


Abstract
Break-induced replication (BIR) is a mechanism used to heal one-ended DNA double-strand breaks, such as those formed at collapsed replication forks or eroded telomeres. Instead of utilizing a canonical replication fork, BIR is driven by a migrating D-loop and is associated with a high frequency of mutagenesis. Here we show that when BIR encounters an interstitial telomere sequence (ITS), the machinery frequently terminates, resulting in the formation of an ectopic telomere. The primary mechanism to convert the ITS to a functional telomere is by telomerase-catalyzed addition of telomeric repeats with homology-directed repair serving as a back-up mechanism. Termination of BIR and creation of an ectopic telomere is promoted by Mph1/FANCM helicase, which has the capacity to disassemble D-loops. Other sequences that have the potential to seed new telomeres but lack the unique features of a natural telomere sequence, do not terminate BIR at a significant frequency in wild-type cells. However, these sequences can form ectopic telomeres if BIR is made less processive. Our results support a model in which features of the ITS itself, such as the propensity to form secondary structures and telomeric protein binding, pose a challenge to BIR and increase the vulnerability of the D-loop to dissociation by helicases, thereby promoting ectopic telomere formation.
INTRODUCTION
DNA double-strand breaks (DSBs) occur frequently during cell growth and division. DSBs can be caused by replication stress, free radicals produced by metabolism, and by external factors such as radiation. If DSBs are left unrepaired they can result in cell death and if repaired incorrectly they can result in mutations ranging from point mutations to chromosome rearrangements. Genomic instability associated with defective DSB repair contributes to many human pathologies, such as neurodegeneration, developmental disorders, aging and cancer (1–3).
Homologous recombination (HR), which involves the use of a homologous DNA sequence as a template for repair, is one of two main pathways to repair DSBs. HR is often considered to be the ‘error free’ repair pathway, because it relies on a homologous template and therefore ensures that sequences are not lost at the break site. However, several studies have documented increased mutagenesis during HR. If strand invasion occurs at a homologous sequence other than the preferred identical sister chromatid, for example a dispersed repeat sequence or the chromosome homolog, then HR can result in deleterious outcomes, including deletion or duplication of DNA sequences, chromosome translocations or loss of heterozygosity (4). In addition to rearrangements, point or frameshift mutations are also increased in DNA that has been synthesized during HR, attributed largely to the limited proofreading abilities of DNA polymerases outside the typical replication fork, inefficient mismatch repair and to template switching during repair synthesis (5,6).
In contrast to DSBs generated by endonucleases, DSBs that arise by erosion of telomeres, replication fork regression, or replication through a single-stranded nick present only one end with homology to a donor template. The initial steps to repair both types of DSBs are similar (7), beginning with end resection to create 3′ single-stranded DNA (ssDNA) overhangs (8). The ssDNA is initially bound by replication protein A (RPA), then Rad52 (or BRCA2 in humans) mediates replacement of RPA with Rad51, the ubiquitous eukaryotic RecA homolog (4). The resulting ssDNA–Rad51 filament searches for a homologous duplex, using the ability of Rad51 to match triplets of DNA (9). Rad51 promotes the exchange of DNA strands creating a displacement loop, or D-loop, in which the ssDNA from the broken chromosome is paired with the complementary strand of the donor duplex. In the simplest form of two-end DSB repair, the invading 3′ end is extended by DNA synthesis, then is displaced from the D-loop by a helicase and anneals to the 3′ ssDNA overhang at the non-invading end (10). Alternatively, the displaced strand of the D-loop pairs with the other 3′ end of the broken DNA molecule so that both ends of the break are interacting with the homologous duplex (11). This creates a double Holliday junction (dHJ) intermediate and can result in crossover or non-crossover products depending on how the junctions are resolved or dissolved. However, in the case of a one-ended break, there is no second end of the break to complete the repair process so DNA synthesis can continue in a process called break-induced replication (BIR) to the end of the chromosome (12), or until a converging replication fork collides with the D loop (13).
BIR has been most extensively studied in the budding yeast Saccharomyces cerevisiae (7,12). Two hallmarks of BIR are the dependency on Pol32 (14–16), a non-essential subunit of the DNA polymerase delta (Pol δ) complex that increases its processivity (17), and to a lesser degree, its dependence on the Pif1 helicase (18–21). A similar process has been identified in human cells, where it was shown to be responsible for certain duplications found in cancer cell lines (22), and for replication of fragile sites during early mitosis (23). This mammalian BIR-like process is dependent on the POLD3, the human homologue of Pol32, and Rad52 (22,24,25). Rad52 is also essential for BIR in yeast (26,27). In addition, a Pol32 and Pif1-dependent BIR-like mechanism to heal one-ended chromosome breaks arising from the breakage of a dicentric chromosome has been identified in Drosophila melanogaster (28).
In yeast, DNA synthesis during BIR occurs via a migrating D loop and is catalyzed by DNA Pol δ (19,21,29). This type of DNA synthesis is conservative, where the newly synthesized DNA is associated with the formerly broken ‘recipient’ chromosome and the template or ‘donor’ chromosome remains unchanged (19,30). One outcome of this type of DNA synthesis is increased point and frameshift mutations (31). While this increase in mutations also occurs during two-ended DSB repair (5,6), BIR synthesizes significantly more DNA than what is synthesized during repair of a two-ended DSB. Furthermore, the newly synthesized ssDNA is released behind the D loop, without the opportunity of reannealing to the other end of the break where mismatch repair can occur (32). In the case of BIR, mutations that accumulate during synthesis of the invading strand are made permanent by the synthesis of the second strand.
In addition to the increase of point and frameshift mutations, BIR can also result in deletions, duplications, and loss of heterozygosity if a template other than the identical sister chromatid is chosen for repair. However, the initial invasion step is not the only opportunity for BIR to choose the wrong template. BIR products can also be templated by DNA tracts from multiple chromosomes, a process termed template switching (33). Template switching involves the dissociation of the D loop by the Mph1 helicase and the reinvasion and continued synthesis at a new site (34).
Due to the extreme mutagenic potential of BIR, its atypical mode of DNA synthesis, and its use by cells during times of replication stress, it is essential to understand the mechanisms and factors influencing the mutagenicity of BIR. It may be that these characteristics cause BIR to be exceptionally sensitive to difficult-to-replicate DNA sequences, and in turn, increase the mutagenic potential of these sequences. This becomes relevant to human health in many ways, particularly as DNA replication stress and genomic instability are associated with tumorigenesis and other diseases. Of note, BIR has been suggested to play a role in the pathogenic expansions of CAG microsatellites found in Huntington's and other degenerative diseases (35,36).
To further understand BIR-associated mutagenesis, we investigate here the role BIR may play in the instability of interstitial telomere sequences (ITS). ITS are telomeric sequences found outside the telomere region. ITS are abundant in yeast, plants, and vertebrates (37,38). Like traditional telomeres, ITS can form secondary structures such as G quadruplexes in the G-rich strand (39), and even certain structures in the C-rich strand (40,41). They are also found to bind at least some of the proteins that typically bind telomeres such as Rap1 in yeast or TRF2 in humans (38,42). ITS are sites of instability and rearrangements (37,42–46), and evidence suggests that Rap1 binding and the ability to form G quadruplexes contribute to ITS-induced chromosome fragility in yeast (47,48). Chromosomal breakage at or near an ITS can cause an ectopic telomere to form at the site instead of correct repair of the DSB (49–51). Here, we show that if BIR encounters an ITS placed in its path, BIR terminates at the ITS 12% of the time, with the formation of an ectopic telomere at this location. Ectopic telomere formation is driven by Mph1 helicase and can occur by either de novo synthesis by telomerase or HR. Our findings have implications for understanding the role ITS play in genome instability, as well as the influence of different DNA sequences on the accuracy of BIR.
MATERIALS AND METHODS
Yeast strains and plasmids
All strains described here (Supplementary Table S1) are derivatives of strain LSY2689-12A, previously used to detect BIR (30). This strain contains a recipient lys-HO-KanMX integrated 32 kb from the left telomere of chromosome (Chr) V, and the donor TRP1-ys2 integrated 70 kb from the left telomere of Chr XI and has the MATa-inc allele to prevent HO cleavage at the MAT locus. In addition, the endogenous LYS2 gene was replaced with NatMX and a GAL-HO cassette was integrated at the ade3 locus. Strain LSY3881 was generated from LSY2689-12A by replacing lys2::NatMX with lys2::LEU2, and the HML and HMR loci were replaced with oripRS, and ampR respectively to prevent aberrant HO cutting (52). To insert an ITS on the donor chromosome of strain LSY3881, we PCR amplified (TGTGTGGG)8 and K. lactis URA3 using a forward primer with 20 bp homology to target area of Chr XI + ITS + 5′ end of URA3 and a reverse primer with 20 bp homology to target area of Chr XI + reverse primer to 3′ end of URA3 (Supplementary Table S2). The resulting PCR product contained the desired insert followed by the URA3 cassette. Homology to the target sequence in Chr XI was extended to 40 bp on both sides by a second round of PCR. These fragments were then integrated 2.5 kb centromere distal to the ys2 donor, generating strain LSY3944. Successful integration of the ITS was confirmed by PCR using primers F insert 2.5 and R insert 2.5 followed by DNA sequencing. A control strain (LSY3945) containing the URA3 cassette but no insert was created the same way. Strains containing the ITS 48 kb away from the ys2 donor, and the strains containing Y’ element inserts, and GT repeat inserts, were generated using the same method, but with different sets of primers (Supplementary Table S2).
To delete MPH1, POL32, RAD10, SGS1 and TLC1, the HphMX gene was amplified from pAG32 with primers containing homology upstream and downstream of the gene to be deleted (53). Genetic crosses were used to generate yeast strains with the pif1-m2 or TetO2-TLC1 allele and haploid spores with the correct genotype were identified by PCR followed by restriction endonuclease digestion or DNA sequencing (54). The senescence phenotype of the BIR strain harboring the TetO2-TLC1 allele was confirmed by growing cells in the absence or presence of doxycycline for four days.
A plasmid containing MPH1 under the control of a galactose-inducible promoter, and a KanMX marker was received as a gift from the lab of Brian Luke (55). The KanMX marker was switched to HphMX by PCR amplification of HphMX from pAG32 with primers overlapping KanMX. The full plasmid was then PCR amplified without the KanMX gene and assembled with the HphMX gene using Gibson Assembly (New England Biolabs). The resulting plasmid was sequenced to confirm correct assembly.
BIR assay
Cells were grown to exponential phase in 1% yeast extract, 2% peptone, 2% lactate (YPL), then diluted and plated on rich medium containing either 2% glucose (YPD) or 2% galactose (YPG). For strains containing the TetO2-TLC1 allele, 30 μg/ml doxycycline was added to the YPL cultures and also to the YPD and YPG plates. Colonies were counted after 3–4 days and were then replica plated onto synthetic complete (SC) medium lacking lysine or YPD containing geneticin (G418). Cell viability after HO induction was determined by dividing the number of CFUs on YPG by that on YPD. Repair by BIR was confirmed by comparing the number of Lys+ to the number of YPG CFU. Greater than 99% of cells repaired via BIR, while <1% remained Lys− G418r following growth on YPG, presumably because of repair by non-homologous end joining (NHEJ). BIR frequencies shown are averages of at least three independent trials for each strain. The numbers of full-length and truncated BIR products were normalized to the BIR frequencies in Figures 1–5.
Figure 1.

An interstitial telomere sequence can disrupt BIR. (A) Schematic of experimental BIR system showing site of the HO-induced DSB, strand invasion (black bent arrow), centromeres (filled circles), telomere tracts (horizontal carets) and possible BIR outcomes with the predicted sizes of the repaired Chr V:XI translocation chromosome and truncated product. Note that the left arms of chromosomes V and XI are shown in inverted orientation. (B) Percent repair by strains +/-ITS with percent of colonies found to have truncations illustrated with darker shading. Error bars represent mean BIR frequencies from at least 14 trials ± standard deviations (SD). No truncations were seen in 57 -ITS colonies screened, but were found in 14 out of 115 +ITS colonies (P < 0.01). (C) Representative pulsed field gels showing chromosome sizes before and after induction of HO with galactose. Note that chromosomes V and VIII co-migrate under the conditions used. Gel of +ITS (lower panel) shows two clones with truncated BIR products. The un-cropped images are shown in Supplementary Figure S1. D. PFGE and Southern blot of a parental uninduced strain (lane 1) and four representative colonies containing full length (lanes 2 and 3) or truncated (lanes 4 and 5) BIR products. Southern blotting shows that as expected, the chromosome V probe targeting a sequence centromeric to the break site hybridizes to the repair products in all four colonies, while the probe targeting a site on chromosome XI telomeric to the ITS only hybridizes to the donor chromosome and the full-length BIR products.
Figure 5.
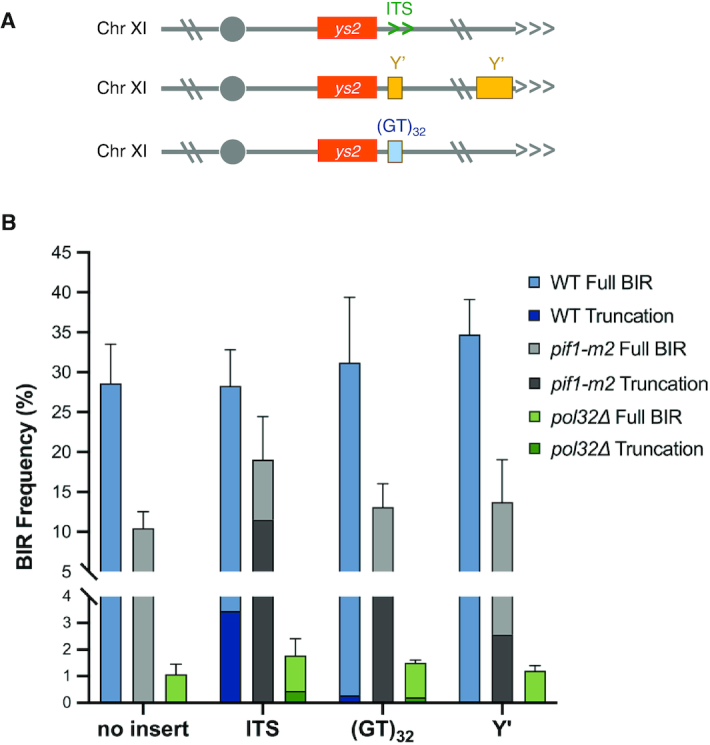
Unique features of the ITS contribute to formation of truncated chromosomes. (A) Schematic of the alternate sequences used in place of the ITS. (B) Percent BIR for the indicated strains with truncated products indicated by darker shading; error bars represent mean BIR frequencies from at least three trials ± SD. BIR completion was significantly higher in the pif1-m2 +ITS strain than pif1-m2 -ITS (P < 0.001). Overall BIR frequencies for WT, pif1-m2 and pol32 strains were significantly different from each other.
Chromosome size determination by pulsed field gel electrophoresis (PFGE)
Lys+ colonies were suspended in YPD and grown to saturation. Cells from 4 ml of each culture were harvested by centrifugation, and pellets were weighed and re-suspended in low melting point agarose and molded in Bio-Rad plug molds. Per 50 mg of cell pellet the following amounts were used: 0.5% low melting point agarose in 450 ul of 100 mM EDTA (pH 8.0); and 20 ul 25 mg/ml zymolyase 20T in 10 mM KPO4 (pH 7.5). The resulting plugs were incubated overnight in 1 ml 500 mM EDTA, 10 mM Tris–HCl (pH 7.5) at 37°C. The next day 400 ul of 5% sarcosyl, 5 mg/ml proteinase K in 500 mM EDTA (pH 8.0) was added to the plug buffer and incubated at 50°C for >5 h with occasional mixing. TE washes (four washes, >1 h each, nutating) were then done at 4°C. Chromosomes were separated by electrophoresis through 1% agarose at 6 V in 0.5× Tris–borate–EDTA at 14°C for 36 h (initial switch = 45 s, final switch = 95 s) using a CHEF-DR II Pulsed-Field Electrophoresis system (Bio-Rad) and visualized by SYBR Gold staining. Truncation frequencies were determined by counting the number of lanes of a PFG showing the truncated size chromosome and dividing by the total number of samples tested by PFGE.
Southern blots were performed by overnight transfer of DNA from the PFGE to nylon membrane (Amersham Hybond-N). The membrane was then hybridized with a radio-labeled probe generated by amplification of either a region of the COS9 gene for Chr XI, or a region of the PCM1 gene for Chr V, and visualized by phosphorimaging.
Telomere sequencing
To sequence the region near the ITS and the potential new telomere, DNA was extracted from three clones with a truncated BIR chromosome that had been identified by PFGE. C-tailing was performed on genomic DNA isolated from each clone using terminal deoxynucleotidyl transferase according to the manufacturer's protocol (New England Biolabs). C-tailed DNA was used in a PCR reaction with a poly-G primer and a primer just upstream of the ITS insert (Supplementary Table S2, F insert 2.5). The resulting PCR product was gel purified and sequenced.
Generation of telomerase-negative survivors
To generate type II telomerase survivors, tlc1Δ cells were grown in 25 ml YPD to saturation followed by a dilution of 1:200 or greater. This procedure was repeated at least four times. Cells with a faster growth rate, indicative of type II survivors (56,57), were identified by streaking onto a YPD plate for single colonies.
PCR analysis of chromosome truncations associated with the Y’ insert
DNA was extracted from 10 clones with a truncated BIR chromosome that had been identified by PFGE. Standard PCR was performed with primers F insert 2.5 located upstream of the 80 bp Y’ insertion and R Y’ primer that anneals to native Y’ sequence, telomere proximal to the Y’ sequence inserted on the donor chromosome (Supplementary Table S2). An additional PCR with F insert 2.5 and R insert 2.5 primers was a control to amplify the Y’ insert on the donor chromosome common to full-length and truncated BIR products.
Statistical methods
Statistical analyses were performed using PRISM software. For BIR frequencies, trials were performed at least three times and averages were compared using unpaired t tests. The mean values and standard deviations (SD) for total BIR are presented in Table 1. Differences in the numbers of chromosome truncations were compared using a Fisher's exact test.
Table 1.
Percent BIR and truncated chromosomes.
| Strain | BIR frequency (%)a | Number of Truncations | % Truncationsb | Truncation frequencyc | Nd |
|---|---|---|---|---|---|
| WT −ITS | 28.6 ± 4.9 (17) | 0 | 0 | 0 | 57 |
| WT +ITS | 28.3 ± 4.5 (14) | 14 | 12.2 | 3.45 | 115 |
| WT +(GT)32 | 31.2 ± 8.2 (5) | 1 | 0.89 *** | 0.28 | 112 |
| WT +Y′ | 34.7 ± 4.3 (6) | 0 | 0 *** | 0 | 78 |
| WT −ITS (48 kb) | 19.51 ± 2.6 (4) | ND | |||
| WT +ITS (48 kb) | 18.39 ± 2.5 (4) | 1 | 1.79 * | 0.33 | 56 |
| TetO2-TLC1 +ITS + dox | 22.9 ± 6.8 (6) | 9 | 5.33 * | 1.22 | 169 |
| tlc1Δ +ITS | 17.4 ± 6.2 (3) | 9 | 9.68 | 1.68 | 93 |
| pif1-m2 −ITS | 10.4 ± 2.1 (8) | 0 | 0 | 0 | 81 |
| pif1-m2 +ITS | 19.0 ± 5.4 (8) | 49 | 60.5 *** | 11.5 | 81 |
| pif1-m2 +(GT)32 | 13.1 ± 2.9 (4) | 42 | 34.1 ^^^ | 4.47 | 123 |
| pif1-m2 +Y′ | 13.7 ± 5.3 (4) | 17 | 18.7 ### | 2.56 | 91 |
| pif1-m2 tlc1Δ +ITS | 15.5 ± 3.1 (3) | 5 | 8.77 | 1.36 | 57 |
| mph1Δ −ITS | 60.5 ± 7.7 (6) | ND | |||
| mph1Δ +ITS | 59.6 ± 8.5 (6) | 0 | 0 ** | 0 | 74 |
| MPH1 OE −ITS | 1.02 ± 0.25 (3) | 0 | 0 | 0 | 24 |
| MPH1 OE +ITS | 2.25 ± 0.19 (3) | 17 | 70.8 *** | 1.59 | 24 |
| sgs1Δ +ITS | 37.4 ± 10.2 (12) | 12 | 7.41 | 2.77 | 162 |
| rad10Δ +ITS | 27.4 ± 3.7 (3) | 11 | 6.63 | 1.82 | 166 |
| pol32Δ −ITS | 1.07 ± 0.38 (4) | 0 | 0 | 0 | 36 |
| pol32Δ +ITS | 1.78 ± 0.63 (4) | 16 | 24.6 * | 0.44 | 65 |
| pol32Δ +(GT)32 | 1.51 ± 0.1 (4) | 12 | 14.1 ^^^ | 0.21 | 85 |
| pol32Δ +Y′ | 1.2 ± 0.2 (3) | 1 | 1.45 | 0.02 | 69 |
aMean BIR frequency with SD. The number of independent trials is shown in parentheses.
bPercent of BIR events analyzed by PFGE that terminate at the ITS or other insert. Significance values when compared with WT +ITS: *P < 0.05; **P < 0.01; ***P < 0.001. Significance values when compared with WT +(GT)32 or +Y’ are shown with ^ or # symbols, respectively.
cPercent truncations adjusted to the BIR frequency.
dNumber of Lys+ colonies analyzed by PFGE.
ND, not determined.
RESULTS
An interstitial telomere sequence can disrupt BIR
To investigate whether and how the BIR machinery is affected by an ITS, we created a variation of a previously described system to monitor BIR (30). In this system, an HO endonuclease recognition site (HOcs) is integrated 32 kb from the left telomere of chromosome (Chr) V in a strain expressing a galactose-inducible HO gene (Figure 1A). The HOcs is directly adjacent to a 3′ truncated LYS2 gene (‘lys’) on the centromeric side, and a KanMX gene providing resistance to gentamycin (G418) on the telomeric side. A 5′ truncated LYS2 gene (‘ys2’) inserted 70 kb from the left telomere of Chr XI provides homology for repair. These two lys2 fragments share 2.2 kb homology, and are non-functional before recombination since they lack the complete ORF. After transferring cells to galactose-containing medium to induce the DSB, the centromeric side of the break containing ‘lys’ invades the homologous ‘ys2’ site and copies to the end of donor Chr XI by BIR. This heals the break and creates a functional LYS2 gene on the recipient chromosome (Figure 1A). The distal end of Chr V contains no essential genes and is lost due to lack of available homology. More than 99% of cells that grow on galactose-containing medium are Lys+ and have lost the KanMX marker located centromere-distal to the DSB on the recipient chromosome. Thus, the BIR frequency is derived from the ratio of colony-forming units on galactose and glucose-containing media.
To generate a context in which the BIR machinery must encounter an ITS, we inserted a 64 bp tract of telomere sequence (TGTGTGGG)8 and a K. lactis URA3 marker on the donor chromosome into our BIR reporter strain, 2.5 kb centromere-distal to the ys2 cassette (‘+ITS’ strain). The insertion site and size were based on previous studies showing that BIR is unstable over the first few kb of synthesis (33) and a 64 bp ITS is sufficient to induce genomic instability (42,58). As a control, we generated a strain with just the URA3 marker inserted at the same site as the +ITS strain (‘-ITS’ strain). Following strand invasion, DNA synthesis in the +ITS strain must traverse the ITS to complete BIR. If cells complete BIR, the resulting BIR product would be ∼620 kb, whereas termination of BIR at the ITS would result in a truncated BIR product of ∼552 kb (Figure 1A).
We first investigated whether an ITS affects BIR completion. The rates of BIR for the two strains, with or without the ITS, were not significantly different: 28.3% and 28.6% respectively (Figure 1B, Table 1), suggesting that BIR synthesis is not impaired by the ITS. Next we asked whether the ITS changes the outcome of BIR by causing a telomere to be formed at the ITS, effectively terminating BIR DNA synthesis and generating a truncated chromosome. To address this question, we analyzed the size of Chr V from independent colonies that completed BIR (Lys+ G418s colonies after HO induction) by pulsed field gel electrophoresis (PFGE), which allows separation and visualization of intact chromosomes. In a strain without the ITS, all colonies screened contained the expected size Chr V:XI translocation chromosome (Figure 1C). We found that 12% of colonies derived from the strain containing the ITS had BIR products ∼68 kb shorter than full-length BIR products (Figure 1C, Supplementary Figure S1, Table 1). Southern blot hybridization using probes specific to Chr V or Chr XI confirmed the identity of the full length and truncated chromosomes (Fig 1D). Since this is the size of product that would be created by termination of BIR at the ITS, we PCR amplified and sequenced the region from three independent colonies containing this size BIR product and found a full length (200–300 bp) telomere to have been added (Supplementary Figure S2).
To further understand the mechanism for disruption of BIR by the ITS, we analyzed BIR outcomes of a strain containing an ITS in a different location. It has been shown that template switching during BIR occurs more frequently closer to the site of strand invasion than further away (33). If creation of ectopic telomeres functions similarly, it would be expected to occur more frequently when the ITS is close to the site of strand invasion. To test this idea, we created a BIR strain containing an ITS 48 kb centromere distal to the site of strand invasion. As for the 2.5 kb ITS strain, no difference was seen in overall BIR completion between the strain with no ITS and the strain containing the ITS (Table 1). By contrast to the 2.5 kb ITS strain, only one truncation was seen among 56 colonies screened (P = 0.023) (Table 1) indicating that similar to template switching during BIR, truncated chromosome products are more likely to occur closer to the start of BIR. Whether this difference is due to the 2.5 kb ITS capturing BIR products that would otherwise have failed, a change in stability of the D-loop over time, or other factors such as approaching replication forks is not yet clear.
Telomere synthesis is driven by de novo synthesis by telomerase or by HR
We envisioned two mechanisms by which ectopic telomeres could be generated: first, telomerase could act directly at the ITS to synthesize a telomere; and second, the ssDNA intermediate formed when the migrating D-loop traverses the ITS could invade a natural telomere and complete synthesis by homology-directed repair (Figure 2A). To distinguish between these two possibilities, we tested the ability of cells lacking the RNA template for telomerase (TLC1) to form truncated BIR products.
Figure 2.
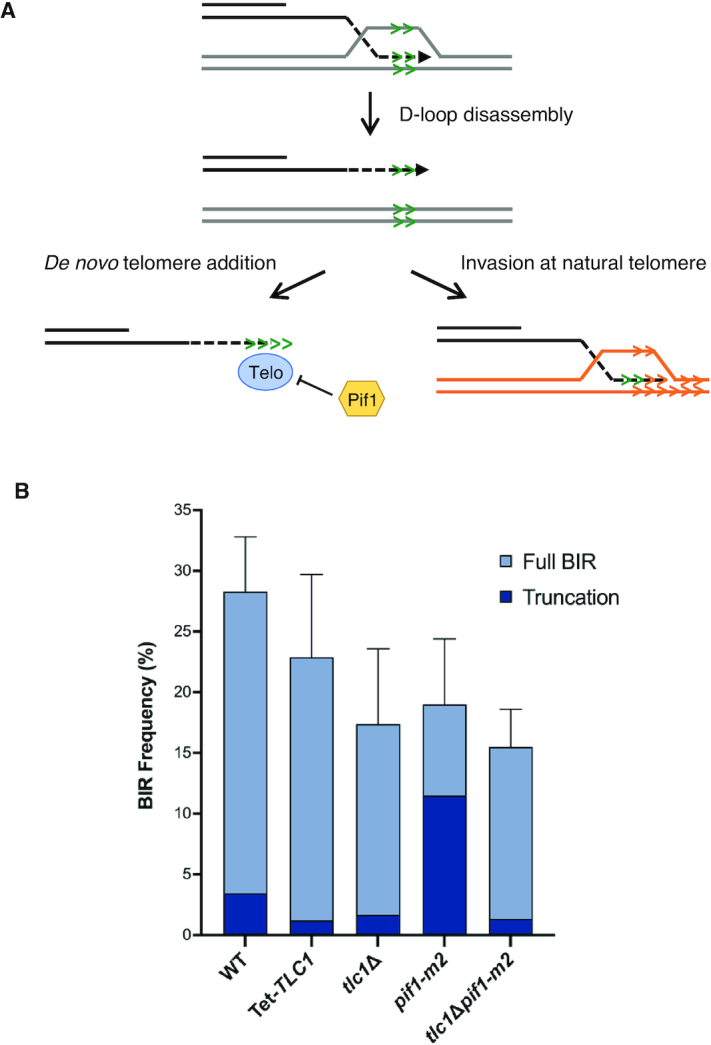
Telomere synthesis is driven by de novo synthesis by telomerase or HR. (A) Schematic of possible mechanisms of telomere creation, showing de novo telomere addition by telomerase (left panel) and HR using natural telomeres (right panel). Telo refers to telomerase. (B) Percent BIR for each of the indicated +ITS strains with truncated events indicated by darker shading. Error bars represent mean BIR frequencies from at least three trials ± SD (see Table 1). Differences in truncation numbers were significant between pif1-m2 and all other strains (P values: < 0.0001 for all comparisons).
Yeast cells lacking telomerase senesce after ca. 40 generations (59,60). To acutely deplete TLC1 during the time course of DSB induction and repair by BIR, we generated a tetracycline-regulated TLC1 allele in the +ITS BIR strain (60). Cells were grown for 16 h in YPL medium containing doxycycline and then plated on YPD or YPG plates with doxycycline. After generation of BIR products, cells were grown without doxycycline to restore telomerase function. We found a small but significant decrease in truncated products, from 12.2% in WT to 5.3% in the Tet-TLC1 cells grown with doxycycline (P < 0.05) (Figure 2B), suggesting that telomerase is the main mechanism for ectopic telomere formation.
Since truncated chromosomes could still be recovered from cells with acute loss of TLC1 we asked whether telomerase-negative survivors that arise by a BIR-like mechanism more readily generate ectopic telomeres at the ITS. In yeast, telomerase-negative survivors arise by amplification of the sub-telomeric Y′ elements (type I survivors) or by expanding the telomeric repeats (type II survivors) (57). Haploid tlc1Δ cells were first allowed to adapt in liquid culture to select for the faster growing type II telomerase-negative survivors. Although the overall BIR frequency was lower in the tlc1Δ mutant than in WT (P<0.005) (Figure 2B), truncated chromosomes were formed in 9.7% of the BIR clones tested by PFGE, similar to WT (P = 0.66). Thus, cells lacking telomerase can employ HR to generate an ectopic telomere and this is likely favored in type II survivors which have long heterogeneous telomeres as well as extrachromosomal telomeric circles (57,61).
To further address the role of telomerase in generation of stable truncations we analyzed cells containing the pif1-m2 mutation, which reduces the level of nuclear Pif1, an inhibitor of telomerase activity at DSBs (62), while retaining mitochondrial Pif1 function (54). We reasoned that because Pif1 inhibits telomerase at a DSB, the frequency of truncated chromosomes would be expected to increase in the absence of Pif1. Consistent with previous studies (19,21), the BIR frequencies of the pif1-m2 strains were lower than WT (P < 0.0001 and P = 0.0003 for the -ITS and +ITS versions, respectively), whereas the number of chromosome truncations increased to 60.5% in the pif1-m2 +ITS strain (P < 0.0001) (Figure 2B). While this result supports the idea that telomerase can act directly at the ITS an important caveat is that Pif1 is required for BIR (19,21), and the increase in chromosome truncations could be due to decreased processivity during BIR resulting in premature termination at the ITS. Indeed, the BIR frequency of the pif1-m2 +ITS strain was significantly higher than the pif1-m2 -ITS strain (P < 0.001), suggesting that BIR events aborted at the ITS can be rescued by ectopic telomere formation.
To determine whether ectopic telomere formation in the pif1-m2 mutant is due to telomerase activity, we tested strains lacking both nuclear Pif1 and TLC1. A previous study showed that type II-like survivors with expanded telomere tracts are generated in the pif1-m2 tlc1Δ double mutant (63). Nine percent of the BIR products recovered from the double mutant were truncated chromosomes, significantly lower than the pif1-m2 single mutant (P < 0.0001), but not significantly different from WT or tlc1Δ alone (Figure 2B), indicating that the increased frequency of truncations seen in pif1-m2 strains is mostly due to the activity of telomerase.
Disassembly of the D-loop by Mph1 is critical for generation of ectopic telomeres
Since both methods of telomere formation require a free single stranded 3′ DNA end, we reasoned that a critical step in the formation of chromosome truncations would be disassembly of the D-loop via a helicase. To test this hypothesis, we deleted MPH1, a helicase that disassembles D-loops and is required for template switching during BIR (34,64). We found a higher frequency of BIR in the mph1Δ derivatives than in WT (P < 0.0001), consistent with previous studies (34,55,65) (Figure 3A). We analyzed the sizes of BIR products from 74 independent Lys+mph1Δ +ITS colonies by PFGE and all contained full-length BIR products (Table 1), indicating that disassembly of the D-loop by Mph1 is critical to the formation of an ectopic telomere at the ITS.
Figure 3.
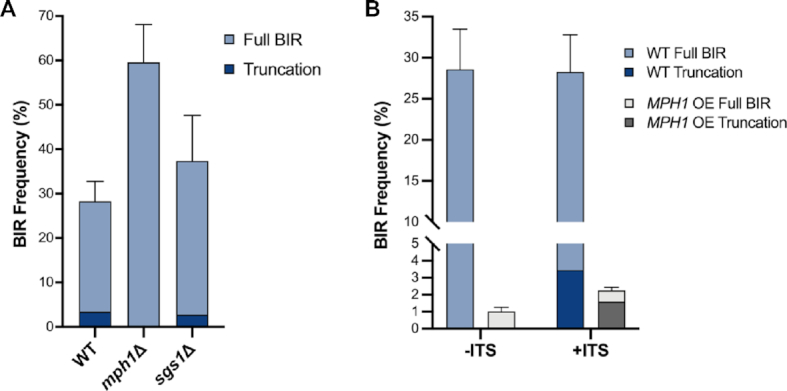
Disassembly of the D-loop by Mph1 is critical for formation of truncated BIR products. (A) Percent BIR repair in WT, mph1Δ and sgs1Δ +ITS strains. (B) Percent BIR in –ITS and +ITS strains over-expressing MPH1. Error bars represent mean BIR frequencies from at least three trials ± SD and truncated products are indicated by darker shading. BIR frequencies were not affected by the presence of the ITS, except for MPH1 OE which has significantly more BIR in the +ITS than the –ITS strain (P = 0.002).
We reasoned that an increased level of Mph1 protein in the cell would disassemble D-loops more frequently leading to fewer BIR events (55), and more truncation events. We investigated this possibility by analyzing strains that overexpress (OE) MPH1. As expected, MPH1 OE reduced BIR to 0.9% and 2.3% for -ITS and +ITS strains, respectively (Figure 3B). Of the rare colonies obtained from cells with MPH1 OE, we found that most were the result of chromosome truncations (71% of Lys+ colonies) indicating that instability of the D-loop during BIR leads to the formation of truncated chromosome products. The significant increase in BIR for the +ITS compared with the -ITS MPH1 OE cells (P < 0.005) suggests that the ITS rescues some aborted BIR events. Importantly, only full-length products were recovered from the -ITS strain following MPH1 OE (Table 1), consistent with the need for a telomere seed sequence for ectopic telomere formation.
The Sgs1-Top3-Rmi1 (STR) complex has also been shown to dismantle D-loops in vitro and in vivo (66,67). Furthermore, sgs1Δ is epistatic to mph1Δ for accumulation of nascent D-loops suggesting they function together in D-loop dissociation (67). Consistent with previous studies (68), we observed a small but significant increase in the BIR frequency in cells lacking Sgs1 (Table 1, P = 0.006). Although we found a decrease in truncated products, from 12.2% in WT to 7.4% in the sgs1Δ mutant (Figure 3A), this reduction is not statistically significant (P = 0.22). However, the number of truncations recovered from sgs1Δ cells is significantly higher than from the mph1Δ mutant (P = 0.02), indicating that Mph1 functions independently of STR to promote ectopic telomere formation at the ITS.
Clipping of heterologous tails by Rad1-Rad10 is not essential for new telomere formation
If DNA synthesis were to proceed past the ITS prior to D-loop disruption by Mph1, then formation of truncated chromosome products should require removal of the heterologous DNA between the ITS and the 3′ end that needs to be extended during HR or by telomerase (Figure 4A). The Rad1–Rad10 complex is responsible for endonucleolytic removal of 3′ heterologous tails during SSA and the strand invasion step of HR, and is important for spontaneous de novo telomere addition in some genetic backgrounds (69–71). Although truncated products were identified in only 6.6% of the rad10Δ BIR clones tested as compared with 12.2% for WT, the difference is not significant (P = 0.14) (Figure 4B). Thus, clipping by Rad1–Rad10 endonuclease is not essential for the formation of truncated products, suggesting that DNA synthesis is likely to dissociate at the ITS rather than to synthesize past it.
Figure 4.
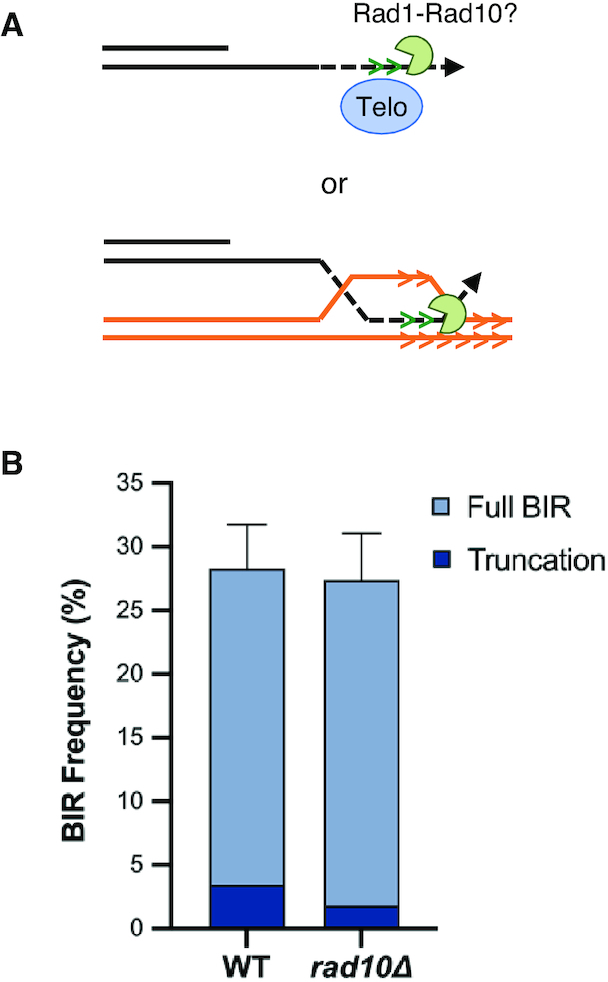
Rad1–Rad10 is not required to form truncated chromosomes. (A) Schematic showing potential role for Rad1–Rad10 clipping of the heterologous tail during telomere addition or HR if the invading strand dissociated after traversing the ITS. (B) BIR frequencies for the indicated +ITS strains with truncated products indicated by darker shading. Error bars represent mean BIR frequencies from at least three trials ± SD.
Unique features of the ITS contribute to formation of truncated chromosomes
Since our data suggest that formation of ectopic telomeres can occur by telomerase-catalyzed addition of telomere repeats or by HR, we wanted to address whether other sequences could promote chromosome truncations. If this were the case we would predict that if the ITS were replaced with another telomere seeding sequence, or any repeated sequence near a telomere, we would recover truncated BIR products. We created two new strains to test this idea (Figure 5A). In one the ITS was replaced with a (GT)32 tract, which retains the ability to seed telomeres but is predicted to not form G quadruplexes and lacks a Rap1 binding site (72). In the other, the ITS was replaced with an 80 bp sequence from the conserved region of the yeast Y’ element, a naturally occurring sub-telomeric repeat present on many yeast chromosomes. Ectopic telomere formation at the Y’ element insert is expected to be restricted to HR. One truncated chromosome was recovered from 112 BIR clones analyzed from the strain with the (GT)32 insert and none were found from 78 tested for the Y’ strain (Table 1, Figure 5B), significantly less than observed for the ITS (P < 0.001).
These data suggest that unique features of the ITS, such as Rap1 binding or G quadruplex formation contribute to the formation of truncations, potentially by disrupting synthesis during BIR. If this is the case, then the alternate sequences, (GT)32 and Y’ element insertions, should be able to form ectopic telomeres if BIR is artificially disrupted. To test this notion, we created strains with pol32Δ and pif1-m2 mutations, which are known to decrease the processivity of DNA synthesis during BIR (14–16,18–21). As expected, the BIR frequency was reduced to 1–2% in the pol32Δ mutant (Table 1, Figure 5B) (15). In the pol32Δ derivative with the ITS insert, 25% of the rare BIR products recovered contained chromosome truncations, while 14% of the clones analyzed from the (GT)32 and 1.4% of the clones analyzed from the Y’ element strain exhibited the shorter BIR product (Table 1, Figure 5B). The increase in ectopic telomere formation at the ITS and (GT)32 sequences in the pol32Δ mutant is significantly higher than observed in the WT strain (P = 0.038 and 0.0002, respectively). However, in contrast to WT, there was no significant difference in ectopic telomere formation between the ITS and (GT)32 inserts in the pol32Δ mutant, consistent with the ITS being disruptive to BIR in the WT strain. In strains lacking nuclear Pif1, we found that 60.5% of the BIR products terminated at the ITS, 34.1% at the (GT)32 insert, and 18.7% of the BIR products derived from the Y’ insert strain had terminated in the vicinity of the Y’ element (Figure 5B). To confirm that chromosome truncations generated at the Y’ sequence were due to HR and not promiscuous telomere addition, we analyzed 10 independent clones by PCR using primers that anneal to sequences upstream of the Y’ insertion on Chr XI and downstream of the Y’ homology region of Y’ elements. All of the clones analyzed were consistent with ectopic telomere formation by HR (Supplementary Figure S3). The increased ectopic telomere formation in the pif1-m2 strain is significantly higher than WT for all inserts (P < 0.0001), as well as the pol32Δ mutant (P ≤ 0.0001, 0.0012 and 0.0006 for ITS, (GT)32 and Y’, respectively). Thus we conclude that the ITS poses a unique challenge to BIR synthesis resulting in premature termination and generation of an ectopic telomere.
DISCUSSION
Here, we show that if BIR encounters an interstitial telomere sequence (ITS) placed in its path, BIR terminates at the ITS 12% of the time, with the formation of a new telomere at this location. The generation of an ectopic telomere during BIR is several orders of magnitude more frequent than spontaneous telomere formation at an 81-bp ITS (47), suggesting that BIR is more sensitive to ‘difficult to replicate’ regions of the genome than normal S-phase synthesis. We found that the ITS is converted to a functional telomere by direct addition of telomeric repeats by telomerase, with HR using natural telomeres to template addition as a back-up mechanism. The termination and creation of an ectopic telomere is promoted by Mph1 helicase, which is known to disassemble D-loops (64). We also found that other sequences with the potential to form new telomeres, but lacking the unique features of a natural telomere sequence, terminate BIR infrequently in WT cells. However, these sequences can cause chromosome truncations if BIR is made less processive by loss of Pol32 or Pif1. Together, these findings indicate that features of the ITS itself such as propensity to form secondary structure and/or activity in telomeric protein binding pose a challenge to BIR. The consequence is an increased vulnerability of the D-loop to dissociation by Mph1 with accompanying telomere formation at the site instead of completion of BIR (see Figure 6 and discussion below).
Figure 6.

Model for ectopic telomere formation. BIR progression is slower at the ITS or when BIR is less processive, facilitating Mph1 disassembly of the D-loop. At the ITS this is shown as the red stop sign, at the (GT)32 or Y’ element inserts this is shown as the effects of the loss of Pif1 or Pol32. Formation of an ectopic telomere at the ITS can occur by either direct action of telomerase or HR, whereas telomere formation at the Y’ element is restricted to HR. The (GT)32 tract is expected to seed an ectopic telomere by the action of telomerase and would be expected to be a poor substrate for HR. The end product is a recipient chromosome with a full-length telomere added at the site of the insertion.
In principle, a new telomere could form at the ITS by either the direct action of telomerase or HR using a natural telomere as a template. We observed a significant decrease in the formation of truncated chromosomes when TLC1 was acutely depleted from cells, consistent with telomerase as the main mechanism. However, truncated chromosomes still form in the absence of telomerase indicating that HR can be used to generate an ectopic telomere at the ITS. Because of the need for recombination proteins to initiate BIR, we cannot evaluate the role of HR in the generation of truncated BIR products. The finding that ectopic telomere formation is significantly increased at the (GT)32 insert relative to the Y’ insert in pif1-m2 and pol32Δ mutants provides additional support for predominant usage of telomerase. While these data do indeed demonstrate that both mechanisms can be used by the cell to form telomeres at the ITS, there are several caveats to this conclusion since none of the experiments done to determine the origin of the new telomeres could be performed in biologically unperturbed cells.
The first caveat is that telomeres would begin to shorten during the time course of the BIR assay when telomerase was depleted from cells resulting in less homology to support HR. In the case of type II survivors derived from the tlc1Δ strain, the long and heterogeneous telomeres, as well as extrachromosomal telomeric circles, could provide additional homologous templates, potentially tipping the balance toward HR. The second caveat is the complication in interpretation posed by the many roles of Pif1 that are relevant to BIR. In addition to its role in repressing telomerase activity at DSB ends, Pif1 also plays a role unwinding G quadruplexes (73,74) as well as in the processivity of DNA polymerase δ during BIR (19,21). Our finding that the increased frequency of truncations in the absence of nuclear Pif1 is dependent on TLC1 supports the suggestion that telomerase can act at the ITS. However, because the pif1-m2 mutation also increases formation of truncated chromosomes in the strain with the Y’ element insertion, which cannot use telomerase to form telomeres, Pif1 must be playing at least two roles in the formation of truncations—increasing polymerase processivity and decreasing the action of telomerase at the DNA end. Disruption of either role can increase the formation of ectopic telomeres during BIR.
The minimal telomere tract for efficient telomere addition at a DSB is ∼34 bp and the threshold is reduced to ∼18 bp in the pif1-m2 mutant (75). The ITS is 64 bp in length and is expected to be efficiently used as a seed sequence by telomerase if displacement of the invading strand occurred after replication of at least half of the telomere repeat tract. The length threshold for HR is substantially greater than for telomere addition. A previous study reported a BIR frequency of ∼1% between sequences with 54 bp of shared homology as compared to 14% when the homology is 108 bp (76). Furthermore, if the invading end has a heterologous tail of >30 nucleotides (nt) then the frequency of BIR is reduced by >4-fold (76). Although we found no significant decrease in the percent of truncated chromosomes in the rad10Δ mutant, a heterologous tail of <30 nt can be removed by the proofreading function of DNA Pol δ in the absence of Rad1-Rad10 (77). Even if displacement of the invading end occurred at the end of the ITS we would predict a low frequency of HR. If displacement occurred before the end of the ITS to reveal much shorter homology, or extended beyond it to create a heterologous tail, then HR would be a rare event. The absolute frequency of chromosome truncations in the tlc1Δ strain is 1.7%, higher than expected based on the length of homology, suggesting that the availability of more homologous templates in type II telomerase-negative survivors facilitates repair by HR.
We also find that Mph1 is required to form ectopic telomeres. This requirement is consistent with previous studies showing that Mph1 (i) dissociates Rad51-generated D-loops in vivo and in vitro (64,67,78), (ii) has a negative effect on BIR (55), and (iii) promotes template switching (34). We propose that Mph1 dissociates the D-loop when it traverses the ITS such that the resulting 3′ ssDNA end containing the telomere repeats can be used to form a telomere, either by the action of telomerase or by invasion at a natural telomere followed by a second round of BIR. Stalling or slowing of the polymerase at the ITS site may render the D-loop vulnerable to Mph1 activity, or it is possible that secondary structures formed at the ITS make it easier for Mph1 disassembly. Both of these scenarios could be caused by tight binding of proteins like Rap1, which has been shown to be disruptive to replication at telomeres, or by G quadruplex formation ahead or behind the polymerase in the G rich displaced strand of the D-loop, or in the newly synthesized G-rich strand. Consistent with our findings, loss of Mph1 was shown to decrease ITS repeat expansions caused by replication fork stalling and recombinational repair (58). Loss of Mph1 also suppresses chromosome truncations resulting from aberrant recombination at a telomeric protein-induced replication fork barrier (79). Although the STR complex has been shown to disassemble D-loop intermediates (66,67), our data indicate that Mph1 is the main activity to dismantle the migrating D-loop during BIR and the role of Sgs1 is minimal.
Recent studies have shown that FANCM, the ortholog of Mph1 in mammalian cells, is essential for viability of telomerase-negative cancer cells that extend their telomeres by the HR-dependent Alternative Lengthening of Telomeres (ALT) mechanism (80–82). ALT cells exhibit increased telomeric replication stress due to a relaxed chromatin structure and elevated levels of R-loops formed by annealing of TERRA to the C-rich telomeric strand (83). FANCM is thought to relieve replication stress by promoting fork reversal, displacing R-loops and through dissociation of the D-loops that are generated by increased HR (84). When FANCM is depleted from ALT cells, replication stress is further increased resulting in hyper BIR activity, cell cycle arrest and eventual cell death (80,82). Although Mph1 is not required for growth of telomerase-negative yeast cells (55), like FANCM it limits the use of BIR by displacing the invading strand of recombination intermediates and prevents the accumulation of R-loops at telomeres (55,65,85).
In WT cells, truncated BIR products are readily detected from the strain with the natural telomere repeat inserted on the donor chromosome. When we tested sequences that share homology to Y’ elements, a class of sub-telomeric repeats, we found these sequences did not cause truncated chromosomes to be formed at a detectable level. We also found that (GT)32 sequences, which can act as telomere seeding sequences but which lack the hallmarks of telomeres, such as the abilities to form G quadruplexes and to bind telomeric proteins like Rap1, only rarely caused chromosome truncations. This finding suggests that telomeric sequences have unique features that influence synthesis during BIR and allow for generation of ectopic telomeres.
By decreasing the processivity of BIR by eliminating Pif1 or Pol32, we were able to recover BIR events that terminated at the Y’ element or (GT)32 tract. The increase in chromosome truncations was greatest in strains containing tracts of GT repeats, which we predict to mainly use telomerase to generate a new telomere because of the sequence divergence between the (GT)32 tract and natural telomeres. The significant reduction in the formation of ectopic telomeres at the (GT)32 tract, as compared to the ITS, in WT and pif1-m2 strains could be due to features of the ITS that are more disruptive to BIR synthesis or to more efficient recognition by telomerase. Of note, Rap1 binding forms a strong barrier to the strand displacement activity of DNA polymerase delta in a reconstituted system that can be overcome by Pif1 (86). Truncations were still seen, though to a lesser degree, in strains containing an insert with homology to Y’ elements, that can only use HR and have a longer stretch of DNA to synthesize before reaching the end of the chromosome. The low frequency of chromosome truncations recovered from the pol32Δ +Y’ strain is presumably because of the requirement for two rounds of BIR and is consistent with the finding that Pol32 is essential in telomerase-negative cells (15). These experiments are in agreement with earlier studies showing more mutagenic outcomes of BIR caused by lack of processivity in pol32Δ and pif1-m2 cells (14,16,19,21). It is important to note that neither the lack of Pif1 or Pol32 nor over expression of MPH1 caused truncations to form at non telomere-seeding locations. This finding shows that truncations are not simply uncontrolled telomere formation at random locations caused by BIR disruption or increased telomerase activity, as a seed sequence or homology near a telomere is still required. The reduced processivity of BIR resulting from loss of nuclear Pif1, or from MPH1 over-expression, results in fewer cells completing BIR, a condition that can be partially mitigated if cells can terminate BIR at the ITS.
While generation of HR-mediated ectopic telomeres seems similar to template switch events, it is problematic to directly compare the frequencies observed here to other template switch events because of the differing amounts of homology in each context. For example, Smith et al. (33) showed template switching in 15–25% of the BIR products analyzed. However, in the chromosome fragment assay system employed there are two copies of the donor chromosome. Moreover, regardless of where D-loop dissociation occurs, there is a homologous template available to switch to, unlike in the case of the ITS system described in the current study where successful switching is limited to the ITS. Interestingly, Smith et al. (33) found template switch events between ectopic sequences. In 11 out of 94 transformants analyzed, the BIR product was an aberrant size, and in all five analyzed in depth a template switch event had occurred between delta elements, which are the LTRs of the Ty family of retroelements that are present in ∼300 copies/haploid genome. Template switch events between delta elements have some similarity with the events triggered by the ITS in the current study, but the extent of homology available for invasion and the number of potential donors is quite different.
Taken together, the findings suggest that the ITS creates the ‘perfect storm’ for generating ectopic telomeres. On the other hand, (GT)32 and Y’ elements lack at least one of the key elements, which include tight protein binding, secondary structure formation, abundance of homologous templates in the cell, and the ability to recruit telomerase. However, the non-ITS sequences can be used if the processivity of BIR is disrupted. Thus, the sequence features of the ITS likely favor Mph1-mediated disassembly of the D-loop intermediate at the ITS, creating a substrate for telomere addition or HR. Our findings support the idea that DNA sequences which are more challenging for normal DNA replication are particularly problematic during BIR, and raise the possibility that BIR contributes to fragility of ITS in human cells.
Supplementary Material
ACKNOWLEDGEMENTS
We thank S. Mirkin, B. Luke, M.T. Teixeira and R. Rothstein for gifts of plasmids and yeast strains, and L. Berchowitz, W. Holloman and members of the Symington lab for review of the manuscript and helpful discussions.
Contributor Information
Elizabeth A Stivison, Program in Nutritional and Metabolic Biology, Columbia University Irving Medical Center, New York, NY 10032, USA; Department of Microbiology and Immunology, Columbia University Irving Medical Center, New York, NY 10032, USA.
Kati J Young, Department of Microbiology and Immunology, Columbia University Irving Medical Center, New York, NY 10032, USA.
Lorraine S Symington, Department of Microbiology and Immunology, Columbia University Irving Medical Center, New York, NY 10032, USA; Department of Genetics and Development, Columbia University Irving Medical Center, New York, NY 10032, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health [R35 GM126997]. Funding for open access charge: NIH.
Conflict of interest statement. None declared.
REFERENCES
- 1. Colnaghi R., Carpenter G., Volker M., O’Driscoll M.. The consequences of structural genomic alterations in humans: genomic disorders, genomic instability and cancer. Semin. Cell Dev. Biol. 2011; 22:875–885. [DOI] [PubMed] [Google Scholar]
- 2. Moynahan M.E., Jasin M.. Mitotic homologous recombination maintains genomic stability and suppresses tumorigenesis. Nat. Rev. Mol. Cell Biol. 2010; 11:196–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Pinto R.M., Dragileva E., Kirby A., Lloret A., Lopez E., St Claire J., Panigrahi G.B., Hou C., Holloway K., Gillis T. et al.. Mismatch repair genes Mlh1 and Mlh3 modify CAG instability in Huntington's disease mice: genome-wide and candidate approaches. PLoS Genet. 2013; 9:e1003930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Symington L.S., Rothstein R., Lisby M.. Mechanisms and regulation of mitotic recombination in Saccharomyces cerevisiae. Genetics. 2014; 198:795–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hicks W.M., Kim M., Haber J.E.. Increased mutagenesis and unique mutation signature associated with mitotic gene conversion. Science. 2010; 329:82–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Strathern J.N., Shafer B.K., McGill C.B.. DNA synthesis errors associated with double-strand-break repair. Genetics. 1995; 140:965–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Llorente B., Smith C.E., Symington L.S.. Break-induced replication: what is it and what is it for. Cell Cycle. 2008; 7:859–864. [DOI] [PubMed] [Google Scholar]
- 8. Mimitou E.P., Symington L.S.. DNA end resection: many nucleases make light work. DNA Repair (Amst.). 2009; 8:983–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lee J.Y., Terakawa T., Qi Z., Steinfeld J.B., Redding S., Kwon Y., Gaines W.A., Zhao W., Sung P., Greene E.C.. DNA RECOMBINATION. Base triplet stepping by the Rad51/RecA family of recombinases. Science. 2015; 349:977–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ferguson D.O., Holloman W.K.. Recombinational repair of gaps in DNA is asymmetric in Ustilago maydis and can be explained by a migrating D-loop model. Proc. Natl. Acad. Sci. U.S.A. 1996; 93:5419–5424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Szostak J.W., Orr-Weaver T.L., Rothstein R.J., Stahl F.W.. The double-strand-break repair model for recombination. Cell. 1983; 33:25–35. [DOI] [PubMed] [Google Scholar]
- 12. Kramara J., Osia B., Malkova A.. Break-induced replication: the where, the why, and the how. Trends Genet. 2018; 34:518–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mayle R., Campbell I.M., Beck C.R., Yu Y., Wilson M., Shaw C.A., Bjergbaek L., Lupski J.R., Ira G.. DNA REPAIR. Mus81 and converging forks limit the mutagenicity of replication fork breakage. Science. 2015; 349:742–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Deem A., Barker K., Vanhulle K., Downing B., Vayl A., Malkova A.. Defective break-induced replication leads to half-crossovers in Saccharomyces cerevisiae. Genetics. 2008; 179:1845–1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lydeard J.R., Jain S., Yamaguchi M., Haber J.E.. Break-induced replication and telomerase-independent telomere maintenance require Pol32. Nature. 2007; 448:820–823. [DOI] [PubMed] [Google Scholar]
- 16. Smith C.E., Lam A.F., Symington L.S.. Aberrant double-strand break repair resulting in half crossovers in mutants defective for Rad51 or the DNA polymerase delta complex. Mol. Cell. Biol. 2009; 29:1432–1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Burgers P.M., Gerik K.J.. Structure and processivity of two forms of Saccharomyces cerevisiae DNA polymerase delta. J. Biol. Chem. 1998; 273:19756–19762. [DOI] [PubMed] [Google Scholar]
- 18. Buzovetsky O., Kwon Y., Pham N.T., Kim C., Ira G., Sung P., Xiong Y.. Role of the Pif1-PCNA complex in pol delta-Dependent strand displacement DNA synthesis and Break-Induced replication. Cell Rep. 2017; 21:1707–1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Saini N., Ramakrishnan S., Elango R., Ayyar S., Zhang Y., Deem A., Ira G., Haber J.E., Lobachev K.S., Malkova A.. Migrating bubble during break-induced replication drives conservative DNA synthesis. Nature. 2013; 502:389–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Vasianovich Y., Harrington L.A., Makovets S.. Break-induced replication requires DNA damage-induced phosphorylation of Pif1 and leads to telomere lengthening. PLos Genet. 2014; 10:e1004679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wilson M.A., Kwon Y., Xu Y., Chung W.H., Chi P., Niu H., Mayle R., Chen X., Malkova A., Sung P. et al.. Pif1 helicase and Poldelta promote recombination-coupled DNA synthesis via bubble migration. Nature. 2013; 502:393–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Costantino L., Sotiriou S.K., Rantala J.K., Magin S., Mladenov E., Helleday T., Haber J.E., Iliakis G., Kallioniemi O.P., Halazonetis T.D.. Break-induced replication repair of damaged forks induces genomic duplications in human cells. Science. 2014; 343:88–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Minocherhomji S., Ying S., Bjerregaard V.A., Bursomanno S., Aleliunaite A., Wu W., Mankouri H.W., Shen H., Liu Y., Hickson I.D.. Replication stress activates DNA repair synthesis in mitosis. Nature. 2015; 528:286–290. [DOI] [PubMed] [Google Scholar]
- 24. Bhowmick R., Minocherhomji S., Hickson I.D.. RAD52 facilitates mitotic DNA synthesis following replication stress. Mol. Cell. 2016; 64:1117–1126. [DOI] [PubMed] [Google Scholar]
- 25. Sotiriou S.K., Kamileri I., Lugli N., Evangelou K., Da-Re C., Huber F., Padayachy L., Tardy S., Nicati N.L., Barriot S. et al.. Mammalian RAD52 functions in Break-Induced replication repair of collapsed DNA replication forks. Mol. Cell. 2016; 64:1127–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Davis A.P., Symington L.S.. RAD51-dependent break-induced replication in yeast. Mol. Cell. Biol. 2004; 24:2344–2351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Malkova A., Ivanov E.L., Haber J.E.. Double-strand break repair in the absence of RAD51 in yeast: a possible role for break-induced DNA replication. Proc. Natl. Acad. Sci. USA. 1996; 93:7131–7136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bhandari J., Karg T., Golic K.G.. Homolog-dependent repair following dicentric chromosome breakage in Drosophila melanogaster. Genetics. 2019; 212:615–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Donnianni R.A., Zhou Z.X., Lujan S.A., Al-Zain A., Garcia V., Glancy E., Burkholder A.B., Kunkel T.A., Symington L.S.. DNA polymerase delta synthesizes both strands during Break-Induced replication. Mol. Cell. 2019; 76:371–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Donnianni R.A., Symington L.S.. Break-induced replication occurs by conservative DNA synthesis. Proc. Natl. Acad. Sci. U.S.A. 2013; 110:13475–13480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Deem A., Keszthelyi A., Blackgrove T., Vayl A., Coffey B., Mathur R., Chabes A., Malkova A.. Break-induced replication is highly inaccurate. PLoS Biol. 2011; 9:e1000594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hum Y.F., Jinks-Robertson S.. Mismatch recognition and subsequent processing have distinct effects on mitotic recombination intermediates and outcomes in yeast. Nucleic Acids Res. 2019; 47:4554–4568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Smith C.E., Llorente B., Symington L.S.. Template switching during break-induced replication. Nature. 2007; 447:102–105. [DOI] [PubMed] [Google Scholar]
- 34. Stafa A., Donnianni R.A., Timashev L.A., Lam A.F., Symington L.S.. Template switching during break-induced replication is promoted by the Mph1 helicase in Saccharomyces cerevisiae. Genetics. 2014; 196:1017–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kim J.C., Harris S.T., Dinter T., Shah K.A., Mirkin S.M.. The role of break-induced replication in large-scale expansions of (CAG)n/(CTG)n repeats. Nat. Struct. Mol. Biol. 2017; 24:55–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Leffak M. Break-induced replication links microsatellite expansion to complex genome rearrangements. Bioessays. 2017; 39:doi:10.1002/bies.201700025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Aksenova A.Y., Mirkin S.M.. At the beginning of the end and in the middle of the beginning: structure and maintenance of telomeric DNA repeats and interstitial Telomeric Sequences. Genes (Basel). 2019; 10:doi:10.3390/genes10020118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Simonet T., Zaragosi L.E., Philippe C., Lebrigand K., Schouteden C., Augereau A., Bauwens S., Ye J., Santagostino M., Giulotto E. et al.. The human TTAGGG repeat factors 1 and 2 bind to a subset of interstitial telomeric sequences and satellite repeats. Cell Res. 2011; 21:1028–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lam E.Y., Beraldi D., Tannahill D., Balasubramanian S.. G-quadruplex structures are stable and detectable in human genomic DNA. Nat. Commun. 2013; 4:1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Day H.A., Pavlou P., Waller Z.A.. i-Motif DNA: structure, stability and targeting with ligands. Bioorg. Med. Chem. 2014; 22:4407–4418. [DOI] [PubMed] [Google Scholar]
- 41. Gehring K., Leroy J.L., Gueron M.. A tetrameric DNA structure with protonated cytosine.cytosine base pairs. Nature. 1993; 363:561–565. [DOI] [PubMed] [Google Scholar]
- 42. Aksenova A.Y., Greenwell P.W., Dominska M., Shishkin A.A., Kim J.C., Petes T.D., Mirkin S.M.. Genome rearrangements caused by interstitial telomeric sequences in yeast. Proc. Natl. Acad. Sci. U.S.A. 2013; 110:19866–19871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ashley T., Ward D.C.. A “hot spot” of recombination coincides with an interstitial telomeric sequence in the Armenian hamster. Cytogenet. Cell Genet. 1993; 62:169–171. [DOI] [PubMed] [Google Scholar]
- 44. Bertoni L., Attolini C., Tessera L., Mucciolo E., Giulotto E.. Telomeric and nontelomeric (TTAGGG)n sequences in gene amplification and chromosome stability. Genomics. 1994; 24:53–62. [DOI] [PubMed] [Google Scholar]
- 45. Kilburn A.E., Shea M.J., Sargent R.G., Wilson J.H.. Insertion of a telomere repeat sequence into a mammalian gene causes chromosome instability. Mol. Cell. Biol. 2001; 21:126–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mondello C., Pirzio L., Azzalin C.M., Giulotto E.. Instability of interstitial telomeric sequences in the human genome. Genomics. 2000; 68:111–117. [DOI] [PubMed] [Google Scholar]
- 47. Goto G.H., Zencir S., Hirano Y., Ogi H., Ivessa A., Sugimoto K.. Binding of multiple rap1 proteins stimulates chromosome breakage induction during DNA replication. PLos Genet. 2015; 11:e1005283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Piazza A., Serero A., Boule J.B., Legoix-Ne P., Lopes J., Nicolas A.. Stimulation of gross chromosomal rearrangements by the human CEB1 and CEB25 minisatellites in Saccharomyces cerevisiae depends on G-quadruplexes or Cdc13. PLoS Genet. 2012; 8:e1003033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Diede S.J., Gottschling D.E.. Telomerase-mediated telomere addition in vivo requires DNA primase and DNA polymerases alpha and delta. Cell. 1999; 99:723–733. [DOI] [PubMed] [Google Scholar]
- 50. Obodo U.C., Epum E.A., Platts M.H., Seloff J., Dahlson N.A., Velkovsky S.M., Paul S.R., Friedman K.L.. Endogenous hot spots of de novo telomere addition in the yeast genome contain proximal enhancers that bind Cdc13. Mol. Cell. Biol. 2016; 36:1750–1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Putnam C.D., Pennaneach V., Kolodner R.D.. Chromosome healing through terminal deletions generated by de novo telomere additions in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. U.S.A. 2004; 101:13262–13267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zierhut C., Diffley J.F.. Break dosage, cell cycle stage and DNA replication influence DNA double strand break response. EMBO J. 2008; 27:1875–1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Goldstein A.L., McCusker J.H.. Three new dominant drug resistance cassettes for gene disruption in Saccharomyces cerevisiae. Yeast. 1999; 15:1541–1553. [DOI] [PubMed] [Google Scholar]
- 54. Schulz V.P., Zakian V.A.. The saccharomyces PIF1 DNA helicase inhibits telomere elongation and de novo telomere formation. Cell. 1994; 76:145–155. [DOI] [PubMed] [Google Scholar]
- 55. Luke-Glaser S., Luke B.. The Mph1 helicase can promote telomere uncapping and premature senescence in budding yeast. PLoS One. 2012; 7:e42028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Chen Q., Ijpma A., Greider C.W.. Two survivor pathways that allow growth in the absence of telomerase are generated by distinct telomere recombination events. Mol. Cell. Biol. 2001; 21:1819–1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Teng S.C., Zakian V.A.. Telomere-telomere recombination is an efficient bypass pathway for telomere maintenance in Saccharomyces cerevisiae. Mol. Cell. Biol. 1999; 19:8083–8093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Aksenova A.Y., Han G., Shishkin A.A., Volkov K.V., Mirkin S.M.. Expansion of interstitial telomeric sequences in yeast. Cell Rep. 2015; 13:1545–1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Lundblad V., Blackburn E.H.. An alternative pathway for yeast telomere maintenance rescues est1- senescence. Cell. 1993; 73:347–360. [DOI] [PubMed] [Google Scholar]
- 60. Xu Z., Fallet E., Paoletti C., Fehrmann S., Charvin G., Teixeira M.T.. Two routes to senescence revealed by real-time analysis of telomerase-negative single lineages. Nat. Commun. 2015; 6:7680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Cesare A.J., Griffith J.D.. Telomeric DNA in ALT cells is characterized by free telomeric circles and heterogeneous t-loops. Mol. Cell. Biol. 2004; 24:9948–9957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Boule J.B., Vega L.R., Zakian V.A.. The yeast Pif1p helicase removes telomerase from telomeric DNA. Nature. 2005; 438:57–61. [DOI] [PubMed] [Google Scholar]
- 63. Hu Y., Tang H.B., Liu N.N., Tong X.J., Dang W., Duan Y.M., Fu X.H., Zhang Y., Peng J., Meng F.L. et al.. Telomerase-null survivor screening identifies novel telomere recombination regulators. PLoS Genet. 2013; 9:e1003208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Prakash R., Satory D., Dray E., Papusha A., Scheller J., Kramer W., Krejci L., Klein H., Haber J.E., Sung P. et al.. Yeast Mph1 helicase dissociates Rad51-made D-loops: implications for crossover control in mitotic recombination. Genes Dev. 2009; 23:67–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Mehta A., Beach A., Haber J.E.. Homology requirements and competition between gene conversion and Break-Induced replication during double-strand break repair. Mol. Cell. 2017; 65:515–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Fasching C.L., Cejka P., Kowalczykowski S.C., Heyer W.D.. Top3-Rmi1 dissolve Rad51-mediated D loops by a topoisomerase-based mechanism. Mol. Cell. 2015; 57:595–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Piazza A., Shah S.S., Wright W.D., Gore S.K., Koszul R., Heyer W.D.. Dynamic processing of displacement loops during recombinational DNA repair. Mol. Cell. 2019; 73:1255–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Lydeard J.R., Lipkin-Moore Z., Jain S., Eapen V.V., Haber J.E.. Sgs1 and exo1 redundantly inhibit break-induced replication and de novo telomere addition at broken chromosome ends. PLoS Genet. 2010; 6:e1000973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Fishman-Lobell J., Haber J.E.. Removal of nonhomologous DNA ends in double-strand break recombination: the role of the yeast ultraviolet repair gene RAD1. Science. 1992; 258:480–484. [DOI] [PubMed] [Google Scholar]
- 70. Ivanov E.L., Haber J.E.. RAD1 and RAD10, but not other excision repair genes, are required for double-strand break-induced recombination in Saccharomyces cerevisiae. Mol. Cell. Biol. 1995; 15:2245–2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Hwang J.Y., Smith S., Myung K.. The Rad1-Rad10 complex promotes the production of gross chromosomal rearrangements from spontaneous DNA damage in Saccharomyces cerevisiae. Genetics. 2005; 169:1927–1937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Lustig A.J. Hoogsteen G-G base pairing is dispensable for telomere healing in yeast. Nucleic Acids Res. 1992; 20:3021–3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Dahan D., Tsirkas I., Dovrat D., Sparks M.A., Singh S.P., Galletto R., Aharoni A.. Pif1 is essential for efficient replisome progression through lagging strand G-quadruplex DNA secondary structures. Nucleic Acids Res. 2018; 46:11847–11857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Paeschke K., Bochman M.L., Garcia P.D., Cejka P., Friedman K.L., Kowalczykowski S.C., Zakian V.A.. Pif1 family helicases suppress genome instability at G-quadruplex motifs. Nature. 2013; 497:458–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Strecker J., Stinus S., Caballero M.P., Szilard R.K., Chang M., Durocher D.. A sharp Pif1-dependent threshold separates DNA double-strand breaks from critically short telomeres. Elife. 2017; 6:e23783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Anand R., Beach A., Li K., Haber J.. Rad51-mediated double-strand break repair and mismatch correction of divergent substrates. Nature. 2017; 544:377–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Paques F., Haber J.E.. Two pathways for removal of nonhomologous DNA ends during double-strand break repair in Saccharomyces cerevisiae. Mol. Cell. Biol. 1997; 17:6765–6771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Sebesta M., Burkovics P., Haracska L., Krejci L.. Reconstitution of DNA repair synthesis in vitro and the role of polymerase and helicase activities. DNA Repair (Amst.). 2011; 10:567–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Jorgensen S.W., Liberti S.E., Larsen N.B., Lisby M., Mankouri H.W., Hickson I.D.. Esc2 promotes telomere stability in response to DNA replication stress. Nucleic Acids Res. 2019; 47:4597–4611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Lu R., O’Rourke J.J., Sobinoff A.P., Allen J.A.M., Nelson C.B., Tomlinson C.G., Lee M., Reddel R.R., Deans A.J., Pickett H.A.. The FANCM-BLM-TOP3A-RMI complex suppresses alternative lengthening of telomeres (ALT). Nat. Commun. 2019; 10:2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Pan X., Drosopoulos W.C., Sethi L., Madireddy A., Schildkraut C.L., Zhang D.. FANCM, BRCA1, and BLM cooperatively resolve the replication stress at the ALT telomeres. Proc. Natl. Acad. Sci. U.S.A. 2017; 114:E5940–E5949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Silva B., Pentz R., Figueira A.M., Arora R., Lee Y.W., Hodson C., Wischnewski H., Deans A.J., Azzalin C.M.. FANCM limits ALT activity by restricting telomeric replication stress induced by deregulated BLM and R-loops. Nat. Commun. 2019; 10:2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Arora R., Lee Y., Wischnewski H., Brun C.M., Schwarz T., Azzalin C.M.. RNaseH1 regulates TERRA-telomeric DNA hybrids and telomere maintenance in ALT tumour cells. Nat. Commun. 2014; 5:5220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Domingues-Silva B., Silva B., Azzalin C.M.. ALTernative functions for human FANCM at telomeres. Front. Mol. Biosci. 2019; 6:84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Lafuente-Barquero J., Luke-Glaser S., Graf M., Silva S., Gomez-Gonzalez B., Lockhart A., Lisby M., Aguilera A., Luke B.. The Smc5/6 complex regulates the yeast Mph1 helicase at RNA-DNA hybrid-mediated DNA damage. PLoS Genet. 2017; 13:e1007136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Koc K.N., Singh S.P., Stodola J.L., Burgers P.M., Galletto R.. Pif1 removes a Rap1-dependent barrier to the strand displacement activity of DNA polymerase delta. Nucleic Acids Res. 2016; 44:3811–3819. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.