

Abstract
Growth hormone (GH) promotes postnatal human growth primarily by regulating insulin-like growth factor (IGF)-I production through activation of the GH receptor (GHR)-JAK2-signal transducer and activator of transcription (STAT)-5B signaling pathway. Inactivating STAT5B mutations, both autosomal recessive (AR) and dominant-negative (DN), are causal of a spectrum of GH insensitivity (GHI) syndrome, IGF-I deficiency and postnatal growth failure. Only AR STAT5B defects, however, confer additional characteristics of immune dysfunction which can manifest as chronic, potentially fatal, pulmonary disease. Somatic activating STAT5B and JAK2 mutations are associated with a plethora of immune abnormalities but appear not to impact human linear growth. In this review, molecular defects associated with STAT5B deficiency is highlighted and insights towards understanding human growth and immunity is emphasized.
Keywords: STAT5B deficiency, IGF-I deficiency, growth hormone insensitivity, JAK2
1. Introduction
The human growth hormone receptor (GHR) relies on the cytosolic JAK2 (Janus kinase 2)-STAT5B (signal transducer and activator of transcription 5B) signaling pathway to transduce signals from the circulating growth hormone (GH), to the IGF1 (insulin-like growth factor-I) gene important for normal human growth. Multiple other pathways are activated by the GHR-JAK2 system, including STAT1, STAT3, STAT5A, the MAPK (mitogen-activated protein kinase) and the PI3K (phosphoinositide-3 kinase) pathways. GHR belongs to the Type 1 cytokine receptor superfamily which includes the prolactin receptor, the erythropoietin receptor, leptin receptor and a number of interleukin receptors. Type 1 cytokine receptors lack intrinsic kinase activities and require the recruitment of one or a combination of the 4-membered JAK family of kinases for initiating signal transduction. For the homo-dimeric GHR, conformational changes are induced upon binding of GH, leading to the trans-activation of the two associated JAK2, one per GHR monomer (1,2). The activated JAK2 phosphorylates the 7 tyrosines located within the intracellular human GHR (3), permitting cytosolic components such as STAT5B, to dock to GHR.
Congenital mutations in many signaling components downstream of the GHR-JAK2 system have been identified, expedited, in part, by increased accessibility of next-generation sequencing in clinical settings. Clinical phenotype associated with inactivating STAT5B mutations (MIM245590) support STAT5B signaling as a key GHR signaling pathway for normal human growth. Patients with molecular STAT5B defects, similar to GHR deficient patients, present with growth hormone insensitivity (GHI) syndrome (MIM262500), severe insulin-like growth factor (IGF)-1 deficiency (IGFD), and profound postnatal growth failure (4). An additional co-morbidity of STAT5B deficiency absent in patients with GHR defects, is immune dysfunction which can potentially be fatal (4). In this review, the spectrum of STAT5B deficiency and insights gained will be discussed. JAK2 defects, which, to date, have been predominantly somatic and associated with immune disorders, will be briefly summarized.
2. JAK2 gain-of-function (GOF) defects are not associated with growth failure
The human JAK2 gene, located on chromosome 9p24.1, encodes for a large multi-domain protein of 1132 amino acid residues. JAK2 carries a N-terminal FERM (4.1, ezrin, radixin, moesin) domain which binds to GHR and other receptors, a SH2 domain that binds STATs, a pseudokinase inhibitory domain and the C-terminal tyrosine kinase domain (Figure 1). Through its FERM domain, JAK2 interacts with the intracellular GHR Box 1 region (Leu298 - Pro304). Insights into how JAK2 is activated by GHR was elegantly presented in studies from Brooks et al (2), who provided evidence supporting pre-formed human homo-dimers of GHR. Brooks and colleagues proposed that, in the absence of the GH ligand, the parallel configuration of the dimeric intracellular GHR domains are “locked” together at the Box 1 regions via the two associated inactivated JAK2. The two JAK2 trans-inhibit each other through apposition of pseudokinase inhibitory domain of one JAK2 with the kinase domain of the other (2). The binding of GH induces conformational changes that force the two JAK2 apart and, with the removal of the trans-inhibition, the juxtaposition of the kinase domains triggers JAK2 auto trans-phosphorylation and subsequent downstream signaling (2). Of note, activated JAK2, depending on the receptor system and ligand, is known to phosphorylate all members of STAT family except for STAT6 (5). In contrast to the human GHR-JAK2 interaction, a recent study suggest, interestingly, that rabbit GHR undergo ligand induced dimerization and associated JAK2 dimerizes through its pseudokinase domain (6).
Figure 1.
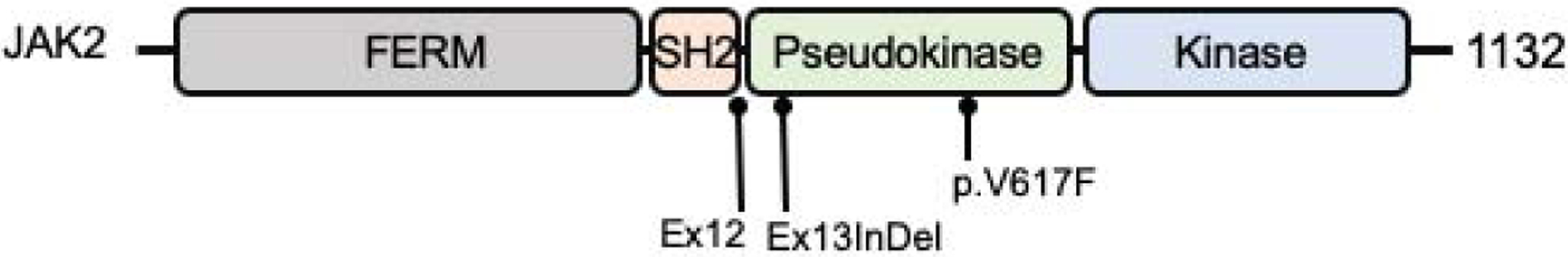
Schematic of the human JAK2 protein. The three most common gain-of-function mutations are indicated. Ex12, exon 12; Ex13InDel, exon13 insertion/deletion; p.V617F, p.Val617Phe. FERM, amino acids 37–380; SH2, amino acids 401–482; Pseudokinase, amino acids 545–808; Kinase, amino acids 849–1124.
To date, somatic and germline dominantly inherited gain-of-function (GOF) JAK2 mutations are well established causes of a number of hematologic disorders (myeloproliferative neoplasma, thrombocythemia-3, thrombocytopenia, polycythemia vera). Recurrent somatic heterozygous mutations include Val617Phe (V617F) located in the pseudokinase domain (exon 14) which was first described in 2005 (7–10), mutations in exon 12 (11), and, more recently, a 4 amino-acid-deletion and variable 1-amino-acid insertion in exon 13 (12) (Figure 1). The V617F is the most prevalent of the JAK2 defects and has also been reported in pediatric cases of essential thrombocythemia (13–15). Described germline inherited GOF variants, similarly located in the pseudokinase domain, include V617I mutation, associated with hereditary thrombocytosis (16), H608N with essential thrombocythemia (17), G571S with clonal hematopoiesis (18) and compound heterozygous E846D and R1063H with hereditary erythrocytosis (19). Stature was not reported in affected patients.
3. STAT5B gene and protein
The human GHR-JAK2 system activates STAT1, STAT3, STAT5A, and STAT5B of the 7 membered family of mammalian STAT proteins, a well-established family of cytosolic proteins that mediates biological actions of multiple growth factors and cytokines in many cell types (20). Availability of next-generation genomic sequencing in clinical settings has led to identification of pathological molecular defects, germline and somatic, in all but the STAT5A genes (21–25). All STAT defects are associated with distinct immuno-deficiencies but only STAT5B deficiency consistently confer GHI, IGF-I deficiency and postnatal growth failure (4).
The human STAT5B gene, on chromosome 17q21.2, is within a ~204 kb DNA region that includes the STAT5A and STAT3 genes. The STAT5B and STAT5A genes (77.23 kb and 24.4 kb, respectively) are only ~11 kb apart (26), suggesting evolutionary duplication of an ancestral gene. The STAT5B protein, 787 amino acid residues, is encoded by 19 exons, while the STAT5A is slightly larger (794 amino acids). The two proteins share a remarkable ~96% amino acid identity, are often considered interchangeable and collectively referred as STAT5. The identification of pathological inactivating STAT5B mutations clearly indicate STAT5B and STAT5A have non-redundant as well as redundant roles. STAT5A, furthermore, is unable to compensate for loss of STAT5B despite their high degree of identity. Human mutations in STAT5A, as noted above, have yet to be identified.
Human STAT5B, typical of members of the STAT family (20), carries discrete protein modules (Figure 2A). The N-terminal domain (ND) and coiled-coiled domain (CCD) mediate protein-protein interactions, with the 4 alpha-helixes of CCD also serving as an unconventional nuclear localization signal (27). The common modular src-homology 2 (SH2) domain is necessary for binding phosphorylated tyrosines (Y), thus permitting STAT5B to dock on phospho-Y of activated receptors and to homo-dimerize when STAT5B itself is phosphorylated at Tyr699 (Y699) by JAK2 (Figure 2B). In the nucleus, the DNA binding domain (DBD) is essential for binding DNA elements and the transcriptional activation domain (TAD) drives transcriptional activities. In addition to Y699, the phosphorylation of S128 and S193 and acetylation of L701, have been reported as other mechanisms for regulating STAT5B activities (28–31). Pathophysiological mutations have been described in all domains except ND (Figure 3).
Figure 2.
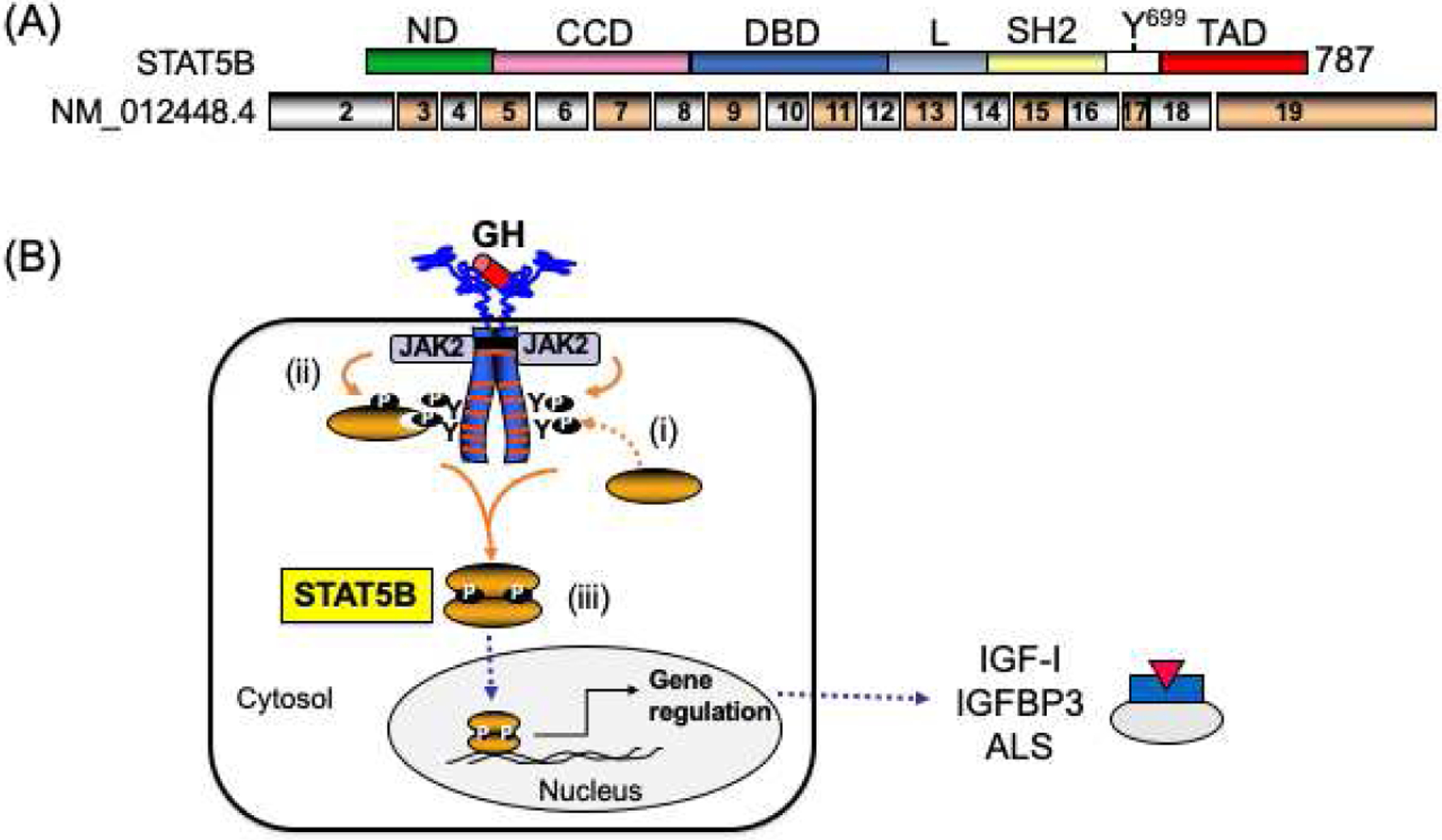
Schematic of the human GH-induced activation of STAT5B. (A) Schematic of the human STAT5B peptide (upper schematic) and encoding exons (lower schematic). The domains indicated: ND, N-terminal domain; CCD, coil-coiled domain; DBD, DNA binding domain; L, linker domain; SH2, Src-homology 2 domain; TAD, transactivation domain. Tyrosine 699 (Y699) that can be phosphorylated by JAK2 and other kinases. (B) Recruitment and activation of STAT5B to the human growth hormone receptor (GHR): (i) Upon GH-GHR interactions, STAT5B docks to 3 of the 7 JAK2-phosphorylated tyrosines (Y) on activated intracellular domain of GHR; (ii) the recruited STAT5B is Tyr699 (Y699) phosphorylated by JAK2; (iii) Phosphorylated STAT5B homo-dimerize, translocate to the nucleus where it binds DNA and transcriptionally regulated genes such as IGF1, IGFBP3 and IGFALS. Broken arrows, translocation process.
Figure 3.
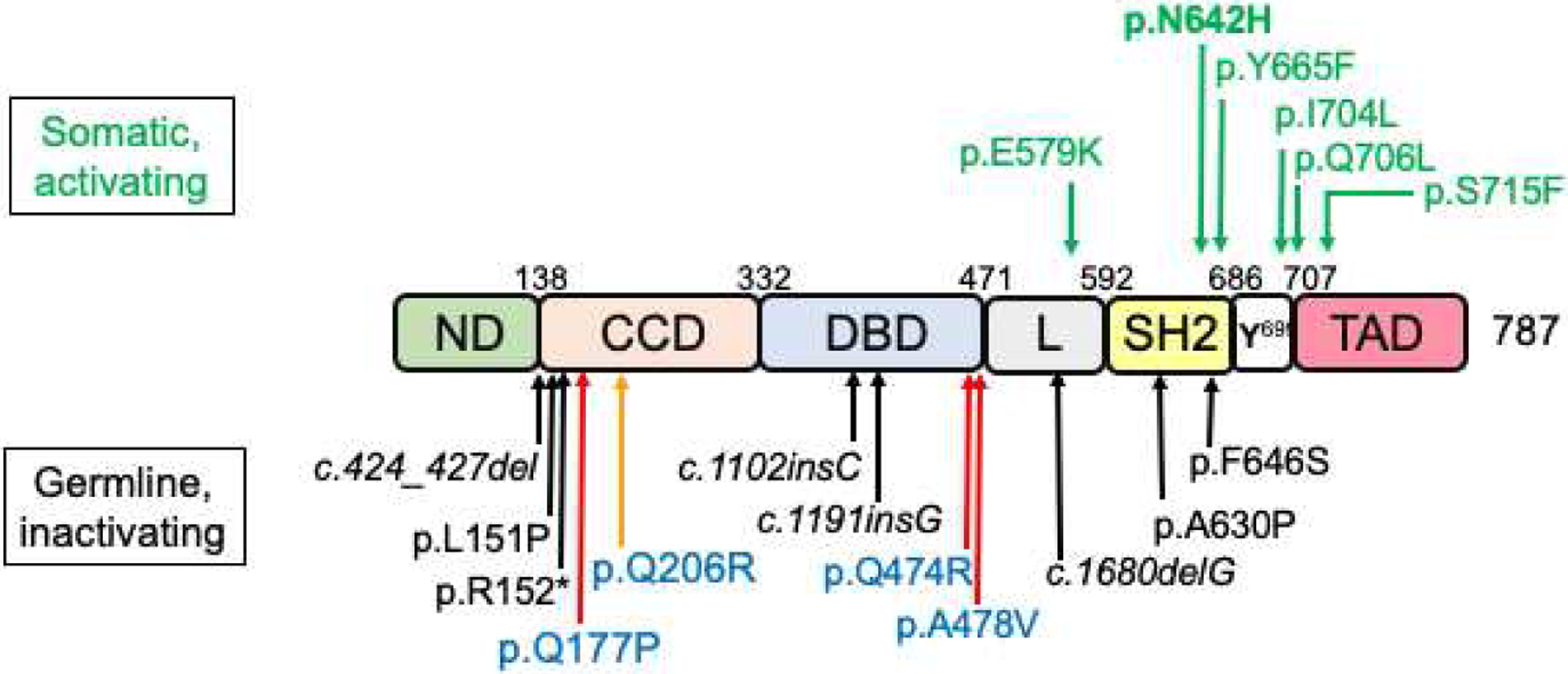
Pathophysiological STAT5B mutations. Schematic of the human STAT5B protein (see Figure 2A for definition of the protein modules). Amino acid numbering is based on recent crystal structure of human STAT5B (47). Germline, inactivating, mutations are indicated below and somatic, activating, mutations above the schematic of STAT5B structure. Black arrows, homozygous; red arrow, dominant-negative; orange arrow, dominant-negative associated with immunity only; green arrows, gain-of-function mutations.
4. STAT5B molecular defects
The binding of GH to cell surface homo-dimeric human GHR activates the associated JAK2 (1,2), which initiate signaling cascades by phosphorylating the 7 tyrosines within the GHR intracellular domain. The docking of STAT5B to any one of three specific phosphorylated GHR tyrosines (Y534, Y566, Y627) suggest a redundancy in GHR tyrosine selection by STAT5B (32). This redundancy may explain why damaging GHR mutations in the intracellular domain, including dominant-negative GHR mutations (33–38), frequently involve frameshifts that abrogate the JAK2 binding site and/or the three critical GHR tyrosines (39–42). The sequence of events, upon STAT5B docking to GHR, involves STAT5B Y699 phosphorylation by JAK2, homo-dimerization of phosphorylated STAT5B which then mobilize to the nucleus to drive transcriptional activities (Figure 2B). Mutations in STAT5B disrupting this sequence of events are described below.
(a). Homozygous Loss-of-Function (LOF) STAT5B defects
All loss of function (LOF) mutations in STAT5B described to date are germline. The identification of these congenital inactivating STAT5B mutations associated with GHI, IGF-I deficiency and severe growth failure, provided definitive evidence that STAT5B signaling is key for normal GH-GHR-JAK2 mediated postnatal growth (Table 1; (43,44)). The first homozygous inactivating STAT5B mutation was a missense mutation, p.Ala630Pro (p.A630P), reported in 2003. The 16 year old female subject, from a consanguineous pedigree, had normal GHR but had clinical features that were reminiscent of patients with GHR defects (45). Additional features of chronic pulmonary disease were reported (45). The autosomal recessive (AR) p.A630P mutation, located in the SH2 domain of STAT5B, disrupted the core anti-parallel ß-sheets that forms the pocket for binding phosphate groups on activated receptors and other proteins (46–48). The amino acid substitution caused thermodynamic instability of both the SH2 domain and the entire protein (49,50). Mutant STAT5B p.A630P was thus very poorly detected and could not be activated in either primary cells from the patient (45,51) or in reconstituted systems (52). Conclusions drawn from these cell-based analyses were consistent with in vivo clinical IGF deficiency and GHI presentations, supporting the hypothesis that the presence of functional STAT5A (52) could not compensate for loss of functional STAT5B.
Table 1.
Genetic and clinical phenotype of STAT5B deficient patients
| STAT5B Exon | cDNA | Protein | Domain | Sex | Age (yr) | Height SDS | Birth | GHI | IGFD | Prolactin elevated | Hypergamma-globulinemia | T-cell lymphopenia | Pulmonary Disease | Reference |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Homozygous | ||||||||||||||
| 5 | c.424_427del | p.Leu142Argfs*20 | CCD | M | 6 | −5.6 | AGA | +++ | +++ | +++ | + | +++ | +++ | (54)# |
| 5 | c.424_427del | p.Leu142Arqfs*20 | CCD | M | 2 | −3.0 | AGA | +++ | +++ | +++ | No | +++ | +++ | (54)# |
| 5 | c.452T>C | p.Leu151 Pro | CCD | M | 8 | −3.2 | AGA | +++ | +++ | Normal | No | No | No | (60) |
| 5 | c.454C>T | p.Arg152* | CCD | F | 15.3 | −9.9 | NA | ND | ND | +++ | +++ | +++ | +++ | (57) |
| 5 | c.454C>T | p.Arq152* | CCD | F | 12 | −5.3 | SGA | +++ | +++ | Normal | N0 | No | +++ | (58) |
| 9 | c.1102insC | p.Gln368Profs*9 | DBD | M | 31 | −5.9 | AGA | +++ | +++ | +++ | No | No | No | (56) |
| 10 | c.1191insG | p.Asn398Glufs*16 | DBD | F | 16.4 | −7.8 | AGA | +++ | +++ | ND | ND | ND | +++ | (55) |
| 13 | c.1680delG | p.Glu561Arqfs*17 | Linker | F | 4 | −5.6 | AGA | +++ | +++ | ND | ND | ND | + | (53)$ |
| 13 | c.1680delG | p.Glu561Argfs*17 | Linker | F | 2 | −5.8 | AGA | +++ | +++ | ND | ND | ND | + | (53)$ |
| 15 | c.1888G>C | p.Ala630Pro | SH2 | F | 16.5 | −7.5 | AGA | +++ | +++ | +++ | +++ | +++ | +++ | (45) |
| 16 | c.1937T>C | p.Phe646Ser | SH2 | F | 14.8 | −5.95 | NA | +++ | +++ | +++ | +++ | +++ | No | (59) |
| Dominant-negative | ||||||||||||||
| 5 | c.530A>C | p.Gln177Pro | CCD | M | 14.5 | −5.3 | AGA | ++ | ++ | Normal | No | No | No | (65) |
| 12 | c.1421A>G | p.Gln474Arq | DBD | F | 12.8 | −4.5 | AGA | ++ | No | +++ | No | No | No | (65) |
| 12 | c.1433C>T | p.Ala478Val | DBD | M | 1.8 | −2.9 | AGA | ++ | +++ | Normal | No | No | No | (65) |
+ to +++, increasing severity of indications.
AGA, appropriate for gestational age; ND, not determined
siblings
siblings
Seven other homozygous inactivating STAT5B mutations have since been reported, involving 4 male and 6 female GHI patients and includes 2 sets of siblings (53–60). These rare AR STAT5B mutations suggest that STAT5B haploinsufficiency minimally perturbed IGF-I expression, growth, and immune functions (43). The molecular defects, located in different domains of the STAT5B protein (Table 1; Figure 3). The one nonsense (57) and 4 frameshift (53–56) mutations are predicted to be pathogenic as a result of early protein termination. In addition to p.A630P, two other missense mutations, p.F646S in the SH2 domain (45,59) and p.L151P in CCD (60), were functionally compromised, consistent with clinical phenotype. All STAT5B LOF mutations were ultimately unable to drive transcription. It remains to be determined if predicted truncated peptides are expressed in vivo.
In humans, the inability of STAT5A to compensate for loss of STAT5B is of note and is in contrast to mouse germline Stat5b−/− knock-out models (61,62). Sexual dimorphic growth patterns were observed in Stat5b−/− knock-out mice, with both male Stat5b−/− and female Stat5b−/− mice comparable to size of wild-type Stat5b+/+ females, all of whom were smaller than wild-type Stat5b+/+ males. Growth of Stat5a−/− mice were consistently normal (62,63). Only Stat5a−/− and Stat5b−/− double knock-out mouse models (62) conferred the severity of growth failure, IGF-I deficiency, and immune dysregulation observed in the human STAT5B deficiency state. Interestingly, a recent study provided evidence that, in mice, Stat5b was the dominant paralog over Stat5a, and, further, that this was likely a consequence of higher expression of Stat5b compared to Stat5a rather than functional differences (64), a hypothesis first proposed over 20 years ago (62). Whether similar asymmetric expressions could account for the severe co-morbidities in clinical STAT5B deficiency remain unresolved. Clearly, the loss of functional STAT5B has significant pathophysiological consequences for humans not observed in mouse Stat5b−/− models.
(b). Dominant-negative LOF STAT5B defects
STAT5B deficiency is established as an autosomal recessive disorder. The first germline dominant-negative inactivating STAT5B mutations were recently reported in 3 unrelated patients who exhibited GHI, IGF-I deficiency and post-natal growth deficits less severe than those with “classical” STAT5B deficiency (65). Whole exome sequencing analysis confirmed that, in each case, the heterozygous STAT5B variant was the top candidate. Each of the LOF mutation was a missense variant, with the two familial mutations, p.Q474R (p.Gln474Arg, exon 12) and p.A478V (p.Ala478Val) located in the DBD, and the one de novo mutation, p.Q177P (p.Gln177Pro, exon 5) located in the CCD (Table 1, Figure 3).
In vitro functional studies demonstrated that all three expressed mutants were robustly tyrosine phosphorylated upon GH-stimulation. However, the two DBD mutations were unable to bind DNA, while the p.Q177P was impaired in nuclear localization, the first reported STAT variant with a nuclear localization defect (65). The Q177P substitution in the first alpha-helix of the CCD, surprisingly, did not disrupt protein structure although prolines are known to be incompatible with alpha-helix structures. Instead, the functional integrity of the 4-helix bundle of CCD was disrupted (27). The p.Q474R and p.A478V mutations, both in highly conserved amino acid residues, are in positions analogous to dominant-negative STAT3 DBD missense mutations (66–68) associated with hyper-IgE syndrome (MIM 147060) (68,69). Importantly, each of the STAT5B mutant retained the capability to interact with wild-type STAT5B and inhibit normal dimeric STAT5B functions (65).
One dominant-negative STAT5B mutation, p.Gln206Arg (p.Q206R, exon 5, alpha-helix 2, CCD) was recently reported in a 33 yr old male patient with severe autoimmune lymphoproliferative syndrome-like features from childhood (70) but whose height was not reported. The mutation was inherited from his severely immunocompromised mother. The p.Q206R was shown to translocate to the nucleus upon IL-2 stimulation and exhibited dominant negative effects on IL-2-stimulated T-cell functions. It remains unclear if the severity of immune dysregulation could be due solely to the heterozygous STAT5B p.Q206R.
It is of note that three AR STAT5B mutations (Table 1), similar to p.Q177P, disrupt the first alpha-helix of CCD (exon 5). The frameshift c.424_427del (p.L142Rfs*20) (54) and nonsense p.R152* (57) mutations are clearly pathogenic and likely result in total STAT5B deficiency. The recently described missense p.L151P mutation was found to be normally expressed in patient B cells but IL2-induced phosphorylation of peripheral blood mononuclear cells was blunted (60). Whether the STAT5B p.L151P can translocate to the nucleus is unknown nor is potential dominant-negative effects known, as the growth phenotype of the carrier parents and the one sibling who was wild-type for STAT5B, were not reported. Nevertheless, it is striking that the first alpha-helix of the CCD, encoded by exon 5, appears to be vulnerable to molecular disruptions.
The rarity of pathological defects in STAT5B has heighten awareness of heterozygous STAT5B variants identified in genomic screens of patients with milder GHI and diagnosed with idiopathic short stature (ISS). Two such variants, however, were proven to be functionally benign (p.V498M, exon 13, Linker, (71); and p.E315A, exon 8, CCD (72)). A third recently reported heterozygous variant, p.K632N (exon 15, SH2) was functionally inert when evaluated in homozygous state but did not display dominant-negative effects, raising questions of the contribution of this heterozygous variant to clinical phenotype (73). As part of a genetic analysis of a cohort of 89 patients with ISS, a heterozygous p.T479A (exon 12, DBD) was reported in one patient with a height SDS of −3.56 but pathogenicity remains unclear (74). In each of these cases, it is possible that digenic or oligogenic effects may explain the short stature phenotype of the patient.
(c). Somatic Gain-of-Function (GOF) STAT5B defects
To date, GOF STAT5B mutations have been somatic and are associated with hematological malignancies, predominantly of T-cell origin, with recurring mutational “hot spots” in the SH2 and TAD modules (47). The p.N642H mutation, first reported in 2015 (75–77), is the most common GOF STAT5B mutation and has now been detected in over 150 patients. Recent crystallography studies of the human STAT5B core regions (CCD-DBD-Linker-SH2, amino acid residues A136 – Q703), comparing wild-type and p.N642H structures, suggested the STAT5BN642H can adopt a hyper-activated and hyper-inactivated state which enhanced affinity for self-dimerization and is resistance to dephosphorylation (47,48). Other reported GOF are schematically shown in Figure 3.
5. STAT5B deficiency: spectrum of GHI and severe postnatal short stature
The post-natal growth impairment associated with STAT5B deficiency is remarkably similar to those of patients carrying GHR defects (65). Severe height deficits as consequences of AR STAT5B deficiency are comparable to AR GHR deficiency (78) (Figure 4). For the 11 subjects carrying DN STAT5B mutations (65), the mean height SDS strikingly resembled subjects carrying dominant-negative GHR mutations (33,35–38,79) and subjects carrying GHR pseudo-exon 6 (80–82).
Figure 4.
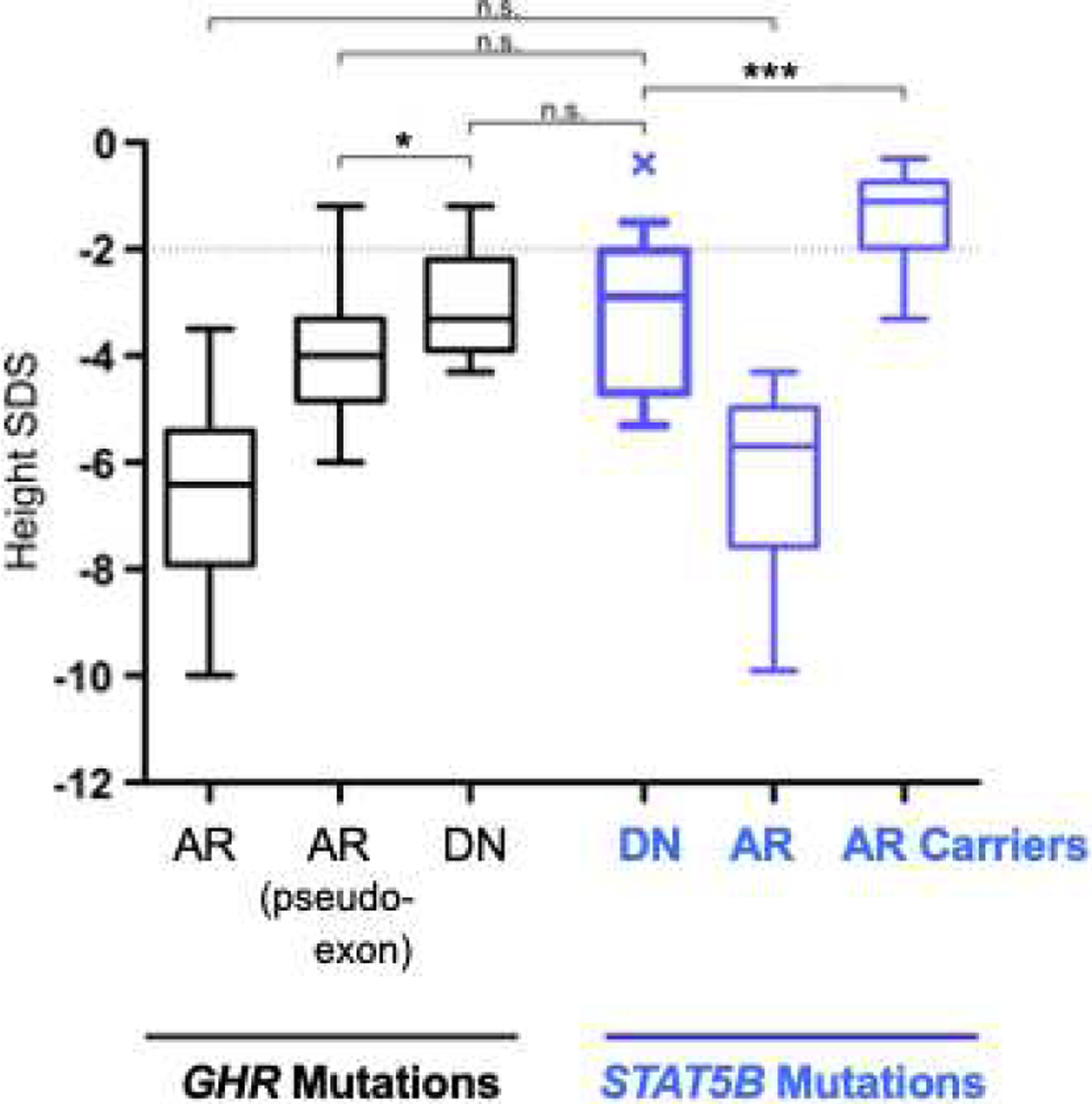
Height of STAT5B deficient patients are comparable to GHR deficient patients. Height SDS values of GHI patients with GHR mutations (black) were compared with height SDS values of STAT5B mutation carriers (blue). Growth Hormone Receptor (GHR) Defects: AR, Autosomal Recessive GHR mutations (n=100); AR pseudoexon, GHR pseudoexon 6Ψ mutations (n=21); DN, Dominant Negative GHR mutations (n=16). STAT5B Defects: DN, Dominant Negative STAT5B mutations (n=11); AR, Autosomal Recessive STAT5B mutations (n=10); AR Carriers, STAT5B mutation carriers (n=14). Box (median, 25th and 75th percentiles) and whiskers (minimum and maximum values) plots; statistical analysis by Student’s t-test, * P<0.05; *** P<0.001; n.s., not significant. (Reproduced with permission from Klammt J, et al, Nat Commun 2018; 9:2105)
Birth size, where documented (both AR and DN STAT5B), was normal for gestation (Table 1), indicating that STAT5B, similar to GHR, is not essential for in utero growth. It was notable, however, that 6 out of 9 subjects carrying homozygous STAT5B, and 1 out 2 carrying DN-STAT5B, were documented to be born before 37 weeks of gestation. Postnatal growth failure, as noted (Table 1), was significant and consistent with IGF deficiency. At first report, height SDS of reported probands ranged from −2.9 SDS to −9.9 (Table 1). Bone age, when measured, was significantly delayed (54,57,65). Puberty was also consistently delayed (Table 1), reflective of the low levels of circulating IGF-I. Mild facial dysmorphic features, such as a prominent forehead, depressed nasal bridge and high-pitched voice, were noted for some of the subjects carrying AR STAT5B mutations (45,54,57).
For subjects carrying AR STAT5B mutations, basal GH levels were normal (ranging from 0.1 to 17.6ng/ml), and when stimulated, GH concentrations were often elevated (4.0 – 53.8 ng/ml). Serum IGF-I, IGFBP-3 and ALS concentrations in all cases were abnormally low, and remained low after GH treatment, during IGF-I generation tests (45,55,83) or during GH therapy (54,56). Some of the subjects underwent growth hormone therapy (1yr to 4yr), but growth response was uniformly poor (45,54). Recombinant human IGF-I, a therapeutic option to by-pass blockage in GH signaling, had limited efficacy in one treated AR STAT5B proband (54) and two of the DN STAT5B probands (65). Interestingly, serum prolactin levels, when recorded, were abnormally high in majority of AR STAT5B subjects (Table 1). It is likely that the STAT5B mutations disrupted the negative feedback loop for PRL production, although the mechanisms remain to be clarified.
Collectively, clinical observations of AR and DN STAT5B deficient patients fully supported the critical importance of the GHR-STAT5B signaling pathway for post-natal linear growth and IGF-I production in response to GH. The identification of DN STAT5B mutations, moreover, has expanded the spectrum of growth impairments from severe (AR mutations) to mild (DN STAT5B).
6. Impaired Immune Functions associated with STAT5B deficiency
A distinguishing feature in the patients carrying STAT5B mutations is symptoms of immune dysfunction with potential fatal pulmonary involvement (Table 1). Shared symptoms amongst the 11 AR STAT5B deficient probands include severe eczema (9 out of 9), auto-immune thyroiditis (7 out of 11), chronic pulmonary disease (8 out of 11) manifesting as early as the first year of life (54,57,58), and confirmed lung fibrosis and/or lymphoid interstitial pneumonia (LIP) (7 out of 9), a condition of unknown etiology that is rare in children and associated with autoimmune disease (84). In the severest cases, corticosteroid and oxygen treatments temporarily stabilize worsening pulmonary functions. Only one patient, a 17.5 year old male, has been reported to undergo a lung transplantation which appeared to have alleviated impaired pulmonary function and the requirement for oxygen (54). Overall, however, long-term prognosis for AR STAT5B deficiency is poor. The first described patient, carrying p.Ala630Pro (45), succumbed and died before the age of 30 years as consequences of progressive pulmonary fibrosis and respiratory failure (85); not officially reported, four other AR STAT5B deficient patients have since succumbed.
In subjects carrying DN STAT5B mutations, eczema was milder and other extrapulmonary disease was absent or transient, except in one proband diagnosed with thyroiditis and celiac disease (65). In general, immune dysregulation was milder, with elevated IgE concentrations detected in 8 of 9 DN STAT5B deficient patients. The immune profiles of the 3 probands were otherwise normal (65). Elevated IgE concentrations was also detected in 5 of 8 evaluated AR STAT5B deficient patients (4,60,85,86).
STAT5B mutations has been accepted by the International Union of Immmunological Societies as causal of human inborn error of immunity, with the most recent update in 2019 (87). Unlike the endocrine profiles which showed an absolute association between STAT5B mutations and IGF deficiency, abnormalities in the immunological profiles, where evaluated, were variable, even between siblings who carried the same homozygous STAT5B mutation (54,88).
The immune complications of AR STAT5B deficiency have been attributed, in part, to T-lymphopenia, with both CD4+ and CD8+ T-cells often below normal (51,54,57,59,85). Although multiple cytokines are known to activate STAT5 (89), specific roles of STAT5B and STAT5A are not always delineated. STAT5B defects have highlighted some key pathways in which STAT5B play a more significant role (51). Notably, in STAT5B deficiency, dysregulated IL-2 signaling significantly reduced T-regulatory cells (Treg) (45,51,90), which are a subset of CD4+ cells essential for the propagation and homeostasis of T-cell populations (91). In human Tregs, STAT5B was shown to be important for regulating expression of CD25, the α-subunit of the heterotrimeric IL-2 receptor (IL2R) complex, and for regulating the transcription factor, FOXP3 (51). In AR STAT5B deficient patients, Treg+FOXP3+ are abnormally low (45,51,90). Specifically, peripheral, naïve Treg subsets (CD45RA+) were noted to be low, while dividing memory Treg subsets (CD45RO+) were abnormally elevated and had reduced suppressive functionality. This skewing of Treg sub-populations likely contributed to the autoimmune phenotypes of STAT5B deficient patients (90). Further, the increased susceptibility to opportunistic infections experienced by STAT5B deficient patients could also be related to decreased CD25 on all T cells, as has been reported in patients with severe CD25 deficiency (92).
Recent evidence indicated the association of two of the AR STAT5B mutations, c.1680delG and p.F646S, with impaired maturation and cytolytic actions of NK (natural killer) cells (88,93). NK cells are early effectors against viral infections, pathogens and malignant cells, and dysregulation could, therefore, contribute to the susceptibility of STAT5B patients to infections, including specific viral infections such as Herpes Zoster (reported in a number of the patients). The majority of AR STAT5B deficient patients display low-normal NK cell numbers, although functional integrity has yet to be analyzed.
A recent follow-up report of siblings carrying AR STAT5B c.1680delC (Table 1), first reported in 2007 (53), demonstrated that initial indications of pulmonary disease in early childhood, has progressed in severity (88). It remains unclear how immune dysfunction led to pulmonary insufficiencies observed in AR STAT5B deficiency. Intriguingly, three of the reported AR STAT5B patients lacked severe immune and/or pulmonary problems (56,59,60). Immune dysfunction were variably detected, with the male patient carrying STAT5B c.1102insC (56) presenting Treg+FOXP3+ that were ~75% of normal healthy control (90), but, otherwise, appeared relatively healthy at 31 yrs when first reported (56). The female patient carrying STAT5B p.F646S (59) who had autoimmune thyroiditis, psoriasis, alopecia, and was diagnosed with Celiac disease at age 20 yr, presented with progressive immune dysregulation but no pulmonary disease at the time of report (85). The AR STAT5B p.L151P patient, a 15 year old male, who similarly has severe eczema and autoimmune disease, lacked pulmonary issues (60). An explanation for the lack of chronic pulmonary disease in these three patients remains to be elucidated.
Perspective and Future Directions
The continued identification of patients with unequivocal defects in the GH-induced STAT5B signaling has advanced our understanding of the molecular basis of growth failure associated with primary IGF deficiency. In addition to AR inactivating STAT5B mutations, recent reports of DN STAT5B has broadened the spectrum of STAT5B deficiency. Collectively, in humans, the presence of STAT5A cannot compensate for the loss of STAT5B in terms of GH-dependent growth and IGF-I production. The GHI and IGF-I deficiency but mild immune dysregulation in DN STAT5B deficient subjects, furthermore, support the hypothesis that STAT5B-mediated growth functions can be delineated from its immune functions. Mechanism of how STAT5B regulate IGF-I production in humans still need to be fully elucidated. Currently, there is a lack of a unifying mechanism to explain the complex immune-related pathologies observed in STAT5B deficiency, although dysregulated Treg+FOXP3+ likely contributes to autoimmunity and increased susceptibility to infections. New evidence of dysregulated NK cells (88,93) highlight the importance of investigating the contribution of different immune cell populations to clinical immune pathology associated with STAT5B deficiency. Understanding the mechanisms leading to progressive immune dysfunction and whether this process precede fatal pulmonary insufficiency, is essential to preventing mortality associated with STAT5B deficiency.
Therapeutic options to improve poor linear growth and immunodeficiency for STAT5B deficiency remain extremely limited. For immune dysregulation, a combination of steroids with either cyclosporine A (cell cycle inhibitor), tacrolimus (calcineurin inhibitor) or sirolimus (mTOR inhibitor) may provide temporary sparing from T-cell dysregulation as have been shown for patients with IPEX (Immune Dysregulation, Enteropathy, X-linked) and IPEX-like syndrome (94). A lung transplantation appeared to have, at least temporarily, alleviated problems caused by chronic pulmonary disease (54). Bone marrow transplants is currently being considered for STAT5B deficiency. For linear growth, efficacy of recombinant human IGF-I treatment remains uncertain, as linear growth response was poor in one treated AR STAT5B deficient patient (54) and two DN STAT5B deficient subjects (65).
The current application of next-generation genomic sequencing will likely expedite identification of new STAT5B defects, both germline and somatic. Early genetic detection of germline LOF STAT5B mutations and functional evaluation of the defects are essential towards improving understanding the pathophysiology of STAT5B deficiency and crucial for improving patient management.
Acknowledgement:
Funding from Eunice Kennedy Shriver National Institute of Child Health & Human Development of the National Institutes of Health R21HD098417 (V.H.)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Brown RJ, Adams JJ, Pelekanos RA, Wan Y, Mckinstry WJ, Palethorpe K, Seeber RM, Monks TA, Eidne KA, Parker MW, Waters MJ. Model for growth hormone receptor activation based on subunit rotation within a receptor dimer. Nat Struct Mol Biol. 2005;12(9):814–821. [DOI] [PubMed] [Google Scholar]
- 2.Brooks AJ, Dai W, O’Mara ML, Abankwa D, Chhabra Y, Pelekanos RA, Gardon O, Tunny KA, Blucher KM, Morton CJ, Parker MW, Sierecki E, Gambin Y, Gomez GA, Alexandrov K, Wilson IA, Doxastakis M, Mark AE, Waters MJ. Mechanism of activation of protein kinase JAK2 by the growth hormone receptor. Science. 2014;344(6185):1249783. [DOI] [PubMed] [Google Scholar]
- 3.Derr MA, Fang P, Sinha SK, Ten S, Hwa V, Rosenfeld RG. A novel Y332C missense mutation in the intracellular domain of the human growth hormone receptor (GHR) does not alter STAT5b signaling: redundancy of GHR intracellular tyrosines involved in STAT5b signaling. Horm Res. 2011;75(3):187–199. [DOI] [PubMed] [Google Scholar]
- 4.Hwa V, Nadeau K, Wit JM, Rosenfeld RG. STAT5b deficiency: lessons from STAT5b gene mutations. Best Pract Res Clin Endocrinol Metab. 2011;25:61–75. [DOI] [PubMed] [Google Scholar]
- 5.Saltzman A, Stone M, Franks C, Searfoss G, Munro R, Jaye M, Ivashchenko Y. Cloning and characterization of human Jak-2 kinase: high mRNA expression in immune cells and muscle tissue. Biochemical and biophysical research communications. 1998;246(3):627–633. [DOI] [PubMed] [Google Scholar]
- 6.Wilmes S, Hafer M, Vuorio J, Tucker JA, Winkelmann H, Lochte S, Stanly TA, Pulgar Prieto KD, Poojari C, Sharma V, Richter CP, Kurre R, Hubbard SR, Garcia KC, Moraga I, Vattulainen I, Hitchcock IS, Piehler J. Mechanism of homodimeric cytokine receptor activation and dysregulation by oncogenic mutations. Science. 2020;367(6478):643–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kralovics R, Passamonti F, Buser AS, Teo S-S, Tiedt R, Passweg JR, Tichelli A, Cazzola M, Skoda RC. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med. 2005;352:1779–1790. [DOI] [PubMed] [Google Scholar]
- 8.Baxter EJ, Scott LM, Campbell PJ, East C, Fourouclas N, Swanton S, Vassiliou GS, Bench AJ, Boyd EM, Curtin N, Scott MA, Erber WN, Project tCG, Green AR. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet. 2005;365:1054–1061. [DOI] [PubMed] [Google Scholar]
- 9.James C, Ugo V, le Couedic J-P, Staerk J, Delhommeau F, Lacout C, Garcon L, Raslova H, Berger R, Bennaceur-Griscelli A, Villeval JL, Constantinescu SN, Casadevall N, Vainchenker W. A unique cloncal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005;434:1144–1148. [DOI] [PubMed] [Google Scholar]
- 10.Tefferi A, Lasho TL, Gilliland G. JAK2 mutations in myeloproliferative disorders. N Engl J Med. 2005;353(13):1416–1417; author reply 1416–1417. [DOI] [PubMed] [Google Scholar]
- 11.Scott LM, Tong W, Levine RL, Scott MA, Beer PA, Stratton MR, Futreal PA, Erber WN, McMullin MF, Harrison CN, Warren AJ, Gilliland DG, Lodish HF, Green AR. JAK2 exon 12 mutations in polycythemia vera and idiopathic erythrocytosis. N Engl J Med. 2007;356(5):459–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Patel AB, Franzini A, Leroy E, Kim SJ, Pomicter AD, Genet L, Xiao M, Yan D, Ahmann JM, Agarwal AM, Clair P, Addada J, Lambert J, Salmon M, Gleich GJ, Cross NCP, Constantinescu SN, O’Hare T, Prchal JT, Deininger MW. JAK2 ex13InDel drives oncogenic transformation and is associated with chronic eosinophilic leukemia and polycythemia vera. Blood. 2019;134(26):2388–2398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nakatani T, Imamura T, Ishida H, Wakaizumi K, Yamamoto T, Otabe O, Ishigami T, Adachi S, Morimoto A. Frequency and clinical features of the JAK2 V617F mutation in pediatric patients with sporadic essential thrombocythemia. Pediatr Blood Cancer. 2008;51(6):802–805. [DOI] [PubMed] [Google Scholar]
- 14.Randi ML, Geranio G, Bertozzi I, Micalizzi C, Ramenghi U, Tucci F, Notarangelo LD, Ladogana S, Menna G, Giordano P, Consarino C, Farruggia P, Zanazzo GA, Fiori GM, Burnelli R, Russo G, Jankovich M, Peroni E, Duner E, Basso G, Fabris F, Putti MC. Are all cases of paediatric essential thrombocythaemia really myeloproliferative neoplasms? Analysis of a large cohort. Br J Haematol. 2015;169(4):584–589. [DOI] [PubMed] [Google Scholar]
- 15.Randi ML, Putti MC, Scapin M, Pacquola E, Tucci F, Micalizzi C, Zanesco L, Fabris F. Pediatric patients with essential thrombocythemia are mostly polyclonal and V617FJAK2 negative. Blood. 2006;108(10):3600–3602. [DOI] [PubMed] [Google Scholar]
- 16.Mead AJ, Rugless MJ, Jacobsen SE, Schuh A. Germline JAK2 mutation in a family with hereditary thrombocytosis. N Engl J Med. 2012;366(10):967–969. [DOI] [PubMed] [Google Scholar]
- 17.Rumi E, Harutyunyan AS, Casetti I, Pietra D, Nivarthi H, Moriggl R, Cleary C, Bagienski K, Astori C, Bellini M, Berg T, Passamonti F, Kralovics R, Cazzola M. A novel germline JAK2 mutation in familial myeloproliferative neoplasms. Am J Hematol. 2014;89(1):117–118. [DOI] [PubMed] [Google Scholar]
- 18.Milosevic Feenstra JD, Nivarthi H, Gisslinger H, Leroy E, Rumi E, Chachoua I, Bagienski K, Kubesova B, Pietra D, Gisslinger B, Milanesi C, Jager R, Chen D, Berg T, Schalling M, Schuster M, Bock C, Constantinescu SN, Cazzola M, Kralovics R. Whole-exome sequencing identifies novel MPL and JAK2 mutations in triple-negative myeloproliferative neoplasms. Blood. 2016;127(3):325–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kapralova K, Horvathova M, Pecquet C, Fialova Kucerova J, Pospisilova D, Leroy E, Kralova B, Milosevic Feenstra JD, Schischlik F, Kralovics R, Constantinescu SN, Divoky V. Cooperation of germ line JAK2 mutations E846D and R1063H in hereditary erythrocytosis with megakaryocytic atypia. Blood. 2016;128(10):1418–1423. [DOI] [PubMed] [Google Scholar]
- 20.Levy DE, Darnell JE, Jr. STATs: transcriptional control and biological impact. Nat Rev Mol Cell Biol. 2002;3:651–662. [DOI] [PubMed] [Google Scholar]
- 21.Casanova J-L, Holland SM, Notarangelo LD. Inborn Errors of Human JAKs and STATs. Immunity. 2012;36:515–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.O’Shea JJ, Holland SM, Staudt LM. JAKs and STATs in immunity, immunodeficiency, and cancer. N Engl J Med. 2013;368(2):161–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hambleton S, Goodbourn S, Young DF, Dickinson P, Mohamad SM, Valappil M, McGovern N, Cant AJ, Hackett SJ, Ghazal P, Morgan NV, Randall RE. STAT2 deficiency and susceptibility to viral illness in humans. Proc Natl Acad Sci U S A. 2013;110(8):3053–3058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yildiz M, Li H, Bernard D, Amin NA, Ouillette P, Jones S, Saiya-Cork K, Parkin B, Jacobi K, Shedden K, Wang S, Chang AE, Kaminski MS, Malek SN. Activating STAT6 mutations in follicular lymphoma. Blood. 2015;125(4):668–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schimke LF, Hibbard J, Martinez-Barricarte R, Khan TA, de Souza Cavalcante R, Borges de Oliveira Junior E, Takahashi Franca T, Iqbal A, Yamamoto G, Arslanian C, Feriotti C, Costa TA, Bustamante J, Boisson-Dupuis S, Casanova JL, Marzagao Barbuto JA, Zatz M, Poncio Mendes R, Garcia Calich VL, Ochs HD, Torgerson TR, Cabral-Marques O, Condino-Neto A. Paracoccidioidomycosis Associated With a Heterozygous STAT4 Mutation and Impaired IFN-gamma Immunity. J Infect Dis. 2017;216(12):1623–1634. [DOI] [PubMed] [Google Scholar]
- 26.Ambrosio R, Fimiani G, Monfregola J, Sanzari E, De Felice N, Salerno MC, Pignata C, D’Urso M, Ursini MV. The structure of human STAT5A and B genes reveals two regions of nearly identical sequence and an alternative tissue specific STAT5B promoter. Gene. 2002;285(1–2):311–318. [DOI] [PubMed] [Google Scholar]
- 27.Shin HY, Reich NC. Dynamic trafficking of STAT5 depends on an unconventional nuclear localization signal. J Cell Sci. 2013;126(Pt 15):3333–3343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Beausoleil SA, Villen J, Gerber SA, Rush J, Gygi SP. A probability-based approach for high-throughput protein phosphorylation analysis and site localization. Nat Biotechnol. 2006;24:1285–1292. [DOI] [PubMed] [Google Scholar]
- 29.Daub H, Olsen JV, Bairlein M, Gnad F, Oppermann FS, Korner R, Greff Z, Keri G, Stemmann O, Mann M. Kinase-selective enrichment enables quantitative phosphoproteomics of the kinome across the cell cycle. Mol Cell. 2008;31:438–448. [DOI] [PubMed] [Google Scholar]
- 30.Mayya V, Lundgren DH, Hwang S-J, Rezaul K, Wu L, Eng JK, Rodionov V, Han DK. Quantitative phosphoproteomic analysis of T cell receptor signaling reveals system-wide modulation of protein-protein interactions. Sci Signal. 2009;2:RA46–RA46. [DOI] [PubMed] [Google Scholar]
- 31.Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther T, Olsen JV, Mann M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009;325:834–840. [DOI] [PubMed] [Google Scholar]
- 32.Derr MA, Fang P, Sinha SK, Ten S, Hwa V, Rosenfeld RG. A novel Y332C missense mutation in the intracellular domain of the human growth hormone receptor (GHR) does not alter STAT5b signaling: redundancy of GHR intracellular tyrosines involved in STAT5b signaling. Horm Res. 2010;In press. [DOI] [PubMed] [Google Scholar]
- 33.Ayling RM, Ross R, Towner P, Von Laue S, Finidori J, Moutoussamy S, Buchanan CR, Clayton PE, Norman MR. A dominant-negative mutation of the growth hormone receptor causes familial short stature. Nature genetics. 1997;16(1):13–14. [DOI] [PubMed] [Google Scholar]
- 34.Iida K, Takahashi Y, Kaji H, Nose O, Okimura Y, Abe H, Chihara K. Growth hormone (GH) insensitivity syndrome with high serum GH-binding protein levels caused by a heterozygous splice site mutation of the GH receptor gene producing a lack of intracellular domain. J Clin Endocrinol Metab. 1998;83(2):531–537. [DOI] [PubMed] [Google Scholar]
- 35.Iida K, Takahashi Y, Kaji H, Takahashi MO, Okimura Y, Nose O, Abe H, Chihara K. Functional characterization of truncated growth hormone (GH) receptor- (1–277) causing partial GH insensitivity syndrome with high GH-binding protein. J Clin Endocrinol Metab. 1999;84(3):1011–1016. [DOI] [PubMed] [Google Scholar]
- 36.Aisenberg J, Auyeung V, Pedro JF, Sugalski R, Chartoff A, Rothenberg R, Derr MA, Hwa V, Rosenfeld RG. Atypical growth hormone insensitivity syndrome (GHIS) and severe insulin-like growth factor-I deficiency (IGFD) resulting from compound heterozygous mutations of the GH receptor (GHR), including a novel frameshift mutation affecting the intracellular domain. Horm Res Paediatr. 2010;74(6):406–411. [DOI] [PubMed] [Google Scholar]
- 37.Takagi M, Shinohara H, Nagashima Y, Hasegawa Y, Narumi S, Hasegawa T. A novel dominant negative mutation in the intracellular domain of GHR is associated with growth hormone insensitivity. Clinical endocrinology. 2016;85(4):669–671. [DOI] [PubMed] [Google Scholar]
- 38.Vairamani K, Merjaneh L, Casano-Sancho P, Sanli ME, David A, Metherell LA, Savage MO, Sanchez del Pozo J, Backeljauw P, Rosenfeld RG, Aisenberg J, Dauber A, Hwa V. Novel Dominant-Negative GH Receptor Mutations Expands the Spectrum of GHI and IGF-I Deficiency. J Endocr Soc. 2017;1(4):345–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.David A, Hwa V, Metherell LA, Netchine I, Camacho-Hubner C, Clark AJ, Rosenfeld RG, Savage MO. Evidence for a continuum of genetic, phenotypic, and biochemical abnormalities in children with growth hormone insensitivity. Endo Rev. 2011;32(4):472–497. [DOI] [PubMed] [Google Scholar]
- 40.Hwa V Growth Hormone Receptor in Growth In: Ho K, ed. Growth Hormone Related Diseases and Therapy: a molecular and physiological perspective for the clinician. New York: Humana Press; 2011:3–16. [Google Scholar]
- 41.Savage MO, Attie KM, David A, Metherell LA, Clark AJ, Camacho-Hubner C. Endocrine assessment, molecular characterization and treatment of growth hormone insensitivity disorders. Nat Clin Pract Endocrinol Metab. 2006;2:395–407. [DOI] [PubMed] [Google Scholar]
- 42.Milward A, Metherell L, Maamra M, Barahona MJ, Wilkinson IR, Camacho-Hubner C, Savage MO, Bidlingmaier CM, Clark AJL, Ross RJM, Webb SM. Growth hormone (GH) insensitivity syndrome due to a GH receptor truncated after Box1, resulting in isolated failure of STAT5 signal transduction. J Clin Endocrinol Metab. 2004;89:1259–1266. [DOI] [PubMed] [Google Scholar]
- 43.Rosenfeld RG, Belgorosky A, Camacho-Hubner C, Savage MO, Wit JM, Hwa V. Defects in growth hormone receptor signaling. Trends Endocrinol Metab. 2007;18(4):134–141. [DOI] [PubMed] [Google Scholar]
- 44.Rosenfeld RG, Hwa V. The growth hormone cascade and its role in mammalian growth. Horm Res. 2009;71(Suppl 2):36–40. [DOI] [PubMed] [Google Scholar]
- 45.Kofoed EM, Hwa V, Little B, Woods KA, Buckway CK, Tsubaki J, Pratt KL, Bezrodnik L, Jasper H, Tepper A, Heinrich J, Rosenfeld RG. Growth-hormone insensitivity (GHI) associated with a STAT5b mutation. N Engl J Med. 2003;349:1139–1147. [DOI] [PubMed] [Google Scholar]
- 46.Chen X, Vinkemeier U, Zhao Y, Jerzalmi D, Darnell JEJ, Kuriyan J. Crystal Structure of a tyrosine phosphorylated STAT-1 dimer bound to DNA. Cell. 1998;93:827–839. [DOI] [PubMed] [Google Scholar]
- 47.de Araujo ED, Erdogan F, Neubauer HA, Meneksedag-Erol D, Manaswiyoungkul P, Eram MS, Seo HS, Qadree AK, Israelian J, Orlova A, Suske T, Pham HTT, Boersma A, Tangermann S, Kenner L, Rulicke T, Dong A, Ravichandran M, Brown PJ, Audette GF, Rauscher S, Dhe-Paganon S, Moriggl R, Gunning PT. Structural and functional consequences of the STAT5B(N642H) driver mutation. Nat Commun. 2019;10(1):2517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.de Araujo ED, Orlova A, Neubauer HA, Bajusz D, Seo HS, Dhe-Paganon S, Keseru GM, Moriggl R, Gunning PT. Structural Implications of STAT3 and STAT5 SH2 Domain Mutations. Cancers (Basel). 2019;11(11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chia DJ, Subbian E, Buck TM, Hwa V, Rosenfeld RG, Skach WR, Shinde U, Rotwein P. Aberrant folding of a mutant STAT5b causes growth hormone insensitivity and proteasomal dysfunction. J Biol Chem. 2006;281:6552–6558. [DOI] [PubMed] [Google Scholar]
- 50.Fang P, Kofoed EM, Little BM, Wang X, Ross RJM, Frank SJ, Hwa V, Rosenfeld RG. A mutant signal transducer and activator of transcription 5B, associated with growth hormone insensitivity and insulin-like growth factor-I deficiecny, cannot function as a signal transducer or transcription factor. J Clin Endocrinol & Metab. 2006;91:1526–1534. [DOI] [PubMed] [Google Scholar]
- 51.Cohen AC, Nadeau KC, Tu W, Hwa V, Dionis K, Bezrodnik L, Teper A, Gaillard M, Heinrich J, Krensky AM, Rosenfeld RG, Lewis DB. Cutting Edge: decreased accumulation and regulatory function of CD4+CD25high T cells in human STAT5b deficiency. J Immunol. 2006;177:2770–2774. [DOI] [PubMed] [Google Scholar]
- 52.Hwa V, Little B, Kofoed EM, Rosenfeld RG. Transcriptional regulation of insulin-like growth factor-I (IGF-I) by interferon-gamma (IFN-g) requires Stat-5b. J Biol Chem. 2004;279:2728–2736. [DOI] [PubMed] [Google Scholar]
- 53.Hwa V, Camacho-Hubner C, Little BM, David A, Metherell LA, El-Khatib N, Savage MO, Rosenfeld RG. Growth hormone insensitivity and severe short stature in siblings: a novel mutation at the exon13-intron 13 junction of the STAT5b gene. Horm Res. 2007;68(5):218–224. [DOI] [PubMed] [Google Scholar]
- 54.Pugliese-Pires PN, Tonelli CA, Dora JM, Silva PCA, Czepielewski M, Simoni G, Arnhold IJ, Jorge AAL. A novel STAT5B mutation causing GH insenstivity syndrome associated with hyperprolactinemia and immune dysfunction in two male siblings. . Eur J Endocrinol. 2010;163:349–355. [DOI] [PubMed] [Google Scholar]
- 55.Hwa V, Little B, Adiyaman P, Kofoed EM, Pratt KL, Ocal G, Berberoglu M, Rosenfeld RG. Severe growth hormone insensivity resulting from total absence of signal transducer and activator of transcription 5b. J Clin Endocrinol & Metab. 2005;90:4260–4266. [DOI] [PubMed] [Google Scholar]
- 56.Vidarsdottir S, Walenkamp MJE, Pereira AM, Karperien M, van Doorn J, van Duyvenvoorde HA, White S, Breuning MH, Roelfsema F, Femke Kruithof M, van Dissel J, Janssen R, Wit JM, Romijn JA. Clinical and biochemical characteristics of a male patient with a novel homozygous STAT5b mutation. J Clin Endocrinol & Metab. 2006;91:3482–3485. [DOI] [PubMed] [Google Scholar]
- 57.Bernasconi A, Marino R, Ribas A, Rossi J, Ciaccio M, Oleastro M, Ornani A, Paz R, Rivarola M, Zelazko M, Belgorosky A. Characterization of immunodeficiency in a patient with growth hormone insensitivity secondary to a novel STAT5b gene mutation. . Pediatrics. 2006;118:e1584–e1592. [DOI] [PubMed] [Google Scholar]
- 58.Boyanovsky A, Lozano A, Testa G, Munoz L, Marino R, Bernasconi A, Belgorosky A, Miras M. Growth hormone insensitivity and immunodeficiency: mutation in the STAT5B gene. 8th Joint Meeting of the Lawson Wilkins Pediatric Endocrine Society/European Society for Paediatric Endocrinology. 2009:P01–067. [Google Scholar]
- 59.Scaglia PA, Martinez AS, Feigerlová E, Bezrodnik L, Gaillard MI, Di Giovanni D, Ballerini MG, Jasper HG, Heinrich JJ, Fang P, Domené HM, Rosenfeld RG, Hwa V. A novel missense mutation in the SH2 domain of the STAT5B gene results in a transcriptionally inactive STAT5b associated with severe IGF-I deficiency, immune dysfunction, and lack of pulmonary disease. J Clin Endocrinol & Metab. 2012;97:E830–E839. [DOI] [PubMed] [Google Scholar]
- 60.Acres MJ, Gothe F, Grainger A, Skelton AJ, Swan DJ, Willet JDP, Leech S, Galcheva S, Iotova V, Hambleton S, Engelhardt KR. Signal transducer and activator of transcription 5B deficiency due to a novel missense mutation in the coiled-coil domain. J Allergy Clin Immunol. 2019;143(1):413–416 e414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Udy GB, Towers RP, Snell RG, Wilkins RJ, Park SH, Ram PA, Waxman DJ, Davey HW. Requirement of STAT5b for sexual dimorphism of body growth rates and liver gene expression. Proceedings of the National Academy of Sciences of the United States of America. 1997;94(14):7239–7244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Teglund S, McKay C, Schuetz E, van Deursen JM, Stravopodis D, Wang D, Brown M, Bodner S, Grosveld G, Ihle JN. Stat5a and Stat5b proteins have essential and nonessential, or redundant, roles in cytokine responses. Cell. 1998;93(5):841–850. [DOI] [PubMed] [Google Scholar]
- 63.Liu X, Robinson GW, Wagner KU, Garrett L, Wynshaw-Boris A, Hennighausen L. Stat5a is mandatory for adult mammary gland development and lactogenesis. Genes Dev. 1997;11:179–186. [DOI] [PubMed] [Google Scholar]
- 64.Villarino A, Laurence A, Robinson GW, Bonelli M, Dema B, Afzali B, Shih HY, Sun HW, Brooks SR, Hennighausen L, Kanno Y, O’Shea JJ. Signal transducer and activator of transcription 5 (STAT5) paralog dose governs T cell effector and regulatory functions. Elife. 2016;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Klammt J, Neumann D, Gevers EF, Andrew SF, Schwartz ID, Rockstroh D, Colombo R, Sanchez MA, Vokurkova D, Kowalczyk J, Metherell LA, Rosenfeld RG, Pfaffle R, Dattani MT, Dauber A, Hwa V. Dominant-negative STAT5B mutations cause growth hormone insensitivity with short stature and mild immune dysregulation. Nat Commun. 2018;9(1):2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Holland SM, DeLeo FR, Elloumi HZ, Hsu AP, Uzel G, Brodsky N, Freeman AF, Demidowich A, Davis J, Turner ML, Anderson VL, Darnell DN, Welch PA, Kuhns DB, Frucht DM, Malech HL, Gallin JI, Kobayashi SD, Whitney AR, Voyich JM, Musser JM, Woellner C, Schaffer AA, Puck JM, Grimbacher B. STAT3 mutations in the hyper-IgE syndrome. N Engl J Med. 2007;357(16):1608–1619. [DOI] [PubMed] [Google Scholar]
- 67.Minegishi Y, Saito M, Tsuchiya S, Tsuge I, Takada H, Hara T, Kawamura N, Ariga T, Pasic S, Stojkovic O, Metin A, Karasuyama H. Dominant-negative mutations in the DNA-binding domains of STAT3 cause hyper-IgE syndrome. Nature. 2007;448:1058–1062. [DOI] [PubMed] [Google Scholar]
- 68.Renner ED, Rylaarsdam S, Anover-Sombke S, Rack AL, Reichenbach J, Carey JC, Zhu Q, Jansson AF, Barboza J, Schimke LF, Leppert MF, Getz MM, Seger RA, Hill HR, Belohradsky BH, Torgerson TR, Ochs HD. Novel signal transducer and activator of transcription 3 (STAT3) mutations, reduced T(H)17 cell numbers, and variably defective STAT3 phosphorylation in hyper-IgE syndrome. J Allergy Clin Immunol. 2008;122(1):181–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Schimke LF, Sawalle-Belohradsky J, Roesler J, Wollenberg A, Rack A, Borte M, Rieber N, Cremer R, Maass E, Dopfer R, Reichenbach J, Wahn V, Hoenig M, Jansson AF, Roesen-Wolff A, Schaub B, Seger R, Hill HR, Ochs HD, Torgerson TR, Belohradsky BH, Renner ED. Diagnostic approach to the hyper-IgE syndromes: immunologic and clinical key findings to differentiate hyper-IgE syndromes from atopic dermatitis. J Allergy Clin Immunol. 2010;126(3):611–617 e611. [DOI] [PubMed] [Google Scholar]
- 70.Majri SS, Fritz JM, Villarino AV, Zheng L, Kanellopoulou C, Chaigne-Delalande B, Gronholm J, Niemela JE, Afzali B, Biancalana M, Pittaluga S, Sun A, Cohen JL, Holland SM, O’Shea JJ, Uzel G, Lenardo MJ. STAT5B: A Differential Regulator of the Life and Death of CD4(+) Effector Memory T Cells. J Immunol. 2018;200(1):110–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wit JM, van Duyvenvoorde HA, Scheltinga SA, de Bruin S, Hafkenscheid L, Kant SG, Ruivenkamp CA, Gijsbers ACJ, van Doorn J, Feigerlová E, Noordam C, Walenkamp MJ, Claahsen-van de Grinten H, Stouthart P, Bonapart IE, Pereira AM, Gosen J, Delemaare-van de Waal HA, Hwa V, Breuning MH, Domene HM, Oostdijk W, Losekoot M. Genetic analysis of short children with apparent growth hormone insensitivity. Horm Res Paediatr. 2012;77:320–333. [DOI] [PubMed] [Google Scholar]
- 72.Walenkamp MJ, Klammt J, Feigerlova E, Losekoot M, van Duyvenvoorde HA, Hwa V, Pfaffle R, Wit JM. Genetic analysis of GHR should contain sequencing of all coding exons and specific intron sequences, and screening for exon deletions. Horm Res Paediatr. 2013;80(6):406–412. [DOI] [PubMed] [Google Scholar]
- 73.Ramirez L, Sanguineti N, Scaglia P, Keselman A, Ballerini MG, Karabatas L, Landi E, Castro J, Domene S, Pennisi P, Jasper H, Rey RA, Vazquez M, Domene H, Bergada I, Gutierrez M. A novel heterozygous STAT5B variant in a patient with short stature and partial growth hormone insensitivity (GHI). Growth Horm IGF Res. 2020;50:61–70. [DOI] [PubMed] [Google Scholar]
- 74.Hattori A, Katoh-Fukui Y, Nakamura A, Matsubara K, Kamimaki T, Tanaka H, Dateki S, Adachi M, Muroya K, Yoshida S, Ida S, Mitani M, Nagasaki K, Ogata T, Suzuki E, Hata K, Nakabayashi K, Matsubara Y, Narumi S, Tanaka T, Fukami M. Next generation sequencing-based mutation screening of 86 patients with idiopathic short stature. Endocrine journal. 2017;64(10):947–954. [DOI] [PubMed] [Google Scholar]
- 75.Kiel MJ, Sahasrabuddhe AA, Rolland DC, Velusamy T, Chung F, Schaller M, Bailey NG, Betz BL, Miranda RN, Porcu P, Byrd JC, Medeiros LJ, Kunkel SL, Bahler DW, Lim MS, Elenitoba-Johnson KS. Genomic analyses reveal recurrent mutations in epigenetic modifiers and the JAK-STAT pathway in Sezary syndrome. Nat Commun. 2015;6:8470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kucuk C, Jiang B, Hu X, Zhang W, Chan JK, Xiao W, Lack N, Alkan C, Williams JC, Avery KN, Kavak P, Scuto A, Sen E, Gaulard P, Staudt L, Iqbal J, Zhang W, Cornish A, Gong Q, Yang Q, Sun H, d’Amore F, Leppa S, Liu W, Fu K, de Leval L, McKeithan T, Chan WC. Activating mutations of STAT5B and STAT3 in lymphomas derived from gammadelta-T or NK cells. Nat Commun. 2015;6:6025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Jiang L, Gu ZH, Yan ZX, Zhao X, Xie YY, Zhang ZG, Pan CM, Hu Y, Cai CP, Dong Y, Huang JY, Wang L, Shen Y, Meng G, Zhou JF, Hu JD, Wang JF, Liu YH, Yang LH, Zhang F, Wang JM, Wang Z, Peng ZG, Chen FY, Sun ZM, Ding H, Shi JM, Hou J, Yan JS, Shi JY, Xu L, Li Y, Lu J, Zheng Z, Xue W, Zhao WL, Chen Z, Chen SJ. Exome sequencing identifies somatic mutations of DDX3X in natural killer/T-cell lymphoma. Nature genetics. 2015;47(9):1061–1066. [DOI] [PubMed] [Google Scholar]
- 78.Laron Z, Lilos P, Klinger B. Growth curves for Laron syndrome. Arch Dis Child. 1993;68:768–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Derr MA, Aisenberg J, Fang P, Tenenbaum-Rakover Y, Rosenfeld RG, Hwa V. The growth hormone receptor (GHR) c.899dupC mutation functions as a dominant negative: insights into the pathophysiology of intracellular GHR defects. J Clin Endocrinol Metab. 2011;96(11):E1896–1904. [DOI] [PubMed] [Google Scholar]
- 80.Maamra M, Milward A, Esfahani HZ, Abbot LP, Metherell LA, Savage MO, Clark AJ, Ross RJ. A 36 residues insertion in the dimerization domain of the growth hormone receptor results in defective trafficking rather than impaired signaling. J Endocrinol. 2006;188(2):251–261. [DOI] [PubMed] [Google Scholar]
- 81.Metherell LA, Akker SA, Munroe PB, Rose SJ, Caulfield M, Savage MO, Chew SL, Clark AJ. Pseudoexon activation as a novel mechanism for disease resulting in atypical growth-hormone insensitivity. Am J Hum Genet. 2001;69(3):641–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.David A, Camacho-Hubner C, Bhangoo A, Rose SJ, Miraki-Moud F, Akker SA, Butler GE, Ten S, Clayton PE, Clark AJ, Savage MO, Metherell LA. An intronic growth hormone receptor mutation causing activation of a pseudoexon is associated with a broad spectrum of growth hormone insensitivity phenotypes. J Clin Endocrinol & Metab. 2007;92(2):655–659. [DOI] [PubMed] [Google Scholar]
- 83.Walenkamp MJE, Vidarsdottir S, Pereira AM, Karperien M, van Doorn J, van Duyvenvoorde HA, Breuning MH, Roelfsema F, Kruithof MF, van Dissel J, Janssen R, Wit JM, Romijn JA. Growth hormone secretion and immunological function of a male patient with a homozygous STAT5b mutation. Eur J Endocrinol. 2007;156:155–165. [DOI] [PubMed] [Google Scholar]
- 84.Rubinstein A, Bernstein LJ, Charytan M, Krieger BZ, Ziprkowski M. Corticosteroid treatment for pulmonary lymphoid hyperplasia in children with the acquired immunoe deficiency syndrome. Pediatr Pulmonol. 1988;4:13–17. [DOI] [PubMed] [Google Scholar]
- 85.Bezrodnik L, Di Giovanni D, Caldirola MS, Azcoiti ME, Torgerson T, Gaillard MI. Long-term follow-up of STAT5B deficiency in three argentinian patients: clinical and immunological features. Journal of clinical immunology. 2015;35(3):264–272. [DOI] [PubMed] [Google Scholar]
- 86.Nadeau K, Hwa V, Rosenfeld RG. STAT5b deficiency: an unsuspected cause of growth failure, immunodeficiency, and severe pulmonary disease. J Pediatr. 2011;158:701–708. [DOI] [PubMed] [Google Scholar]
- 87.Tangye SG, Al-Herz W, Bousfiha A, Chatila T, Cunningham-Rundles C, Etzioni A, Franco JL, Holland SM, Klein C, Morio T, Ochs HD, Oksenhendler E, Picard C, Puck J, Torgerson TR, Casanova JL, Sullivan KE. Human Inborn Errors of Immunity: 2019 Update on the Classification from the International Union of Immunological Societies Expert Committee. Journal of clinical immunology. 2020;40(1):24–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Vargas-Hernandez A, Witalisz-Siepracka A, Prchal-Murphy M, Klein K, Mahapatra S, Al-Herz W, Mace EM, Carisey AF, Orange JS, Sexl V, Forbes LR. Human signal transducer and activator of transcription 5b (STAT5b) mutation causes dysregulated human natural killer cell maturation and impaired lytic function. J Allergy Clin Immunol. 2020;145(1):345–357 e349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Schepers H, Wierenga AT, Vellenga E, Schuringa JJ. STAT5-mediated self-renewal of normal hematopoietic and leukemic stem cells. JAKSTAT. 2012;1(1):13–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Jenks JA, Seki S, Kanai T, Huang J, Morgan AA, Scalco RC, Nath R, Bucayu R, Wit JM, Al-Herz W, Ramadan D, Jorge AA, Bacchetta R, Hwa V, Rosenfeld R, Nadeau KC. Differentiating the roles of STAT5B and STAT5A in human CD4+ T cells. Clin Immunol. 2013;148:227–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Burchill MA, Yang J, Vang KB, Farrar MA. Interleukin-2 receptor signaling in regulatory T cell development and homeostasis. . Immunol Lett. 2007;114(1):1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Torgerson TR. Immune dysregulation in primary immunodeficiency disorders. Immunol Allergy Clin N Am. 2008;28:315–327. [DOI] [PubMed] [Google Scholar]
- 93.Caldirola MS, Rodriguez Broggi MG, Gaillard MI, Bezrodnik L, Zwirner NW. Primary Immunodeficiencies Unravel the Role of IL-2/CD25/STAT5b in Human Natural Killer Cell Maturation. Front Immunol. 2018;9:1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Gambineri E, Ciullini Mannurita S, Hagin D, Vignoli M, Anover-Sombke S, DeBoer S, Segundo GRS, Allenspach EJ, Favre C, Ochs HD, Torgerson TR. Clinical, Immunological, and Molecular Heterogeneity of 173 Patients With the Phenotype of Immune Dysregulation, Polyendocrinopathy, Enteropathy, X-Linked (IPEX) Syndrome. Front Immunol. 2018;9:2411. [DOI] [PMC free article] [PubMed] [Google Scholar]