

Abstract
Symptoms of attention deficit hyperactivity disorder (ADHD) run a variable course through adolescence. While most affected individuals show some improvement, particularly of hyperactivity-impulsivity, symptoms of inattention are more persistent, and some individuals may meet diagnostic criteria for the first time during adolescence. Genetic factors impact adolescent symptom trajectories; those showing persistence likely carry a greater burden of common risk alleles. Rare structural genomic variants, such as copy number variants and point mutations, might also play a role. While psychostimulant medication is associated with better functional outcomes, an impact on underlying adolescent symptom trajectories has been hard to demonstrate. At a neural level, several studies report that adolescents whose childhood ADHD symptoms have remitted are indistinguishable from neurotypical individuals. This finding could reflect the ‘carrying forward’ of relatively typical childhood neural features among those destined for adolescent remission or the correction of early childhood anomalies with a convergence towards typical dimensions. Other studies have noted unique, possibly compensatory patterns of neural activity among adolescents whose ADHD has improved. Finally, different neural processes might occur in different brain regions. Thus, some functional imaging studies find that subcortical anomalies reflect the onset of ADHD, and remain lifelong, regardless of symptom change, whereas the variable clinical course of adolescent ADHD is determined by plasticity of the cerebral cortex. Integrating an understanding of the neural processes with genomic risk could elucidate the mechanisms underlying the complex course of adolescent ADHD.
Keywords: Attention Deficit Hyperactivity Disorder, Brain function, Brain structure, Recovery, Adult outcome, Prognosis
Introduction.
One of the most fascinating aspects of ADHD is its highly variable course through adolescence. During the teenage years, some youth will shed their childhood symptoms of ADHD, while others have persistent, even worsening symptoms. Here, we review the factors that drive different adolescent symptom trajectories and propose different neural models to explain why some children ‘grow out’ of ADHD during adolescence while others do not.
Section 1: The course of adolescent ADHD.
First, we summarize the literature on the adolescent course of ADHD, based on a literature search, supplemented by papers of theoretical importance- details in Supplemental material.
(i). Categorical outcomes and symptom trajectories.
The DSM-5 views outcome categorically, contrasting those who continue to meet diagnostic criteria against either those showing either full or partial remission. Remission is achieved with the resolution of both symptoms and impairment and partial remission is attained when full criteria have not been met for at least 6 months, but symptoms persist that cause impairment. Using this categorical approach, the reported rates of persistence of childhood ADHD into adulthood vary from 4% to 77%. The wide range reflects differences in definition (e.g. insistence on impairment from symptoms), measurement (questionnaires vs clinical interview), rater (persistence is higher in parent vs youth self-report), baseline severity (studies including only combined vs all presentations) and sampling strategy (population vs clinical cohorts) (1). While limited by the heterogeneity in prevalence estimates, a meta-analysis of studies up to 2005 found 15-20% of children with ADHD show persistence of the full syndrome into adulthood, with a further 50% showing persistence of subthreshold symptoms with impairment (2).
Several groups have used data driven methods such as growth mixture modelling to characterize groups with different adolescent symptom trajectories, rather than considering shifts in diagnostic status – see Table 1 (3-10). Most such studies find a subgroup with clear age-related improvement, ranging from between 33% to 66% of affected children, with more improvement in hyperactivity-impulsivity than inattention (5, 6, 9). The studies also point to a subgroup with worsening symptoms, which reached 22-24% in clinic-based studies of youth at risk for conduct disorder or bipolar affective disorder (6, 9). There is evidence that the worsening symptom group may include individuals who pass the threshold for diagnosis for the first time during adolescence. For example, a population survey that found around 14% of children who had subthreshold symptoms of ADHD at 9 years of age met diagnostic criteria 4 years later (11). The possibility of a de novo onset of ADHD in adolescence, which lacks any childhood symptoms, is beyond the scope of this paper but reviewed elsewhere (12-16).
Table 1.
Studies that included assessments of ADHD symptoms during adolescence. Studies confined to childhood are excluded. HI=hyperactivity-impulsivity.
| Study | Design | Age range covered |
Measurement | Findings |
|---|---|---|---|---|
| Pingault et al. (4) | Twin cohort (N=8395 pairs) | 8 to 16 years (4 assessments) | Mother-reported DSM symptoms scales | 2 trajectories: - HI: marked decrease (6 to 2.9 symptoms) - Inattention: modest decrease (5.8 to 4.9 symptoms) |
| Swanson et al (5) | ADHD and community controls (N = 485) | 7 to 10 years (3 assessments over 22 months) | Combined teacher- and parent-reported DSM symptoms via the SNAP | 3 trajectories: - Linear decreasing (34%) - Large initial symptom decrease followed by a sustained lower level (52%) - Initial increase followed by a return to baseline (14%) |
| Riglin et al (6) | Population cohort (N=9757 children) | 4 to 17 years (7 assessments) | Parent-reported Strength and Difficulties Questionnaire (SDQ) | 4 trajectories (total ADHD symptoms): - Low/stable (~83%) - Intermediate (~8%) - Childhood-limited (~6%) - Persistent (~4%) |
| Tandon et al. (7) | High risk cohort (ADHD, bipolar 1 or unaffected controls) (N=251 individuals) | 6 assessments over 10 years | Clinician-ascertained symptoms (K-SADS) | 4 trajectories (total ADHD symptoms): - Persisting low (~38%) - Rapid improvement (~16%) - Gradual improvement (~24%) - Persisting high (~22%) |
| Döpfner et al. (8) | Combined 11 population cohorts: (N~2500) | 7 to 19 years (variable N of assessments) | Parent-reported ADHD Symptom Checklist | 3 trajectories for inattention and HI: - High (3%) - Moderate (13-19%) - Low (78-83%) - Moderate symptom and high HI trajectories improved - High inattention did not improve |
| Murray et al (9) | Population cohort (N=1571) | 7 to 15 years (8 measurements) | Teacher-reported Social Behavior Questionnaire (8 DSM symptoms) | 4 trajectory groups for HI: - High/stable (24% of males, 9% of females) - Low/stable (63% of males, 81% of females) - High/increasing (13% of males) - Concave (10% of females) 3 trajectory groups for inattention: - High/stable (39% of males, 10% of females) - Low/stable (61% of males, 59% of females) - Moderate/stable (31% of females) - Gender differences noted: females showed large HI symptom increases in early adolescence; males elevated from childhood |
| Malone et al. (10) | Cohort: at-risk for conduct problems (N=754) | ~9 to 15 years (3 observations) | Parent completed computer version of the Diagnostic Interview Schedule for Children | 3 symptom trajectories: - Low/stable (58%) - Convex (increase, then decrease) (18%) - Concave (decrease then increase) (24%) |
| Larsson et al. (11) | Population twin (N=1450 pairs) | 8 to 17 years (3 assessments) | Parent-reported DSM symptoms checklist | 2 trajectories: - Low (91% for HI, 86% for inattention) - High/decreasing (9% for HI, 14% for inattention) |
(ii). ADHD as a risk factor for adolescent-onset disorders.
ADHD acts a risk factor for the emergence of many disorders during adolescence: here we focus on substance use and mood disorders. ADHD doubles the risk of nicotine dependence [O.R=2.36, 95%CI 1.71 to 3.27) and shows weaker associations with alcohol misuse (OR 1.35, CI 1.11 to 1.64) (17). ADHD also increases the risk for adolescent depression particularly among females (hazard ratio 4.32 in (18), and some, but not all, studies find an increased risk for bipolar affective disorder (19)(20). Genetic pleiotropy could partly explain these co-occurring disorders as there is an overlap between the genetic variants that confer risk for ADHD, substance misuse and mood disorders (21, 22). Likewise, environmental factors, such as prenatal substance exposure, lead exposure and socioeconomic disadvantage, act as risk factors for many disorders, including ADHD (23). Large cohorts will be needed to parse such transdiagnostic etiological factors, considering both gene-environment interactions and correlations (24).
In conclusion, most with childhood-onset ADHD show some symptom improvement during adolescence, particularly of hyperactivity-impulsivity. A large minority will retain the full syndrome into adulthood and/or show worsening symptoms. Childhood ADHD is also a major risk factor for adolescent-onset substance and mood disorders.
Section 2. Markers and mechanisms of adolescent symptom trajectories.
Can we predict and explain the variable course of ADHD symptoms in adolescence?
(i). Markers of adolescent outcomes.
A recent meta-analysis of sixteen prospective studies found that several baseline clinical features increased the risk of adult persistence, specifically symptom severity (OR, 2.33 CI 1.6 to 3.39), comorbid conduct disorder (OR 1.85, CI 1.06 t0 3.24) and depressive disorder (OR 1.8, CI 1.1 to 2.95) (13). By contrast, demographic features, such as gender, socio-economic status and family structure were not associated with persistence. Preliminary associations have been found between the adolescent course of ADHD and earlier measures of neuroanatomy (specifically dimensions of the anterior cingulate cortex and superior cerebellar vermis), and functional connectivity (coupling between activity in the medial and dorsolateral prefrontal cortex) (25-27). Some, but not all studies, find childhood cognitive measures, specifically intact working memory and low response time variability, are associated with later symptom improvement (28-30). In summary, some childhood clinical features are modestly associated with adolescent symptom course, and there is preliminary evidence for the prognostic utility of neural, and perhaps cognitive features.
(ii). Mechanisms: genomics, social contexts and medication.
What mechanisms might determine adolescent symptom trajectories? Genetic factors are pivotal in onset of ADHD but to what extent do genes contribute to the adolescent course of ADHD symptoms(31)? A seminal twin study addressed this question by following 8395 pairs of twins from ages 8 to 16 (3). It found that 81% of the change in hyperactivity-impulsivity and 50% of the change in inattention was attributable to genetic factors. There were modest but significant contributions from non-shared and shared environmental factors.
Other have asked if the genetic loading for ADHD is greater among those with the persisting form. In support, a population study found that the risk of having ADHD among relatives was higher among those whose ADHD had persisted beyond age 18 (hazard ratio of 11.4, CI 9.97 to 13.25) compared to those whose with a ADHD diagnosis only before age 18 (hazard ratio of 4.6, CI 3.83 to 5.72) (32). This finding is consistent with earlier family studies reporting that ADHD that persists into adulthood is tied to a greater familial aggregation of ADHD, thought to reflect a higher genetic risk (33).
The longitudinal twin study, mentioned above, also found that the genes contributing to the onset of ADHD overlapped substantially but not completely (at around 50%) with those determining outcome (3). The finding of shared genetic effects between ADHD onset and course is consistent with the high genetic correlation of around 0.8 reported between childhood ADHD and adult ADHD (34). Combined, these studies suggest that those who harbor a higher common variant (polygenic) risk for the onset of ADHD might show symptom persistence. One study confirmed this prediction: those whose childhood ADHD persisted through adolescence had a higher polygenic risk for ADHD than those with childhood limited symptoms (5). The genetic factors unique to outcome will also play a role, with different genetic factors coming into play at different developmental stages. Finally, there is also evidence that rare structural genetic variants are enriched in ADHD, including an excess of copy number variants (CNVs) in the alpha-7 nicotinic acetylcholine receptor (CHRNA7), immune function and metabotropic glutamate receptor genes (35, 36). It is unclear if a greater burden of copy number variants denotes a more severe phenotype, which may have a worse prognosis (37, 38).
What role is played by the environment in shaping symptom trajectories? Twin studies suggest little contribution from the shared environment as a main effect but these studies do not exclude an interaction between the environment and genes (3). It has also been argued that low estimates of shared environmental factors reflect underpowered studies and analytic limitations, such as the inability to include dominant genetic and shared environmental components in the same twin model (39). Links between social contextual factors and the adolescent course of ADHD symptoms are rarely explored. A pilot longitudinal study of 190 children with ADHD found those living in relatively affluent neighborhoods showed improvement with age in inattention, largely independent of variation in a wide range of familial factors (40). By contrast, children living in less affluent neighborhood showed clinical deterioration only if the family had high levels of conflict or if the parents were of lower economic/educational status.
The final environmental factor we consider is treatment. Systematic reviews conclude that psychostimulant treatment (the most widely prescribed medication) is often but not invariably associated with better real-life outcomes in adolescence, such as fewer accidents, improved academic performance, and better social relationships (41, 42). However, studies that directly map medication use onto symptom trajectories find little evidence of symptom resolution tied to psychostimulant medication (43). Of course, causal effects cannot be inferred from such observational studies. Among the many possible confounding factors could be severity: those who have more severe symptoms may have a worse long-term course and be more likely to receive medication.
What can be concluded about the etiological factors driving ADHD symptoms during adolescence? Genetic factors play a major role: those destined to have persistence may carry more risk alleles, shared with the onset of the disorder. Additionally, novel genetic factors may come into play during adolescence, shaping trajectories. While psychostimulant medication might improve functional outcomes, a clear impact on symptom trajectories has been hard to detect, and greater consideration is needed of other environmental factors, particularly their interplay with genomic risk.
Section 3: Developmental models.
Finally we consider developmental models of the neurobiology that could underpin different adolescent ADHD trajectories, building on decades of developmental psychopathology research by others in our earlier review of adult ADHD (44). The models and their differing predictions on neural substrates in the childhood, adolescent and adult brains are illustrated in Figure 1.
Figure 1:
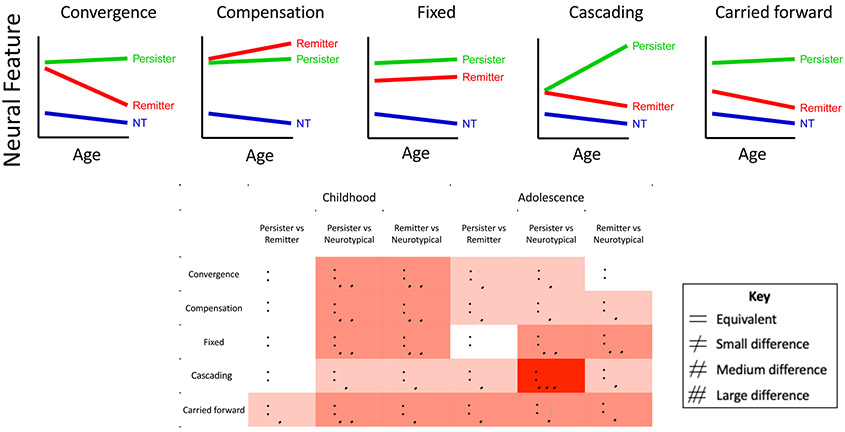
Upper Panel: Graphs illustrating the neural trajectories of remitters, persisters and neurotypical (NT) individuals predicted by the model. Lower panel: The table illustrates the pattern of difference in the childhood and late adolescent brain predicted by each model.
The first model (‘convergence’) views adolescent symptom improvement as the result of the convergence of neural features from an atypical childhood baseline towards more typical brain function and structure. By contrast, those with persistent adolescent symptoms will show persisting neural anomalies. This model encompasses ideas of late maturation in ADHD, in which remission is underpinned by correction of an early developmental delay. A second model (‘compensation’) postulates that symptom improvement is driven by the recruitment of new brain systems that compensate for core ADHD symptoms. Hence ‘improvers’ show unique compensatory neural features, that become more marked during adolescence, and are not seen in either persisters or those who are never affected. A third model (‘carried forward’) holds that different adolescent neural trajectories originate in childhood. Those destined for improvement and eventual remission have more typical neural features in childhood which they carry forward during adolescence. By contrast, the persistence of symptoms during adolescence is held to reflect childhood anomalies in neural features that are also carried forward. A fourth model (‘cascading anomalies’) posits that early symptoms of ADHD per se may exacerbate or even create downstream neural anomalies. By this reckoning, the presence of ADHD symptoms during adolescence engenders neural dysfunction that in turn exacerbates future symptoms. This model could encompass instances of neurodevelopmental ‘arrest’, whereby an initially (near) typical trajectory is halted, and thus diverges further from the neurotypical range with age (45). A final model (‘fixed anomalies’) holds that a history of childhood ADHD anomalies will leave an indelible neural imprint that persists regardless of clinical course during adolescence.
Three points warrant mention. First, these models are compatible: different processes might occur in different brain regions. For example, it has been argued that while ADHD is caused by subcortical anomalies which persist throughout the lifespan, its variable clinical course is determined by plasticity of the cerebral cortex (46). Secondly, given evidence that different genes may contribute to the onset and course of ADHD symptoms, it is possible that different models may be more prominent at different developmental stages within the same individual. Thirdly, the ability to map neurodevelopmental trajectories will partly depend on the metrics used (47). For example, some neural measures may have such a wide range of individual variation, or contain so much error in measurement, that it may prove difficult to discern the ‘true’ underlying developmental trajectory.
The five models each predict different neural trajectories during adolescence among those showing symptom improvement. In the convergence model, ‘improvers’ will have neural features that converge towards the typical range, whereas the compensation model predicts a unique trajectory among improvers, that localizes to compensatory neural substrates. In the ‘carried forward’ model, the main difference is in the childhood starting point, which will lie near, if not within the typical range among those who will later improve symptomatically. Finally, improvers and persisters will follow the same neural trajectory in brain regions that show fixed anomalies. Turning the focus on persisters, the models generally predict that those with ADHD persisting through adolescence will have a fixed difference compared to the never affected (a ‘parallel’ trajectory that does not converge to the typical range). The only exception is the model of ‘cascading anomalies’ which entails a trajectory that diverges increasingly with age from typical ranges among those with persisting symptoms.
We now review the literature through the lens of these models–Table 2. Most studies have considered the neural substrates underlying either ‘top-down’, effortful cognitive processes or ‘bottom-up’, more automatic processes; we retain this division. Two overarching themes emerge. Firstly, there are almost no studies that prospectively acquired both imaging and clinical data at multiple points during adolescence. Rather, imaging was mostly conducted only at endpoints in late adolescence or early adulthood. As a result, findings will often be compatible with more than one model. For example, the demonstration that those who attain remission by adolescence are similar to the never affected is compatible with both the model of convergence and the model of relatively typical childhood neural substrates being ‘carried forward’. Secondly, the proposition that different neural processes can occur at different brain regions has received considerable empirical support.
Table 2:
Neuroimaging studies that included adolescents or young adults with variable outcomes from their childhood ADHD. There were no examples of the ‘emergent’ model among the literature of adolescent ADHD. Note that cross-sectional imaging studies conducted in adolescence that find remitters are similar to the never affected are consistent with both the convergence or ‘carried forward’ models.
| Reference | Persistent ADHD / Remitted / Never Affected |
Entry Age (years mean and SD) |
Follow- up |
Modality and task |
Convergence OR Carried forward (remitters= typicals) |
Compensation (remitters show unique features) |
Fixed anomaly (persisters= remitters) |
|---|---|---|---|---|---|---|---|
| Michelini et al. (58) | 87/23/169 | 13 | 19 (3) | EEG: Eriksen Flanker task | Error related negativity (remitters=never-affected) Conflict monitoring (remitters lie intermediate to persisters and never affected) |
- | - |
| Cheung et al. (57) | 87/23/169 | 13 | 19 (3) | EEG: Cued flanker continuous performance task | Contingent negative variation during preparation-vigilance (remitters=never affected) Executive, processes (remitters lie intermediate to persisters and never affected) |
- | - |
| Michelini et al (67) | 87/23/169 | 13 | 19 (3) | EEG: arrow flanker task | - | - | Hyper-connectivity in beta band pre and post-stimulus and reduced pre-post stimulus change in theta connectivity. |
| Schulz et al. (51) | 11/16/28 | 7-11 | 24 (2) | fMRI: Stimulus and response conflict task | Fronto-parietal activation during high cognitive demand | - | - |
| Szekely et al. (53) | fMRI: 24/40/84 MEG: 25/26/46 |
9 (1) | 24 (4) | MEG and fMRI: Response inhibition | Prefrontal inhibitory processing | - | Striatal inhibitory activity |
| Sudre et al. (54) | fMRI: 41/35/71 MEG: 32/35/58 |
9 (1) | 24 (4) | MEG and fMRI: Default mode and its interconnections | Default mode network internal and external connectivity | - | |
| Francx et al. (55) | 59 (persistent symptoms)/42 (improving symptoms) /40 | 12 | 18 (3) | Diffusion tensor imaging | Voxel level measures of white matter microstructure | - | - |
| Shaw et al. (44) | 32/43/74 | 10 (3) | 23 (4) | Diffusion tensor imaging | Tract level measures of white matter tract microstructure | - | - |
| Shaw et al. (50) | 37/55/184 | 10 (3) | 24 (3) | Anatomic imaging | Thickness of cortical regions involved in cognitive control (the finding is only consistent with convergence). | - | - |
| Luo et al (56) | 16/16/32 | 9(1) | 24(2) | Diffusion tensor imaging | Tract level measures of white matter tract microstructure (caudate to cortical regions) | Microstructure of L parieto-insular tract | |
| Clerkin et al (66) | 16/19/32 | 9(1) | 24(2) | fMRI: cued response preparation | - | Thalamo-cortical connectivity during response preparation | Thalamic activity |
| Francx et al. (65) | 74 (persistent symptoms)/55 (improving symptoms; 19 reached remission)/100 | 12 | 18 (3) | Resting state fMRI: Cognitive control network | - | Hyper- connectivity in anterior regions of cognitive control network | - |
Convergence:
The convergence model derives support from studies examining neural substrate of ‘top-down’ processes, such as response inhibition and conflict monitoring. One study prospectively acquired neuroanatomic images in tandem with clinical assessments from childhood though adolescence into early adulthood on 92 children with ADHD and 184 never affected controls (48). It found a link between early adult symptom outcomes and the developmental trajectory of cortical regions that comprise the cognitive control network–specifically, change in the thickness of the posterior cingulate, right inferior parietal and dorsolateral prefrontal cortex. Here, more symptoms of inattention in adulthood were tied to higher rates of cortical thinning during adolescence. These trajectory differences meant that as inattention resolved throughout adolescence, there was a significant convergence towards typical cortical dimensions, rectifying early anomalies. Persisting inattention was tied to anomalies in these regions that persisted throughout adolescence. The findings held when those with comorbid disorders or on regular psychostimulant medication were excluded. By combining clinical and neuroanatomic observations, the study suggests convergence of cortical anatomy of the cognitive control network occurring in tandem with improvements in inattention.
Several other studies of ‘top-down’ processes examined cohorts followed clinically from childhood who had their neuroimaging conducted for the first-time in adolescence or early adulthood. Using fMRI, one study found that under conditions of high cognitive demand (with combined stimulus and response conflicts), patterns of brain activity in a group of young adults who had remitted did not differ from never-affected controls (49). Similar findings emerge from two fMRI study of response inhibition (50, 51). Both studies found that atypical activation was confined to those who had persisting symptoms; those showing symptoms resolution were indistinguishable from the never-affected. This finding extends to the microstructure of some white matter tracts that connect the cortical region regions involved in motor inhibition control, such as the superior longitudinal fasciculus, and tracts connecting the caudate to multiple cortical regions (52-54).
Symptom improvement during adolescence may also be underpinned by convergence towards more typical dimensions of the neural substrates supporting 'bottom-up', automatic processes. One study found that those who had remitted by eighteen years of age did not differ from the never-affected on electrophysiological measures of 'bottom-up' processes of vigilance and response preparation, such as the contingent negative variation (the cerebral potential that follows a warning stimulus preparing the individual to respond to an imperative stimulus) (55, 56). Using the same electrophysiological data, markers of ‘top-down’ cognitive processing were also extracted, such as conflict monitoring activity following an incongruent stimulus in the flanker test (the N2 signal). In these ‘top-down’ cognitive indices, remitters generally occupied a position intermediate to the never affected and persistent groups. This raises the interesting possibility that these adolescent remitters were heading towards normalization, which may eventually be reached by adulthood.
Carried forward.
Indistinguishable neural substrates in adolescent remitters and the never-affected are usually interpreted as the result of the correction of childhood anomalies, in keeping with the convergence model. However, as noted above, all of the functional imaging studies were conducted at an adolescent/early adulthood endpoint only. Thus, it is possible that improvers during adolescence had more typical brain function in childhood than persisters that is simply carried forward into adolescence, promoting symptom improvement.
These considerations are pertinent to role of the default mode network (DMN) in adolescent ADHD (57, 58). The DMN refers to the neural systems that become most prominent during task free periods that evoke introspective processing, and intrusions of the DMN into task-oriented processing have been tied to key ADHD features such as deficient sustained attention (59). Might DMN anomalies also impact symptom trajectories? Two studies reported that atypical activity within the DMN was confined to young adults with persistent ADHD; those with remitted ADHD showed typical DMN activity (60, 61). However, as these studies examined the adult ‘endpoint’ only, the typical DMN in remitters could reflect either a correction of childhood DMN anomalies or a more typical childhood DMN carried forward by those destined to remit.
Cascading anomalies.
The lack of longitudinal imaging means there is also little direct evidence of symptoms of ADHD per se exacerbating or causing downstream neural anomalies. One relevant study, albeit of childhood, used cross-lag panel analyses to show that higher ratings for externalizing symptoms–including ADHD–at age 8 predicted smaller subcortical volumes and atypical white matter microstructure two years later (62). The reverse relationships did not hold: brain measures did not predict later symptoms. These findings suggest symptoms of ADHD were causally predominant and led to ‘downstream’ neural anomalies. A similar chain of causal predominance might hold in adolescence; a possibility that only longitudinal imaging and clinical data can address.
Compensation.
Next we consider examples of neural compensation, in which adolescents showing symptom improvement exhibit unique patterns of neuronal reorganization that might compensate for core symptoms. One study used resting state fMRI to define the cognitive control network at an average of 18 years of age among youth with and without a childhood history of ADHD (63). The study showed that improvement in hyperactivity-impulsivity during adolescence was associated with increased connectivity between anterior regions of the cognitive control network. This pattern of anterior hyper-connectivity was unique to those who showed improvement–not shared by either the persisters or never affected–and may represent the substrate for improved cognitive control and resolution of impulsivity. A second example consistent with compensation comes from a fMRI study of response preparation (64). It found that young adult remitters showed enhanced functional connectivity between the thalamus and the prefrontal cortex during the preparation of responses to a cue: a connectivity feature that was not prominent in either persisters or the never-affected.
Fixed anomalies.
Finally, there are several examples of fixed neural anomalies that linger, regardless of adolescent symptom course. Interestingly, many instances of fixed anomalies occur at subcortical levels, or pertain to connectivity between subcortical and cortical regions. For example, one study mapped the neural substrate of response preparation–a 'bottom-up' process–and found atypical hypo-activation in thalamo-striato-prefrontal cortical regions during response preparation in both those with childhood ADHD remitted by late adolescence and persisters (64). A separate study of inhibitory processes, mentioned earlier, likewise found that atypical activity in the caudate during motor inhibition was shared by those with a childhood history of ADHD (51). That is, remitters and persisters both differed significantly from the never-affected. Finally, applying network analytic techniques to EEG data acquired during an arrow-flanker task, atypical patterns of functional hyperconnectivity in several bands were found in both persisters and remitters (65). Such findings are compatible with the concept of fixed anomalous brain function that is carried through adolescence regardless of symptomatic improvement. Fixed anomalies, which are by definition unrelated to ADHD outcomes, tell us where not to look for mechanisms driving clinical course: these must be occurring at other neural levels. It is thus notable that studies reporting fixed anomalies mostly also found dynamic processes such as convergence occurring in other neural processes among remitters.
To summarize, a longitudinal neuroanatomic study found direct evidence of convergence, with adolescent improvement in symptoms underpinned by the correction of childhood cortical anatomic anomalies. Functional imaging and white matter tract microstructural studies conducted in late adolescence also mostly find that remitters do not differ from the never-affected. However, as noted earlier, the absence of imaging in childhood in these studies means these findings are compatible with models of either convergence or relatively typical childhood neural features being ‘carried forward’. The paucity of longitudinal data also make it difficult to assess whether neural anomalies may arise partly as a consequence of symptoms, as posited by the model of emergence. Unique, possibly compensatory patters of neural activity have been noted in some studies among adolescents whose ADHD has improved. Finally, several studies note that different processes may be occurring at different neural levels.
Future directions.
There are several key areas for future research. First, how do genomic factors relate to neurodevelopmental trajectories and ADHD symptom course? It is appealing to assume that the neurodevelopmental trajectories lie along the causal pathway between genetic risk and clinical course (47). However, while plausible, a demonstration of such a mediating role for neurodevelopmental trajectories is needed and could be attained through the use of longitudinal, familial (ideally twin) observations or the experimental manipulation of trajectories (e.g. accelerating the neural changes of convergence or compensation through treatment) (66). It is also possible that overlapping genetic factors are associated independently with both neurodevelopmental trajectories and ADHD outcomes, implying that the trajectories indicate likely clinical course but do not drive outcomes
Some initial genetic hypotheses can be made concerning the different neurodevelopmental models. Fixed neural anomalies might reflect the genomic factors that underpin the onset rather than the course of ADHD symptoms (5). Similarly, as different levels of genetic risk for ADHD are ‘carried forward’ from childhood into adulthood, so a smaller polygenic or rare variant burden may predispose to remission. The neural reorganization that underpins compensation could reflect the adolescent emergence of ‘new’ genetic factors, not shared with the onset of the disorder (3). Conversely, convergence might be underpinned by genes with deleterious neural effects going ‘off-line’. Partly heritable cognitive factors may also be pertinent: for example are those with greater cognitive reserve, reflected in a higher IQ, more likely to show neural reorganization and compensation? Finally, in considering the ‘cascading’ model, we note that ADHD symptoms can lead to many adverse exposures- such as suboptimal interactions with teachers, peers and parents- and the accumulation of such exposures could exacerbate symptoms. It is also possible that cascading anomalies could reflect new genetic risk factors coming ‘on-line’ with age, or an interaction of such genetic susceptibility with suboptimal environmental exposures (67, 68). A further goal is to also map the impact of pharmacological and behavioral treatments on these neurodevelopmental trajectories, determining if efficacy is underpinned by similar neural bases and influenced by similar genomic factors.
A mechanistic understanding of the complex course of adolescent ADHD could be attained by delineating the underlying neural processes and how they are impacted by genomic risk. Such research might help individualize treatment based on prognosis and drive the development of new treatment strategies.
Supplementary Material
Acknowledgments.
The authors are funded by the intramural programs of the National Human Genome Research Institute and the National Institute of Mental Health.
Footnotes
Disclosures. The authors declare no biomedical financial interests or potential conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Sibley MH, Mitchell JT, Becker SP (2016): Method of adult diagnosis influences estimated persistence of childhood ADHD: a systematic review of longitudinal studies. The Lancet Psychiatry. 3:1157–1165. [DOI] [PubMed] [Google Scholar]
- 2.Faraone SV, Biederman J, Mick E (2006): The age-dependent decline of attention deficit hyperactivity disorder: a meta-analysis of follow-up studies. Psychological Medicine. 36:159–165. [DOI] [PubMed] [Google Scholar]
- 3.Pingault JB, Viding E, Galera C, Greven C, Zheng Y, R. P, et al. (2015): Genetic and environmental influences on the developmental course of attention-deficit/hyperactivity disorder symptoms from childhood to adolescence. JAMA Psychiatry. doi: 10.1001/jamapsychiatry.2015.0469 T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Swanson JM, Hinshaw SP, Arnold LE, Gibbons RD, Marcus S, Hur K, et al. (2007): Secondary evaluations of MTA 36-month outcomes: propensity score and growth mixture model analyses. Journal of the American Academy of Child & Adolescent Psychiatry. 46:1003–1014. [DOI] [PubMed] [Google Scholar]
- 5.Riglin L, Collishaw S, Thapar AK, Dalsgaard S, Langley K, Smith GD, et al. (2016): Association of genetic risk variants with attention-deficit/hyperactivity disorder trajectories in the general population. JAMA psychiatry. 73:1285–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tandon M, Tillman R, Agrawal A, Luby J (2016): Trajectories of ADHD severity over 10 years from childhood into adulthood. ADHD Attention Deficit and Hyperactivity Disorders. 8:121–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Döpfner M, Hautmann C, Görtz-Dorten A, Klasen F, Ravens-Sieberer U, Group BS (2015): Long-term course of ADHD symptoms from childhood to early adulthood in a community sample. European Child & Adolescent Psychiatry. 24:665–673. [DOI] [PubMed] [Google Scholar]
- 8.Murray AL, Booth T, Eisner M, Auyeung B, Murray G, Ribeaud D (2019): Sex differences in ADHD trajectories across childhood and adolescence. Developmental science. 22:e12721. [DOI] [PubMed] [Google Scholar]
- 9.Malone PS, Van Eck K, Flory K, Lamis DA (2010): A mixture-model approach to linking ADHD to adolescent onset of illicit drug use. Developmental psychology. 46:1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Larsson H, Dilshad R, Lichtenstein P, Barker ED (2011): Developmental trajectories of DSM-IV symptoms of attention-deficit/hyperactivity disorder: Genetic effects, family risk and associated psychopathology. Journal of Child Psychology and Psychiatry. 52:954–963. [DOI] [PubMed] [Google Scholar]
- 11.Lecendreux M, Silverstein M, Konofal E, Cortese S, Faraone SV (2019): A 9-Year Follow-Up of Attention-Deficit/Hyperactivity Disorder in a Population Sample. J Clin Psychiatry. 80. [DOI] [PubMed] [Google Scholar]
- 12.Agnew-Blais JC, Polanczyk GV, Danese A, Wertz J, Moffitt TE, Arseneault L (2016): Evaluation of the persistence, remission, and emergence of attention-deficit/hyperactivity disorder in young adulthood. JAMA psychiatry. 73:713–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Caye A, Spadini AV, Karam RG, Grevet EH, Rovaris DL, Bau CH, et al. (2016): Predictors of persistence of ADHD into adulthood: a systematic review of the literature and meta-analysis. European Child & Adolescent Psychiatry. 25:1151–1159. [DOI] [PubMed] [Google Scholar]
- 14.Sibley MH, Rohde LA, Swanson JM, Hechtman LT, Molina BS, Mitchell JT, et al. (2017): Late-onset ADHD reconsidered with comprehensive repeated assessments between ages 10 and 25. American Journal of Psychiatry.appi.ajp.2017.17030298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Caye A, Sibley MH, Swanson JM, Rohde LA (2017): Late-Onset ADHD: Understanding the Evidence and Building Theoretical Frameworks. Current psychiatry reports. 19:106. [DOI] [PubMed] [Google Scholar]
- 16.Asherson P, Agnew-Blais J (2019): Annual Research Review: Does late-onset attention-deficit/hyperactivity disorder exist? Journal of Child Psychology and Psychiatry. 60:333–352. [DOI] [PubMed] [Google Scholar]
- 17.Charach A, Yeung E, Climans T, Lillie E (2011): Childhood attention-deficit/hyperactivity disorder and future substance use disorders: comparative meta-analyses. Journal of the American Academy of Child & Adolescent Psychiatry. 50:9–21. [DOI] [PubMed] [Google Scholar]
- 18.Chronis-Tuscano A, Molina BS, Pelham WE, Applegate B, Dahlke A, Overmyer M, et al. (2010): Very early predictors of adolescent depression and suicide attempts in children with attention-deficit/hyperactivity disorder. Archives of general psychiatry. 67:1044–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Meier SM, Pavlova B, Dalsgaard S, Nordentoft M, Mors O, Mortensen PB, et al. (2018): Attention-deficit hyperactivity disorder and anxiety disorders as precursors of bipolar disorder onset in adulthood. The British Journal of Psychiatry. 213:555–560. [DOI] [PubMed] [Google Scholar]
- 20.Duffy A (2012): The Nature of the Association Between Childhood ADHD and the Development of Bipolar Disorder: A Review of Prospective High-Risk Studies. American Journal of Psychiatry. 169:1247–1255. [DOI] [PubMed] [Google Scholar]
- 21.Cole J, Ball HA, Martin NC, Scourfield J, McGuffin P (2009): Genetic overlap between measures of hyperactivity/inattention and mood in children and adolescents. Journal of the American Academy of Child & Adolescent Psychiatry. 48:1094–1101. [DOI] [PubMed] [Google Scholar]
- 22.Pettersson E, Larsson H, Lichtenstein P (2016): Common psychiatric disorders share the same genetic origin: a multivariate sibling study of the Swedish population. Molecular psychiatry. 21:717–721. [DOI] [PubMed] [Google Scholar]
- 23.Froehlich TE, Anixt JS, Loe IM, Chirdkiatgumchai V, Kuan L, Gilman RC (2011): Update on environmental risk factors for attention-deficit/hyperactivity disorder. Current psychiatry reports. 13:333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Uher R, Zwicker A (2017): Etiology in psychiatry: embracing the reality of poly-gene-environmental causation of mental illness. World Psychiatry. 16:121–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mackie S, Shaw P, Lenroot R, Pierson R, Greenstein DK, Nugent TF 3rd, et al. (2007): Cerebellar development and clinical outcome in attention deficit hyperactivity disorder.[see comment]. American Journal of Psychiatry. 164:647–655. [DOI] [PubMed] [Google Scholar]
- 26.Shaw P, Lerch J, Greenstein D, Sharp W, Clasen L, Evans A, et al. (2006): Longitudinal mapping of cortical thickness and clinical outcome in children and adolescents with attention deficit/hyperactivity disorder. Archives of General Psychiatry. 63:540–549. [DOI] [PubMed] [Google Scholar]
- 27.Whitfield-Gabrieli S, Wendelken C, Nieto-Castañón A, Bailey SK, Anteraper SA, Lee YJ, et al. (2019): Association of Intrinsic Brain Architecture With Changes in Attentional and Mood Symptoms During Development. JAMA psychiatry. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.van Lieshout M, Luman M, Twisk JWR, Faraone SV, Heslenfeld DJ, Hartman CA, et al. (2017): Neurocognitive Predictors of ADHD Outcome: a 6-Year Follow-up Study. Journal of Abnormal Child Psychology. 45:261–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sjöwall D, Bohlin G, Rydell A-M, Thorell LB (2017): Neuropsychological deficits in preschool as predictors of ADHD symptoms and academic achievement in late adolescence. Child Neuropsychology. 23:111–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Karalunas SL, Gustafsson HC, Dieckmann NF, Tipsord J, Mitchell SH, Nigg JT (2017): Heterogeneity in development of aspects of working memory predicts longitudinal attention deficit hyperactivity disorder symptom change. Journal of abnormal psychology. 126:774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Faraone SV, Larsson H (2018): Genetics of attention deficit hyperactivity disorder. Molecular psychiatry.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen Q, Brikell I, Lichtenstein P, Serlachius E, Kuja-Halkola R, Sandin S, et al. (2017): Familial aggregation of attention-deficit/hyperactivity disorder. Journal of Child Psychology and Psychiatry. 58:231–239. [DOI] [PubMed] [Google Scholar]
- 33.Biederman J, Faraone S, Milberger S, Curtis S, Chen L, Marrs A, et al. (1996): Predictors of Persistence and Remission of ADHD into Adolescence: Results from a Four-Year Prospective Follow-up Study. Journal of the American Academy of Child & Adolescent Psychiatry. 35:343–351. [DOI] [PubMed] [Google Scholar]
- 34.Rovira P, Demontis D, Sánchez-Mora C, Zayats T, Klein M, Mota NR, et al. (2020): Shared genetic background between children and adults with attention deficit/hyperactivity disorder. Neuropsychopharmacology.1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Elia J, Glessner JT, Wang K, Takahashi N, Shtir CJ, Hadley D, et al. (2012): Genome-wide copy number variation study associates metabotropic glutamate receptor gene networks with attention deficit hyperactivity disorder. Nat Genet. 44:78–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Thapar A, Martin J, Mick E, Vásquez AA, Langley K, Scherer SW, et al. (2016): Psychiatric gene discoveries shape evidence on ADHD’s biology. Molecular psychiatry. 21:1202–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Langley K, Martin J, Agha SS, Davies C, Stergiakouli E, Holmans P, et al. (2011): Clinical and cognitive characteristics of children with attention-deficit hyperactivity disorder, with and without copy number variants. The British Journal of Psychiatry. 199:398–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Akutagava-Martins GC, Salatino-Oliveira A, Genro JP, Contini V, Polanczyk G, Zeni C, et al. (2014): Glutamatergic copy number variants and their role in attention-deficit/hyperactivity disorder. American Journal of Medical Genetics Part B: Neuropsychiatric Genetics. 165:502–509. [DOI] [PubMed] [Google Scholar]
- 39.Wood AC, Buitelaar J, Rijsdijk F, Asherson P, Kuntsi J (2010): Rethinking shared environment as a source of variance underlying attention-deficit/hyperactivity disorder symptoms: comment on Burt (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sharp W, Mangalmurti A, Hall C, Choudhury S, Shaw P (2019): Associations between neighborhood, family factors and symptom change in childhood attention deficit hyperactivity disorder. Social Science & Medicine.112203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shaw M, Hodgkins P, Caci H, Young S, Kahle J, Woods AG, et al. (2012): A systematic review and analysis of long-term outcomes in attention deficit hyperactivity disorder: effects of treatment and non-treatment. BMC medicine. 10:99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dalsgaard S, Østergaard SD, Leckman JF, Mortensen PB, Pedersen MG (2015): Mortality in children, adolescents, and adults with attention deficit hyperactivity disorder: a nationwide cohort study. The Lancet. 385:2190–2196. [Google Scholar]
- 43.Swanson JM, Arnold LE, Molina BS, Sibley MH, Hechtman LT, Hinshaw SP, et al. (2017): Young adult outcomes in the follow-up of the multimodal treatment study of attention-deficit/hyperactivity disorder: symptom persistence, source discrepancy, and height suppression. Journal of Child Psychology and Psychiatry. 58:663–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sudre G, Mangalmurti A, Shaw P (2018): Growing out of attention deficit hyperactivity disorder: insights from the ‘remitted’brain. Neuroscience & Biobehavioral Reviews. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Di Martino A, Fair DA, Kelly C, Satterthwaite TD, Castellanos FX, Thomason ME, et al. (2014): Unraveling the miswired connectome: a developmental perspective. Neuron. 83:1335–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Halperin JM, Schulz KP (2006): Revisiting the role of the prefrontal cortex in the pathophysiology of attention-deficit/hyperactivity disorder. Psychological Bulletin. 132:560–581. [DOI] [PubMed] [Google Scholar]
- 47.Kendler KS, Neale MC (2010): Endophenotype: a conceptual analysis. Molecular Psychiatry. 15:789–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shaw P, Malek M, Watson B, Greenstein D, de Rossi P, Sharp W (2013): Trajectories of Cerebral Cortical Development in Childhood and Adolescence and Adult Attention- Deficit/Hyperactivity Disorder. Biological Psychiatry. 74:599–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schulz KP, Li X, Clerkin SM, Fan J, Berwid OG, Newcorn JH, et al. (2017): Prefrontal and parietal correlates of cognitive control related to the adult outcome of attention-deficit/hyperactivity disorder diagnosed in childhood. Cortex. 90:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schneider MF, Krick CM, Retz W, Hengesch G, Retz-Junginger P, Reith W, et al. (2010): Impairment of fronto-striatal and parietal cerebral networks correlates with attention deficit hyperactivity disorder (ADHD) psychopathology in adults: a functional magnetic resonance imaging (fMRI) study. Psychiatry Research: Neuroimaging. 183:75–84. [DOI] [PubMed] [Google Scholar]
- 51.Szekely E, Sudre GP, Sharp W, Leibenluft E, Shaw P (2017): Defining the neural substrate of the adult outcome of childhood ADHD: A multimodal neuroimaging study of response inhibition. American Journal of Psychiatry. 174:867–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sudre G, Shaw P, Wharton A, Weingart D, Sharp W, Sarlls J (2015): White matter microstructure and the variable adult outcome of childhood Attention Deficit Hyperactivity Disorder. Neuropsychopharmacology. 40:746–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Francx W, Zwiers MP, Mennes M, Oosterlaan J, Heslenfeld D, Hoekstra PJ, et al. (2015): White matter microstructure and developmental improvement of hyperactive/impulsive symptoms in Attention,ÄêDeficit/Hyperactivity Disorder. Journal of Child Psychology and Psychiatry. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Luo Y, Halperin JM, Li X (2020): Anatomical substrates of symptom remission and persistence in young adults with childhood attention deficit/hyperactivity disorder. European Neuropsychopharmacology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cheung CH, Rijsdijk F, McLoughlin G, Brandeis D, Banaschewski T, Asherson P, et al. (2016): Cognitive and neurophysiological markers of ADHD persistence and remission. The British Journal of Psychiatry. 208:548–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Michelini G, Kitsune GL, Cheung CH, Brandeis D, Banaschewski T, Asherson P, et al. (2016): Attention-deficit/hyperactivity disorder remission is linked to better neurophysiological error detection and attention-vigilance processes. Biological psychiatry. 80:923–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Castellanos FX, Aoki Y (2016): Intrinsic functional connectivity in attention-deficit/hyperactivity disorder: A science in development. Biological psychiatry: cognitive neuroscience and neuroimaging. 1:253–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Konrad K, Eickhoff SB (2010): Is the ADHD brain wired differently? A review on structural and functional connectivity in attention deficit hyperactivity disorder. Human Brain Mapping. 31:904–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wen X, Yao L, Liu Y, Ding M (2012): Causal interactions in attention networks predict behavioral performance. The Journal of neuroscience. 32:1284–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mattfeld AT, Gabrieli JD, Biederman J, Spencer T, Brown A, Kotte A, et al. (2014): Brain differences between persistent and remitted attention deficit hyperactivity disorder. Brain. awu137. [DOI] [PubMed] [Google Scholar]
- 61.Sudre G, Szekely E, Sharp W, Kasparek S, Shaw P (2017): Multimodal mapping of the brain’s functional connectivity and the adult outcome of attention deficit hyperactivity disorder. Proceedings of the National Academy of Sciences 114:11787–11792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Muetzel RL, Blanken LM, van der Ende J, El Marroun H, Shaw P, Sudre G, et al. (2018): Tracking brain development and dimensional psychiatric symptoms in children: A longitudinal population-based neuroimaging study. American Journal of psychiatry. 175:54–62. [DOI] [PubMed] [Google Scholar]
- 63.Francx W, Oldehinkel M, Oosterlaan J, Heslenfeld D, Hartman CA, Hoekstra PJ, et al. (2015): The executive control network and symptomatic improvement in attention-deficit/hyperactivity disorder. Cortex. 73:62–72. [DOI] [PubMed] [Google Scholar]
- 64.Clerkin SM, Schulz KP, Berwid OG, Fan J, Newcorn JH, Tang CY, et al. (2013): Thalamo-Cortical Activation and Connectivity During Response Preparation in Adults With Persistent and Remitted ADHD. American Journal of Psychiatry. 170:1011–1019. [DOI] [PubMed] [Google Scholar]
- 65.Michelini G, Jurgiel J, Bakolis I, Cheung CH, Asherson P, Loo SK, et al. (2019): Atypical functional connectivity in adolescents and adults with persistent and remitted ADHD during a cognitive control task. Translational psychiatry. 9:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kendler KS, Neale MC, Kessler RC, Heath AC, Eaves LJ (1993): A longitudinal twin study of personality and major depression in women. Archives of general psychiatry. 50:853–862. [DOI] [PubMed] [Google Scholar]
- 67.Boardman JD, Daw J, Freese J (2013): Defining the environment in gene–environment research: Lessons from social epidemiology. American journal of public health. 103:S64–S72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Belsky J, Jonassaint C, Pluess M, Stanton M, Brummett B, Williams R (2009): Vulnerability genes or plasticity genes? Molecular psychiatry. 14:746–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.