

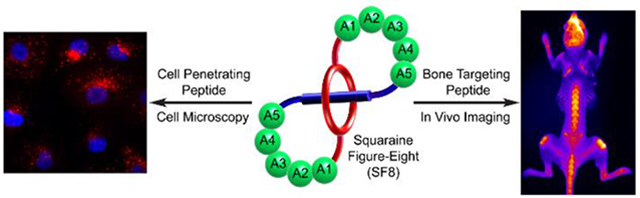
Abstract
A generalizable synthetic method creates a new class of covalently connected, self-threaded fluorescent molecular probes with figure-eight topology, an encapsulated deep-red fluorophore, and two peripheral peptide loops. The globular molecular shape and rigidified peptide loops enhance imaging performance by promoting water solubility, eliminating probe self-aggregation, and increasing probe stability. Moreover, the peptide loops determine the affinity and selectivity for targets within complex biological samples such as cell culture, tissue histology slices, or living subjects. For example, a probe with cell penetrating peptide loops targets the surface of cell plasma membranes; whereas, a probe with bone targeting peptide loops selectively stains the skeleton within a living mouse. The unique combination of bright deep-red fluorescence, high stability, and predictable peptide-based targeting is ideal for photon intense fluorescence microscopy and biological imaging.
Keywords: dyes/pigments, fluorescent probes, imaging agents, molecular recognition, peptides
Graphical Abstract
Where to hide a fluorophore: bury it inside a self-threaded peptide probe which will promote high water solubility, lower the propensity for probe self-aggregation, and greatly enhance probe stability. The peptide loops determine the affinity and selectivity for targets within complex biological samples such as cell culture, tissue histology slices, or living subjects.
Introduction
Synthetic peptides are popular choices as targeting molecules for bioimaging or diagnostic applications.[1] Unfortunately, broad utilization of linear, naturally occurring peptides is severely limited by their, (a) inherent flexibility which often lowers targeting capability, (b) charged N- and C-terminal groups which inhibit cell permeation, and (c) sensitivity to proteolytic degradation.[2] Efforts to obviate these drawbacks have led to a wide range of molecular design strategies to create peptidomimetics with improved properties.[3] One approach is to design structures that incorporate amino acids with unnatural side chains or opposite chirality, or alternatively use an unnatural polymer backbone.[4,5] However, the synthesis of unnatural peptidomimetics is often a laborious task compared to standard peptide synthesis methodology using naturally occurring amino acids. Thus, there is high interest in developing new peptide design strategies that incorporate sequences of naturally occurring amino acids as crucial building blocks within structural topologies that enhance the molecular properties. In this regard, peptide modifications such as macrocyclization, internal crosslinking, or physical entanglement have been shown to produce peptide derivatives with improved targeting, permeability, and stability.[6-9] The specific focus here is on fluorescently labeled peptidyl structures as targeted molecular probes for fluorescence imaging.[1] It is widely known that imaging performance is enhanced by using bright fluorophore labels that absorb and emit light with deep-red or near-infrared wavelengths.[10] Deep-red fluorescent probes are especially valuable because they can be used effectively in several different but complementary fluorescence imaging techniques.[11] Image contrast in deep-red cell microscopy is inherently high because there is a low amount of background absorption and autofluorescence from endogenous cell biomolecules; likewise, mesoscale deep-red imaging of living subjects is favored by deep penetration and lower scattering of the light through skin and tissue.[12] Moreover, the detectors and silicon based CCD cameras in modern microscopes and in vivo imaging stations detect deep-red light with high quantum efficiency. Indeed, the quantum efficiency for common back-illuminated silicon based CCD cameras is higher for deep-red light than near-infrared light.[13] But an inherent problem with deep-red and near-infrared fluorophores is the large hydrophobic surface areas of the π-extended molecules which can overwhelm the targeting and pharmacokinetic properties of the attached peptide (Scheme 1).[14,15] At present, the standard solution is to simply decorate the fluorophore structure with multiple charged functional groups[16] but this approach can be problematic because a highly charged fluorescent label can alter a small peptide’s targeting properties.[17]
Scheme 1.
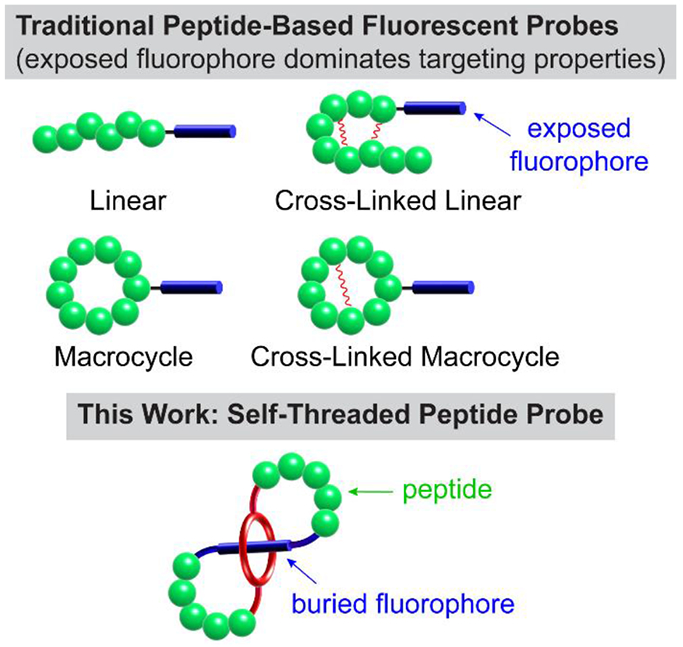
Comparison of traditional peptide-based fluorescent probes with self-threaded probe architecture.
Here we describe a conceptually new way to create a deep-red fluorescent peptide for high-performance bioimaging. The key design element is a self-threaded molecular figure-eight architecture with a buried fluorophore. The literature contains a small scattered collection of publications describing figure-eight molecules,[18-24] and a few of them are self-threaded structures,[25-27] although none of the latter incorporate peptides or were designed for a practical application such as fluorescence imaging. The buried fluorophore is a squaraine dye that exhibits narrow and intense deep-red absorption and emission bands (ex: 640–660 nm, em: 660–680 nm).[28,29] More than a decade ago we discovered that the chemical and fluorescence properties of a water-soluble squaraine dye can be greatly enhanced by encapsulating the dye within a protective tetralactam macrocycle and creating a mechanically interlocked squaraine rotaxane.[30] Over the intervening years, we and others have successfully converted squaraine rotaxanes into targeted fluorescence imaging probes for use in cell microscopy and in vivo imaging of living subjects. Notably, independent labs have reported data suggesting that molecular probes with appended squaraine rotaxane dyes exhibit unsurpassed photostability for deep-red single-fluorescent-molecule tracking and extremely high brightness for one and two-photon fluorescence imaging.[31-33] Squaraine rotaxane dyes are now commercially available as reagents for bioconjugation and they have been used successfully to label antibodies and large proteins.[31,34] However, the relatively large size and polyanionic charge of commercial squaraine rotaxanes means they are less useful for labeling small peptides. We reasoned that these problems could be solved by a structural innovation that blended the peptide and squaraine rotaxane components together and created a single hybrid molecule that we call SF8 (Squaraine Figure-Eight). These compounds have two distinctive structural features, (a) an extremely bright and highly stable deep-red squaraine fluorochrome that is buried deep inside the molecule and thus, does not influence its targeting or pharmacokinetic properties, and (b) easy synthetic incorporation of external peptide loops as rigidified recognition motifs for selective targeting of biological interfaces. We disclose a versatile and general synthetic method to prepare SF8 molecules and a detailed structural characterization by X-ray diffraction and NMR spectroscopy. We also report our findings that these versatile peptide-based SF8 molecules are very effective as high performance deep-red fluorescent molecular probes for cell microscopy and fluorescence imaging of living subjects.
Results and Discussion
Synthesis
Shown in Scheme 2 is a summary of the general synthetic sequence that is used to convert a mechanically interlocked squaraine [2]rotaxane into a covalently connected self-threaded SF8 molecule. The key starting compound is squaraine rotaxane SR1 which is easily prepared in a few steps using well-established synthetic chemistry. A crucial structural feature within SR1 is an N-propyl chain at each end of the encapsulated squaraine which is sterically large enough to keep SR1 threaded during its purification and subsequent amide bond coupling reaction. The descriptor we use for these figure-eight molecules is SF8 followed by an acronym for the two loop components. Thus, the prototype compound is called SF8(C6)2 where C6 refers to the two identical hexyl-containing loops (Figure 1). This compound was prepared in 53% isolated yield by conducting a two-step sequence that started with amide bond coupling of SR1 and two molar equivalents of 6-azidohexanoic acid, followed by a double macrocyclization step that employed a copper catalyzed azide/alkyne cycloaddition (CuCAAC) reaction.
Scheme 2.
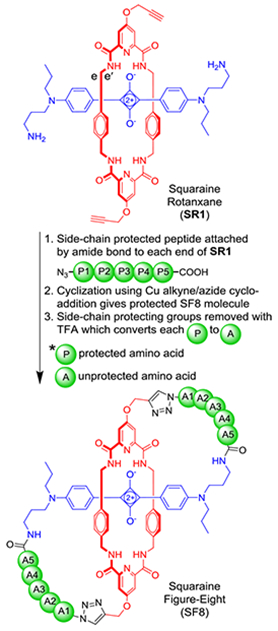
General synthetic scheme showing conversion of Squaraine Rotaxane, SR1, into a peptidyl Squaraine Figure-Eight (SF8) with two pentapeptide loops.
Figure 1.

A) Two views of the X-ray crystal structure of SF8(C6)2. The green dotted lines indicate internal hydrogen bonds. B) Partial 1H NMR spectra of SF8(C6)2 at two temperatures reflecting hindered spinning of aryl rings.
The next synthetic goal was to prepare the three SF8 structures in Scheme 3 that each have two identical peptidyl loops. First, standard solid phase peptide synthesis methodology was employed to prepare a precursor peptide with protected side chains, a free C-terminus carboxyl, and an N-terminus capped with a short azido-containing chain. Then, as shown in Scheme 2, amide bond coupling was used to attach a copy of the protected peptide to each end of SR1 followed by double macrocyclization using CuCAAC chemistry to give the desired organic soluble, side-chain protected peptidyl SF8 structure (27–53% isolated yield for the double macrocyclization step). Facile removal of the side-chain protecting groups using TFA provided the water-soluble fluorescent SF8 peptide probes SF8(DDDDG)2, SF8(RRRG)2, and SF8(RADG)2.[35]
Scheme 3.

Peptidyl SF8 probes and linear control probes.
Structure and Molecular Motion of Prototype SF8(C6)2
Single crystals of the prototype molecule, SF8(C6)2, were analyzed by X-ray diffraction and provided in Figures 1A and S1 are two views of the solid-state structure which has Ci symmetry. The self-threaded squaraine/tetralactam core of the SF8 molecule retains the same threaded co-conformation adopted by the parent squaraine rotaxane. Notably, there is bifurcated hydrogen bonding of each squaraine oxygen to the two proximal amide NH residues in the surrounding tetralactam component. There is also cofacial stacking of the two tetralactam phenylene sidewalls over both faces of the electron deficient C4O2 ring within the encapsulated squaraine fluorophore, with a centroid-to-centroid distance between the two phenylenes of 6.72 Å. The hexyl chain within each loop of the SF8 molecule includes some C-C bonds in a gauche conformation which allows the loop to turn. The amide functional group within each loop is hydrogen bonded to a crystal lattice water molecule that acts as a solid-state connection point for three SF8(C6)2 molecules (Figure S2).
The synthetic conversion of precursor rotaxane SR1 into SF8(C6)2 eliminates two orthogonal planes of symmetry which alters the 1H NMR spectral patterns in diagnostic ways. For example, the chemical shift equivalent tetralactam methylene protons e and e’ in rotaxane SR1 are chemically inequivalent in SF8(C6)2 and they differ by ~1 ppm in chemical shift (see Scheme 2 and Figure 1B for atom labels). Variable temperature NMR studies of SF8(C6)2 in organic solvent revealed two internal molecular motions that have been previously documented in related squaraine rotaxanes.[36] One motion is hindered spinning of the two phenylene sidewalls within the central tetralactam component that surrounds the squaraine fluorochrome, a process that exchanges the sites of chemically inequivalent phenylene protons f and f’ (Figures 1B and S4-S6). There is also hindered spinning of the encapsulated squaraine aryl rings, a process that exchanges the sites of chemically inequivalent protons g/g’. Measurements of the coalescence temperature and limiting chemical shifts enabled the free energy of activation for phenylene spinning within SF8(C6)2 to be determined as 14.6 kcal/mol, which is very close to value of 15.4 kcal/mol for the same phenylene spinning process in a structurally related squaraine rotaxane.[36] These energy barriers correspond to a rate constant of ~100 s−1 at room temperature.
Studies of Peptidyl SF8 Molecules
The three peptidyl SF8 probes selected for preliminary study were SF8(DDDDG)2 (polyanionic), SF8(RRRG)2 (polycationic) and SF8(RADG)2 (charge-balanced). The wide dispersity in molecular charge, while keeping the same fluorescent core and very similar molecular size, enabled us to evaluate the impact of peptide electrostatics on fundamental probe performance properties such as solubility, stability and self-aggregation. We also wanted to assess the inherent probe affinity for various biological interfaces such as the surfaces of proteins, membranes, and bone.
Each peptidyl SF8 probe is a chiral molecule with C2 symmetry. The 1H NMR spectrum of each molecule showed the same line broadening exhibited by the organic prototype, SF8(C6)2, reflecting hindered aryl ring spinning within the constrained squaraine/tetralactam core of the molecules. An important early question was to determine if the SF8 architecture provided any major improvement in imaging-related molecular properties. To gain insight, we compared the peptidyl SF8 probes, SF8(DDDDG)2 and SF8(RRRG)2, with their linear squaraine rotaxane structural isomers SR(azido-DDDDG)2 and SR(azido-RRRG)2, respectively (Scheme 3).
In the case of oligo-aspartate SF8(DDDDG)2, its high water-solubility contrasted to the low solubility of linear isomer SR(azido-DDDDG)2. In addition, the absorption spectrum of SF8(DDDDG)2 in water exhibited a narrow peak at 660 nm indicating no molecular self-aggregation; whereas, the absorption spectrum of linear SR(azido-DDDDG)2 showed substantial peak broadening due to the formation of non-fluorescent H-aggregates (Figure S7). As a result, the overall fluorescence brightness of SF8(DDDDG)2 was substantially higher than its linear isomer SR(azido-DDDDG)2, especially at increased probe concentration which favored SR(azido-DDDDG)2 aggregation. A likely reason for the difference in water solubility and propensity for self-aggregation is the change in molecular shape; the globular structure of SF8(DDDDG)2 with its outward directed carboxylate side chains is better solvated in water than isomeric SR(azido-DDDDG)2 whose linear triblock structure (polar-nonpolar-polar) favors intermolecular packing (Scheme S9). Both compounds exhibited high chemical stability in biological media, even in the presence of excess amounts of strong nucleophiles such as glutathione (Figure S8 and S9). A related stability attribute of peptidyl SF8 molecules is enhanced resistance to proteolytic degradation. This favorable property was apparent in experiments that incubated separate 10 μM samples of SF8(DDDDG)2 probe or the linear pentapeptide azido-DDDDG with Asp-N, an endoproteinase that hydrolyzes peptide bonds on the N-terminal side of aspartic acid residues. LCMS analysis of the two mixtures after 2 hours revealed that 87% of SF8(DDDDG)2 was still intact, whereas, only 51% of azido-DDDDG remained (Figure S15). In other words, proteolytic degradation of the peptide sequence was reduced by a factor of 3.8.
The comparison of oligo-arginine SF8(RRRG)2 and its linear structural isomer SR(azido-RRRG)2 found that both molecules were soluble as unaggregated molecules in water and they possessed the same high fluorescence brightness (Figure S10). But there was a major difference in chemical stability of the encapsulated squaraine fluorophore. While the fluorescence profile of a solution containing SF8(RRRG)2 in PBS, pH 7.4 was unchanged over 15 hours, the linear SR(azido-RRRG)2 was almost completely bleached after a few hours (Figure S11A). It is well-established that squaraine bleaching is due to nucleophilic attack at its C4O2 core.[30] Thus, the difference in chemical stability is attributed to extra lateral mobility of the surrounding macrocycle in mechanically bonded rotaxane SR(azido-RRRG)2 which transiently exposes the core of the encapsulated squaraine to nucleophilic attack. Moreover, the results in Figures S11-S14 suggest that the squaraine core is attacked intramolecularly by a nucleophilic nitrogen atom on the appended arginine side chains, which promotes hydrolytic fragmentation (Scheme S10).[37] In contrast, the entire structure of SF8(RRRG)2 is a single covalently bonded molecule, which means the two peptidyl loops strongly secure the surrounding macrocycle over the internal squaraine component leading to more robust squaraine protection and greatly increased probe chemical stability.
Listed in Table 1 is a summary of absorption and fluorescence emission properties in water. The fluorescence quantum yields for SF8(DDDDG)2, SF8(RRRG)2, and SF8(RADG)2 were measured to be 0.19, 0.10, and 0.15, respectively, which are all quite high compared to the typical values for deep-red fluorescent dyes in water.[11] The high chemical stability of SF8(DDDDG)2 in biological media prompted us to compare its photostability relative to samples of the pentamethine cyanine dyes, Cy5 (Scheme S8) and Alex Fluor 647, two common benchmark deep-red fluorophores.[38] Separate cuvettes containing each dye sample (3 μM in PBS) were irradiated with a 150 W Xenon lamp with a > 495 nm band pass filter. The intensity of the absorption maxima band for the SF8(DDDDG)2 sample was essentially unchanged after one hour of constant photon-intense irradiation whereas absorption intensities for the Cy5 and Alexa Fluor 647 samples were reduced to 33% and 77% respectively (Figure S16). The enhanced photostability of the SF8 probe compared to Alexa Fluor 647 is notable and consistent with a previous report that a squaraine rotaxane was 4.2 times more photostable than Alexa Fluor 647.[32]
Table 1.
Photophysical data for SF8 molecules and squaraine rotaxanes.
| Compound | λabs (nm) | λem (nm) | Log ε | Φf |
|---|---|---|---|---|
| SF8(C6)2 | 645 | 660 | 5.49 | 0.64[a] |
| SF8(RRRG)2 | 660 | 678 | 5.12 | 0.10[b] |
| SR(azido-RRRG)2 | 660 | 678 | 5.06 | 0.14[b] |
| SF8(DDDDG)2 | 660 | 677 | 5.44 | 0.19[b] |
| SR(azido-DDDDG)2 | 660 | 676 | 4.94 | 0.07[b] |
| SF8(RADG)2 | 660 | 678 | 5.54 | 0.15[b] |
In CHCl3, measurements of quantum yield (Φf) (±5%) relative to 4,4-[bis(N,N-dimethylamino)phenyl]squaraine (Φf = 0.70). ε is molar absorptivity.
In H2O, quantum yield measurements (±5%) relative to methylene blue (Φf = 0.02).
Taken together, the physical data shows that covalently bonded peptidyl SF8 molecules have more favorable solubility, aggregation, fluorescence or stability properties than their mechanically bonded squaraine rotaxane predecessors. The high water solubility of peptidyl SF8 fluorescent probes, and low propensity for probe self-aggregation, derives from their relatively rigid globular shape with a buried deep-red fluorophore and outward directed polar side chains. The covalently connected peptide loops make it harder to disrupt the self-threaded squaraine/tetralactam core and thus ensure that high fluorescent probe performance is a more general outcome.
The next step in the project was to determine if peptidyl SF8 molecules have enhanced utility for fluorescence bioimaging, and we chose to evaluate them as targeted probes for specific biological interfaces that are commonly found within complex biomedical samples such as cell culture, histological sections, and living subjects. As a prelude to cell studies we evaluated the capabilities of the three peptidyl SF8 probes to interact with biomimetic phospholipid bilayer membranes. This was achieved by conducting liposome leakage studies that measured escape of a fluorescent dye (carboxyfluorescein) from two sets of liposomes: charge-balanced liposomes comprised entirely of 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (100% POPC) and anionic liposomes that included 5% of 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-L-serine (95:5, POPC:POPS) (Figures S17-S21). We found that addition of charge-balanced SF8(RADG)2 did not induce any liposome leakage (Figure 2A). In contrast, polycationic hexa-arginine SF8(RRRG)2 was quite membrane active and promoted substantial liposome leakage, a common property exhibited by oligo-arginine derivatives.[39] Although SF8(RRRG)2 is membrane active at low micromolar concentration it is not acutely toxic to mammalian cells, as determined by standard methyl thiazolyl tetrazolium (MTT) assays which showed that probe concentrations up to 10 μM produced no loss in cell viability over 24 hours (Figure S23). Fluorescence cell microscopy revealed evidence for rapid accumulation of the SF8(RRRG)2 probe on the surface of living mammalian cells followed by probe uptake within punctate intracellular structures (Figure 2B) that colocalization microscopy experiments indicated to be primarily endosomes and lysosomes (Figure S24). We hypothesize that the polycationic SF8(RRRG)2 probe first associates with the abundant polyanionic glycosaminoglycans that decorate the cell surface, followed by probe partitioning into the plasma membrane and subsequent endocytosis. The probe is retained in the membrane by ion-pairing with anionic polar lipids.[40] Supporting this hypothesis are the results of water/octanol partitioning experiments which showed that the polycationic SF8(RRRG)2 is quite hydrophilic with a log P = −2.0, but a 1:1 mixture of SF8(RRRG)2 and the anionic phospholipid POPS forms a lipophilic ion-pair complex with log P = 1.3 (Figure S22).
Figure 2.
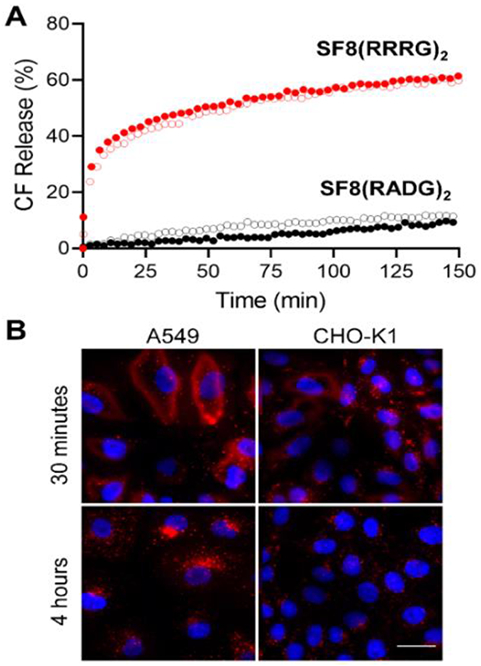
A) Leakage of carboxyfluorescein (CF) from charge-balanced liposomes (empty circles; 100% POPC) or anionic liposomes (filled circles; 95% POPC, 5% POPS) induced by addition of SF8 probe (1 μM), in 20 mM HEPES buffer with 150 mM NaCl, pH 7.4. B) Representative deep-red fluorescence micrographs showing uptake of SF8(RRRG)2 (5 μM) by live cells after 4 hour incubation. Blue fluorescence shows Hoechst 33342 nuclear stain. Scale bar = 30 μm.
Microscopy studies of mammalian cells mixed with polyanionic SF8(DDDDG)2 showed no probe permeation into cells (Figure S24A), consistent with the liposome results indicating no measurable probe interaction with bilayer membranes. But a series of bioimaging studies found that SF8(DDDDG)2 has high avidity for the mineral matrix of bone, a known targeting property of oligo-aspartate peptides.[41,42] Bone targeting was first tested by conducting a set of cuvette experiments using bulk samples of powdered mouse tibia and femur bone. An aqueous solution of SF8(DDDDG)2 probe was treated with powdered bone and after extended shaking the bone powder was separated from the supernatant by centrifugation. Inspection of the pelleted bone powder revealed strong staining by the blue-colored probe (Figure 3A), and deep-red fluorescence analysis of the supernatant indicated a commensurate loss of the SF8(DDDDG)2 (Figure 3B). In contrast, a control experiment using the probe SF8(RADG)2, which has a charge-balanced and net-neutral molecular structure, found negligible affinity for the bone powder. We next conducted histology experiments using thin sections of mouse tibia bone and observed the same trend. The highly fluorescent SF8(DDDDG)2 strongly stained the mineral segments within the bone slices (Figure 3C), whereas there was no bone staining by the charge-balanced control probe SF8(RADG)2 (Figure S25). Proof that the SF8(DDDDG)2 was targeting the Ca2+ ions in the bone matrix was gained by observing no staining of bone slices that had been demineralized with a pre-treatment of EDTA (Figure S26).
Figure 3.

Targeting of SF8(DDDDG)2 or SF8(RADG)2 probe to mouse tibia and femur bone. Microcentrifuge tubes containing powdered mouse tibia and femur bone (50 mg) and a solution of fluorescent probe (4 μM) were shaken and centrifuged at 2,000 g for 5 minutes. The photographs in (A) show that SF8(DDDDG)2 produced blue-colored bone powder but SF8(RADG)2 did not, and the graph in (B) shows the commensurate loss of probe fluorescence from the supernatant of probe solutions that were treated with powdered bone. The representative micrographs in (C) show histology slices of mouse bone after treatment with a solution of SF8(DDDDG)2 (5 μM) for 30 minutes; scale bar = 0.15 mm. D) Representative deep-red fluorescent dorsal image of a skinless mouse (N = 3) that had been dosed with 10 nmol of SF8(DDDDG)2 and euthanized 3 hours later. E) Graph of mean pixel intensity (MPI) for the deep-red fluorescence whole-body skeletal images of skinless mice (normalized to thigh muscle from the same animal) that had been dosed with 10 nmol of SF8(DDDDG)2 and euthanized 3 hours (N = 3) or 48 hours (N = 2) later. F) Biodistribution of mice injected with SF8(DDDDG)2. Mice received intravenous injection of SF8(DDDDG)2 (10 nmol) and were sacrificed at either 3 h (N = 3) or 48 h (N = 2). The mean pixel intensity (MPI) for each resected organ is relative to the MPI for thigh muscle from the same animal; error bars indicate ±SEM.
These favorable bone staining results prompted us to conduct a live mouse fluorescence imaging study using a protocol approved by our Institutional Animal Care and Use Committee. A cohort of live mice was dosed intravenously with 10 nmol of SF8(DDDDG)2 and subsequently imaged at 0, 1.5, and 3 hours (Figure S27). At 3 hours, the mice were sacrificed, the skin was removed and the skeleton imaged using a standard Cy5 filter set (ex: 640 nm, em: 710 nm). The representative fluorescence image of a skinless mouse in Figure 3D shows that SF8(DDDDG)2 targeted the exposed mouse skeletal locations that were undergoing bone turnover.[43] Moreover, bone avidity was essentially irreversible as indicated by a comparison of two cohorts of mice that had received the same dose of SF8(DDDDG)2 but had been sacrificed at 3 or 48 hours after probe dosing. As shown in Figure 3E, there was no reduction in the amount of skeletal staining at the longer time point. This result also highlights the high chemical and biochemical stability of the SF8(DDDDG)2 probe at the bone target site. Probe biodistribution throughout the mice was determined by harvesting the mouse organs and acquiring deep-red fluorescence images as a measure of probe accumulation. The fluorescence of each organ was quantified as mean pixel intensity and the results indicated that probe clearance from the blood was rapid and primarily through the kidney (Figure 3F).
The high affinity of SF8(DDDDG)2 for the mineral matrix of exposed bone is consistent with the bone-seeking behavior of oligo-aspartate peptides.[41,42,44] From a practical perspective, there is substantial ongoing interest in developing fluorescent bone-seeking probes for both preclinical research and clinical imaging of diseases such as osteoporosis and bone cancer.[45,46] Most of the literature probes are based on bisphosphonate structures which have the drawback of eliciting strong pharmaceutical activity;[43] thus, there is need for alternative fluorescent probes with different types of bone targeting groups.[47] [48] The biocompatibility of oligo-aspartate probes makes them an attractive option; however, the susceptibility of linear L-aspartate sequences to proteolytic degradation diminishes in vivo targeting performance and this problem has motivated efforts to devise protease resistant analogues such as linear D-aspartate sequences,[42,49] or dendritic structures.[50] In this regard, the remarkable ability of deep-red fluorescent SF8(DDDDG)2, with its protease resistant tetra L-aspartate loops, to produce bright and extremely stable staining of bone within a living subject is a notable finding and demonstrates the great promise of peptidyl SF8 molecules as a new class of high performance, targeted fluorescence imaging agents.
Conclusion
A generalizable synthetic method enables convenient preparation of self-threaded, peptide Squaraine Figure-Eight (SF8) fluorescent probes. SF8 probes possess a favorable combination of molecular and deep-red fluorescence properties that makes them highly suitable for photon intense microscopy and biological imaging. The side chains on the peptide loops of an SF8 probe determine the selective affinity for biological interfaces within complex biomedical samples such as cell culture, tissue histology slices, or living subjects. Fluorescence imaging experiments showed that SF8(RRRG)2 with two oligo-arginine loops targets the cell plasma membrane; SF8(DDDDG)2 with two oligo-aspartate loops does not interact with cell membranes but has high and selective affinity for the mineral matrix in bone, and SF8(RADG)2 with two charge-balanced peptide loops does not associate with either of these biological interfaces. Mouse imaging studies with SF8(DDDDG)2 demonstrated that SF8 probes have high chemical and biochemical stability in complex physiological environments. The next step in the research is to determine if peptidyl SF8 probes can be used for selective, high affinity targeting of specific biomedically relevant biomolecules such as cell surface receptors.[51] The longest peptidyl loop within the current set of SF8 probes is a pentapeptide,[35] but there is no reason why the loops cannot be longer. Future synthetic work in this direction will be facilitated by exploiting the large number of known synthetic peptide turn mimetics and hairpin nucleators.[4] It should also be possible to modify the synthetic methodology and make additional classes of self-threaded squaraine peptide architectures including unsymmetrical figure-eight structures with each loop comprised of a different peptide sequence, and single loop systems that are close mimics of physically entangled peptides such as lasso peptides and knottins.[8,9,52]
Supplementary Material
Acknowledgements
We are grateful for funding support from the US NIH (R01GM059078, R35GM136212 and T32GM075762), the University of Notre Dame (Berry Family Foundation Fellowship, ERRP grant to acquire a Bruker impact II mass spectrometer), and the US NSF Graduate Research Fellowship Program (DGE-1841556). We warmly thank Dr. M. Lee for valuable analytical chemistry assistance.
Footnotes
Conflict of interest
The authors declare no conflict of interest.
References
- [1].Wang W, Hu Z, Adv. Mater 2019, 31, 1–8. [DOI] [PubMed] [Google Scholar]
- [2].Ellert-Miklaszewska A, Poleszak K, Kaminska B, Future Med. Chem 2017, 9, 199–221. [DOI] [PubMed] [Google Scholar]
- [3].Walport LJ, Obexer R, Suga H, Curr. Opin. Biotechnol 2017, 48, 242–250. [DOI] [PubMed] [Google Scholar]
- [4].Nair RV, Baravkar SB, Ingole TS, Sanjayan GJ, Chem. Commun 2014, 50, 13874–13884. [DOI] [PubMed] [Google Scholar]
- [5].Angell Y, Holford M, Moos WH, Protein Pept. Lett 2018, 25, 1044–1050. [DOI] [PubMed] [Google Scholar]
- [6].Gang D, Kim DW, Park HS, Genes 2018, 9, 557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Zhang RY, Thapa P, Espiritu MJ, Menon V, Bingham JP, Bioorg. Med. Chem 2018, 26, 1135–1150. [DOI] [PubMed] [Google Scholar]
- [8].Kintzing JR, Cochran JR, Curr. Opin. Chem. Biol 2016, 34, 143–150. [DOI] [PubMed] [Google Scholar]
- [9].Martin-Gómez H, Tulla-Puche J, Org. Biomol. Chem 2018, 16, 5065–5080. [DOI] [PubMed] [Google Scholar]
- [10].Gao M, Yu F, Lv C, Choo J, Chen L, Chem. Soc. Rev 2017, 46, 2237–2271. [DOI] [PubMed] [Google Scholar]
- [11].Ahn H-Y, Yao S, Wang X, Belfield KD, ACS Appl. Mater. Interfaces 2012, 4, 2847–2854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Specht EA, Braselmann E, Palmer AE, Annu. Rev. Physiol 2016, 79, 93–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Mangold K, Shaw JA, Vollmer M, Eur. J. Phys 2013, 34, S51–S71. [Google Scholar]
- [14].Li D, Schreiber C, Smith BD, Angew. Chem. Int. Ed 2020, 59, 12154–12161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Mendive‐Tapia L, Wang J, Vendrell M, Pept. Sci 2020, 1–13. [Google Scholar]
- [16].Bernardin A, Chipon B, Bouteiller C, Clavé G, Massonneau M, Romieu A, Renard P-Y, Bioconjug. Chem 2007, 18, 1303–1317. [DOI] [PubMed] [Google Scholar]
- [17].Choi HS, Gibbs SL, Lee JH, Kim SH, Ashitate Y, Liu F, Hyun H, Park G, Xie Y, Bae S, Henary M, V Frangioni J, Nat. Biotechnol 2013, 31, 148–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Lei SN, Xiao H, Zeng Y, Tung CH, Wu LZ, Cong H, Angew. Chem. Int. Ed 2020, 59, 10059–10065. [DOI] [PubMed] [Google Scholar]
- [19].Wanner MJ, Steemers L, Uiterweerd MT, Klijn RS, Ehlers AW, van Maarseveen JH, Chem. Eur. J 2018, 24, 13114–13117. [DOI] [PubMed] [Google Scholar]
- [20].Prusty S, Podh MB, Purohit CS, ChemistrySelect 2018, 3, 9690–9693. [Google Scholar]
- [21].Niess F, Duplan V, Sauvage JP, J. Am. Chem. Soc 2014, 136, 5876–5879. [DOI] [PubMed] [Google Scholar]
- [22].Steemers L, Wanner MJ, Lutz M, Hiemstra H, Van Maarseveen JH, Nat. Commun 2017, 8, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Saikawa M, Nakamura T, Uchida J, Yamamura M, Nabeshima T, Chem. Commun 2016, 52, 10727–10730. [DOI] [PubMed] [Google Scholar]
- [24].Niess F, Duplan V, Diercks CS, Sauvage JP, Chem. Eur. J 2015, 21, 14393–14400. [DOI] [PubMed] [Google Scholar]
- [25].Reuter C, Wienand W, Schmuck C, Vögtle F, Chem. Eur. J 2001, 7, 1728–1733. [DOI] [PubMed] [Google Scholar]
- [26].Boyle MM, Forgan RS, Friedman DC, Gassensmith JJ, Smaldone RA, Stoddart JF, Sauvage JP, Chem. Commun 2011, 47, 11870–11872. [DOI] [PubMed] [Google Scholar]
- [27].Boyle MM, Gassensmith JJ, Whalley AC, Forgan RS, Smaldone RA, Hartlieb KJ, Blackburn AK, Sauvage JP, Stoddart JF, Chem. Eur. J 2012, 18, 10312–10323. [DOI] [PubMed] [Google Scholar]
- [28].Johnson JR, Fu N, Arunkumar E, Leevy WM, Gammon ST, Piwnica-Worms D, Smith BD, Angew. Chem. Int. Ed 2007, 46, 5528–5531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Avirah RR, Jayaram DT, Adarsh N, Ramaiah D, Org. Biomol. Chem 2012, 10, 911–920. [DOI] [PubMed] [Google Scholar]
- [30].Gassensmith JJ, Baumes JM, Smith BD, Chem. Commun 2009, 6329–6338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Tsunoyama TA, Watanabe Y, Goto J, Naito K, Kasai RS, Suzuki KGN, Fujiwara TK, Kusumi A, Nat. Chem. Biol 2018, 14, 497–506. [DOI] [PubMed] [Google Scholar]
- [32].Duggal D, Nagwekar J, Rich R, Midde K, Fudala R, Gryczynski I, Borejdo J, Am. J. Physiol. Regul. Integr. Comp. Physiol 2019, 222–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Podgorski K, Terpetschnig E, Klochko OP, Obukhova OM, Haas K, PLoS One 2012, 7, e51980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Perez Bay AE, Schreiner R, Benedicto I, Paz Marzolo M, Banfelder J, Weinstein AM, Rodriguez-Boulan EJ, Nat. Commun 2016, 7, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Our early research focus is on SF8 probes with relative short tetra- or pentapeptide loops that favor cell permeation for fluorescence microscopy or rapid blood extravasation for in vivo fluorescence imaging. Within the set of three probes reported here, the yield of the key double macrocyclization step was lowest for the precursor of tetrapeptide-based SF8(RRRG)2 (27%). We attributed this to steric hindrance caused by the large size of the protected Arg side chains and thus we never attempted to make the homologous pentapeptide version. But as part of ongoing unpublished studies we have prepared a number of SF8 probes with pentapeptide or hexapeptide loops and in all cases the yields for the key double macrocyclization step are in the range 25 – 65% with no isolated larger cyclic oligomers as byproducts. For a useful review of peptide cyclization using the copper-catalyzed azide–alkyne cycloaddition reaction, see: Li X, Chem. Asian J 2011, 6, 2606 – 2616.22043498 [Google Scholar]
- [36].Murgu I, Baumes JM, Eberhard J, Gassensmith JJ, Arunkumar E, Smith BD, J. Org. Chem 2011, 76, 688–691. [DOI] [PubMed] [Google Scholar]
- [37].Spence GT, Lo SS, Ke C, Destecroix H, Davis AP, Hartland GV, Smith BD, Chem. Eur. J 2014, 20, 12628–12635. [DOI] [PubMed] [Google Scholar]
- [38].Gorka AP, Nani RR, Schnermann MJ, Acc. Chem. Res 2018, 51, 3226–3235. [DOI] [PubMed] [Google Scholar]
- [39].Peraro L, Kritzer JA, Angew. Chem. Int. Ed 2018, 57, 11868–11881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Gasparini G, Bang EK, Montenegro J, Matile S, Chem. Commun 2015, 51, 10389–10402. [DOI] [PubMed] [Google Scholar]
- [41].Kasugai S, Fujisawa R, Waki Y, Miyamoto K-I, Ohya K, J. Bone Miner. Res 2010, 15, 936–943. [DOI] [PubMed] [Google Scholar]
- [42].Sekido T, Sakura N, Higashi Y, Miya K, Nitta Y, Nomura M, Sawanishi H, Morito K, Masamune Y, Kasugai S, Yokogawa K, Miyamoto KI, J. Drug Target 2001, 9, 111–121. [DOI] [PubMed] [Google Scholar]
- [43].Cawthray J, Wasan E, Wasan K, Drug Deliv. Transl. Res 2017, 7, 466–481. [DOI] [PubMed] [Google Scholar]
- [44].Wang M, Park S, Nam Y, Nielsen J, Low SA, Srinivasarao M, Low PS, Bioconjug. Chem 2018, 29, 3800–3809. [DOI] [PubMed] [Google Scholar]
- [45].Jung JS, Jo D, Jo G, Hyun H, Tissue Eng. Regen. Med 2019, 16, 443–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Cho N, Shokeen M, J. Bone Oncol 2019, 17, 100249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Harmatys KM, Cole EL, Smith BD, Mol. Pharm 2013, 10, 4263–4271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Surender EM, Comby S, Cavanagh BL, Brennan O, Lee TC, Gunnlaugsson T, Chem 2016, 1, 438–455. [Google Scholar]
- [49].Ogawa K, Ishizaki A, Takai K, Kitamura Y, Makino A, Kozaka T, Kiyono Y, Shiba K, Odani A, Sci. Rep 2017, 7, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Zhao Z, Chen C, Xie C, Zhao Y, Chem. Phys. Lipids 2020, 226, 104832. [DOI] [PubMed] [Google Scholar]
- [51].Hegemann JD, De Simone M, Zimmermann M, Knappe TA, Xie X, Di Leva FS, Marinelli L, Novellino E, Zahler S, Kessler H, Marahiel MA, J. Med. Chem 2014, 57, 5829–5834. [DOI] [PubMed] [Google Scholar]
- [52].Saito F, Bode JW, Chem. Sci 2017, 8, 2878–2884. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.