This suggests that pathways active within the TdLN are important for mediating the therapeutic effects of ICB mAbs. In addition, a significant reduction in mAb dose used with locoregional (i.d. and i.t.) administration routes was achieved without loss of therapeutic efficacy and led to far less liver toxicity compared to systemic i.p. administration [199].
The promising therapeutic immunomodulatory effects of locoregional ICB mAb administration [197–199] was further augmented by employing a Pluronic F127-based hydrogel to allow prolonged retention of therapeutic mAb at the injection site and enhanced mAb accumulation in the TdLN compared to free (saline-formulated) mAb [199]. Incorporation of the hydrogel DDS to enhance delivery of ICB mAb to the TdLN from an i.d. injection into the i.l. limb led to decreased tumor volume in a murine melanoma model compared to treatment either with free mAb i.d. in the i.l. limb or hydrogel-formulated mAb administered i.d. in the c.l. limb, demonstrating the potential for controlled release strategies to improve the efficacy of ICB and the potential for TdLN targeting to mediate these effects [199]. Other DDSs have been developed that allow prolonged release and retention of mAb compared to locoregional bolus administration [201, 202], or stimulus-triggered release [203–205], though not in the context of LN delivery. ICB mAb delivery strategies have been developed, including large porous microparticles [201], modified platelets [203], and depot-forming hydrogel [202] and microneedle [204, 205] approaches, which have been previosuly reviewed elsewhere [3]. An additional strategy involved the engineering of an ICB mAb to include the ECM binding domain of placenta growth factor-2, which allowed enhanced peritumoral retention and subsequent therapeutic efficacy of ICB mAb [206]. Though many of these ICB mAb delivery platforms are currently effective in the context of i.t. delivery, in future work they could be adapted to mediate extended or triggered release of ICB mAb that results in their accumulation specifically in TdLNs after locoregional administration.
7. Current and future strategies for modulating the tumor lymphatic vasculature
Aside from delivery of immune-modulating therapeutics to lymphatic tissues, precise control over the immune response to cancer may also be achieved by direct modulation of the presence and function of the lymphatic vasculature itself, particularly tumor-associated lymphatic vessels, due to the complex and direct role of lymphatic vessels in pro- and anti-tumor immune processes (Figure 6a). While the effects of direct therapeutic lymphatic vessel modulation on the resulting anti-tumor immune response has yet to be comprehensibly elucidated, drug delivery strategies have been developed to deliver factors in order to induce or inhibit lymphangiogenesis or ablate lymphatic vessels in the context of anti-metastasis or in non-cancer disease settings.
Figure 6.
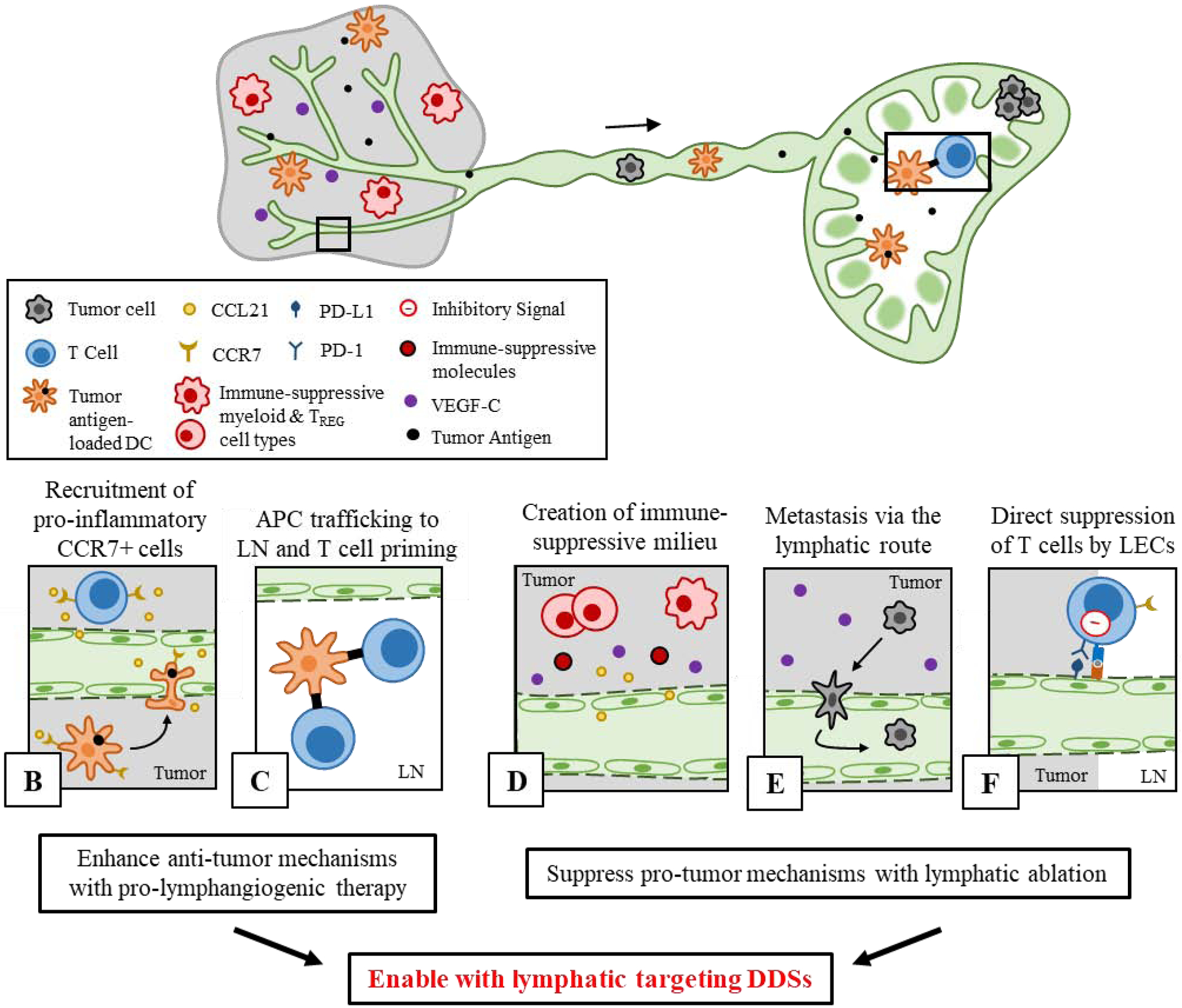
Pro- and anti-tumor effects of lymphatics on cancer immunity, and future directions of DDS-enabled lymphatic targeting for therapeutic lymphangiogenesis or lymphatic ablation. A) Tumor-associated lymphatic vessels and TdLNs have complex involvement in both anti- and pro-tumor immune processes. B-C) Role of lymphatics in anti-tumor immunity. B) In the tumor, local CCL21 secretion by tumor lymphatics promotes infiltration of CCR7+ APCs which C) migrate to LNs in order to prime tumor antigen-specific T cells. D-F) Lymphatic tissues also support pro-tumor immune processes. D) In the tumor, presence of lymphatic vessels and local secretion of VEGF-C and CCL21 are linked to an immune-suppressive molecular milieu and infiltration of regulatory T cells (TREG) and myeloid cells, and E) promote lymphatic metastasis. F) In both the LN and tumor, LECs can cross present tumor antigen to T cells and provide inhibitory signals in order to delete or suppress T cell function (pro-tumor). Established principles of DDS design can enable targeted modulation of lymphatics in order to support or suppress these functions.
Lymphatic vessels and the complex signaling events associated with lymphangiogenesis directly mediate a variety of immune processes relevant to the mounting of anti-tumor immunity, and promotion of a pro-lymphangiogenic environment has demonstrated promise in augmenting the success of immunotherapeutics. Secretion of CCL21 by LECs, promoted by tumor lymphangiogenesis and the presence of VEGF-C [207], is required for the migration of CCR7+ peripheral DCs through lymphatic vessels toward LNs (Figure 6b) upon activation in order to prime T cells and induce adaptive immunity (Figure 6c) [208–210]. DC transmigration into lymphatic vessels is also directly mediated by LECs by several mechanisms [211]. In murine glioblastoma, a pathology relatively devoid of lymphangiogenic signals, induction of lymphangiogenesis via adeno-associated viral vector transfection of VEGF-C led to enhanced lymphatic drainage from tumors to dLNs [34]. This enabled immunosurveillance of drained tumor antigen and induction of a protective T cell response followed by tumor rejection, which was reversed when tumor-draining afferent lymphatic vessels were ligated, and potentiated ICB therapy [34]. Additionally, tumor lymphangiogenesis has been shown to potentiate the therapeutic efficacy of cancer vaccines [32]. Using several cancer vaccine modalities, therapeutic efficacy was consistently mitigated with the use of a VEGFR-3 blocking antibody that inhibits tumor lymphangiogenesis. This effect was suggested to be a result of enhanced recruitment of CCR7+ T cells to the tumor [32]. In the absence of VEGF-C/VEGFR-3 signaling, which promotes lymphatic secretion of the CCR7 ligand CCL21 [207], CCR7+ T cells exhibit impaired tumor infiltration, causing a poorer response to cancer vaccine therapy [32]. Thus, promotion of lymphangiogenesis could have positive implications for potentiating other cancer treatment modalities, such as cancer vaccines and ICB mAbs. To this end, DDSs have been developed to deliver pro-lymphangiogenic factors in order to direct lymphatic vasculature formation, albeit in other disease contexts.
In order to enhance the retention of the pro-lymphangiogenic factor VEGF-C at tissues of interest, in this case at a wound site, a VEGF-C variant fused to a fibrin-binding domain (FB-VEGF-C) has been developed [212]. Using this protein variant, fibrin bound VEGF-C could be released from FB-VEGF-C via fibrin degradation, resulting in enhanced retention of FB-VEGF-C to ten days post-injection in vivo in fibrin gels. Additionally, released VEGF-C was bioactive and induced LEC proliferation in vitro. In a cartilage replacement model in vivo, FB-VEGF-C implanted in fibrin gels allowed extensive lymphangiogenesis to occur in comparison to free VEGF-C or blank gels, which had no deleterious effects on lymphatic capillary morphology and did not have a remodeling effect on downstream collecting lymphatic vessels. Importantly, lymphangiogenesis induced by FB-VEGF-C enhanced the local migration of exogenous DCs towards the downstream dLN, and moreover promoted CCL21 expression and leukocyte interaction with formed lymphatic vasculature [212] demonstrating the potential applicability of this system in the context of cancer immunotherapy.
A similar approach was employed for the induction of lymphangiogenesis to relieve inflammation in a chronic colitis model, wherein VEGF-C was fused to an F8 antibody that targets a fibronectin domain common in inflamed tissues (F8-VEGF-C) [213]. In a chronic colitis model, the F8-VEGF-C fusion protein accumulated specifically in the inflamed colon from an i.v. injection compared to non-targeted VEGF-C, and additionally induced expansion of the lymphatic vasculature in the inflamed tissue and promoted resolution of colitis-related inflammation [213]. In another study, the same F8-VEGF-C fusion construct demonstrated similar lymphangiogenic properties in a model of skin inflammation, which enhanced lymphatic function and resolved edema [214].
Another popular strategy for induction of therapeutic lymphangiogenesis involves the use of adenoviral transduction of VEGF-C into target tissues [215], which has been successful in a variety of in vivo applications [34, 216–218]. However, the hazards associated with adenoviral transfection, including potential for transfection of off-target tissues [219], identifies DDS-enabled delivery of VEGF adenoviral vectors as a possible future application of DDS for therapeutic lymphangiogenesis.
Contrary to what was discussed above, a large body of work has amassed evidence that lymphatic vasculature can also mediate cancer metastasis [220–222] and directly suppress anti-tumor immune responses, including supporting a suppressive local cell infiltrate and cytokine milieu (Figure 6d) [187, 188, 223], promoting metastasis (Figure 6e) [222, 224, 225], and mediating the direct deletion of tumor antigen-specific T cells (Figure 6f) [188, 193–195]. Inhibition of lymphangiogenesis has been achieved for applications in cancer and other pathologies using therapeutic strategies that trap secreted pro-lymphangiogenic factors such as VEGF-C and VEGF-D [226, 227], and small molecule inhibitors or antibodies that block or impair some aspect of lymphangiogenic signaling (anti-lymphangiogenic therapy) [220], reviewed previously in the indicated references. Besides inhibition of further expansion of lymphatic vasculature, photodynamic ablation of existing lymphatic vessels to prevent metastasis or destroy transiting cancer cells has also been employed [228], which utilizes a liposomal delivery system optimally sized for lymphatic uptake in order to concentrate a photosensitizer in tumor lymphatic vessels [228, 229]. Unlike systemically administered small molecule lymphangiogenic inhibitors or blocking antibodies that distribute in off-target tissues, photodynamic ablation using a lymphatic-targeting delivery system allows for highly specific disruption of tumor associated lymphatic vessel [228] and can potentially avoid unwanted side effects in healthy (non-target) tissues.
Despite the successful employment of anti-lymphangiogenic or lymphatic-ablating therapeutics, their effect on the direct involvement of tumor lymphatics in pro-tumor immune processes or ability to potentiate the efficacy other treatment modalities has not yet been comprehensively established. Future work to further elucidate the kinetics and conditions under which tumor lymphatics exert a net pro- or anti-tumor immune effect will guide the design of lymphangiogenic potentiating or depleting therapeutic strategies, which certainly have the potential to benefit from the incorporation of engineered DDS platforms to bias their accumulation into lymphatics.
8. Conclusions
Lymphatic vessels and LNs play important roles in mediating or potentiating the therapeutic efficacy of several cancer treatment modalities, including cancer vaccines, immune-modulatory chemotherapies, ICB and other cancer immunotherapy modalities. The potential of engineered drug delivery platforms in a variety of forms to enable the accumulation of immune-modulating vaccines and chemotherapeutics in LNs and resident immune cells has now been widely demonstrated to confer enhanced protective immunity that is specific to tumors and capable of impeding disease progression. Furthermore, direct modulation of lymphatic tissues, in particular tumor-associated lymphatic vessels, has the potential to allow modulation of the anti-cancer immune response by virtue of the involvement of lymphatics in pro- and anti-tumor immune processes. Drug delivery strategies to therapeutically modulate the presence and function of the lymphatic vasculature have been investigated in the context of cancer metastasis and other disease settings but is in a nascent stage of exploration in the field of cancer immune-modulation. Promising technological advancements in engineered DDSs that enable the delivery of immune-modulating therapeutics into lymphatic tissues thus have high potential contribute to the increasing clinical successes of cancer immunotherapies [230].
Article Highlights.
Lymphatic tissues are heavily involved in anti-cancer immunity.
Drug delivery systems enable immunomodulation of lymphatic tissues and resident immune cells.
Delivery of immunotherapy to lymph nodes enhances anti-tumor responses.
Modulation of lymphatic vessels may support or suppress pro- or anti-tumor-immune processes.
DDSs allow improved safety of delivered therapeutics by reducing off-target accumulation.
DDS technologies offer the ability to improve the state of the art in cancer therapy.
Financial Support:
This work was supported by National Institutes of Health (NIH) Grants R01CA207619 and R01CA247484, Department of Defense Grant CA150523, and the Curci Foundation. M.P.M. was supported by a National Science Foundation Graduate Research Fellowship.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reprinted from “Zhong, X. et al. An aluminum adjuvant-integrated nano-MOF as antigen delivery system to induce strong humoral and cellular immune responses. J. Control. Release 300, 81–92 (2019)” with permission from Elsevier.
Reprinted from “Schudel, A. et al. Programmable multistage drug delivery to lymph nodes. Nature Nanotechnology 15, 491–499 (2020)” with permission from Nature Publishing Group.
Reprinted from “Thomas, S. N., et al. Targeting the tumor-draining lymph node with adjuvanted nanoparticles reshapes the anti-tumor immune response. Biomaterials 35, 814–824 (2014)” with permission from Elsevier.
Reprinted from “Kim, J.,et al. Poly(cyclodextrin)-Polydrug Nanocomplexes as Synthetic Oncolytic Virus for Locoregional Melanoma Chemoimmunotherapy. Adv. Funct. Mater. 1908788 (2020) doi:10.1002/adfm.201908788.
References
- [1].Swartz MA, Hirosue S & Hubbell JA Engineering Approaches to Immunotherapy. Sci. Transl. Med 4, (2012). [DOI] [PubMed] [Google Scholar]
- [2].Kim J, Manspeaker MP & Thomas SN Augmenting the synergies of chemotherapy and immunotherapy through drug delivery. Acta Biomaterialia (2019) doi: 10.1016/j.actbio.2019.02.012. [DOI] [PubMed] [Google Scholar]
- [3].Francis DM & Thomas SN Progress and opportunities for enhancing the delivery and efficacy of checkpoint inhibitors for cancer immunotherapy. Advanced Drug Delivery Reviews (2017) doi: 10.1016/j.addr.2017.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Okazaki T & Honjo T PD-1 and PD-1 ligands: from discovery to clinical application. Int. Immunol 19, 813–824 (2007). [DOI] [PubMed] [Google Scholar]
- [5].Krummel MF & Allison JP CD28 and CTLA-4 have opposing effects on the response of T ceils to stimulation. J. Exp. Med 182, 459–465 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Kantoff PW et al. Sipuleucel-T Immunotherapy for Castration-Resistant Prostate Cancer. N. Engl. J. Med 363, 411–422 (2010). [DOI] [PubMed] [Google Scholar]
- [7].Andtbacka RHI et al. Talimogene laherparepvec improves durable response rate in patients with advanced melanoma. J. Clin. Oncol 33, 2780–2788 (2015). [DOI] [PubMed] [Google Scholar]
- [8].Maude SL et al. Chimeric Antigen Receptor T Cells for Sustained Remissions in Leukemia. N. Engl. J. Med 371, 1507–1517 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Yee C et al. Adoptive T cell therapy using antigen-specific CD8+ T cell clones for the treatment of patients with metastatic melanoma: In vivo persistence, migration, and antitumor effect of transferred T cells. Proc. Natl. Acad. Sci. U. S. A 99, 16168–16173 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Negrier S et al. Recombinant Human Interleukin-2, Recombinant Human Interferon Alfa-2a, or Both in Metastatic Renal-Cell Carcinoma. N. Engl. J. Med 338, 1272–1278 (1998). [DOI] [PubMed] [Google Scholar]
- [11].Berraondo P et al. Cytokines in clinical cancer immunotherapy. Br. J. Cancer 120, 6–15 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Kirkwood JM et al. Interferon alfa-2b adjuvant therapy of high-risk resected cutaneous melanoma: The Eastern Cooperative Oncology Group trial EST 1684. J. Clin. Oncol 14, 7–17 (1996). [DOI] [PubMed] [Google Scholar]
- [13].Kantarjian H et al. Blinatumomab versus Chemotherapy for Advanced Acute Lymphoblastic Leukemia. N. Engl. J. Med 376, 836–847 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Goebeler M-E & Bargou RC T cell-engaging therapies — BiTEs and beyond . Nat. Rev. Clin. Oncol (2020) doi: 10.1038/s41571-020-0347-5. [DOI] [PubMed] [Google Scholar]
- [15].von Minckwitz G et al. Trastuzumab Emtansine for Residual Invasive HER2-Positive Breast Cancer. N. Engl. J. Med 380, 617–628 (2019). [DOI] [PubMed] [Google Scholar]
- [16].Leung D et al. Antibody Conjugates-Recent Advances and Future Innovations. Antibodies 9, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Pan C et al. Next-generation immuno-oncology agents: Current momentum shifts in cancer immunotherapy. J. Hematol. Oncol 13, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Hegde PS & Chen DS Top 10 Challenges in Cancer Immunotherapy. Immunity vol. 52 17–35 (2020). [DOI] [PubMed] [Google Scholar]
- [19].Schadendorf D et al. Pooled Analysis of Long-Term Survival Data From Phase II and Phase III Trials of Ipilimumab in Unresectable or Metastatic Melanoma. J. Clin. Oncol 33, 1889–94 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Eun Y et al. Risk factors for immune-related adverse events associated with anti-PD-1 pembrolizumab. Sci. Rep 9, 1–8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].De Martin E et al. Characterization of liver injury induced by cancer immunotherapy using immune checkpoint inhibitors. J. Hepatol 68, 1181–1190 (2018). [DOI] [PubMed] [Google Scholar]
- [22].Milling L, Zhang Y & Irvine DJ Delivering safer immunotherapies for cancer. Adv. Drug Deliv. Rev 114, 79–101 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Fenton OS, Olafson KN, Pillai PS, Mitchell MJ & Langer R Advances in Biomaterials for Drug Delivery. Adv. Mater 30, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Uhrich KE, Cannizzaro SM, Langer RS & Shakesheff KM Polymeric Systems for Controlled Drug Release. Chem. Rev 99, 3181–3198 (1999). [DOI] [PubMed] [Google Scholar]
- [25].Mehtani D et al. Biomaterials for Sustained and Controlled Delivery of Small Drug Molecules in Biomaterials and Bionanotechnology (ed. Tekade RK.) 89–152 (Academic Press, 2019). [Google Scholar]
- [26].Senapati S, Mahanta AK, Kumar S & Maiti P Controlled drug delivery vehicles for cancer treatment and their performance. Signal Transduct. Target. Ther 3, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Soppimath KS, Aminabhavi TM, Kulkarni AR & Rudzinski WE Biodegradable polymeric nanoparticles as drug delivery devices. J. Control. Release 70, 1–20 (2001). [DOI] [PubMed] [Google Scholar]
- [28].Alitalo A & Detmar M Interaction of tumor cells and lymphatic vessels in cancer progression. Oncogene vol. 31 4499–4508 (2012). [DOI] [PubMed] [Google Scholar]
- [29].Thomas SN, Rohner NA & Edwards EE Implications of Lymphatic Transport to Lymph Nodes in Immunity and Immunotherapy. Annu. Rev. Biomed. Eng 18, 207–233 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Randolph GJ, Ivanov S, Zinselmeyer BH & Scallan JP The Lymphatic System: Integral Roles in Immunity. Annu. Rev. Immunol 35, 31–52 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Fransen MF et al. Tumor-draining lymph nodes are pivotal in PD-1/PD-L1 checkpoint therapy. JCI insight 3, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Fankhauser M et al. Tumor lymphangiogenesis promotes T cell infiltration and potentiates immunotherapy in melanoma. Sci. Transl. Med 9, (2017). [DOI] [PubMed] [Google Scholar]
- [33].Spitzer MH et al. Systemic Immunity Is Required for Effective Cancer Immunotherapy. Cell 168, 502 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Song E et al. VEGF-C-driven lymphatic drainage enables immunosurveillance of brain tumours. Nature 577, 689 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Thomas SN & Schudel A Overcoming transport barriers for interstitial-, lymphatic-, and lymph node-targeted drug delivery. Current Opinion in Chemical Engineering (2015) doi: 10.1016/j.coche.2014.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Schudel A, Francis DM & Thomas SN Material design for lymph node drug delivery. Nature Reviews Materials vol. 4 415–428 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Trevaskis NL, Kaminskas LM & H Porter CJ From sewer to saviour — targeting the lymphatic system to promote drug exposure and activity. Nat. Publ. Gr 14, (2015). [DOI] [PubMed] [Google Scholar]
- [38].Maisel K, Sasso MS, Potin L & Swartz MA Exploiting lymphatic vessels for immunomodulation: Rationale, opportunities, and challenges. Advanced Drug Delivery Reviews vol. 114 43–59 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Riley RS, June CH, Langer R & Mitchell MJ Delivery technologies for cancer immunotherapy. Nat. Rev. Drug Discov 18, 175–196 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Cueni LN & Detmar M The Lymphatic System in Health and Disease. Lymphat Res Biol 6, 109–122 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Padera TP, Meijer EFJ & Munn LL The Lymphatic System in Disease Processes and Cancer Progression. Annu. Rev. Biomed. Eng 18, 125–158 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Swartz MA The physiology of the lymphatic system. Advanced Drug Delivery Reviews vol. 50 3–20 (2001). [DOI] [PubMed] [Google Scholar]
- [43].Alitalo K The lymphatic vasculature in disease. Nat. Med 17, (2011). [DOI] [PubMed] [Google Scholar]
- [44].Wiig H & Swartz MA Interstitial fluid and lymph formation and transport: Physiological regulation and roles in inflammation and cancer. Physiol. Rev 92, 1005–1060 (2012). [DOI] [PubMed] [Google Scholar]
- [45].Randolph GJ, Angeli V & Swartz MA Dendritic-cell trafficking to lymph nodes through lymphatic vessels. Nat. Rev. Immunol 5, 617–628 (2005). [DOI] [PubMed] [Google Scholar]
- [46].Lund AW & Swartz MA Role of Lymphatic Vessels in Tumor Immunity: Passive Conduits or Active Participants? J Mammary Gland Biol Neoplasia 15, 341–352 (2010). [DOI] [PubMed] [Google Scholar]
- [47].Witte MH et al. Structure function relationships in the lymphatic system and implications for cancer biology. Cancer Metastasis Rev 25, 159–184 (2006). [DOI] [PubMed] [Google Scholar]
- [48].Chaplin DD Overview of the immune response. J. Allergy Clin. Immunol 125, S3–S23 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Rohner NA & Thomas SN Melanoma growth effects on molecular clearance from tumors and biodistribution into systemic tissues versus draining lymph nodes. J. Control. Release 223, 99–108 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Rohner NA & Thomas SN Flexible Macromolecule versus Rigid Particle Retention in the Injected Skin and Accumulation in Draining Lymph Nodes Are Differentially Influenced by Hydrodynamic Size. ACS Biomater. Sci. Eng 3, 153–159 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Kourtis IC et al. Peripherally Administered Nanoparticles Target Monocytic Myeloid Cells, Secondary Lymphoid Organs and Tumors in Mice. PLoS One 8, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Worley DR, Hansen RJ, Wittenburg LA, Chubb LS & Gustafson DL Docetaxel Accumulates in Lymphatic Circulation Following Subcutaneous Delivery Compared to Intravenous Delivery in Rats. Anticancer Res. 36, 5071–5078 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Reddy ST, Rehor A, Schmoekel HG, Hubbell JA & Swartz MA In vivo targeting of dendritic cells in lymph nodes with poly(propylene sulfide) nanoparticles. J. Control. Release 112, 26–34 (2006). [DOI] [PubMed] [Google Scholar]
- [54].Manolova V et al. Nanoparticles target distinct dendritic cell populations according to their size. Eur. J. Immunol 38, 1404–1413 (2008). [DOI] [PubMed] [Google Scholar]
- [55].Cabral H et al. Systemic Targeting of Lymph Node Metastasis through the Blood Vascular System by Using Size-Controlled Nanocarriers. ACS Nano 9, 4957–4967 (2015). [DOI] [PubMed] [Google Scholar]
- [56].Proulx ST et al. Use of a PEG-conjugated bright near-infrared dye for functional imaging of rerouting of tumor lymphatic drainage after sentinel lymph node metastasis. Biomaterials 34, 5128–5137 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Nathanson SD & Mahan M Sentinel lymph node pressure in breast cancer. Ann. Surg. Oncol 18, 3791–3796 (2011). [DOI] [PubMed] [Google Scholar]
- [58].Mahinroosta M, Jomeh Farsangi Z, Allahverdi A & Shakoori Z Hydrogels as intelligent materials: A brief review of synthesis, properties and applications. Mater. Today Chem 8, 42–55 (2018). [Google Scholar]
- [59].Li J & Mooney DJ Designing hydrogels for controlled drug delivery. Nat. Rev. Mater 1, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].O’Brien FJ Biomaterials & scaffolds for tissue engineering. Mater. Today 14, 88–95 (2011). [Google Scholar]
- [61].Deb P, Deoghare AB, Borah A, Barua E & Das Lala S Scaffold Development Using Biomaterials: A Review . Mater. Today Proc 5, 12909–12919 (2018). [Google Scholar]
- [62].Ma G & Wu C Microneedle, bio-microneedle and bio-inspired microneedle: A review. J. Control. Release 251, 11–23 (2017). [DOI] [PubMed] [Google Scholar]
- [63].Prausnitz MR Microneedles for transdermal drug delivery. Adv. Drug Deliv. Rev 56, 581–587 (2004). [DOI] [PubMed] [Google Scholar]
- [64].Schwendeman SP, Shah RB, Bailey BA & Schwendeman AS Injectable controlled release depots for large molecules. J. Control. Release 190, 240–253 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Chen W, Yung BC, Qian Z & Chen X Improving long-term subcutaneous drug delivery by regulating material-bioenvironment interaction. Adv. Drug Deliv. Rev 127, 20–34 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Chaudhary S, Garg T, Murthy RSR, Rath G & Goyal AK Recent approaches of lipid-based delivery system for lymphatic targeting via oral route. J. Drug Target 22, 871–882 (2014). [DOI] [PubMed] [Google Scholar]
- [67].Ahn H & Park JH Liposomal delivery systems for intestinal lymphatic drug transport. Biomater. Res 20, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Trevaskis NL, Charman WN & Porter CJH Lipid-based delivery systems and intestinal lymphatic drug transport: A mechanistic update. Advanced Drug Delivery Reviews vol. 60 702–716 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Wu L, Shan W, Zhang Z & Huang Y Engineering nanomaterials to overcome the mucosal barrier by modulating surface properties. Adv. Drug Deliv. Rev 124, 150–163 (2018). [DOI] [PubMed] [Google Scholar]
- [70].Hearnden V et al. New developments and opportunities in oral mucosal drug delivery for local and systemic disease. Adv. Drug Deliv. Rev 64, 16–28 (2012). [DOI] [PubMed] [Google Scholar]
- [71].Bailon P & Won C-Y PEG-modified biopharmaceuticals. Expert Opin. Drug Deliv 6, 1–16 (2009). [DOI] [PubMed] [Google Scholar]
- [72].Siram K et al. A brief perspective on the diverging theories of lymphatic targeting with colloids. Int. J. Nanomedicine 11, 2872 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Owens III DE & Peppas NA Opsonization, biodistribution, and pharmacokinetics of polymeric nanoparticles. Int. J. Pharm 307, 93–102 (2006). [DOI] [PubMed] [Google Scholar]
- [74].Kaminskas LM et al. PEGylation of polylysine dendrimers improves absorption and lymphatic targeting following SC administration in rats. J. Control. Release 140, 108–116 (2009). [DOI] [PubMed] [Google Scholar]
- [75].Longmire M, Choyke PL & Kobayashi H Clearance properties of nano-sized particles and molecules as imaging agents: Considerations and caveats. Nanomedicine 3, 703–717 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Suk JS, Xu Q, Kim N, Hanes J & Ensign LM PEGylation as a strategy for improving nanoparticle-based drug and gene delivery. Adv. Drug Deliv. Rev 99, 28–51 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Dixon JB Lymphatic lipid transport: Sewer or subway? Trends Endocrinol. Metab 21, 480–487 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Lee G et al. Lymphatic Uptake of Liposomes after Intraperitoneal Administration Primarily Occurs via the Diaphragmatic Lymphatics and is Dependent on Liposome Surface Properties. Mol. Pharm 16, 4987–4999 (2019). [DOI] [PubMed] [Google Scholar]
- [79].Oussoren C et al. Lymphatic uptake and biodistribution of liposomes after subcutaneous injection IV. Fate of liposomes in regional lymph nodes. Biochim. Biophys. Acta - Biomembr 1370, 259–272 (1998). [DOI] [PubMed] [Google Scholar]
- [80].Kim C-K, Lee M-K, Han J-H & Lee B-J Pharmacokinetics and tissue distribution of methotrexate after intravenous injection of differently charged liposome-entrapped methotrexate to rats. Int. J. Pharm 108, 21–29 (1994). [Google Scholar]
- [81].Charman WN Lipids, lipophilic drugs, and oral drug delivery—Some emerging concepts. J. Pharm. Sci 89, 967–978 (2000). [DOI] [PubMed] [Google Scholar]
- [82].Porter CJH & Charman WN Uptake of drugs into the intestinal lymphatics after oral administration. Adv. Drug Deliv. Rev 25, 71–89 (1997). [Google Scholar]
- [83].Caliph SM et al. The impact of lymphatic transport on the systemic disposition of lipophilic drugs. J. Pharm. Sci 102, 2395–2408 (2013). [DOI] [PubMed] [Google Scholar]
- [84].Porter CJH, Trevaskis NL & Charman WN Lipids and lipid-based formulations: optimizing the oral delivery of lipophilic drugs. Nat. Rev. Drug Discov 6, 231–248 (2007). [DOI] [PubMed] [Google Scholar]
- [85].Goyal AK, Rath G, Faujdar C & Malik B Application and Perspective of pH-Responsive Nano Drug Delivery Systems in Applications of Targeted Nano Drugs and Delivery Systems 15–33 (Elsevier, 2019). doi: 10.1016/b978-0-12-814029-1.00002-8. [DOI] [Google Scholar]
- [86].Nembrini C et al. Nanoparticle conjugation of antigen enhances cytotoxic T-cell responses in pulmonary vaccination. Proc. Natl. Acad. Sci. U. S. A 108, E989–E997 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Shen H et al. Enhanced and prolonged cross-presentation following endosomal escape of exogenous antigens encapsulated in biodegradable nanoparticles. Immunology 117, 78–88 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Tong X & Yang F Recent Progress in Developing Injectable Matrices for Enhancing Cell Delivery and Tissue Regeneration . Adv. Healthc. Mater 7, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Canal T & Peppas NA Correlation between mesh size and equilibrium degree of swelling of polymeric networks. J. Biomed. Mater. Res 23, 1183–1193 (1989). [DOI] [PubMed] [Google Scholar]
- [90].Martinez AW, Caves JM, Ravi S, Li W & Chaikof EL Effects of crosslinking on the mechanical properties, drug release and cytocompatibility of protein polymers. Acta Biomater. 10, 26–33 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Wang H & Mooney DJ Biomaterial-assisted targeted modulation of immune cells in cancer treatment. Nature Materials vol. 17 761–772 (2018). [DOI] [PubMed] [Google Scholar]
- [92].Irvine DJ, Hanson MC, Rakhra K & Tokatlian T Synthetic Nanoparticles for Vaccines and Immunotherapy. Chem. Rev 115, 11109–11146 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Koshy ST & Mooney DJ Biomaterials for enhancing anti-cancer immunity. Curr. Opin. Biotechnol 40, 1–8 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Singh A & Peppas NA Hydrogels and scaffolds for immunomodulation. Adv. Mater 26, 6530–6541 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Foged C, Brodin B, Frokjaer S & Sundblad A Particle size and surface charge affect particle uptake by human dendritic cells in an in vitro model. Int. J. Pharm 298, 315–322 (2005). [DOI] [PubMed] [Google Scholar]
- [96].Chauhan VP et al. Fluorescent Nanorods and Nanospheres for Real-Time In Vivo Probing of Nanoparticle Shape-Dependent Tumor Penetration. Angew. Chemie Int. Ed 50, 11417–11420 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Kumar S, Anselmo AC, Banerjee A, Zakrewsky M & Mitragotri S Shape and size-dependent immune response to antigen-carrying nanoparticles. J. Control. Release 220, 141–148 (2015). [DOI] [PubMed] [Google Scholar]
- [98].Bonifaz LC et al. In Vivo Targeting of Antigens to Maturing Dendritic Cells via the DEC-205 Receptor Improves T Cell Vaccination. J. Exp. Med 199, 815–824 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Cruz LJ et al. Targeting nanoparticles to CD40, DEC-205 or CD11c molecules on dendritic cells for efficient CD8+ T cell response: A comparative study. J. Control. Release 192, 209–218 (2014). [DOI] [PubMed] [Google Scholar]
- [100].Guo Q, Liu Y, Xu K, Ren K & Sun WG Mouse lymphatic endothelial cell targeted probes: Anti-LYVE-1 antibody-based magnetic nanoparticles. Int. J. Nanomedicine 8, 2273–2284 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Yang H et al. Feasibility of MR imaging in evaluating breast cancer lymphangiogenesis using Polyethylene glycol-GoldMag nanoparticles. Clin. Radiol 68, 1233–1240 (2013). [DOI] [PubMed] [Google Scholar]
- [102].Azzi J et al. Targeted Delivery of Immunomodulators to Lymph Nodes. Cell Rep. 15, 1202–1213 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Kim J et al. Injectable, spontaneously assembling, inorganic scaffolds modulate immune cells in vivo and increase vaccine efficacy. Nat. Biotechnol 33, 64–72 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Dellacherie MO, Li AW, Lu BY & Mooney DJ Covalent Conjugation of Peptide Antigen to Mesoporous Silica Rods to Enhance Cellular Responses. Bioconjug. Chem 29, 733–741 (2018). [DOI] [PubMed] [Google Scholar]
- [105].Bencherif SA et al. Injectable cryogel-based whole-cell cancer vaccines. Nat. Commun 6, 1–13 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Bellis SL Advantages of RGD peptides for directing cell association with biomaterials. Biomaterials (2011) doi: 10.1016/j.biomaterials.2011.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Ali OA, Huebsch N, Cao L, Dranoff G & Mooney DJ Infection-mimicking materials to program dendritic cells in situ. Nat. Mater 8, 151–158 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Sánchez-Sánchez N, Riol-Blanco L, Luis J & Fernández R The Multiple Personalities of the Chemokine Receptor CCR7 in Dendritic Cells. J. Immunol 176, 5153–5159 (2006). [DOI] [PubMed] [Google Scholar]
- [109].Kim NW et al. Enhanced Cancer Vaccination by in Situ Nanomicelle-Generating Dissolving Microneedles. ACS Nano (2018) doi: 10.1021/acsnano.8b04146. [DOI] [PubMed] [Google Scholar]
- [110].Patra JK et al. Nano based drug delivery systems: Recent developments and future prospects. J. Nanobiotechnology 16, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Hans ML & Lowman AM Biodegradable nanoparticles for drug delivery and targeting. Curr. Opin. Solid State Mater. Sci 6, 319–327 (2002). [Google Scholar]
- [112].Chenthamara D et al. Therapeutic efficacy of nanoparticles and routes of administration. Biomater. Res 23, 1 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Naz S et al. Advances in therapeutic implications of inorganic drug delivery nano-platforms for cancer. Int. J. Mol. Sci 20, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Zhong X et al. An aluminum adjuvant-integrated nano-MOF as antigen delivery system to induce strong humoral and cellular immune responses. J. Control. Release 300, 81–92 (2019). [DOI] [PubMed] [Google Scholar]
- [115].Minchin RF & Martin DJ Minireview: Nanoparticles for Molecular Imaging—An Overview. Endocrinology 151, 474–481 (2010). [DOI] [PubMed] [Google Scholar]
- [116].Ruiz-De-Angulo A, Zabaleta A, Gómez-Vallejo V, Llop J & Mareque-Rivas JC Microdosed lipid-coated 67Ga-magnetite enhances antigen-specific immunity by image tracked delivery of antigen and cpg to lymph nodes. ACS Nano 10, 1602–1618 (2016). [DOI] [PubMed] [Google Scholar]
- [117].Xiang J et al. Antigen-Loaded Upconversion Nanoparticles for Dendritic Cell Stimulation, Tracking, and Vaccination in Dendritic Cell-Based Immunotherapy. ACS Nano 9, 6401–6411 (2015). [DOI] [PubMed] [Google Scholar]
- [118].Elsabahy M & Wooley KL Strategies toward well-defined polymer nanoparticles inspired by nature: Chemistry versus versatility. J. Polym. Sci. Part A Polym. Chem 50, 1869–1880 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Park JH et al. Polymeric nanomedicine for cancer therapy. Prog. Polym. Sci 33, 113–137 (2008). [Google Scholar]
- [120].Parveen S & Sahoo SK Polymeric nanoparticles for cancer therapy. J. Drug Target 16, 108–123 (2008). [DOI] [PubMed] [Google Scholar]
- [121].Reddy ST et al. Exploiting lymphatic transport and complement activation in nanoparticle vaccines. Nat. Biotechnol 25, 1159–1164 (2007). [DOI] [PubMed] [Google Scholar]
- [122].De Titta A et al. Nanoparticle conjugation of CpG enhances adjuvancy for cellular immunity and memory recall at low dose. doi: 10.1073/pnas.1313152110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Jeanbart L et al. Enhancing efficacy of anticancer vaccines by targeted delivery to tumor-draining lymph nodes. Cancer Immunol. Res 2, 436–447 (2014). [DOI] [PubMed] [Google Scholar]
- [124].Thomas SN, Vokali E, Lund AW, Hubbell JA & Swartz MA Targeting the tumor-draining lymph node with adjuvanted nanoparticles reshapes the anti-tumor immune response. Biomaterials 35, 814–824 (2014). [DOI] [PubMed] [Google Scholar]
- [125].Rehor A, Hubbell JA & Tirelli N Oxidation-Sensitive Polymeric Nanoparticles. Langmuir 21, 411–417 (2005). [DOI] [PubMed] [Google Scholar]
- [126].Van Der Vlies AJ, Oneil CP, Hasegawa U, Hammond N & Hubbell JA Synthesis of pyridyl disulfide-functionalized nanoparticles for conjugating thiol-containing small molecules, peptides, and proteins. Bioconjug. Chem 21, 653–662 (2010). [DOI] [PubMed] [Google Scholar]
- [127].Hirosue S, Kourtis IC, van der Vlies AJ, Hubbell JA & Swartz MA Antigen delivery to dendritic cells by poly(propylene sulfide) nanoparticles with disulfide conjugated peptides: Cross-presentation and T cell activation. Vaccine (2010) doi: 10.1016/j.vaccine.2010.09.077. [DOI] [PubMed] [Google Scholar]
- [128].Stano A et al. PPS nanoparticles as versatile delivery system to induce systemic and broad mucosal immunity after intranasal administration. Vaccine 29, 804–812 (2011). [DOI] [PubMed] [Google Scholar]
- [129].Schudel A et al. Programmable multistage drug delivery to lymph nodes. Nat. Nanotechnol 15, 491–499 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Kislukhin AA, Higginson CJ, Hong VP & Finn MG Degradable conjugates from oxanorbornadiene reagents. J. Am. Chem. Soc 134, 6491–6497 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Higginson CJ, Kim SY, Peláez-Fernández M, Fernández-Nieves A & Finn MG Modular degradable hydrogels based on thiol-reactive oxanorbornadiene linkers. J. Am. Chem. Soc 137, 4984–4987 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Luo M et al. A STING-activating nanovaccine for cancer immunotherapy. Nat. Nanotechnol 12, 648–654 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Yang R et al. Cancer Cell Membrane-Coated Adjuvant Nanoparticles with Mannose Modification for Effective Anticancer Vaccination. ACS Nano 12, 5121–5129 (2018). [DOI] [PubMed] [Google Scholar]
- [134].Carrillo-Conde B et al. Mannose-Functionalized ‘Pathogen-like’ Polyanhydride Nanoparticles Target C-Type Lectin Receptors on Dendritic Cells. Mol. Pharm 8, 1877–1886 (2011). [DOI] [PubMed] [Google Scholar]
- [135].Conniot J et al. Immunization with mannosylated nanovaccines and inhibition of the immune-suppressing microenvironment sensitizes melanoma to immune checkpoint modulators. Nat. Nanotechnol 14, 891–901 (2019). [DOI] [PubMed] [Google Scholar]
- [136].Neek M, Kim T Il & Wang SW Protein-based nanoparticles in cancer vaccine development. Nanomedicine Nanotechnology, Biol. Med 15, 164–174 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Molino NM & Wang SW Caged protein nanoparticles for drug delivery. Curr. Opin. Biotechnol 28, 75–82 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Choi B, Kim H, Choi H & Kang S Protein cage nanoparticles as delivery nanoplatforms in Advances in Experimental Medicine and Biology vol. 1064 27–43 (Springer New York LLC, 2018). [DOI] [PubMed] [Google Scholar]
- [139].Smith MT, Hawes AK & Bundy BC Reengineering viruses and virus-like particles through chemical functionalization strategies. Curr. Opin. Biotechnol 24, 620–626 (2013). [DOI] [PubMed] [Google Scholar]
- [140].Mohsen MO, Speiser DE, Knuth A & Bachmann MF Virus-like particles for vaccination against cancer. Wiley Interdiscip. Rev. Nanomedicine Nanobiotechnology 12, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Mohsen MO et al. Delivering adjuvants and antigens in separate nanoparticles eliminates the need of physical linkage for effective vaccination. J. Control. Release (2017) doi: 10.1016/j.jconrel.2017.02.031. [DOI] [PubMed] [Google Scholar]
- [142].Mohsen MO et al. Targeting Mutated Plus Germline Epitopes Confers Pre-clinical Efficacy of an Instantly Formulated Cancer Nano-Vaccine. Front. Immunol 10, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Lee BR et al. Engineered Human Ferritin Nanoparticles for Direct Delivery of Tumor Antigens to Lymph Node and Cancer Immunotherapy. Sci. Rep 6, 1–12 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Sercombe L et al. Advances and challenges of liposome assisted drug delivery. Front. Pharmacol 6, 286 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Olusanya TOB, Ahmad RRH, Ibegbu DM, Smith JR & Elkordy AA Liposomal drug delivery systems and anticancer drugs. Molecules 23, 907 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Pathak K, Pattnaik S & Swain K Application of Nanoemulsions in Drug Delivery in Nanoemulsions: Formulation, Applications, and Characterization 415–433 (Elsevier Inc., 2018). doi: 10.1016/B978-0-12-811838-2.00013-8. [DOI] [Google Scholar]
- [147].Singh Y et al. Nanoemulsion: Concepts, development and applications in drug delivery. Journal of Controlled Release vol. 252 28–49 (2017). [DOI] [PubMed] [Google Scholar]
- [148].Vargas KM & Shon Y-S Hybrid lipid-nanoparticle complexes for biomedical applications Journal of Materials Chemistry B Materials for biology and medicine Hybrid lipid-nanoparticle complexes for biomedical applications. J. Mater. Chem B 7, 708 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Shukla T, Upmanyu N, Prakash Pandey S & Gosh D Lipid nanocarriers in Lipid Nanocarriers for Drug Targeting 1–47 (Elsevier, 2018). doi: 10.1016/b978-0-12-813687-4.00001-3. [DOI] [Google Scholar]
- [150].Yingchoncharoen P, Kalinowski DS & Richardson DR Lipid-Based Drug Delivery Systems in Cancer Therapy: What Is Available and What Is Yet to Come. Pharmacol. Rev 68, 701–787 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Hoogenboezem EN & Duvall CL Harnessing albumin as a carrier for cancer therapies. Adv. Drug Deliv. Rev 130, 73–89 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Kim H, Kim Y & Lee J Liposomal formulations for enhanced lymphatic delivery. Asian J. Pharm. Sci 8, 100–109 (2013). [Google Scholar]
- [153].Cai S, Yang Q, Bagby TR & Forrest ML Lymphatic drug delivery using engineered liposomes and solid lipid nanoparticles. Adv. Drug Deliv. Rev 63, 901–908 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Oussoren C & Storm G Liposomes to target the lymphatics by subcutaneous administration. Adv. Drug Deliv. Rev 50, 143–156 (2001). [DOI] [PubMed] [Google Scholar]
- [155].Oussoren C, Zuidema J, Crommelin DJA & Storm G Lymphatic uptake and biodistribution of liposomes after subcutaneous injection. II. Influence of liposomal size, lipid composition and lipid dose. Biochim. Biophys. Acta - Biomembr 1328, 261–272 (1997). [DOI] [PubMed] [Google Scholar]
- [156].Khan AA, Mudassir J, Mohtar N & Darwis Y Advanced drug delivery to the lymphatic system: Lipid-based nanoformulations. Int. J. Nanomedicine 8, 2733–2744 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Videira MA et al. Lymphatic uptake of pulmonary delivered radiolabelled solid lipid nanoparticles. Journal of Drug Targeting vol. 10 607–613 (2002). [DOI] [PubMed] [Google Scholar]
- [158].Kwong B, Gai SA, Elkhader J, Wittrup KD & Irvine DJ Localized Immunotherapy via Liposome-Anchored Anti-CD137 þ IL-2 Prevents Lethal Toxicity and Elicits Local and Systemic Antitumor Immunity. (2013) doi: 10.1158/0008-5472.CAN-12-3343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Kuai R, Ochyl LJ, Bahjat KS, Schwendeman A & Moon JJ Designer vaccine nanodiscs for personalized cancer immunotherapy. Nat. Mater 16, 489–496 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Qian Y et al. Targeting dendritic cells in lymph node with an antigen peptide-based nanovaccine for cancer immunotherapy. Biomaterials 98, 171–183 (2016). [DOI] [PubMed] [Google Scholar]
- [161].Yu X et al. Melittin-lipid nanoparticles target to lymph nodes and elicit a systemic anti-tumor immune response. Nat. Commun 11, 1110 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Tsopelas C & Sutton R Why certain dyes are useful for localizing the sentinel lymph node. J. Nucl. Med 43, 1377–1382 (2002). [PubMed] [Google Scholar]
- [163].Faries MB et al. Active Macromolecule Uptake by Lymph Node Antigen-Presenting Cells: A Novel Mechanism in Determining Sentinel Lymph Node Status. Ann. Surg. Oncol 7, 98–105 (2000). [DOI] [PubMed] [Google Scholar]
- [164].Liu H et al. Structure-based programming of lymph-node targeting in molecular vaccines. Nature 507, 519–522 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Zhu EF et al. Synergistic Innate and Adaptive Immune Response to Combination Immunotherapy with Anti-Tumor Antigen Antibodies and Extended Serum Half-Life IL-2 Cancer Cell Article Synergistic Innate and Adaptive Immune Response to Combination Immunotherapy with Anti-Tumor Antigen Antibodies and Extended Serum Half-Life IL-2. Cancer Cell 27, 489–501 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Moynihan KD et al. Eradication of large established tumors in mice by combination immunotherapy that engages innate and adaptive immune responses. Nat. Med 22, 1402–1410 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Ma L et al. Enhanced CAR-T cell activity against solid tumors by vaccine boosting through the chimeric receptor. Science (80-.). 365, 162–168 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [168].Galluzzi L, Senovilla L, Zitvogel L & Kroemer G The secret ally: Immunostimulation by anticancer drugs. Nat. Rev. Drug Discov 11, 215–233 (2012). [DOI] [PubMed] [Google Scholar]
- [169].Galluzzi L, Buqué A, Kepp O, Zitvogel L & Kroemer G Immunogenic cell death in cancer and infectious disease. Nat. Rev. Immunol 17, 97–111 (2017). [DOI] [PubMed] [Google Scholar]
- [170].Galluzzi L, Buqué A, Kepp O, Zitvogel L & Kroemer G Immunological Effects of Conventional Chemotherapy and Targeted Anticancer Agents. Cancer Cell 28, 690–714 (2015). [DOI] [PubMed] [Google Scholar]
- [171].Alizadeh D & Larmonier N Chemotherapeutic targeting of cancer-induced immunosuppressive cells. Cancer Res. 74, 2663–2668 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [172].Shurin GV, Tourkova IL, Kaneno R & Shurin MR Chemotherapeutic Agents in Noncytotoxic Concentrations Increase Antigen Presentation by Dendritic Cells via an IL-12-Dependent Mechanism. J. Immunol 183, 137–144 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [173].John J et al. Differential effects of Paclitaxel on dendritic cell function. BMC Immunol. 11, (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [174].Zhang L et al. Differential impairment of regulatory T cells rather than effector T cells by paclitaxel-based chemotherapy. Clin. Immunol (2008) doi: 10.1016/j.clim.2008.07.013. [DOI] [PubMed] [Google Scholar]
- [175].Pfannenstiel LW, Lam SSK, Emens LA, Jaffee EM & Armstrong TD Paclitaxel enhances early dendritic cell maturation and function through TLR4 signaling in mice. Cell. Immunol (2010) doi: 10.1016/j.cellimm.2010.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [176].Kim J, Sestito LF, Im S, Kim WJ & Thomas SN Poly(cyclodextrin)-Polydrug Nanocomplexes as Synthetic Oncolytic Virus for Locoregional Melanoma Chemoimmunotherapy. Adv. Funct. Mater 1908788 (2020) doi: 10.1002/adfm.201908788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [177].Bogdan C Nitric oxide synthase in innate and adaptive immunity: an update. Trends Immunol. 36, 161–178 (2015). [DOI] [PubMed] [Google Scholar]
- [178].Zhang XY & Lu WY Recent advances in lymphatic targeted drug delivery system for tumor metastasis. Cancer Biol. Med 11, 247–254 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [179].Xie Y, Bagby TR, Cohen MS & Forrest ML Drug delivery to the lymphatic system: Importance in future cancer diagnosis and therapies. Expert Opin. Drug Deliv 6, 785–792 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [180].Kato S, Shirai Y, Sakamoto M, Mori S & Kodama T Use of a Lymphatic Drug Delivery System and Sonoporation to Target Malignant Metastatic Breast Cancer Cells Proliferating in the Marginal Sinuses. Sci. Rep 9, 1–12 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [181].Tada A, Horie S, Mori S & Kodama T Therapeutic effect of cisplatin given with a lymphatic drug delivery system on false-negative metastatic lymph nodes. Cancer Sci. 108, 2115–2121 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [182].Cheng J et al. Novel Multifunctional Nanoagent for Visual Chemo/Photothermal Therapy of Metastatic Lymph Nodes via Lymphatic Delivery. ACS Omega 5, 3194–3206 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [183].Alishekevitz D et al. Macrophage-Induced Lymphangiogenesis and Metastasis following Paclitaxel Chemotherapy Is Regulated by VEGFR3. Cell Rep. 17, 1344–1356 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [184].Harris AR, Perez MJ & Munson JM Docetaxel facilitates lymphatic-tumor crosstalk to promote lymphangiogenesis and cancer progression. BMC Cancer 18, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [185].Zamora A et al. Paclitaxel induces lymphatic endothelial cells autophagy to promote metastasis. Cell Death Dis. 10, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [186].Tacconi C et al. Activation of the VEGFC/VEGFR3 pathway induces tumor immune escape in colorectal cancer. Cancer Res. 79, 4196–4210 (2019). [DOI] [PubMed] [Google Scholar]
- [187].Bordry N et al. Lymphatic vessel density is associated with CD8 + T cell infiltration and immunosuppressive factors in human melanoma. (2018) doi: 10.1080/2162402X.2018.1462878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [188].Lund AW et al. VEGF-C Promotes Immune Tolerance in B16 Melanomas and Cross-Presentation of Tumor Antigen by Lymph Node Lymphatics. Cell Rep. 1, 191–199 (2012). [DOI] [PubMed] [Google Scholar]
- [189].Špirić Z, Eri Ž & Erić M Lymphatic vessel density and VEGF-C expression as independent predictors of melanoma metastases. J. Plast. Reconstr. Aesthetic Surg 70, 1653–1659 (2017). [DOI] [PubMed] [Google Scholar]
- [190].Filipe V, Que I, Carpenter JF, Löwik C & Jiskoot W In Vivo Fluorescence Imaging of IgG1 Aggregates After Subcutaneous and Intravenous Injection in Mice. Pharm Res. 31, 216–227 (2014). [DOI] [PubMed] [Google Scholar]
- [191].Kijanka G et al. Fate of Multimeric Oligomers, Submicron, and Micron Size Aggregates of Monoclonal Antibodies Upon Subcutaneous Injection in Mice. J. Pharm. Sci (2016) doi: 10.1016/j.xphs.2016.02.034. [DOI] [PubMed] [Google Scholar]
- [192].Yip V et al. Quantitative cumulative biodistribution of antibodies in mice Effect of modulating binding affinity to the neonatal Fc receptor. MAbs 6, 689–696 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [193].Dieterich LC et al. Tumor-Associated Lymphatic Vessels Upregulate PDL1 to Inhibit T-Cell Activation. Front. Immunol 8, 66 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [194].Lane RS et al. IFNγ-activated dermal lymphatic vessels inhibit cytotoxic T cells in melanoma and inflamed skin. J. Exp. Med 215, 3057–3074 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [195].Tewalt EF et al. Lymphatic endothelial cells induce tolerance via PD-L1 and lack of costimulation leading to high-level PD-1 expression on CD8 T cells. Blood 120, 4772–4782 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [196].Fransen MF, van der Sluis TC, Ossendorp F, Arens R & Melief CJ Controlled Local Delivery of CTLA-4 Blocking Antibody Induces CD8+ T-Cell-Dependent Tumor Eradication and Decreases Risk of Toxic Side Effects. Clin Cancer Res 19. [DOI] [PubMed] [Google Scholar]
- [197].Fransen MF, Sluijter M, Morreau H, Arens R & Melief CJM Local Activation of CD8 T Cells and Systemic Tumor Eradication without Toxicity via Slow Release and Local Delivery of Agonistic CD40 Antibody. (2011) doi: 10.1158/1078-0432.CCR-10-2888. [DOI] [PubMed] [Google Scholar]
- [198].van Hooren L et al. Local checkpoint inhibition of CTLA-4 as a monotherapy or in combination with anti-PD1 prevents the growth of murine bladder cancer. Eur. J. Immunol 47, 385–393 (2017). [DOI] [PubMed] [Google Scholar]
- [199].Francis DM et al. Blockade of immune checkpoints in lymph nodes through locoregional delivery augments cancer immunotherapy. Sci. Transl. Med 12, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [200].Kähäri L et al. Transcytosis route mediates rapid delivery of intact antibodies to draining lymph nodes. J. Clin. Invest (2019) doi: 10.1172/JCI125740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [201].Lei C et al. Local release of highly loaded antibodies from functionalized nanoporous support for cancer immunotherapy. J. Am. Chem. Soc 132, 6906–6907 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [202].Li Y et al. Hydrogel dual delivered celecoxib and anti-PD-1 synergistically improve antitumor immunity. Oncoimmunology 5, e1074374 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [203].Wang C et al. In situ activation of platelets with checkpoint inhibitors for post-surgical cancer immunotherapy. Nat. Biomed. Eng 1, (2017). [Google Scholar]
- [204].Wang C, Ye Y, Hochu GM, Sadeghifar H & Gu Z Enhanced Cancer Immunotherapy by Microneedle Patch-Assisted Delivery of Anti-PD1 Antibody. Nano Lett 16, 2334–2340 (2016). [DOI] [PubMed] [Google Scholar]
- [205].Ye Y et al. Synergistic Transcutaneous Immunotherapy Enhances Antitumor Immune Responses through Delivery of Checkpoint Inhibitors. ACS Nano 10, 8956–8963 (2016). [DOI] [PubMed] [Google Scholar]
- [206].Ishihara J et al. Matrix-binding checkpoint immunotherapies enhance antitumor efficacy and reduce adverse events. Sci. Transl. Med 9, (2017). [DOI] [PubMed] [Google Scholar]
- [207].Issa A, Le TX, Shoushtari AN, Shields JD & Swartz MA Vascular endothelial growth factor-C and C-C chemokine receptor 7 in tumor cell-lymphatic cross-talk promote invasive phenotype. Cancer Res. 69, 349–357 (2009). [DOI] [PubMed] [Google Scholar]
- [208].Russo E et al. Intralymphatic CCL21 Promotes Tissue Egress of Dendritic Cells through Afferent Lymphatic Vessels. Cell Rep. 14, 1723–1734 (2016). [DOI] [PubMed] [Google Scholar]
- [209].Vaahtomeri K et al. Locally Triggered Release of the Chemokine CCL21 Promotes Dendritic Cell Transmigration across Lymphatic Endothelia. Cell Rep. 19, 902–909 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [210].Martín-Fontecha A et al. Regulation of dendritic cell migration to the draining lymph node: Impact on T lymphocyte traffic and priming. J. Exp. Med 198, 615–621 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [211].Johnson LA et al. Dendritic cells enter lymph vessels by hyaluronan-mediated docking to the endothelial receptor LYVE-1. Nat. Immunol 18, 762–770 (2017). [DOI] [PubMed] [Google Scholar]
- [212].Güç E et al. Local induction of lymphangiogenesis with engineered fibrin-binding VEGF-C promotes wound healing by increasing immune cell trafficking and matrix remodeling. Biomaterials 131, 160–175 (2017). [DOI] [PubMed] [Google Scholar]
- [213].Tacconi C et al. Antibody-Mediated Delivery of VEGFC Ameliorates Experimental Chronic Colitis. (2019) doi: 10.1021/acsptsci.9b00037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [214].Schwager S et al. Antibody-mediated delivery of VEGF-C potently reduces chronic skin inflammation. JCI insight 3, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [215].Norrmén C, Tammela T, Petrova TV & Alitalo K Biological Basis of Therapeutic Lymphangiogenesis. Circulation 123, 1335–1351 (2011). [DOI] [PubMed] [Google Scholar]
- [216].Tammela T et al. Therapeutic differentiation and maturation of lymphatic vessels after lymph node dissection and transplantation. Nat. Med 13, 1458–1466 (2007). [DOI] [PubMed] [Google Scholar]
- [217].Yoon Y. sup et al. VEGF-C gene therapy augments postnatal lymphangiogenesis and ameliorates secondary lymphedema. J. Clin. Invest 111, 717–725 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [218].Lähteenvuo M et al. Growth factor therapy and autologous lymph node transfer in lymphedema. Circulation 123, 613–620 (2011). [DOI] [PubMed] [Google Scholar]
- [219].Colella P, Ronzitti G & Mingozzi F Emerging Issues in AAV-Mediated In Vivo Gene Therapy. Mol. Ther. - Methods Clin. Dev 8, 87–104 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [220].Stachura J et al. The dual role of tumor lymphatic vessels in dissemination of metastases and immune response development. Oncoimmunology 5, e1182278 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [221].Achen MG, Mann GB & Stacker SA Targeting lymphangiogenesis to prevent tumour metastasis. Br. J. Cancer 94, 1355–1360 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [222].Shields JD et al. Autologous Chemotaxis as a Mechanism of Tumor Cell Homing to Lymphatics via Interstitial Flow and Autocrine CCR7 Signaling. Cancer Cell 11, 526–538 (2007). [DOI] [PubMed] [Google Scholar]
- [223].Lund AW et al. Lymphatic vessels regulate immune microenvironments in human and murine melanoma. J. Clin. Invest 126, 3389–3402 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [224].Shi M, Chen D, Yang D & Liu X-Y CCL21-CCR7 promotes the lymph node metastasis of esophageal squamous cell carcinoma by up-regulating MUC1. J. Exp. Clin. Cancer Res 34, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [225].Ma Q et al. Unexpected contribution of lymphatic vessels to promotion of distant metastatic tumor spread. Sci. Adv 4, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [226].Zheng W, Aspelund A & Alitalo K Lymphangiogenic factors, mechanisms, and applications. J. Clin. Invest 124, 878–887 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [227].Sestito LF & Thomas SN Biomaterials for Modulating Lymphatic Function in Immunoengineering. ACS Pharmacol. Transl. Sci 2, 293–310 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [228].Tammela T et al. Photodynamic Ablation of Lymphatic Vessels and Intralymphatic Cancer Cells Prevents Metastasis. Sci. Transl. Med 3, (2011). [DOI] [PubMed] [Google Scholar]
- [229].Kilarski WW et al. Optimization and regeneration kinetics of lymphatic-specific photodynamic therapy in the mouse dermis. Angiogenesis 17, 347–357 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [230].U.S. National Library of Medicine. Dendritic Cell Activating Scaffold in Melanoma - ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/study/NCT01753089.