

Summary
Microglia are brain-resident immune cells and regulate mechanisms essential for cognitive functions. Down syndrome (DS), the most frequent cause of genetic intellectual disability, is caused by a supernumerary chromosome 21, containing also genes related to the immune system. In the hippocampus of the Dp(16) mouse model of DS and DS individuals, we found activated microglia, as assessed by their morphology; activation markers; and, for DS mice, electrophysiological profile. Accordingly, we found increased pro-inflammatory cytokine levels and altered interferon signaling in Dp(16) hippocampi. DS mice also showed decreased spine density and activity of hippocampal neurons and hippocampus-dependent cognitive behavioral deficits. Depletion of defective microglia or treatment with a commonly used anti-inflammatory drug rescued the neuronal spine and activity impairments and cognitive deficits in juvenile Dp(16) mice. Our results suggest an involvement of microglia in Dp(16)-mouse cognitive deficits and identify a new potential therapeutic approach for cognitive disabilities in DS individuals.
Keywords: Down syndrome, neurodevelopmental disorders, microglia, Dp(16), cognitive defects, acetaminophen, dendritic spines, neuroinflammation, Ts65Dn, proteomics
Graphical Abstract
Highlights
-
•
DS mice display microglia alterations and cognitive impairment
-
•
Depletion of microglia rescues cognitive impairment in DS mice
-
•
Acetaminophen treatment rescues microglia and cognitive impairments in DS mice
-
•
Brain samples of DS people recapitulate microglia alterations observed in DS mice
Pinto, Morelli et al. identify a critical role for activated microglia in cognitive impairments of Down syndrome mouse models that can be ameliorated by either depleting microglia or using anti-inflammatory drugs to reduce microglia activation. In this work, microglia activation is also revealed in brains of people with Down syndrome.
Introduction
Down syndrome (DS) is the main genetic cause of intellectual disability (Dierssen, 2012), and it is caused by a third copy of the human chromosome 21 (HSA21). In addition to cognitive disability mostly in hippocampal-related functions, individuals with DS often suffer from dysfunction of immune responses, such as an increased risk of infections, hematological/autoimmune disorders, and hyperactivation of the interferon (IFN) signaling (Sullivan et al., 2016). Interestingly, peripheral or brain inflammation has been extensively linked to decreased cognition in the young and physiologically aging brain. This suggests a possible regulation of inflammatory states in learning and memory (Shobin et al., 2017).
The resident immunological cells of the CNS are microglia, the brain macrophages (Ginhoux et al., 2013). In adults under physiological conditions, these cells show a ramified morphology, and they constantly surveil the brain parenchyma (Tremblay et al., 2011). In the case of injury and in the presence of pro-inflammatory molecules, microglia shift morphology to a mobile, more amoeboid form with increased branching movements (Arcuri et al., 2017; Hanisch and Kettenmann, 2007). During inflammation, microglia also present depolarized membrane potential, upregulation of activation and phagocytosis markers (MHCII, Iba1, and CD68, Lamp1, respectively), and downregulation of Gi-coupled receptor for adenosine diphosphate (ADP) P2Y12 and secrete large amount of cytokines (Sipe et al., 2016; Kierdorf and Prinz, 2013). Interestingly, microglia also play key roles during early development and in juvenile animals (Tremblay et al., 2011). In particular, they shape the synaptic landscape of the developing brain through phagocytosis of neural progenitors and dendritic synaptic pruning in experience-dependent brain plasticity (Kaur et al., 2017; Wu et al., 2015). Remarkably, dendritic spine deficits have been extensively described in DS persons and murine models (Dierssen, 2012; Haas et al., 2013).
An increased immunological activity with a subsequent increase in microglial activation and excessive spine pruning could therefore play a role in the cognitive deficits in DS. To test this hypothesis, we used the Dp(16) mouse model of DS. Dp(16) mice bear a duplication orthologous to the region q11–q22.3 of HSA21 on one chromosome 16, and they recapitulate many of the characteristics of the human syndrome, including cognitive deficits (Goodliffe et al., 2016; Li et al., 2007).
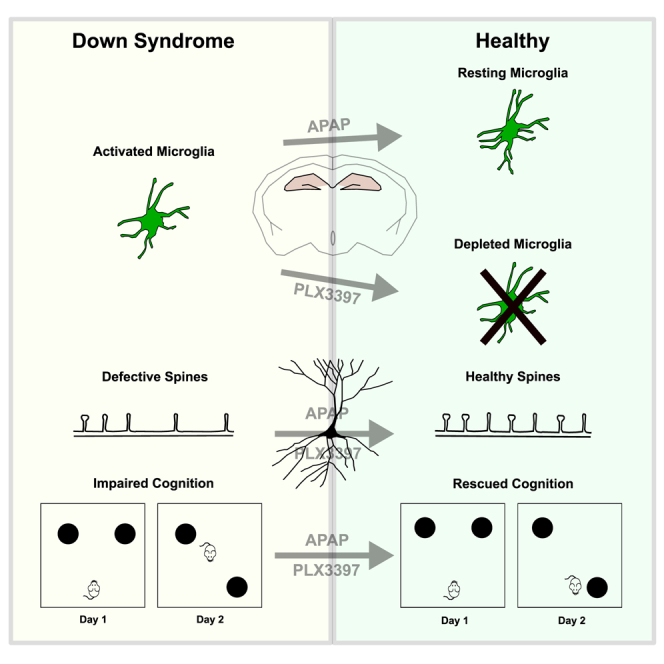
Here, we show that hippocampal Dp(16) microglia display an activated state characterized by morphofunctional alterations; increased levels of Iba1, CD68, and Lamp1; decreased levels of P2Y12; and increased IFN signaling. Microglial alterations are accompanied by a reduction of neuronal dendritic spines and activity together with downregulation of drebrin (DBN), a protein essential for correct spine functioning and dysregulated in DS. The depletion of microglia or the treatment with the anti-inflammatory drug acetaminophen rescues microglia alterations, the neuronal spine-associated phenotypes, and cognitive deficits in juvenile Dp(16) mice. Finally, post mortem brains of DS subjects show dysregulation of immunological pathways with enrichment of dysregulated genes in microglial cells and an activated microglial phenotype similar to that of Dp(16) mice.
Results
Dp(16) Mice Have Hyperactive Microglia and Increased Hippocampal Inflammation
To start our investigation on the role of microglia in DS, we counted the number and analyzed the morphology of microglia (defined as Iba1+ cells; Ginhoux et al., 2013) in hippocampal brain slices (stratum pyramidalis of the CA (cornus ammonis)1, CA3, and DG (dentate gyrus) regions) from postnatal day (P)22 animals of the Dp(16) mouse model of DS and wild-type (WT) littermates (Figures 1A and 1B). We found no difference in the number of microglia between the two strains (WT: 341.14 ± 15.28 Iba1+ cells/mm2, n = 7; Dp(16): 338.3 ± 9.26 Iba1+ cells/mm2, n = 4). However, when compared to WT microglia, Dp(16) microglia presented enlarged cell bodies (Figures 1B and 1C), branching impairment (Figures 1D–1F). A specific skeleton analysis on the same cells revealed a significant decrease in branch numbers (Figures 1G and 1H), with no changes in average branch length (WT 3.53 ± 0.35 μm, n = 8; Dp(16): 3.45 ± 0.13 μm, n = 7). We observed no significant gender difference within genotypes.
Figure 1.

Microglial Morphology and Activity Are Altered in Dp(16) Hippocampi
(A) Experimental protocol.
(B) Iba1-stained hippocampal slices from P22 WT and Dp(16) animals. Scale bar: 10 μm.
(C) Quantification of the microglial cell body area. Bars represent the average of microglial cell body areas in all analyzed animals ± SEM, and symbols (circles, males; triangles, females) represent data points for each animal (38–73 cells/animal; 1 slice per animal). ∗p < 0.05; unpaired two-tailed Student’s t test; t = 2.8; df = 14.
(D) Binary images of selected cells from the fields in (B). Scale bar: 10 μm.
(E) Sholl analysis of microglial cells. Data are expressed as average number of intersections at each distance from cell bodies of all analyzed cells ± SEM. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; two-way ANOVA; FStrain (33, 7,650) = 317.5; p < 0.001; Holm-Sidak post hoc test. In parenthesis: analyzed cells, animals (1 slice per animal).
(F) Quantification of the average number of intersections from the Sholl analysis. Bars represent the average number of intersections in all analyzed animals ± SEM, and symbols (circles, males; triangles, females) represent data points for each animal (12–21 cells/animal; 1 slice per animal). ∗∗p < 0.01; unpaired two-tailed Student’s t test; t = 3.47; df = 11.
(G) Representative skeleton images of binary images. Scale bar: 10 μm.
(H) Quantification of the number of branches per microglial cell. Bars represent the average number of branches per microglial cell ± SEM in all analyzed animals; symbols (circles, males; triangles, females) represent data points for each animal (12–21 cells/animal; 1 slice per animal). ∗p < 0.05; unpaired two-tailed Student’s t test; t = 4.03; df = 11.
(I) Voltage-activated current profile in GFP+ microglia of acute hippocampal slices from WTCX3CR1-GFP and Dp(16)CX3CR1-GFP animals (left inset). Voltage currents were analyzed using ramps ranging from −100 to 80 mV (right inset). Data are expressed as average current for each mV step of all analyzed cells ± SEM. ∗∗p < 0.01; ∗∗∗p < 0.001; two-way repeated measures (RM)ANOVA; FInteraction (9, 900) = 13.35; p < 0.001; Holm-Sidak post hoc test. In parenthesis: analyzed cells, animals.
(J) Confocal and binary example images of a P22 WTCX3CR1-GFP hippocampal slice in time-lapse experiments at different time points. The arrow points to the merge of the t0 and the t1 image used for quantifying pixel intensity changing over time. Scale bar: 20 μm.
(K) Quantification of the mobility index of microglial cells. Bars represent the average mobility index in all analyzed fields ± SEM, and circles represent data points for each field (3 animals per genotype). ∗∗∗p < 0.001; unpaired two-tailed Student’s t test; t = 3.98; df = 22.
(L) Representative images of GFP+ moving cells (4.5 min apart). Scale bar: 20 μm (left). Quantification of process speed of moving microglial cells is shown (right). Bars represent the average process speed in all analyzed cells ± SEM, and circles represent data points for each cell (3 animals per genotype). ∗∗∗p < 0.001; two-tailed Mann-Whitney test; U = 701.5.
Microglia distribution and transcriptional identity is regionally heterogeneous in the brain (Masuda et al., 2019). In the somatosensory cortex, we found a cell-body enlargement in trisomic microglia similar to that observed in the hippocampus (Figures S1B and S1C) but no differences in branch number and distribution between the two strains (Figures S1D–S1H). However, we observed a significant increase in the average length of the branches developed by trisomic microglia (Figure S1I).
Next, we investigated whether the morphological rearrangement of the Dp(16) hippocampal microglia corresponded to a baseline activation state. Thus, we crossed Dp(16) animals with CX3CR1-GFP animals. This resulted in GFP+ microglia in brains from WTCX3CR1-GFP and Dp(16)CX3CR1-GFP mice. We then performed patch-clamp experiments on GFP+ cells in the hippocampus of acute brain slices at P22. Trisomic microglia presented an input current/voltage (I/V) profile with larger outward currents typical of activated microglia (Figure 1I; Carrillo-de Sauvage et al., 2013). Moreover, Dp(16) microglia presented increased branch mobility and branch movement speed (Figures 1J–1L) by ex vivo live imaging. When we analyzed microglial activity and phagocytosis markers via immunohistochemistry and western blotting, we found increased Iba1, CD68, and Lamp1 levels; no difference in MHCII expression; and decreased P2Y12 levels in Dp(16) microglia (Figure S2).
We then measured 40 cytokines using an antibody array in hippocampal lysates from Dp(16) and WT littermates and found a significant increase in the levels of 15 cytokines typical of activated microglia (Figures S3A and S3B). Finally, we defined the activated state of DS microglia by profiling their protein-expression pattern. We ran proteomic analysis on microglia homogenates obtained by fluorescence-activated cell sorting (FACS) of hippocampal cellular suspensions from WTCX3CR1-GFP and Dp(16)CX3CR1-GFP mice. We found 44 differentially expressed proteins (Table S1). When we ran a gene ontology for biological processes (GOBP) analysis on the differentially expressed proteins, the cellular response to IFN-alpha (IFNα) resulted significantly dysregulated from our analysis restricted to microglia (Figure S4A). This suggests that the hyperactive IFN signaling may contribute to the hyperactivation of microglia in Dp(16) animals (Papageorgiou et al., 2016). Of note, the remaining 6 out of 7 biological processes significantly dysregulated in DS microglia were involved in overall DNA and RNA processing (Figure S4A).
These data point to a state of activation of microglia in Dp(16) mice.
Depletion of Microglia Recovers the Cognitive Deficits in Dp(16) Mice
Next, we tested whether the activated microglia could play a role in cognitive deficits of Dp(16) mice. Because stress per se may cause microglial activation (Delpech et al., 2016), we subjected DS animals and their WT littermates to two mildly stressful hippocampus-dependent cognitive tests: the novel object recognition (NOR) and object location test (OLT). DS animals presented cognitive deficits in NOR and OLT compared to their WT littermates (Figures 2A, 2E, and 2F). Interestingly, when we ran a proteomic analysis on the hippocampi of P22 Dp(16) animals and WT littermates followed by GOBP analysis on the 169 differentially expressed proteins (Table S1), we found the term “memory” among the first hits (Figures S5A and S5B; Table S2). To directly address whether defective microglia play a role in the low performance of Dp(16) mice in NOR and OLT, we took advantage of PLX3397. PLX3397 is a drug that decreases the microglial population, without causing significant cognitive impairment in WT animals (Elmore et al., 2014). We thus treated P16 Dp(16) mice and their WT littermates with PLX3397 in their food supply (445 mg/kg of food available ad libitum) for 1 week and then evaluated the impact of microglial depletion on cognitive performance at P22 (Figure 2A). As expected, PLX3397 treatment sharply decreased the microglia count and Iba1 protein levels in the hippocampus of WT and Dp(16) mice (Figures 2B–2D). PLX3397 caused a trend to decreased levels of oligodendrocyte progenitors (NG2) in Dp(16) mice (Figure 2D), as previously shown (Liu et al., 2019). However, this reduction, in our hands, was not statistically significant. Remarkably, PLX3397 treatment in Dp(16) mice fully rescued their cognitive deficits in the NOR and OLT tests (Figures 2E and 2F).
Figure 2.

PLX3397 Treatment Depletes Microglia and Rescues Cognitive Deficits in Dp(16) Mice
(A) Experimental protocol.
(B) Iba1-stained hippocampal slices and highlighted regions from P22 WT and Dp(16) animals treated with either vehicle (top) or PLX3397 (bottom) in the chow. Scale bar: 100 μm.
(C) Quantification of the density of microglial cells. Bars represent the average density of Iba1+ cells of all the analyzed animals ± SEM, and symbols represent data points for each animal (1 slice per animal). ∗∗∗p < 0.001; two-way ANOVA; FTreatment (1, 29) = 642.6; p < 0.001; Holm-Sidak post hoc test.
(D) Immunoblots on protein extracts from lysates of hippocampi obtained from P22 WT and Dp(16) mice treated with vehicle or PLX3397 (left). Quantification of Iba1 (middle) and NG2 (right) levels normalized to GADPH is shown. Bars represent the average percentage of Iba1 or NG2 over WT vehicle of all analyzed animals ± SEM, and symbols represent data points for each animal. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001; for Iba1: two-way ANOVA, FTreatment (1, 50) = 54.53, p < 0.0001, Holm-Sidak post hoc test. For NG2: two-way ANOVA; FTreatment (1, 46) = 6.774; p = 0.0126; Holm-Sidak post hoc test.
(E and F) Quantification of the discrimination index in the NOR and OLT tests in P22 WT and Dp(16) mice following vehicle or PLX3397 treatment. Bars represent the average discrimination index of all analyzed animals ± SEM, and symbols (circles, males; triangles, females) represent data points for each animal.
(E) ∗p < 0.05; ∗∗p < 0.01; two-way ANOVA; FInteraction (1, 61) = 13.04; p = 0.0006; Holm-Sidak post hoc test.
(F) ∗∗∗∗p < 0.0001; two-way ANOVA; FInteraction (1, 44) = 24.54; p < 0.0001; Holm-Sidak post hoc test.
These data indicate that depletion of activated microglia ameliorates cognitive performance in DS mice.
Microglial Depletion Restores the Levels of Neuronal Dendritic-Spine Protein DBN and Rescues Dendritic Spine Number and Function in Dp(16) Mice
We then investigated whether the overactive microglia of the Dp(16) mice could impair their cognitive performance by affecting the number and/or the level of maturation of neuronal spines. We first focused on the dendritic spine marker DBN. We observed significantly decreased levels of DBN in the hippocampi of P22 Dp(16) mice in comparison to WT mice (Figure 3A). Accordingly, when we ran a targeted proteomic protocol on hippocampi from Dp(16) and WT littermates, we found a sharp decrease in the levels of 4 different peptide fragments ascribed to DBN in trisomic mice (Figures S5C and S5D). Interestingly, PLX3397 treatment completely rescued the DBN levels in Dp(16) mice (Figure 3A). In agreement with our hypothesis, we found a decreased number of spines in CA1 hippocampal excitatory pyramidal neurons from Dp(16) mice counted after Golgi-Cox staining, which was rescued by the PLX3397 treatment (Figures 3B and 3C). In particular, PLX3397 rescued the density of motile functional spines (thin, stubby, and mushroom type) without affecting the immature filamentous (Figures S6A–S6C). To investigate whether defective trisomic microglia also impinge on the correct functionality of neuronal dendritic spines, we recorded miniature excitatory post-synaptic currents (mEPSCs) in WT and Dp(16) neurons from acute brain slices of animals treated with PLX3397. PLX3397 treatment fully rescued the frequency and amplitude of mEPSCs, which we found affected in Dp(16) in comparison to WT mice (Figures 3D and 3E). Finally, we performed electron microscopy and immunostaining to further support our hypothesis for the role of the overactive microglia in DS animals in regulating the number of neuronal spines. In P22 Dp(16) mice, we observed an increased number of contacts between microglia and pre-/post-synaptic elements compared to WT animals (Figures 3F and 3G) and an increase of excitatory synaptic marker (Vglut1 and Psd-95) puncta engulfed by microglia (Figures 3H–3J).
Figure 3.

Drebrin Levels, Dendritic-Spine Density, and Miniature Post-synaptic Events Are Altered and Rescued by PLX3397 Treatment in Dp(16) Mice
(A) Immunoblots on protein lysates of P22 WT and Dp(16) hippocampi (left). Quantification of Drebrin levels normalized to GADPH is shown (right). Bars represent the average percentage of drebrin in Dp(16) over WT hippocampi for all analyzed animals ± SEM, and symbols represent data points for each animal. ∗∗p < 0.01; ∗∗∗p < 0.001; two-way ANOVA; FInteraction (1, 39) = 7.166; p = 0.0108; Holm-Sidak post hoc test. Data were averaged across 6 independent experiments.
(B) Dendritic spines from Golgi-Cox-stained slices of hippocampi from P22 WT and Dp(16) mice treated with vehicle or PLX3397. Scale bar: 4 μm.
(C) Quantification of the spine density. Bars represent the average spine density of all analyzed cells ± SEM, and symbols represent data points for each cell (3 animals per condition). ∗∗∗p < 0.001; two-way ANOVA; FInteraction (1, 51) = 20.84; p < 0.0001; Holm-Sidak post hoc.
(D) Representative traces of mEPSCs in CA1 hippocampal pyramidal neurons from P22 WT and Dp(16) mice treated with vehicle or PLX3397. Scale bars: 10 pA and 1 s.
(E) Quantification of the mEPSCs frequency and amplitude. Bars represent the average frequency and the average amplitude for all analyzed cells ± SEM, and symbols represent data points for each cell (3 WT vehicle-treated animals and 4 animals per each remaining conditions). ∗∗∗p < 0.001; frequency: two-way ANOVA, FInteraction (2, 86) = 14.09, p < 0.0001; amplitude: two-way ANOVA, FInteraction (2, 86) = 20.61, p < 0.0001; Holm-Sidak post hoc test.
(F) Electron microscopy images of hippocampal slices from P22 WT and Dp(16) mice. Scale bar: 500 nm. Arrows point to synaptic elements; a, astrocytes; d, dendrite shaft; m, microglia; s, post-synaptic element; t, pre-synaptic element.
(G) Quantification of the interaction between microglia and pre-/post-synaptic sites. Bars represent the average number of contact points between microglia and spines of all animals analyzed ± SEM, and circles represent the average of all analyzed fields for each animal (20–44 fields/animal). (Left) ∗p < 0.05; unpaired two-tailed Student’s t test, t = 3.14, df = 5; (right) ∗p < 0.05; unpaired two-tailed Student’s t test, t = 3.09, df = 5.
(H) Confocal images of Iba1 (green), Psd-95 (red), and Vglut1 (blue)-stained hippocampal slices from P22 WT and Dp(16) animals. Scale bar: 5 μm.
(I and J) Quantification of engulfed Psd-95 or Vglut1 puncta in Iba1-labeled microglia of P22 WT and Dp(16) animals normalized to the volume of the microglia. Bars represent the average for all analyzed animals ± SEM, and symbols represent the average of all analyzed microglia for each animal (10–20 cells/animal; 1 slice per animal). ∗∗∗∗p < 0.0001; (I) two-way ANOVA; Finteraction (1, 26) = 26.23; p < 0.0001; Holm-Sidak post hoc test.
(J) Two-way ANOVA; Finteraction (1, 26) = 29.47; p < 0.0001; Holm-Sidak post hoc test.
These data are consistent with a causal role of overactive microglia for altered neuronal dendritic-spine turnover and function in Dp(16) mice.
Acetaminophen Treatment Rapidly Rescues Microglial Activation State through TrpV1 Channels in Dp(16) Mice
Recent evidence in brains from the Ts65Dn mouse model of DS and from subjects with DS indicates increased expression of the inducible form of the cyclo-oxygenase enzymes (Cox2) in microglia and neurons (Mulet et al., 2017). This is a sign of increased immunological activity, because activated microglia produce a large amount of the pro-inflammatory prostaglandin E2 (PGE2) (Zhang et al., 2009). In Dp(16) hippocampal microglia, we also found increased Cox2 and PGE2 expression (Figures S7A–S7D). Therefore, we considered acetaminophen (APAP), a main Cox2 inhibitor that reduces PGE2 production, as a potential candidate to rescue the microglia and cognitive impairments in Dp(16) mice. APAP readily crosses the blood-brain barrier, has a very safe profile with a few contraindications, and might downregulate the expression of IFN-induced genes (Farnsworth et al., 2010). Thus, we injected Dp(16) mice and WT littermates with 1 dose of APAP (intraperitoneally [i.p.], 100 mg/kg/day) or vehicle (i.p., DMSO, 2%; Figure 4A) during 3 consecutive days. APAP treatment significantly rescued the levels of Cox2 and PGE2 in Dp(16) microglia (Figures S7A–S7D). Moreover, APAP treatment rescued microglial morphology (Figures 4B, 4C, and S7E–S7I) and the levels of Iba1 (Figure S2B) in the hippocampi of Dp(16) mice and the microglia cell body area in the somatosensory cortex (Figures S1B and S1C). Finally, treatment with APAP in vivo was also able to rescue the ex vivo electrophysiological signature of activated microglia (Figure 4D). Notably, acute bath application of APAP (1 h, 300 μM) to brain slices from naive WTCX3CR1-GFP and Dp(16)CX3CR1-GFP animals was sufficient to rescue the electrophysiological signature of activated DS microglia together with the hyper-motility and the speed movement of DS microglial branches (Figures 5A–5D).
Figure 4.

APAP Treatment Rescues Microglial Morphology and Activity, Dendritic Spine Deficits, and Cognitive Impairment in Dp(16) Mice
(A) Experimental protocol.
(B) Iba1-stained hippocampal slices from WT and Dp(16) animals treated with either vehicle or APAP. Scale bar: 10 μm.
(C) Quantification of the cell body area of microglial cells. Bars represent the average of microglial cell body areas in all analyzed animals ± SEM, and circles represent single data points of the cell averages for each animal (14–73 cells/animal; 1 slice per animal). ∗p < 0.05; two-way ANOVA; FInteraction (1, 23) = 14.88; p = 0.0008; Holm-Sidak post hoc test. Dotted circles represent data are from Figure 1C for comparison.
(D) Voltage-activated current profile of microglia in WTCX3CR1-GFP and Dp(16)CX3CR1-GFP acute hippocampal slices treated with vehicle or APAP in vivo. Voltage currents were analyzed using ramps ranging from −100 to 80 mV (inset). Data are expressed as average current (picoAmpere,pA) normalized for the capacitance (picoFarad,pF) for each mV step of all analyzed cells ± SEM. ∗ represents the statistical difference between Dp(16) and WT vehicle; ∗∗p < 0.01; ∗∗∗p < 0.001; § represents the statistical difference between Dp(16) vehicle and Dp(16) APAP; §§p < 0.01; §§§p < 0.001. Two-way RM ANOVA; FInteraction (27, 1,890) = 9.261; p < 0.001; Holm-Sidak post hoc test. In parenthesis: analyzed cells, animals. The vehicle-treated data are from Figure 1I for comparison.
(E) Dendritic spines from Golgi-Cox-stained slices of hippocampi from P22 WT and Dp(16) mice treated with vehicle or APAP. Scale bar: 4 μm.
(F) Quantification of the spine density. Bars represent the average spine density of all analyzed cells ± SEM, and circles represent single data points for each cell (3 animals per condition). ∗∗∗p < 0.001; two-way ANOVA; FInteraction (1, 56) = 42.99; p < 0.001; Holm-Sidak post hoc test. The vehicle-treated cell data (dotted circles) are from Figure 3C for comparison.
(G and H) Quantification of the discrimination index in the NOR and OLT tests in P22 WT and Dp(16) mice following vehicle or APAP treatment. Bars represent the average discrimination index of all analyzed animals ± SEM, and symbols (circles, males; triangles, females) represent single data points for each animal. ∗p < 0.05; ∗∗p < 0.01; (G) two-way ANOVA, FInteraction (1, 51) = 23.76, p < 0.001; Holm-Sidak post hoc test; (H) two-way ANOVA, FInteraction (1, 35) = 11.24, p < 0.002; Holm-Sidak post hoc test. The vehicle-treated data (dotted circles and triangles) are from Figures 2E or 2F for comparison.
Figure 5.

Acute APAP Treatment Influences Microglia Activity, and TrpV1 Channels Acutely Regulate Microglia Activation in Dp(16) Animals
(A) Experimental protocol.
(B) Voltage-activated current profile in microglia of WTCX3CR1-GFP and Dp(16)CX3CR1-GFP hippocampal slices (left inset) treated acutely with vehicle or APAP. Voltage currents were analyzed using ramps ranging from −100 to 80 mV (right inset). Data are expressed as average current (picoAmpere,pA) normalized for the capacitance (picoFarad,pF) for each mV step of all analyzed cells ± SEM. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; two-way RM ANOVA; FInteraction (9, 315) = 10.55; p < 0.001; Holm-Sidak post hoc test. In parenthesis: analyzed cells, animals.
(C and D) Quantification of the mobility index and process speed of microglial cells. Bars represent the average mobility index and process speed in all analyzed fields ± SEM, and circles represent data points for each field (3 animals per genotype). ∗∗∗p < 0.001; (C) two-way ANOVA, FInteraction (1, 43) = 15.07, p < 0.001; Holm-Sidak post hoc test; (D) two-way ANOVA, FInteraction (1, 225) = 31.25, p < 0.001; Holm-Sidak post hoc test. The vehicle-treated cell data (dotted circles) are from Figures 1K and 1L for comparison.
(E–G) Voltage-activated current profile in microglia of WTCX3CR1-GFP and Dp(16)CX3CR1-GFP hippocampal slices treated with vehicle or the indicated drug (OLDA, 2 μM; AM-251, 1 μM; AM-630, 1 μM; capsazepine, 10 μM). Voltage currents were analyzed using ramps ranging from −100 to 80 mV, as in the inset. Data are expressed as average current (picoAmpere,pA) normalized for the capacitance (picoFarad,pF) for each mV step of all analyzed cells ± SEM. ∗ represents the statistical difference between Dp(16) and WT vehicle, ∗p < 0.05, ∗∗∗p < 0.001; § represents the statistical difference between Dp(16) vehicle and Dp(16) treated, §p < 0.05, §§p < 0.01, §§§p < 0.001; (E) two-way RM ANOVA, FInteraction (27, 1,341) = 14.35, p < 0.001; Holm-Sidak post hoc test; (F) two-way RM ANOVA, FInteraction (27, 1,449) = 20.34, p < 0.001; Holm-Sidak post hoc test; (G) two-way RM ANOVA, FInteraction (27, 1,580) = 18,89, p < 0.001; Holm-Sidak post hoc test. In parenthesis: analyzed cells, animals.
(H) Cartoon depicting the putative mechanism of action of APAP. FAAH, fatty acid amide hydrolase.
Thus, APAP’s action may be mediated not only by the inhibition of COX enzymes—which is slow because it requires a first decrease in PGE2 synthesis and a consequent change in gene expression (Ajmone-Cat et al., 2010)—but also by another faster unknown mechanism. Therefore, we characterized the possible mediators of the fast action of APAP on microglia. In particular, literature suggests that it is not APAP tout-court but its metabolite AM404 that is able to inhibit microglial activation (Sharma et al., 2017). Among the targets of AM404 on microglia (other than COX enzymes) is the transient receptor potential cation channel subfamily V member 1 (TrpV1). This receptor channel is highly expressed in microglia, where it inhibits microglia activation and production of PGE (Bhatia et al., 2017). To mimic the effect of APAP, we bath applied the most potent and selective endogenous agonist of TrpV1, N-oleoyldopamine (OLDA), in ex vivo slices from WTCX3CR1-GFP and Dp(16)CX3CR1-GFP animals during electrophysiological recordings (Figure 5E). In agreement with our hypothesis, OLDA fully rescued microglial activation (Figure 5E). Although OLDA has a very high affinity to TrpV1, it can also bind to the cannabinoid receptors CB1/CB2 (Zhong and Wang, 2008). Hence, to tease apart the role of these two pathways on microglia activation, we performed the same electrophysiology experiment with bath application of OLDA together with AM-251 (inhibitor of CB1) and AM-630 (inhibitor of CB2). Bath application of the three drugs rescued the membrane potential of Dp(16) microglia (Figure 5F), indicating that receptors CB1/CB2 may be dispensable for microglia activation. Finally, we bath applied APAP together with a potent blocker of Trpv1, capsazepine. This was able to fully prevent the rescuing action of APAP on microglia (Figures 5G and 5H).
These data suggest that APAP’s action on Dp(16) microglia is mediated not only by its action on COX enzymes but also by a faster activation of Trpv1 channels.
APAP Treatment Influences Microglial State through IFN Signaling and Rescues Cytokine Levels and Neuronal Spine Phenotype in Dp(16) Mice
To further probe the impact of APAP treatment on DS microglia in vivo, we performed a proteomic analysis on FACS-sorted microglial cells from WTCX3CR1-GFP and Dp(16)CX3CR1-GFP animals treated for 3 days (P20–P22) with either APAP or vehicle (Figure 6A). On these samples, we ran a principal-component analysis (PCA) on the proteomic data. The PCA evidenced an extensive resemblance between Dp(16) microglia after the treatment with APAP and WT microglia after vehicle treatment. Conversely, microglia from Dp(16) mice treated with vehicle fell far apart (Figure 6B). Next, we ran a weighted correlation network analysis (WGCNA) to define biologically meaningful groups of proteins (clusters) based on their co-expression patterns (Langfelder and Horvath, 2008). The WGCNA helped us identify which protein clusters would be mostly influenced by the APAP treatment in Dp(16) microglia. We reasoned that those proteins could be the early mediators of the overall positive outcomes of APAP treatment on Dp(16) mice. We identified 26 diverse clusters, with proteins significantly correlated among them within each cluster (Figure 6C, colors). We focused our further analysis on the brown cluster (Figure 6C; Table S1), because its proteins were indeed the most influenced by APAP treatment in Dp(16) microglia. GOBP analysis performed on the proteins belonging to the brown cluster revealed that they were related to overall immune response to infections, mainly IFNα/β- and IFNγ-mediated responses, and to the overall response to cytokines (Figure 6D, red nodes). This suggested that the early effect of APAP on microglia may be IFN dependent. Thus, we reasoned that the chief proteins responsible for the APAP positive effect on Dp(16) mice would be proteins that, upon treatment with APAP, would regain correlations among them similar to that of WT mice. Interestingly, among the 5 clusters of proteins in Dp(16) mice that regained similar correlation to WT mice by treatment with APAP, the GOBP and a gene ontology molecular functions (GOMF) analysis revealed proteins involved in cytoskeleton and cell movement (Figures S4B and S4C, dark red and orange clusters; Table S1). This is in agreement with our findings on the rescue of the aberrant morphology and branch movement in Dp(16) microglia by APAP treatment. In agreement with our GOBP analysis (Figure S4A), 12 out of the remaining 20 clusters contained protein related to DNA replication and gene expression.
Figure 6.

APAP Treatment Influences IFN Signaling in Dp(16) Mice
(A) Experimental protocol.
(B) Graph representing the component 2 and 3 deriving from PCA of proteomic data from FACS-isolated microglia of WTCX3CR1-GFP and Dp(16)CX3CR1-GFP animals treated with vehicle or APAP. The percentages indicate the amount of variability explained by each plotted component. Dots represent single animals; dotted circles separate the groups.
(C) Heatmap of the protein clusters obtained after WGNCA clustering, using the proteins identified from the experiment in (A). Proteins were grouped based on the correlation of their expression and cluster. Each color represents a cluster. The degree of correlation is color coded by the scale bar.
(D) Fisher test enrichment of the GOBP annotation from the protein brown cluster in (C) from Uniprot with a significance threshold of Benjamini-Hochberg false discovery rate (FDR) < 0.02. The output file is converted into a small network, where the node size is proportional to −log10 (Benjamini-Hochberg FDR) and the edge width to the enrichment factor. Red nodes: pathways of interest for our work.
Next, we investigated whether the effect of APAP would be sufficient to influence also the general level of inflammation in the brains of Dp(16) mice. When we performed the antibody array for the detection of cytokines in brain lysates from Dp(16) and WT littermates treated with APAP or vehicle, we found that the drug treatment rescued the levels of all overrepresented cytokines in the Dp(16) mouse hippocampi (Figure S3C). Finally, we investigated whether the rescue of microglia activation by APAP treatment would also positively affect neuronal spines. APAP treatment significantly rescued neuronal spine density, with a full rescue of the mature mushroom type and a partial rescue of thin-type spines in Dp(16) mice (Figures 4E and S6A–S6C). Accordingly, APAP treatment also rescued engulfment of Psd-95 and Vglut1 puncta within the microglia (Figures S6D–S6F) and the frequency and amplitude of mEPSCs in Dp(16) neurons (Figures S7J and S7K).
These results indicate that APAP treatment decreases the inflammation state of the trisomic brain and is able to rescue neuronal spine density and function.
APAP Treatment Rescues Cognitive Deficits in Dp(16) and Ts65Dn Mice
Our results on the rescue of cognitive impairment in Dp(16) mice upon depletion of microglia strongly suggest that these cells regulate learning and memory in trisomic animals. We then investigated whether rescue of microglia activation state by APAP treatment would be sufficient to restore cognitive abilities in DS mice. We treated Dp(16) and WT littermates with APAP or vehicle (P20–P22) and performed the NOR and OLT cognitive tests (Figure 4A). APAP treatment rescued cognitive deficits in Dp(16) animals (Figures 4G and 4H). Notably, in WT animals, the treatment with APAP caused a slight decrease in cognitive performance in both the NOR and OLT tests (Figures 4G and 4H). This was accompanied by a more amoeboid microglial morphology and a significant decrease in the number of neuronal spines and in mEPSCs amplitude but no significant effect on cytokine levels (Figures 4B–4F, S3D, and S7K). The same effects of APAP on Dp(16) and WT animals were confirmed in another set of animals tested in the OLT before (P20–P22) as naive (no treatment) and retested 1 week later (P29–P31) in another OLT—using different objects located in a different position in recording arena—after drug (APAP or vehicle) treatment (Figures S8A–S8C). Moreover, the administration of an inhibitor of the JAK1/2 pathway (ruxolitinib), whose activation is important for microglial and IFN pathway activation (Qin et al., 2016), significantly rescued microglial branching together with cognitive performance in Dp(16) mice (Figure S9). This is in agreement with the hindering role of activated microglia in the cognitive abilities of Dp(16) mice and the hyper-activation of IFN-mediated response in trisomic microglia. Next, to assess whether the effect of APAP on cognitive performance was long lasting, we performed drug withdrawal experiments treating Dp(16) and WT littermates daily for 3 days (P20–P22) with APAP or vehicle and assessing microglial morphology and cognitive performance (NOR test) 16 days after the last APAP administration (P38–P40; Figure S10A). After APAP withdrawal, Dp(16) animals displayed microglial morphology and cognitive performance similar to vehicle-treated Dp(16) mice (Figure S10).
Then, we investigated whether APAP treatment for 3 days (P59–P61) would have positive effects also in fully adult animals (Figure S8D) and found a significant positive effect on microglial ramifications and a slight, but not significant, trend versus improved cognitive performances in Dp(16) mice (Figures S8E–S8J).
Finally, to strengthen the translational potential of our findings on juvenile mice, we resorted also to another model of DS, the Ts65Dn mouse. In P22 Ts65Dn animals, we observed microglial branching deficits similar to those found in Dp(16) animals but no significant increase in the cell body area (Figures S11A–S11G). Treatment of Ts65Dn mice with APAP was able to rescue the microglial branching phenotype together with the cognitive performance (OLT; Figure S11).
These data indicate that APAP treatment in juvenile (but not fully adult) animals rescues cognitive impairment in two diverse mouse models of DS, although the effect is not long lasting.
Microglial Defects Are Also Present in the Brains of Persons with DS
For translational readout, we assessed the mRNA transcript levels (RNA sequencing [RNA-seq]) in post mortem hippocampi from individuals with DS (13–39 years old) and age-matched controls (13–35 years old) used in (Deidda et al., 2015; Table S3). We found 702 mRNAs with significantly different expression levels in DS versus control brains. On those differentially expressed mRNAs, we ran a cell-enrichment analysis. Upregulated mRNAs were significantly enriched in glial cells (including microglia) and not in neurons (Figure 7A). Interestingly, Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis on the RNA-seq data revealed that, out of 38 pathways significantly altered in the hippocampi of DS people, 20 were related to the immune system (Table S4). To confirm our RNA-seq findings also from independent sources, we ran the same analysis on two published microarray datasets from freshly frozen prefrontal cortex tissue from people with DS (Lockstone et al., 2007; Olmos-Serrano et al., 2016). Remarkably, we found 16 pathways that were common to both studies and to our RNA-seq dataset, and ten of those were immunological pathways (Table S4). We confirmed the upregulation of 6 microglial proteins involved in microglia activation also by quantitative polymerase chain reaction (qPCR) (Figure 7B) in our samples.
Figure 7.

Microglial Alterations Are Present in Brains from DS Individuals
(A) Bootstrapping tests performed using the expression weighted celltype enrichment (EWCE) method. Cell-type enrichment of upregulated mRNA in DS brains (n = 4) shows enrichment in glial cells, including microglia (green), when compared to controls (n = 5). Bars represent the average number of standard deviations from the mean. Asterisks: Benjamini-Hochberg corrected p < 0.05.
(B) Quantification of the qPCR ran on human hippocampus from the same controls and DS samples in (A). Bars represent the average fold-change expression versus controls of all analyzed samples ± SEM, and circles represent data points for each sample. ∗p < 0.05; independent unpaired two-tailed t tests. For CD22, t(7) = −2.8393; for FCGR1A, t(7) = −3.095; for IL4R, t(7) = −3.8841; for MARCKSL1, t(7) = −2.6033; for NKFBIA, t(7) = −2.7274; and for PECAM1, t(7) = −2.9214.
(C) Immunoblots of hippocampal protein lysates from age- and sex-matched controls and DS people (left). Quantification of Iba1 normalized to actin and P2Y12 normalized to GAPDH. Bars represent the average percentage of Iba1 or P2Y12 levels in DS over control hippocampi in all individuals ± SEM, and circles represent data points for each person. Data were averaged across 3 independent experiments. ∗p = 0.027 for Iba1; unpaired one-tailed Mann Whitney test U = 3; and ∗p = 0.0317 for P2Y12; unpaired two-tailed Mann Whitney test U = 2.
(D) Iba1-stained hippocampal slices from a control and a DS person with no signs of amyloid deposition or diagnosis of Alzheimer’s disease (individual 13202 in Table S3). Scale bar: 20 μm.
(E) Quantification of microglial cell-body area. Bars represent average of the microglia cell-body areas of all analyzed individuals ± SEM, and circles represent data points of the cell averages for each individual (24–60 cells; 3–7 slices per individual). Red circles represent people with hippocampal signs or diagnosis of Alzheimer’s disease, as defined in Table S3. ∗p = 0.0423; unpaired one-tailed Student’s t test; t = 1.83; df = 17.
(F) Sholl analysis of microglial cells. Data are expressed as average number of intersections at each distance from the cell bodies of all analyzed cells ± SEM. The statistical analysis revealed a significant effect of the diagnosis of DS repeated-measure two-way ANOVA; FDiagnosis (1, 35) = 4.663; p < 0.0378. Numbers in parenthesis: cells (19 versus 20), analyzed individuals (2 versus 2; 4–10 slices per individual) for 51 different radius points. Slices from individuals 13149, 13202, 13235, and 13250 were analyzed.
(G and H) Quantification of the number and average length of branches per microglial cell. Bars represent the average number or average length of branches per microglial cell ± SEM in all analyzed individuals, and circles represent data points of the cell averages for each individual (9–10 cell/individual; 4–10 slices per individual from the same individuals in E).
(G) ∗p < 0.05; unpaired one-tailed Mann-Whitney test, U = 115; (H) ∗∗∗p < 0.01; unpaired two-tailed t test, t = 4.69, df = 35.
Next, we analyzed the state of microglial activation in the same brain samples of the RNA-seq analysis using Iba1 as a marker of activation by western blot. We found increased expression in DS individuals compared to controls (Figure 7C), reflecting our findings in the Dp(16) mice. We then performed immunohistochemistry from a second set of individuals with DS (36–67 years old) and age-matched controls (40–86 years old; Table S3) with an Iba1 antibody. Although this second set of human samples may be influenced by age-related effects on microglia, it allowed us to perform the same analysis of microglial morphology performed with Dp(16) animals. Similarly to Dp(16) microglia, human DS microglia had enlarged cell bodies (Figures 7D and 7E) and ramification deficits in human DS microglia (Figure 7F). A subsequent skeleton analysis revealed a decreased number of branches (Figure 7G), which were, however, longer on average (Figure 7H).
These results suggest that the microglial dysfunction observed in DS mice is also present in persons with DS.
Discussion
Recent evidence indicated an increased level of circulating pro-inflammatory molecules (Wilcock and Griffin, 2013) and high incidence of hematological and autoimmune diseases in DS (Alexander et al., 2016). Signs of high inflammation and activated microglial morphology were described also in DS murine models but almost exclusively in adult/aging animals (Guedj et al., 2015; Gally et al., 2018). As a DS animal model to investigate microglia and their role in cognition in DS during development, we preferred the Dp(16) mouse to the more commonly used Ts65Dn mouse because Dp(16) mice lack marked prenatal signs of defective neuronal-precursor proliferation and neurogenesis (Goodliffe et al., 2016). These are per se a cause of cognitive defects in Ts65Dn mice (Parrini et al., 2017) and could be a confounding factor when investigating the role of microglia in cognitive performance. Moreover, we observed a milder phenotype in Ts65Dn mice (even when comparing the WT control strains). In Dp(16) mice, we initially restricted our investigation to the third postnatal week of life (P22). We considered this period the one with the main translational potentials for a safe—but still effective—treatment, well before the onset of the precocious aging and the AD (Alzheimer's disease) phenotype typical of DS. At P22, the microglia in the Dp(16) (but also Ts65Dn) hippocampus showed morphological signs of activation. In agreement with the idea that the peculiar morphology of the trisomic microglia was reflective of an activated state due to chronic inflammation in DS, we found an increase in channel openings via an I/V curve in Dp(16) mice. This is considered a sign of microglial activation due to the opening of inward-rectifying potassium channels activated by inflammatory signals (Chung et al., 1999). Moreover, in the Dp(16) hippocampus, we found increased levels of Iba1, a microglial protein whose expression is increased in concomitance with cytoskeleton-dependent changes in cell morphology that occur during microglial activation (Bosco et al., 2011; Nilsson et al., 2008). Iba1 upregulation was accompanied by increased levels of pro-inflammatory cytokines normally secreted by activated microglia. Interestingly, we found upregulated in the hippocampi of Dp(16) mice also two anti-inflammatory cytokines (interleukin-4 [IL-4] and IL-10). Notably, an increased production of IL-10, as a reaction to a chronic inflammatory state, has been described in subjects with DS (Broers et al., 2014). Although we found high levels of the CD68 marker of activated microglia, we failed to detect any expression of the classical marker MHCII in Dp(16) mice. The same has been reported during the microglia-dependent synaptic pruning necessary for critical-period plasticity or the microglial non-canonical activation by increased neuronal firing (Sipe et al., 2016). Interestingly, GABA signaling contributes to increased hippocampal excitability in Ts65Dn mice, which could possibly add to their microglial changes (Deidda et al., 2015; Savardi et al., 2020). In agreement with increase in CD68 expression, we also found increased levels of Lamp1, a protein highly expressed in lysosomes and upregulated in microglia (and other cell types) during phagocytosis and in DS (Colacurcio et al., 2018).
Microglia play a pivotal role in remodeling neuronal dendritic spines and regulate their activity in brain development, memory, and plasticity (Salter and Stevens, 2017; Kaur et al., 2017). Moreover, individuals with DS and DS animal models (Haas et al., 2013; Belichenko et al., 2009) present altered spine density, possibly due to increased synaptic pruning (Phillips and Pozzo-Miller, 2015). This experimental evidence prompted us to hypothesize that the link between the microglial changes observed in Dp(16) mice and their cognitive deficits may be the action of the hyperactive microglia on neuronal dendritic spines. Remarkably, the depletion of defective microglia by PLX3397 treatment rescued the neuronal dendritic spine number of the functional subtypes (thin and mushroom; Xu et al., 2018; Majewska and Sur, 2003) and the amplitude and frequency of mEPSCs together with the cognitive performance in Dp(16) mice. Notably, microglia activation and deficit in neuronal dendritic spines and mEPSCs have been described in other animal models of neurodevelopmental disorders (e.g., autism, Rett’s syndrome, and schizophrenia; Blank and Prinz, 2013; Cowan and Petri, 2018). As a proxy to study spine density, we used DBN—an actin-binding protein playing a pivotal role in spine maintenance and maturation (Weitzdoerfer et al., 2001). DBN levels are decreased in DS people (Shim and Lubec, 2002) and during inflammation and microglia activation (Rao et al., 2012). The level of expression of DBN together with the spine density and the cognitive impairment were rescued upon depletion of the aberrant microglia by treatment of Dp(16) mice with PLX3397. All the above results (together with increased interaction between microglia and spine elements) sustain our hypothesis that the higher activation state of microglia leads to abnormal pruning of dendritic spines that in turn sustains cognitive deficits in DS animals. Finally, the hippocampal proteomic analysis in Dp(16) and WT mice supports the idea that their difference in cognitive behavior may depend on altered spine pruning. Indeed, among the differentially expressed proteins, we singled out 4 significant proteins that fall into the gene ontology (GO) memory category (integrin alpha 3, sodium/calcium exchanger member 3, sortilin-related VPS10 domain containing receptor 3, and kalirin) and are all involved in dendritic spine function and structure (Kerrisk et al., 2013; Herring and Nicoll, 2016; Minelli et al., 2007; Christiansen et al., 2017).
In post mortem brains of adult human subjects with DS, our analysis suggested microglial activation similar to DS animals. In particular, our RNA-seq data from DS subjects showed dysregulation in immunological pathways overrepresented in microglia. Moreover, we found increased Iba1, decreased P2Y12 expression, and morphological alterations. Only a limited number of studies have investigated microglia in individuals with DS, but they mostly focused on the effect of AD on microglia, as most individuals with DS develop signs of AD by the age of 40 (Dekker et al., 2017). In these studies, microglia were often found in a senescent state (dystrophic and less responsive to stimuli) similar to an AD phenotype (Streit et al., 2014). Conversely, only 3 of out of 15 of our DS brain samples presented a diagnosis of AD (no indication of the progression of the disease), and 3 presented only some anatomical signs of AD (low burden of Aβ or fibrillary tau), as indicated by the biobank providers. Notably, the presence or absence of AD or signs of AD did not influence the morphological abnormalities we observed in our DS samples. The fact that we could not find any difference between the microglia of the 5 healthy controls and the 3 controls with some signs of AD seems to suggest that, unless AD is in a very late progression, microglia do not show signs of activation in DS individuals. Finally, we found that upregulated mRNAs from human hippocampus were enriched not only in microglia but also in astrocytes and oligodendrocytes. Interestingly, astrocyte activation can be triggered by microglia activation (Rothhammer et al., 2018), leading to alterations in cognitive processes (Tchessalova et al., 2018; Illouz et al., 2019). On the other hand, defective oligodendrocytes and consequent transcriptome abnormalities have been already described in DS (Olmos-Serrano et al., 2016). The oligodendrocyte transcriptome abnormalities that we found may thus be unrelated to the immunological defects we are describing here but may still play a role in DS cognitive impairment (Olmos-Serrano et al., 2016).
How can microglial activation be reconducted to chromosome 21 triplication in DS? DS is caused by the triplication of a very long stretch of DNA, comprising 110–150 genes in humans, many of which are expressed by microglia (Mrak and Griffin, 2004). In the Dp(16) mice, 113 genes are triplicated. Thus, it is very difficult to pinpoint the primary cause of the overall DS immunological dysfunction and the microglial defects we observed in this study. Nevertheless, at least 16 genes that are triplicated in both DS and Dp(16) mice are known to play a direct role in immune system regulation (e.g., the IFN receptors IFNAR1, IFNAR2, and IFNGR2; Table S4; Li et al., 2007). In agreement, we found that, in Dp(16) microglia, the response to IFN is dysregulated and affected by APAP treatment. Interestingly, IFNβ can influence TrpV1 channels activation (Stampanoni Bassi et al., 2019). In addition to IFN receptors, the RUNX-1 gene is also triplicated in DS and Dp(16) mice. RUNX-1 encodes a transcription factor that plays key roles in microglia differentiation, proliferation, and activation (Kierdorf and Prinz, 2013).
In this study, we showed that APAP treatment rescued the activation state of trisomic microglia, the release of pro-inflammatory cytokines, the neuronal spine density and function, and cognitive performance in Dp(16) animals. Remarkably, APAP treatment had a similar outcome also in the Ts65Dn mouse model of DS. These results were strictly restricted to young animals, indicating a critical period of brain development more sensitive to this therapeutic approach.
Although APAP is well known for being a potential hepatotoxic drug in the case of overdose or long-lasting chronic treatment, it is extremely safe even at relatively high doses (up to 4 g daily for adults) for acute treatments (Ajmone-Cat et al., 2010). Thus, APAP is a good candidate for safely assessing the role of inflammation in young people with DS by drug repurposing in future clinical trials (Strittmatter, 2014). In addition, APAP is considered so safe that it is routinely prescribed as an anti-inflammatory treatment to pregnant women and newborns. Therefore, APAP could potentially also be a candidate for later possible further clinical studies at early stages of development. The treatment of DS subjects during gestation or early postnatal life could potentially lead to even more positive (or long-lasting) outcomes on their cognitive performance, because DS is a neurodevelopmental disorder and microglia play a fundamental role in brain development. Nevertheless, our study only focused on late postnatal development, and there is just scanty knowledge on microglia state in DS fetal development (Guedj et al., 2015; Wierzba-Bobrowicz et al., 1999). Moreover, recent studies on WT animals and humans have raised concerns about the safety of APAP during gestation and perinatal stages because it can influence physiological brain development (Good, 2018). These detrimental effects on CNS development may be due nevertheless to specific actions of APAP on WT embryos and may not apply to DS. Indeed, in our current study, APAP treatment had a mild opposite effect on the WT microglia, neuronal dendritic spines, and mEPSCs in comparison to Dp(16) mice and consistently decreased cognitive performance in WT mice. In line with our results, administration of APAP to WT rats can cause a significant decrease in cognitive behavior (Blecharz-Klin et al., 2018; Ishida et al., 2007).
The mild impairments in microglia state and cognitive behavior upon APAP treatment on WT animals call for some considerations on the current methods of validation of the efficacy of new pharmacological therapies (i.e., clinical trials). Indeed, according to the Food and Drug Administration (FDA), approximately 30% of new drugs are discontinued after phase I clinical trials (https://www.fda.gov/patients/drug-development-process/step-3-clinical-research), which involves an initial testing of the safety, side effects, best dose, and formulation method for the drug in healthy volunteers. Based on our observations with APAP in Dp(16) mice, drug treatments could have completely different outcomes based on the presence or absence of morbidity. This could mean that potentially valuable drugs for the treatment of certain conditions are erroneously discarded during phase I clinical trials because of the appearance of adverse effects in healthy volunteers, which might not be reproduced in patients.
In summary, our study proposes microglial dysregulation as one of the possible causes of the cognitive deficits in DS mice. In particular, we hypothesize that the hyperactive state of microglia, on a background of chronic neuroinflammation, causes a profound alteration in the functionality of neuronal circuits via aberrant synaptic pruning, thus leading to cognitive deficits. Remarkably, the use of APAP (an over-the-counter anti-inflammatory drug) is able to recover the aforementioned deficits in DS mice. Thus, although by no means our results should encourage the unsupervised assumption of APAP by DS individuals, they do open up to the possibility of safely testing in clinical trials the role of inflammation in DS by repurposing anti-inflammatory drugs for the possible amelioration of DS cognitive deficits in the near future.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-Iba1 | WAKO | Cat# 019-19741, RRID:AB_839504 |
| Rat anti-Lamp1 | Santa Cruz Biotechnology | Cat# sc-19992, RRID:AB_2134495 |
| Rat anti-MHCII | Santa Cruz Biotechnology | Cat# sc-59322, RRID:AB_831551 |
| Rabbit anti-P2Y12 | Thermo Fisher Scientific | Cat# PA5-77671, RRID:AB_2736305 |
| Rat anti-CD68 | Abcam | Cat# ab53444, RRID:AB_869007 |
| Rabbit anti-Iba1 | WAKO | Cat# 016-20001, RRID:AB_839506 |
| Mouse anti-Drebrin | Enzo Lifesciences | Cat# ADI-NBA-110-E, RRID:AB_2039073 |
| Chicken anti-GAPDH | Merck Millipore | Cat# AB2302, RRID:AB_10615768 |
| Rabbit anti-Actin | Sigma-Aldrich | Cat# A2066, RRID:AB_476693 |
| Mouse monoclonal IgG anti-Iba1 | Millipore | Cat# MABN92, RRID:AB_10917271 |
| Rabbit anti-NG2 Chondroitin Sulfate Proteoglycan | Millipore | Cat# ab5320, RRID:AB_91789 |
| Rabbit anti-Cox2 | Abcam | Cat# ab62331, RRID:AB_942121 |
| Rabbit anti-PGE | Abcam | Cat# ab2318, RRID:AB_302974) |
| Guinea pig anti-Vglut1 | Synaptic Systems | Cat# 135 304, RRID:AB_887878 |
| Mouse anti-Psd-95 | Thermo Fisher Scientific | Cat# MA1-045, RRID:AB_325399 |
| Biological Samples | ||
| Human Samples used for Western Blotting and RNA sequencing | Table S3 | N/A |
| Human Samples used for Immunohistochemistry | Table S3 | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| PLX3397 (Pexidartinib) | Selleckem | Cat#S7818 |
| Dimethyl Sulfoxide (DMSO) | Sigma-Aldrich | Cat#276855 |
| Acetaminophen (APAP) | Sigma-Aldrich | Cat#A7085 |
| Ruxolitinib | Sigma-Aldrich | Cat# S1378 |
| Hoechst | Sigma-Aldrich | Cat#H6024 |
| Paraformaldehyde | Sigma-Aldrich | Cat#158127 |
| VECTASHIELD Mounting Medium | Vector Laboratories | Cat# H-1000, RRID:AB_2336789 |
| Potassium dichromate | Sigma-Aldrich | Cat#207802 |
| Mercuric chloride | Sigma-Aldrich | Cat#215465 |
| Potassium chromate | Sigma-Aldrich | Cat#216615 |
| Ammonium hydroxide | Sigma-Aldrich | Cat#221228 |
| DPX-Mounting medium | Sigma-Aldrich | Cat#06522 |
| NaCl | Sigma-Aldrich | Cat#71376 |
| HEPES | Sigma-Aldrich | Cat# H3375 |
| NP40 | Sigma-Aldrich | Cat#74385 |
| Sodiumdeoxycholate | Sigma-Aldrich | Cat#D6750 |
| Pierce BCA assay | Thermo-Fisher | Cat#23225 |
| NuPAGE LDS Sample Buffer | Thermo-Fisher | Cat#NP0007 |
| 1,4-Dithiothreitol | Sigma-Aldrich | Cat#DTT-RO Roche |
| NuPAGE 4-12% Bis-Tris Midi Protein gels | Thermo-Fisher | Cat#WG1402BOX |
| Nitrocellulose membrane | Sigma-Aldrich | Cat#GE10600001 |
| Trizma | Sigma-Aldrich | Cat#T6066 |
| Tween 20 | Sigma-Aldrich | Cat# P7949 |
| Nonfat dry milk | Euroclone | Cat#EMR180500 |
| Sucrose | Sigma-Aldrich | Cat#S7903 |
| Magnesium chloride hexadrydate | Sigma-Aldrich | Cat#M9272 |
| Potassium chloride | Sigma-Aldrich | Cat#P9333 |
| Sodium phosphate monobasic dihydrate | Sigma-Aldrich | Cat#71505 |
| Calcium chloride dihydrate | Sigma-Aldrich | Cat#C3306 |
| Sodium bicarbonate | Sigma-Aldrich | Cat#S5761 |
| D(+) Glucose | Sigma-Aldrich | Cat#G7021 |
| Ethylene glycol-bis(2-aminoethylether)-N,N,N,N-tetraacetic acid | Sigma-Aldrich | Cat#E4378 |
| Adenosine 5′-triphosphate magnesium salt | Sigma-Aldrich | Cat#A9178 |
| Potassium D-gluconate | Sigma-Aldrich | Cat#G4500 |
| Guanosine 5′-triphosphate sodium salt hydrate | Sigma-Aldrich | Cat#G8877 |
| Phosphocreatine disodium salt hydrate | Sigma-Aldrich | Cat#P7936 |
| Capsazepine | Hello Bio | Cat#E0282-2-3 |
| N-Oleoyldopamine (OLDA) | Tocris | Cat# 1641 |
| AM-251 | Sigma-Aldrich | Cat# A6226 |
| AM630 | Sigma-Aldrich | Cat# SML0327 |
| Bicuculline methiodide | Hello Bio | Cat# HB0893 |
| Tetradotoxin | Hello Bio | Cat# HB1035 |
| Glutaraldehyde | Sigma-Aldrich | Cat#G5882 |
| Bovine Serum Albumin | Sigma-Aldrich | Cat#A2153 |
| Sodium Azide | Sigma-Aldrich | Cat#SX0299 |
| Saponin | Sigma-Aldrich | Cat#47036 |
| 3,3′-Diaminobenzidine | Sigma-Aldrich | Cat#D8001 |
| Hydrogen peroxide-18O2 solution | Sigma-Aldrich | Cat#609978 |
| Osmium tetroxide | Sigma-Aldrich | Cat#O5500 |
| Uranyl Acetate | Polysciences | Cat#21447-25 |
| EPON resin | Sigma-Aldrich | Cat#45359 |
| Percoll | Sigma-Aldrich | Cat#GE17-0891-01 |
| Sodium deoxycholate | Sigma-Aldrich | Cat#D6750 |
| Chloroacetamide | Sigma-Aldrich | Cat#C0267 |
| Tris(2-carboxyethyl)phosphine hydrochloride | Sigma-Aldrich | Cat#C4706 |
| Dako fluorescent medium | Dako North America | Cat#S3023 |
| Critical Commercial Assays | ||
| SuperSignal West Pico PLUS Chemiluminescent Substrate detection system | Thermo-Fisher Scientific | Cat#34578 |
| HRP/DAB (ABC) Detection IHC Kit | Abcam | Cat#AB64259 |
| QuantSeq 3′ mRNA-Seq Library Prep Kit FWD | Lexogen | Cat#015 |
| QuantiTect Reverse Transcription Kit | QIAGEN | Cat#205310 |
| RayBio Mouse Inflammation Array 1 antibody | RayBiotech | Cat# AAM-INF-1-8, RRID:AB_10181099 |
| Pierce BCA assay | Thermo-Fisher Scientific | Cat# 23225 |
| FASP Protein Digestion Kit | Expedeon Inc | Cat# 44250 |
| Deposited Data | ||
| Proteomics Dataset for microglia | This paper | PRIDE: PXD015113; |
| Proteomics Dataset for hippocampal tissues | This paper | PRIDE: PXD015148 |
| RNaseq datasets | This paper | GEO Series accession number: GEO: GSE137939 |
| Uncropped blot, qPCR information and results of cognitive performance regarding total distance traveled, total object exploration time and preference during acquisition trials | This paper | https://data.mendeley.com/datasets/gkbrjc8rmd/draft?a=4c602482-bb3a-442a-8a90-c43e8efa9f00 |
| Experimental Models: Organisms/Strains | ||
| Mouse: B6.129S7-Dp(16Lipi-Zbtb21)1Yey/JB6.129S7-Dp(16Lipi-Zbtb21)1Yey/J | The Jackson Laboratory | Cat # JAX:013530; RRID:IMSR_JAX:013530 |
| Mouse: C57BL/6J | The Jackson Laboratory | Cat# JAX:000664, RRID:IMSR_JAX:000664 |
| Mouse: B6.129P2(Cg)-Cx3cr1tm1Litt/J | The Jackson Laboratory | Cat# JAX:005582, RRID:IMSR_JAX:005582 |
| Mouse:B6EiC3Sn a/A-Ts(1716)65Dn/J | The Jackson Laboratory | Cat# JAX:001924, RRID:IMSR_JAX:001924 |
| Mouse: (C57BL/6JEiJ x C3H/HeSnJ)F1/J | The Jackson Laboratory | Cat# JAX:001875, RRID:IMSR_JAX:001875 |
| Oligonucleotides | ||
| primer: GAPDH forward: ATGAAGGGGTCA TTGATGG |
Sigma-Aldrich | N/A |
| primer: GAPDH reverse: AGGTGAAGGTCG GAGTCAA |
Sigma-Aldrich | N/A |
| primer: PPIA forward: TTCTGCTGTCTTTG GGACCT |
Sigma-Aldrich | N/A |
| primer: PPIA reverse: CACCGTGTTCTTCG ACATTG |
Sigma-Aldrich | N/A |
| primer:STAT3 forward: TGGTGTCTCCAC TGGTCTATC |
Sigma-Aldrich | N/A |
| primer: STATE reverse: TCCGAATGCCTC CTCCTTGG |
Sigma-Aldrich | N/A |
| primer:FCGR1A forward: GCGATACCATA GAGTCCAGATC |
Sigma-Aldrich | N/A |
| primer: FCGR1A reverse: CCAAGTATGA GAGCAGCGTATC |
Sigma-Aldrich | N/A |
| primer:IL4R forward: AGCCGAGCCTAGA AACTAACAC |
Sigma-Aldrich | N/A |
| primer: IL4R reverse: TGACCACCCTCC CTGAAGT |
Sigma-Aldrich | N/A |
| primer: MARCKSL1 forward:AACTGTGC TGTCCTTGTG |
Sigma-Aldrich | N/A |
| primer: MARCKSL1 reverse: GAACCAG ATGGCTGATG |
Sigma-Aldrich | N/A |
| primer: PECAM1 forward: AGCGTCATT GGCGTGTTG |
Sigma-Aldrich | N/A |
| primer: PECAM1 reverse:GAGACATCT CCTTCCCGTTTACCT |
Sigma-Aldrich | N/A |
| Software and Algorithms | ||
| Fiji ImageJ | Schindelin et al., 2012 | https://fiji.sc/; RRID: SCR_002285 |
| GraphPad PRISM | GraphPad Software Inc. | https://www.graphpad.com/scientific-software/prism/ |
| Anymaze | Anymaze | http://www.anymaze.co.uk/index.htm |
| MaxQuant software | Cox and Mann, 2008 | https://www.maxquant.org/ |
| Uniprot mouse database | Uniprot Consortium | https://www.uniprot.org/ |
| Perseus software | Tyanova et al., 2016 | https://maxquant.net/perseus/ |
| FastQC v0.11.3 | Babraham Bioinformatics group | http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ |
| Trim Galore software | Babraham Bioinformatics group | https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/ |
| STAR v2.7 | UCSC Genome Browser for human (hg38) genome build | https://genome.ucsc.edu/ |
| EdgeR v3.26.8 | Bioconductor | https://bioconductor.org/packages/release/bioc/html/edgeR.html |
| EWCE R package | Bioconductor | https://bioconductor.riken.jp/packages/3.4/bioc/html/EWCE.html |
| ClusterProfiler v3.0.4 | Yu et al., 2012 | https://www.rdocumentation.org/packages/clusterProfiler/versions/3.0.4 |
| Beacon Designer software | Premier Biosoft | http://www.premierbiosoft.com/molecular_beacons/ |
| qBasePlus software | Biogazelle | https://services.biogazelle.com/ |
| Picky software | Zauber et al., 2018 | N/A |
| Skyline software | MacCoss Lab Software | https://skyline.ms/project/home/begin.view? |
| Xcalibur Software | Thermo Fisher Scientific | OPTON-30965 |
| Neurolucida | Mbf Bioscience | https://www.mbfbioscience.com/neurolucida |
| Other | ||
| TCS SP5 microscope | Leica Microsystems | https://www.leica-microsystems.com |
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Laura Cancedda (laura.cancedda@iit.it).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
The datasets of this work are available https://data.mendeley.com/datasets/gkbrjc8rmd/draft?a=4c602482-bb3a-442a-8a90-c43e8efa9f00. Uncropped blot, qPCR information and results of cognitive performance regarding total distance traveled, total object exploration time and preference during acquisition trials are available in the same repository. The accession number for proteomic datasets are available in the PRIDE archive as PRIDE: PXD015113 for microglial cells and PRIDE: PXD015148 for hippocampal tissues and drebrin protein described in STAR Methods. The accession number forthe RNaseq datasets in this study have been deposited in NCBI’s Gene Expression Omnibus (Pinto, Morelli et al., 2020) and are accessible through GEO Series accession number GEO: GSE137939 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE137939).
Experimental Model and Subject Details
Mice
All care of animals and experimental procedures were conducted in accordance with IIT licensing and were approved by the Italian Ministry of Health. All animals were housed in a climate-controlled animal facility (22 ± 2°C) and maintained on a 12 h light/dark cycle with ad libitum access to food and water. Animal housing rooms are maintained at MP14 barrier (pathogen and opportunistic free) in the animal facility prior experiments. The Dp(16)1Yey/+ animals were purchased from The Jackson Laboratory (B6.129S7-Dp(16Lipi-Zbtb21)1Yey/J, catalog #013530) and were used to create a colony by mating Dp(16) males with C57BL6/J females. Females homozygous CX3CR1-GFP (B6.129P2(Cg)-Cx3cr1tm1Litt/J) were purchased from the Jackson Laboratory (catalog #005582) and crossed with Dp(16) males to generate WTCX3CR1-GFPand Dp(16)CX3CR1-GFP animals. Ts65Dn and their respective WT mice were generated by repeated backcrossing of Ts65Dn females (obtained from the Jackson Laboratory, catalog #001924) to C57BL/6JEi × C3SnHeSnJ (B6EiC3) F1 males. In this study, both male and female mice were used for all experiments.
Human Subjects
For RNA sequencing and western blotting: hippocampal samples from adult humans with Down syndrome and age- and sex-matched controls were obtained from the Brain and Tissue Bank for Developmental Disorders at the University of Maryland, Baltimore, MD. Information on the samples are reported in Table S3. For immunohistochemistry: tissue from adult humans with Down syndrome and age- and sex-matched controls were obtained from Neurobiobank (USA) and Biobank (Spain). Information on the samples are reported in Table S3. No difference were observed between genders.
Method Details
Drug treatments
PLX3397 (Pexidartinib) was purchased from Selleckem (catalog #S7818). The powder was dissolved in Dimethyl Sulfoxide (DMSO; Sigma-Aldrich, catalog #276855). The solution was added to pulverized mouse chow at a concentration of 445 mg/kg and given to animals ad libitum from P15 to P22. The final dosage of around 4.7 mg total of drug/mouse over the 7 days of treatment was calculated on an average of 1.5 g of chow consumed by each mouse/day (Bachmanov et al., 2002). Acetaminophen (APAP) was purchased from Sigma-Aldrich (catalog #A7085). The powder was dissolved in DMSO and subsequently diluted in saline (NaCl 0.9% w/v) to generate a 10 mg/mL (66.15 mM) solution. This solution was injected intraperitoneally (IP) to animals with a dosage of 100mg/kg once a day from P20 to P22, or from P29 to P31 or P59 to P61 depending on the experiment. Ruxolitinib was purchased from Selleckem (catalog #S1378). The powder was dissolved in DMSO and subsequently diluted in saline (NaCl 0.9% w/v) to generate a 1.25mg/mL (4.08 mM) solution. This solution was injected subcutaneously to animals with a dosage of 50mg/kg twice a day from P20 to P22 (Bhagwat et al., 2014). A light sonication was applied to obtain full solubility of the Ruxolitinib-DMSO solution in saline. The solution was kept in ice during sonication to avoid overheating. All injected drugs were dissolved in the same vehicle (2% DMSO v/v in Saline).
To decrease the number of utilized animals in agreement with ethic directions from the Italian Ministry of Health, all vehicle-treated animals from all experiments from P20 to P22 were pulled together. PLX3397-treated animals were compared to the 2% DMSO treated animals as well (refer to the Statistical methods section for further information on how these data were analyzed). For every acetaminophen or ruxolitinib-treated litter, at least one animal of each strain was treated with vehicle (except for 3 cases out of a total of 19 where the number of animals in a given litter did not allowed for such precaution no controls were assigned to the litter).
Slice histology, immunostaining, image acquisition and analysis
Mouse brains were fixed by transcardial perfusion with PBS 1X followed by 12-24 hours PFA fixation (4% w/v in PB). Then, brains were cryopreserved in sucrose (30% w/v) and sectioned coronally in 50 μm-thick slices with a microtome (Microm HM 450 Sliding Microtome equipped with Freezing Unit Microm KS34, Thermo Scientific). For immunostaining, an antigen retrieval protocol was applied on free-floating slices. In brief, slices were incubated for 30 minutes in Sodium citrate buffer (10 mM Sodium citrate, 0.05% Tween 20, pH 6.0) at 90°C and subsequently cooled down to room temperature and washed in PBS. Slices were then permeabilized and blocked with PBS containing 0.3% Triton X-100 and 10% NGS. Primary antibodies were incubated in PBS containing 0.3% Triton X-100 and 5% NGS overnight [rabbit anti-Iba1, 1:300 (WAKO, catalog #019-19741); rat anti-Lamp1, 1:300; rat anti-MHCII, 1:100; rabbit anti-P2Y12,1:300; rat anti-CD68, 1:250; rabbit anti-Cox2, 1:200; rabbit anti-PGE2, 1:100; guinea pig anti-Vglut1,1:1000; mouse anti-Psd-95, 1:100)]. Immunostaining was detected using Alexa 488 (for Iba1) 1:1000 or Alexa 543 (for Lamp1, MHCII, CD68, Cox2, Psd-95 and P2Y12) and Alexa 647 (for Vglut1) fluorescent secondary antibodies (Invitrogen), 1:600, incubated in PBS containing 0.3% Triton X-100 and 5% NGS for 1 hour. Slices were counterstained with Hoechst (2,5 μg/μL; Sigma). All slices were mounted in Vectashield Mounting Medium (Vector Laboratories, Burlingame, CA).
For microglial cell count and morphology analysis, a stack of images spanning the whole thickness of the slice (1 μm thick z stacks) from Iba1 stained hippocampal slices were acquired using a confocal laser-scanning microscope (TCS SP5; Leica Microsystems, Milan, Italy) equipped with a 40x (NA 1.25) immersion objective. One randomly chosen slice per animal was acquired and cells from DG, CA1 and CA3 areas from the hippocampus were chosen in a balanced manner (20-36 cells for each brain area). For the somatosensory cortex, cells from all layers were selected in a balanced manner (10-15 cells for each cortical layer). For cell count, the number of cells was divided by the total area of the acquired field to represent cell density. For the calculation of the cell-body area, the measurements were performed using the freehand selection tool in ImageJ. Sholl analysis was performed as previously described (Sipe et al., 2016). Briefly, cells were cropped and thresholded to generate a binary (black and white) image. The background was manually cleaned for each cell and the ImageJ plugin ShollAnalysis (Ferreira et al., 2014) was used to perform the analysis (starting radius: 4 μm, ending radius: 70 μm, radius step size: 2 μm, radius span: 0 μm). For skeleton analysis, the same binary image created for Sholl Analysis was used. The image was skeletonized using the ImageJ Plugin Skeletonize3D (Lee et al., 1994) and was subsequently analyzed using the plugin AnalyzeSkeleton.
For the analysis of Lamp1/Iba1, CD68/Iba1, Cox2/GFP, PGE2/GFP or P2Y12/GFP co-localization area a stack of images spanning the whole thickness 0.5 μm thick z stacks of stained hippocampal slices were acquired using a confocal laser-scanning microscope equipped with a 20x −40x immersion objective and projected on a 2D image. Each channel (Iba1, Lamp1, CD68, PGE2, Cox2 or P2Y12, GFP) was thresholded (“Default Threshold” in ImageJ) to generate a binary (black and white) image using ImageJ. The background of binary images only was manually cleaned to ensure that only one cell was present. Subsequently, the two images were merged, converted to 32-bit and thresholded again to generate a binary image of only the pixels that came from both images. The area of the double positive pixels was calculated and divided by the area of the Iba1 or GFP positive pixels only for normalization. One randomly chosen slice per animal was acquired.
Microglia phagocytosis was quantified in the triple-stained Iba1/Psd-95/Vglut1 images in a modified manner from Bellesi et al. (2017). To optimize the detection of the positive puncta engulfed within the microglia, green (Iba1), red (Psd-95) and blue (Vglut1) channels were processed separately. The background noise of the green channel was reduced by using the function Subtract Background (rolling ball radius, 50 pixels) in Fiji ImageJ. The image was subsequently filtered using a 3D maximal filter. The background noise of the red and blue channel was diminished using the function Subtract Background (rolling ball radius, 2 pixels) and Despeckle in FIJI. The image was subsequently filtered through a 3D Maximum Filter (radius, 3 pixels in every dimension), automatically thresholded (“Yen Threshold”). Green, red and magenta channels were then remerged. Only Vglut1 and Psd-95 positive puncta bigger than 100 pixels (∼0.03 μm3) in xyz, and showing 100% overlap with the processed Iba1 signal, were quantified.
Golgi-Cox staining, image acquisition and analysis
The animals were perfused transcardially with 0.9% saline. Whole brains were dissected and immersed in the Golgi-Cox solution (5% potassium dichromate, 5% mercuric chloride and 5% potassium chromate) for 35 days. Then, the brains were transferred to a 30% sucrose solution and stored in the dark at 4°C. 200 μm-thick coronal slices were cut at room temperature with a microtome (Microm HM 450 Sliding Microtome) and transferred onto 1% gelatin-coated slides to initiate the staining process in humidified chambers. Ammonium hydroxide was applied for 10 minutes. Next, the slices were incubated in an increasing grade of ethanol (50%, 75%, 95% and 100%), then in xylene for 30 minutes, and finally mounted in DPX-Mounting medium (Sigma-Aldrich, Catalog #06522). A stack of 80-150 focal images (1 μm -hick z stacks) per cell (for a total of 5 cells per animal) were acquired from randomly chosen cortical slices at the level of the somatosensory cortex on a Neurolucida microscope equipped with the software Neurolucida (MicroBrightField) and a 100x objective (NA 1.3). On each image stack, the apical dendrite of a neuron was visually identified. Spines were counted on the whole visible length of 3 collateral dendrites for each image and divided by this length using the Reconstruct software, which allows for the measurement of spine length and width for an unbiased classification of spine types (Risher et al., 2014). The densities of spines for each dendrite were averaged per each image stack. All the chemicals were purchased from Sigma-Aldrich, unless otherwise specified.
Western Blotting
All hippocampus samples were lysed in RIPA buffer (150 mM NaCl, 50 mM HEPES, 0.5% NP40, 1% Sodiumdeoxycholate). After 30 minutes on ice, the lysates were clarified by centrifugation for 20 minutes at 16,000 g. The protein samples were measured via Pierce BCA assay (Thermo-Fisher, Catalog #23225). For protein identification and relative quantification via western blotting, a proper volume of sample containing an equal amount of proteins was diluted with 25% NuPAGE LDS Sample Buffer (4X; Thermo-Fisher, Catalog #NP0007) and 5% 1,4-Dithiothreitol (DTT; equivalent of 50 mM, Sigma-Aldrich, Catalog #DTT-RO Roche) and loaded onto NuPAGE 4%–12% Bis-Tris Midi Protein gels (Thermo-Fisher, Catalog # WG1402BOX). The proteins were then transferred onto nitrocellulose membrane (Sigma-Aldrich, catalog #GE10600001) at 200 mA overnight at 4◦C. The primary antibodies (rabbit anti-Iba1,1:1000 (WAKO, catalog #016-20001); rabbit anti-NG2 Chondroitin Sulfate Proteoglycan, 1:1000; mouse anti-Drebrin, 1:600; chicken anti-GAPDH, 1:2000; rabbit anti-Actin, 1:2500) were then applied overnight in a blocking buffer (20 mM Tris, pH 7.4, 150 mM NaCl, 0.1% Tween 20, and 5% nonfat dry milk). The HRP-conjugated secondary antibodies [Thermo-Fisher, anti-mouse (catalog #31430), anti-rabbit (catalog #31460), anti-rat (catalog #31470) or anti-chicken (catalog #PA1-28798)] were all used in a ratio of 1:2500. The signal was detected using the SuperSignal West Pico PLUS Chemiluminescent Substrate detection system (Thermo-Fisher, catalog #34578). Images were acquired by the Image Quant LAS 4000 mini image system (General-Electrics Healthcare) and protein quantification was performed by measuring the optical density of the specific bands with the Image Quant TL software (General-Electrics Healthcare). Samples from all experimental groups were loaded on each experiment and normalized to the WT treated with vehicle. For statistical analysis, normalized data across independent experiments were used together. Uncropped blots are available in the repository in “Data and Code Availability.”
Microglia and neuronal In vitro electrophysiology
Patch-clamp experiments were performed in P22-P30 mice. For microglial experiments, WTCX3CR1-GFPand Dp(16)CX3CR1-GFP mice were used. For experiments in neurons, WT and Dp(16) mice were used. In both cases, animals were anesthetized with isoflurane and transcardially perfused with ice-cold cutting solution containing: 220 mM sucrose, 7 mM MgCl2, 2.5 mM KCl, 1.25 mM NaH2PO4, 0.5 mM CaCl2, 25 mM NaHCO3 and 25 mM D-glucose (∼300 mOsm, pH 7.4, oxygenated with 95% O2 and 5% CO2). For all experiments, brains were removed and immersed in cutting solution. 270 μm-thick coronal slices (cut with VT1000S Leica Microsystem vibratome) were allowed to recover for 1 hour at 35°C in a solution containing: 117 mM NaCl, 2.5 mM KCl, 1.25 mM NaH2PO4, 3 mM MgCl2, 0.5 mM CaCl2, 25 mM NaHCO3 and 25 mM glucose (∼310 mOsm, pH 7.4, oxygenated with 95% O2 and 5% CO2). All recordings were performed at room temperature in artificial cerebrospinal fluid (ACSF) composed of: 117 mM NaCl, 2.5 mM KCl, 1.25 mM NaH2PO4, 1 mM MgCl2, 2 mM CaCl2, 25 mM NaHCO3 and 25 mM glucose (∼310 mOsm, pH 7.4, oxygenated with 95% O2 and 5% CO2). Patch pipettes were made from thick-wall borosilicate glass capillaries (Sutter Instrument, catalog #B150-86-7.5). In both cases, recordings were performed in voltage-clamp mode.
For microglia: whole-cell patch-clamp recordings were performed on GFP+ cells from WTCX3CR1-GFPand Dp(16)CX3CR1-GFP mice at a series resistance ranging from 10 to 30 mΩ. Pipettes (6-8 mΩ) were filled with intracellular solution containing: (in mM): 130 KCl, 4 MgCl2, 4 Na2ATP, 10 EGTA, and 10 HEPES (∼290 mOsm pH adjusted to 7.2 with KOH). Voltage ramps of 2 s from −100 to 80 mV were applied from a holding potential of −60 mV. An initial voltage step (−100 mV; 100 ms) was applied before the onset of the ramp. Three ramps were applied for each microglia at 2 s intervals. The evoked currents of the three ramps were then averaged for analysis and a single value of current for each voltage step (interval 10 mV) was plotted to compare different genotypes and/or treatments. For acute treatments: 1-hour incubation was performed with 300 μM APAP and 10 μM Capsazepine (Hello Bio, batch E0282-2-3); 15 min with 2 μM N-Oleoyldopamine (OLDA; Tocris, catalog #1641); 15 min with 1 μM AM-251 (A6226) and 1 μM AM-630 (SML0327) or equivalent amount of DMSO. Data, filtered at 0.1 Hz and 5 kHz and sampled at 10 kHz, were acquired by a patch-clamp amplifier (Multiclamp 700B, Molecular Devices) and analyzed using MiniAnalysis (Synaptosoft) for mEPSCs. All chemicals were purchased from Sigma-Aldrich, unless otherwise specified. For neurons: whole-cell patch-clamp recordings were made from visually identified CA1 neurons at a series resistance ranging from 10 to 25 mΩ.
For recording of neuronal mEPSCs, pipettes (4-6 mΩ) were filled with intracellular solution containing (mM): 130 K-gluconate, 10 HEPES, 7 KCl, 0.6 EGTA, 4 Mg2ATP, 0.3 NaGTP, 10 Phosphocreatine. The pH was adjusted to 7.3 with HCl. Three-minute recordings of neuronal mEPSCs were performed after a 10-minute bath-application of 20 μM of Bicuculline methiodide (HelloBio, catalog # HB0893) and 1 μM Tetradotoxin (TTX; HelloBio, catalog # HB1035) to isolate excitatory events.
Microglia time-lapse analysis
Brain slices from P22-P30 WTCX3CR1-GFPand Dp(16)CX3CR1-GFP animals were prepared and perfused as in ex vivo electrophysiology. Images were acquired using a confocal laser-scanning microscope (TCS SP5; Leica Microsystems, Milan, Italy) equipped with a 25x (NA 0.75) immersion objective and a bath perfusion system. Stacks of images spanning 100 μm of the slice (1 μm thick z stacks) were acquired every 2.5 minutes. Image analysis and data quantification were performed as previously described (Sipe et al., 2016), with slight modifications. Briefly, images were stabilized using the StackReg plugin in ImageJ. Then they were despeckled, projected onto a 2D image, and finally thresholded to create binary images. Each binary projection from each time point t(x) was color-merged with a t(x+1) image projection to generate images containing red pixels (that represent pixels that were white in time point x but are black in time point x+1, called retraction pixels), green pixels (that represent pixels that were black in time point x but are white in time point x+1, called extension pixels) and yellow pixels (that represent pixels that were white in time point x and are still white in time point x+1, called stable pixels). The number of pixels of each color was counted using the Color Counter (by Wayne Rasband) plugin in ImageJ. For each pair of images (t(x) and t(x+1), we calculated a mobility index as follows: (green+red)/yellow . However, in our experimental case, the dimensions of the cell body varied among groups. Because the cell body pixels remained stable during the whole time-lapse, they could have hidden the movement quantifications. Thus, the mobility index was multiplied by the percentage of pixels occupied with GFP fluorescence (the whole area of microglia) in the whole merged binary image as follows: (green+red+yellow)/total pixel count. The mobility index of each pair of time points (e.g., t0,t1; t1,t2) were then averaged for the whole time-lapse and the average was used for statistical analysis. The velocity of retracting and extending branches of microglia was calculated by following a single branch for the whole length of the recording using MTrackJ plugin of Fiji ImageJ.
Electron microscopy
Brains for electron microscopy analysis, were subjected to pre-embedding immuno-labeling as previously described (Hebert-Chatelain et al., 2014). Briefly, P22 brains were fixed by transcardial perfusion with PBS 1X followed by PFA and Glutaraldehyde mixed solution (4% PFA, 0,2% Glutaraldehyde w/v in PB). Next, 70 μm-thick coronal sections were cut using a vibratome (VT1000S Leica Microsystem). After a pre-incubation in blocking solution (10% BSA, 0.1% sodium azide and 0.02% saponin in Tris-buffered saline (TBS), the sections were incubated with rabbit anti-Iba1, 1:300 (WAKO, catalog # 019-19741) in blocking solution with 0.004% saponin overnight at 4°C. After several washes with 1% BSA in TBS, the sections were processed using the mouse specific HRP/DAB (ABC) Detection IHC Kit (Abcam, catalog #ab64259). In brief, the sections were incubated for 2 hours with a goat anti-rabbit biotinylated secondary antibody, then with avidin-biotin complex for 2 hours, and finally incubated in 0.05% diamibenzidine (DAB) and 0.01% hydrogen peroxide mixed solution. After 10 minutes fixation with 1% glutaraldehyde solution, the sections were post-fixed in Osmium tetroxide 1% for 20 minutes, and stained 2h in aqueous 0.5% uranyl acetate solution. Then, the samples were washed several times in distilled water, dehydrated in a graded ethanol series (50%, 75%, 95% and 100%) and then embedded in EPON resin. 70nm-thick sections of the CA1 hippocampal region were collected with a Leica EMU C6 ultra-microtome. Transmission electron microscopy (TEM) micrographs were collected with Jeol JEM 1011 (Jeol, Tokyo, Japan) electron microscope (Electron Microscopy Facility – Fondazione Istituto Italiano di Tecnologia, Genoa, Italy), operating at an acceleration voltage of 100 kV, and recorded with a 11 Mp fiber optical charge-coupled device (CCD) camera (Gatan Orius SC-1000, Gatan, USA) at 10,000x. The images were then quantified as previously described (Sipe et al., 2016). The post-synaptic elements were identified primarily by the presence of electron-dense accumulations (postsynaptic densities) when contacting pre-synaptic elements, together with the absence of mitochondria and rounded morphologies. The pre-synaptic elements were primarily identified by the presence of ∼40nm-diameter synaptic vesicles, but also by synaptic contacts with dendritic shafts and post-synaptic elements and the often-present mitochondria. The interactions were calculated by dividing the number of contacts (per Iba1-labeled process) by the area of the microglial processes (expressed in μm2) and averaging across processes (40-55 per animal).
Behavioral experiments
The behavioral experiments were performed on both male and female animals in the dark. Different sets of animals were used for the novel object recognition and the object location tests. The NOR was performed as previously described (Deidda et al., 2015). In brief, a gray acrylic arena (44 × 44 cm) was used. On the day before the NOR test, the mice were allowed to become habituated to the apparatus by freely exploring the open arena for 15 min. During the acquisition sessions, three different objects different in shape, color, size and material were placed into the arena, and the mice were allowed to explore for 15 min. The object exploration and preference were evaluated during these sessions to rule out any eventual intrinsic bias. The testing occurred 24 h later in the same arena. In the test, one of the objects used in the acquisition session was replaced by a novel object, and the mice were allowed to explore freely for 15 min. The objects were counterbalanced between the sessions and were cleaned with 70% ethanol after each trial as well as the arena. The exploratory behavior toward an object was defined as direct contact with the object by the animal’s mouth, nose or paws or as an instance when the animal approached the object so that its nose was within 1 cm of the object. Any indirect or accidental contact with the objects was not included in the scoring. The time spent exploring each object, expressed as a percentage of the total exploration time, was measured for each trial. The discrimination index was calculated as the difference between the percentage of time spent investigating the novel object and that of time spent investigating the familiar objects: Discrimination Index = ((Novel Object Exploration Time/Total Exploration Time) × 100) − ((Familiar Object Exploration Time/Total Exploration Time) × 100). The OLT was performed as previously described (Deidda et al., 2015). In brief, a gray acrylic arena (44 × 44 cm) was used. On the day before the OLT test, the mice were allowed to become habituated to the apparatus by freely exploring the open arena for 15 min. The next day, the mice were exposed to two identical objects for 15 min during the acquisition phase. The object preference was evaluated during this session to rule out any eventual intrinsic bias. The testing occurred 24 h later in the same arena. During the test session, one of the objects was moved to a novel location, and the mice were allowed to explore the objects for 15 min. The objects and the arena were cleaned with 70% ethanol after each trial. For the experiment of pre and post-APAP treatment, different location for different objects was used, and the two testing sessions were done 1 week apart from one another. The time mice spent exploring each object was measured. The exploratory behavior toward an object was defined as direct contact with the object by the animal’s mouth, nose or paws or as an instance when the animal approached the object so that its nose was within 1 cm of the object. Any indirect or accidental contact with the objects was not included in the scoring. The time spent exploring each object, expressed as a percentage of the total exploration time, was measured for each trial. The discrimination index was calculated as the percentage of time spent investigating the object in the new location minus the percentage of time spent investigating the object in the old location: Discrimination Index = ((New Object Location Exploration Time/Total Exploration Time) × 100) − ((Old Object Location Exploration Time/Total Exploration Time) × 100). The WT animals treated with APAP showed a small intrinsically preference for object 1 (on average animals explored 5.04% more object 1 than object 2 during acquisition). The bias did not affect the results on the testing day. The DI obtained from APAP treated WT animals in which the object in the new location was object 1 as opposed to object 2 (3.43 ± SEM with N = 5 versus 7.73 ± SEM with N = 7), was not significantly different. All parameters used as control for the tests such as total objects exploration time, object acquisition preferences and total distance traveled are available in the repository in “Data and Code Availability.”
Flow Cytometry
Microglia were sorted from P20-P30 brains of CX3CR1-GFPWT and CX3CR1-GFPDp(16) animals, as previously described (Bennett et al., 2016). All buffers were prepared as previously described (Bennett et al., 2016). Briefly, brains were homogenized in dounce buffer with a tissue–in-glass homogenizer on ice. The suspension was then applied onto a 70 μm cell-strainer (Falcon) on ice and centrifuged at 1500 RPM at 4°C. Next, the pellet was resuspended in 40% Percoll (GE Healthcare) in PBS, and centrifuged for at 1500 RPM at 4°C to remove myelin. The cell pellet was then resuspended in FACS buffer and kept on ice until sorting. Sorting was carried out using a BD FACSAria III sorter (BD bioscience) equipped with a 100μm nozzle.
Microglial proteomics
Sample preparation
FACS-sorted microglia from WTCX3CR1-GFPand Dp(16)CX3CR1-GFP mice were solubilized in 15μl 2% sodium deoxycholate (SDC), 40 mM Chloroacetamide, 10mM tris(2-carboxyethyl)phosphine (TCEP) and 100mM Tris HCl, pH 8.0 at 100°C, and sonicated with an Ultrasonic Processor UP200St (Hielscher). Lysate samples were digested with 0.3 μg Trypsin and 0.1 μg Endoproteinase Lys-C over night at 37°C. Digested samples were then processed using the iST protocol as previously described (Kulak et al., 2014).
NanoLC setup
The samples obtain from the previous step were loaded from the sample loop directly into a 75-μm ID × 50 cm 2 μm, 100 Å C18 column mounted in the thermostated column compartment and the peptides were separated with increasing organic solvent at a flow rate of 250 nl/min using with a non-linear gradient of 5%–45% solution B (80% CAN and 20% H2O, 5% DMSO, 0.1% FA) in 140 min.
Mass Spectrometer setup
Eluting peptides from the previous step were analyzed using an Orbitrap Fusion Tribrid mass spectrometer (Thermo Scientific Instruments, Bremen, Germany). MS scans (MS1) were acquired at a resolution of 240 000 between 375 and 1500 m/z and an AGC target of 4.0E5, while Ion-Trap detection was used for MS/MS (MS2) measurements with Rapid Ion Trap Scan Rate. Advanced Peak Detection was enabled for MS measurements. Data-dependent MS2 analysis was performed in top-speed mode with a 2 s. cycle time and, the quadruple isolation setting was 1.4 m/z and dynamic exclusion was enabled for 25 s. Automatic gain-control targets were 4 × 105 for MS1 and 3 × 104 for MS2, with 50 and 35ms maximum injection times, respectively. The signal intensity threshold for MS2 was 5 × 103. High collision energy dissociation (HCD) fragmentation was performed using 28% normalized collision energy and 1 microscan was used for both MS1 and MS2 events.
Data analysis and statistics
The MaxQuant software (Cox and Mann, 2008), version 1.6.6.0, was used to process the raw data, setting a false discovery rate (FDR) of 0.01 for the identification of proteins, peptides and peptide-spectrum match (PSM). For peptide identification, a minimum length of 6 amino acids was required. Andromeda engine, incorporated into MaxQuant software, was used to search MS2 spectra against Uniprot mouse database (release UP000000589 10090 April 2018). In the processing, the variable modifications are Acetyl (Protein N-Term), Oxidation (M), Deamidation (NQ). The Carbamidomethyl (C) was in fact selected as fixed modification. The intensity values were extracted and statistically evaluated using the ProteinGroup Table and Perseus software. Algorithm MaxLFQ was chosen for the protein quantification with the activated option “match between runs” to reduce the number of the missing proteins. After filtering for “reverse,” “only identified by site” and “potential contaminants,” we filtered the dataset for 100% data completeness in at least one experimental group, replacing the missing values by a normal distribution (downshift 2.0 and width 0.3). Weighted correlation network analysis (WGCNA) analysis was performed in the Perseus platform (Tyanova et al., 2016). To this aim we used the PluginCoExpression, the PerseusR and the PluginInterop, which interface directly with the established WGCNA library (Rudolph and Cox, 2019). In order to obtain a co-expression network, a power parameter of 20 was selected by a “Soft-threshold” activity set on a scale-free fit index of 0.9.
Human samples RNA sequencing
The mRNA-Seq library preparation was performed using QuantSeq 3′ mRNA-Seq Library Prep Kit FWD on Illumina platform (Lexogen). Single end reads underwent quality check using FastQC (v0.11.3; from http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Sequence reads were trimmed using Trim Galore software (from https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/) to remove adaptor sequences and low-quality end bases. Alignment was performed with STAR (v2.7; on the indication provided by the UCSC Genome Browser for human (hg38) genome build (Kent et al., 2002). Only uniquely mapped reads were used for gene expression analysis and all genes multiple mapping (> 20%) were removed. Genes wit < 1 cpm (counts per million) in less than n-1 samples were also removed. The expression level of genes were determined with htseq-count (v0.10.0; (Anders et al., 2015)). Differential expression analysis was performed using edgeR (v3.26.8; 147, a statistical package based on generalized linear models where the normalization was performed using Trimmed means of M values (TMM) method (Robinson and Oshlack, 2010).
Cell-type enrichment analysis method and KEGGs Pathways
The existing single-cell RNA data from Zeisel et al. (2015) was used for mouse hippocampus. We integrated the Zeisel et al. data with cell-type specific transcriptome signatures with the bulk RNA-sequencing data for human hippocampus that we generated. The EWCE R package (Skene and Grant, 2016) was used to obtain this integration. Two ranked gene lists, one for significantly upregulated genes and another for significantly downregulated genes were ordered according to their Log2 Fold Change (FC). In EWCE, random samples were obtained by reordering the ranked list 100,000 times. We controlled for transcript length and GC content. Data were represented as Z-scores i.e., standard deviations from the mean. Values below 0 indicate depletion in expression, which have been assigned the value of 0. For the Gene Set Enrichment Analysis (GSEA) for KEGG pathways, and for all Gene Ontology analysis performed in this manuscript, the ClusterProfiler v3.0.4 package in R was used 59.
Human samples Quantitative PCR
qPCR was performed as previously described (Pozzi et al., 2013). Briefly, RNA was extracted with QIAzol reagent and purified on RNeasy spin columns (QIAGEN). RNA samples were quantified at 260 nm with an ND1000 Nanodrop spectrophotometer (Thermo Scientific). RNA purity was also determined by absorbance at 280 and 230 nm. All samples showed A260/280 and A260/230 ratios greater than 1.9. RNA quality and integrity were verified by microfluidic assay with Agilent 2100 Bioanalyzer. The RNA quality indicator (RQI) ranged between 5.5 and 8.0. Reverse transcription was performed according to the manufacturer’s recommendations on 1 μg of RNA with the QuantiTect Reverse Transcription Kit (QIAGEN), which includes a genomic DNA–removal step. SYBR green qRT-PCR was performed in triplicate with 10 ng of template cDNA using QuantiTect Master Mix (QIAGEN) on a 7900-HT Fast Real-time System (Applied Biosystems) and using the following universal conditions: 5 min at 95°C, 40 cycles of denaturation at 95°C for 15 s, and annealing/extension at 60°C for 30 s. Product specificity and occurrence of primer dimers were verified by melting-curve analysis. Primers were designed with Beacon Designer software (Premier Biosoft) to avoid template secondary structure and significant cross-homology with other genes by BLAST search. For each target gene, primers were designed to target all possible transcript variants annotated in the RefSeq database (https://www.ncbi.nlm.nih.gov/refseq). In each experiment, no-template controls and RT-minus controls were run in parallel to the experimental samples. The PCR reaction efficiency for each primer pair was calculated via the standard curve method with four serial-dilution points for cDNA (32, 8, 2 and 0.5 ng). The PCR efficiency calculated for each primer set was used for subsequent analysis. All experimental samples were detected within the linear range of the assay. Gene-expression data were normalized via the multiple-internal-control-gene method (Vandesompele et al., 2002). To determine an accurate normalization factor for data analysis, we evaluated the expression stability of different control genes with the GeNorm algorithm available in qBasePlus software (Biogazelle). The tested control genes were GAPDH (glyceraldehyde-3-phosphate dehydrogenase) and PPIA (peptidylprolyl isomerase A). On the basis of the relative expression stability of the control genes calculated via GeNorm analysis, expression data for the different samples were normalized with GAPDH and PPIA.SYBR Green primer sequences, calibration curve parameters, PCR reaction efficiency and amplicon information are reported in the repository link in “Data and Code Availability.”
Human samples western Blotting
All hippocampus samples were lysed in RIPA buffer (150 mM NaCl, 50 mM HEPES, 0.5% NP40, 1% Sodiumdeoxycholate). After 30 minutes on ice, the lysates were clarified by centrifugation for 20 minutes at 16,000 g. The proteins were measured via Pierce BCA assay (Thermo-Fisher, Catalog #23225). For protein identification and relative quantification via western blotting, a proper volume of sample containing an equal amount of proteins was diluted with 25% NuPAGE LDS Sample Buffer (4X; Thermo-Fisher, Catalog #NP0007) and 5% 1,4-Dithiothreitol (DTT; equivalent of 50mM, Sigma-Aldrich, Catalog #. DTT-RO Roche), and loaded onto NuPAGE 4%–12% Bis-Tris Midi Protein gels (Thermo-Fisher, Catalog #WG1402BOX). The proteins were then transferred onto nitrocellulose membrane that was previously incubated with Transfer Buffer for 15 minutes (Sigma-Aldrich, catalog #. GE10600001) at 200mA overnight at 4◦C. The primary antibodies [rabbit anti-Iba,1 1:1000 (WAKO, catalog #016-20001), and rabbit anti-P2Y12,1:1000] were then applied overnight in a blocking buffer (20 mM Tris, pH 7.4, 150 mM NaCl, 0.1% Tween 20, and 5% nonfat dry milk). The HRP-conjugated secondary antibodies [Thermo-Fisher, anti-rabbit (catalog #31460)] was used in a ratio of 1:2500. The signal was detected using the SuperSignal West Pico PLUS Chemiluminescent Substrate detection system (Thermo-Fisher, catalog #34578). Images were acquired by the Image Quant LAS 4000 mini image system (General-Electrics Healthcare) and protein quantification was performed by measuring the optical density of the specific bands with the Image Quant TL software (General-Electrics Healthcare). The samples were loaded in three independent gels and averaged across experiments.
Human samples slice histology, immunostaining, image acquisition and analysis
Hippocampi were fixed (24h, PFA 4% (w/v) in PBS 1X), cryoprotected (sucrose 30%, w/v) and stored at −80°C. Tissue blocks were cut using a vibratome (VT1000S Leica Microsystem vibratome) into 8-10 μm thick (samples from HCB-IDIBAPS) or 25 μm thick (samples from NeuroBioBank) sections. Sections were then incubated with citrate buffer (0.01M, pH 6.0) for 1 minute at 90°C. Next, sections were treated for 1 h with 5% normal donkey serum (NDS, Jackson Immuno-Research Laboratories, catalog #017-000-121) in PBS 1X with 0.2% Triton- X100 and incubated overnight at room temperature with mouse monoclonal IgG anti-Iba1 1:500 (Chemicon, Catalog #MABN92). After washing, sections were incubated with secondary antibody (Alexa 488 donkey anti-mouse IgG 1:200 (Invitrogen, Catalog #A-21202). After further washing, sections were then mounted using Dako fluorescent medium (Dako North America, Catalog #S3023). For microglial morphology analysis, a stack of images spanning the whole thickness of the slice (1.15 μm-thick z stacks) from stained hippocampal slices were acquired using a confocal laser-scanning microscope (TSC-SPE; Leica Microsystems) equipped with a 40x (NA 1.25) immersion objective. For calculation of the cell-body area, the measurements were performed using the freehand selection tool in ImageJ. Sholl analysis for microglial branching was performed as previously described (Sipe et al., 2016) in only two samples per group due to tissue quality problems that would not allow a reliable quantification. Briefly, cells were cropped and thresholded to generate a binary (black and white) image. The background was manually cleaned for each cell and the ImageJ plugin Sholl Analysis (Ferreira et al., 2014) was used to perform the analysis (starting radius: 4 μm, ending radius: 70 μm, radius step size: 2 μm, radius span: 0 μm). For skeleton analysis, the same binary image created for Sholl Analysis was used. The image was skeletonized using the ImageJ Plugin Skeletonize3D (Lee et al., 1994) and was subsequently analyzed using the plugin AnalyzeSkeleton (Arganda-Carreras et al., 2010).
Cytokine assay
The mouse Inflammation Array C1 was purchased from RayBiotech. Mice were perfused with PBS. The hippocampal samples were lysed in RIPA buffer in the same way as for western-blotting. The protein quantification was performed by Pierce BCA assay, following manufacturer’s instructions. The arrays were incubated with the hippocampal lysate over night at 4°C, then incubated with biotinylated secondary antibodies and HRP. The bioluminescence emitted by the arrays were acquired by the Image Quant LAS 4000 mini image system (General-Electrics Healthcare) and dot quantification was performed by measuring the optical density of each dot using the oval tool in ImageJ. The same area was used to quantify all the arrays. The data were analyzed following manufacturer’s instructions. In brief, the arrays were analyzed in pairs where one of the arrays was used as reference (array X below) and the other was expressed as fold change (array Y below). For each dot, the following formula was used: X(Ny) = X(y)∗P1/P(y), where P1 is the mean signal for the positive control on the reference array (here called array X), P(y) is the mean signal for the positive control on the other array of the pair (here called array Y), X(y) is the raw quantification of a dot being analyzed and X(Ny) is the normalized quantification of the same dot. X(Ny) was then divided by the same dot’s signal on array X giving a fold change quantification for each single dot.
Mass-Spectrometry Based Proteomics
Sample preparation
Samples of hippocampi Dp(16) and WT littermates were lysed by FASP protocol (Wiśniewski et al., 2009) using the FASP Protein Digestion Kit.Briefly, the samples were mixed with 0.3ml of 8M Urea in 0.1M Tris/HCl pH 8.5 (UA solution), loaded into the filtration devices, alkylated in 0.1ml of 50mM iodoacetamide in UA solution for 1 h in darkness at RT. The samples were digested using sequentially 1 μg of LysC and 1μg of Trypsin in 50 mM NaHCO3 solution at 37°C overnight. The obtained peptides were collected by centrifugation of the filter units for 10 min and the filter devices were rinsed with two 40 μl washes 50 mM NaHCO3 and 50μl 0.5M NaCl to eliminate the hydrophobic interactions.
NanoLC and mass spectrometer setup
The prepared samples were loaded directly into the separation column and the peptides were eluted with a non-linear gradient of 5%–65% solution B (80% CAN and 20% H2O, 5% DMSO, 0.1% FA) in 180min at a flow rate of 250nl/min. The peptide separations were carried out at 55°C by a 75-μm ID × 50cm 2 μm, 100 Å C18 column mounted in the thermostatic column compartment of the machine. Eluting peptides were electrosprayed and analyzed by tandem mass spectrometry on an Orbitrap Fusion Tribrid (Thermo Fischer Scientific) operating in positive ionization mode. The precursors were ionized using an EASY-spray source held at +2.2kV and the inlet capillary temperature was held at 300°C. Single MS survey scans were performed in the Orbitrap, recording a mass window between 375 –1500 m/z with an AGC target of 250,000, at maximum injection time of 50ms, and a resolution of 120,000 at 200 m/z. Monoisotopic precursor selection was enabled for peptide isotopic distributions, precursors of z = 2-5 were selected for 2 s of cycle time, and dynamic exclusion was set to 45 s with a ± 10 ppm window set around the precursor. HCD (High-energy Collisional Dissociation) was performed with a target value of 10,000 ions in the linear ion trap, a maximal ion injection time of 45ms, normalized collision energy of 30%, a Q-value of 0.25 and the possibility to inject ions for all available parallelizable time was enabled.
Proteomic data analysis
The raw data were processed with MaxQuant software (Cox and Mann, 2008). The database used by the software was the Mouse database (release 2016_02). For the search trypsin allowing for cleavage N-terminal to proline was chosen as enzyme specificity. A contaminants database by the Andromeda search engine (Cox et al., 2011), with cysteine carbamidomethylation as fixed modification and acetylation protein N-terminal methionine oxidation and deamidation (N, Q) as variable modifications, were selected. For the identification of proteins, a false discovery rate (FDR) of 0.01 was requested. For peptide identification, peptides and peptide-spectrum match (PSM) and a minimum length of 6 amino acids were requested. Quantification in MaxQuant was performed using the built-in label-free quantification algorithm (Luber et al., 2010), enabling the “Match Between Runs” (Nagaraj et al., 2012). All proteins and peptides matching to the reversed database were filtered out. Label-free protein quantitation (LFQ) was performed with a minimum ratio count of 1 (Cox et al., 2014).
Proteomic bioinformatic analysis
All bioinformatics analyses were performed with the Perseus software of the MaxQuant computational platform. Protein groups were filtered to require 100% valid values in at least one experimental group. The label-free intensities were expressed as base log2 and empty values were imputed with random numbers from a normal distribution for each column, to best simulate low abundance values close to the noise level. For each sample, a Student’s t test with permutation-based FDR statistics was run. 250 permutations were performed, with an s0 of 0.1 and required an FDR of 0.05.
Targeted Proteomics
Targeted Peptides for drebrin protein were selected based on the data-dependent analysis (DDA), experimental results and on the theoretical scores obtained by the Picky software (Zauber et al., 2018). Peptide samples were prepared using the same FASP protocol used for the discovery study and analyzed with an Orbitrap Fusion Tribrid mass spectrometer (Thermo Scientific) coupled to a Thermo/Dionex Ultimate 3000 Rapid Separation UPLC system and EasySpray nano source. Samples were separated on an EasySpray PepMap RSLC, C18, 2 μm particle, 75 μm x 50 cm column at a 250 nl/min flow rate. Solvent A was water and solvent B was a solution of acetonitrile, water and DMSO (80/20 at 5%, final v/v), each containing 0.1% (v/v) formic acid. After loading at 2% B for 5 min, peptides were separated using a 100-min gradient from 7%–30% B and a 20-min gradient from 30%–50% B, 5-min at 80% B, followed by a 20 min re-equilibration at 2% B. Peptides were analyzed using the targeted MS2 mode of the Xcalibur software in which the doubly or triply charged precursor ion corresponding to each peptide was isolated in the quadrupole, fragmented by HCD, and full m/z 300-1200 scans of fragment ions at 30,000 resolution collected in the Orbitrap. Targeted MS2 parameters included an isolation width of 1.8 m/z for each precursor of interest, collision energy of 28%, AGC target of 5 × 10e4, maximum ion injection time of 64 ms, spray voltage of 2200 V, and ion transfer temperature of 300°C. A number of 10 precursors were targeted in each run and no scheduling was used. We used the software package Skyline (MacLean et al., 2010) to generate precursor isolation lists for all peptides of interest and export them into the Orbitrap. Skyline was used to analyze targeted MS/MS data. Chromatographic and spectral data from RAW files were loaded into Skyline and manually analyzed to identify fragment ion peaks corresponding to each peptide. RAW files were also processed using MaxQuant software in order to match MS/MS spectra to the drebrin proteotype sequence peptide. Peak areas for all selected precursors and transitions in Skyline were combined for quantitation. For transitions refinement, we checked if all transitions were of good quality and reproducible over the samples. If certain transitions were of low quality (low intensity, high CV (%)) or irreproducible over runs, we removed them from the analysis.
Quantification and Statistical Analysis
All behavioral, electrophysiological and biochemical analyses were performed blinded by the operator. For statistical analysis, the Graph Pad Prism 7 software (GraphPad Software, USA) was used. The statistical analysis was performed by Student’s t test (comparisons between two-groups), one-way analysis of variance (ANOVA) followed by Holm-Sidak post hoc test (comparisons among three or more groups) or ordinary and/or repeated-measure two-way analysis of variance (ANOVA) followed by Holm-Sidak post hoc test (comparisons among four or more groups with two different variables (i.e., Strain and Treatment). For heteroskedastic datasets and/or for datasets of non-normal distribution, Mann-Whitney test comparison (comparisons between two-groups) or Kruskal-Wallis test followed by Dunn’s post hoc test (for comparison between three or more groups) were used. For datasets in Figures S3B–S3D; independent One-Sample t test against 0 for each cytokine was used. Outliers were excluded only from the final pool of data by a Grubb’s test run iteratively until no outliers were found. In all cases, where the datasets from vehicle-treated animals were reused for comparison in different graphs, the statistical significance depicted in the graphs derives from one single two-way ANOVA including all groups across all figures. The T or F statistics reported in the figure legends derive from statistical tests including only the groups shown in each individual graph.
Acknowledgments
We thank Silvia Ghezzi for inspiring the work. We acknowledge the animal facility staff, M.M. Chellali for experimental help, and The Spanish Brain Biobank and NeuroBioBank (USA) for human samples. This project has received funding from the European Research Council (ERC) (grant 725563) and partial support from Telethon-Italy (grant TCP15021).
Author Contributions
B.P., G.M., L.E.P., and L.C. designed the experiments and wrote the manuscript. B.P. and G.M. performed most of the experiments. A.S. performed behavior on Ts65Dn mice. A.F. performed western blotting. M.R. and A.C. performed the RNA-seq and qRT-PCR. E.V. performed human sample histology. M.B. and A.P. performed the proteomic analysis. T.C. prepared the electron microscopy samples. All of the authors revised the manuscript.
Declaration of Interests
L.C. and A.C. are co-inventors on patent application IT 102019000004929.
Published: October 6, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.neuron.2020.09.010.
Supplemental Information
Brown, dark-red and orange cluster in Figures 6C–6E.
Sample 5227 was identified as outlier by Grubb’s test.
Highlighted in red are immunological pathways common among Our Study, Lockstone et al. (2007), and Olmos-Serrano (2016). Triplicated genes in Dp(16) mice that play a role in the function of the immune system (red) modified from Li et al. (2007) in accordance with our study.
References
- Ajmone-Cat M.A., Bernardo A., Greco A., Minghetti L. Non-steroidal anti-inflammatory drugs and brain inflammation: effects on microglial functions. Pharmaceuticals (Basel) 2010;3:1949–1965. doi: 10.3390/ph3061949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander M., Petri H., Ding Y., Wandel C., Khwaja O., Foskett N. Morbidity and medication in a large population of individuals with Down syndrome compared to the general population. Dev. Med. Child Neurol. 2016;58:246–254. doi: 10.1111/dmcn.12868. [DOI] [PubMed] [Google Scholar]
- Anders S., Pyl P.T., Huber W. HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arcuri C., Mecca C., Bianchi R., Giambanco I., Donato R. The pathophysiological role of microglia in dynamic surveillance, phagocytosis and structural remodeling of the developing CNS. Front. Mol. Neurosci. 2017;10:191. doi: 10.3389/fnmol.2017.00191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arganda-Carreras I., Fernández-González R., Muñoz-Barrutia A., Ortiz-De-Solorzano C. 3D reconstruction of histological sections: application to mammary gland tissue. Microsc. Res. Tech. 2010;73:1019–1029. doi: 10.1002/jemt.20829. [DOI] [PubMed] [Google Scholar]
- Bachmanov A.A., Reed D.R., Beauchamp G.K., Tordoff M.G. Food intake, water intake, and drinking spout side preference of 28 mouse strains. Behav. Genet. 2002;32:435–443. doi: 10.1023/a:1020884312053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belichenko N.P., Belichenko P.V., Kleschevnikov A.M., Salehi A., Reeves R.H., Mobley W.C. The “Down syndrome critical region” is sufficient in the mouse model to confer behavioral, neurophysiological, and synaptic phenotypes characteristic of Down syndrome. J. Neurosci. 2009;29:5938–5948. doi: 10.1523/JNEUROSCI.1547-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellesi M., de Vivo L., Chini M., Gilli F., Tononi G., Cirelli C. Sleep loss promotes astrocytic phagocytosis and microglial activation in mouse cerebral cortex. J. Neurosci. 2017;37:5263–5273. doi: 10.1523/JNEUROSCI.3981-16.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett M.L., Bennett F.C., Liddelow S.A., Ajami B., Zamanian J.L., Fernhoff N.B., Mulinyawe S.B., Bohlen C.J., Adil A., Tucker A. New tools for studying microglia in the mouse and human CNS. Proc. Natl. Acad. Sci. USA. 2016;113:E1738–E1746. doi: 10.1073/pnas.1525528113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhagwat N., Koppikar P., Keller M., Marubayashi S., Shank K., Rampal R., Qi J., Kleppe M., Patel H.J., Shah S.K. Improved targeting of JAK2 leads to increased therapeutic efficacy in myeloproliferative neoplasms. Blood. 2014;123:2075–2083. doi: 10.1182/blood-2014-01-547760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatia H.S., Roelofs N., Muñoz E., Fiebich B.L. Alleviation of microglial activation induced by p38 MAPK/MK2/PGE2 axis by capsaicin: potential involvement of other than TRPV1 mechanism/s. Sci. Rep. 2017;7:116. doi: 10.1038/s41598-017-00225-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blank T., Prinz M. Microglia as modulators of cognition and neuropsychiatric disorders. Glia. 2013;61:62–70. doi: 10.1002/glia.22372. [DOI] [PubMed] [Google Scholar]
- Blecharz-Klin K., Wawer A., Jawna-Zboińska K., Pyrzanowska J., Piechal A., Mirowska-Guzel D., Widy-Tyszkiewicz E. Early paracetamol exposure decreases brain-derived neurotrophic factor (BDNF) in striatum and affects social behaviour and exploration in rats. Pharmacol. Biochem. Behav. 2018;168:25–32. doi: 10.1016/j.pbb.2018.03.004. [DOI] [PubMed] [Google Scholar]
- Bosco A., Steele M.R., Vetter M.L. Early microglia activation in a mouse model of chronic glaucoma. J. Comp. Neurol. 2011;519:599–620. doi: 10.1002/cne.22516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broers C.J., Gemke R.J., Morré S.A., Weijerman M.E., van Furth A.M. Increased production of interleukin-10 in children with Down syndrome upon ex vivo stimulation with Streptococcus pneumoniae. Pediatr. Res. 2014;75:109–113. doi: 10.1038/pr.2013.173. [DOI] [PubMed] [Google Scholar]
- Carrillo-de Sauvage M.A., Maatouk L., Arnoux I., Pasco M., Sanz Diez A., Delahaye M., Herrero M.T., Newman T.A., Calvo C.F., Audinat E. Potent and multiple regulatory actions of microglial glucocorticoid receptors during CNS inflammation. Cell Death Differ. 2013;20:1546–1557. doi: 10.1038/cdd.2013.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christiansen G.B., Andersen K.H., Riis S., Nykjaer A., Bolcho U., Jensen M.S., Holm M.M. The sorting receptor SorCS3 is a stronger regulator of glutamate receptor functions compared to GABAergic mechanisms in the hippocampus. Hippocampus. 2017;27:235–248. doi: 10.1002/hipo.22689. [DOI] [PubMed] [Google Scholar]
- Chung S., Jung W., Lee M.Y. Inward and outward rectifying potassium currents set membrane potentials in activated rat microglia. Neurosci. Lett. 1999;262:121–124. doi: 10.1016/s0304-3940(99)00053-1. [DOI] [PubMed] [Google Scholar]
- Colacurcio D.J., Pensalfini A., Jiang Y., Nixon R.A. Dysfunction of autophagy and endosomal-lysosomal pathways: roles in pathogenesis of Down syndrome and Alzheimer’s Disease. Free Radic. Biol. Med. 2018;114:40–51. doi: 10.1016/j.freeradbiomed.2017.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowan M., Petri W.A., Jr. Microglia: immune regulators of neurodevelopment. Front. Immunol. 2018;9:2576. doi: 10.3389/fimmu.2018.02576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J., Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008;26:1367–1372. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- Cox J., Neuhauser N., Michalski A., Scheltema R.A., Olsen J.V., Mann M. Andromeda: a peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 2011;10:1794–1805. doi: 10.1021/pr101065j. [DOI] [PubMed] [Google Scholar]
- Cox J., Hein M.Y., Luber C.A., Paron I., Nagaraj N., Mann M. Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol. Cell. Proteomics. 2014;13:2513–2526. doi: 10.1074/mcp.M113.031591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deidda G., Parrini M., Naskar S., Bozarth I.F., Contestabile A., Cancedda L. Reversing excitatory GABAAR signaling restores synaptic plasticity and memory in a mouse model of Down syndrome. Nat. Med. 2015;21:318–326. doi: 10.1038/nm.3827. [DOI] [PubMed] [Google Scholar]
- Dekker A.D., Fortea J., Blesa R., De Deyn P.P. Cerebrospinal fluid biomarkers for Alzheimer’s disease in Down syndrome. Alzheimers Dement. (Amst.) 2017;8:1–10. doi: 10.1016/j.dadm.2017.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delpech J.C., Wei L., Hao J., Yu X., Madore C., Butovsky O., Kaffman A. Early life stress perturbs the maturation of microglia in the developing hippocampus. Brain Behav. Immun. 2016;57:79–93. doi: 10.1016/j.bbi.2016.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dierssen M. Down syndrome: the brain in trisomic mode. Nat. Rev. Neurosci. 2012;13:844–858. doi: 10.1038/nrn3314. [DOI] [PubMed] [Google Scholar]
- Elmore M.R., Najafi A.R., Koike M.A., Dagher N.N., Spangenberg E.E., Rice R.A., Kitazawa M., Matusow B., Nguyen H., West B.L., Green K.N. Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron. 2014;82:380–397. doi: 10.1016/j.neuron.2014.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farnsworth A., Flaman A.S., Prasad S.S., Gravel C., Williams A., Yauk C.L., Li X. Acetaminophen modulates the transcriptional response to recombinant interferon-beta. PLoS ONE. 2010;5:e11031. doi: 10.1371/journal.pone.0011031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira T.A., Blackman A.V., Oyrer J., Jayabal S., Chung A.J., Watt A.J., Sjöström P.J., van Meyel D.J. Neuronal morphometry directly from bitmap images. Nat. Methods. 2014;11:982–984. doi: 10.1038/nmeth.3125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gally F., Rao D.M., Schmitz C., Colvin K.L., Yeager M.E., Perraud A.L. The TRPM2 ion channel contributes to cytokine hyperproduction in a mouse model of Down Syndrome. Biochim. Biophys. Acta Mol. Basis Dis. 2018;1864:126–132. doi: 10.1016/j.bbadis.2017.09.025. [DOI] [PubMed] [Google Scholar]
- Ginhoux F., Lim S., Hoeffel G., Low D., Huber T. Origin and differentiation of microglia. Front. Cell. Neurosci. 2013;7:45. doi: 10.3389/fncel.2013.00045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Good P. Evidence the U.S. autism epidemic initiated by acetaminophen (Tylenol) is aggravated by oral antibiotic amoxicillin/clavulanate (Augmentin) and now exponentially by herbicide glyphosate (Roundup) Clin. Nutr. ESPEN. 2018;23:171–183. doi: 10.1016/j.clnesp.2017.10.005. [DOI] [PubMed] [Google Scholar]
- Goodliffe J.W., Olmos-Serrano J.L., Aziz N.M., Pennings J.L., Guedj F., Bianchi D.W., Haydar T.F. Absence of prenatal forebrain defects in the Dp(16)1Yey/+ mouse model of Down syndrome. J. Neurosci. 2016;36:2926–2944. doi: 10.1523/JNEUROSCI.2513-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guedj F., Pennings J.L., Ferres M.A., Graham L.C., Wick H.C., Miczek K.A., Slonim D.K., Bianchi D.W. The fetal brain transcriptome and neonatal behavioral phenotype in the Ts1Cje mouse model of Down syndrome. Am. J. Med. Genet. A. 2015;167A:1993–2008. doi: 10.1002/ajmg.a.37156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas M.A., Bell D., Slender A., Lana-Elola E., Watson-Scales S., Fisher E.M., Tybulewicz V.L., Guillemot F. Alterations to dendritic spine morphology, but not dendrite patterning, of cortical projection neurons in Tc1 and Ts1Rhr mouse models of Down syndrome. PLoS ONE. 2013;8:e78561. doi: 10.1371/journal.pone.0078561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanisch U.K., Kettenmann H. Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat. Neurosci. 2007;10:1387–1394. doi: 10.1038/nn1997. [DOI] [PubMed] [Google Scholar]
- Hebert-Chatelain E., Reguero L., Puente N., Lutz B., Chaouloff F., Rossignol R., Piazza P.V., Benard G., Grandes P., Marsicano G. Cannabinoid control of brain bioenergetics: Exploring the subcellular localization of the CB1 receptor. Mol. Metab. 2014;3:495–504. doi: 10.1016/j.molmet.2014.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herring B.E., Nicoll R.A. Kalirin and Trio proteins serve critical roles in excitatory synaptic transmission and LTP. Proc. Natl. Acad. Sci. USA. 2016;113:2264–2269. doi: 10.1073/pnas.1600179113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Illouz T., Madar R., Biragyn A., Okun E. Restoring microglial and astroglial homeostasis using DNA immunization in a Down syndrome mouse model. Brain Behav. Immun. 2019;75:163–180. doi: 10.1016/j.bbi.2018.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishida T., Sato T., Irifune M., Tanaka K., Nakamura N., Nishikawa T. Effect of acetaminophen, a cyclooxygenase inhibitor, on Morris water maze task performance in mice. J. Psychopharmacol. (Oxford) 2007;21:757–767. doi: 10.1177/0269881107076369. [DOI] [PubMed] [Google Scholar]
- Kaur C., Rathnasamy G., Ling E.A. Biology of microglia in the developing brain. J. Neuropathol. Exp. Neurol. 2017;76:736–753. doi: 10.1093/jnen/nlx056. [DOI] [PubMed] [Google Scholar]
- Kent W.J., Sugnet C.W., Furey T.S., Roskin K.M., Pringle T.H., Zahler A.M., Haussler D. The human genome browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerrisk M.E., Greer C.A., Koleske A.J. Integrin α3 is required for late postnatal stability of dendrite arbors, dendritic spines and synapses, and mouse behavior. J. Neurosci. 2013;33:6742–6752. doi: 10.1523/JNEUROSCI.0528-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kierdorf K., Prinz M. Factors regulating microglia activation. Front. Cell. Neurosci. 2013;7:44. doi: 10.3389/fncel.2013.00044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulak N.A., Pichler G., Paron I., Nagaraj N., Mann M. Minimal, encapsulated proteomic-sample processing applied to copy-number estimation in eukaryotic cells. Nat. Methods. 2014;11:319–324. doi: 10.1038/nmeth.2834. [DOI] [PubMed] [Google Scholar]
- Langfelder P., Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee T.C., Kashyap R.L., Chu C.N. Building skeleton models via 3-D medial surface axis thinning algorithms. CVGIP Graph. Models Image Process. 1994;56:462–478. [Google Scholar]
- Li Z., Yu T., Morishima M., Pao A., LaDuca J., Conroy J., Nowak N., Matsui S., Shiraishi I., Yu Y.E. Duplication of the entire 22.9 Mb human chromosome 21 syntenic region on mouse chromosome 16 causes cardiovascular and gastrointestinal abnormalities. Hum. Mol. Genet. 2007;16:1359–1366. doi: 10.1093/hmg/ddm086. [DOI] [PubMed] [Google Scholar]
- Liu Y., Given K.S., Dickson E.L., Owens G.P., Macklin W.B., Bennett J.L. Concentration-dependent effects of CSF1R inhibitors on oligodendrocyte progenitor cells ex vivo and in vivo. Exp. Neurol. 2019;318:32–41. doi: 10.1016/j.expneurol.2019.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lockstone H.E., Harris L.W., Swatton J.E., Wayland M.T., Holland A.J., Bahn S. Gene expression profiling in the adult Down syndrome brain. Genomics. 2007;90:647–660. doi: 10.1016/j.ygeno.2007.08.005. [DOI] [PubMed] [Google Scholar]
- Luber C.A., Cox J., Lauterbach H., Fancke B., Selbach M., Tschopp J., Akira S., Wiegand M., Hochrein H., O’Keeffe M., Mann M. Quantitative proteomics reveals subset-specific viral recognition in dendritic cells. Immunity. 2010;32:279–289. doi: 10.1016/j.immuni.2010.01.013. [DOI] [PubMed] [Google Scholar]
- MacLean B., Tomazela D.M., Shulman N., Chambers M., Finney G.L., Frewen B., Kern R., Tabb D.L., Liebler D.C., MacCoss M.J. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics. 2010;26:966–968. doi: 10.1093/bioinformatics/btq054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majewska A., Sur M. Motility of dendritic spines in visual cortex in vivo: changes during the critical period and effects of visual deprivation. Proc. Natl. Acad. Sci. USA. 2003;100:16024–16029. doi: 10.1073/pnas.2636949100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuda T., Sankowski R., Staszewski O., Böttcher C., Amann L., Sagar, Scheiwe C., Nessler S., Kunz P., van Loo G. Spatial and temporal heterogeneity of mouse and human microglia at single-cell resolution. Nature. 2019;566:388–392. doi: 10.1038/s41586-019-0924-x. [DOI] [PubMed] [Google Scholar]
- Minelli A., Castaldo P., Gobbi P., Salucci S., Magi S., Amoroso S. Cellular and subcellular localization of Na+-Ca2+ exchanger protein isoforms, NCX1, NCX2, and NCX3 in cerebral cortex and hippocampus of adult rat. Cell Calcium. 2007;41:221–234. doi: 10.1016/j.ceca.2006.06.004. [DOI] [PubMed] [Google Scholar]
- Mrak R.E., Griffin W.S. Trisomy 21 and the brain. J. Neuropathol. Exp. Neurol. 2004;63:679–685. doi: 10.1093/jnen/63.7.679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulet M., Blasco-Ibáńez J.M., Crespo C., Nácher J., Varea E. Early increased density of cyclooxygenase-2 (COX-2) immunoreactive neurons in Down syndrome. Folia Neuropathol. 2017;55:154–160. doi: 10.5114/fn.2017.68582. [DOI] [PubMed] [Google Scholar]
- Nagaraj N., Kulak N.A., Cox J., Neuhauser N., Mayr K., Hoerning O., Vorm O., Mann M. System-wide perturbation analysis with nearly complete coverage of the yeast proteome by single-shot ultra HPLC runs on a bench top Orbitrap. Mol. Cell Proteomics. 2012;11 doi: 10.1074/mcp.M111.013722. M111.013722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson I., Lindfors C., Fetissov S.O., Hökfelt T., Johansen J.E. Aberrant agouti-related protein system in the hypothalamus of the anx/anx mouse is associated with activation of microglia. J. Comp. Neurol. 2008;507:1128–1140. doi: 10.1002/cne.21599. [DOI] [PubMed] [Google Scholar]
- Olmos-Serrano J.L., Kang H.J., Tyler W.A., Silbereis J.C., Cheng F., Zhu Y., Pletikos M., Jankovic-Rapan L., Cramer N.P., Galdzicki Z. Down syndrome developmental brain transcriptome reveals defective oligodendrocyte differentiation and myelination. Neuron. 2016;89:1208–1222. doi: 10.1016/j.neuron.2016.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papageorgiou I.E., Lewen A., Galow L.V., Cesetti T., Scheffel J., Regen T., Hanisch U.K., Kann O. TLR4-activated microglia require IFN-γ to induce severe neuronal dysfunction and death in situ. Proc. Natl. Acad. Sci. USA. 2016;113:212–217. doi: 10.1073/pnas.1513853113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrini M., Ghezzi D., Deidda G., Medrihan L., Castroflorio E., Alberti M., Baldelli P., Cancedda L., Contestabile A. Aerobic exercise and a BDNF-mimetic therapy rescue learning and memory in a mouse model of Down syndrome. Sci. Rep. 2017;7:16825. doi: 10.1038/s41598-017-17201-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips M., Pozzo-Miller L. Dendritic spine dysgenesis in autism related disorders. Neurosci. Lett. 2015;601:30–40. doi: 10.1016/j.neulet.2015.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pozzi D., Lignani G., Ferrea E., Contestabile A., Paonessa F., D’Alessandro R., Lippiello P., Boido D., Fassio A., Meldolesi J. REST/NRSF-mediated intrinsic homeostasis protects neuronal networks from hyperexcitability. EMBO J. 2013;32:2994–3007. doi: 10.1038/emboj.2013.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin H., Buckley J.A., Li X., Liu Y., Fox T.H., 3rd, Meares G.P., Yu H., Yan Z., Harms A.S., Li Y. Inhibition of the JAK/STAT pathway protects against α-synuclein-induced neuroinflammation and dopaminergic neurodegeneration. J. Neurosci. 2016;36:5144–5159. doi: 10.1523/JNEUROSCI.4658-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao J.S., Kellom M., Kim H.W., Rapoport S.I., Reese E.A. Neuroinflammation and synaptic loss. Neurochem. Res. 2012;37:903–910. doi: 10.1007/s11064-012-0708-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risher W.C., Ustunkaya T., Singh Alvarado J., Eroglu C. Rapid Golgi analysis method for efficient and unbiased classification of dendritic spines. PLoS ONE. 2014;9:e107591. doi: 10.1371/journal.pone.0107591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson M.D., Oshlack A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010;11:R25. doi: 10.1186/gb-2010-11-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothhammer V., Borucki D.M., Tjon E.C., Takenaka M.C., Chao C.C., Ardura-Fabregat A., de Lima K.A., Gutiérrez-Vázquez C., Hewson P., Staszewski O. Microglial control of astrocytes in response to microbial metabolites. Nature. 2018;557:724–728. doi: 10.1038/s41586-018-0119-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolph J.D., Cox J. A network module for the Perseus software for computational proteomics facilitates proteome interaction graph analysis. J. Proteome Res. 2019;18:2052–2064. doi: 10.1021/acs.jproteome.8b00927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salter M.W., Stevens B. Microglia emerge as central players in brain disease. Nat. Med. 2017;23:1018–1027. doi: 10.1038/nm.4397. [DOI] [PubMed] [Google Scholar]
- Savardi A., Borgogno M., Narducci R., La Sala G., Ortega J.A., Summa M., Armirotti A., Bertorelli R., Contestabile A., De Vivo M. Discovery of a small molecule drug candidate for selective NKCC1 inhibition in brain disorders. Chem. 2020;6:2073–2096. doi: 10.1016/j.chempr.2020.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma C.V., Long J.H., Shah S., Rahman J., Perrett D., Ayoub S.S., Mehta V. First evidence of the conversion of paracetamol to AM404 in human cerebrospinal fluid. J. Pain Res. 2017;10:2703–2709. doi: 10.2147/JPR.S143500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shim K.S., Lubec G. Drebrin, a dendritic spine protein, is manifold decreased in brains of patients with Alzheimer’s disease and Down syndrome. Neurosci. Lett. 2002;324:209–212. doi: 10.1016/s0304-3940(02)00210-0. [DOI] [PubMed] [Google Scholar]
- Shobin E., Bowley M.P., Estrada L.I., Heyworth N.C., Orczykowski M.E., Eldridge S.A., Calderazzo S.M., Mortazavi F., Moore T.L., Rosene D.L. Microglia activation and phagocytosis: relationship with aging and cognitive impairment in the rhesus monkey. Geroscience. 2017;39:199–220. doi: 10.1007/s11357-017-9965-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sipe G.O., Lowery R.L., Tremblay M.E., Kelly E.A., Lamantia C.E., Majewska A.K. Microglial P2Y12 is necessary for synaptic plasticity in mouse visual cortex. Nat. Commun. 2016;7:10905. doi: 10.1038/ncomms10905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skene N.G., Grant S.G. Identification of vulnerable cell types in major brain disorders using single cell transcriptomes and expression weighted cell type enrichment. Front. Neurosci. 2016;10:16. doi: 10.3389/fnins.2016.00016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stampanoni Bassi M., Gentile A., Iezzi E., Zagaglia S., Musella A., Simonelli I., Gilio L., Furlan R., Finardi A., Marfia G.A. Transient receptor potential vanilloid 1 modulates central inflammation in multiple sclerosis. Front. Neurol. 2019;10:30. doi: 10.3389/fneur.2019.00030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streit W.J., Xue Q.S., Tischer J., Bechmann I. Microglial pathology. Acta Neuropathol. Commun. 2014;2:142. doi: 10.1186/s40478-014-0142-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strittmatter S.M. Overcoming drug development bottlenecks with repurposing: old drugs learn new tricks. Nat. Med. 2014;20:590–591. doi: 10.1038/nm.3595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan K.D., Lewis H.C., Hill A.A., Pandey A., Jackson L.P., Cabral J.M., Smith K.P., Liggett L.A., Gomez E.B., Galbraith M.D. Trisomy 21 consistently activates the interferon response. eLife. 2016;5:e16220. doi: 10.7554/eLife.16220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tchessalova D., Posillico C.K., Tronson N.C. Neuroimmune activation drives multiple brain states. Front. Syst. Neurosci. 2018;12:39. doi: 10.3389/fnsys.2018.00039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremblay M.E., Stevens B., Sierra A., Wake H., Bessis A., Nimmerjahn A. The role of microglia in the healthy brain. J. Neurosci. 2011;31:16064–16069. doi: 10.1523/JNEUROSCI.4158-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyanova S., Temu T., Sinitcyn P., Carlson A., Hein M.Y., Geiger T., Mann M., Cox J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods. 2016;13:731–740. doi: 10.1038/nmeth.3901. [DOI] [PubMed] [Google Scholar]
- Vandesompele J., De Preter K., Pattyn F., Poppe B., Van Roy N., De Paepe A., Speleman F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002;3 doi: 10.1186/gb-2002-3-7-research0034. research0034.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weitzdoerfer R., Dierssen M., Fountoulakis M., Lubec G. Fetal life in Down syndrome starts with normal neuronal density but impaired dendritic spines and synaptosomal structure. J. Neural Transm. Suppl. 2001:59–70. doi: 10.1007/978-3-7091-6262-0_5. [DOI] [PubMed] [Google Scholar]
- Wierzba-Bobrowicz T., Lewandowska E., Schmidt-Sidor B., Gwiazda E. The comparison of microglia maturation in CNS of normal human fetuses and fetuses with Down’s syndrome. Folia Neuropathol. 1999;37:227–234. [PubMed] [Google Scholar]
- Wilcock D.M., Griffin W.S. Down’s syndrome, neuroinflammation, and Alzheimer neuropathogenesis. J. Neuroinflammation. 2013;10:84. doi: 10.1186/1742-2094-10-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiśniewski J.R., Zougman A., Mann M. Combination of FASP and StageTip-based fractionation allows in-depth analysis of the hippocampal membrane proteome. J. Proteome Res. 2009;8:5674–5678. doi: 10.1021/pr900748n. [DOI] [PubMed] [Google Scholar]
- Wu Y., Dissing-Olesen L., MacVicar B.A., Stevens B. Microglia: dynamic mediators of synapse development and plasticity. Trends Immunol. 2015;36:605–613. doi: 10.1016/j.it.2015.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu B., Sun A., He Y., Qian F., Xi S., Long D., Chen Y. Loss of thin spines and small synapses contributes to defective hippocampal function in aged mice. Neurobiol. Aging. 2018;71:91–104. doi: 10.1016/j.neurobiolaging.2018.07.010. [DOI] [PubMed] [Google Scholar]
- Yu G., Wang L.-G., Han Y., He Q.-Y. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16:284–287. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zauber H., Kirchner M., Selbach M. Picky: a simple online PRM and SRM method designer for targeted proteomics. Nat. Methods. 2018;15:156–157. doi: 10.1038/nmeth.4607. [DOI] [PubMed] [Google Scholar]
- Zeisel A., Muñoz-Manchado A.B., Codeluppi S., Lönnerberg P., La Manno G., Juréus A., Marques S., Munguba H., He L., Betsholtz C. Brain structure. Cell types in the mouse cortex and hippocampus revealed by single-cell RNA-seq. Science. 2015;347:1138–1142. doi: 10.1126/science.aaa1934. [DOI] [PubMed] [Google Scholar]
- Zhang D., Hu X., Qian L., Wilson B., Lee C., Flood P., Langenbach R., Hong J.S. Prostaglandin E2 released from activated microglia enhances astrocyte proliferation in vitro. Toxicol. Appl. Pharmacol. 2009;238:64–70. doi: 10.1016/j.taap.2009.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong B., Wang D.H. N-oleoyldopamine, a novel endogenous capsaicin-like lipid, protects the heart against ischemia-reperfusion injury via activation of TRPV1. Am. J. Physiol. Heart Circ. Physiol. 2008;295:H728–H735. doi: 10.1152/ajpheart.00022.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Brown, dark-red and orange cluster in Figures 6C–6E.
Sample 5227 was identified as outlier by Grubb’s test.
Highlighted in red are immunological pathways common among Our Study, Lockstone et al. (2007), and Olmos-Serrano (2016). Triplicated genes in Dp(16) mice that play a role in the function of the immune system (red) modified from Li et al. (2007) in accordance with our study.
Data Availability Statement
The datasets of this work are available https://data.mendeley.com/datasets/gkbrjc8rmd/draft?a=4c602482-bb3a-442a-8a90-c43e8efa9f00. Uncropped blot, qPCR information and results of cognitive performance regarding total distance traveled, total object exploration time and preference during acquisition trials are available in the same repository. The accession number for proteomic datasets are available in the PRIDE archive as PRIDE: PXD015113 for microglial cells and PRIDE: PXD015148 for hippocampal tissues and drebrin protein described in STAR Methods. The accession number forthe RNaseq datasets in this study have been deposited in NCBI’s Gene Expression Omnibus (Pinto, Morelli et al., 2020) and are accessible through GEO Series accession number GEO: GSE137939 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE137939).