

Abstract
The clinical phenotype of Gaucher disease type 3 (GD3), a neuronopathic lysosomal storage disorder, encompasses a wide array of neurological manifestations including neuro-ophthalmological findings, developmental delay, and seizures including progressive myoclonic epilepsy. Electroencephalography (EEG) is a widely available tool used to identify abnormalities in cerebral function, as well as epileptiform abnormalities indicating an increased risk of seizures. We characterized the EEG findings in GD3, reviewing 67 patients with 293 EEGs collected over nearly 50 years. Over 93% of patients had some form of EEG abnormality, most consisting of background slowing (90%), followed by interictal epileptiform discharges (IEDs) (54%), and photoparoxysmal responses (25%). The seven patients without background slowing were all under age 14 (mean 6.7 years). There was a history of seizures in 37% of this cohort; only 30% of these had IEDs on EEG. Conversely, only 56% of patients with IEDs had a history of seizures. These observed EEG abnormalities document an important aspect of the natural history of GD3 and could potentially assist in identifying neurological involvement in a patient with subtle clinical findings. Additionally, this comprehensive description of longitudinal EEG data provides essential baseline data for understanding central nervous system involvement in neuronopathic GD.
Keywords: neuronopathic Gaucher disease, electroencephalography, epileptiform activity, progressive myoclonus epilepsy, glucocerebrosidase
1. Introduction
Gaucher disease (GD) is a rare, inherited, lysosomal storage disorder caused by homozygous pathogenic variants in GBA1, the gene encoding the enzyme glucocerebrosidase (GCase). The functional deficiency of GCase causes an accumulation of glucosylceramide and glucosylsphingosine in lysosomes. The clinical phenotype associated with GD is remarkably heterogeneous and includes neuronopathic and non-neuronopathic forms of the disease. Non-neuronopathic GD, or GD type 1 (GD1), is the most common form of GD with a frequency of roughly 1 in 40,000 in the general population. The disease can present at any age and manifestations include hepatosplenomegaly, anemia, thrombocytopenia, and/or skeletal manifestations. There are two types of neuronopathic GD, type 2 or acute neuronopathic (GD2) and type 3 or chronic neuronopathic (GD3). GD2 is the most rare and most severe form of GD in which affected infants present prenatally, at birth, or in the first months of life with a rapidly progressive neurodegenerative course [1]. In contrast, patients with GD3 can have a later presentation with a wide range of associated neurological involvement, ranging from isolated slowing of horizontal saccadic eye movements to myoclonic epilepsy or developmental delay[1]. In many regions of the world, including parts of Asia and Africa, GD3 is the more frequent type encountered.
The diagnosis of GD3 can be challenging because many of the associated signs and/or symptoms of central nervous system (CNS) dysfunction can be subtle, overlooked, or may not be present until later in the disease course. The neuro-ophthalmologic finding of slowed horizontal saccadic eye movements is one of the most common unifying features of patients with GD3, but it is also difficult to quantify and may appear later in the disease course [2]. Although there are some genotype-phenotype correlations in GD, the identified pathological variants in GBA1 cannot always be relied on to determine the type of GD. For this reason, it is important to identify other clinical signs of neurologic involvement in this patient population.
Since the discovery and implementation of enzyme replacement therapy as a treatment, the prognosis for patients with GD has greatly improved[3]. While none of the currently approved and widely used therapies cross the blood-brain-barrier, patients with neuronopathic GD are living longer and with fewer systemic manifestations [4]. Currently, new brain-penetrant therapies to correct neurological involvement are being developed to address this unmet need [5]. Because of the vast heterogeneity in associated manifestations seen among patients, it is important to identify clinical features that may serve to establish parameters indicative of CNS involvement in this patient population.
Electroencephalography (EEG) is a widely used non-invasive modality permitting a unique window into underlying brain function. Epilepsy and epileptiform activity on EEG recordings have been previously reported in patients with neuronopathic GD [6]. Background slowing, particularly of the posterior dominant rhythm (PDR), has also been associated with neuronopathic forms of GD [4], although it is a non-specific indicator of cerebral dysfunction and can be seen in patients with a host of neurodegenerative conditions including Parkinson disease and progressive dementias, as well as metabolic/toxic encephalopathies, infections, and medications [7, 8].
This study evaluates EEG findings in a large cohort of patients with GD3 (n=67) followed for nearly 50 years. The aim of this study was to characterize the frequency and types of EEG abnormalities in this cohort. The availability of longitudinal data, at times spanning several decades, also enabled us to further evaluate the frequency and pattern of EEG abnormalities in patients with GD3 over time.
2. Material and methods
2.1. Patient population, protocols, studies, and consent
A retrospective chart review was performed examining the records of 67 patients with a clinical and molecular diagnosis of GD3, enrolled in studies approved by the National Human Genome Research Institute or the National Institute of Neurologic Disorders and Stroke Institutional Review Boards, at the National Institutes of Health (NIH) between 1972 and 2019 (Table 1). Written informed consents/assents were obtained as appropriate. Each patient record was evaluated by the study team and included official EEG reports generated by neurologists from the National Institute of Neurological Disorders and Stroke (NINDS) EEG Section. Each patient included completed at least one EEG study at the NIH between 1972 and 2019.
Table 1.
Cohort characteristics
| Subject | Allele A | Allele B | Sex | Age at first EEG | Age at last EEG | Number of EEGs | Seizure history (or myoclonus) | Anti-Convulsant therapy |
|---|---|---|---|---|---|---|---|---|
| 1 | V394L | RecNcil | M | 8 | 10 | 3 | Yes (myoclonus) | Yes |
| 2 | L444P | F213I | F | 6 | 6 | 1 | Yes (myoclonus) | Yes |
| 3 | L444P | L444P | F | 2 | 6 | 4 | No | |
| 4 | L444P | L444P | M | 8 | 10 | 3 | Yes | Yes |
| 5 | G377S | Y205C | F | 29 | 33 | 5 | Yes (myoclonus) | Yes |
| 6 | P122S | P122S | M | 14 | 15 | 4 | Yes | Yes |
| 7 | L444P | L444P | M | 6 | 8 | 2 | No | |
| 8 | L444P | L444P | F | 9 | 27 | 6 | No | |
| 9 | R463C | c.84insG | M | 12 | 14 | 3 | No | |
| 10 | NA | NA | F | 33 | 34 | 2 | No | |
| 11 | R463C | R257Q | F | 9 | 12 | 3 | Yes | Yes |
| 12 | R463C | RecNcil | M | 9 | 12 | 4 | No | |
| 13 | R463C | RecNcil | F | 5 | 9 | 4 | No | |
| 14 | L444P | L444P | M | 10 | 15 | 3 | No | |
| 15 | L444P | L444P | F | 11 | 13 | 3 | No | |
| 16 | D409H | RecNcil | M | 5 | 6 | 2 | Yes (myoclonus) | |
| 17 | L444P | L444P | F | 2 | 3 | 2 | No | |
| 18 | G377S | R463H | M | 8 | 10 | 4 | Yes (myoclonus) | |
| 19 | L444P | L444P | M | 7 | 7 | 1 | No | |
| 20 | L444P | L444P | M | 3 | 13 | 4 | Yes | Yes |
| 21 | NA | NA | F | 11 | 11 | 1 | No | |
| 22 | R463C | c.84insG | M | 7 | 13 | 2 | No | |
| 23 | L444P | L444P | F | 2 | 12 | 4 | No | |
| 24 | L444P | L444P | M | 8 | 12 | 4 | No | |
| 25 | L444P | L444P | M | 1 | 8 | 2 | No | |
| 26 | L444P | L444P | M | 2 | 10 | 7 | Yes | Yes |
| 27 | L444P | L444P | M | 8 | 14 | 5 | No | |
| 28 | L444P | L444P | F | 15 | 15 | 1 | No | |
| 29 | NA | NA | M | 7 | 9 | 3 | No | Yes |
| 30 | L444P | L444P | M | 6 | 16 | 13 | Yes | Yes |
| 31 | R463C | A176D | M | 4 | 9 | 5 | No | |
| 32 | L444P | L444P | M | 5 | 15 | 10 | No | |
| 33 | L444P | L444P | M | 2 | 2 | 1 | No | |
| 34 | L444P | L444P | F | 24 | 24 | 1 | Yes | Yes |
| 35 | L444P | L444P | F | 4 | 4 | 1 | Yes | Yes |
| 36 | L444P | L444P | M | 3 | 17 | 7 | No | |
| 37 | L444P | D409H+Rec7 | F | 50 | 53 | 2 | No | Yes |
| 38 | L444P | L444P | F | 8 | 10 | 2 | No | |
| 39 | N188S+Rec7 | Unknown | M | 7 | 10 | 4 | Yes (myoclonus) | |
| 40 | R463C | Y304X | M | 3 | 10 | 3 | No | |
| 41 | L444P | D409H | F | 3 | 3 | 1 | No | |
| 42 | R463C+Rec7 | RecNcil | F | 13 | 31 | 5 | No | |
| 43 | L444P | L444P | M | 1 | 16 | 12 | No | |
| 44 | D409H | D409H | F | 3 | 3 | 1 | No | |
| 45 | N188S | RecNcil | M | 19 | 25 | 9 | Yes (myoclonus) | Yes |
| 46 | V458G | RecNcil | M | 4 | 4 | 2 | Yes | |
| 47 | K157Q | D140H+E326K* | M | 20 | 27 | 7 | Yes (myoclonus) | Yes |
| 48 | L444P | L444P | M | 12 | 14 | 4 | No | |
| 49 | L444P | L444P | M | 6 | 10 | 4 | No | |
| 50 | L444P | L444P | F | 2 | 23 | 14 | Yes | |
| 51 | L444P | L444P | M | 3 | 17 | 20 | No | |
| 52 | L444P | L444P | F | 1 | 28 | 21 | No | |
| 53 | R463C | D399N | F | 7 | 14 | 6 | No | |
| 54 | c.del55bp | F216Y | M | 28 | 28 | 1 | Yes (myoclonus) | Yes |
| 55 | V394L | RecNcil | F | 3 | 5 | 3 | Yes | Yes |
| 56 | N188S | S107L | M | 11 | 11 | 1 | Yes (myoclonus) | |
| 57 | L444P | L444P | F | 25 | 25 | 1 | Yes | Yes |
| 58 | L444P | L444P | F | 19 | 20 | 2 | Yes | Yes |
| 59 | L444P | L444P | F | 19 | 21 | 2 | Yes | Yes |
| 60 | L444P | L444P | F | 9 | 19 | 5 | No | Yes |
| 61 | L444P | L444P | F | 1 | 13 | 2 | No | |
| 62 | R463C | IVS2+1G>A | M | 3 | 6 | 3 | No | |
| 63 | L444P | L444P | F | 1 | 15 | 13 | No | |
| 64 | L444P | L444P | M | 1 | 2 | 2 | No | |
| 65 | R463C | RecNcil | F | 8 | 9 | 2 | No | |
| 66 | G377S | c.102delT | M | 6 | 7 | 2 | Yes (myoclonus) | |
| 67 | L444P | L444P | M | 9 | 23 | 7 | No |
This patient also has a mutation in SCARB-218
Medications were recorded in the table if the patient had ever received the medication. Seizure medications include: Ativan, carbamazepine, clonazepam, depakene, Depakote, dilantin, diphenylydantoin, divalproex, frisium, gapapentin, klonopin, Lamictal, lamotrigine, levetiracetam, librax, mysoline, phenobarbital, Prilosec, primidone, Sinemet, Tegretol, tiracetam, Topamax, tranxene, Trileptal, urbanyl, valium, valproic acid/valproate/VPA, zarontin, and zonisamide.
2.2. Molecular analyses
Genotypic analyses were performed on 64 of the participants by examining all exons of the GBA1 gene by Sanger sequencing as previously described [9]. The remaining three subjects had biochemical testing and clinical evaluations consistent with a GD3 diagnosis.
2.3. Electroencephalogram evaluations
Study participants had EEGs consistently performed as part of a battery of tests administered at each visit, and EEG evaluations were not prompted by a suspicion of epilepsy. Each patient had 1 to 21 individual EEG evaluations performed at the NIH Clinical Center using at least 18 and typically 21 EEG channels. Electrodes were placed according to the International 10/20 system. EEGs were typically recorded for at least 25 minutes. Intermittent photic stimulation at multiple flash frequencies was performed at least once in 65 of the subjects, and studies with hyperventilation, performed when the patient was able to cooperate, were obtained in 54 patients at least once. EEGs were read using at least three standard montages by a board-certified clinical neurophysiologist at the NIH Clinical Center at the time that they were performed. Data was retrospectively extracted from these reports and was again reviewed for the purposes of this study by a board-certified clinical neurophysiologist in the NINDS EEG Section at the NIH Clinical Center (SI).
Background slowing while awake was identified if specifically noted in the interpretation or when a slow PDR for age or excessive delta or theta activity were described in the body of the report. Epileptiform activity was determined to be present if sharp wave, spike or polyspike and wave activity was reported in the body of the report or in the interpretation. Photoparoxysmal responses were noted as present if epileptiform activity was elicited during photic stimulation; positive responses were noted in patients with epileptiform activity during the rest of the recording only if present at a significantly higher rate during photic stimulation.
2.4. Statistical analyses and visualization
R (version 4.0.0) in RStudio (version 1.2.5042; R Project for Statistical Computing) was used for statistical analysis to demonstrate within-group correlation for the PDR values. Descriptive statistics were calculated from each individual’s maximum PDR (Hz) at every EEG recording date. For each EEG, a binary “yes” or “no” categorical value was recorded for background slowing, presence of epileptiform activity, photoparoxysmal responses, seizure history, and overall abnormal EEG designation. These values were determined by the EEG rater who originally summarized the results as well as the reviewing epileptologist. A binomial proportion of individuals who received a “yes” to each of the abnormalities on at least one EEG date was calculated.
3. Results
A summary of the patients included is provided in Table 1. Our retrospective analysis of reported EEG findings specifically identified mention of background slowing, epileptiform abnormalities, and photoparoxysmal responses. The frequency of these findings is noted at different ages in Table 2. The most consistent abnormality found was background slowing. Although only 205 of the total 293 EEGs were classified as having background slowing, 90% of the patients had background slowing in at least one EEG during the course of their participation in the study (Figure 1). 56% of patients also had at least one EEG with a slow maximal posterior dominant rhythm frequency compared to normative values for age (Figure 2, Table 2). Although not specific, background slowing in the absence of other known etiologies is a widely described finding suggestive of cerebral dysfunction, and may provide supportive evidence of neurological involvement in this cohort.
Table 2.
Percentage of patients with EEG abnormalities and average maximum posterior dominant rhythms by age category.
| Age range | Patients per age (%) | EEGs Total | EEGs with background slowing (%) | Patients with background slowing (%) | EEGs with IEDs (%) | Patients with IEDs (%) | EEGs with PPR (%) | Patients with PPR (%) | Maximum PDR (average) |
|---|---|---|---|---|---|---|---|---|---|
| 1-5 yrs | 26 | 57 | 28 | 50 | 21 | 35 | 5 | 4 | 7.1 |
| 6-10 yrs | 38 | 105 | 73 | 84 | 36 | 47 | 10 | 18 | 7.7 |
| 11-15 yrs | 29 | 76 | 87 | 97 | 30 | 41 | 8 | 21 | 8.1 |
| 16-20 yrs | 12 | 18 | 67 | 67 | 28 | 25 | 17 | 17 | 8.6 |
| 21+ | 14 | 37 | 92 | 86 | 51 | 36 | 43 | 29 | 6.9 |
| Total | 67 | 293 | 70 | 90 | 33 | 54 | 13 | 25 | 7.6 |
| % male | 55 | ||||||||
| % female | 45 |
EEG= electroencephalogram; IED= interictal epileptiform activity; PPR= photoparoxysmal response; PDR= posterior dominant rhythm
Figure 1:
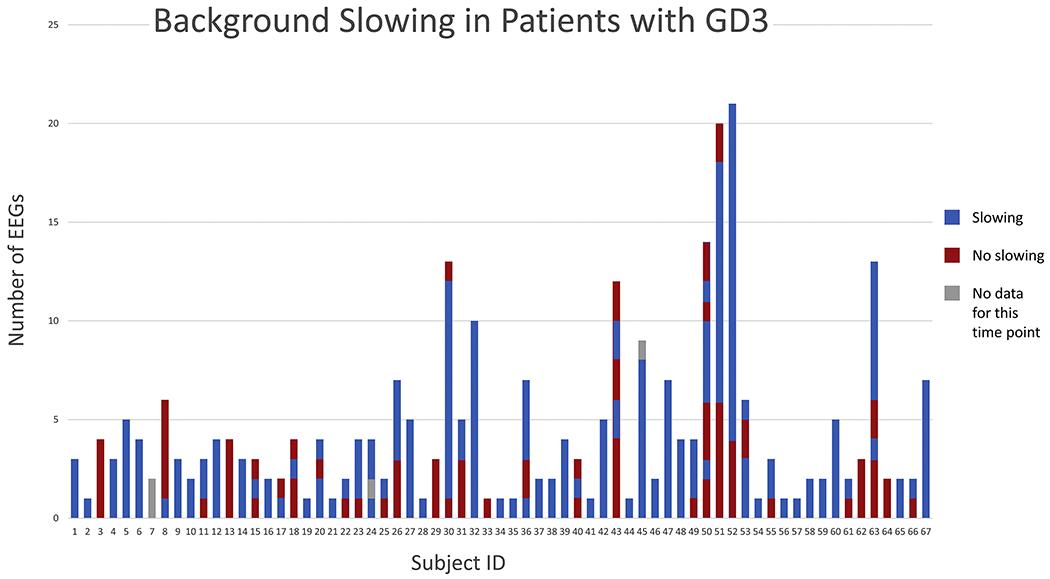
Background slowing in patients with GD3 over time. From bottom to top, The EEGs are ordered from bottom to top for each individual by time with the top-most EEG being the most recent data point taken from the patient.
Figure 2:
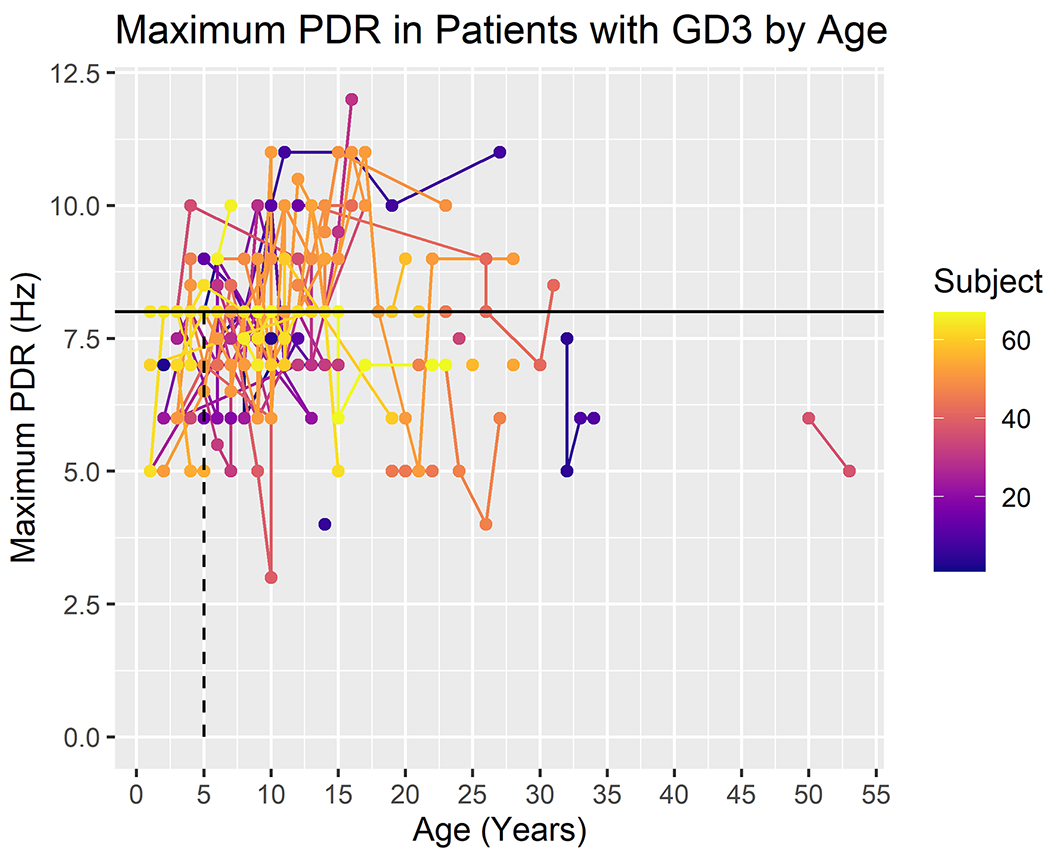
Maximum posterior dominant rhythm (PDR) in patients with GD3 by age (in years). The expected lower limit of normal frequency for the PDR increases with age, reaching alpha frequency by approximately age 3. For adults, a maximal PDR of at least 8.5 Hz is considered to be normal. The color scale reflects subject number.
Both seizures and interictal epileptiform activity were also frequently reported in our cohort. Over one-third (27/67) of patients had clinical evidence of some form of epilepsy, predominantly consisting of myoclonic seizures or generalized tonic-clonic seizures, although focal seizures with impaired awareness were reported in several patients. Myoclonus was noted in the history in eleven patients, mostly in the context of progressive myoclonus epilepsy (PME). Generalized tonic-clonic seizures, myoclonus, as well as PME have been described in GD3. In patients with PME, the age at presentation and rate of progression varied widely, ranging from relatively early onset of myoclonus, along with severe developmental and/or cognitive deficits, visceral involvement, and early death, to a more mild neurologic phenotype, with later onset of myoclonus, slower progression of neurological symptoms, and less visceral involvement. Adult patients tend to have generalized seizures in addition to myoclonus[6].
Interictal epileptiform activity is considered a marker of an increased risk of seizures. In this GD3 cohort, 54% were noted to have such activity in the form of polyspikes, spike-and-slow waves, and/or photoparoxysmal responses. Of 36 GD3 patients with interictal epileptiform activity on EEG, approximately 40% had focal or multifocal discharges, while the remainder had generalized or a combination of multifocal and generalized epileptiform activity. Of the patients demonstrating epileptiform activity, only 56% had a known history of clinical seizures. Thus, while the presence of epileptiform activity does suggest neurologic involvement and an overall increased risk of seizures, a significant proportion of patients in this cohort with IEDs did not have clinically apparent seizures.
In comparison, Zivin and Ajmone-Marsan found an overall incidence of epileptiform discharges of only 2.2% in EEGs performed on 6947 non-epileptic subjects; of these 15% eventually had seizures[10]. In a similar large cohort of children (age 6-13) without a history of seizures, 3.5% had interictal epileptiform activity, and 5% went on to develop clinical seizures[11]. Finally, in patients with known clinical seizures and no epileptiform findings on EEG, additional prolonged studies including recordings during sleep may have a higher yield for detecting abnormalities.
In addition to spontaneous interictal epileptiform activity, 25% of the patients in this cohort had an increased frequency of epileptiform activity during photic stimulation, most often consisting of generalized spike or polyspike and wave activity, and at times accompanied by myoclonus. Such photoparoxysmal responses (PPRs) are rare in the general population. A prospective study in patients newly diagnosed with epilepsy in Great Britain found PPRs reported in 2% of EEGs overall and 10% of EEG in subjects aged 17-19 years, suggesting that they are more common in this age group[12]. PPRs have been described in patients with photosensitive occipital lobe epilepsy[13], as well as in patients with generalized epilepsy syndromes. One study found PPRs in 40% of patients with absence seizures and 20% with generalized tonic-clonic seizures[14]. PPRs are also common in juvenile myoclonic epilepsy [15], as well as in multiple forms of PME, including Unverricht-Lundborg Disease, Myoclonus Epilepsy with Ragged-Red-Fibers and Lafora Disease [16].
4. Discussion
To our knowledge, this is the largest cohort of patients with GD3 evaluated longitudinally by EEG over the course of almost 50 years. Fifty-five patients had more than one EEG done, and 19 had 5 or more studies. An analysis of data chronologically for each patient with >1 evaluation did not provide any specific pattern or evolution of abnormalities, but the repeated evaluations did ultimately uncover additional relevant findings. Especially with young patients, serial evaluations at regular intervals may provide additional evidence for neurological involvement in this disease.
EEG examinations are a widely accessible non-invasive study that provide brief snapshots of brain function over time. Identification of EEG abnormalities, such as background slowing or interictal epileptiform discharges, may be useful when discerning between a diagnosis of GD1 versus GD3. However, an abnormal EEG does not provide a definitive diagnosis of GD3, since patients with GD1 could have epilepsy from other causes. Several patients in this cohort were originally diagnosed with GD1, but changes in either their neuro-ophthalmologic or neurological evaluation over time resulted in a subsequent reclassification. Thus, the observation of background slowing, epileptiform activity, or the presence of seizures could potentially be used as an additional finding to identify CNS involvement in a patient with GD with subtle or absent neurological clinical signs. In many of the patients, these EEG findings were not observed at the first evaluation but were captured in subsequent studies.
The abnormalities identified in this study, namely background slowing and interictal epileptiform activity, tend to occur intermittently. They are therefore subject to sampling error given the limited duration of a routine EEG study. The presence of abnormalities may also be affected by many factors not related to the severity of CNS pathology, including the state of arousal, medications, and time of day, This variability is apparent in our cohort, as the presence of background slowing was present consistently in 36 of the 60 patients with background slowing on any EEG recording. The percentage of the total patients with background slowing, epileptiform activity, and PPRs at different age ranges is shown in Figure 3. All three findings were less frequent in children under 5 years of age. The EEG background remained normal during the duration of the study in 7 patients (Figure 1). However, all of the 7 were children, aged 2-14 at their last assessment, and 4 were under age 6, so further follow-up is warranted. Each of the 7 had slow horizontal saccades, and none had clinical seizures or myoclonus. Twelve (18%) of the patients who initially had a normal EEG ultimately demonstrated epileptiform activity during subsequent studies. Thus, repeated studies helped to establish the finding over time, even after several normal EEGs.
Figure 3:
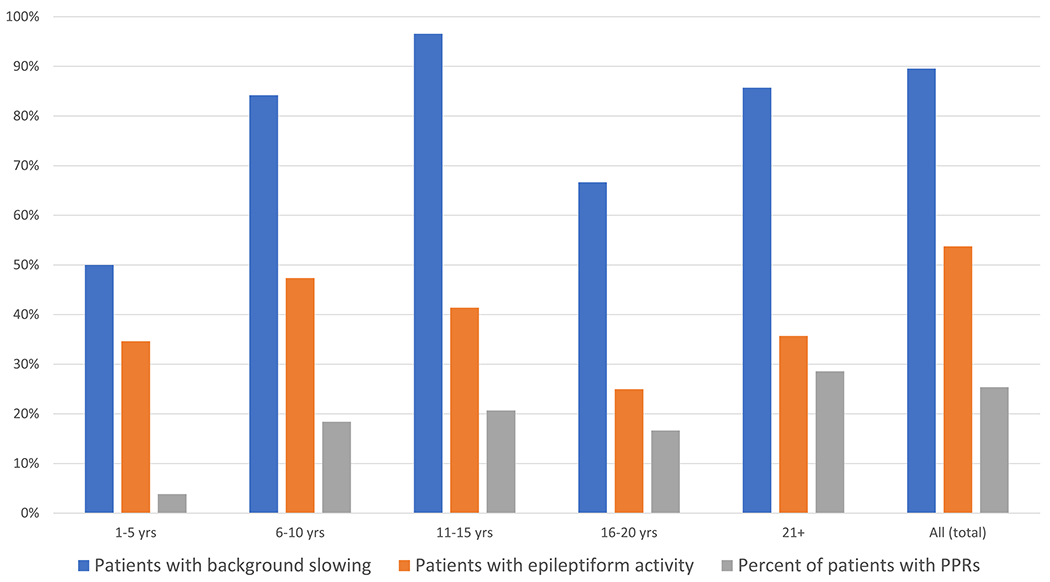
Percentage of total patients in the GD3 cohort with background slowing, epileptiform activity, and photoparoxysmal responses (PPRs) shown at different age intervals.
In this patient cohort, the most common GBA1 mutations identified were L444P(p.L483C), R463C(p.R502C,) and RecNcil. Of the 64 patients genotyped in this study, 36 (56%) were confirmed to be L444P homozygotes. While most patients confirmed to have genotype L444P/L444P without other evidence of other pseudogene-derived sequence tend to have GD3, several exceptions have been reported. Patients with genotype L444P/L444P have a range of associated phenotypes, spanning from severe developmental delays to highly functional college graduates[17]. Twelve patients had an allele with R463C, a mutation associated with all three types of GD. Additionally, 8 individuals had one copy of the recombinant allele RecNcil, but never in combination with a second L444P allele. As previously reported [6] mutations G377S (p.G416S) and N188S (p.G427S) were often associated with myoclonus (Table 1). As expected, none of the patients carried a N370S (p.N409S) allele.
5. Conclusion
As new therapies are developed for the neuronopathic forms of GD, it will be important to identify any associated disease parameters or diagnostic features that could be monitored over time. Diffuse background slowing was the most common EEG abnormality in our cohort. Because this finding is non-specific, it would not be uniquely predictive of a GD3 phenotype, but in concert with other suggestive findings, the presence of EEG abnormalities might have diagnostic utility in confirming neurologic involvement. A normal study is likely of less utility, as identified abnormalities were not always constant across EEG studies in the same patient. This variability also suggests that the presence or absence of EEG abnormalities would not necessarily serve as an efficacy parameter for future therapeutic trials. However, consistent changes in EEG patterns in an individual patient after therapeutic intervention could be important to capture in future clinical trials. One weakness of this study is that the frequency of EEG abnormalities in patients with non-neuronopathic GD has not been well-documented, and might be addressed in future studies. However, our longitudinal findings provide essential baseline data describing CNS involvement in patients with GD3 and might serve as a prognostic guide when counseling patients with GD and their caregivers.
Highlights.
Gaucher disease(GD) type 3 is a lysosomal disorder with neurological manifestations.
GD type 3 is phenotypically heterogenous.
Neurological signs and symptoms may be subtle at early stages of the disease.
EEG is a non-invasive clinical tool to characterize abnormalities in cerebral function.
EEGs have utility in the evaluation of Gaucher disease.
Acknowledements and Funding Support:
This work was supported by the Intramural Research Programs of the National Institutes of Health, National Human Genome Research Institute and National Institute of Neurological Disorders and Stroke.
Abreviations used:
- GD
Gaucher disease
- EEG
electroencephalogram
- IED
interictal epileptiform activity
- PPR
photoparoxysmal response
- PDR
posterior dominant rhythm
- CNS
central nervous system
- GCase
glucocerebrosidase
- PME
Progressive myoclonic epilepsy
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Schiffmann R, Sevigny J, Rolfs A, et al. The definition of neuronopathic Gaucher disease. J Inherit Metab Dis. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Benko W, Ries M, Wiggs EA, Brady RO, Schiffmann R, Fitzgibbon EJ. The saccadic and neurological deficits in type 3 Gaucher disease. PLoS One. 2011. 6: e22410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Weinreb NJ, Charrow J, Andersson HC, et al. Effectiveness of enzyme replacement therapy in 1028 patients with type 1 Gaucher disease after 2 to 5 years of treatment: a report from the Gaucher Registry. Am J Med. 2002. 113: 112–119. [DOI] [PubMed] [Google Scholar]
- [4].Altarescu G, Hill S, Wiggs E, et al. The efficacy of enzyme replacement therapy in patients with chronic neuronopathic Gaucher’s disease. J Pediatr. 2001. 138: 539–547. [DOI] [PubMed] [Google Scholar]
- [5].Stirnemann J, Belmatoug N, Camou F, et al. A Review of Gaucher Disease Pathophysiology, Clinical Presentation and Treatments. Int J Mol Sci. 2017. 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Park JK, Orvisky E, Tayebi N, et al. Myoclonic epilepsy in Gaucher disease: genotype-phenotype insights from a rare patient subgroup. Pediatr Res. 2003. 53: 387–395. [DOI] [PubMed] [Google Scholar]
- [7].Neufeld MY, Inzelberg R, Korczyn AD. EEG in demented and non-demented parkinsonian patients. Acta Neurol Scand. 1988. 78: 1–5. [DOI] [PubMed] [Google Scholar]
- [8].Smith SJ. EEG in neurological conditions other than epilepsy: when does it help, what does it add? J Neurol Neurosurg Psychiatry. 2005. 76 Suppl 2: ii8–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Koprivica V, Stone DL, Park JK, et al. Analysis and classification of 304 mutant alleles in patients with type 1 and type 3 Gaucher disease. Am J Hum Genet. 2000. 66: 1777–1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Zivin L, Marsan CA. Incidence and prognostic significance of “epileptiform” activity in the eeg of non-epileptic subjects. Brain. 1968. 91: 751–778. [DOI] [PubMed] [Google Scholar]
- [11].Cavazzuti GB, Cappella L, Nalin A. Longitudinal study of epileptiform EEG patterns in normal children. Epilepsia. 1980. 21: 43–55. [DOI] [PubMed] [Google Scholar]
- [12].Quirk JA, Fish DR, Smith SJ, Sander JW, Shorvon SD, Allen PJ. Incidence of photosensitive epilepsy: a prospective national study. Electroencephalogr Clin Neurophysiol. 1995. 95: 260–267. [DOI] [PubMed] [Google Scholar]
- [13].Guerrini R, Dravet C, Genton P, et al. Idiopathic photosensitive occipital lobe epilepsy. Epilepsia. 1995. 36: 883–891. [DOI] [PubMed] [Google Scholar]
- [14].Gastaut H, Gastaut Y. Electroencephalographic and clinical study of anoxic convulsions in children; their location within the group of infantile convulsions and their differenciation from epilepsy. Electroencephalogr Clin Neurophysiol. 1958. 10: 607–620. [DOI] [PubMed] [Google Scholar]
- [15].Cavazos JE. Epilepsy, A Comprehensive Textbook, Volume Three. Journal of Clinical Neurophysiology. 1998. 15: 279–280. [Google Scholar]
- [16].Berkovic SF, Mulley JC, Scheffer IE, Petrou S. Human epilepsies: interaction of genetic and acquired factors. Trends Neurosci. 2006. 29: 391–397. [DOI] [PubMed] [Google Scholar]
- [17].Goker-Alpan O, Wiggs EA, Eblan MJ, et al. Cognitive outcome in treated patients with chronic neuronopathic Gaucher disease. J Pediatr. 2008. 153: 89–94. [DOI] [PubMed] [Google Scholar]