


Abstract
Nucleotide excision repair (NER) in eukaryotes is orchestrated by the core form of the general transcription factor TFIIH, containing the helicases XPB, XPD and five ‘structural’ subunits, p62, p44, p34, p52 and p8. Recent cryo-EM structures show that p62 makes extensive contacts with p44 and in part occupies XPD’s DNA binding site. While p44 is known to regulate the helicase activity of XPD during NER, p62 is thought to be purely structural. Here, using helicase and adenosine triphosphatase assays we show that a complex containing p44 and p62 enhances XPD’s affinity for dsDNA 3-fold over p44 alone. Remarkably, the relative affinity is further increased to 60-fold by dsDNA damage. Direct binding studies show this preference derives from p44/p62’s high affinity (20 nM) for damaged ssDNA. Single molecule imaging of p44/p62 complexes without XPD reveals they bind to and randomly diffuse on DNA, however, in the presence of UV-induced DNA lesions these complexes stall. Combined with the analysis of a recent cryo-EM structure, we suggest that p44/p62 acts as a novel DNA-binding entity that enhances damage recognition in TFIIH. This revises our understanding of TFIIH and prompts investigation into the core subunits for an active role during DNA repair and/or transcription.
INTRODUCTION
Cell survival relies on accurate replication and transcription of genetic material. When DNA is damaged, rapid, efficient repair is essential to ensure genome integrity. Deficiencies in these repair processes have severe consequences as exemplified by defects in the nucleotide excision repair (NER) machinery leading to diseases such as xeroderma pigmentosum (XP), which is phenotypically characterized by UV hyper-sensitivity and increased incidence of skin cancers (1). Mutations found in many components of the NER pathway, including TFIIH can lead to XP. The core structure of TFIIH comprises two helicases; of these, XPB is enzymatically involved in both transcription and repair, whereas XPD’s activity is only essential for repair (2,3). In addition, TFIIH possesses five other core subunits: p44, p62, p34, p8 and p52 (4–13). While it has been shown that p44 and p52/p8 can stimulate XPD and XPB’s helicase activities respectively (8,14,15), most of the core subunits remain relatively unstudied. A recent cryo-EM structure of TFIIH reveals significant interactions between p44, p62 and XPD (16). Part of p62 is located in XPD’s DNA binding site, suggesting that p62 might assume a role in modulating XPD’s ability to interact with DNA. This would expand p62’s function beyond stabilizing TFIIH and acting as a hub for protein interactions in transcription and crucially with the initial damage sensor XPC-HR23B for NER (14,17–20).
Previous attempts to purify full-length p44 have been hampered by protein stability issues, however, a truncated form of p44 (N-p44) has been expressed and has been shown to activate the adenosine triphosphatase (ATPase) of XPD (21). In this study, we successfully purified full length p44 through co-expression with p62. We show that the complex formed by both proteins enhances XPD’s affinity for dsDNA. However, the presence of damaged dsDNA dramatically increases the relative affinity for the ternary XPD-p44/p62 complex 60-fold versus XPD-N-p44. Remarkably, in the absence of XPD, we observe that p44/p62 also preferentially binds to damaged single-stranded DNA compared to undamaged ssDNA, likely explaining the enhanced selectivity for damage in the presence of XPD-p44/p62. Using single molecule fluorescence microscopy, we show that p44/p62 alone binds and randomly diffuses on non-damaged dsDNA, but this complex is stalled in the presence of UV lesions, suggesting a direct response to damage. This hypothesis is supported by the in-depth analysis of a recent TFIIH cryo-EM structure (22), where we placed p62 fragments into unassigned regions of the EM maps to propose that p62 is located proximally to XPD’s DNA binding pocket when bound to single-stranded DNA (ssDNA). Our data thus reveal that p44/p62 acts as a damage-specific DNA binding entity within TFIIH and we provide a structural model for how this may be mediated.
MATERIALS AND METHODS
Purification
The genes encoding p44 and p62 were cloned from Chaetomium thermophilum cDNA. p62 was cloned into the pETM-11 vector (EMBL) without a tag. p44 was cloned into the pBADM-11 vector (EMBL) containing an N-terminal hexa-Histidine tag followed by a TEV cleavage site. p62 and p44 were co-expressed in Escherichia coli BL21 CodonPlus (DE3) RIL cells (Agilent) and were co-purified via immobilized metal affinity chromatography (Ni-TED, Macherey-Nagel), followed by size exclusion chromatography (SEC), and anion exchange chromatography (AEC). SEC was conducted with a HiLoad 16/600 Superdex 200 prep grade column (CYTIVA) in 20 mM HEPES pH 7.5, 250 mM NaCl, and 1 mM TCEP. AEC was conducted with a MonoQ 5/50 GL column (CYTIVA). The proteins were eluted via a salt gradient ranging from 50 to 1000 mM NaCl. AEC buffers were composed of 20 mM HEPES pH 7.5, 50/1000 mM NaCl and 1 mM TCEP. The p44/p62 protein complex was concentrated to ∼20 mg/ml and flash frozen in liquid nitrogen for storage.
XPD and N-p44 (1–285) from C. thermophilum were cloned as described previously (3). XPD was expressed as N-terminally His-tagged proteins in E. coli ArcticExpress (DE3)-RIL cells (Agilent). Cells were grown in TB medium at 37°C until they reached an OD600 of 0.6–0.8. Expression was initiated by the addition of 0.05% L-arabinose and performed at 11°C for 20 h. N-p44 was expressed as N-terminally His-tagged protein in E. coli BL21-CodonPlus (DE3)-RIL cells (Stratagene). Cells were grown as described for ctXPD and expression was started by adding 0.1 mM IPTG at 14°C for 18 h. XPD and N-p44 were purified to homogeneity by metal affinity chromatography (Ni-IDA, Macherey-Nagel) as described previously (3) followed by SEC (20 mM HEPES pH 7.5, 200 mM NaCl) and an additional AEC step in the case of XPD. AEC was performed using a MonoQ 5/50 GL column (CYTIVA) with 20 mM HEPES pH 7.5, 50 mM NaCl and 1 mM TCEP as loading buffer and the same buffer containing 1 M NaCl was used for elution. The final buffer after AEC was 20 mM HEPES pH 7.5, 250 mM NaCl and 1 mM TCEP. The proteins were concentrated to at least 5 mg/ml based on their calculated extinction coefficient using ProtParam (SwissProt) and then flash frozen for storage at −80°C.
Size exclusion chromatography
SEC of XPD, p44/p62 and the XPD/p44/p62 complex was performed in 20 mM HEPES pH 7.5, 250 mM NaCl, 1 mM TCEP at a concentration of 40 μM. XPD was mixed with p44/p62 at an equal molar ratio with the final concentration of 40 μM prior to the SEC analysis. SEC was performed on an Äkta Pure system using a Superdex200 Increase 3.2/300 at a flow rate of 0.05 ml/min.
Fluorescence anisotropy
DNA binding was analyzed by fluorescence anisotropy employing the DNA substrates indicated in the methods section for the ATPase with the addition of a Cy3 label on the 3′ end of F26,50 with and without the modification. In addition, we used a hairpin DNA representing the open fork structure with a Cy3 label on the 3′ end (5′TTT TTT TTT TTT TTTCCC GGC CAT GC *GAA* GC ATG GCC GGG TTT TT3′, the stars (*) mark the unpaired bases within the hairpin). Assays were carried out in 20 mM HEPES pH 7.5, 30 mM KCl, 5 mM MgCl2, 1 mM TCEP and 5 nM DNA at room temperature. The protein was used at concentrations of 2–5000 nM as indicated. Fluorescence was detected at an excitation wavelength of 540 nm and an emission wavelength of 590 nm with a Clariostar plate reader (BMG labtech). The gain was adjusted to a well containing buffer and DNA but no protein. Curves were fitted with GraphPad Prism and represent the averages of at least three different reactions.
ATPase assay
dsDNA substrates used:
F26,50 contains a fluorescein moiety covalently attached to thymine (*);
5′GACTACGTACTGTTACGGCTCCATCT*CTACCGCAATCAGGCCAGATCTGC 3′
The reverse complementary sequence to F26,50;
5′GCAGATCTGGCCTGATTGCGGTAGCGATGGAGCCGTAACAGTACGTAGTC 3′
F26,50 without the fluorescein moiety;
5′GACTACGTACTGTTACGGCTCCATCTCTACCGCAATCAGGCCAGATCTGC 3′
The NADH-coupled ATPase assay was performed as described previously (23) in plate reader format. Imaging buffer containing the NADH-reaction components was supplemented with 1 mM fresh TCEP, protein (50 nM (equimolar concentrations for XPD N-p44 and XPD p44/p62)), and increasing amounts of DNA substrate (4–500 nM). The reaction was started with the addition of 2 mM ATP to each well, and the change in OD340 (NADH) was monitored every 8 seconds/well over 30 min at room temperature in a Clariostar plate reader. The rates of NADH consumption were used to calculate kcat. Reactions were repeated at least three times and include biological replicates, and S.E.M used as errors values.
In vitro helicase assay
Helicase activity was analyzed utilizing a fluorescence-based assay. We used an open fork substrate with a Cy3 label at the 3′ end of the translocated strand where unwinding of the DNA substrate reduces quenching of the Cy3 fluorescence.
5′AGCTACCATGCCTGCACGAATTAAGCAATTCGTAATCATGGTCATAGC-Cy3 3′ and a dabcyl modification on the 5′ end of the opposite strand
5′ Dabcyl-GCTATGACCATGATTACGAATTGCTTGGAATCCTGACGAACTGTAG 3′
Assays were carried out in 20 mM HEPES pH 7.5, 50 mM KCl, 5 mM MgCl2, and 1 mM TCEP. DNA was used at a concentration of 250 nM. Helicase activity was measured with equimolar concentrations of XPD in the presence of N-p44 or p44/p62, respectively. The mix of reagents, were preincubated at 37°C and the reaction was subsequently started with the addition of 5 mM ATP. Kinetics were recorded with a Fluostar Optima plate reader (BMG labtech). Fluorescence was detected at an excitation wavelength of 550 nm (slit width, 2 nm) and an emission wavelength of 570 nm (slit width, 2 nm). Initial velocities were fitted with the MARS software package (BMG labtech) and represent the averages of at least three different reactions and two independent protein batches.
Single molecule DNA tightrope assay
For a detailed protocol see (24). p44/p62 interactions with DNA were studied in imaging buffer (20 mM Tris pH 8.0, 10 mM KCl (100 mM for high salt), 5 mM MgCl2, 1 mM TCEP). Videos for diffusion analysis were collected between 30 s and 5 min at 10 frames per second. Video analysis was performed in ImageJ as described previously (25). Experiments using UV damaged lambda DNA are described elsewhere (23). Briefly, lambda DNA was irradiated to 500 J/m2 using a 254 nm lamp immediately before constructing DNA tightropes. Exposure of DNA to this wavelength and power randomly induces ∼106 CPDs per tightrope (calculation based on (23,26)) which are recognised as damage by NER (27).
Structural model building
The additional p62 fragments were built into the cryo-EM map of pdb entry 6RO4 using manual model building features in coot. For the C-terminal part, the corresponding segments of p62 from the PDB entry 6NMI (residues 397–547) were used and fitted into the cryo-EM map. The N-terminal helical bundle was built de novo and residues were marked as unknown since the assignment of residues in this segment was ambiguous. Final real space refinement was carried out using the Phenix Suite (28).
RESULTS AND DISCUSSION
p44/p62 co-purify as a complex that stimulate XPD’s activity
Both p44 and p62 make extensive contacts with XPD in TFIIH (16). To investigate this interaction, recombinant p44, p62 and XPD from C. thermophilum were utilized. All subunits share high sequence homology with the human TFIIH subunits and we have shown previously that they serve as an ideal model for the human proteins (3,29). Only the N-terminal region (residues 1–285) of p44, containing the von Willebrand domain (N-p44), has been successfully purified in isolation to date, nonetheless this is sufficient to activate XPD’s ATPase and helicase (3). By co-expressing full-length p44 with full-length p62, we purified a tightly associated complex of p44/p62 that also interacts with XPD (Figure 1), thus providing the first successful purification of full length p44 and suggesting that p44 requires p62 to fold correctly.
Figure 1.

SEC analysis of XPD, the dimeric p44/p62 and the ternary XPD/p44/p62 complexes. SEC chromatograms of XPD (92 kDa), p44/p62 (132 kDa) and the XPD/p44/p62 (224 kDa) complex were performed in 20 mM HEPES pH 7.5, 250 mM NaCl and 1 mM TCEP on a Superdex200 Increase 3.2/300. XPD was mixed with p44/p62 at an equal molar ratio (final concentration of 40 μM) prior to SEC. The inset shows the SDS-PAGE analysis of the center fractions from the XPD/p44/p62 elution, the markers on the x-axis correspond to the fractions in the gel.
Recently Greber et al. showed by cryo-EM, that a portion of p62 is located in XPD’s DNA binding site within the DNA-free transcriptional apo-TFIIH structure (16). To understand how this might affect XPD, we measured the helicase activity of XPD-p44/p62 relative to XPD-N-p44. Surprisingly, XPD-p44/p62 possessed a significantly enhanced ability to unwind the DNA substrate compared to XPD-N-p44 (Figure 2A), indicating that p62 enhances rather than blocks DNA binding. This was confirmed using a dsDNA titration of XPD’s ATPase activity in complex with either N-p44 or p44/p62, (Figure 2B and C) revealing a substantial enhancement in the affinity of XPD-p44/p62 for dsDNA (57.8 ± 9.0 nM) versus XPD-N-p44 (149.6 ± 55 nM). Surprisingly, the ATPase kcat rate constant was only slightly affected by the presence of p62 (Figure 2B). Altogether these data suggest that p62 activates XPD by enhancing its affinity for DNA.
Figure 2.

Steady-state ATPase and helicase activity of XPD complexes in the presence of various DNA substrates and core TFIIH proteins. (A) XPD’s helicase activity is stimulated by N-p44 (hashed) and p44/p62 (white) on an open fork substrate with lengths indicated in nucleotides. XPD alone displays no helicase activity (21). Errors are shown as S.E.M from nine repeats. Statistical significance determined using a student's t-test where * = P < 0.05, n.s = not statistically significant. (B) Summary table of the kcat values and KM values obtained from DNA substrate titrations. Errors are S.E.M fit values. (C) Summary of the effect of DNA substrates on XPD-p44/p62 (white) and XPD-N-p44 (hashed) affinities. XPD-p44/p62 shows a 60-fold enhancement in affinity versus XPD-N-p44. (D) XPD-p44/p62’s ATPase is stimulated by the presence of DNA substrates. Damaged dsDNA (squares) binds much more tightly than undamaged DNA (triangles), and approximately equivalent to ssDNA (circles). ATPase rates are normalized to the protein concentration used so that the asymptote reports the kcat. Errors are shown as S.E.M from three or more repeats. In the absence of DNA, XPD possesses a slow ATPase that is unaffected by N-p44, p44/p62 or damaged DNA (Supplementary Table S1) indicating that p44/p62 displays no ATPase activity by itself.
More remarkably, titrating different DNA substrates while measuring the ATPase of XPD-p44/p62 (Figure 2C and D) revealed a substantial enhancement in the affinity for fluorescein-modified (recognized as damage by NER (27)) dsDNA (2.7 ± 0.2 nM) versus undamaged dsDNA (57.8 ± 9.0 nM). In contrast, XPD-N-p44 showed near identical affinity for dsDNA regardless of the presence of DNA damage (Figure 2C). Both XPD-N-p44 and XPD-p44/p62 showed similar affinity for ssDNA (2.5 ± 0.5 versus 2.6 ± 0.2 nM), indicating p44/p62 is not simply displaying a higher affinity for exposed ssDNA (Figure 2B and D). These observations provide the first evidence that p62 in the ternary XPD-p44/p62 complex may enhance the affinity of the complex for DNA damage. Such behavior explains the sensitization to UV irradiation observed for truncations of the yeast p62 homolog (17,30,31).
The p44/p62 complex binds directly to DNA
How p62 enhances the affinity of the ternary complex for DNA is unknown; however, it is conceivable that p62 undergoes a conformational change and is thereby able to interact directly with the DNA. To investigate this hypothesis, we determined p44/p62’s affinity for different DNA substrates relevant to NER by fluorescence polarization (Figure 3). In support of our hypothesis, p44/p62 was found to bind dsDNA with a KD of 1.18 μM. Interestingly, DNA intermediates expected to form during NER showed even stronger binding by p44/p62 than dsDNA (KD (ssDNA) 0.84 μM and KD (open fork) 0.21 μM).
Figure 3.

p44/p62 binding affinities for different DNA substrates. Fluorescence polarization was used to determine the binding of various substrates to increasing concentrations of p44/p62. p44/p62 displayed an intermediate affinity for dsDNA irrespective of the presence of a fluorescein DNA damage, but a higher affinity for open fork and ssDNA. The tightest binding was observed with ssDNA containing a fluorescein DNA damage. Data were plotted and fitted in GraphPad, presented here as normalized values with error bars representing the SD from at least three repeats.
Since p44/p62 enhanced XPD’s affinity for damaged DNA, we also investigated the affinity of p44/p62 for different DNA damage containing substrates independent of XPD. p44/p62 bound dsDNA containing a fluorescein moiety with an affinity similar to that of undamaged DNA (KD 1.23 and 1.18 μM, respectively). In contrast, we observed a striking 40-fold increase in binding affinity to a ssDNA substrate containing a central fluorescein lesion (KD 0.02 μM), compared to the ssDNA substrate without damage. The tighter binding to these NER intermediates strengthens the hypothesis that p62 contacts DNA within TFIIH, and specifically locates to damage within an opened bubble.
The p44/p62 complex independently diffuses on DNA
To directly determine how p44/p62 interacts with DNA, we imaged individual fluorescently tagged proteins binding to DNA tightropes in real time using fluorescence microscopy (25). In this assay, p44/p62 is fluorescently labeled using a quantum dot (Qdot) conjugated to the protein via its poly-histidine purification tag and an anti-His IgG antibody (24). Individual protein molecules are then imaged interacting with single DNA molecules in real time (Figure 4A). We observed substantial binding of p44/p62 complexes to dsDNA, and of these ∼80% diffused randomly (n = 599 total). To investigate the mechanism of p44/p62’s diffusion on the DNA, the diffusion constant and exponent (used to determine if motion is directionally biased) was calculated in high and low salt conditions (100 versus 10 mM KCl). At elevated KCl concentrations fewer p44/p62 complexes bound to DNA, and the calculated diffusion constant using mean-squared displacement analysis (25) showed no significant change (P > 0.05) between salt conditions (10 mM KCl 0.067 μm2/s ± 0.006 versus 100 mM KCl 0.042 μm2/s ± 0.010; Figure 4B), consistent with p44/p62 complexes sliding along the DNA helix (32). The diffusive exponent was found to be ∼0.9 in either salt condition, which indicates that there is no directional bias to the diffusive motion (33). Based on the estimated size of a p44/p62 complex conjugated to a Qdot, the diffusion constant is consistent with rotation-coupled diffusion around the backbone of the DNA helix (25,34,35). Altogether, these data provide the first observation that the p44/p62 complex can bind dsDNA and randomly diffuse, independently of XPD.
Figure 4.

Imaging of p44/p62’s motility on DNA tightropes. (A) Schematic drawing of the DNA tightrope assay: Qdot-labeled proteins are imaged interacting with single molecules of DNA suspended between 5 μm diameter beads. (B) Diffusion constant versus exponent (alpha) at high (circles) and low salt (crosses). The average diffusion constant values are given in the main text. Average alpha exponent values were 0.89 ± 0.04 and 0.91 ± 0.02 in high and low salt, respectively. (C) Single molecule fluorescence imaging reveals that the number of diffusing p44/p62 complexes on dsDNA tightropes decreases in the presence of UV-induced DNA damage. Data are average percentages from 8 (No UV) or 6 (UV) experimental repeats with the S.E.M as errors bars, Statistical significance determined using a student's t-test where * = P < 0.05. (D) Example kymograph of a molecule showing constrained diffusion. The scale-bar represents 10 s and 1 μm.
p44/p62 complexes are stalled by the presence of damaged DNA
To further analyze these observations, we investigated the effect of DNA damage on the behavior of single p44/p62 complexes on DNA tightropes irradiated with 500 J/m2 of 254 nm UV light, which generates cyclobutane pyrimidine dimers and 6–4 photoproducts (23). We observed a striking and significant decrease in the number of diffusing p44/p62 complexes from ∼80% to 50% ((P < 0.05) Figure 4C). These results clearly indicate that p44/p62 independently recognizes DNA damage and stalls at damaged sites. We also observed a change in the mechanism of p44/p62’s diffusion in the presence of UV damage; a population of p44/p62 complexes now appeared limited in their total excursion distance on DNA (Figure 4D), similar to the damage recognition protein XPC, which undergoes constrained diffusion around a lesion (36,37). Although p44/p62 cannot discriminate DNA damage in a dsDNA substrate without XPD (Figure 3), the observed stalling in the DNA tightrope assay suggests that p44/p62 may have located exposed ssDNA regions containing UV lesions.
In summary, we have shown an enhanced affinity of XPD-p44/p62 for damaged dsDNA (Figure 2), enhanced affinity of p44/p62 for damaged ssDNA but not to damaged dsDNA (Figure 3), and an ability to differentiate damage sites on dsDNA tightropes (Figure 4C). Altogether, these observations are consistent with a role for p44/p62 in recognizing DNA lesions in the opened bubble of TFIIH.
The structural basis of p62-DNA contacts within TFIIH
The recent cryo-EM structure of TFIIH bound to DNA shows the path of single-stranded DNA as it is passed from XPB to XPD (22). In this structure, p44 is located in the same position as in apo-TFIIH (16), but p62 is unassigned. However, additional cryo-EM densities can be observed in the structure at a position similar to p62 in apo-TFIIH (Figure 5A) (16). We modeled regions of p62 from the apo-TFIIH into the DNA-bound TFIIH structure and found excellent agreement with the position of p62 (Figure 5B). This model suggests that the cryo-EM density close to XPD could represent N-terminal regions of p62. Closer examination of the DNA binding groove within XPD shows that p62 occupies this site in the absence of DNA (Figure 5C), i.e. in the apo TFIIH structure. Analysis of the same region in the DNA-bound structure suggests that the p62 loop could undergo a conformational change to form a helical bundle that interacts with the Arch domain of XPD and the ssDNA bound to XPD (Figure 5D); however, we cannot entirely exclude the small possibility that another protein (e.g. XPG) also contributes to this density. How these interactions result in damage recognition is an open question since the precise mechanism of damage recognition/verification has not been established. However, it could be postulated that p62’s Arch interaction affects recognition at the FeS cluster pocket (38). Or even more speculative, given its position 5′ from the FeS cluster, it is also conceivable that a second recognition site may exist, explaining the origin of differential damage detection (27). This would be in good agreement with damage recognition site postulated for the XPD homolog DinG that is located directly below the p62 Arch domain junction (39).
Figure 5.
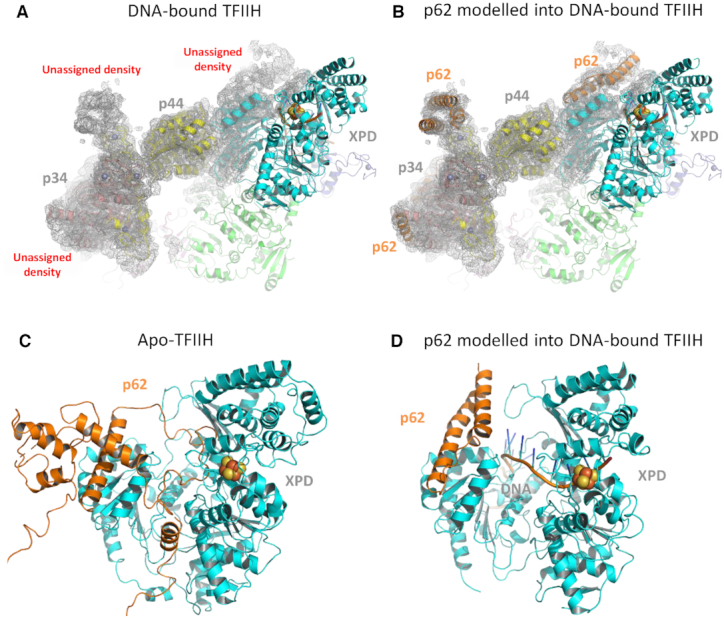
The position of p62 in apo- and DNA bound TFIIH structures. (A) Representation of the Kokic et al. DNA-bound TFIIH structure (PDB ID: 6RO4) shown in cartoon mode with the EM-envelope displayed as gray mesh. (B) The unmodeled cryo-EM density around p44/p34 was fit using p62 from the Greber et al. apo-TFIIH structure (PDB ID: 6NMI) and the three helical bundle (residues 450–547) superimposed with an rmsd of 0.5 Å after rebuilding. Residues 397–450 differ in orientation due the concerted movement of XPD-p44 in 6RO4. The additional part of p62 interacting with XPD was modeled according to the cryo-EM density with no defined amino acid sequence. (C) Close-up view of XPD and p62 based on the apo-TFIIH structure, with the iron sulphur cluster in XPD shown as spheres. (D) Equivalent view of XPD from the DNA-bound TFIIH structure with the additional cryo-EM density modeled as p62. The modeled structure has been deposited in the worldwide protein databank, accession code: 7AD8.
In conclusion, we present the first mechanistic characterization of the non-helicase TFIIH subunits p44/p62. Our biochemical data clearly show that the p44/p62 complex binds to DNA, but displays a profoundly tighter affinity for ssDNA containing damage (Figure 3). Modeling of non-assigned densities in a recent cryo-EM structure indicate p62 to be in a position where it could interact with XPD and DNA in a substrate dependent manner (Figure 5). Taken together, these data suggest that p44/p62 interacts with ssDNA bound to XPD and is capable of modulating its helicase/ATPase activity through interactions with the helicase and/or Arch domains. This provides a structural explanation for the observed DNA damage-dependent increase in XPD’s DNA substrate affinity in the presence of p44/p62 (Figure 2). Furthermore, we have used fluorescence polarization binding studies and single molecule imaging to directly demonstrate that p44/p62 complexes are capable of recognizing UV damage and consequently stalling on DNA (Figures 3 and 4). This behavior could be important for stalling XPD at a lesion and triggering damage verification. Nonetheless, our results clearly show that the p44/p62 complex plays an active role in TFIIH’s recognition of DNA damage and indicates p44/p62 can no longer be thought of simply as ‘structural subunits’.
DATA AVAILABILITY
All data will be made available through the University of Kent academic repository (https://kar.kent.ac.uk/). The structure modeled in Figure 5 has been deposited in the worldwide protein databank, accession code: 7AD8.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank the members of the Kad group for useful discussions.
Author Contributions: Collected data: J.T.B., J.K. and W.K. Designed experiments: J.T.B., J.K., C.K. and N.M.K. Analyzed Data: J.T.B., J.K. and N.M.K.. Wrote paper: J.T.B., J.K., C.K. and N.M.K.
Contributor Information
Jamie T Barnett, School of Biological Sciences, University of Kent, Canterbury CT2 7NH, UK.
Jochen Kuper, Rudolf Virchow Center for Integrative and Translational Bioimaging, Institute for Structural Biology, University of Würzburg, 97080 Würzburg, Germany.
Wolfgang Koelmel, Rudolf Virchow Center for Integrative and Translational Bioimaging, Institute for Structural Biology, University of Würzburg, 97080 Würzburg, Germany.
Caroline Kisker, Rudolf Virchow Center for Integrative and Translational Bioimaging, Institute for Structural Biology, University of Würzburg, 97080 Würzburg, Germany.
Neil M Kad, School of Biological Sciences, University of Kent, Canterbury CT2 7NH, UK.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Biotechnology and Biological Sciences Research Council (BBSRC) [BB/P00847X/1, BB/M019144/1, BB/I003460/1 to N.M.K., BB/M01603X/1 to N.M.K., J.T.B.]; German Research Foundation [KI-562/7-1 to C.K.] Funding for open access charge: University of Kent; BBSRC.
Conflict of interest statement. None declared.
REFERENCES
- 1. Bradford P.T., Goldstein A.M., Tamura D., Khan S.G., Ueda T., Boyle J., Oh K.S., Imoto K., Inui H., Moriwaki S. et al.. Cancer and neurologic degeneration in xeroderma pigmentosum: long term follow-up characterises the role of DNA repair. J. Med. Genet. 2011; 48:168–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Coin F., Oksenych V., Egly J.M.. Distinct roles for the XPB/p52 and XPD/p44 subcomplexes of TFIIH in damaged DNA opening during nucleotide excision repair. Mol. Cell. 2007; 26:245–256. [DOI] [PubMed] [Google Scholar]
- 3. Kuper J., Braun C., Elias A., Michels G., Sauer F., Schmitt D.R., Poterszman A., Egly J.M., Kisker C.. In TFIIH, XPD helicase is exclusively devoted to DNA repair. PLoS Biol. 2014; 12:e1001954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Roy R., Schaeffer L., Humbert S., Vermeulen W., Weeda G., Egly J.M.. The DNA-dependent ATPase activity associated with the class II basic transcription factor BTF2/TFIIH. J. Biol. Chem. 1994; 269:9826–9832. [PubMed] [Google Scholar]
- 5. Greber B.J., Nguyen T.H.D., Fang J., Afonine P.V., Adams P.D., Nogales E.. The cryo-electron microscopy structure of human transcription factor IIH. Nature. 2017; 549:414–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sung P., Bailly V., Weber C., Thompson L.H., Prakash L., Prakash S.. Human xeroderma pigmentosum group D gene encodes a DNA helicase. Nature. 1993; 365:852–855. [DOI] [PubMed] [Google Scholar]
- 7. Schultz P., Fribourg S., Poterszman A., Mallouh V., Moras D., Egly J.M.. Molecular structure of human TFIIH. Cell. 2000; 102:599–607. [DOI] [PubMed] [Google Scholar]
- 8. Luo J., Cimermancic P., Viswanath S., Ebmeier C.C., Kim B., Dehecq M., Raman V., Greenberg C.H., Pellarin R., Sali A. et al.. Architecture of the human and yeast general transcription and DNA repair factor TFIIH. Mol. Cell. 2015; 59:794–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Araujo S.J., Tirode F., Coin F., Pospiech H., Syvaoja J.E., Stucki M., Hubscher U., Egly J.M., Wood R.D.. Nucleotide excision repair of DNA with recombinant human proteins: definition of the minimal set of factors, active forms of TFIIH, and modulation by CAK. Genes Dev. 2000; 14:349–359. [PMC free article] [PubMed] [Google Scholar]
- 10. Svejstrup J.Q., Wang Z., Feave W.J., Wu X., Bushnell D.A., Donahue T.F., Friedberg E.C., Kornberg R.D.. Different forms of TFIIH for transcription and DNA repair: Holo-TFIIH and a nucleotide excision repairosome. Cell. 1995; 80:21–28. [DOI] [PubMed] [Google Scholar]
- 11. Habraken Y., Sung P., Prakash S., Prakash L.. Transcription factor TFIIH and DNA endonuclease Rad2 constitute yeast nucleotide excision repair factor 3: implications for nucleotide excision repair and Cockayne syndrome. PNAS. 1996; 93:10718–10722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sung P., Prakash L., Matson S.W., Prakash S.. RAD3 protein of Saccharomyces cerevisiae is a DNA helicase. Proc. Natl. Acad. Sci. U.S.A. 1987; 84:8951–8955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sung P., Prakash L., Weber S., Prakash S.. The RAD3 gene of Saccharomyces cerevisiae encodes a DNA-dependent ATPase. PNAS. 1987; 84:6045–6049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jawhari A., Laine J.P., Dubaele S., Lamour V., Poterszman A., Coin F., Moras D., Egly J.M.. p52 Mediates XPB function within the transcription/repair factor TFIIH. J. Biol. Chem. 2002; 277:31761–31767. [DOI] [PubMed] [Google Scholar]
- 15. Coin F., Marinoni J.C., Rodolfo C., Fribourg S., Pedrini A.M., Egly J.M.. Mutations in the XPD helicase gene result in XP and TTD phenotypes, preventing interaction between XPD and the p44 subunit of TFIIH. Nat. Genet. 1998; 20:184–188. [DOI] [PubMed] [Google Scholar]
- 16. Greber B.J., Toso D.B., Fang J., Nogales E.. The complete structure of the human TFIIH core complex. eLife. 2019; 8:e44771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gervais V., Lamour V., Jawhari A., Frindel F., Wasielewski E., Dubaele S., Egly J.M., Thierry J.C., Kieffer B., Poterszman A.. TFIIH contains a PH domain involved in DNA nucleotide excision repair. Nat. Struct. Mol. Biol. 2004; 11:616–622. [DOI] [PubMed] [Google Scholar]
- 18. Yokoi M., Masutani C., Maekawa T., Sugasawa K., Ohkuma Y., Hanaoka F.. The xeroderma pigmentosum group C protein complex XPC-HR23B plays an important role in the recruitment of transcription factor IIH to damaged DNA. J. Biol. Chem. 2000; 275:9870–9875. [DOI] [PubMed] [Google Scholar]
- 19. Sugasawa K., Masutani C., Uchida A., Maekawa T., Van Der Spek P., Bootsma D., Hoeijmakers J., Hanaoka F.. HHR23B, a human Rad23 homolog, stimulates XPC protein in nucleotide excision repair in vitro. Mol. Cell. Biol. 1996; 16:4852–4861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Di Lello P., Jenkins L.M.M., Jones T.N., Nguyen B.D., Hara T., Yamaguchi H., Dikeakos J.D., Appella E., Legault P., Omichinski J.G.. Structure of the Tfb1/p53 complex: Insights into the interaction between the p62/Tfb1 subunit of TFIIH and the activation domain of p53. Mol. Cell. 2006; 22:731–740. [DOI] [PubMed] [Google Scholar]
- 21. Kuper J., Braun C., Elias A., Michels G., Sauer F., Schmitt D.R., Poterszman A., Egly J.-M., Kisker C.. In TFIIH, XPD helicase is exclusively devoted to DNA repair. PLoS Biol. 2014; 12:e1001954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kokic G., Chernev A., Tegunov D., Dienemann C., Urlaub H., Cramer P.. Structural basis of TFIIH activation for nucleotide excision repair. Nat. Commun. 2019; 10:2885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Barnett J.T., Kad N.M.. Understanding the coupling between DNA damage detection and UvrA’s ATPase using bulk and single molecule kinetics. FASEB J. 2019; 33:763–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Springall L., Inchingolo A.V., Kad N.M.. DNA-protein interactions studied directly using single molecule fluorescence imaging of quantum dot tagged proteins moving on DNA tightropes. Methods Mol. Biol. 2016; 1431:141–150. [DOI] [PubMed] [Google Scholar]
- 25. Kad N.M., Wang H., Kennedy G.G., Warshaw D.M., Van Houten B.. Collaborative dynamic DNA scanning by nucleotide excision repair proteins investigated by single- molecule imaging of quantum-dot-labeled proteins. Mol. Cell. 2010; 37:702–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jiang Y., Ke C., Mieczkowski P.A., Marszalek P.E.. Detecting ultraviolet damage in single DNA molecules by atomic force microscopy. Biophys. J. 2007; 93:1758–1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Buechner C.N., Heil K., Michels G., Carell T., Kisker C., Tessmer I.. Strand-specific recognition of DNA damages by XPD provides insights into nucleotide excision repair substrate versatility. J. Biol. Chem. 2014; 289:3613–3624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Adams P.D., Afonine P.V., Bunkóczi G., Chen V.B., Davis I.W., Echols N., Headd J.J., Hung L.W., Kapral G.J., Grosse-Kunstleve R.W. et al.. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D. Biol. Crystallogr. 2010; 66:213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Radu L., Schoenwetter E., Braun C., Marcoux J., Koelmel W., Schmitt D.R., Kuper J., Cianferani S., Egly J.M., Poterszman A. et al.. The intricate network between the p34 and p44 subunits is central to the activity of the transcription/DNA repair factor TFIIH. Nucleic. Acids. Res. 2017; 45:10872–10883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gileadi O., Feaver W.J., Kornberg R.D.. Cloning of a subunit of yeast RNA polymerase II transcription factor b and CTD kinase. Science. 1992; 257:1389–1392. [DOI] [PubMed] [Google Scholar]
- 31. Matsui P., DePaulo J., Buratowski S.. An interaction between the Tfb1 and Ssl1 subunits of yeast TFIIH correlates with DNA repair activity. Nucleic Acids Res. 1995; 23:767–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. von Hippel P.H., Berg O.G.. Facilitated target location in biological systems. J. Biol. Chem. 1989; 264:675–678. [PubMed] [Google Scholar]
- 33. Saxton M.J. Anomalous subdiffusion in fluorescence photobleaching recovery: a Monte Carlo study. Biophys. J. 2001; 81:2226–2240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Schurr J.M. The one-dimensional diffusion coefficient of proteins absorbed on DNA. Biophys. Chem. 1979; 9:413–414. [PubMed] [Google Scholar]
- 35. Hughes C.D., Wang H., Ghodke H., Simons M., Towheed A., Peng Y., Van Houten B., Kad N.M.. Real-time single-molecule imaging reveals a direct interaction between UvrC and UvrB on DNA tightropes. Nucleic Acids Res. 2013; 41:4901–4912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kong M., Liu L., Chen X., Driscoll K.I., Mao P., Böhm S., Kad N.M., Watkins S.C., Bernstein K.A., Wyrick J.J.. Single-molecule imaging reveals that Rad4 employs a dynamic DNA damage recognition process. Mol. Cell. 2016; 64:376–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cheon N.Y., Kim H.S., Yeo J.E., Scharer O.D., Lee J.Y.. Single-molecule visualization reveals the damage search mechanism for the human NER protein XPC-RAD23B. Nucleic Acids Res. 2019; 47:8337–8347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wolski S.C., Kuper J., Hanzelmann P., Truglio J.J., Croteau D.L., Van Houten B., Kisker C.. Crystal structure of the FeS cluster-containing nucleotide excision repair helicase XPD. PLoS Biol. 2008; 6:e149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Cheng K., Wigley D.B.. DNA translocation mechanism of an XPD family helicase. eLife. 2018; 7:e42400. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data will be made available through the University of Kent academic repository (https://kar.kent.ac.uk/). The structure modeled in Figure 5 has been deposited in the worldwide protein databank, accession code: 7AD8.