


Abstract
SARS-CoV-2 is a betacoronavirus with a linear single-stranded, positive-sense RNA genome, whose outbreak caused the ongoing COVID-19 pandemic. The ability of coronaviruses to rapidly evolve, adapt, and cross species barriers makes the development of effective and durable therapeutic strategies a challenging and urgent need. As for other RNA viruses, genomic RNA structures are expected to play crucial roles in several steps of the coronavirus replication cycle. Despite this, only a handful of functionally-conserved coronavirus structural RNA elements have been identified to date. Here, we performed RNA structure probing to obtain single-base resolution secondary structure maps of the full SARS-CoV-2 coronavirus genome both in vitro and in living infected cells. Probing data recapitulate the previously described coronavirus RNA elements (5′ UTR and s2m), and reveal new structures. Of these, ∼10.2% show significant covariation among SARS-CoV-2 and other coronaviruses, hinting at their functionally-conserved role. Secondary structure-restrained 3D modeling of these segments further allowed for the identification of putative druggable pockets. In addition, we identify a set of single-stranded segments in vivo, showing high sequence conservation, suitable for the development of antisense oligonucleotide therapeutics. Collectively, our work lays the foundation for the development of innovative RNA-targeted therapeutic strategies to fight SARS-related infections.
INTRODUCTION
RNA viruses encode the information needed to take control of the host cell on two levels (1). On one hand, the linear sequence of their RNA genomes encodes all the proteins needed to take over the host cell machinery and to assemble new viral particles. On the other hand, their single-stranded RNA genome folds back on itself to form intricate secondary and tertiary structures that have been proven essential for viral replication, protein synthesis, packaging, immune system evasion and more. RNA viruses, that are responsible for numerous deadly diseases (e.g. AIDS, Hepatitis C, SARS, Dengue and Ebola), are characterized by higher mutation rates compared to DNA viruses, enabling them to rapidly evolve and adapt (2). As a consequence of their high mutation rates, RNA viruses can rapidly develop resistance towards drugs and vaccines by slightly altering their core proteins (3). In contrast, certain RNA structures formed in the context of viral RNA genomes are well conserved (1), in spite of changes in the underlying encoded amino acid sequence, making them valuable therapeutic targets.
Coronaviruses (CoV) are positive-sense, single-stranded RNA viruses, members of the Coronaviridae family (4). This family consists of four genera, of which two (alpha and beta) can only infect mammals, while the other two (gamma and delta) mostly infect birds, although some of them can also infect mammals. These viruses were not anticipated to be highly pathogenic in humans until the outbreak of the severe acute respiratory syndrome coronavirus (SARS-CoV) in 2002 (5). Since then, two other major outbreaks of coronaviruses occurred, one by the Middle East respiratory syndrome coronavirus (MERS-CoV) in 2012 and recently by the new SARS-related coronavirus SARS-CoV-2 (also known as 2019-nCoV) at the end of 2019 (6). The latter, still ongoing at the time of writing this article, rapidly resulted in a pandemic, with (to date, October 2020) >37 million people infected and over 1 million deaths across the world. The ability to evolve inside different reservoirs and to cross species barriers, infecting humans with high morbidity and mortality, makes this genus a recurrent potential threat for worldwide public health. Indeed, certain SARS-CoV-like viruses from bats have been previously shown to be able to infect human cells without the need for any prior adaptation; thus, suggesting that SARS-related outbreaks can potentially re-emerge at any time (5).
In light of these considerations, identifying new therapeutically-relevant and durable druggable targets for the treatment of coronavirus infections constitutes a key and highly-timely need. In this perspective, RNA structural elements represent attractive targets for drug discovery. Indeed, inhibition of viral replication by RNA-targeting small molecule drugs has already been proven to be feasible for other RNA viruses, such as the human immunodeficiency virus (HIV), hepatitis C virus (HCV), SARS-CoV and influenza A virus (7) (IAV). Additionally, the identification of highly-conserved weakly structured regions within viral RNA genomes might aid the design of oligonucleotide-bases antiviral therapeutics (8).
Coronaviruses bear the largest genomes among RNA viruses, ranging from ∼26–32 kb. A handful of functional cis-regulatory RNA structural elements have been previously identified by phylogenetic analyses and these include structures in the 5′ and 3′ untranslated regions (UTRs) and the ribosomal frameshifting element (9–11) (FSE). In the 5′ UTR of most betacoronaviruses (Beta-CoV), several stem-loops (SL1–5) involved in mediating viral replication have been identified. The ORF1a-ORF1b boundary hosts the FSE (12). This pseudoknotted structure is pivotal for the programmed ribosomal frameshifting, that allows the translation of the ORF1ab polyprotein. A second putative pseudoknot has been proposed to be located in the 3′ UTR (13,14), that includes an extremely conserved octanucleotide within the hypervariable region (15) (HVR), and the stem-loop-like 2 motif (s2m) (16).
Sharing over 79.6% sequence identity to the genome of SARS-CoV (6), SARS-CoV-2 has been predicted to harbor these typical RNA structure elements (17). However, the structural complexity of SARS-CoV-2 and other Beta-CoV genomes has remained largely unexplored so far. Deep comprehension of the structural architecture of the SARS-CoV-2 RNA genome is crucial to identify new key (un)structured elements for the development of innovative therapeutic approaches.
To address this need, in this work we provide the first experimental characterization of the full-length genome of a coronavirus by SHAPE and DMS mutational profiling (SHAPE-MaP and DMS-MaPseq) analyses (18,19), using the novel SARS-CoV-2 virus as a model. After modeling the secondary structure of the entire RNA genome under both in vitro and in vivo conditions, we identified a subset of regions showing a low propensity towards folding in vivo, ideal for the design of antisense oligonucleotide (ASO) therapeutics, as well as a set of stable RNA structures, coherently folded under both in vitro and in vivo conditions, of which at least 10.2% are under selective pressure and show significant covariation. By coupling secondary structure constraints and coarse-grained 3D modeling, we then infer the 3D structure of these elements and identify the most suitable pockets for accommodating the interaction with small molecule drugs. Collectively, our work provides the cornerstone for the development of RNA-targeted therapeutic strategies to fight SARS-related infections.
MATERIALS AND METHODS
Synthesis of 2-methylnicotinic acid imidazolide (NAI)
NAI was synthesized as previously described (20). Briefly, 137.14 mg of 2-methylpyridine-3-carboxylic acid (Sigma Aldrich, cat. 325228) were resuspended in 500 μl DMSO anhydrous (Sigma Aldrich, cat. 276855). ∼162.15 mg of 1,1’-carbonyldiimidazole (Sigma Aldrich, cat. 115533) were resuspended in 500 μl DMSO anhydrous and added dropwise to the 2-methylpyridine-3-carboxylic acid solution while constantly stirring, over a period of 5 minutes. The reaction mixture was then incubated at room temperature with constant stirring for 2 h. This mixture (assumed to represent a 1 M stock) was aliquoted in 50 μl aliquots and stored at −80°C.
Cell culture and SARS-CoV-2 infection
Vero E6 cells were cultured in T-175 flasks in Dulbecco's modified Eagle's medium (DMEM; Lonza, cat. 12-604F), supplemented with 8% fetal calf serum (FCS; Bodinco), 2 mM l-glutamine, 100 U/ml of penicillin, and 100 μg/ml of streptomycin (Sigma Aldrich, cat. P4333-20ML) at 37°C in an atmosphere of 5% CO2 and 95–99% humidity. Cells were infected at an MOI of 1.5 with SARS-CoV-2/Leiden-0002 (GenBank accession: MT510999), a clinical isolate obtained from a nasopharyngeal sample at LUMC, which was passaged twice in Vero E6 cells before use. Infections were performed in Eagle's minimal essential medium (EMEM; Lonza, cat. 12-611F) supplemented with 25 mM HEPES, 2% FCS, 2 mM l-glutamine, and antibiotics. At 16 h post-infection, infected cells were harvested by trypsinization, followed by resuspension in EMEM supplemented with 2% FCS, and then washed with 50 ml 1× PBS.
All experiments with infectious SARS-CoV-2 were performed in a biosafety level 3 facility at the LUMC.
In vivo RNA probing
Infected Vero E6 cells were resuspended in 1× PBS, and NAI from a 1 M stock (in DMSO) was added at a final concentration of 100 mM. A corresponding amount of DMSO was added to the control sample. Samples were then incubated at 37°C with constant shaking for 15 min, followed by quenching of NAI through the addition of DTT at a final concentration of 0.5 M. Following centrifugation at 10 000 × g for 1 min, the supernatant was discarded and cell pellets were resuspended in 2 mL ice-cold TriPure Isolation Reagent (Sigma Aldrich, cat. 11667157001), then stored at −80°C until further use.
Total RNA extraction and in vitro folding
Approximately 5 × 106 of the harvested infected cells were resuspended in 1 ml of TriPure Isolation Reagent and 200 μl of chloroform were added. The sample was vigorously vortexed for 15 s and then incubated for 2 min at room temperature, after which it was centrifuged for 15 min at 12 500 × g (4°C). The upper aqueous phase was collected in a clean 2 ml tube, supplemented with 1 mL (∼2 volumes) of 100% ethanol, and then loaded on an RNA Clean & Concentrator-25 column (Zymo Research, cat. R1017). For in vitro folding, ∼2 μg of total RNA in a volume of 39 μl were denatured at 95°C for 2 min, then transferred immediately to ice and incubated for 1 min. 10 μl of ice-cold 5× RNA Folding Buffer [500 mM HEPES pH 7.9; 500 mM NaCl] supplemented with 20 U of SUPERase•In™ RNase Inhibitor (ThermoFisher Scientific, cat. AM2696) were added. RNA was then incubated for 10 min at 37°C to allow secondary structure formation. Subsequently, 1 μl of 500 mM MgCl2 (pre-warmed at 37°C) was added and RNA was further incubated for 20 min at 37°C to allow tertiary structure formation.
In vitro probing of RNA
For in vitro SHAPE probing of RNA, NAI was added to a final concentration of 50 mM and samples were incubated at 37°C for 10 min. A negative control reaction was also prepared, by adding an equal amount of DMSO. For DMS probing, dimethyl sulfate (Sigma Aldrich, cat. D186309) from a 1:6 dilution in 100% ethanol was added at a final concentration of 150 mM and samples were incubated at 37°C for 2 min. Reactions were then quenched by the addition of 1 volume DTT 1 M and then purified on an RNA Clean & Concentrator-5 column (Zymo Research, cat. R1013).
Extraction of native deproteinized E. coli rRNAs
Deproteinized E. coli RNA was prepared essentially as previously described (21), with minor changes. Briefly, a single colony of E. coli K-12 DH10B was picked and inoculated in LB medium without antibiotics, then grown to exponential phase (OD600 ∼ 0.5). 2 ml aliquots were collected by centrifugation at 1000 × g (4°C) for 5 min. Cell pellets were resuspended in 1 ml of Resuspension Buffer [15 mM Tris–HCl pH 8.0; 450 mM sucrose; 8 mM EDTA], and lysozyme was added to a final concentration of 100 μg/ml. After incubation at 22°C for 5 min and on ice for 10 min, protoplasts were collected by centrifugation at 5000 × g (4°C) for 5 min. Pellets were resuspended in 120 μl Protoplast Lysis Buffer [50 mM HEPES pH 8.0; 200 mM NaCl; 5 mM MgCl2; 1.5% SDS], supplemented with 0.2 μg/μl Proteinase K. Samples were incubated for 5 min at 22°C and for 5 min on ice. SDS was removed by addition of 30 μl SDS Precipitation Buffer [50 mM HEPES pH 8.0; 1 M potassium acetate; 5 mM MgCl2], followed by centrifugation at 17 000 × g (4°C) for 5 min. Supernatant was then extracted 2 times with phenol:chloroform:isoamyl alcohol (25:24:1, pre-equilibrated three times with a buffer containing [50 mM HEPES pH 8.0; 200 mM NaCl; 5 mM MgCl2]), and once with chloroform. 20 U SUPERase•In™ RNase Inhibitor were then added and RNA was equilibrated at 37°C for 20 min prior to probing.
Ex vivo probing of E. coli rRNAs
180 μl of deproteinized E. coli rRNAs, pre-equilibrated at 37°C for 20 min, were mixed with 20 μl of NAI. RNA was then allowed to react at 37°C for 15 min, with moderate shaking, after which 200 μl of 1 M DTT were added, to quench the reaction. Samples were then vortexed briefly and 1 ml of ice-cold QIAzol added to each sample, followed by extensive vortexing. RNA extraction was carried out as already described above for SARS-CoV-2-infected cells.
Multiplex SHAPE-MaP/DMS-MaPseq of in vitro refolded SARS-CoV-2 RNA
For the SHAPE-MaP/DMS-MaPseq analysis of the in vitro refolded SARS-CoV-2 genome, 70 oligonucleotide pairs, tiling the entire length of the genome (29 903 nt), were automatically designed using Primer3 (22) and the following parameters: amplicon size between 480 and 520 bp, maximum poly(N) length between 2 and 3, minimum/optimal/maximum oligonucleotide size of 20/25/30, minimum/optimal/maximum Tm of 56/60/62 degrees, minimum/optimal/maximum GC content of 30/50/65%. Also, pairs were designed in such a way that the target amplicon would include the target region of the reverse primer from the previous set and of the forward primer of the following set. Primers were then searched against the GENCODE v33 human transcriptome, keeping only those with <60% predicted base-pairing or >60% predicted base-pairing and more than two mismatched bases at the 3′ end. For reverse transcription, all reverse primers were equimolarly pooled to yield a 100 μM RT primer mix. An additional anchored oligo-dT primer (TTTTTTTTTTTTTTTTTTTTVN) was added to the mix. For SHAPE-treated samples, two 20 μl reverse transcription reactions were carried out. 3 μg of total RNA were mixed with 0.5 μl RT primer mix and 1 μl dNTPs (10 mM each), incubated at 70°C for 5 min and then transferred immediately to ice for 1 min. Reactions were then supplemented with 4 μl 5× RT Buffer [250 mM Tris–HCl pH 8.3; 375 mM KCl], 2 μl DTT 0.1 M, 1 μl MnCl2 120 mM, 20 U SUPERase•In™ RNase Inhibitor, and 200 U SuperScript II RT (ThermoFisher Scientific, cat. 18064014). Reactions were incubated at 42°C for 2 h and then RT was heat-inactivated at 75°C for 15 min. RNA was then degraded by the addition of 5 U RNase H (New England Biolabs, cat. M0297S) and cDNA from every two reactions was purified on a single RNA Clean & Concentrator-5 column and eluted in 36 μl NF H2O. For DMS-treated samples, two 20 μl reverse transcription reactions were carried out. 3 μg of total RNA were mixed with 0.5 μl RT primer mix and 2 μl dNTPs (10 mM each), incubated at 70°C for 5 min and then transferred immediately to ice for 1 min. Reactions were then supplemented with 4 μl 5× RT Buffer [250 mM Tris–HCl pH 8.3; 375 mM KCl; 15 mM MgCl2], 1 μl DTT 0.1 M, 20 U SUPERase•In™ RNase Inhibitor, and 200 U TGIRT™-III Enzyme (InGex, cat. TGIRT50). Reactions were incubated at 42°C for 5 min, then at 57°C for 2 h. To break the RT-RNA–cDNA complex, 1 μl 5 M NaOH was added, and samples were boiled at 95°C for 3 min. cDNA from every two reactions was purified on a single RNA Clean & Concentrator-5 column and eluted in 36 μl NF H2O.
For targeted amplification, primer pairs were split into 2 non-overlapping sets, odd and even. For each of these sets, pairs were pooled in smaller sets of 3–4 pairs, as follows: 1–19–37–55, 2–20–38–56, 3–21-39–57, 4–22-40–58, 5–23-41–59, 6–24-42–60, 7–25–43–61, 8–26-44–62, 9–27-45–63, 10–28–46–64, 11–29–47–65, 12–30–48–66, 13–31-49–67, 14–32–50–68, 15–33–51–69, 16–34–52–70, 17–35–53, 18–36–54. The complete primer list can be found in Supplementary Table S1. PCR reactions were carried out in 50 μl, using 2 μl of eluted cDNA, 0.3 mM final each dNTP, 0.1 μM final each primer in the set and 2.5 U TaKaRa Taq™ DNA Polymerase (TaKaRa, cat. R001A). Cycling was performed using a touch-down approach. Briefly, for the first 10 cycles, the annealing temperature was lowered by 0.5°C per cycle, starting at 58°C. Then, 20 additional cycles were performed at the lowest temperature. PCR products were purified using 1 volume of NucleoMag NGS Clean-up and Size Select beads (Macherey Nagel, cat. 744970), checked on a 2% agarose gel. Each primer set gave a single specific band of the expected size. PCR products were equimolarly pooled to yield the final odd and even sets. 500 ng of each set were fragmented in a final volume of 20 μl, using 2 μl of NEBNext dsDNA Fragmentase (New England Biolabs, cat. M0348), supplemented with 1 μl 200 mM MgCl2, by incubating at 37°C for 25 min. This yielded fragments in the range of ∼50–200 bp. Reactions were purified using 1 volume of NucleoMag NGS Clean-up and Size Select beads. 5 ng of fragmented DNA were then used as input for the NEBNext® Ultra™ II DNA Library Prep Kit for Illumina® (New England Biolabs, cat. E7645L), as per manufacturer instructions.
SHAPE-MaP of E. coli rRNAs and in vivo probed SARS-CoV-2
Total RNA from infected and probed Vero E6 cells was first depleted of ribosomal RNAs using the RiboMinus™ Eukaryote System v2 (ThermoFisher Scientific, cat. A15026), following manufacturer instructions. Total RNA from E. coli or ribo- RNA from infected and probed Vero E6 cells was fragmented to a median size of 150 nt by incubation at 94°C for 8 min in RNA Fragmentation Buffer [65 mM Tris–HCl pH 8.0; 95 mM KCl; 4 mM MgCl2], then purified with NucleoMag NGS Clean-up and Size Select beads (Macherey Nagel, cat. 744970), supplemented with 10 U SUPERase•In™ RNase Inhibitor, and eluted in 8 μl NF H2O. Eluted RNA was supplemented with 1 μl 50 μM random hexamers and 1 μl dNTPs (10 mM each), then incubated at 70°C for 5 min and immediately transferred to ice for 1 min. Reverse transcription reactions were conducted in a final volume of 20 μl. Reactions were supplemented with 4 μl 5– RT Buffer [250 mM Tris–HCl pH 8.3; 375 mM KCl], 2 μl DTT 0.1 M, 1 μl MnCl2 120 mM, 20 U SUPERase•In™ RNase Inhibitor and 200 U SuperScript II RT. Reactions were incubated at 25°C for 10 min to allow partial primer extension, followed by 2 h at 42°C. SSII was heat-inactivated by incubating at 75°C for 15 min. 6 mM final EDTA was added to chelate Mn2+ ions, followed by 5 min incubation at room temperature and addition of 6 mM final MgCl2. Reverse transcription reactions were then used as input for the NEBNext® Ultra II Non-Directional RNA Second Strand Synthesis Module (New England Biolabs, cat. E6111L). Second strand synthesis was performed by incubating 1 h at 16°C, as per manufacturer instructions. DsDNA was purified using NucleoMag NGS Clean-up and Size Select beads, and used as input for the NEBNext® Ultra™ II DNA Library Prep Kit for Illumina, following manufacturer instructions.
SHAPE-MaP/DMS-MaPseq data analysis
Analysis of SHAPE-MaP data has been conducted using RNA Framework v2.6.9 (23) (https://github.com/dincarnato/RNAFramework). Reads pre-processing and mapping was performed using the rf-map module (parameters: -ctn -cmn 0 -cqo -cq5 20 -b2 -mp ‘–very-sensitive-local’). Reads were trimmed of terminal Ns and low-quality bases (Phred < 20). Reads with internal Ns were discarded. Mapping was performed using the ‘very-sensitive-local’ preset of Bowtie2 (24). The SHAPE mutational signal was then derived using the rf-count module (parameters: -m -rd), enabling the right re-alignment of deletions. For DMS, an additional restraint on the maximum deletion size was imposed (parameter: -md 3). When processing in vitro SARS-CoV-2 data, a mask file containing the sequences of primer pairing regions was passed along (through the -mf parameter). For the in vitro samples, generated RC files, from both the even and odd sets, containing per base mutation counts and coverage, were then combined in a single RC file using the rf-rctools module (mode: merge). Data normalization was performed using the rf-norm module (parameters: -sm <3 (NAI) or 4 (DMS)> -nm 3 -n 500 -mm 1), by using the Siegfried et al. scoring scheme (18) and box-plot normalization of base reactivities. For the normalization of DMS samples, only A and C bases were considered (parameter: -rb AC).
Experimentally-informed SARS-CoV-2 RNA secondary structure modeling
To model the secondary structure of the SARS-CoV-2 genome, we first sought to determine optimal slope/intercept parameters by grid search, using the rf-jackknife module (parameters: -rp ‘-md 600 -nlp’ -x) and ViennaRNA package v2.4.14 (25), with E. coli SHAPE-MaP data and reference E. coli 16S/23S rRNA structures (26). Isolated base-pairs were disallowed, the maximum base-pairing distance was set to 600 nt and the relaxed structure comparison mode (27) was enabled (a base pair i/j was considered to be correctly predicted if any of the following pairs exist in the reference structure: i/j; i–1/j; i+1/j; i/j–1; i/j+1). Optimal slope and intercept parameters were respectively determined to be 0.8 and −0.2 (extremely close to previously determined parameters (28) 1.1 and 0). For DMS, we used the previously determined slope of 2.6 and intercept of −1 (21).
As folding the full SARS-CoV-2 genome as a single entity would be an extremely challenging, and currently unfeasible, computational task, folding was performed using the rf-fold module and a windowed approach previously used by us and other groups (18,21) to model viral RNA genomes (parameters: -sl <slope> -in <intercept> -w -fw 3000 -fo 300 -wt 200 -pw 1500 -po 250 -dp -sh -nlp -md 600). At all stages, SHAPE/DMS data was incorporated in the form of soft constraints. Briefly, in the first stage partition function folding was performed by sliding a window of 1500 nt along the genome, with an offset of 250 nt. For each window, the first and last 100 nt were ignored, to avoid terminal biases. Two additional foldings were computed at both the 5′ and 3′ ends to increase structure sampling (window sizes: 1400 and 1450 nt). Base-pairing probabilities from all windows were then averaged and base-pairs with a probability ≥0.99 were kept and used as a constraint for the next stage. At this stage, base-pairing probabilities were also used to calculate per-base Shannon entropies. In the second stage, the maximum expected accuracy (MEA) structure was predicted by sliding a window of 3000 nt, with an offset of 300 nt along the genome. Four additional foldings were computed at both the 5′ and 3′ ends to increase structure sampling (window sizes: 2900, 2950, 3050 and 3100 nt). Base-pairs appearing in >50% of the windows were then retained to yield the final secondary structure pseudoknot-free model. Pseudoknots were instead introduced during the modeling of the 3D structure.
Identification of candidate RNA structures
For the identification of candidate RNA structures, median Shannon entropies and SHAPE reactivities were first calculated in sliding, centered, 50 nt windows. Then, a window of 50 nt was slid along the genome. Windows in which >75% of the bases were below both the global Shannon and SHAPE median (calculated on the full SARS-CoV-2 genome) were picked and windows residing <10 nt apart were merged. The same procedure was repeated for both the SHAPE-treated in vitro and in vivo samples. In addition, for the in vitro sample, only regions having low Shannon (and showing coherent folding) also in the DMS-treated sample were retained. To only select segments likely to represent well-defined locally-stable RNA structures, we further predicted three additional structures by using the in vitro SHAPE data as a constraint, and by increasing the folding and partition function window sizes to 3000, 5000 or 10000 nt, and removing any restraint on the maximum base-pairing distance. Windows were then slid in 10% increments. Structures showing coherent folding in all three predictions, as well as between in vitro and in vivo structure models, were selected. Coherence between structures was calculated as the geometric mean of PPV and sensitivity, with a stringent cutoff of 0.8. Only structure elements having >50% of the segment they spanned falling within a low Shannon – low SHAPE region were kept for downstream analyses.
Identification of low Shannon—high SHAPE regions
For the identification of low Shannon—high SHAPE regions, instead, a window of 25 nt was slid along the genome. Windows in which >75% of the bases were below the global Shannon median and above the global SHAPE median (calculated on the full SARS-CoV-2 genome) and >50% of the bases were predicted to be single-stranded in the MEA structure, were picked and windows residing less than 10 nt apart were merged.
Identification of conserved RNA structure elements
To identify conserved low Shannon—low SHAPE RNA structure elements, we implemented an automated pipeline (cm-builder; https://github.com/dincarnato/labtools) built on top of Infernal 1.1.3 (29). Briefly, we first built a covariance model (CM) from a Stockholm file containing only the SARS-CoV-2 sequence and the structure of the selected elements, using the cmbuild module. After calibrating the CM using the cmcalibrate module, it was used to search for RNA homologs in a database composed of all the non-redundant coronavirus complete genome sequences from the ViPR database (30) (https://www.viprbrc.org/brc/home.spg?decorator = corona), as well as a set of representative coronavirus genomes from NCBI database, using the cmsearch module. Only matches from the sense strand were kept and a very relaxed E-value threshold of 10 was used at this stage to select potential homologs. Three additional filtering criteria were used. First, we took advantage of the extremely conserved architecture of coronavirus genomes (31) and restricted the selection to matches falling at the same relative position within their genome, with a tolerance of 3.5% (roughly corresponding to a maximum allowed shift of 1050 nt in a 30 kb genome). Through this more ‘conservative’ selection, we only kept matches likely to represent true structural homologs, although at the cost of probably losing some true matches. Second, we filtered out matches retaining <55% of the canonical base-pairs from the original structure elements. Third, truncated hits covering <50% of the structure were discarded. The resulting set of homologs was then aligned to the original CM using the cmalign module and the resulting alignment was used to build a new CM. The whole process was repeated for a total of three times. The alignment was then refactored, removing gap-only positions and including only bases spanning the first to the last base-paired residue. The alignment file was then analyzed using R-scape 1.4.0 (32) and APC-corrected G-test statistics to identify motifs showing significantly covarying base-pairs. Only structures with at least 1 covarying pair at E-value <0.05, or two covarying pairs at E-value <0.1 were retained. Stockholm files for these structures were then used to build the final CMs and to re-search the entire database with the default stringent E-value cutoff of 0.01.
Determination of low Shannon—high SHAPE regions’ conservation
To assess the sequence conservation of the identified low Shannon—high SHAPE regions, we computed four multiple sequence alignments using MAFFT v7.429 (33) (parameters: –maxiterate 100 –auto), the reference SARS-CoV-2 sequence and one of the following datasets: (i) SARS-CoV (243 sequences); (ii) MERS-CoV (281 sequences); (iii) other Beta-CoV (excluding SARS-CoV/SARS-CoV-2/MERS-CoV, 681 sequences); (iv) other CoV (excluding Beta-CoV, 1657 sequences). Sequences were obtained from the ViPR database and 100% identical sequences were collapsed. Region conservation was calculated as the average of the conservation of each nucleotide in that region (using SARS-CoV-2 as the reference).
RNA 3D structure modeling
3D structures were predicted for the aforementioned segments using SimRNA (34), a method for simulations of RNA folding, which uses a coarse-grained representation, relies on the Monte Carlo method for sampling the conformational space, and employs a statistical potential to approximate the energy of the studied RNA molecule (34,35). The patterns of base-pairs inferred to be common to in vitro and in vivo conditions were used as soft restraints. It is important to emphasize that SimRNA interprets the secondary restraints only in a positive sense (i.e. it attempts to pair the restrained residues, while other residues are allowed to sample different conformations). As a result, residues not restrained to be paired, may be eventually found to be paired (canonically or non-canonically) in the resulting low-energy models.
By default, for each RNA segment analyzed, we carried out eight independent simulations with SimRNA, each comprising 10 replicas, with 16 million Monte Carlo steps in each run. Each simulation resulted in a trajectory, comprising up to 10 000 conformations (hence 80 000 conformations total), broadly representing local energy minima in the whole conformational space sampled by SimRNA for a given molecule. For each segment, the 1000 best-scored (according to SimRNA energy criterion) conformations in a coarse-grained SimRNA representation were selected to represent the low-energy regions of the conformational landscape and approximate the distribution of most common conformations existing in solution. These 1000 conformations were clustered at the RMSD threshold equal to 0.1 times the length of the sequence (a default parameter used in SimRNA), yielding ensembles of similar conformations (up to 10 clusters for a given RNA). A single lowest-energy representative of each cluster was retained to represent a given ensemble of structurally similar low-energy conformations.
For the 1000 best-scored conformations for each segment, all-atom models were generated and their conformational details were refined by running 5000 steps of a steepest-descent minimization with QRNAS (36) which is an extension of the AMBER simulation method (37) with additional restraints. For each of the 1000 models, the secondary structure was extracted by running ClaRNA (38) and the set of 1000 secondary structures was used to generate the consensus structure for each segment, to aid in the data visualization and for comparison with the secondary structure used as restraints.
Prediction of potential ligand binding sites in RNA 3D structures (druggable pockets)
To identify potential ligand-binding pockets in RNA 3D structures, we used Fpocket (39), originally developed for the identification of ligand-binding pockets in proteins.
We conducted a benchmarking study on a small subset of known viral RNA structures bound to their small molecule or peptide ligands (PDB codes: 1ETG [Rev response element with Rev peptide], 1LVJ [HIV-1 TAR with PMZ], 1UTS [HIV-1 TAR with P13], 2KMJ [HIV-2 TAR with a pyrimidinylpeptide], 2KTZ [HCV IRES with ISH], and 2L94 [HIV-1 FSE with the frameshifting inhibitor DB213]). These RNAs were folded using SimRNA (one simulation with 10 replicas and 16 million Monte Carlo steps). For each RNA, 1000 decoys with the lowest SimRNA energy were identified, their all-atom conformations were generated, and analyzed with Fpocket. In each analyzed model, a ribonucleotide residue with at least two atoms within the pocket was considered as a potential ligand-contacting residue. For each residue in each RNA, a normalized score was assigned based on the total number of contacts predicted in the 1000 low-energy conformations. The normalized score, Si for a residue i in an RNA sequence of length L was calculated as follows:
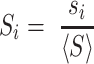 |
where si is the number of contacts for residue i, and <S> is the average number of contacts, calculated as:
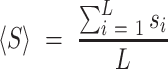 |
To aid in the visualization of the 3D models, the B-factor field in the PDB files was changed to the relative ligand-binding score S from the F-pocket analysis, using the programs we previously developed for the RNAProbe web server (40). The residues with score S of 1.2 or above were selected and extended by one residue on either side to include the neighbors at the sequence level. After the extension, the stretches with six nucleotides or more were retained. The shorter stretches were added, if any of their constituent residues were paired with the longer ones, based on the consensus secondary structure derived from 1000 lowest-energy decoys. Additional residues forming Watson–Crick base pairs with the nucleotides in these stretches were also included. The resulting set of residues was considered a candidate pocket, reflecting the structural features of all 1000 models per RNA analyzed.
To retrieve the potentially druggable pockets in complex 3D motifs from the SARS-CoV-2 models, we applied the same procedure to the sets of 1000 best-scored models generated for conserved and stably structured regions of the viral RNA described above. The pockets were evaluated visually in the context of the secondary structure as well as in 3D models (representatives of largest clusters), and in some cases, larger pockets were subdivided.
RESULTS
In vitro and in vivo mapping of the SARS-CoV-2 genome RNA structure
To characterize the secondary structure landscape of SARS-CoV-2, we performed both SHAPE and dimethyl sulfate (DMS) mutational profiling (SHAPE-MaP and DMS-MaPseq) analyses of the entire viral genome (∼30 kb) following in vitro refolding, as well as SHAPE-MaP analysis in living infected Vero E6 cells following 2-methylnicotinic acid imidazolide (NAI) probing. For in vitro experiments, total RNA from Vero E6 cells infected with SARS-CoV-2 was refolded and probed with NAI, or with DMS to provide orthogonal validation. This approach has a considerable advantage, compared to in vitro transcription, of allowing the analysis of high amounts of full-length SARS-CoV-2 RNA, bearing post-transcriptional RNA modifications that could occur in vivo during natural infection, and that might affect local RNA folding. For all conditions, we obtained two independent replicates. Mutational signatures showed high correlations for all experiments (in vitro SHAPE R2 = 0.984; in vitro DMS R2 = 0.987; in vivo SHAPE R2 = 0.876; Figure 1A), hence replicates were combined for downstream analyses.
Figure 1.

Genome-wide SHAPE-MaP analysis of SARS-CoV-2. (A) Heat scatter plot of normalized reactivities across two biological replicates for the in vitro SHAPE (left) and DMS (center) datasets, and the in vivo SHAPE dataset (right). (B) Normalized in vivo SHAPE reactivities superimposed on the 5′ UTR structure. Highly (red) and moderately (yellow) reactive residues from in vitro SHAPE (circles) and DMS (triangles) experiments are also indicated. A novel putative stem-loop 8 (pSL8), coherently predicted in all our datasets is also reported.
We observed good agreement between known Beta-CoV secondary structure elements (5′ UTR and 3′ stem-loop II-like motif) and reactivities measured both in vitro and in vivo (Figure 1B and Supplementary Figure S1), with on average ∼91.3% of highly reactive bases (≥0.7) not being involved in canonical base-pairing, or residing adjacent to helices termini or bulges/loops (in vitro SHAPE: 88.5%; in vitro DMS: 96.1%; in vivo SHAPE: 89.3%). Comparison between in vitro and in vivo SHAPE reactivities showed unexpectedly high correlation (R2 = 0.552; Figure 2A), as compared, for example, to a previous study conducted in Influenza A virus (21), suggesting that, at least in part, sequence and thermodynamics alone are able to drive SARS-CoV-2 RNA folding. Nonetheless, as previously described for other RNAs (21,41,42), significant differences exist when looking at reactivity distributions, with the in vivo structure being skewed towards higher reactivity values, suggesting a slightly lower degree of structuring as compared to the in vitro structure (median SHAPE in vivo: 0.25; median SHAPE in vitro: 0.20; P < 2.2e–16, Wilcoxon rank sum test; Figure 2B). This can be, at least partly, a consequence of the ribosome-mediated unfolding of the SARS-CoV-2 genome during translation (43). In agreement with this hypothesis, the Pearson correlation calculated only over the first and last 500 nt of the SARS-CoV-2 genome, roughly corresponding to the UTR regions, showed a markedly higher correlation (R2 = 0.8). Nonetheless, we cannot rule out the possibility that the observed structural differences might arise from the presence of multiple alternative conformations within high Shannon entropy regions. Accordingly, the distribution of Gini indexes, a measure of the evenness of a distribution (41,42,44), calculated in 50 nucleotide sliding windows, showed significantly higher Gini index values for the SARS-CoV-2 genome in vitro (median Gini in vivo: 0.55; median Gini in vitro: 0.63; P < 2.2e–16, Wilcoxon rank sum test; Figure 2C), indicating an overall higher degree of structuring as compared to the genome in vivo.
Figure 2.

Comparison between in vitro and in vivo structure of the SARS-CoV-2 genome. (A) Heat scatter plot of normalized SHAPE reactivities for the in vitro refolded SARS-CoV-2 genome versus the in vivo-probed one. (B) Distribution of SHAPE reactivities for the in vitro refolded SARS-CoV-2 genome, versus the in vivo-probed one. (C) Violin plot depicting the distribution of Gini indexes calculated in 50 nt windows slid along the SARS-CoV-2 genome for the in vitro refolded sample versus the in vivo-probed one (sliding offset: 25 nt).
Next, we used SHAPE-derived constraints to model the secondary structure of the SARS-CoV-2 genome (see Methods). This approach has been extensively validated and previously applied by us and other groups to successfully map viral RNA structures (18,21). To convert NAI reactivities into pseudo-free energy contributions, a complementary SHAPE-MaP dataset was produced by probing E. coli rRNAs with NAI under ex vivo deproteinized conditions and determined optimal slope and intercept to be 0.8 and −0.2 by grid search, respectively (Supplementary Figure S2). For DMS, previously determined parameters were used (21). By incorporating these parameters into our RNA Framework algorithm for probing-directed RNA structure prediction (23), we performed the experimentally-guided modeling of the SARS-CoV-2 RNA genome secondary structure, under both in vitro (Figure 3) and in vivo (Figure 4) conditions. Previously described RNA structure elements typical of Sarbecoviruses and other Beta-CoV (11), such as 5′ UTR helices S1 to S7 and the 3′ UTR stem-loop II-like motif (s2m) were successfully modeled in the context of the full genome, without the need to impose any hard constraints, supporting the high accuracy and reliability of our analysis. Interestingly, the recently proposed stem-loops SL8 to SL10 (17) (position: ∼400–450 nt) are not supported by our data. Instead, the region spanning these three short hairpins appears to be involved in the formation of a large stem-loop-like structure, spanning nucleotides 407–478 (putative stem–loop 8, pSL8; Figure 1).
Figure 3.

Structure map of the full SARS-CoV-2 genome in vitro. Map of the SARS-CoV-2 genome depicting (top to bottom): median SHAPE reactivity (in 50 nt centered windows, with respect to the median reactivity across the whole genome), median DMS reactivity (in 50 nt centered widows, with respect to the median reactivity across the whole genome), Shannon entropy and base-pairing probabilities for both the SHAPE (up) and DMS (down) samples, set of low SHAPE – low Shannon helices coherently predicted in both datasets. Regions with both low Shannon and low SHAPE, likely identifying RNA segments with well-defined foldings, are marked in grey. Two regions of low sequencing coverage are marked in red.
Figure 4.

Structure map of the full SARS-CoV-2 genome in vivo. Map of the SARS-CoV-2 genome depicting (top to bottom): median SHAPE reactivity (in 50 nt centered widows, with respect to the median reactivity across the whole genome), Shannon entropy, base-pairing probabilities, Pearson correlation coefficient (in 50 nt centered windows; PCC) between in vitro and in vivo SHAPE data, set of low SHAPE – low Shannon under both in vitro and in vivo conditions (blue), set of helices coherently predicted independently of the window size employed (3000, 5000 or 10 000 nt; magenta), set of helices coherently predicted under both in vitro and in vivo conditions (aqua green). Selected structure elements are marked in light blue. Two short regions of low sequencing coverage are marked in red.
To only select high confidence RNA structure elements, to be used for the development of new RNA-targeted therapeutic strategies, we implemented a number of filtering steps. First, we identified regions having both low SHAPE and low Shannon entropy in all structure models. The Shannon entropy provides an estimate of the likelihood of a given RNA region to fold into multiple alternative conformations (18). We therefore selected regions showing low Shannon entropies both in vitro and in vivo. Starting from the in vitro SHAPE-guided model, we then selected only structures falling within low SHAPE—low Shannon regions, and that were predicted to fold coherently in all the analyzed datasets. This filtering aimed at selecting only highly thermodynamically-favorable RNA structure elements, folding into a single well-defined, and unambiguously modeled, conformation. To further rule out the possibility that some of these elements have been predicted only as a consequence of the chosen analysis parameters, such as folding and partition function window size and offset, we further selected only those structures coherently predicted with increasingly larger window sizes (3000, 5000 and 10 000 nucleotides) in the absence of any constraint on the maximum base-pair distance. Notably, most of the identified structures passed this filtering step, indicating that they represent bona fide locally-stable RNA structure elements. This workflow resulted in the selection of 87 highly structured regions. Additionally, we compared the folding free energy of these structures to that of 1000 shuffled sequences, obtained by keeping either the same nucleotide or dinucleotide content (Supplementary Figure S3). Independently of the shuffling strategy employed, the majority of these elements showed folding free energies significantly lower than it would be expected by chance (84/87, 96.6%) for simple nucleotide shuffling; 81/87 (93.1%) for dinucleotide shuffling; Z-test, P < 0.05). Overall, these regions encompassed ∼18.9% of the SARS-CoV-2 genome (5653 bases), and included the 5′ UTR. Due to the high stringency of our analysis, this subset of structures represents a conservative estimate.
The FSE and the 3′ UTR show unexpected folds in vivo
While, as previously stated, the 5′ UTR region folded as expected under both in vitro and in vivo conditions, the entire 3′ UTR region did not pass our stringent filtering, possibly because of the higher reactivity observed at the level of the HVR in vivo (data not shown), suggesting that part of this structure might be unfolded inside the cell. Nonetheless, the upper portion of the 3′ UTR region (position: 29 695–29 809), consisting of a four-way junction encompassing the s2m and the conserved octanucleotide, showed reactivities in agreement with the phylogenetically-determined structure in all of the analyzed samples. Hence, we decided to manually include the full 3′ UTR (position: 29 622–29 867) in our analyses (in addition to the originally selected 87 fragments).
Furthermore, our analysis did not identify the SARS-CoV-2 FSE as having a low SHAPE signal and low Shannon entropy. While this element may be structured under in vitro conditions, as found by other studies, and as demonstrated by structure determination by cryo-EM (45), we found that the cryo-EM-derived model of the FSE structure (PDB: 6XRZ) is not fully consistent with the observed SHAPE reactivities in vivo. In particular, mapping of in vivo SHAPE reactivities onto that model, corresponding to an in vitro folded local structure (Supplementary Figure S4), revealed high reactivities of some residues expected to be base-paired, suggesting that this functional element might be partially unfolded in vivo, and that its conformation may significantly change in the context of the full SARS-CoV-2 genome.
A subset of structures shows significant conservation across coronaviruses
To further prioritize the structured segments of SARS-CoV-2 RNA more likely to represent functional elements, we devised an automatic strategy for the identification of segments showing significant base-pair covariation across coronaviruses (see Materials and Methods). By building a first covariance model (CM) using Infernal (29) and the sole SARS-CoV-2 sequence, together with the secondary structure inferred from probing data, we searched (with a relaxed E-value cutoff) for homologous sequences in a non-redundant CoV database, discarding those matches in which <55% of the canonical base-pairs from the SARS-CoV-2 structure were supported. Furthermore, by taking advantage of the extremely conserved architecture of CoV genomes (31), we further discarded those matches whose relative position in the genome changed by >3.5% with respect to that of SARS-CoV-2. The resulting alignment was then used to refine the CM and the whole procedure was repeated three times.
This analysis identified a total of nine RNA elements (out of the originally selected 87 structures plus the 3′ UTR, 10.2%) for which at least two base-pairs showed significant covariation (average significant base-pairs, 16.1% at E-value < 0.05, 17.7% at E-value < 0.1; Figure 5), as determined by the R-scape software (32). Among these, our pipeline successfully recovered both the Sarbecovirus 5′ UTR SL5 (segment 150–294) and the 3′ UTR (segment 29 622–29 867; Supplementary Figure S5). Identified RNA structures show variable degrees of conservation across different CoV genera, with 6 out of 9 (66.7%) being conserved also in Alpha-CoV, 7 out of 9 (77.8%) in Gamma-CoV, and 5 out of 9 (55.6%) in Delta-CoV (Supplementary Figure S6). These nine conserved segments folding into stable structures are likely to be involved in the mechanistic and structural aspects of the viral RNA function.
Figure 5.

Structure of conserved elements in SARS-CoV-2 RNA. Structure models for segments 7924–8128 (A), 22 826–22 913 (B) and 23 970–24 098 (C). All three segments consist of conserved three-way junction structures. Structure models have been generated using the R2R software. Base-pairs showing significant covariation (as determined by R-scape) are boxed in green (E-value < 0.05) and violet (E-value < 0.1) respectively. Alongside, the structure of each segment in SARS-CoV-2 (as inferred from in vitro SHAPE-MaP data) with superimposed in vitro SHAPE-MaP (left) or DMS-MaPseq (center) reactivities, or in vivo SHAPE-MaP reactivities (right) is shown, together with the putative conserved structure in other CoV as derived from the alignment (manually optimized with the ViennaRNA package).
Secondary structure-guided 3D structure modeling identifies druggable pockets within conserved structure elements
Previous analyses have shown that complex structured RNA molecules often exhibit pockets with physical properties suitable for small molecule binding, which can be identified in RNA 3D structure models (46). Thus, the discovery of multiple structured segments in SARS-CoV-2 genomic RNA offered an opportunity to test their potential to be targeted by small molecules.
Towards this goal, we modeled the 3D folding of the structured segments, by using the SimRNA method we previously developed (34). As the 3D structure for an RNA molecule may be flexible even under the constraints of a particular secondary structure, our goal was not to predict just a single 3D conformation for each segment, but to sample the conformational space in search of local and global energy minima, that are most populated by the folded structures.
We used secondary structure information derived from the SHAPE-MaP/DMS-MaPseq experiments to restrain the folding trajectories that sampled the 3D conformational space. Soft restraints were used to allow conformational flexibility while maximizing the sampling of the conformational space in the vicinity of the inferred secondary structure.
For each 3D-folded segment, we generated 1000 lowest-energy models to represent the most energetically favorable 3D conformations (according to the statistical potential of SimRNA). Then, as in the standard SimRNA protocol (47), we clustered these structures to identify lowest-energy representatives of the most frequently sampled 3D conformations (i.e. corresponding to the main local energy minima). As expected, while the patterns of base pairs agreed very well with those inferred from experimental probing, the 3D structures exhibited significant overall conformational variability. Nonetheless, the 3D modeling revealed a number of persistent complex spatial motifs recurring in structures from many spatial clusters, often comprising junctions and structured bulges and loops.
Next, we used the Fpocket software (39) to score the putative druggable sites in 1000 lowest-energy models for each RNA segment, based on the criteria we established on a set of experimentally determined 3D structures of small viral RNAs complexed with small molecule ligands (Supplementary Figure S7; see Methods). We mapped the predicted druggable pockets on the RNA sequence to identify groups of adjacent residues that formed potential ligand-binding sites in the largest fraction of best-energy models (Figure 6 and Supplementary Figures S8-S13).
Figure 6.

3D modeling of SARS-CoV-2 RNA structured segments and identification of druggable pockets. 3D structure of the most abundant cluster for segments 7924–8128 (A), 22826–22913 (B) and 23970–24098 (C), as derived from the 1000 lowest energy 3D structure structures modeled by SimRNA (using the consensus MEA secondary structure, inferred from in vitro and in vivo SHAPE-MaP analyses, as a restraint). Residues composing the identified druggable pockets are shaded.
A variety of potentially druggable pockets were found within the nine analyzed segments. Segment 150–294 (Supplementary Figure S8) has two druggable pockets, one involving a four-way junction (4WJ) and three of its constituting helices, and the other one lying within a bulged duplex region. Segment 3462–3616 (Supplementary Figure S9) has one pocket involving a three-way junction (3WJ), as well as two of the helices that constitute it, containing 2-nt bulges in the vicinity of the junction. Segment 7924–8128 (Figure 6A) has two pockets, the first one corresponds to a 3WJ, while the second one is formed through tertiary contacts between a bulge and a distal duplex. Segment 18 071–18 329 (Supplementary Figure S10) has two pockets, one encompassing a 4WJ and another one comprising a bulged helical structure. Segment 20 241–20 411 (Supplementary Figure S11) has two pockets, one encompassing a 3WJ, and another one encompassing a bulged helix. Segment 22 826–22 913 (Figure 6B) has a pocket encompassing a 3WJ as well as the three helices constituting it. Segment 23 970–24 098 (Figure 6C) has a pocket comprising a 3WJ along with two of its constituting helices, one of them containing several bulges. Segment 28 065–28 118 (Supplementary Figure S12) has a structurally simpler pocket if compared to multiway junctions, which comprises a helix with a single-residue bulge. Segment 29 622–29 867 (Supplementary Figure S13) has three pockets: two of them comprise 4WJs, while the third one corresponds to a bulged helical region. This last segment includes the 3′ stem–loop II-like motif, a highly conserved motif found at the 3′-UTRs of astrovirus, coronavirus, and equine rhinovirus genomes (48) and has been proposed to affect host translation by either interacting with ribosomal proteins or by getting processed into a mature microRNA (16,49 ). Overall, the majority of targetable pockets were found in high-information-content structures such as multiway junctions (50) (Figure 6), although they were also very often found in asymmetrical bulges. On the other hand, terminal loops and symmetrical bulges were rarely identified as druggable sites in SARS-CoV-2 RNA segments.
In addition to analyzing the conserved, structurally stable segments, we carried out an additional analysis for the FSE element (segment 13 458–13 545), which is known to be functionally important, and which we found to be structurally variable (and therefore excluded from the selected set of candidate RNA structures). As restraints we used secondary structure derived from in vivo SHAPE probing, which partially agrees with only one of the helices in the cryo-EM-derived structure (45), and leaves unpaired a large part of the FSE sequence (Supplementary Figure S14A). Interestingly, despite these differences, simulations with SimRNA have led to models in which the pseudoknot was largely recapitulated. Models representing the largest clusters from SimRNA simulation show similar architecture to the one observed in the cryo-EM-derived model, even though the global RMSD values are moderate (∼17.7 Å). By analyzing 1000 low-energy conformations generated by SimRNA, we found two tentative druggable pockets in the FSE (Supplementary Figure S14B). The larger pocket is found in the pseudoknotted region. Importantly, this pocket successfully recapitulates a previously reported ligand-binding region for the SARS-CoV FSE (51) (Supplementary Figure S14C). The analysis of FSE simulations provided additional validation of the methodology for prediction of druggable pockets described in this work, and suggests that it may be applicable not only to RNAs with well-defined 3D structure, but also to shape-shifting molecules.
Mapping of persistently single-stranded regions in vivo identifies potential targets for antisense oligonucleotide therapeutics
To further aid the development of antisense oligonucleotide (ASO) therapeutics, segments were also identified that showed both low structural variability (low Shannon) and paucity of pairing (high SHAPE) in vivo. Such segments are likely to persistently expose long stretches of Watson–Crick edges of multiple residues available for base-pairing with a complementary sequence, thus representing preferred targets for ASOs. Our analysis (see Materials and Methods) identified 80 such segments, at least 25 nucleotides long, spanning a total of 3756 bases (∼12.6% of the SARS-CoV-2 genome) matching these criteria (Supplementary Figure S15 and Table S2). We next evaluated the sequence conservation of these segments, by multiple sequencing alignment with different sets of coronavirus sequences (Supplementary Figure S16), specifically: (i) SARS-CoV; (ii) MERS-CoV; (iii) other Beta-CoV; (iv) all other CoV (alpha, gamma, and delta). Notably, ∼7.5% of these segments, accounting for roughly 361 bases (∼1.2% of the SARS-CoV-2 genome), showed over 78% conservation in SARS-CoV (average ∼86.2%) and over 60% conservation in the other three datasets (average MERS-CoV: ∼75.6%; average other Beta-CoV: ∼71.9%; average all other CoV: ∼64.6%). The exceptional conservation of these segments suggests that some essential regulatory RNA elements might reside here and be under strong selection.
DISCUSSION
In this study, we describe for the first time the RNA structural landscape of the entire SARS-CoV-2 virus genome. By using SHAPE and DMS mutational profiling (MaP) analyses in living infected host cells, as well as on the in vitro refolded SARS-CoV-2 RNA, we obtained a single-base resolution map of base reactivities that allowed us to model the secondary structure of 87 well-defined structure elements throughout the entire SARS-CoV-2 genome.
Our approach has two main limitations. From an experimental point of view, it cannot discriminate between the probing signal coming from the genomic RNA and the subgenomic mRNAs (sgRNAs). Nonetheless, the identification of low SHAPE—low Shannon regions, corresponding to highly-structured elements having a high-probability of folding into a single well-defined conformation (18,21), identifies locally-stable RNA structure elements that are coherently present in both the gRNA and the sgRNAs. From a computational point of view, modeling the secondary structure of the full-length SARS-CoV-2 virus RNA (∼30 kb) is not feasible, and the use of a windowed approach hampers our ability to predict long-range interactions. This limitation can only be addressed by using alternative experimental strategies, such as those based on in vivo RNA crosslinking coupled to proximity ligation (28,52–56).
Despite the aforementioned limitations, our analysis enabled us to identify a set of RNA structure elements, supported by multiple sources of evidence. Firstly, the folding free energies of these structures are significantly lower than those of random sequences of matching nucleotide (or dinucleotide) composition. Secondly, these structures show coherent folding under both in vivo and in vitro conditions. Thirdly, at least 10% of these structures show significant covariation support. Altogether, these observations suggest that a strong selective pressure acts on the selected elements, such that sequence and thermodynamics alone are able to drive the proper folding of these structures. While the presence of covariation can support the presence of functional RNA structures, the lack of covariation is not evidence for the lack of function. Therefore, although we did not find sufficient significant covariation support for many of the identified structures (possibly also due to the stringency of our filtering criteria), they can anyway represent either recently evolved RNA structures or RNA cis-regulatory elements residing within regions whose sequence is constrained by the underlying amino-acid sequence.
Unexpectedly, the presence of previously described structure elements is not fully supported by our data. For instance, the proposed three-stem pseudoknotted structure of the FSE (17,45) does not appear to be the prominent conformation in vivo (nor in vitro). Moreover, although the 3′ UTR region folds into the expected conformation in vitro, it shows high SHAPE reactivity at the level of the HVR in vivo. Two main possibilities can explain the observed differences. Firstly, prior structural studies have been mostly conducted on isolated RNA elements, outside of the context of the full-length genomic RNA. Secondly, these regions might exhibit high structural dynamicity in vivo, possibly folding into multiple mutually-exclusive conformations. This is in agreement with a recent report suggesting that the FSE can switch between two conformations, none of which compatible with the previously described pseudoknotted structure (57).
The identification of thermodynamically-favored, conserved RNA structure elements in the SARS-CoV-2 genome, constitutes a unique opportunity for the development of RNA-targeted small molecule drugs. To this end, we sampled the 3D conformational space of conserved SARS-CoV-2 RNA structure elements and produced 3D models that allowed us to identify putative druggable pockets in these structures. Many of these structures presented high-information-content geometries (50), such as multiway junctions, that represent attractive RNA 3D targets for further analyses.
Additionally, our analysis identified conserved sites of persistent single-strandedness in the SARS-CoV-2 genome in vivo. These regions might represent ideal targets for the design of antisense oligonucleotide therapeutics (ASO), already proven to represent a promising approach for the treatment of infections by other RNA viruses (58–61).
Overall, our analyses reveal structures of the SARS-CoV-2 virus RNA that may turn out to be its weak spots. Not only our data will provide a fundamental resource for the development of innovative RNA-targeted therapeutic strategies, but also it will help elucidating still unknown aspects of the life cycle of coronaviruses once the functional role of the structured elements in the coronavirus RNA genome here identified will be characterized.
DATA AVAILABILITY
SHAPE-MaP data has been deposited to the Gene Expression Omnibus (GEO) database, under the accession GSE151327. RNA Count (RC) files for use with RNA Framework, as well as XML files with normalized SHAPE-MaP reactivities are provided, together with the full secondary structure in dot-bracket notation and the selected low Shannon - low SHAPE structures. Stockholm alignment files and the derived covariance models (CMs) for the structure elements, showing significant covariation support, are also provided. For each structured segment in the SARS-CoV-2 genome, we further provide the representative 3D structure models for up to 10 of the largest clusters, illustrating the most typical conformational classes, as well as the 1000 best-energy models used to predict ligand-binding modes. In all models, the B-factor field presents the relative ligand-binding score obtained from the Fpocket analysis. These additional processed files (RNA Framework RC and XML files, dot-bracket structures, CMs, Stockholm alignments, 3D models of SARS-CoV-2 segments and druggable pockets) are available at http://www.incarnatolab.com/datasets/SARS_Manfredonia_2020.php and on Mendeley at http://dx.doi.org/10.17632/8gj97c4kgv.1.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to further acknowledge Dr Diana Spierings, Dr Nancy Halsema, and all the staff at the sequencing facility of ERIBA (European Research Institute for the Biology of Ageing, University Medical Center Groningen). Computational resources for calculations done by the Bujnicki group were provided by the Poznań Supercomputing and Networking Center at the Institute of Bioorganic Chemistry, Polish Academy of Sciences through the Polish Grid Infrastructure (grants: rnpmd, simcryox, and plgnithinaneesh2019a) and the Interdisciplinary Centre for Mathematical and Computational Modelling at the University of Warsaw (grants: G73-4, GB76-30, and G81-5).
Contributor Information
Ilaria Manfredonia, Department of Molecular Genetics, Groningen Biomolecular Sciences and Biotechnology Institute (GBB), University of Groningen, Nijenborgh 7, 9747 AG, Groningen, the Netherlands.
Chandran Nithin, Laboratory of Bioinformatics and Protein Engineering, International Institute of Molecular and Cell Biology in Warsaw, ul. Ks. Trojdena 4, 02-109 Warsaw, Poland.
Almudena Ponce-Salvatierra, Laboratory of Bioinformatics and Protein Engineering, International Institute of Molecular and Cell Biology in Warsaw, ul. Ks. Trojdena 4, 02-109 Warsaw, Poland.
Pritha Ghosh, Laboratory of Bioinformatics and Protein Engineering, International Institute of Molecular and Cell Biology in Warsaw, ul. Ks. Trojdena 4, 02-109 Warsaw, Poland.
Tomasz K Wirecki, Laboratory of Bioinformatics and Protein Engineering, International Institute of Molecular and Cell Biology in Warsaw, ul. Ks. Trojdena 4, 02-109 Warsaw, Poland.
Tycho Marinus, Department of Molecular Genetics, Groningen Biomolecular Sciences and Biotechnology Institute (GBB), University of Groningen, Nijenborgh 7, 9747 AG, Groningen, the Netherlands.
Natacha S Ogando, Molecular Virology Laboratory, Department of Medical Microbiology, Leiden University Medical Center, Leiden, the Netherlands.
Eric J Snijder, Molecular Virology Laboratory, Department of Medical Microbiology, Leiden University Medical Center, Leiden, the Netherlands.
Martijn J van Hemert, Molecular Virology Laboratory, Department of Medical Microbiology, Leiden University Medical Center, Leiden, the Netherlands.
Janusz M Bujnicki, Laboratory of Bioinformatics and Protein Engineering, International Institute of Molecular and Cell Biology in Warsaw, ul. Ks. Trojdena 4, 02-109 Warsaw, Poland.
Danny Incarnato, Department of Molecular Genetics, Groningen Biomolecular Sciences and Biotechnology Institute (GBB), University of Groningen, Nijenborgh 7, 9747 AG, Groningen, the Netherlands.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Nederlandse Organisatie voor Wetenschappelijk Onderzoek (NWO) [OCENW.XS3.044 to D.I.]; National Science Centre [2020/01/0/NZ1/00232 to J.M.B., 2017/26/A/NZ1/01083 to J.M.B., 2017/25/B/NZ2/01294 to J.M.B., 2018/31/D/NZ2/01883 to A.P.S.]; Foundation for Polish Science [TEAM/2016-3/18 to J.M.B.]; Groningen Biomolecular Sciences and Biotechnology Institute (GBB, University of Groningen) [personal funding to D.I.]; Leiden University Fund (LUF), Bontius Foundation and donations from the crowdfunding initiative ‘wake up to corona’. Funding for open access charge: Grant fundings (to D.I. and J.M.B.).
Conflict of interest statement. J.M.B. is an Executive Editor of Nucleic Acids Research.
This paper is linked to: doi:10.1093/nar/gkaa1013.
REFERENCES
- 1. Boerneke M.A., Ehrhardt J.E., Weeks K.M.. Physical and functional analysis of viral RNA genomes by SHAPE. Ann. Rev. Virol. 2019; 6:93–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sanju′n R., Nebot M.R., Chirico N., Mansky L.M., Belshaw R.. Viral mutation rates. J. Virol. 2010; 84:9733–9748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Irwin K.K., Renzette N., Kowalik T.F., Jensen J.D.. Antiviral drug resistance as an adaptive process. Virus Evol. 2016; 2:vew014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cui J., Li F., Shi Z.-L.. Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol. 2018; 17:181–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. de Wit E., van Doremalen N., Falzarano D., Munster V.J.. SARS and MERS: recent insights into emerging coronaviruses. Nat. Rev. Microbiol. 2016; 14:523–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhou P., Yang X.-L., Wang X.-G., Hu B., Zhang L., Zhang W., Si H.-R., Zhu Y., Li B., Huang C.-L. et al.. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020; 579:270–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hermann T. Small molecules targeting viral RNA. Wiley Interdiscipl. Rev.: RNA. 2016; 7:726–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bennett C.F. Therapeutic antisense oligonucleotides are coming of age. Annu. Rev. Med. 2019; 70:307–321. [DOI] [PubMed] [Google Scholar]
- 9. Madhugiri R., Karl N., Petersen D., Lamkiewicz K., Fricke M., Wend U., Scheuer R., Marz M., Ziebuhr J.. Structural and functional conservation of cis-acting RNA elements in coronavirus 5′-terminal genome regions. Virology. 2018; 517:44–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Madhugiri R., Fricke M., Marz M., Ziebuhr J.. Chapter four coronavirus cis-acting RNA elements. Adv. Virus Res. 2016; 96:127–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yang D., Leibowitz J.L.. The structure and functions of coronavirus genomic 3′ and 5′ ends. Virus Res. 2015; 206:120–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Brierley I., Digard P., Inglis S.C.. Characterization of an efficient coronavirus ribosomal frameshifting signal: Requirement for an RNA pseudoknot. Cell. 1989; 57:537–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Goebel S.J., Hsue B., Dombrowski T.F., Masters P.S.. Characterization of the RNA components of a putative molecular switch in the 3′ untranslated region of the murine coronavirus genome. J. Virol. 2004; 78:669–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Stammler S.N., Cao S., Chen S.-J., Giedroc D.P.. A conserved RNA pseudoknot in a putative molecular switch domain of the 3′-untranslated region of coronaviruses is only marginally stable. RNA. 2011; 17:1747–1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Goebel S.J., Miller T.B., Bennett C.J., Bernard K.A., Masters P.S.. A hypervariable region within the 3′ cis-Acting element of the murine coronavirus genome is nonessential for RNA synthesis but affects pathogenesis▿. J. Virol. 2007; 81:1274–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Robertson M.P., Igel H., Baertsch R., Haussler D., Ares M., Scott W.G.. The structure of a rigorously conserved RNA element within the SARS virus genome. PLoS Biol. 2005; 3:e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rangan R., Zheludev I.N., Das R.. RNA genome conservation and secondary structure in SARS-CoV-2 and SARS-related viruses: a first look. RNA. 2020; doi:10.1261/rna.076141.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Siegfried N.A., Busan S., Rice G.M., Nelson J.A.E., Weeks K.M.. RNA motif discovery by SHAPE and mutational profiling (SHAPE-MaP). Nat. Methods. 2014; 11:959–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zubradt M., Gupta P., Persad S., Lambowitz A.M., Weissman J.S., Rouskin S.. DMS-MaPseq for genome-wide or targeted RNA structure probing in vivo. Nat. Methods. 2017; 14:75–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Spitale R.C., Crisalli P., Flynn R.A., Torre E.A., Kool E.T., Chang H.Y.. RNA SHAPE analysis in living cells. Nat. Chem. Biol. 2013; 9:18–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Simon L.M., Morandi E., Luganini A., Gribaudo G., Martinez-Sobrido L., Turner D.H., Oliviero S., Incarnato D.. In vivo analysis of influenza A mRNA secondary structures identifies critical regulatory motifs. Nucleic. Acids. Res. 2019; 47:7003–7017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Untergasser A., Cutcutache I., Koressaar T., Ye J., Faircloth B.C., Remm M., Rozen S.G.. Primer3—new capabilities and interfaces. Nucleic. Acids. Res. 2012; 40:e115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Incarnato D., Morandi E., Simon L.M., Oliviero S.. RNA framework: an all-in-one toolkit for the analysis of RNA structures and post-transcriptional modifications. Nucleic Acids Res. 2018; 46:e97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Langmead B., Salzberg S.L.. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012; 9:357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lorenz R., Bernhart S.H., Siederdissen C.H.Z., Tafer H., Flamm C., Stadler P.F., Hofacker I.L.. ViennaRNA Package 2.0. Algorithms Mol. Biol.: AMB. 2011; 6:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cannone J.J., Subramanian S., Schnare M.N., Collett J.R., D’Souza L.M., Du Y., Feng B., Lin N., Madabusi L.V., Müller K.M. et al.. The comparative RNA web (CRW) site: an online database of comparative sequence and structure information for ribosomal, intron, and other RNAs. BMC Bioinformatics. 2002; 3:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Deigan K.E., Li T.W., Mathews D.H., Weeks K.M.. Accurate SHAPE-directed RNA structure determination. Proc. Natl. Acad. Sci. U.S.A. 2009; 106:97–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Huber R.G., Lim X.N., Ng W.C., Sim A.Y.L., Poh H.X., Shen Y., Lim S.Y., Sundstrom K.B., Sun X., Aw J.G. et al.. Structure mapping of dengue and Zika viruses reveals functional long-range interactions. Nat. Commun. 2019; 10:1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nawrocki E.P., Eddy S.R.. Infernal 1.1: 100-fold faster RNA homology searches. Bioinform Oxf. Engl. 2013; 29:2933–2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pickett B.E., Sadat E.L., Zhang Y., Noronha J.M., Squires R.B., Hunt V., Liu M., Kumar S., Zaremba S., Gu Z. et al.. ViPR: an open bioinformatics database and analysis resource for virology research. Nucleic Acids Res. 2011; 40:D593–D598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lauber C., Goeman J.J., Parquet M., del C., Nga P.T., Snijder E.J., Morita K., Gorbalenya A.E.. The footprint of genome architecture in the largest genome expansion in RNA viruses. PLoS Pathog. 2013; 9:e1003500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rivas E., Clements J., Eddy S.R.. A statistical test for conserved RNA structure shows lack of evidence for structure in lncRNAs. Nat. Methods. 2017; 14:45–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Katoh K., Standley D.M.. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 2013; 30:772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Boniecki M.J., Lach G., Dawson W.K., Tomala K., Lukasz P., Soltysinski T., Rother K.M., Bujnicki J.M.. SimRNA: a coarse-grained method for RNA folding simulations and 3D structure prediction. Nucleic. Acids. Res. 2015; 44:e63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Magnus M., Boniecki M.J., Dawson W., Bujnicki J.M.. SimRNAweb: a web server for RNA 3D structure modeling with optional restraints. Nucleic Acids Res. 2016; 44:W315–W319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Stasiewicz J., Mukherjee S., Nithin C., Bujnicki J.M.. QRNAS: software tool for refinement of nucleic acid structures. BMC Struct. Biol. 2019; 19:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Salomon‐Ferrer R., Case D.A., Walker R.C.. An overview of the Amber biomolecular simulation package. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2013; 3:198–210. [Google Scholar]
- 38. Waleń T., Chojnowski G., Gierski P., Bujnicki J.M.. ClaRNA: a classifier of contacts in RNA 3D structures based on a comparative analysis of various classification schemes. Nucleic. Acids. Res. 2014; 42:e151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Guilloux V.L., Schmidtke P., Tuffery P.. Fpocket: an open source platform for ligand pocket detection. BMC Bioinformatics. 2009; 10:168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wirecki T.K., Merdas K., Bernat A., Boniecki M.J., Bujnicki J.M., Stefaniak F.. RNAProbe: a web server for normalization and analysis of RNA structure probing data. Nucleic Acids Res. 2020; 48:W292–W299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Spitale R.C., Flynn R.A., Zhang Q.C., Crisalli P., Lee B., Jung J.-W., Kuchelmeister H.Y., Batista P.J., Torre E.A., Kool E.T. et al.. Structural imprints in vivo decode RNA regulatory mechanisms. Nature. 2015; 519:486–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rouskin S., Zubradt M., Washietl S., Kellis M., Weissman J.S.. Genome-wide probing of RNA structure reveals active unfolding of mRNA structures in vivo. Nature. 2014; 505:701–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Beaudoin J.-D., Novoa E.M., Vejnar C.E., Yartseva V., Takacs C.M., Kellis M., Giraldez A.J.. Analyses of mRNA structure dynamics identify embryonic gene regulatory programs. Nat. Struct. Mol. Biol. 2018; 25:677–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Incarnato D., Morandi E., Anselmi F., Simon L.M., Basile G., Oliviero S.. In vivo probing of nascent RNA structures reveals principles of cotranscriptional folding. Nucleic Acids Res. 2017; 45:9716–9725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zhang K., Zheludev I.N., Hagey R.J., Wu M.T.-P., Haslecker R., Hou Y.J., Kretsch R., Pintilie G.D., Rangan R., Kladwang W. et al.. Cryo-electron microscopy and exploratory antisense targeting of the 28-kDa frameshift stimulation element from the SARS-CoV-2 RNA genome. 2020; bioRxiv doi:20 July 2020, preprint: not peer reviewed 10.1101/2020.07.18.209270. [DOI] [PMC free article] [PubMed]
- 46. Hewitt W.M., Calabrese D.R., Schneekloth J.S.. Evidence for ligandable sites in structured RNA throughout the Protein Data Bank. Bioorg. Med. Chem. 2019; 27:2253–2260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wirecki T.K., Nithin C., Mukherjee S., Bujnicki J.M., Boniecki M.J.. Protein structure prediction. Methods Mol. Biol. Clifton NJ. 2020; 2165:103–125. [DOI] [PubMed] [Google Scholar]
- 48. Jonassen C.M., Jonassen T.O., Grinde B.. A common RNA motif in the 3′ end of the genomes of astroviruses, avian infectious bronchitis virus and an equine rhinovirus. J. Gen. Virol. 1998; 79:715–718. [DOI] [PubMed] [Google Scholar]
- 49. Tengs T., Kristoffersen A.B., Bachvaroff T.R., Jonassen C.M.. A mobile genetic element with unknown function found in distantly related viruses. Virol J. 2013; 10:132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Warner K.D., Hajdin C.E., Weeks K.M.. Principles for targeting RNA with drug-like small molecules. Nat. Rev. Drug Discov. 2018; 17:547–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Park S.-J., Kim Y.-G., Park H.-J.. Identification of RNA pseudoknot-binding ligand that inhibits the -1 ribosomal frameshifting of SARS-coronavirus by structure-based virtual screening. J. Am. Chem. Soc. 2011; 133:10094–10100. [DOI] [PubMed] [Google Scholar]
- 52. Lu Z., Zhang Q.C., Lee B., Flynn R.A., Smith M.A., Robinson J.T., Davidovich C., Gooding A.R., Goodrich K.J., Mattick J.S. et al.. RNA duplex map in living cells reveals higher-order transcriptome structure. Cell. 2016; 165:1267–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Nguyen T.C., Cao X., Yu P., Xiao S., Lu J., Biase F.H., Sridhar B., Huang N., Zhang K., Zhong S.. Mapping RNA-RNA interactome and RNA structure in vivo by MARIO. Nat. Commun. 2016; 7:12023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Aw J.G.A., Shen Y., Wilm A., Sun M., Lim X.N., Boon K.-L., Tapsin S., Chan Y.-S., Tan C.-P., Sim A.Y.L. et al.. In vivo mapping of eukaryotic RNA interactomes reveals principles of higher-order organization and regulation. Mol. Cell. 2016; 62:603–617. [DOI] [PubMed] [Google Scholar]
- 55. Ziv O., Gabryelska M.M., Lun A.T.L., Gebert L.F.R., Sheu-Gruttadauria J., Meredith L.W., Liu Z.-Y., Kwok C.K., Qin C.-F., MacRae I.J. et al.. COMRADES determines in vivo RNA structures and interactions. Nat. Methods. 2018; 15:785–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ziv O., Price J., Shalamova L., Kamenova T., Goodfellow I., Weber F., Miska E.A.. The short- and long-range RNA-RNA Interactome of SARS-CoV-2. 2020; bioRxiv doi:20 July 2020, preprint: not peer reviewed 10.1101/2020.07.19.211110. [DOI] [PMC free article] [PubMed]
- 57. Lan T.C.T., Allan M.F., Malsick L.E., Khandwala S., Nyeo S.S.Y., Bathe M., Griffiths A., Rouskin S.. Structure of the full SARS-CoV-2 RNA genome in infected cells. 2020; bioRxiv doi:30 June 2020, preprint: not peer reviewed 10.1101/2020.06.29.178343. [DOI]
- 58. Lenartowicz E., Nogales A., Kierzek E., Kierzek R., Martínez-Sobrido L., Turner D.H.. Antisense oligonucleotides targeting influenza a segment 8 genomic RNA inhibit viral replication. Nucleic Acid Ther. 2016; 26:277–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Spurgers K.B., Sharkey C.M., Warfield K.L., Bavari S.. Oligonucleotide antiviral therapeutics: antisense and RNA interference for highly pathogenic RNA viruses. Antivir Res. 2008; 78:26–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Laxton C., Brady K., Moschos S., Turnpenny P., Rawal J., Pryde D.C., Sidders B., Corbau R., Pickford C., Murray E.J.. Selection, optimization, and pharmacokinetic properties of a novel, potent antiviral locked nucleic acid-based antisense oligomer targeting hepatitis C virus internal ribosome entry site. Antimicrob. Agents Ch. 2011; 55:3105–3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Takahashi M., Li H., Zhou J., Chomchan P., Aishwarya V., Damha M.J., Rossi J.J.. Dual mechanisms of action of self-delivering, anti-HIV-1 FANA oligonucleotides as a potential new approach to HIV therapy. Mol. Ther. - Nucleic Acids. 2019; 17:615–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
SHAPE-MaP data has been deposited to the Gene Expression Omnibus (GEO) database, under the accession GSE151327. RNA Count (RC) files for use with RNA Framework, as well as XML files with normalized SHAPE-MaP reactivities are provided, together with the full secondary structure in dot-bracket notation and the selected low Shannon - low SHAPE structures. Stockholm alignment files and the derived covariance models (CMs) for the structure elements, showing significant covariation support, are also provided. For each structured segment in the SARS-CoV-2 genome, we further provide the representative 3D structure models for up to 10 of the largest clusters, illustrating the most typical conformational classes, as well as the 1000 best-energy models used to predict ligand-binding modes. In all models, the B-factor field presents the relative ligand-binding score obtained from the Fpocket analysis. These additional processed files (RNA Framework RC and XML files, dot-bracket structures, CMs, Stockholm alignments, 3D models of SARS-CoV-2 segments and druggable pockets) are available at http://www.incarnatolab.com/datasets/SARS_Manfredonia_2020.php and on Mendeley at http://dx.doi.org/10.17632/8gj97c4kgv.1.