

Abstract
Identification of proteins that interact with Cx43 has been instrumental in the understanding of gap junction (GJ) regulation. An in vitro phosphorylation screen identified that Protein tyrosine kinase 2 beta (Pyk2) phosphorylated purified Cx43CT and this led us to characterize the impact of this phosphorylation on Cx43 function. Mass spectrometry identified Pyk2 phosphorylates Cx43 residues Y247, Y265, Y267, and Y313. Western blot and immunofluorescence staining using HeLaCx43 cells, HEK 293T cells, and neonatal rat ventricular myocytes (NRVMs) revealed Pyk2 can be activated by Src and active Pyk2 interacts with Cx43 at the plasma membrane. Overexpression of Pyk2 increases Cx43 phosphorylation and knock-down of Pyk2 decreases Cx43 phosphorylation, without affecting the level of active Src. In HeLaCx43 cells treated with PMA to activate Pyk2, a decrease in Cx43 GJ intercellular communication (GJIC) was observed when assayed by dye transfer. Moreover, PMA activation of Pyk2 could be inhibited by the small molecule PF4618433. This partially restored GJIC, and when paired with a Src inhibitor, returned GJIC to the no PMA control-level. The ability of Pyk2 and Src inhibitors to restore Cx43 function in the presence of PMA was also observed in NRVMs. Additionally, an animal model of myocardial infarction induced heart failure showed a higher level of active Pyk2 activity and increased interaction with Cx43 in ventricular myocytes. Src inhibitors have been used to reverse Cx43 remodeling and improve heart function after myocardial infarction; however, they alone could not fully restore proper Cx43 function. Our data suggest that Pyk2 may need to be inhibited, in addition to Src, to further (if not completely) reverse Cx43 remodeling and improve intercellular communication.
Keywords: Cx43, Pyk2, Src, phosphorylation, gap junction
1. Introduction
Connexin43 (Cx43), the most abundant cardiac gap junction (GJ) protein [1], mediates ventricular propagation of cardiac action potentials and maintenance of a regular beating rhythm. The importance of Cx43 GJs in the heart is well established: closure, dysregulation or mislocation disrupts propagation, and lethal arrhythmias can ensue [1–4]. Following a myocardial infarction (MI; early stage-remodeling), in the surviving ventricular myocytes from the epicardial border zone (EBZ; adjacent to the necrosing area [5]), the cardiac muscle sarcoplasm experiences a number of abnormalities that includes an increase in Ca2+, decrease in pH, reduced PKA activity, and a drop in ATP [6–9]. Additionally, there is reduced Cx43 GJ intercellular communication (GJIC) due to changes in Cx43 expression, phosphorylation state, and membrane localization (i.e. from the intercalated disc (ID) to the lateral membrane) [10, 11]. An important consequence of cellular uncoupling is an increased dispersion of action potential duration and refractory period, which is pronounced in the EBZ [12]. While lateralization of Cx43 is an almost ubiquitous response to cardiac pathology [12–14], mechanisms of remodeling are unknown.
Left ventricle hypertrophy (LVH) following a MI (late-stage remodeling) is an adaptive response to increased biomechanical stress [15]. Initially, heart mass increases to normalize wall stress and retain normal cardiovascular function, accompanied by a transient increase in Cx43 expression and phosphorylation [16, 17]. However, the increased cardiac mass and sustained overload eventually lead to contractile dysfunction and heart failure (HF) through poorly understood mechanisms [18]. As LVH becomes severe, propagation velocity decreases. This correlates with reduced Cx43 expression and ID localization, altered phosphorylation of Cx43, increased Cx45 expression, and extensive Cx45 co-localization with the remaining Cx43 at the ID (seen in both ischemia and dilated cardiomyopathy [19–22]).
A number of studies have attempted to reverse Cx43 remodeling in the infarct EBZ and LVH. Unfortunately, they have not been sufficient to restore normal conduction and prevent arrhythmias. For example, Macia et al. [23] found that in the EBZ after MI the peptide rotigaptide partially reversed the loss of Cx43 expression but did not affect the increase in S368 phosphorylation (pS368 decreases GJ conductance [23, 24]), nor reverse Cx43 lateralization. The partial reversal of Cx43 remodeling was not sufficient to restore normal conduction or prevent arrhythmias. Greener et al. [25] used gene transfer to show that increased Cx43 expression in the infarct EBZ could improve conduction velocity and lessen arrhythmia susceptibility. Interestingly, while an overall increase of Cx43 was observed at the ID, the ratio of ID/lateral membrane Cx43 was the same between control and animals receiving adenovirus Cx43. The data strongly suggest that any targeted mechanism that can increase the Cx43 ID/lateral membrane ratio under endogenously expressing conditions would be therapeutically beneficial. Finally, Rutledge et al. [26] addressed if Src inhibitors would reverse Cx43 remodeling and improve heart function after MI. Of note, studies by our lab and others have linked Src to dysregulation of Cx43 in the heart [11, 27, 28]. In the EBZ, Src inhibitors PP1 and AZD0530 (in clinical trials to treat cancer [29, 30]) decreased Src activity back to sham levels, raised Cx43 expression by ~69%, and had a higher level of the Cx43 P2 phosphorylation isoform (correlates with GJ localization/promoting GJIC [9]). Although no change in Cx43 lateralization was observed, this still led to ~50% improvement in conduction velocity and lowered arrhythmia inducibility. While affecting the Cx43 phosphorylation state can explain the beneficial effects after Src inhibition, their data indicate that a second undiscovered process prevented full restoration of proper Cx43 function (protein level, phosphorylation state, ID localization).
Src-induced phosphorylation of Cx43 residues Y247, Y265, and Y313 leads to down regulation of GJIC and GJ disassembly [31–39]. Another target of Src is the Ca2+-dependent, nonreceptor tyrosine kinase of the FAK family, Protein tyrosine kinase 2 beta (Pyk2) [40]. Pyk2 activation initiates after auto-phosphorylation at Y402, which facilitates enhanced kinase activity after Src binding (via SH2 domain) and phosphorylation at Y579 and Y580 [41, 42]. A commonality exists between these kinases in that increased expression and activity is observed in animal models of HF and in the left ventricle of failing human hearts [36, 43–48]. Inhibition of Src or Pyk2 (individually) reduces arrhythmias, slows ventricular remodeling, improves ventricle function, and prevents sudden cardiac death [26, 28, 44]. In relation to Src inhibition, the level of Cx43 protein at the plaque and intercellular communication was improved [26, 28, 44]. Based upon these commonalities and that both Src (SH4 domain-myristoylation) and Pyk2 (FERM domain-PIP2) are plasma membrane associated, we will test the hypothesis that inhibition of both Pyk2 and Src phosphorylation is necessary to prevent GJ channel closure and remodeling of Cx43 under conditions that stimulate heart disease.
2. Materials and Methods
2.1. Molecular biology
Cx43 WT and mutant construct (hCx43 Y247/265F) were generated as described previously [39]. v-Src was a gift from Dr. Joan Brugge (Addgene plasmid #14578) and human c-Src plasmid was a gift from Dr. Robert Lefkowitz (Addgene plasmid # 42202) [49]. Pyk2 WT and CT domain (Cell Adhesion Kinase-β-Related Non-kinase, CRNK) were PCR amplified from a pCMV-HA plasmid containing the hPyk2 sequence using primers (Supplemental Methods) suitable for Gibson assembly into the pD2529 vector (Atum). Purified PCR product and vector were assembled using NEBuilder HIFI DNA Assembly Master Mix (NEB) per manufacturer protocol. Pyk2 kinase dead (KD; K457A), constitutively active c-Src (Y530F), SH3-deficient (W121K) or SH2-deficient (R178L) c-SrcY530F constructs were made by QuikChange Lightning Site-Directed Mutagenesis Kit (Agilent) and confirmed by DNA sequencing (ACGT, Inc.).
2.2. Cell line culture
Parental HeLa, MCF-7, and HEK 293T cells (gift from Dr. Myron Toews, University of Nebraska Medical Center) were cultured in Dulbecco’s modified Eagle medium (Corning) at 37°C in a humidified 5% CO2 atmosphere. Stable HeLa cell lines were generated as described previously [39]. Western blots and immunofluorescence were used to screen clones. HeLa or HEK 293T cells at ~80% confluence were transfected using the X-tremeGENE HP (Roche) reagent at 2:1 (reagent:plasmid), diluted in OptiMem under antibiotic free conditions. Cells were analyzed 24 hr after transfection. Protocols for Dulbecco’s modified Eagle medium, Western blot, and immunofluorescence are located in the Supplemental Methods.
2.3. Antibodies
Detection reagents, manufacturers, applications, and concentration ranges for all primary and secondary antibodies utilized in this study are summarized in Supplemental Table 1.
2.4. Pyk2 siRNA transfection
Pyk2 siRNA (ThermoFisher) and a scrambled Pyk2 siRNA (does not target any gene product, negative control; ThermoFisher) were reverse transfected (24 hr) into HeLa Cx43 WT cells using Lipofectamine RNAiMAX (Invitrogen) according to the manufacturer’s protocols. HeLa Cx43 WT cells were then transfected with active c-Src for 24 hr. Protein levels were detected by Western blot.
2.5. Primary cardiomyocyte isolation and contractile analysis
Neonatal rat ventricular myocytes (NRVMs) were isolated from day 1–3 rat hearts using the Pierce Primary Cardiomyocyte Isolation Kit (>90% pure). Isolated NRVMs were plated at a seeding density of 2.5 × 105 cells/cm2 in complete DMEM supplemented with 10% FBS and 1% pen-strep at 37°C in a 5% CO2 incubator for 24 hr. The medium was then replaced with complete DMEM containing Cardiomyocyte Growth Supplement (1000X). After 3 days, NRVMs were pre-treated with Saracatinib, PF4618433, or both for 3 hr and then treated with PMA for 60 min. Videos of NRVMs beating were recorded through the Leica DMi1 inverted microscope. The contraction of NRVMs expressed as beat per minutes and the total number of NRVMs contributing to the beats were calculated.
2.6. Confocal imaging
All cell immunofluorescence images were acquired on a Zeiss LSM 800 Confocal system using appropriate numerical aperture objectives and appropriate filter sets.
2.7. Kinase Screen (Eurofins KinaseProfiler)
Rat Cx43CT236–382 was expressed and purified as described previously [50]. Purified protein was diluted in 1x PBS pH 7.4 to a final concentration of 200 μM in a final volume of 1 mL, flash frozen on dry ice and shipped on dry ice to Eurofins Scientific for a custom Kinase Profiler Assay testing the Cx43CT as substrate for the indicated kinases.
2.8. Mass spectrometry
Purified rat Cx43CT236–382 was incubated with 1.5 μg of active Pyk2 (recombinant human residues 360–690, SignalChem) and 500 μM ATP in reaction buffer for 16 hr at 30°C. The reaction was stopped on ice. 10 nmol of protein was run on a SDS-PAGE gel and stained with Coomassie blue. Once the Cx43CT bands were cut out and washed, the sample was shipped on dry ice to the Harvard Beth Israel Deaconess Medical Center Mass Spectrometry Core Facility for post-translational mapping. For negative control the kinase was substituted with reaction buffer.
2.9. Glutathione S-transferase (GST) Pull-down Assay
The GST pull down assay was modified from [51] and has been described previously [39] (for additional details, see Supplemental Methods).
2.10. Dye-transfer assay
HeLaCx43 cells were treated with Saracatinib, PF4618433, U0126, or both inhibitors 3 hr prior to PMA (100 nM) treatment. 1 hr after PMA treatment, cell imaging dishes with a 100% confluent monolayer were removed from the incubator. All buffer and medium used for Dye-transfer assay were pre-warmed at 37°C. After two washes with 1x PBS, cells were overlaid with 1x PBS containing Lucifer yellow. Cells were scrape-loaded with a fine edged micro-scalpel by three longitudinal scratches and then incubated at room temperature for 5 min. Cells were rinsed twice with 1x PBS and incubated in cell culture medium at 37°C for 5 min. After incubation, cells were washed twice with 1x PBS containing 1 mM CaCl2 and 1 mM MgCl2 to stop dye spread and fixed with 4% Paraformaldehyde for 20 min. After stained with DAPI for 10 min, cells were mounted by adding several drops of SlowFade anti-fade (Invitrogen). Cells were imaged using confocal microscopy.
2.11. 3-(4,5-Dimethylthiazol-2-yl)-2,5-Diphenyltetrazolium Bromide (MTT) assay
NRVMs were seeded on each well of a 24-well plate and cultured in complete DMEM containing Cardiomyocyte Growth Supplement at 37°C in a humidified incubator containing 5% CO2 for 3 days. Saracatinib and PF4618433 in a series of concentrations were individually prepared in the culture media and added to each well of cells followed by 48 hr incubation. The control group contained no kinase inhibitors. Each concentration and control group had 4 replicates. After that, the drug cytotoxicity was determined using MTT (Sigma) assay. Absorbance at 540 nm was measured with a microplate reader. The absorbance value from each group was normalized to the average absorbance value in the control group.
2.12. Left anterior descending artery (LAD) ligation and tissue collection
Rats (Sprague Dawley; 200–250g) were purchased from Sasco Breeding Laboratories. All surgical procedures and animal care protocols were approved by the University of Nebraska Medical Center Institutional Animal Care and Use Committee (IACUC) and conducted according to National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals (National Academic Press, 2011). Rats were assigned into two groups randomly: sham control and HF group. HF was generated by Left anterior descending artery (LAD) ligation as previously described [52]. Briefly, Rats were orally intubated and ventilated with 2–2.5% isoflurane during the surgical procedure. To produce heart failure, the left coronary artery was ligated 1–2 mm below its origin from the aorta. The sham rats had the thoracotomy, the manipulation of the heart and lidocaine with no ligations of coronary artery. HF and the left ventricular dysfunction degree was determined by the hemodynamic and morphological change extent at the end of each experiment (six weeks post-surgery). Rats were anesthetized by one injection of urethane (0.75 g/kg i.p.) and α-chloralose (70 mg/kg i.p.). Body temperature was maintained 36–38°C by a heated stage. A Mikro-Tip catheter (Millar Instruments) was used to measure the left ventricular end-diastolic pressure (LVEDP). After measurement, rats were euthanized by pentobarbital (150 mg/kg, i.p.). Hearts were quickly removed and dissected. The infarct size ratio was determined by dividing the infarct area to the total left ventricle area with the SigmaScan Pro software (Aspire Software International) [53]. Rats with both LVEDP ≥15 mmHg and infarct size ratio ≥30% were considered as HF. The left ventricles of surgical hearts (samples are distal to the infarction; hypertrophy area) were collected for Western blot or immunofluorescence staining.
3. Statistical Analysis
All data were analyzed by using GraphPad Prism 8.0 and presented as the mean ± SD. Statistical analysis performed in GraphPad Prism 8.0 were either one-way ANOVA with a Neuman-Keuls post-hoc analysis or Student’s t-test where appropriate. P-values <0.05 were considered statistically significant.
4. Results
4.1. Pyk2 directly interacts and phosphorylates the Cx43 carboxyl terminal (CT) domain
An in vitro tyrosine phosphorylation screen performed by Eurofins Scientific (KinaseProfiler) discovered that Pyk2 phosphorylates purified Cx43CT236–382 (Supplemental Figure 1A). FAK, the other FAK family member, had significantly less ability to phosphorylate the Cx43CT domain (Tyk2, positive control [54]; SYK and ZAP70, examples with little-to-no Cx43CT236–382 phosphorylation). To confirm the interaction between Cx43 and Pyk2, purified GST-tagged Cx43CT236–382 immobilized on glutathione-Sepharose beads and lysate from MCF-7 cells that express Pyk2 were used in a pull-down assay (Supplemental Figure 1B). Pyk2 could be pulled-down by GST-Cx43CT but not GST. To identify the Cx43CT tyrosine residue(s) phosphorylated by Pyk2, purified Cx43CT236–382 was incubated in vitro with active Pyk2 (SignalChem). After trypsin digestion, Tandem MS/MS identified phosphorylation at Y247, Y265, Y267, and Y313 (Supplemental Figure 1C and Supplemental Table 2). The identical Cx43CT residues were identified by MS/MS to be phosphorylated when the experiment was performed with active c-Src [39].
4.2. Pyk2 phosphorylates Cx43 residues Y247, Y265, and Y313 in HeLa and HEK 293T cells.
To determine if Pyk2 phosphorylation of Cx43 occurs in cells, we initially tested in Cx-deficient HeLa cells stably transfected with Cx43 (HeLaCx43) if Cx43 and active Pyk2 (endogenously expressed) colocalize (Figure 1). We also used a phosphorylation eliminating Cx43 Y247/265F mutant (HeLaCx43Y247/265F) with the goal of enhancing the interaction by trapping Pyk2 on a non-phosphorylatable Cx43. Transient transfection of v-Src activated Pyk2 (increased pY402 and pY579/Y580) and increased the Cx43 P1/P2 ratio that is consistent with closed channels [55] (Figure 1A). The images of HeLaCx43 without v-Src (control, Ctr) show a small level of active Pyk2 that increases in the presence of v-Src (Figure 1B). Active Pyk2 colocalizes with Cx43 (antibody recognizes total Cx43) at the plasma membrane and within intracellular compartments. Of the remaining plasma membrane localized Cx43, the presence of active Pyk2 and v-Src causes a significant decrease in number of plaques and plaque length (Supplemental Figure 2A; statistics from images using an antibody (IF1) that recognizes junctional Cx43 [56]). When the same experiment was performed with HeLaCx43Y247/265F, the Cx43 and Pyk2 colocalization is predominately at the plasma membrane, suggesting Pyk2 phosphorylation occurs at the plasma membrane before the internalization of Cx43 (Figure 1C and Supplemental Figure 2B). A Z-stack image of the Cx43Y247/265F indicates that Pyk2 colocalization occurs in the center of the GJ plaque (Figure 1D). This observation is consistent with previous studies that revealed newly synthesized channels accrue along the plaque edges and removal of channels from plaque centers [57, 58]. Of note, we speculate that the large gap junctions observed for Cx43Y247/265F in the presence of v-Src is a function of β-tubulin remaining attached to the Cx43CT (unable to phosphorylate Y247, [59]) and the inability of AP2 to interact with the Cx43CT (Y265F eliminates the Tyr-based motif YXXΦ, [60]).
Figure 1: Active Pyk2 interacts with Cx43 in HeLa cells.
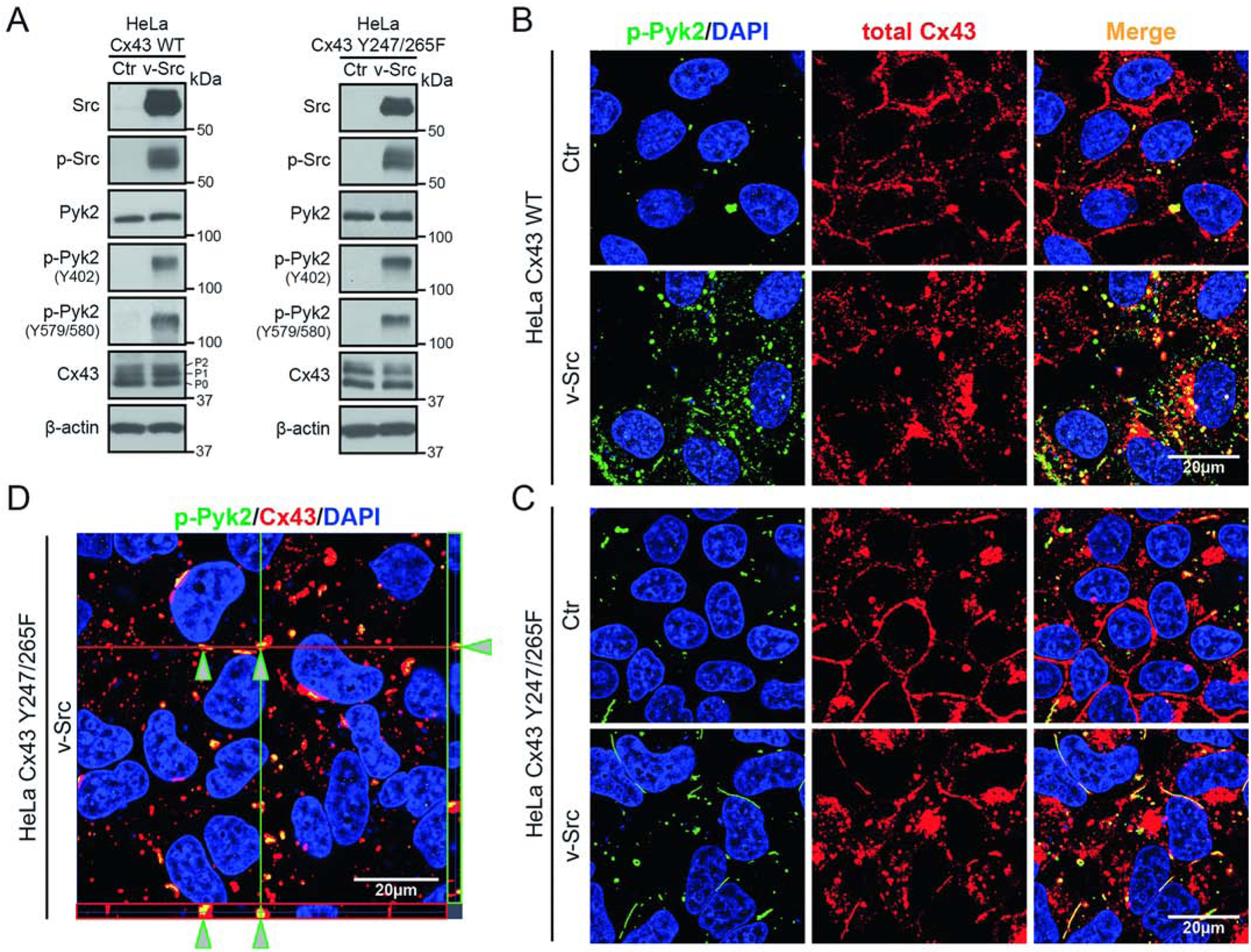
(A) Western blot of lysate from HeLa cells stably expressing Cx43 WT or Cx43 Y247/265F ± v-Src (24 h). Antibodies used are labeled on the left of each panel. The Cx43 P0, P1, and P2 isoforms have been labeled. Cellular localization of (B) Cx43 WT or (C) Cx43 Y247/265F ± v-Src (24 h) in HeLa cells detected by immunofluorescence (green, p-Pyk2Y579/580; blue, DAPI-stained DNA; red, total Cx43; yellow, p-Pyk2/Cx43 colocalization). (D) Z-stack imaging of HeLa Cx43 Y247/265F cells after v-Src transfection. Arrows point to Cx43 colocalization with active Pyk2.
The ability to directly characterize the impact of Pyk2 phosphorylation on Cx43 is challenged by the knowledge that Pyk2 requires c-Src for “enhanced” activation and that they both phosphorylate the same Cx43CT tyrosine residues [39]. To initially address this issue, Cx43 tyrosine phosphorylation levels were evaluated in the HeLaCx43 cells after decreasing the activation of Pyk2 (i.e., “loss of function” study) and probing with the only available Cx43 tyrosine phospho-specific antibodies pY247, pY265, and pY313 (Figure 2). The first “loss of function” experiment used siRNA to knockdown the expression of Pyk2 in the presence and absence of constitutively active c-Src (c-SrcY530F; Figure 2A). In the absence of c-SrcY530F (control, Ctr), Pyk2 has no effect on the phosphorylation of Cx43 residues Y247, Y265, and Y313. In the presence of c-SrcY530F, there is an increase in the level of active Pyk2 (pY402 and pY579/pY580) as well as Cx43 pY247, pY265, and pY313. siRNA knockdown of Pyk2 significantly decreased the phosphorylation of Cx43 residues Y247, Y265, and Y313. Importantly, knockdown of Pyk2 did not affect the level of total or active c-Src.
Figure 2: Inhibition of active Pyk2 decreases Cx43 phosphorylation at residues Y247, Y265, and Y313.
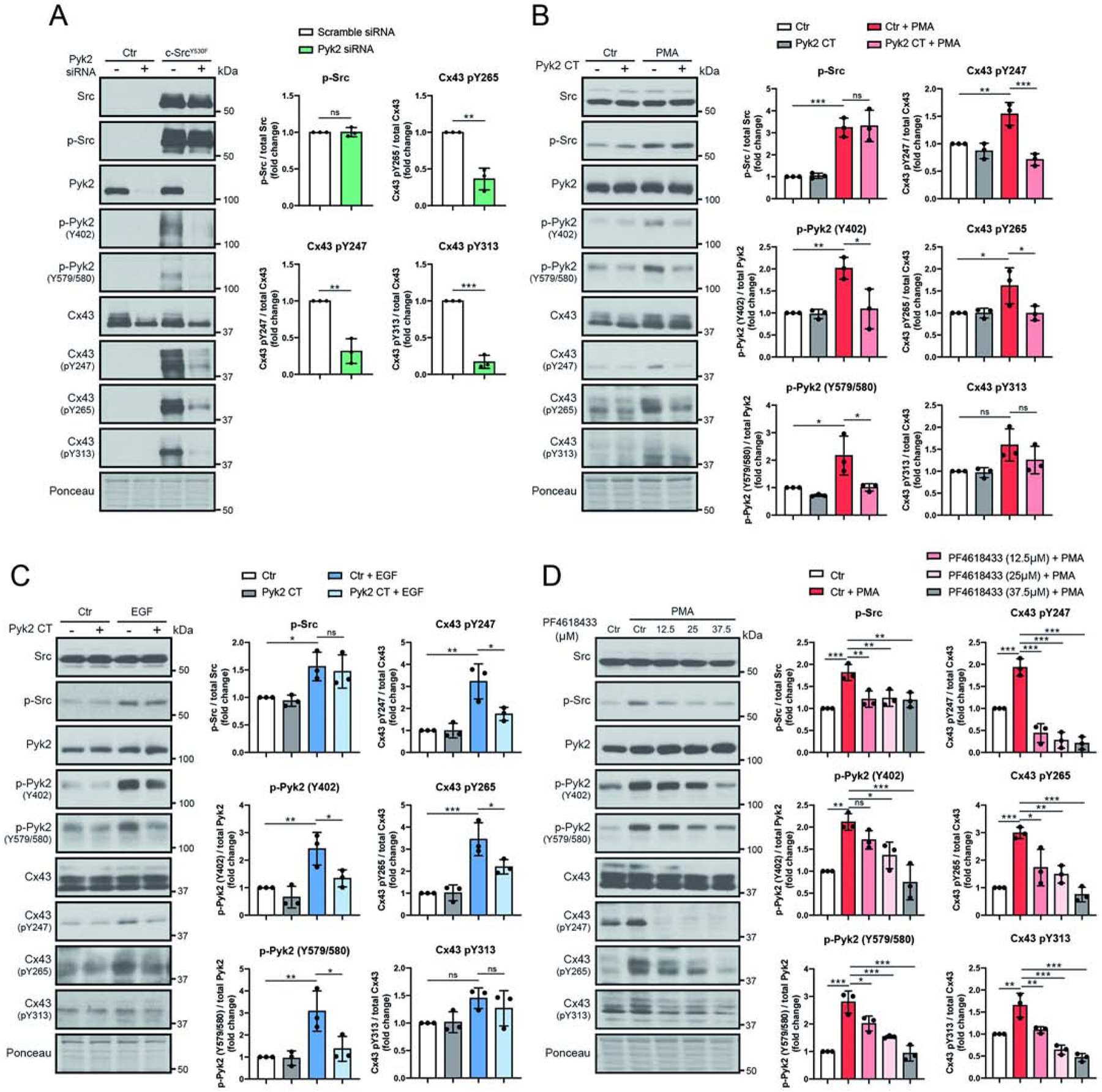
Lysate from HeLaCx43 cells treated with (A) Pyk2 siRNA and c-SrcY530F (active c-Src, 24 h), (B) Pyk2 CT and PMA (100 nM, 1 hr), (C) Pyk2 CT and EGF (100 ng/mL 1 hr), or (D) Pyk2 inhibitor PF4618433 and PMA (100 nM, 1 hr) were Western blotted. Antibodies used are labeled on the left of each panel. Protein levels were quantified by analyzing scanned blots using ImageJ software, with normalization of protein expression to the control lane (value set arbitrarily as 1). Phosphorylated protein levels were normalized to total protein. Data are representative of three independent experiments (one-way ANOVA, *P<0.05, **P<0.01, ***P<0.001).
The next “loss of function” experiment used expression of the Cell Adhesion Kinase-β-Related Non-kinase (CRNK, i.e. Pyk2 CT domain; endogenously expressed inhibitor regulates the function of Pyk2 by blocking auto-phosphorylation at Y402 [44, 61]) to inhibit the activation of Pyk2 in the presence and absence of the indirect Src-activators phorbol 12-myristate 13-acetate (PMA) or epidermal growth factor (EGF) (both lead to decreased Cx43 GJIC [62, 63]; Figure 2B,C). PMA and EGF were utilized so Cx43 tyrosine phosphorylation could be evaluated under conditions of endogenous Src and Pyk2 expression. Although, PMA and EGF also activate a number of serine kinases (see Supplemental Figure 3) that phosphorylate different Cx43 residues [55, 64, 65], we are focusing on the Src/Pyk2 axis using the Cx43 phospho-specific tyrosine antibodies. In the absence of PMA or EGF, the presence of the Pyk2 CT domain had little-to-no effect on the level of active Pyk2 (pY402 and pY579/pY580) as well as phosphorylation of Cx43 residues Y247, Y265, and Y313. The addition of PMA or EGF increased the level of active Src, active Pyk2, and phosphorylation of Cx43 residues Y247 and Y265. Conversely, expression of the Pyk2 CT domain in the presence of PMA or EGF reduced the level of active Pyk2 and phosphorylation of Cx43 residues Y247 and Y265 without affecting the level of total or active c-Src. Of note, although not significant, the (de)phosphorylation of Y313 had the same trend as Y247 and Y265 in the presence of PMA or EGF experiments with and without the Pyk2 CT domain.
The final “loss of function” experiment used a small molecule Pyk2 inhibitor (PF4618433) in the presence of PMA (Figure 2D). Increasing the concentration of PF4618433 to 37.5 μM counteracted the effect of PMA by returning active Pyk2 (pY402 and pY579/pY580) to the pre-PMA treated level. The inhibitor had the additional effect of causing activated c-Src to also return to the pre-PMA treated level; this occurred at a lower concentration of PF4618433 (12.5 μM). Noteworthy is that while additional concentrations of PF4618433 (25 and 37.5 μM) had no further effect on the level of activated c-Src, unlike Pyk2, the level of phosphorylation of Cx43 residues Y247, Y265, and Y313 also continued to decrease with additional PF4618433. Altogether, three different techniques to inhibit active Pyk2 were consistent in suggesting that Pyk2 plays a direct role in the phosphorylation of Cx43 residues Y247, Y265, and Y313.
To complement the “loss of function” study we used HEK 293T cells to investigate the contribution of Pyk2 via a “gain of function” study. HEK 293T cells were utilized because they endogenously express Cx43, contain a low level of endogenous Pyk2 and c-Src, and they are highly efficient for double transfection. HEK 293T cells were transiently transfected with c-SrcY530F and Pyk2 WT or a kinase dead (KD) mutant and evaluated for phosphorylation of Cx43 residues Y247, Y265, and Y313 (Figure 3A; two exposure levels are provided to better visualize the bands). Consistent with our previous study, the transfection of c-SrcY530F alone caused an increase in the phosphorylation of Cx43 residues Y247, Y265, and Y313 (control, Ctr) [39]. However, a significant increase is observed in phosphorylation of Cx43 residues Y247, Y265, and Y313 when Pyk2 is co-expressed with c-SrcY530F (Figure 3B). Importantly, the presence of Pyk2 did not affect the level of active c-SrcY530F. This increase in Cx43 phosphorylation is not seen with expression of Pyk2 KD version (Figure 3A,B). Interestingly, upon examination of the long exposure control and expression of Pyk2 WT without expression of c-SrcY530F, there is still an increase in phosphorylation of Cx43 residues Y247, Y265, and Y313. In total, the “loss” and “gain” of function data are consistent in demonstrating that Pyk2 contributes to the level of Cx43 Y247, Y265, and Y313 phosphorylation.
Figure 3: Overexpression of Pyk2 increases Cx43 phosphorylation at Y247, Y265, and Y313.
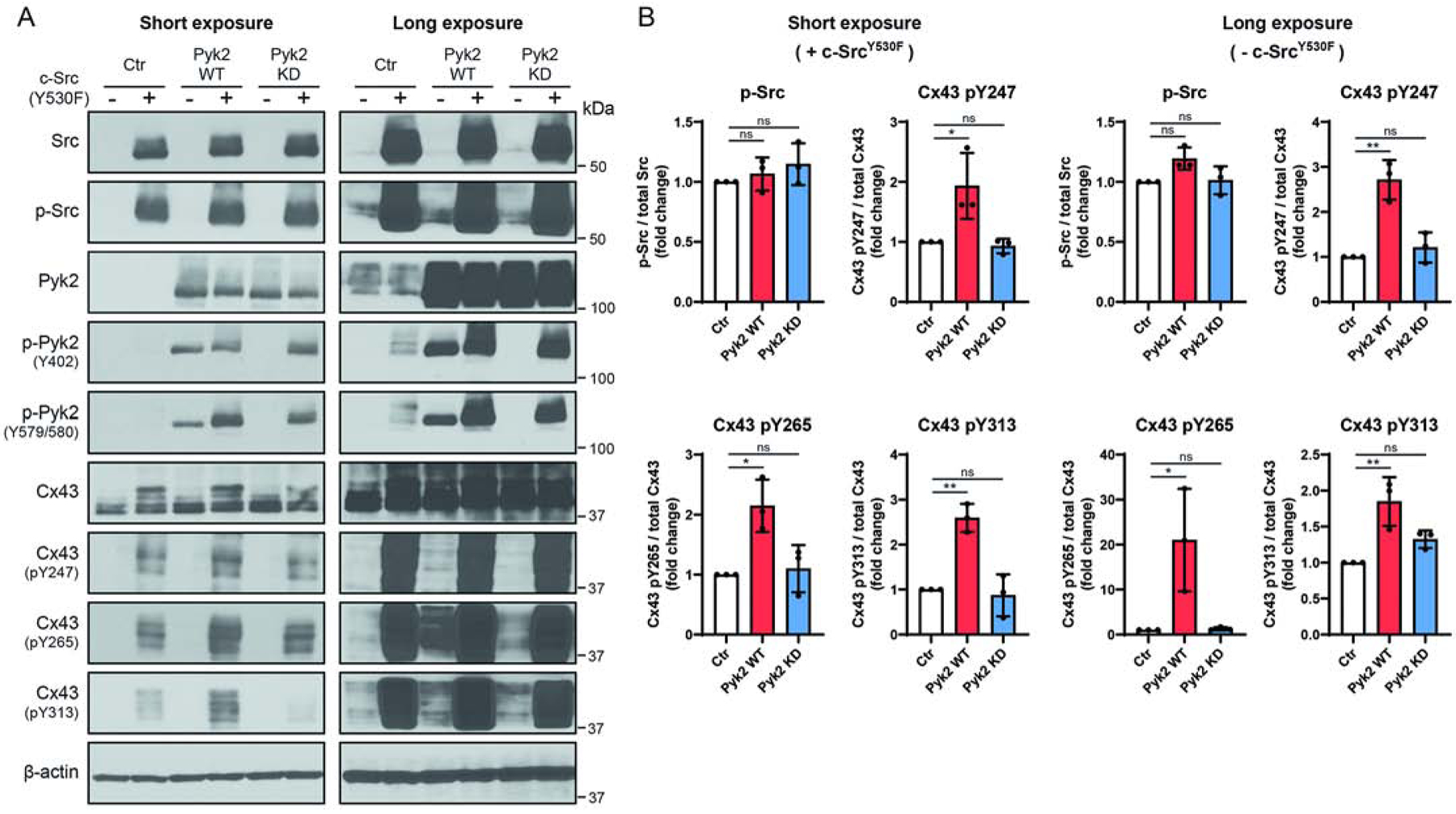
(A) HEK 293T cells were co-transfected with c-SrcY530F (active c-Src; 24 hr) and Pyk2 wild type (WT) or a Pyk2 kinase dead (KD) mutant (K457A) and lysate was Western blotted. Antibodies used are labeled on the left of each panel. (B) Protein levels were quantified by analyzing scanned blots using ImageJ software, with normalization of protein expression to the control lane (value set arbitrarily as 1). Phosphorylated protein levels were normalized to total protein. Data are representative of three independent experiments (one-way ANOVA, *P<0.05, **P<0.01).
4.3. Pyk2 has a greater impact on Cx43 phosphorylation than Src
A working model for Src phosphorylation of Cx43 includes: 1) association initiates between Cx43 residues A276-S282 and the Src SH3 domain [11, 35, 66]; 2) SH3 domain binding causes Cx43CT residues T275-P284 to adopt a left-handed type II helix [11]; 3) Src kinase domain phosphorylates Cx43 residue Y265 [35, 67]; and 4) Src SH2 domain binds pY265, which stabilizes the Src-Cx43 interaction to promote phosphorylation at Y247 and Y313 [35, 39, 66, 68]. Src regulation of Pyk2 includes the association of the Src SH2 domain with Pyk2 pY402 and subsequent Src kinase domain phosphorylation of Pyk2 residues Y579/Y580 to enhance activity [40]. Based upon this information, another method was employed to dissect the impact of Pyk2 and Src phosphorylation on Cx43, the use of a SH3- and SH2-deficient active c-Src (c-SrcY530F). The idea is that a SH3-deficient c-SrcY530F (point mutation disrupts binding, W121K) could still activate Pyk2, but not bind Cx43CT residues T275-P284 to initiate phosphorylation of Y265 and a SH2-deficient c-SrcY530F (point mutation disrupts binding, R178L) would not activate Pyk2, but could still bind Cx43CT residues T275-P284 and phosphorylate Y265. WT, SH3-deficient, and SH2-deficient c-SrcY530F constructs were transiently transfected into HeLaCx43 cells (Figure 4). WT c-SrcY530F increased the level of active Pyk2 and phosphorylation of Cx43 residues Y247, Y265, and Y313 as previously observed. The SH3-deficient c-SrcY530F was able to activate Pyk2 and while there was not a statistically significant decrease in phosphorylation of Cx43 residues Y247, Y265, and Y313 when compared to WT, the trend was lower. Conversely, SH2-deficient c-SrcY530F was unable to activate Pyk2 and phosphorylation only occurred on Cx43 residues Y265, which is significantly lower than in the presence of active Pyk2. The data suggest that the overall phosphorylation level of Cx43 residues Y247, Y265, and Y313 is higher in the presence of active Pyk2 than c-Src. This result is consistent with the PF4618433 data (25 and 37.5 μM; active Src at basal level) which showed that the decrease in phosphorylation of Cx43 residues Y247, Y265, and Y313 correlated with the decrease in active Pyk2.
Figure 4: Src mutations demonstrate the contribution of Pyk2 to Cx43 phosphorylation at Y247, Y265, and Y313.
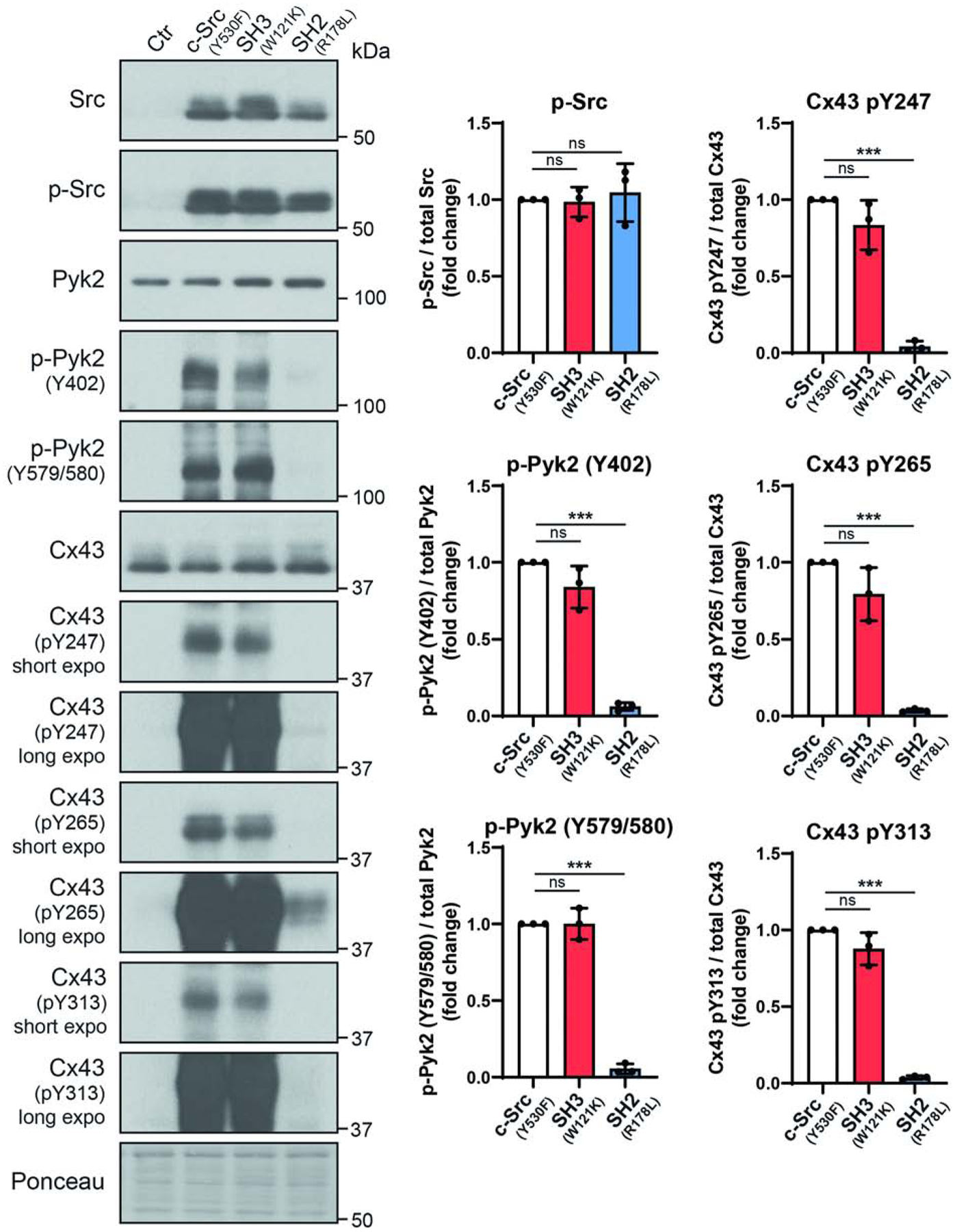
(A) HeLaCx43 cells were transfected with empty vector, c-SrcY530F (active c-Src), SH3W121K (disables SH3 domain from interacting with protein partners), or SH2R178L (disables SH2 domain from interacting with protein partners) for 24 h and lysate was Western blotted. Antibodies used are labeled on the left of each panel. (B) Protein levels were quantified by analyzing scanned blots using ImageJ software, with normalization of protein expression to the control lane (value set arbitrarily as 1). Phosphorylated protein levels were normalized to total protein. Data are representative of three independent experiments (one-way ANOVA,**P<0.01, ***P<0.001).
4.4. Inhibition of Pyk2 is necessary to maintain GJIC
To determine if Pyk2 phosphorylation affects cell-to-cell communication, junctional transfer of the tracer Lucifer Yellow (anionic) was measured in a scrape-loading assay using HeLaCx43 cells treated with PMA in the presence of Pyk2 (PF4618433) and/or c-Src (Saracatinib) inhibitors. Western blot data demonstrates that 60 min of PMA treatment activates Pyk2 (Figure 5A). In addition, while PF4618433 and Saracatinib inhibit the activation of Pyk2 and c-Src, respectively, the inhibitors also inhibit the activation of each other. Similar to previously published studies, Cx43 WT expressing HeLa cells (no PMA) were extensively coupled with respect to Lucifer Yellow (Figure 5B) [69]. PMA reduced coupling by ~81%, which was partially reversed by the addition of Saracatinib (~53% reduction) or PF4618433 (~41% reduction). Only in the presence of both Saracatinib and PF4618433 was the effect of PMA on coupling completely reversed (no reduction). Immunofluorescence from an in situ Triton X-100 extraction assay, used to quantify the junctional (insoluble) and non-junctional (soluble) pools [70, 71], identified that the increase in coupling (seen in Figure 5B) with the inhibitors alone and together correlated with the increase of Cx43 in the junctional fraction at the plasma membrane (Figure 5C). Altogether, the data indicates that inhibition of both Pyk2 and Src is needed to counteract the effect of PMA on GJIC.
Figure 5: PMA mediated Cx43 gap junction closure is reversed by inhibiting active Pyk2 and c-Src.
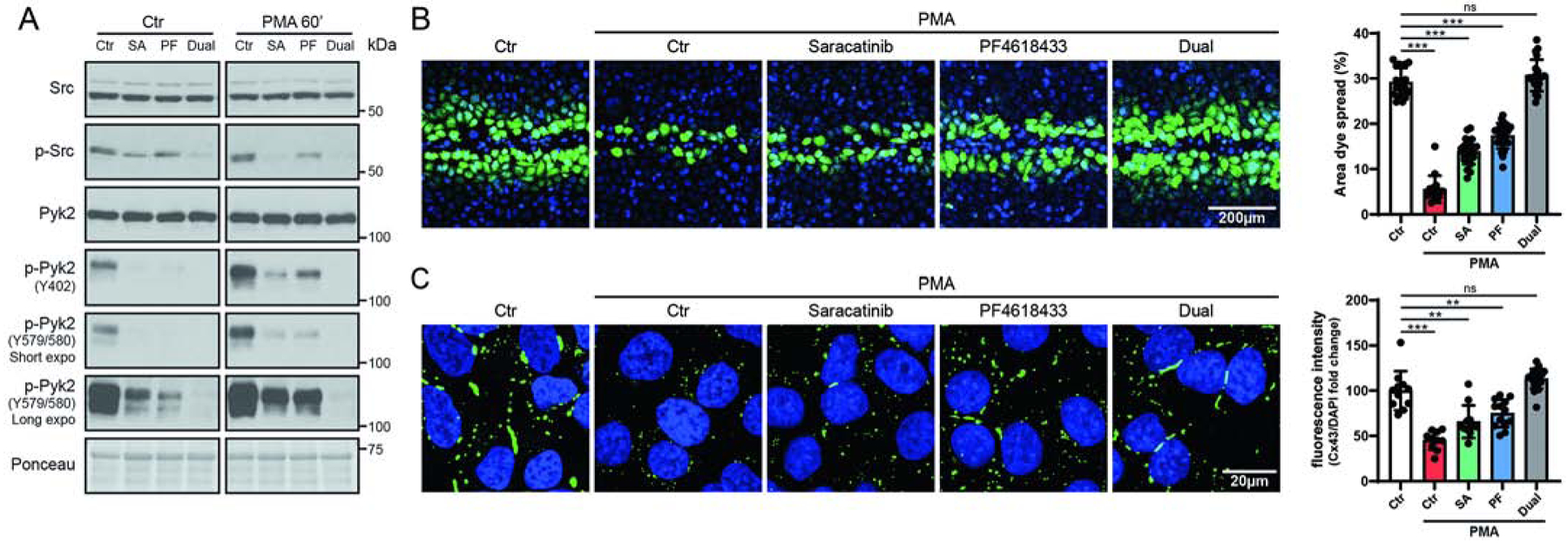
(A) Lysate from PMA treated (100 nM, 60 min) HeLaCx43 ± Src (Saracatinib, SA, 5μM) and/or Pyk2 (PF4619433, PF, 50μM) inhibitors was Western blotted. Antibodies used are labeled on the left of each panel. (B) Level of gap junction intercellular communication in HeLaCx43 cells pre-treated with Saracatinib, PF4618433 or both and then treated with PMA (60 min) was determined using the scrape loading dye transfer assay. Provided are representative immunofluorescence images (green, Lucifer yellow; blue, DAPI). Quantification of the area of Lucifer Yellow transfer shows the effect of inhibiting Src and/or Pyk2 phosphorylation of Cx43. Immunofluorescence spread (area) of Lucifer Yellow transfer was measured by ImageJ software. (C) Representative fluorescent images and quantification of in situ TX-100 extracted Cx43 (green, Cx43; blue, DAPI). Quantification was determined by normalizing the mean fluorescent intensity of insoluble Cx43 to DAPI. All data are representative of three independent experiments (one-way ANOVA, **P<0.01, ***P<0.001).
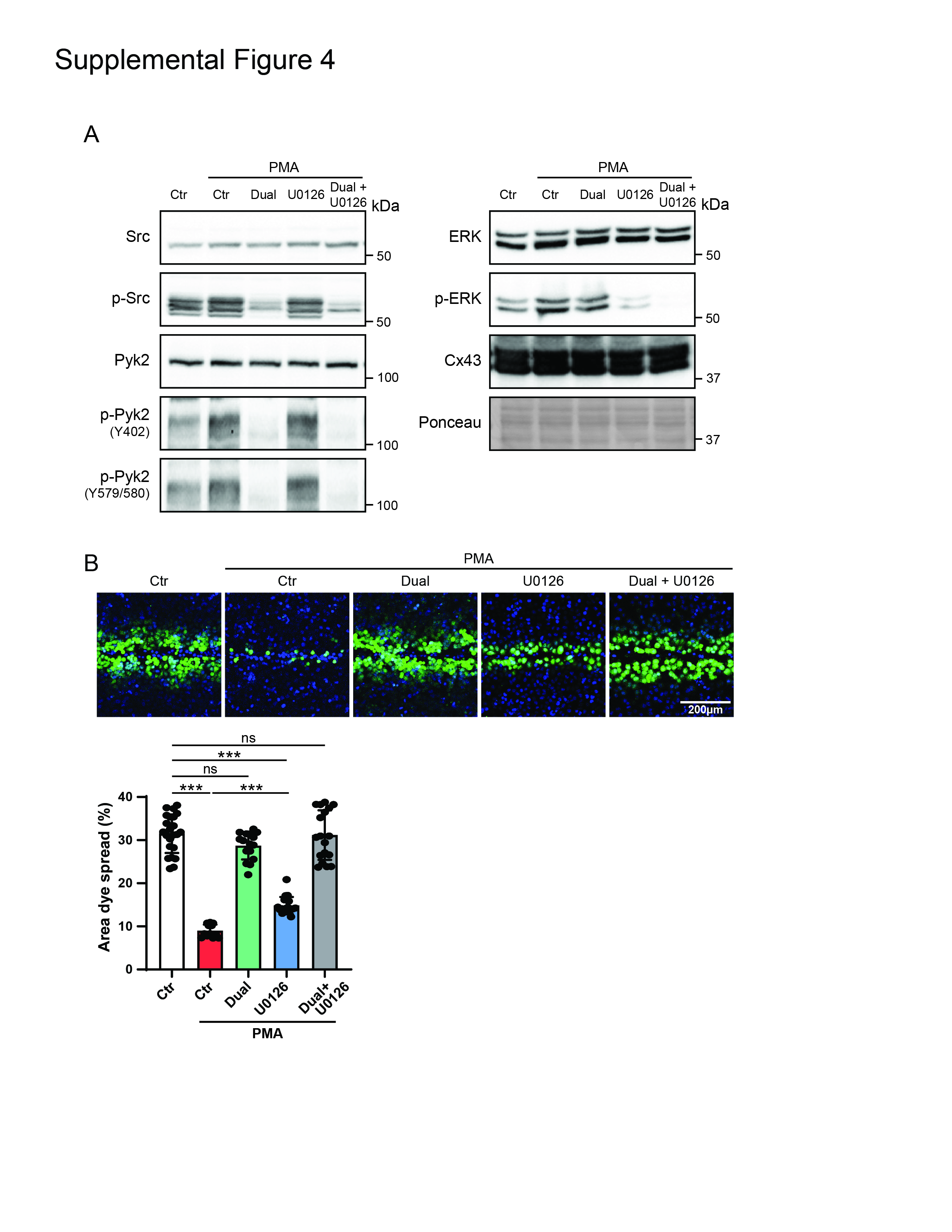
Zhou et al 1999 [37] put forth the hypothesis that MAP kinase (or a related kinase) is necessary for v-Src induced Cx43 gating. A follow-up study presented that chronic gating appears to be connected with tyrosine phosphorylation, while acute gating appears to work through ERK 1/2 [72]. Therefore, we repeated the scrape-loading assay using HeLaCx43 cells treated with PMA in the presence of the specific inhibitor of ERK 1/2 activation U0126 (same as in [72]) alone and combined with the Pyk2 (PF4618433) and c-Src (Saracatinib) inhibitors. Western blot data demonstrates that 60 min of PMA treatment activates ERK (Supplemental Figure 4A). Treatment with U0126 inhibits the activation of ERK, but has no effect on the activation of Pyk2 and c-Src. PMA reduced coupling was only partially reversed by the addition of U0126 alone (~25% reduction). When U0126 was combined with the Pyk2 and c-Src inhibitors, coupling was similar to using the Pyk2 and Src inhibitors alone. The data suggests that while serine phosphorylation can contribute to reducing GJIC, in this experimental system, the larger driving force for reducing GJIC is tyrosine phosphorylation.
Next, we addressed if inhibition of active Pyk2 and Src would contribute to maintaining Cx43-mediated GJIC in a more biologically relevant cell system. Thus, neonatal rat ventricular myocytes (NRVMs) were purified (Supplemental Figure 5A) and found viable in the presence of the c-Src (Saracatinib) or Pyk2 (PF4618433) inhibitors (Supplemental Figure 5B). Western blot data demonstrates that 30 min of PMA treatment activates Src and Pyk2, PF4618433 and Saracatinib partially inhibit the activation of Pyk2 and c-Src, respectively, and the presence of both inhibitors is the most effective in inhibiting active Pyk2 (Figure 6A). Importantly, the increased level of phosphorylation on Cx43 residues Y247 and Y265 seen in the presence of only PMA (similar to PMA treated HeLaCx43, Figure 2B) is reduced to a level below that of even the untreated NRVMs in the presence of both PF4618433 and Saracatinib. These data would suggest that Cx43 in the presence of both inhibitors would have a beneficial effect on impulse propagation (i.e. decreasing tyrosine phosphorylations involved with channel closure and turnover). To test this possibility, we evaluated NRVM contraction in the presence of PMA and the inhibitors. When NRVMs are treated with PMA, only 8% of the cell population is beating and the number of contractions is ~43% of control (Figure 6B). Pre-treatment with Saracatinib or PF4618433 restore the number of beating cells to the control level. Interestingly, Saracatinib only improved the number of contractions to ~63% of control, while PF4618433 completely reversed the effect of PMA (i.e. same as the no PMA control). Pre-treatment with both Saracatinib and PF4618433 resembled with that of PF4618433 alone. Immunofluorescence of the NRVMs with and without PMA revealed that the advantageous effects observed in the presence of the Pyk2 and Src inhibitors was the result of an increase in the level of Cx43 in the GJ plaque (Figure 7).
Figure 6: Inhibiting active Pyk2 reverses PMA mediated decrease in myocyte contraction by decreasing Cx43 tyrosine phosphorylation.
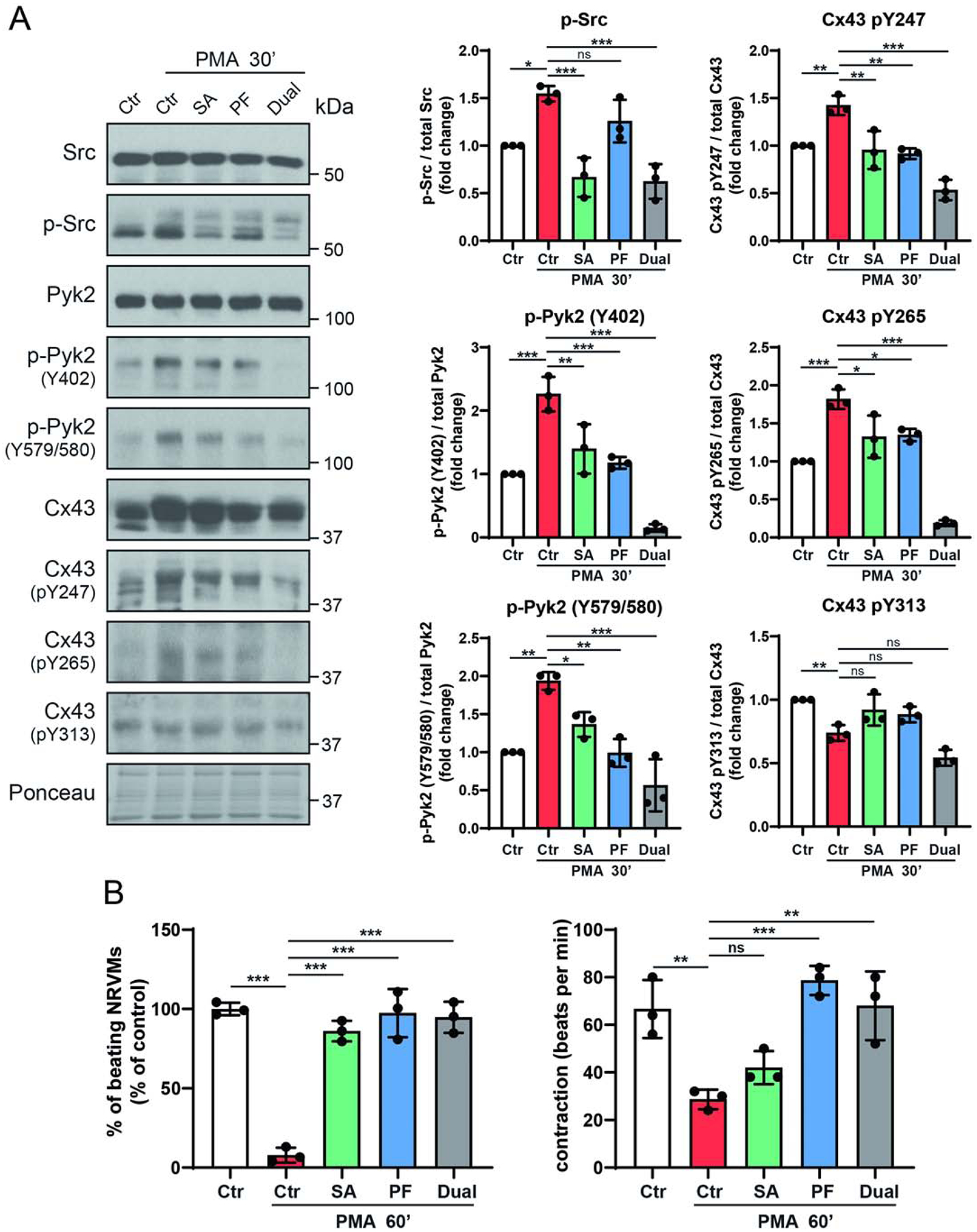
(A) NRVMs were pre-treated with Saracatinib (SA, 0.5μM), PF4618433 (PF, 5μM) or both (3 h) and then treated with PMA (300 nM, 30 min). Lysate from each group was Western blotted. Antibodies used are labeled on the left of each panel. Protein levels were quantified by analyzing scanned blots using ImageJ software, with normalization of protein expression to the control lane (value set arbitrarily as 1). Phosphorylated protein levels were normalized to total protein. (B) NRVMs were pre-treated with Saracatinib, PF4618433 or both (3 h) and then treated with PMA (60 min). Calculated were the total number of NRVMs contributing to the beats (left) and the contraction of NRVMs expressed as beat per minutes (right). Data are representative of three independent experiments (one-way ANOVA, *P<0.05, **P<0.01, ***P<0.001).
Figure 7. Inhibiting active Pyk2 reverses PMA mediated decrease in myocyte contraction by increasing Cx43 at the gap junction (GJ) plaque.
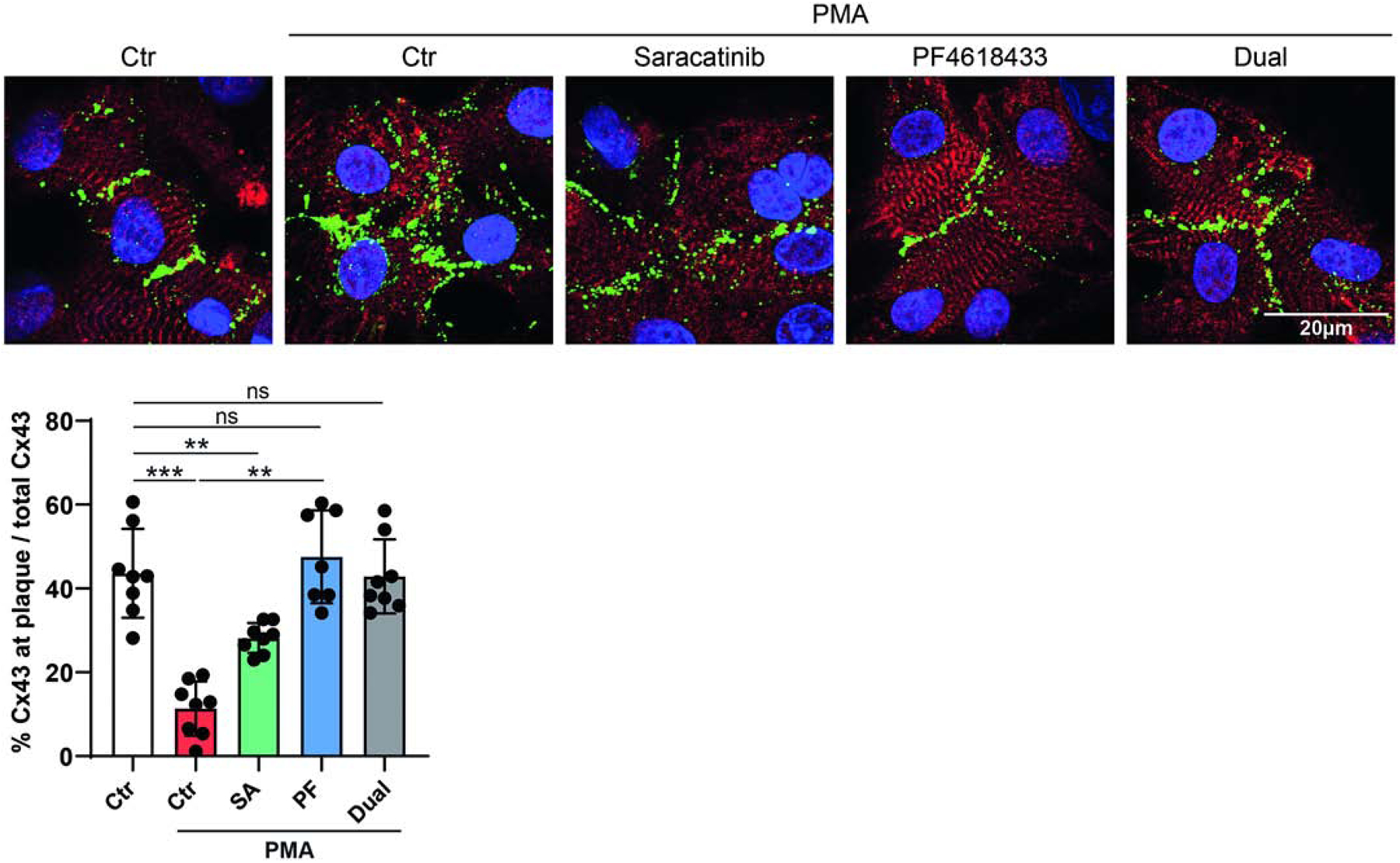
NRVMs were pre-treated with (SA, 0.5μM), PF4618433 (PF, 5μM), or both (3 h) and then treated with PMA (300 nM, 60 min). A representative fluorescent image is shown for each group (red, cardiac marker sarcomeric α-actinin; green, Cx43; blue, DAPI). Quantification was the percentage of total fluorescent intensity of Cx43 at GJ plaque to total Cx43. (one-way ANOVA, **P<0.01, ***P<0.001).
4.5. Pyk2 activity and co-localization with Cx43 increase in the left ventricle during heart failure (HF)
The starting point to determine if Pyk2 could become a target for therapeutic intervention, to restore proper GJIC in HF, would require the interaction with Cx43 under disease conditions. MI leads to a loss in mass of the cardiomyocytes (hypertrophy) and a progression to pathological ventricular remodeling, which can lead to ventricular dysfunction and HF. This is achieved by occluding the left anterior descending artery (LAD) in an animal model [73]. Western blot analysis of left ventricle lysate at six weeks post LAD ligation surgery shows an increase in the hypertrophy and HF marker atrial natriuretic peptide (ANP) validating the HF model (Figure 8A). Immunofluorescence images also confirm HF by the observation of Cx43 at the lateral membrane and within intracellular compartment (longitudinal sections from samples distal to the infraction (hypertrophy area; Figure 8B). LAD ligated animals also have an increase in active Src, active Pyk2, and phosphorylation of Cx43 residue Y265. While there was no statistical difference for the level of total Pyk2 and Cx43, the trend was higher and lower, respectively. The increase in active Pyk2 is associated with an increased localization with Cx43. Interestingly, the Pyk2 and Cx43 colocalization was observed at the ID, lateral membrane, and intracellularly.
Figure 8: Pyk2 activity increases and co-localizes with Cx43 in a heart failure (HF) rat model.
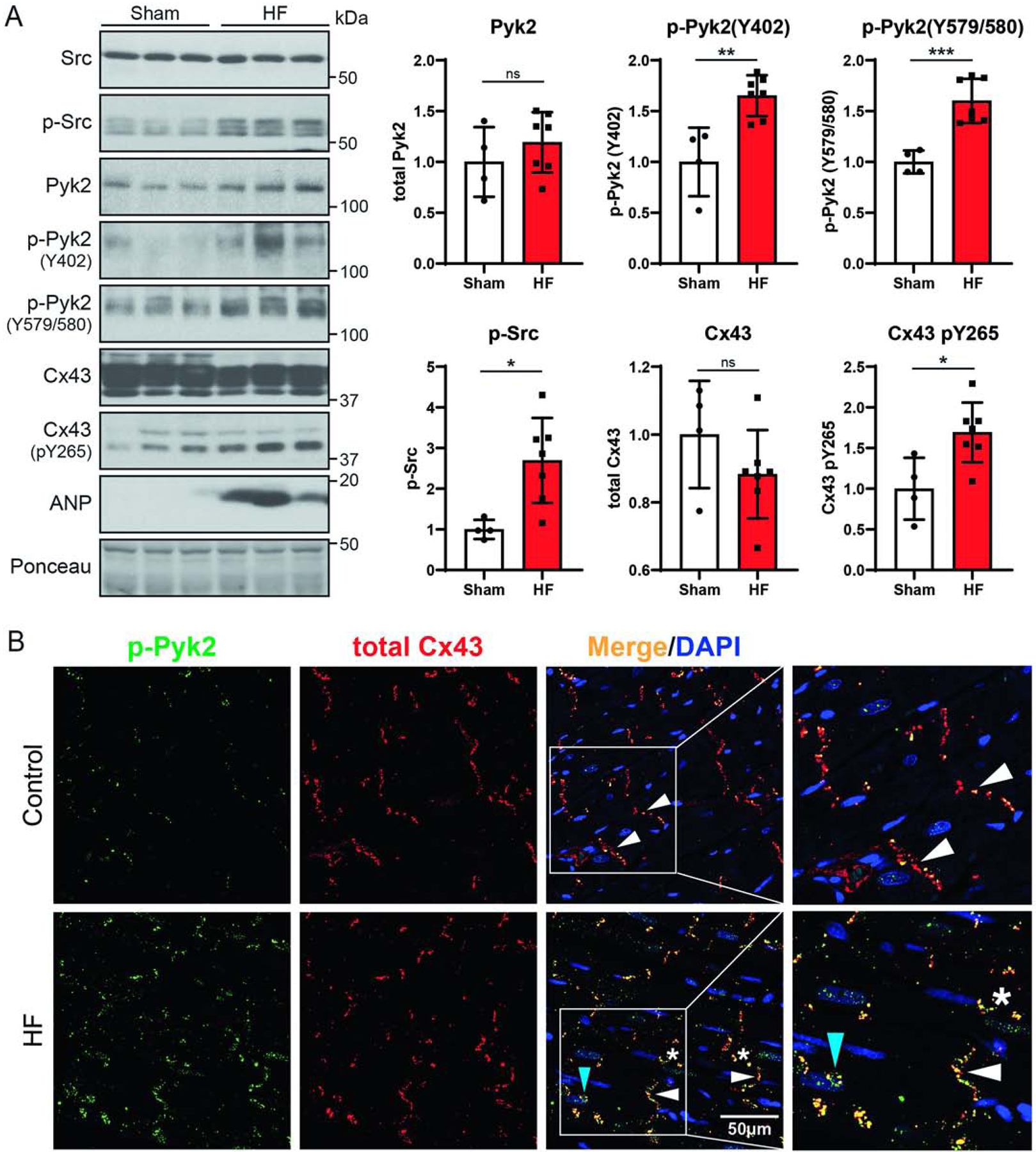
Six weeks after sham or LAD ligation surgery, left ventricle tissue (distal to the infarction, hypertrophy area) was harvested for (A) Western blotting and (B) immunofluorescence imaging (green, enhanced active Pyk2, p-Pyk2Y579/580; red, Cx43; yellow, co-localization; blue, DAPI; white arrows, intercalated disc; stars, lateral membrane; blue arrow, intracellular). All rats with HF had an average infarct area ≥30% of the left ventricle and left ventricular end-diastolic pressure (LVEDP) ≥15 mmHg (Sham, n=4; HF, n=7, t-test, *P<0.05, ***P<0.001).
5. Discussion
Cx43CT phosphorylation is a key regulator of GJ assembly, stability, degradation, channel gating, and selectivity properties. Phosphorylation may alter the structure of the transmembrane α-helices to influence pore size, or modify the binding affinities of regulatory protein partners. Notably, when phosphorylation alters the kinetics of channel assembly or degradation (e.g., via interaction with Nedd4 [51, 74, 75]), cell-to-cell communication is altered. A wealth of data exists about the kinases involved in phosphorylation of Cx43 [54, 76–79]. For example, PKA is associated with enhanced trafficking of Cx43 to the plasma membrane [80–84], while movement of Cx43 into the plaque involves casein kinase 1 [9]. Under disease conditions (e.g., HF), growth factors and other stimuli including coordinated Cx43 phosphorylation by several kinases lead to its trafficking away from the GJ [79, 85]. Sequential phosphorylation by Akt, MAPK, PKC, and Src results in an initial increase and then decrease in GJIC followed by internalization [79, 85]. In this study, we identified that Pyk2 is another kinase involved in the direct phosphorylation and regulation of Cx43.
Pyk2 functions to transduce signals from Ca2+, integrins, and G protein-coupled receptor to downstream MAPK and Akt signaling pathways [42, 86]. Pyk2 activation occurs from an interaction with calmodulin causing homodimerization and auto-phosphorylation at Y402 which facilitates Src binding [41]. Subsequent Src phosphorylation at multiple Pyk2 sites enhances kinase activity and binding of several other adaptor proteins [42]. In cardiomyocytes, intracellular Ca2+ and PKCƐ also regulate Pyk2 activation [87, 88]. This observation may explain why Pyk2 was still “enhanced” activated without expression of c-SrcY530F in HEK 293T cells (albeit to a much lower level, Figure 3) and why the Src inhibitor Saracatinib alone is unable to fully restore Cx43 function (Figures 5 and 6). Pyk2 regulates a wide range of physiological effects, including cell proliferation, differentiation, apoptosis, and cytoskeletal regulation. In the heart, Pyk2 expression is much greater in neonatal, than in adult ventricular myocytes [87]. However, in vivo studies using animal models of HF (e.g., pressure overload) identified an increase in Pyk2 expression and activity [43, 46]. Furthermore, expression and activity of Pyk2 in the LV of failing human hearts is markedly increased and related to lethal arrhythmias [47]. Importantly, the endogenously expressed inhibitor CRNK improves survival, increases LV function, and alters expression of proteins characteristic of LVH in models of MI and dilated cardiomyopathy [44, 61]. Molecular mechanisms responsible for the adverse effects of Pyk2 dysregulation that accompany LVH and HF have remained unclear.
We propose one molecular mechanism by which Pyk2 exerts its adverse effects in the heart is to contribute to the process of Cx43 lateralization and degradation. First, low intracellular pH that results from the MI causes translocation of Src from the perinuclear region to gap junctions at the ID using the cellular actin network [89, 90]. Since Src activation is thought to occur after recruitment to endosomes [89, 91] and Pyk2 also uses the actin cytoskeleton to translocate to the plasma membrane [92], at some point along the journey to the ID, Src interacts and activates Pyk2. At the GJ plaque, they bind directly to Cx43 (Src SH3 domain with Cx43CT residues T275-P284 [11]; currently unknown for Pyk2, although with no SH3 domain, will be different than Src) causing phosphorylation of CT residues Y247, Y265, Y267, and Y313 [35, 39]. pY247 inhibits the Cx43 interaction with β-tubulin [59], pY265 and pY313 inhibit the Cx43 interaction with Drebrin (maintains Cx43 GJs in their functional state at the plasma membrane, binds F-actin) [39, 93], and the Src SH3 domain interacts with the ZO-1 PDZ-2 domain causing displacement of ZO-1 (binds F-actin) from the Cx43CT [11]. Once ZO-1 unhooks from Cx43, the plaque size begins to increase [94, 95], but as it is no longer anchored at the ID (also caused by loss of interaction with tubulin and Drebrin), the entire plaque translocates from the region of high concentration at the ID to the lateral membrane [11]. Consistent with this hypothesis is the lateralized GJs appear larger-than normal plaques [96]. We speculate that Pyk2 inhibition, in addition to Src, would further reverse Cx43 remodeling and improve intercellular communication as seen from inhibiting Src alone [26]. As mentioned above, the reduced phosphorylation at Y247, Y265, and Y313 would correlate with increased binding to Drebrin and tubulin. Additionally, because Src activates ERK1/2, PKC, and Akt [67, 72, 97], Pyk2 activates MAPK and Akt [42, 86], and PKC can activate Pyk2 [87, 88], benefits of inhibiting Pyk2 and Src would also include decreased phosphorylation of S279/282 (MAPK sites), S368 (PKC site), and S373 (Akt site). Consistent with reversing S279/282, S368, and S373 phosphorylation is the observation that in the infarct EBZ, Src inhibition (Tyr phosphorylation) led to higher levels of the Cx43 P2 phosphorylation isoform (Ser phosphorylation at S325, S328, and S330 by casein kinase 1 that promotes GJIC) [9, 26, 98].
GJ channels composed of Cx43 mediate electrical coupling and impulse propagation in the normal working myocardium. In the failing heart caused by a MI, Cx43 remodeling (decreased expression, loss at IDs, and increased presence at lateral membranes) contributes to ventricular arrhythmias; activated Src has been linked to this Cx43 dysregulation. Rutledge et al. demonstrated that inhibiting Src following MI increases Cx43 expression, improves conduction velocity, reduces arrhythmia inducibility, and may represent a new approach to arrhythmia reduction in ischemic cardiomyopathy [26]. However, while increased Cx43 expression was sufficient to explain the beneficial effects after Src inhibition, their data indicate there is an “undiscovered second process” preventing full restoration of Cx43 levels [26]. Our preliminary data suggest we may have identified this second process (i.e., Pyk2) and mechanism that affects Cx43 regulation (e.g., Drebrin [39]). Thus, future studies will inhibit the Pyk2/Src axis in vivo as a therapeutic approach to heart disease.
Supplementary Material
Supplemental Figure 1. Pyk2 phosphorylates Cx43CT tyrosine residues. (A) An in vitro tyrosine kinase screen was performed using the catalytic domain of FAK, Pyk2, SYK, ZAPP70, and Tyk2 to phosphorylate the purified Cx43CT (V236-I382) domain. The amount of Cx43CT phosphorylation was compared with a positive control peptide for each kinase (100% signal). (B) Purified GST or GST-Cx43CT bound on glutathione-agarose beads were incubated ± MCF-7 cell lysate and the pull-down product was analyzed by Western blot using an anti-Pyk2 antibody. (C) Sequence of the Cx43CT domain. Underlined are the phosphorylated tyrosine residues identified via mass spectroscopy from the in vitro kinase assay using the Cx43CT incubated with the Pyk2 kinase domain.
{kind=link}
Supplemental Figure 2. Active Pyk2 and v-Src cause internalization of Cx43. Cellular localization of Cx43 (IF1 antibody, recognizes plaque localized Cx43) ± Src-v (24 h) in HeLa Cx43 WT or (B) HeLa Cx43 Y247/265F cells detected by immunofluorescence (green, p-Pyk2Y579/580; blue, DAPI; red, total Cx43; yellow, p-Pyk2/Cx43 colocalization). Colocalization of Cx43 and p-Pyk2 at GJ plaque was analyzed based on 20 images from three independent experiments. Immunofluorescence intensities of DAPI and Cx43 plaque were measured by ImageJ software. Cx43 plaque immunofluorescence intensity was normalized by DAPI (t-test, ***P<0.001).
{kind=link}
Supplemental Figure 3. Effect of PMA and EGF on selected serine kinases. Lysate from HeLaCx43 cells treated with PMA (100 nM, 1 hr), EGF (100 ng/mL 1 hr), or AngII (100 nM, 1 hr) were Western blotted. Antibodies used are labeled on the left of each panel. SS: Serum starving.
{kind=link}
Supplemental Figure 4: Inhibiting active MAPK only partially reverses PMA mediated closure of Cx43 gap junctions. (A) Lysate from PMA treated HeLaCx43 ± MAPK (U0126, 10μM) and/or Dual (Saracatinib, 5μM and PF4619433, 50μM) inhibitors was Western blotted. Antibodies used are labeled on the left of each panel. (B) Level of gap junction intercellular communication in HeLaCx43 cells pre-treated with MAPK (U0126) and/or Dual (Saracatinib and PF4619433) inhibitors and then treated with PMA was determined using the scrape loading dye transfer assay. Provided are representative immunofluorescence images (green, Lucifer yellow; blue, DAPI). Quantification of the area of Lucifer Yellow transfer shows the effect of inhibiting MAPK and/or Src+Pyk2 phosphorylation of Cx43. Immunofluorescence spread (area) of Lucifer Yellow transfer was measured by ImageJ software. (one-way ANOVA, ***P<0.001).
{kind=link}
Supplemental Figure 5. Pyk2 and Src inhibitors have little-to-no toxicity to NRVMs. (A) Immunofluorescence image illustrating the purity of isolated and cultured NVCMs (>90% pure; red, cardiac marker sarcomeric α-actinin; green, Cx43; blue, DAPI). (B) NVCMs were treated with different concentrations of c-Src (Saracatinib) or Pyk2 (PF4618433) inhibitors (48 hr), and cell viability measured using a MTT assay.
{kind=link}
Highlights.
Protein tyrosine kinase 2 beta (Pyk2) phosphorylates Cx43CT residues Y247, Y265, Y267, and Y313.
Pyk2 phosphorylation of Cx43 leads to decreased GJIC and internalization.
An animal model of myocardial infarction induced heart failure also showed an increase in Pyk2 activity and interaction with Cx43.
Inhibition of both Pyk2 and Src phosphorylation may be necessary to prevent GJ channel closure and remodeling of Cx43 under conditions that stimulate heart disease.
Sources of Funding
This work was supported by grants from the National Institutes of Health (GM072631 and GM131092). Support for the UNMC Advanced Microscopy Core Facility was provided by the Nebraska Research Initiative and an Institutional Development Award (IDeA) from the NIGMS of the NIH (P30GM106397). The following NIH SIG funded instruments were used: LSM 800 Zeiss Confocal Microscope (NIH S10RR027301).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures
The authors declare that they have no conflicts of interest with the contents of this article.
References
- [1].Severs NJ, Bruce AF, Dupont E, Rothery S. Remodelling of gap junctions and connexin expression in diseased myocardium. Cardiovasc Res. 2008;80(1):9–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Reaume AG, de Sousa PA, Kulkarni S, Langille BL, Zhu D, Davies TC, et al. Cardiac malformation in neonatal mice lacking connexin43. Science. 1995;267(5205):1831–4. [DOI] [PubMed] [Google Scholar]
- [3].Gutstein DE, Morley GE, Vaidya D, Liu F, Chen FL, Stuhlmann H, et al. Heterogeneous expression of Gap junction channels in the heart leads to conduction defects and ventricular dysfunction. Circulation. 2001;104(10):1194–9. [DOI] [PubMed] [Google Scholar]
- [4].Gutstein DE, Morley GE, Tamaddon H, Vaidya D, Schneider MD, Chen J, et al. Conduction slowing and sudden arrhythmic death in mice with cardiac-restricted inactivation of connexin43. Circ Res. 2001;88(3):333–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Krijnen PA, Nijmeijer R, Meijer CJ, Visser CA, Hack CE, Niessen HW. Apoptosis in myocardial ischaemia and infarction. J Clin Pathol. 2002;55(11):801–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Jeyaraman M, Tanguy S, Fandrich RR, Lukas A, Kardami E. Ischemia-induced dephosphorylation of cardiomyocyte connexin-43 is reduced by okadaic acid and calyculin A but not fostriecin. Mol Cell Biochem. 2003;242(1–2):129–34. [PubMed] [Google Scholar]
- [7].Turner MS, Haywood GA, Andreka P, You L, Martin PE, Evans WH, et al. Reversible connexin 43 dephosphorylation during hypoxia and reoxygenation is linked to cellular ATP levels. Circ Res. 2004;95(7):726–33. [DOI] [PubMed] [Google Scholar]
- [8].Matsumura K, Mayama T, Lin H, Sakamoto Y, Ogawa K, Imanaga I. Effects of cyclic AMP on the function of the cardiac gap junction during hypoxia. Exp Clin Cardiol. 2006;11(4):286–93. [PMC free article] [PubMed] [Google Scholar]
- [9].Lampe PD, Cooper CD, King TJ, Burt JM. Analysis of Connexin43 phosphorylated at S325, S328 and S330 in normoxic and ischemic heart. J Cell Sci. 2006;119(Pt 16):3435–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Kleber AG, Rudy Y. Basic mechanisms of cardiac impulse propagation and associated arrhythmias. Physiol Rev. 2004;84(2):431–88. [DOI] [PubMed] [Google Scholar]
- [11].Kieken F, Mutsaers N, Dolmatova E, Virgil K, Wit AL, Kellezi A, et al. Structural and molecular mechanisms of gap junction remodeling in epicardial border zone myocytes following myocardial infarction. Circ Res. 2009;104(9):1103–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Peters NS, Coromilas J, Severs NJ, Wit AL. Disturbed connexin43 gap junction distribution correlates with the location of reentrant circuits in the epicardial border zone of healing canine infarcts that cause ventricular tachycardia. Circulation. 1997;95(4):988–96. [DOI] [PubMed] [Google Scholar]
- [13].Akar FG, Nass RD, Hahn S, Cingolani E, Shah M, Hesketh GG, et al. Dynamic changes in conduction velocity and gap junction properties during development of pacing-induced heart failure. Am J Physiol Heart Circ Physiol. 2007;293(2):H1223–30. [DOI] [PubMed] [Google Scholar]
- [14].Kaplan SR, Gard JJ, Carvajal-Huerta L, Ruiz-Cabezas JC, Thiene G, Saffitz JE. Structural and molecular pathology of the heart in Carvajal syndrome. Cardiovasc Pathol. 2004;13(1):26–32. [DOI] [PubMed] [Google Scholar]
- [15].Frey N, Katus HA, Olson EN, Hill JA. Hypertrophy of the heart: a new therapeutic target? Circulation. 2004;109(13):1580–9. [DOI] [PubMed] [Google Scholar]
- [16].Jin H, Chemaly ER, Lee A, Kho C, Hadri L, Hajjar RJ, et al. Mechanoelectrical remodeling and arrhythmias during progression of hypertrophy. Faseb J. 2010;24(2):451–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Formigli L, Ibba-Manneschi L, Perna AM, Pacini A, Polidori L, Nediani C, et al. Altered Cx43 expression during myocardial adaptation to acute and chronic volume overloading. Histol Histopathol. 2003;18(2):359–69. [DOI] [PubMed] [Google Scholar]
- [18].Shiojima I, Sato K, Izumiya Y, Schiekofer S, Ito M, Liao R, et al. Disruption of coordinated cardiac hypertrophy and angiogenesis contributes to the transition to heart failure. J Clin Invest. 2005;115(8):2108–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Yamada KA, Rogers JG, Sundset R, Steinberg TH, Saffitz J. Up-regulation of connexin45 in heart failure. J Cardiovasc Electrophysiol. 2003;14(11):1205–12. [DOI] [PubMed] [Google Scholar]
- [20].Sato T, Ohkusa T, Honjo H, Suzuki S, Yoshida MA, Ishiguro YS, et al. Altered expression of connexin43 contributes to the arrhythmogenic substrate during the development of heart failure in cardiomyopathic hamster. Am J Physiol Heart Circ Physiol. 2008;294(3):H1164–73. [DOI] [PubMed] [Google Scholar]
- [21].Cooklin M, Wallis WR, Sheridan DJ, Fry CH. Changes in cell-to-cell electrical coupling associated with left ventricular hypertrophy. Circ Res. 1997;80(6):765–71. [DOI] [PubMed] [Google Scholar]
- [22].Peters NS. New insights into myocardial arrhythmogenesis: distribution of gap-junctional coupling in normal, ischaemic and hypertrophied human hearts. Clin Sci (Lond). 1996;90(6):447–52. [DOI] [PubMed] [Google Scholar]
- [23].Macia E, Dolmatova E, Cabo C, Sosinsky AZ, Dun W, Coromilas J, et al. Characterization of gap junction remodeling in epicardial border zone of healing canine infarcts and electrophysiological effects of partial reversal by rotigaptide. Circ Arrhythm Electrophysiol. 2011;4(3):344–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Ek-Vitorin JF, King TJ, Heyman NS, Lampe PD, Burt JM. Selectivity of connexin 43 channels is regulated through protein kinase C-dependent phosphorylation. Circ Res. 2006;98(12):1498–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Greener ID, Sasano T, Wan X, Igarashi T, Strom M, Rosenbaum DS, et al. Connexin43 gene transfer reduces ventricular tachycardia susceptibility after myocardial infarction. J Am Coll Cardiol. 2012;60(12):1103–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Rutledge CA, Ng FS, Sulkin MS, Greener ID, Sergeyenko AM, Liu H, et al. c-Src kinase inhibition reduces arrhythmia inducibility and connexin43 dysregulation after myocardial infarction. J Am Coll Cardiol. 2014;63(9):928–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Sorgen PL, Duffy HS, Sahoo P, Coombs W, Delmar M, Spray DC. Structural changes in the carboxyl terminus of the gap junction protein connexin43 indicates signaling between binding domains for c-Src and zonula occludens-1. J Biol Chem. 2004;279(52):54695–701. [DOI] [PubMed] [Google Scholar]
- [28].Sovari AA, Iravanian S, Dolmatova E, Jiao Z, Liu H, Zandieh S, et al. Inhibition of c-Src tyrosine kinase prevents angiotensin II-mediated connexin-43 remodeling and sudden cardiac death. J Am Coll Cardiol. 2011;58(22):2332–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Gubens MA, Burns M, Perkins SM, Pedro-Salcedo MS, Althouse SK, Loehrer PJ, et al. A phase II study of saracatinib (AZD0530), a Src inhibitor, administered orally daily to patients with advanced thymic malignancies. Lung cancer. 2015;89(1):57–60. [DOI] [PubMed] [Google Scholar]
- [30].Reddy SM, Kopetz S, Morris J, Parikh N, Qiao W, Overman MJ, et al. Phase II study of saracatinib (AZD0530) in patients with previously treated metastatic colorectal cancer. Investigational new drugs. 2015;33(4):977–84. [DOI] [PubMed] [Google Scholar]
- [31].Swenson KI, Piwnica-Worms H, McNamee H, Paul DL. Tyrosine phosphorylation of the gap junction protein connexin43 is required for the pp60v-src-induced inhibition of communication. Cell Regul. 1990;1(13):989–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Cottrell GT, Lin R, Warn-Cramer BJ, Lau AF, Burt JM. Mechanism of v-Src- and mitogen-activated protein kinase-induced reduction of gap junction communication. Am J Physiol Cell Physiol. 2003;284(2):C511–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Giepmans BN, Hengeveld T, Postma FR, Moolenaar WH. Interaction of c-Src with gap junction protein connexin-43. Role in the regulation of cell-cell communication. J Biol Chem. 2001;276(11):8544–9. [DOI] [PubMed] [Google Scholar]
- [34].Lin R, Warn-Cramer BJ, Kurata WE, Lau AF. v-Src-mediated phosphorylation of connexin43 on tyrosine disrupts gap junctional communication in mammalian cells. Cell Commun Adhes. 2001;8(4–6):265–9. [DOI] [PubMed] [Google Scholar]
- [35].Lin R, Warn-Cramer BJ, Kurata WE, Lau AF. v-Src phosphorylation of connexin 43 on Tyr247 and Tyr265 disrupts gap junctional communication. J Cell Biol. 2001;154(4):815–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Toyofuku T, Yabuki M, Otsu K, Kuzuya T, Tada M, Hori M. Functional role of c-Src in gap junctions of the cardiomyopathic heart. Circ Res. 1999;85(8):672–81. [DOI] [PubMed] [Google Scholar]
- [37].Zhou L, Kasperek EM, Nicholson BJ. Dissection of the molecular basis of pp60(v-src) induced gating of connexin 43 gap junction channels. J Cell Biol. 1999;144(5):1033–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Loo LW, Berestecky JM, Kanemitsu MY, Lau AF. pp60src-mediated phosphorylation of connexin 43, a gap junction protein. J Biol Chem. 1995;270(21):12751–61. [DOI] [PubMed] [Google Scholar]
- [39].Zheng L, Li H, Cannon A, Trease AJ, Spagnol G, Zheng H, et al. Phosphorylation of Cx43 residue Y313 by Src contributes to blocking the interaction with Drebrin and disassembling gap junctions. J Mol Cell Cardiol. 2019;126:36–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Zhao M, Finlay D, Zharkikh I, Vuori K. Novel Role of Src in Priming Pyk2 Phosphorylation. PLoS One. 2016;11(2):e0149231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Dikic I, Tokiwa G, Lev S, Courtneidge SA, Schlessinger J. A role for Pyk2 and Src in linking G-protein-coupled receptors with MAP kinase activation. Nature. 1996;383(6600):547–50. [DOI] [PubMed] [Google Scholar]
- [42].Blaukat A, Ivankovic-Dikic I, Gronroos E, Dolfi F, Tokiwa G, Vuori K, et al. Adaptor proteins Grb2 and Crk couple Pyk2 with activation of specific mitogen-activated protein kinase cascades. J Biol Chem. 1999;274(21):14893–901. [DOI] [PubMed] [Google Scholar]
- [43].Bayer AL, Heidkamp MC, Patel N, Porter MJ, Engman SJ, Samarel AM. PYK2 expression and phosphorylation increases in pressure overload-induced left ventricular hypertrophy. Am J Physiol Heart Circ Physiol. 2002;283(2):H695–706. [DOI] [PubMed] [Google Scholar]
- [44].Koshman YE, Chu M, Kim T, Kalmanson O, Farjah M, Kumar M, et al. Cardiomyocyte-specific expression of CRNK, the C-terminal domain of PYK2, maintains ventricular function and slows ventricular remodeling in a mouse model of dilated cardiomyopathy. J Mol Cell Cardiol. 2014;72:281–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Fukai K, Nakamura A, Hoshino A, Nakanishi N, Okawa Y, Ariyoshi M, et al. Pyk2 aggravates hypoxia-induced pulmonary hypertension by activating HIF-1alpha. Am J Physiol Heart Circ Physiol. 2015;308(8):H951–9. [DOI] [PubMed] [Google Scholar]
- [46].Melendez J, Welch S, Schaefer E, Moravec CS, Avraham S, Avraham H, et al. Activation of pyk2/related focal adhesion tyrosine kinase and focal adhesion kinase in cardiac remodeling. J Biol Chem. 2002;277(47):45203–10. [DOI] [PubMed] [Google Scholar]
- [47].Lang D, Glukhov AV, Efimova T, Efimov IR. Role of Pyk2 in cardiac arrhythmogenesis. Am J Physiol Heart Circ Physiol. 2011;301(3):H975–83. [DOI] [PubMed] [Google Scholar]
- [48].Callera GE, Antunes TT, He Y, Montezano AC, Yogi A, Savoia C, et al. c-Src Inhibition Improves Cardiovascular Function but not Remodeling or Fibrosis in Angiotensin II-Induced Hypertension. Hypertension. 2016;68(5):1179–90. [DOI] [PubMed] [Google Scholar]
- [49].Luttrell LM, Ferguson SS, Daaka Y, Miller WE, Maudsley S, Della Rocca GJ, et al. Beta-arrestin-dependent formation of beta2 adrenergic receptor-Src protein kinase complexes. Science. 1999;283(5402):655–61. [DOI] [PubMed] [Google Scholar]
- [50].Duffy HS, Sorgen PL, Girvin ME, O’Donnell P, Coombs W, Taffet SM, et al. pH-dependent intramolecular binding and structure involving Cx43 cytoplasmic domains. J Biol Chem. 2002;277(39):36706–14. [DOI] [PubMed] [Google Scholar]
- [51].Leykauf K, Salek M, Bomke J, Frech M, Lehmann WD, Durst M, et al. Ubiquitin protein ligase Nedd4 binds to connexin43 by a phosphorylation-modulated process. J Cell Sci. 2006;119(Pt 17):3634–42. [DOI] [PubMed] [Google Scholar]
- [52].Patel KP, Zhang PL, Carmines PK. Neural influences on renal responses to acute volume expansion in rats with heart failure. Am J Physiol. 1996;271(4 Pt 2):H1441–8. [DOI] [PubMed] [Google Scholar]
- [53].Kleiber AC, Zheng H, Schultz HD, Peuler JD, Patel KP. Exercise training normalizes enhanced glutamate-mediated sympathetic activation from the PVN in heart failure. Am J Physiol Regul Integr Comp Physiol. 2008;294(6):R1863–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Li H, Spagnol G, Zheng L, Stauch KL, Sorgen PL. Regulation of Connexin43 function and expression by Tyrosine kinase 2. J Biol Chem. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Solan JL, Lampe PD. Spatio-temporal regulation of connexin43 phosphorylation and gap junction dynamics. Biochim Biophys Acta Biomembr. 2018;1860(1):83–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Sosinsky GE, Solan JL, Gaietta GM, Ngan L, Lee GJ, Mackey MR, et al. The C-terminus of connexin43 adopts different conformations in the Golgi and gap junction as detected with structure-specific antibodies. Biochem J. 2007;408(3):375–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Falk MM, Baker SM, Gumpert AM, Segretain D, Buckheit RW 3rd. Gap junction turnover is achieved by the internalization of small endocytic double-membrane vesicles. Mol Biol Cell. 2009;20(14):3342–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Gaietta G, Deerinck TJ, Adams SR, Bouwer J, Tour O, Laird DW, et al. Multicolor and electron microscopic imaging of connexin trafficking. Science. 2002;296(5567):503–7. [DOI] [PubMed] [Google Scholar]
- [59].Saidi Brikci-Nigassa A, Clement MJ, Ha-Duong T, Adjadj E, Ziani L, Pastre D, et al. Phosphorylation controls the interaction of the connexin43 C-terminal domain with tubulin and microtubules. Biochemistry. 2012;51(21):4331–42. [DOI] [PubMed] [Google Scholar]
- [60].Fong JT, Kells RM, Falk MM. Two tyrosine-based sorting signals in the Cx43 C-terminus cooperate to mediate gap junction endocytosis. Mol Biol Cell. 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Hart DL, Heidkamp MC, Iyengar R, Vijayan K, Szotek EL, Barakat JA, et al. CRNK gene transfer improves function and reverses the myosin heavy chain isoenzyme switch during post-myocardial infarction left ventricular remodeling. J Mol Cell Cardiol. 2008;45(1):93–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Martinez AD, Hayrapetyan V, Moreno AP, Beyer EC. Connexin43 and connexin45 form heteromeric gap junction channels in which individual components determine permeability and regulation. Circ Res. 2002;90(10):1100–7. [DOI] [PubMed] [Google Scholar]
- [63].Leithe E, Rivedal E. Epidermal growth factor regulates ubiquitination, internalization and proteasome-dependent degradation of connexin43. J Cell Sci. 2004;117(Pt 7):1211–20. [DOI] [PubMed] [Google Scholar]
- [64].Schulz R, Gorge PM, Gorbe A, Ferdinandy P, Lampe PD, Leybaert L. Connexin 43 is an emerging therapeutic target in ischemia/reperfusion injury, cardioprotection and neuroprotection. Pharmacol Ther. 2015;153:90–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Xie Y, Liu S, Hu S, Wei Y. Cardiomyopathy-Associated Gene 1-Sensitive PKC-Dependent Connexin 43 Expression and Phosphorylation in Left Ventricular Noncompaction Cardiomyopathy. Cell Physiol Biochem. 2017;44(2):828–42. [DOI] [PubMed] [Google Scholar]
- [66].Kanemitsu MY, Loo LW, Simon S, Lau AF, Eckhart W. Tyrosine phosphorylation of connexin 43 by v-Src is mediated by SH2 and SH3 domain interactions. J Biol Chem. 1997;272(36):22824–31. [DOI] [PubMed] [Google Scholar]
- [67].Solan JL, Lampe PD. Connexin 43 in LA-25 cells with active v-src is phosphorylated on Y247, Y265, S262, S279/282, and S368 via multiple signaling pathways. Cell Commun Adhes. 2008;15(1):75–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Toyofuku T, Akamatsu Y, Zhang H, Kuzuya T, Tada M, Hori M. c-Src regulates the interaction between connexin-43 and ZO-1 in cardiac myocytes. J Biol Chem. 2001;276(3):1780–8. [DOI] [PubMed] [Google Scholar]
- [69].Valiunas V, Beyer EC, Brink PR. Cardiac gap junction channels show quantitative differences in selectivity. Circ Res. 2002;91(2):104–11. [DOI] [PubMed] [Google Scholar]
- [70].Trease AJ, Capuccino JMV, Contreras J, Harris AL, Sorgen PL. Intramolecular signaling in a cardiac connexin: Role of cytoplasmic domain dimerization. J Mol Cell Cardiol. 2017;111:69–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Li H, Spagnol G, Zheng L, Stauch KL, Sorgen PL. Regulation of Connexin43 Function and Expression by Tyrosine Kinase 2. J Biol Chem. 2016;291(30):15867–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Mitra SS, Xu J, Nicholson BJ. Coregulation of multiple signaling mechanisms in pp60v-Src-induced closure of Cx43 gap junction channels. J Membr Biol. 2012;245(8):495–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Zheng H, Liu X, Sharma NM, Patel KP. Renal denervation improves cardiac function in rats with chronic heart failure: Effects on expression of beta-adrenoceptors. Am J Physiol Heart Circ Physiol. 2016;311(2):H337–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Girao H, Catarino S, Pereira P. Eps15 interacts with ubiquitinated Cx43 and mediates its internalization. Exp Cell Res. 2009;315(20):3587–97. [DOI] [PubMed] [Google Scholar]
- [75].Spagnol G, Kieken F, Kopanic JL, Li H, Zach S, Stauch KL, et al. Structural Studies of the Nedd4 WW Domains and their Selectivity for the Cx43 Carboxyl-terminus. J Biol Chem. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Axelsen LN, Calloe K, Holstein-Rathlou NH, Nielsen MS. Managing the complexity of communication: regulation of gap junctions by post-translational modification. Frontiers in pharmacology. 2013;4:130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Solan JL, Lampe PD. Connexin phosphorylation as a regulatory event linked to gap junction channel assembly. Biochim Biophys Acta. 2005;1711(2):154–63. [DOI] [PubMed] [Google Scholar]
- [78].Lampe PD, Lau AF. The effects of connexin phosphorylation on gap junctional communication. Int J Biochem Cell Biol. 2004;36(7):1171–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Solan JL, Lampe PD. Specific Cx43 phosphorylation events regulate gap junction turnover in vivo. FEBS Lett. 2014;588(8):1423–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Lampe PD. Analyzing phorbol ester effects on gap junctional communication: a dramatic inhibition of assembly. J Cell Biol. 1994;127(6 Pt 2):1895–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Atkinson MM, Lampe PD, Lin HH, Kollander R, Li XR, Kiang DT. Cyclic AMP modifies the cellular distribution of connexin43 and induces a persistent increase in the junctional permeability of mouse mammary tumor cells. J Cell Sci. 1995;108 (Pt 9):3079–90. [DOI] [PubMed] [Google Scholar]
- [82].TenBroek EM, Lampe PD, Solan JL, Reynhout JK, Johnson RG. Ser364 of connexin43 and the upregulation of gap junction assembly by cAMP. J Cell Biol. 2001;155(7):1307–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Sosinsky GE, Solan JL, Gaietta GM, Ngan L, Lee GJ, Mackey MR, et al. The C-terminus of Connexin43 adopts different conformations in the golgi and gap junction as detected with structure specific antibodies. Biochem J. 2007;408(3):375–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Solan JL, Marquez-Rosado L, Sorgen PL, Thornton PJ, Gafken PR, Lampe PD. Phosphorylation at S365 is a gatekeeper event that changes the structure of Cx43 and prevents down-regulation by PKC. J Cell Biol. 2007;179(6):1301–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Thevenin AF, Kowal TJ, Fong JT, Kells RM, Fisher CG, Falk MM. Proteins and mechanisms regulating gap-junction assembly, internalization, and degradation. Physiology (Bethesda). 2013;28(2):93–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Guo J, Sabri A, Elouardighi H, Rybin V, Steinberg SF. Alpha1-adrenergic receptors activate AKT via a Pyk2/PDK-1 pathway that is tonically inhibited by novel protein kinase C isoforms in cardiomyocytes. Circ Res. 2006;99(12):1367–75. [DOI] [PubMed] [Google Scholar]
- [87].Bayer AL, Ferguson AG, Lucchesi PA, Samarel AM. Pyk2 expression and phosphorylation in neonatal and adult cardiomyocytes. J Mol Cell Cardiol. 2001;33(5):1017–30. [DOI] [PubMed] [Google Scholar]
- [88].Bayer AL, Heidkamp MC, Howes AL, Heller Brown J, Byron KL, Samarel AM. Protein kinase C epsilon-dependent activation of proline-rich tyrosine kinase 2 in neonatal rat ventricular myocytes. J Mol Cell Cardiol. 2003;35(9):1121–33. [DOI] [PubMed] [Google Scholar]
- [89].Timpson P, Jones GE, Frame MC, Brunton VG. Coordination of cell polarization and migration by the Rho family GTPases requires Src tyrosine kinase activity. Curr Biol. 2001;11(23):1836–46. [DOI] [PubMed] [Google Scholar]
- [90].Zhang SS, Shaw RM. Trafficking highways to the intercalated disc: new insights unlocking the specificity of connexin 43 localization. Cell Commun Adhes. 2014;21(1):43–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Reinecke JB, Katafiasz D, Naslavsky N, Caplan S. Regulation of Src trafficking and activation by the endocytic regulatory proteins MICAL-L1 and EHD1. J Cell Sci. 2014;127(Pt 8):1684–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Kodama H, Fukuda K, Takahashi T, Sano M, Kato T, Tahara S, et al. Role of EGF Receptor and Pyk2 in endothelin-1-induced ERK activation in rat cardiomyocytes. J Mol Cell Cardiol. 2002;34(2):139–50. [DOI] [PubMed] [Google Scholar]
- [93].Ambrosi C, Ren C, Spagnol G, Cavin G, Cone A, Grintsevich EE, et al. Connexin43 Forms Supramolecular Complexes through Non-Overlapping Binding Sites for Drebrin, Tubulin, and ZO-1. PLoS One. 2016;11(6):e0157073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Hunter AW, Barker RJ, Zhu C, Gourdie RG. Zonula occludens-1 alters connexin43 gap junction size and organization by influencing channel accretion. Mol Biol Cell. 2005;16(12):5686–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Dunn CA, Lampe PD. Injury-triggered Akt phosphorylation of Cx43: a ZO-1-driven molecular switch that regulates gap junction size. J Cell Sci. 2014;127(Pt 2):455–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Cabo C, Yao J, Boyden PA, Chen S, Hussain W, Duffy HS, et al. Heterogeneous gap junction remodeling in reentrant circuits in the epicardial border zone of the healing canine infarct. Cardiovasc Res. 2006;72(2):241–9. [DOI] [PubMed] [Google Scholar]
- [97].Jiang T, Qiu Y. Interaction between Src and a C-terminal proline-rich motif of Akt is required for Akt activation. J Biol Chem. 2003;278(18):15789–93. [DOI] [PubMed] [Google Scholar]
- [98].Grosely R, Kopanic JL, Nabors S, Kieken F, Spagnol G, Al-Mugotir M, et al. Effects of phosphorylation on the structure and backbone dynamics of the intrinsically disordered connexin43 C-terminal domain. J Biol Chem. 2013;288(34):24857–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. Pyk2 phosphorylates Cx43CT tyrosine residues. (A) An in vitro tyrosine kinase screen was performed using the catalytic domain of FAK, Pyk2, SYK, ZAPP70, and Tyk2 to phosphorylate the purified Cx43CT (V236-I382) domain. The amount of Cx43CT phosphorylation was compared with a positive control peptide for each kinase (100% signal). (B) Purified GST or GST-Cx43CT bound on glutathione-agarose beads were incubated ± MCF-7 cell lysate and the pull-down product was analyzed by Western blot using an anti-Pyk2 antibody. (C) Sequence of the Cx43CT domain. Underlined are the phosphorylated tyrosine residues identified via mass spectroscopy from the in vitro kinase assay using the Cx43CT incubated with the Pyk2 kinase domain.
Supplemental Figure 2. Active Pyk2 and v-Src cause internalization of Cx43. Cellular localization of Cx43 (IF1 antibody, recognizes plaque localized Cx43) ± Src-v (24 h) in HeLa Cx43 WT or (B) HeLa Cx43 Y247/265F cells detected by immunofluorescence (green, p-Pyk2Y579/580; blue, DAPI; red, total Cx43; yellow, p-Pyk2/Cx43 colocalization). Colocalization of Cx43 and p-Pyk2 at GJ plaque was analyzed based on 20 images from three independent experiments. Immunofluorescence intensities of DAPI and Cx43 plaque were measured by ImageJ software. Cx43 plaque immunofluorescence intensity was normalized by DAPI (t-test, ***P<0.001).
Supplemental Figure 3. Effect of PMA and EGF on selected serine kinases. Lysate from HeLaCx43 cells treated with PMA (100 nM, 1 hr), EGF (100 ng/mL 1 hr), or AngII (100 nM, 1 hr) were Western blotted. Antibodies used are labeled on the left of each panel. SS: Serum starving.
Supplemental Figure 4: Inhibiting active MAPK only partially reverses PMA mediated closure of Cx43 gap junctions. (A) Lysate from PMA treated HeLaCx43 ± MAPK (U0126, 10μM) and/or Dual (Saracatinib, 5μM and PF4619433, 50μM) inhibitors was Western blotted. Antibodies used are labeled on the left of each panel. (B) Level of gap junction intercellular communication in HeLaCx43 cells pre-treated with MAPK (U0126) and/or Dual (Saracatinib and PF4619433) inhibitors and then treated with PMA was determined using the scrape loading dye transfer assay. Provided are representative immunofluorescence images (green, Lucifer yellow; blue, DAPI). Quantification of the area of Lucifer Yellow transfer shows the effect of inhibiting MAPK and/or Src+Pyk2 phosphorylation of Cx43. Immunofluorescence spread (area) of Lucifer Yellow transfer was measured by ImageJ software. (one-way ANOVA, ***P<0.001).
Supplemental Figure 5. Pyk2 and Src inhibitors have little-to-no toxicity to NRVMs. (A) Immunofluorescence image illustrating the purity of isolated and cultured NVCMs (>90% pure; red, cardiac marker sarcomeric α-actinin; green, Cx43; blue, DAPI). (B) NVCMs were treated with different concentrations of c-Src (Saracatinib) or Pyk2 (PF4618433) inhibitors (48 hr), and cell viability measured using a MTT assay.