


Abstract
Bacterial retrons consist of a reverse transcriptase (RT) and a contiguous non-coding RNA (ncRNA) gene. One third of annotated retrons carry additional open reading frames (ORFs), the contribution and significance of which in retron biology remains to be determined. In this study we developed a computational pipeline for the systematic prediction of genes specifically associated with retron RTs based on a previously reported large dataset representative of the diversity of prokaryotic RTs. We found that retrons generally comprise a tripartite system composed of the ncRNA, the RT and an additional protein or RT-fused domain with diverse enzymatic functions. These retron systems are highly modular, and their components have coevolved to different extents. Based on the additional module, we classified retrons into 13 types, some of which include additional variants. Our findings provide a basis for future studies on the biological function of retrons and for expanding their biotechnological applications.
INTRODUCTION
Reverse transcriptases (RTs, also known as ‘RNA-dependent DNA polymerases’) were discovered in 1970, by Temin and Baltimore, in tumor viruses (1,2). These enzymes are present in the three domains of life capable of polymerizing cDNA from an RNA template. They are known principally as a key component of eukaryotic mobile retrotransposons (3) and retroviruses (4), but they are also widely distributed among bacterial and archaeal species.
Prokaryotic RTs were not discovered until 1989, when they were identified as components of retrons, responsible for the production of short single-stranded linear DNA fragments (5,6). RTs were subsequently shown to be present in diversity-generating retroelements (DGRs) (7,8), abortive phage infection (Abi) systems (9–11), CRISPR-Cas systems (12–19) and group II introns, this last category being the only one for which autonomous mobility has been demonstrated (20–23). The most recent exhaustive phylogenetic analysis of prokaryotic RTs (17) revealed a huge diversity, in the form of group II introns (47%), retron/retron-like sequences (25%), and DGRs (12%), with the remaining 16% clustering into distinct groups, including CRISPR-Cas associated RTs, Abi-like and other yet uncharacterized RTs, such as the G2L (group II-like) or UG (unknown) groups.
Retrons are defined by their unique ability to produce an unusual satellite DNA known as msDNA (multicopy single-stranded DNA) (24), the function of which remains unknown. They consist of ∼2000 bp of DNA including an RT-coding gene (ret) and contiguous inverted sequences (msr and msd). The msr/msd regions and the ret gene are transcribed as a single RNA, which is folded into a specific secondary structure. Once translated, the RT binds the RNA template downstream from the msd region, initiating reverse transcription of the RNA towards its 5′ end, assisted by the 2′OH group present in a conserved branching G residue that acts as a primer. Reverse transcription halts before reaching the msr region, and the resulting DNA, the msDNA, remains covalently attached to the RNA template via a 2′-5′ phosphodiester bond and base-pairing between the 3′ ends of the msDNA and the RNA template (25–28). The msr/msd transcripts from different retrons have very different sequences, but common structural features, which are thought to be involved in retron function. The external regions, at the 5′ and 3′ ends of the msd/msr transcript (a1 and a2, respectively) are complementary and can hybridize, leaving the structures located in the msr and msd regions in internal positions (see Figure 1). The msr region, which is not reverse transcribed, forms one to three short stem-loops or variable size, ranging from 3 to 10 bp, whereas the msd region folds into a single/double long hairpin with a highly variable long stem of 10–50 bp in length that is also present in the final msDNA form (27–29).
Figure 1.
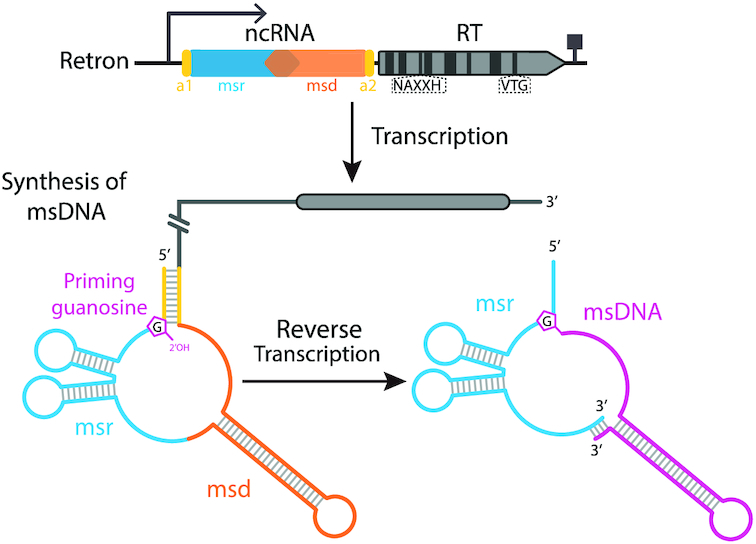
Retron organization and msDNA synthesis process. Retrons comprise a reverse transcriptase (RT) and two non-coding contiguous inverted sequences (named msr and msd) transcribed as a single RNA that is folded into a specific secondary structure. The conserved NAXXH motif and VTG triplet in retron RTs are indicated. The RT binds downstream from the msd region in the RNA, initiating reverse transcription of the RNA template towards its 5′ end, assisted by the 2′OH group present in a conserved branching G residue acting as a primer. Reverse transcription halts before the msr region is reached, and the resulting msDNA remains covalently attached to the RNA template via a 2′-5′ phosphodiester bond and base-pairing of the 3′ ends of the molecules.
Our knowledge of the mechanism of msDNA generation is derived from studies of 38 non-redundant msDNA-producing retrons, only 16 of which contain fully annotated and experimentally validated msr-msd-RT cassettes (28). About one third of all annotated retrons are predicted to have additional open reading frames (ORFs) encoding proteins of unknown function, some of which have been annotated as adenosine-binding proteins or cold-shock proteins (30). The significance of these proteins, and their role in retron biology, remain to be determined.
Despite numerous characterizations in vitro and in vivo, very little is known about the biology of retrons, and their putative function has remained a mystery for almost 30 years (31). Here, we develop a computational pipeline for the systematic prediction of genes specifically associated with retron RTs, and report that most bacterial retrons are tripartite systems including, in addition to the (msr-msd) ncRNA gene and the RT, a primary protein-coding gene or RT-fused domain. The predicted functions of this third component are highly diverse. The three components of the retron systems display an extraordinary modularity, probably expanding their functional and mechanistic diversity, but comparisons between the phylogenies obtained for the ncRNA, the RT and the associated proteins suggest that they have coevolved, providing additional evidence in favor of a functional association. Clustering on the basis of the genes encoding these associated proteins or RT-fused domains groups retron systems into 13 types, some of which include additional subtypes or variants.
MATERIALS AND METHODS
Prokaryotic reverse transcriptase datasets
The dataset of prokaryotic RTs used for the systematic prediction of genes specifically associated with retron RTs reported here consisted of a previously published dataset of 9141 unique representative entries derived from the clustering, at 85% sequence identity, of 198 760 annotated reverse transcriptases obtained from different databases (17). This dataset contains group-II introns (47%), retron/retron-like (25%) and DGRs RT sequences (12%), the remaining 16% clustering into distinct groups including RTs linked to CRISPR-Cas systems, Abi-like RTs and uncharacterized RTs grouped into the G2L (group II-like) and UG groups (17). The specific retron dataset further analyzed in this study comprises 1912 retron/retron-like RT sequences from the dataset described above and 16 RTs from experimentally validated retrons (28) (Supplementary Table S1).
Clustering of neighboring proteins
The ORFs located within ± 30 kb of each RT were retrieved and clustered with the MMseqs2 suite (32). Sequences of <30 amino acids (aa) or >3000 aa were discarded from the analysis due to poor downstream clustering. Connected component clustering was first performed, to cover remote homologs. We then performed deeper iterative cascaded searches until convergence, to merge close clusters and to cover more remote homologs with the mmseqs search and mmseqs result2profile utilities (https://github.com/soedinglab/MMseqs2/wiki). This procedure yielded 62 277 clusters, 5413 of which had more than five members.
Prediction of functional association
We encoded the neighborhood of the 9141 RTs, by constructing a presence/absence matrix, in which the rows corresponded to the RTs ordered by the position on the tree, and the columns corresponded to the various neighboring protein clusters, ordered by size. This matrix was further analyzed by computing two parameters providing an estimate of the non-random distribution of every cluster across the tree.
We first calculated the density of protein occurrence for a given cluster, by calculating the moving average of each column (bandwidth = 50, steps = 1) and normalizing the values obtained with the moving average and moving standard deviation of 10 000 random permutations for each cluster size. We set a cutoff value for statistical significance of the mean value plus four times the standard deviation for the random permutations. In order to normalize different clusters by their size, we calculated a parameter (hereafter referred as phyvalue) consisting of the ratio of the sliding average to the selected cutoff. We only retained clusters for which the maximum ratio was at least 2, and we further divided each cluster into subclusters separated by >10 positions below the cutoff. Using this parameter, we identified 131 protein subclusters associated with retron RTs. We then took into account the background similarity between RTs in the phylogenetic tree due to the underlying speciation events, by calculating sequence identity for each group of RTs associated with a given subcluster. Based on the neighboring clusters found in the vicinity of other groups of RTs, we established a cutoff of 60% for considering this nonrandom colocalization to be probably functional (i.e. groups of RTs with a sequence identity below this value were considered to be different enough for the association to be significant). By adding this second parameter, we were able to decrease the number of protein subclusters predicted to be associated with retron RTs to 71 (Supplementary Table S2).
Reannotation of the genomic neighborhood
For each cluster, a multiple sequence alignment (MSA) was built with MAFFT (33), using the parameters –globalpair –maxiterate 1000 –reorder, and hidden Markov-model protein profiles were built with hmmbuild from the HMMER suite (http://hmmer.org/). Using these profiles, we ran a custom script written in python 3.6 (17) that uses hmmsearch to annotate the ORFs in the vicinity of RTs and retrieves genomic and taxonomic information (Supplementary Table S3).
Analysis and prediction of retron RT-associated protein function
Remote sequence homology was detected on representative sequences from each cluster and subcluster analyzed, with HHpred and HHblits (34,35) for sequence-based predictions of protein function and structure. Protein domains within sequences were identified with InterPro (36), Pfam (37) and SMART (38).
Protein structure prediction
Protein structure modelling was performed on different retron RTs-associated proteins in order to support the sequence-based functional prediction by selecting and submitting representative sequences of each cluster independently to the RaptorX (39), Phyre2 (40), I-TASSER (41,42) and trRosetta (43) prediction servers. Best quality structural models were selected according to the quality criteria of each server for further structure-based functional predictions. Visualization of the models and structural alignment was performed in PyMOL Molecular Graphics System V2 (Schrödinger, LLC). In order to infer plausible functions of the unknown modelled proteins, structural homologs with described functions were identified by performing a structure-based similarity search against all protein structures deposited in the PDB with the DALI server (44) and with the PDBeFold service (45).
Phylogenetic analyses
We used MAFFT software (33) and progressive methods for MSAs. For retron RT phylogeny, a MSA was constructed corresponding to the RT0–7 domain of 1,912 RT sequences from the 9141-entry dataset previously classified as retron/retron-like RTs (17) plus 16 RTs from experimentally validated retrons (28). Representative alignments of associated proteins were obtained with ESPript 3.0 (46). Unless otherwise indicated, phylogenetic trees were constructed with the FastTree program (47) with the WAG evolutionary model, and the discrete gamma model with 20 rate categories. The RT tree was constructed with IQ-TREE v1.6.12, with 1000 ultra-fast bootstraps (UFBoot) and SH-like approximate likelihood ratio test (SH-aLRT) with 1000 replicates (option -bb 1000 -alrt 1000 in IQ-TREE) (48), using the LG+F+R10 model identified as the best model by Modelfinder (49) because it gave the lowest Bayesian Information Criterion (BIC) among 546 protein models available for the tree. The inner nodes of the RT Clades have UFBoot and SH-aLRT support values >85%.
Detection of structured RNAs
We used CMfinder 0.4.1 (50) and R-scape (51) to design a pipeline consisting of several sequential steps for the detection of structured RNAs in the vicinity of retron RT sequences. We first used each experimentally confirmed msr-msd transcript and its close relatives as seeds for CMfinder 0.4.1, an RNA motif prediction tool using statistical measures for structure prediction based on a combination of folding energy and sequence covariation. We then built covariate models with cmbuild from the Infernal suite (52), and used these models to search for similar structures upstream and downstream from the start of the RT gene. In groups in which no validated msr-msd transcripts were previously described, we manually searched for structured regions with features similar to those described for other msr-msd transcripts in the vicinity of RT-coding genes using RNAfold (53), and we built multiple alignments with MAFFT-Q-INS-i (33), with the aim of locating conserved sequences in closely related genomes. We then trimmed the alignments in the a1 and a2 regions, and repeated steps of the process up to the retrieval of upstream and downstream sequences. Finally, we used R-scape (51) to describe covarying base pairs in the proposed consensus structures and to reduce the relative weight of phylogenetic correlations and base composition biases not due to conserved RNA structure.
Coevolution analysis
We studied the coevolution of different components of different retron systems, by building multiple alignments with MAFFT-FFT-NS-i/Q-INS-I and inferring phylogenetic trees with IQ-TREE (48) (default parameters and -B 1000) for the various groups of RTs, clusters and proposed msr-msd transcripts. As a way to measure the co-evolution between the different components of the retrons, we compared the trees distances between the components of the retron tripartite system and that for the corresponding 16S RNAs retrieved from the SILVA database (54). We use 16S RNAs trees to check if the co-evolution is due to a functional association or if the similarity comes from background similarity between the components due to the underlying speciation events themselves. To this end, we computed the cophenetic correlation of the branch length distances (55) using R 3.6 and the package dendextend (56).
A summary of the main software, tools and packages used in this work is shown in Supplementary Table S4.
RESULTS
Phylogeny of retron/retron-like RTs
In a recent survey of 198,760 annotated RTs in prokaryotes clustered into 9141 representative sequences, we found that ∼25% of the dataset could be classified as retron/retron-like RTs (17) widely distributed in the phylum Proteobacteria and to a lesser extent in the Firmicutes, Bacteroidetes, Actinobacteria and Cyanobacteria, and other minor bacterial phyla. The phylogenetic trees inferred from the alignment (domains RT0–7) of 1912 retron/retron-like RT sequences from the above dataset and 16 additional RTs from experimentally validated retrons (28) revealed that they could be grouped into 11 well-supported clades (Figure 2A and Supplementary Table S1). Interestingly, unlike other RTs, such as those encoded by group II introns and DGRs, the clades inferred from the phylogeny of retron RTs displayed strong support even for the inner nodes (Supplementary File S1) probably corresponding to more recent evolution. All these RTs displayed features characteristic of retron-type RTs, including region Y and region X (28). Region Y, which is located at the C-terminus, contains a highly conserved VTG triplet within the RT7 domain (with G the most conserved residue), and directs the RT to its cognate msr. By contrast, region X, located between the RT2 and RT3 domains, presents a characteristic NAXXH motif (with H the most conserved residue) and may be essential for reverse transcription initiation or catalysis (Figure 1). Moreover, 16 experimentally validated retrons (28) were found to cluster in six of the 11 clades identified (Figure 2A, Table 1 and Supplementary Table S1). These data further support the identification of the clustered RTs as retron/retron-like RTs and highlight the wide-ranging genetic diversity of the bacterial retron dataset used in this work.
Figure 2.

Phylogeny of retron/retron-like RTs and associated ncRNA structures. (A) Phylogenetic reconstruction of retron/retron-like RTs. The tree is provided as a newick file in Supplementary File S1. The different colors represent the different types/variants identified as clustering in different phylogenetic groups. The outer lines indicate the phylogenetic clades. RTs belonging to experimentally validated retrons are indicated, and their nomenclature is included. Note that Vc137 correspond in Supplementary Table S1 to entry 1575 (fig|1343738.3.peg.2232) present in the data set of 1,912 retron/retron-like RT sequences. (B) Consensus IA/IIA1 msr/msd transcript structure. The different regions of the msr/msd transcript are highlighted in color: msr (blue), msd (orange), a1/a2 (yellow). The two opposing G residues are highlighted in magenta. An alignment of the msr/msd transcript with 10 representative sequences is shown below, in which positions with >90% gaps were removed. Other consensus ncRNA structures identified are shown in Supplementary Figure S1.
Table 1.
Retron system classification
| RT-Cladea | Structured RNA familya | Retron Type/Subtype/ varianta | Clusterb | Sub-clusterb | Description of domainsc | Described Retronsd |
|---|---|---|---|---|---|---|
| 1 | IA1/IIA1 | I-A | 22 | 1 | N-terminal ATPase module containing Walker A and Walker B motifs of P-loop NTPases (ABC ATPase) | Retron-Eco7 (Ec78); Retron-Eco4 (Ec83); Retron-Vpa1 (Vp96); Retron-Vch1 (Vc95) |
| 39 | 1 | N-terminal HNH endonuclease | ||||
| 1 | IB1 | I-B1 | 22 | 2 | N-terminal ATPase module containing Walker A and Walker B motifs of P-loop NTPases (ABC ATPase) + TOPRIM domain (Motif IV: EDxxL; Motif V: DxD) | |
| 3 | IB2 (Proteobacteria/Firmicutes) | I-B2 | 159 | 1 | N-terminal ATPase module containing Walker A and Walker B motifs of P-loop NTPases (ABC ATPase) + TOPRIM domain (Motif IV: EGxxE; Motif V: DxD) | |
| 8 | IC | I-C1 | NC | RT fused at the C-termini with a TOPRIM domain (Motif IV: EGxxD; Motif V: DxD) | Retron-Eco2 (Ec67) | |
| 8 | IC | I-C2 | NC | RT fused at the C-termini with a TOPRIM domain (Motif IV: EGxxD; Motif V: DxE) | ||
| 8 | Nd | I-C3 | NC | RT fused at the C-termini with a TOPRIM domain (Motif IV: EGxxD; Motif V: DxD) | ||
| 1 | IA1/IIA1 | II-A1 | 42 | 2 | N-terminal Nucleoside deoxyribosyltransferase-like (NDT) + C-terminal DNA binding domain | Retron-Sen2 (St85); Retron-Vch2 (Vc81); Retron-Eco3 (Ec73) |
| 1 | IIA2 | II-A2 | 42 | 3 | N-terminal Nucleoside deoxyribosyltransferase-like (NDT). Some members carries an N-terminal Tryipsine-like serine protease domain; frameshift separating the C-terminal DNA binding domain | |
| 9 | IIA3 (Proteobacteria/Firmicutes) | II-A3 | 42 | 1 | N-terminal Nucleoside deoxyribosyltransferase-like (NDT) + C-terminal DNA binding domain | Retron-Eco1 (Ec86); Retron-Vch3 (Vc137)e |
| 2 | IIIA1 | III-A1 | 113 | 1 | PRTase-like | |
| 742 | 1 | C-terminal winged helix DNA binding domain | ||||
| 9 | IIIA2 | III-A2 | 113 | 4 | PRTase-like | |
| 1121 | 1 | C-terminal winged helix DNA binding domain | ||||
| 9 | IIIA3 | III-A3 | 67 | 3 | N-terminal PRTase-like and RuvB C-terminal winged helix DNA binding domain | |
| 9 | Nd | III-A4 | 67 | 2b | N-terminal PRTase-like and RuvB C-terminal winged helix DNA binding domain | |
| 5,6,9 | nd | III-A5 | 67 | 2a | N-terminal PRTase-like and C-terminal winged helix DNA binding domain | |
| 67 | 1 | PRTase-like | ||||
| 113 | 2 | PRTase-like | ||||
| 113 | 3 | PRTase-like | ||||
| 1689 | 1 | C-terminal winged helix DNA binding domain | ||||
| 3 | IV | IV | 826 | 1 | Integral membrane protein: N-terminal signal peptide and two TM helices | Retron-Eco6 (Ec48) |
| 3 | V | V | 85 | 1 | ‘Cold-Shock’ DNA binding domain | Retron-Sen1 (Se72) |
| 3 | nd | VI | 2 | 1 | Cro/C1-type HTH domain | |
| 2075 | 1 | Accesory protein of unknown function | ||||
| 3272 | 1 | Accesory protein of unknown function | ||||
| 2760 | 1 | Accesory protein of unknown function | ||||
| 4 | nd | VII-A1 | NC | RT fused to DUF3800 at the C-terminus | ||
| 4 | nd | VII-A2 | 2165 | 1 | DUF3800 (Structural homology to RNaseH2) | |
| 4 | nd | VIII | 2446 | 1 | Putative serine esterase (DUF626) | |
| 7 | IX | IX | 1740 | 1 | HEPN domain | |
| 1093 | 1 | C-terminal HTH12; “Winged helix" DNA-binding domain | ||||
| 7 | nd | X | 2273 | 1 | IHF-like DNA-binding proteins and C-terminal VHL-like domain | |
| 8 | nd | XI | NC | RT fused at the C-termini with a Peptidase (Trypsin-like Serine protease) domain | ||
| 8 | nd | XII | NC | RT fused at the C-termini with a C-terminal TIR_2 domain | ||
| 10 | XIII (Firmicutes/Mx162/Mx65/Ne144) | XIII | 114 | 1 | DEDE motif, winged helix domain and C-terminal SWIM Zn-finger domain (CxCx12CxH) | Retron-Nex2 (Ne144); Retron-Mxa1 (Mx162); Retron-Mxa2 (Mx65); Retron-Sau1 (Sa163) |
| 216 | 1 | WGR-like domain and C-terminal HEAT/ARM-like repeat containing protein | ||||
| 259 | 1 | HEAT/ARM-repeat containing protein | ||||
| 1688 | 1 | HEAT/ARM-repeat containing protein | ||||
| 3262 | 1 | HEAT/ARM-repeat containing protein | ||||
| 216 | 2 | HEAT/ARM-repeat containing protein |
aRetron Classification based on RT phylogeny (Clades)/RNA structure/Retron system as specified on Figures 2 and 5.
bSpecific cluster (subcluster) numbers (as specified on Supplementary Table S2) characteristic of every Retron System.
cDescription of protein domains of the associated gene/fusion.
dRetron described in Simon et al. (2019) as ‘experimentally validated retrons’.
eRetron described in Inouye et al. 2011.
The retron ncRNA gene associated with retron/retron-like RT sequences
Retrons are usually defined as contiguous transcriptional cassettes encompassing an ncRNA gene containing the msr and msd regions and a specialized reverse transcriptase (RT) gene (Figure 1). Retron msr-msd transcriptional cassettes have divergent sequences but display substantial structural similarities (28). We, therefore, analyzed the genomic neighborhood of the retron/retron-like RT sequences, searching for conserved RNA secondary structures resembling those of previously characterized msr-msd transcripts. Typically, msr-msd transcripts fold into a characteristic structure in which the inverted repeats at the ends hybridize, leaving the structures located in the msr and msd regions in internal positions. The msr region contains one to three hairpins, whereas the msd region contains a long stable stem-loop (Figure 1). Using covariance models and consensus structure detection, we were able to identify these characteristic structures with a high degree of confidence in eight of the 11 RT clades described here, some of which had no previous experimentally validated representatives (Figure 2B and Supplementary Figure S1). Some of the RT clades had a single consensus structure for the msr-msd transcript (clades 7 and 8), whereas others (clades 1, 2, 3, 9, 10 and 11) displayed greater structural diversity, with two or three different consensus structures, highlighting a modularity of the (msr-msd)-RT cassette. All the proposed structures display the common features described in previous studies, with short hairpins in the msr region and a long stem-loop in the msd region. However, some of the above clades (clades 3, 7, 8 and 9) have large subbranches in which this structure was not found and we were unable to detect significant consensus structures in clades 4, 5 and 6 (Supplementary Table S1). Nevertheless, we noted sequence conservation in the vicinity of these RTs and close relatives (for an example see Supplementary Figure S2). It is not, therefore, possible to rule out the possibility of associated ncRNA transcripts with unusual structures not resembling that of canonical msr-msd transcripts. These results reveal an extraordinary diversity of retron units that could not easily be classified on the basis of the structure of the msr-msd transcript or the phylogenetic origin of the RT. The consensus structures provided in Figure 2B and Supplementary Figure S1 represent a compendium of all the secondary structures from a given group of msd/msr transcripts, and do not, therefore, account for individual variations that could contribute to the biological functions of the retrons concerned. Taken together, these results provide further support for the identification of the retron/retron-like RTs included in this dataset as a component of bona fide retrons.
Prediction of genes functionally associated with retron RTs and classification of retron systems
The increase in the amount of genomic data available from public databases has made it possible to develop and use novel methodologies and analyses, with the aim of predicting functional associations between proteins (57). It is currently thought that only a third of annotated retrons have accessory open reading frames (ORFs) of unknown function (28). We investigated the range of this association and the feasibility of predicting genes functionally associated with retron RTs and separating them from spurious associations without bias related to their hypothetical function, by developing a computational procedure integrating the previously constructed phylogenetic information based on the large prokaryotic RT dataset comprising 9141 RT sequences (17).
This pipeline consisted of several sequential steps summarized in Figure 3 and Supplementary Figure S3. We first retrieved all the aa sequences corresponding to annotated coding sequences in the vicinity (±30 kb) of the 9141 RTs. We clustered these sequences by deep iterative comparison, to ensure that remote homologs were covered. This resulted in in 5413 clusters with more than five representatives. We assessed the likelihood of these clusters being associated with RTs, by calculating two parameters: (i) the non-random distribution of clusters across the tree (phyvalue) and (ii) the percentage sequence identity of the RTs involved. Using these two parameters, we were able to show that the clusters were associated with RTs, including those previously reported to be associated with RT-CRISPR, such as Cas2 (cluster 30) (14,15) (Figure 4), and DGRs such as Avd (cluster 4) (58) (not shown) proteins. Similarly, the clusters predicted to be potentially associated with group II introns (94.56%) on the basis of phylogenetic accumulation did not pass the sequence identity cutoff test, consistent with group II introns being autonomous mobile retroelements (Figure 4). We then analyzed a total of 71 subclusters (Figure 4 and Supplementary Table S2) predicted to be associated with retron RTs further manually and case-by-case, and those considered to be spurious based on inconsistencies in genomic arrangement, irregular presence/absence patterns or poor alignments were removed. The coding sequences in the neighborhood of the retron RTs were then re-annotated with hmmsearch and the custom profiles (Supplementary Table S3). Finally, each cluster was further subdivided into subclusters on the basis of discontinuities in the previously reported phylogenetic tree (17), and multiple alignments and hidden Markov models profiles were built for each (see Materials and Methods).
Figure 3.
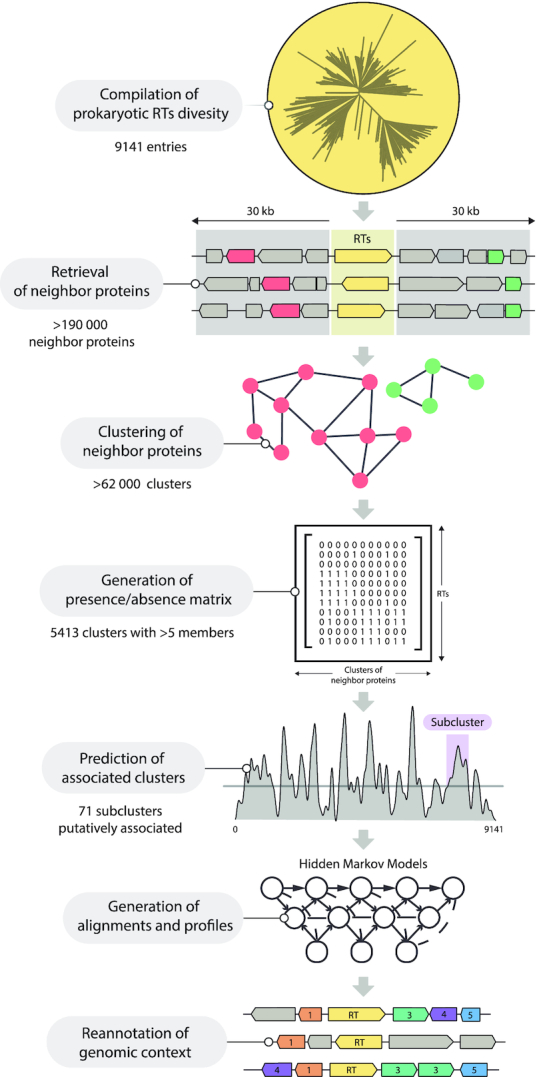
Schematic summary of the different stages of the computational procedure. The different stages of the computational procedure developed for the prediction of genes associated with retron RTs are indicated. See main text and Supplementary Figure S3 for details.
Figure 4.

RT-neighboring protein subclusters patterns. Two-dimensional representation of sequence identity (%) and phyvalues of putative associated subclusters. Subclusters above the selected cutoff for phyvalue are shown. The vertical line at 60% of RT identity represents the sequence identity cutoff for the estimated functional association (see Methods). The size of the dots is proportional to the number of sequences within the corresponding subcluster. (A) Pattern of subclusters found in the neighborhood of group II introns (highlighted in green) were used as a negative control, whereas those found in RT-CRISPR (highlighted in magenta) were used as a positive control. Cas2 cluster dot is indicated. (B) Subclusters found in the vicinity of retrons (highlighted in yellow). Only 71 dots to the right of the vertical line (<60% RT identity) were further considered as functionally associated (Supplementary Table S2).
This sequence similarity clustering and retron RT sequence analysis revealed that most retron RTs have nonrandomly associated specific gene-encoding proteins, whereas sequence analyses showed that other RTs appear to have mostly C-terminal additional domains with diverse predicted functions, many probably related to nucleases, nucleic acid metabolism, DNA/RNA binding, signaling, membrane proteins and proteases. Taken together, these results suggest that retrons may broadly be considered to constitute a tripartite system including a non-coding RNA (msr-msd)-RT cassette for msDNA production, and an associated specific protein or additional domain fused to the RT of potential relevance for determining the functionality of the retron unit. Retron systems can be clustered into 13 types, some of which include additional subtypes or variants, on the basis of the associated protein-coding genes or domains fused to the retron RT (Figure 5 and Table 1).
Figure 5.

Classification of retron systems. Schematic diagram of the genomic organization of the different types/variants of retron systems including the corresponding RT clades as summarized in Table 1. The colors of the different types/variants correlate with those of the RT phylogenetic groups (Figure 2A).
Modularity of retron systems
We found associated proteins or fused domains with distinct predicted functions within a particular RT clade (Table 1). For example, the RTs of clade 1 were associated with NDTs (nucleoside deoxyribosyltransferases) and ATPases; those of clade 3 were linked to distinct DNA-binding and membrane proteins with 2 transmembrane domains (TM); those of clade 4 were associated with esterases and possible RNases; those of clade 7 were associated with possible RNases carrying a HEPN domain and DNA-binding proteins; and those of clade 8 with the RT C-terminus fused to three distinct domains, a Toprim domain, a Toll/interleukin-1 receptor/resistance protein domain, and a peptidase (trypsin-like serine protease) domain. We also found proteins with similar functions associated with distantly related RTs (Table 1), such as the phosphoribosyltransferases (PRTases) found with RTs clustered in clades 2, 5, 6 and 9; or the Toprim domain that appeared to be associated with the RTs of clades 1 and 8. In fact only clade 10 seems to represent a unique RT/putative effector type and this is also true for the associated ncRNA structure (type XIII). Comparisons between RT phylogeny, the distribution of associated protein clusters or domains fused to the C-terminus of the RT and the structural diversity of ncRNA transcripts suggest a modular organization of retron systems with the (msr-msd)-RT cassette as the key module acquiring distinct putative effector proteins. This conclusion may be exemplified by the consensus structure of the ncRNA and RT phylogeny in the cases of retron systems I-A and II-A1 in which a similar (msr-msd)-RT cassette acquired both ATPase and NDT-like putative effector domains. The exchange of putative effector modules may be a consequence of environmental changes to face possibly rapidly evolving phages. The independence of the retron and associated domain modules is also exemplified by the presence of stand-alone (msr-msd)-RT cassette in a branch of the RT clade 2 and clade 11.
Coevolution of various parts of the retron unit
We investigated the evolution of the ncRNA and the RT relative to the associated protein, by performing coevolution analyses taking into account the various parts of the retron unit. We built independent phylogenetic trees for RT, associated proteins and the ncRNA, which we compared, using the 16S rRNA tree for the host genome as a control (see Materials and Methods). We ensured that the analysis had sufficient statistical power, by performing these analyses for the types for which it was possible to find complete retron systems with at least 20 representatives. As examples of retron systems with distinct functionally predicted associated proteins within a particular RT clade, we analyzed types I-A and II-A1, which had a common ncRNA transcript structure (IA/IIA1), in clade 1, and type I-B2 in clade 3. As a representative of retron systems with associated proteins sharing similar functions but linked to distantly related RTs, we analyzed type III-A3 in clade 9. Type XIII in clade 10, the only RT-associated protein system with distinct ncRNA transcript structure, was also analyzed (see Supplementary Figure S4). In all cases, RT tree distances were strongly correlated with the tree distances for the associated protein, providing evidence of co-evolution between these two components of the retron, consistent with probable direct and specific protein-protein interaction. Interestingly, in type XIII, the RT seems to have coevolved with the accessory protein (cluster 216) rather than the associated primary protein (cluster 114), suggesting that the primary protein may be a more recent acquisition. In general, the correlation between the RT and the ncRNA was also strong, although slightly weaker than that between the RT and associated proteins, suggesting a more diverse or wider mechanistical function of the ncRNA transcripts (or msDNA) in the putative retron ribonucleoprotein complexes. Interestingly, all types showed little or no correlation with 16S, with the exception of type XIII, which displayed a moderate correlation with the host 16S (0.78 for cluster114_1), suggesting vertical inheritance. Taken together, these results provide further support that retrons are tripartite systems and suggest that the three components are under mutual selective pressure continuously evolving by either exchanging or acquiring additional putative effector domains, potentially expanding their functional and mechanistic diversity.
Retron systems associated with genes encoding putative nucleases
The retron systems with associated predicted nucleases included type I, type VII, type IX and type XIII systems (Figure 5 and Table 1). Subtype I-A, and the two variants of subtype I-B (B1 and B2) were characterized by associated proteins bearing an N-terminal ATPase module with an additional HNH endonuclease or a Toprim domain (59,60) that is fused to the C-terminus of the retron-encoded RT in subtype I-C and its three variants (C1, C2 and C3). Type VII systems carry an associated protein (subtype VII-A2) or a domain (DUF3800) fused to the RT (subtype VII-A1) structurally similar to RNaseH2 (61). Type IX systems were linked to proteins carrying higher eukaryote and prokaryote nucleotide-binding (HEPN) domains (62), and, finally, type XIII systems were linked to a protein with a central winged-helix DNA-binding domain and a C-terminal SWIM Zn-finger domain (63).
Subtypes I-A and I-B (B1 and B2) were linked to protein members of clusters 22_1, and 22_2 (B1) or 159_1 (B2), all containing an N-terminal ABC ATPase domain fused, in the last two cases to a Toprim domain. The resulting architecture resembled that of overcoming lysogenization defect (OLD) nuclease proteins (59,64,65). Interestingly, the proteins of cluster 22_1 formed an operon with a downstream predicted HNH endonuclease (cluster 39_1), a system reminiscent of the ‘Septu’ antiviral defense system comprising the PtuAB operon (ATPase, HNH endonuclease) (66). Phylogenetic analysis of the ATPase domain (Figure 6A, Supplementary Figure S5 and Supplementary File S2) showed that protein sequences in cluster 22_1 were grouped with other reported PtuA homologs, whereas the N-terminal ATPase domain sequences of clusters 22_2 and 159_1 clustered into two different groups branching off of a common well-supported node more closely related to the ATPase domain of class 1 and class 2 OLD-nucleases. Finally, RaptorX identified the OLD nuclease from Thermus scotoductus (PDB 6p74A) as the best template-protein structure (100% of the residues modeled and a P-value of 2.96e–09) for these proteins. Taken together, these results suggest that the proteins in clusters 22_2 and 159_1 are probably new members of the OLD-protein nuclease family.
Figure 6.

Phylogeny of ATPase and TOPRIM domains associated with type I retrons. (A) The unrooted tree was constructed from an alignment of 1,676 ATPase domain sequences and was obtained with the FastTree program. The tree newick file is provided in Supplementary File S2. The branches corresponding to OLD nucleases, PtuA, and ATPase domains from type I-A, I-B1 and I-B2 retrons are indicated with highlighting in different colors. (B) The unrooted tree was constructed from an alignment of 1167 TOPRIM domain sequences and was obtained with the FastTree program. The newick file is provided as Supplementary File S3. The branches corresponding to OLD nucleases, DnaG primase, topoisomerase II-B (Topo II-B) RNase M5, topoisomerase III (Topo III), topoisomerase I (Topo I), reverse gyrase, RecR, topisomerase II-A (Topo II-A) and GyrB are shown, together with TOPRIM domains from type I-B1, I-B2, I-C1, I-C2 and I-C3 retrons, highlighted in different colors.
The Toprim domain of the protein sequences in clusters 22_2 and 159_1 contained the characteristic aspartate dyad (DxD) of motif V required for activity in all Toprim-containing enzymes, and the N-terminal invariant glutamate of motif IV, all preceded by conserved hydrophobic residues, suggesting that these are catalytically active enzymes (Supplementary Figure S6). The Toprim domain also appears in the three variants of subtype I-C, but fused at the C-terminus of the corresponding RT. The replacement of the second aspartate residue in the DxD motif with a glutamate residue (DxE) is a signature specific to the I-C2 variant. Phylogenetic analyses (Figure 6B and Supplementary File S3) suggested that the Toprim domains of clusters 22_2 (I-B1) and 159_1 (I-B2) were more closely related to that of OLD nucleases, whereas that of subtype I-C arose from a common ancestor and formed a different, presumably new lineage within the Toprim superfamily.
Retron system type VII includes an additional domain (DUF3800) fused at the C-terminus of the RT in the VII-A1 variant, and an associated protein (cluster 2165) in the VII-A2 variant, both containing the DUF3800 domain (Pfam12686). This associated protein of unknown function is characterized by a DE motif at the N-terminus, but lack the conserved Q of the characteristic QxxD motif at the C-terminus (Supplementary Figure S7). Moreover, these protein sequences form two well-differentiated clades (Supplementary Figure S8 and Supplementary File S4), suggesting that they may not result from fission/fusion events. Interestingly, the modeling of cluster 2165 protein sequences with RaptorX delivered, as the best template-protein structure (100% of the residues modeled and a P-value of 1.39e–11), pdb: 3pufA and pdb: 1EKE, corresponding to the human and Methanocaldococcus jannaschii RNase H2, respectively. However, it should be noted that, in the DUF3800 protein/domain associated with the retron type VII system, the second conserved aspartate residue of the four highly conserved carboxylates (DEDD) forming the active site of RNases H is replaced by a glutamate residue (DEED). The putative functional assignment of these enzymes as a type of RNaseH2 therefore requires further verification.
Retron system IX carries two linked genes upstream from the retron module encoding a first protein from cluster 1740_1, predicted by HHpred and HMM searches to be a HEPN domain-containing protein, and a second overlapping (∼8 bp) gene encoding a protein from cluster 1093_1 predicted by HHpred and InterPro to carry a putative HTH12 C-terminal ‘Winged helix’ DNA-binding domain, as found at the N-terminus of ribonuclease R and a number of presumed transcriptional regulatory proteins from archaea. The identification of cluster 1070_1 as corresponding to HEPN domain-containing proteins was also supported by protein structure modeling with RaptorX and Phyre2, which provided, as the best template, pdb:2hsbA (HEPN-domain containing protein from Archaeoglobus fulgidus), with a P-value of 5.09e–05. The proteins of cluster 1740_1 lack the conserved H residue of the characteristic HEPN motif (Rx4–6H), which has repeatedly been shown to be directly involved in RNA cleavage (67–69). Instead, these proteins have a conserved histidine residue located downstream from the arginine residue modeled as close to the R of the HEPN motif, as shown in the predicted structure (Figure 7A and B). It is therefore plausible that this residue is an alternative active-site residue, similar to that present in the HEPN-T family (62). The presence of a putative HEPN-domain active site in the proteins of cluster 1740_1 therefore suggests that they may provide this retron system with RNase activity.
Figure 7.

Sequence alignment and structure of the HEPN-like domain associated with type IX retrons. (A) Sequence alignment of representative HEPN-like domain sequences extracted from a global alignment of 53 sequences, including HEPN-like sequence homologs identified by HMMER. The secondary structure of the HEPN-like domain from Vibrio parahaemolyticus (Vp) is mapped above. Arginine (R) and histidine (H), which may be involved in the catalytic site, are indicated with a star. Sequence shading shows the conservation of the 53 HEPN-like sequence alignment: white text on red background, 100% conserved; boxed red text on white background, 70% conserved. Abbreviations indicating the type of domain are as follows, with accompanying PATRIC IDs: Type IX Vp, Vibrio parahaemolyticus (fig|670.1220.peg.4685), Type IX Db, Desulfobulbaceae bacterium (fig|1961547.3.peg.1368), Type IX Gb, Gammaproteobacteria bacterium (fig|2013797.3.peg.2112), Type IX Ma, Marinobacterium sp. (fig|1714300.3.peg.308), Type IX Kv, Klebsiella variicola (fig|244366.46.peg.3593), Type IX Rb, Rhizobiales bacterium (fig|1909294.17.peg.3454), Type IX Ab, Anaerolineales bacterium (fig|1950192.3.peg.428), Type IX Az, Azotobacter beijerinckii (fig|170623.7.peg.702). (B) Structural alignment of the HEPN domain-containing protein from Archeoglobus fulgidus (PDB: 2HSB), shown in light green, and the structural model of a representative HEPN domain-containing protein from cluster 1740_1, from Vibrio parahaemolyticus, shown in light blue. Putative catalytic arginine and histidine residues from Archeoglobus fulgidus and Vibrio parahaemolyticus are highlighted and labeled in green and blue, respectively. Note that these proteins display the same HEPN-domain fold according to our structural prediction, with R and H in close proximity, consistent with the formation of a putative catalytic site with RNA cleavage activity.
Retron system type XIII has a linked gene upstream or downstream from the RT gene that encodes a cluster 114_1 protein with a C-terminal SWIM Zn-finger domain (CxCx12CxH) (P-value 3e–06) predicted to have DNA-binding and protein-protein interaction functions. Searches for homologous sequences (HMMER) and structural analyses (Phyre2) also predicted the presence of a DprA (DNA-processing protein A) winged helix (Pfam17782) domain upstream from this motif. Moreover, the proteins of cluster 114_1 have a central Dx7[D]x4Ex30–32Dx37E motif, with the second aspartate residue absent or replaced by glutamate in close homologs (Supplementary Figure S9). This DEDE motif has been described in some prokaryotic (IS256 family) mobile elements and in some Mutator transposases from eukaryotes (70), which suggest that the proteins of cluster 114_1 may be derived from unknown transposases. Many, but not all, of the associated 114_1 proteins in type XIII systems are preceded by a gene encoding a predicted HEAT/ARM-like repeat-containing protein a member of cluster 216_1 (the majority), 216_2 or 3262_1 and, in some cases, by two genes, one encoding a cluster 259_1 protein and the other encoding a cluster 1688_1 protein. These classes of proteins with repeated α-helical motifs have an extensive solvent-accessible surface and may interact with proteins and nucleic acids (Supplementary Figure S10).
Retron systems linked to genes encoding proteins predicted to function in nucleotide metabolism
Two retron systems with a large number of members appear to be associated with genes encoding proteins predicted to be involved in the salvage pathway for nucleoside and nucleotide biosynthesis required for DNA synthesis and DNA damage repair. The type II systems are associated with putative NDTs, whereas type III systems are linked to genes encoding putative PRTases (Figure 5 and Table 1).
Retron type II systems are associated with genes encoding proteins from clusters 42_1; 42_2 or 42_3, carrying a putative NDT-like domain (P-value of 9.6e–06, 2.5e–07 and 0.096) and a C-terminal ‘winged helix’ DNA-binding domain, potentially associated with the bacterial membrane as a bitopic protein with only one α-helical transmembrane (TM) domain. These proteins have features similar to those described for members of the NDT family (Supplementary Figure S11). Interestingly, the genes encoding cluster 42_3 proteins have a frameshift separating the two domains of the protein such that the C-terminal DNA-binding domain appears to be fused to the N-terminus of the downstream RT, with some protein sequences also displaying a predicted N-terminal trypsin-like serine protease domain. Phylogenetic analysis of the presumed NDT domain of these protein sequences (Supplementary Figure S12 and Supplementary File S5) showed them to be closely related, but only distantly related to members of the nucleoside 2-deoxyribosyltransferase family (pfam 15891). Together, our data suggest that the cluster 42 proteins may belong to the NDT family, but the catalytic function of these proteins remains uncertain and will require further experimental validation.
Retron type III systems are characterized by association with a predicted PRTase-like protein, which may appear alone (clusters 67_1, 113_2 and 67_2) or in association (clusters 113_1, 113_4 and 113_3) with a downstream-encoded auxiliary protein (clusters 742_1, 1121_1 and 1689_1) with a predicted C-terminal winged helix DNA binding domain. In some clusters (67_2a, 67_2b and 67_3), this auxiliary protein appears to be fused to the C-terminus of the putative PRTase (Table 1). Five variants of this type of system were identified (III-A1 to III-A5), and were supported by the phylogenetic relationships of distinct PRTase-like cluster sequences (Supplementary Figure S13 and Supplementary File S6). Thus, the proteins of clusters 67 and 113 may be new members of the PRTase family, but their precise catalytic function remains to be determined.
Retron systems linked to genes encoding proteins associated with the cell membrane, DNA-binding proteins or proteases
Several retron systems were found to be associated with proteins that bind DNA/ RNA or are associated with the cell membrane. The retron-encoded RTs of clade 3 were associated with distinct protein clusters with apparently diverse functions. The type IV system was linked to a gene encoding a protein of cluster 826_1, corresponding to proteins predicted to be associated with the cell membrane with two TM helices (Figure 5 and Table 1). The loci encoding these proteins were found to be just downstream from or to overlap with the RT gene. These proteins lacked identifiable domains with predicted functions compatible with specific functional prediction. The type V system (Figure 5 and Table 1) is associated with small (67–86 aa) proteins (cluster 85_1) with a predicted cold-shock DNA-binding domain (CSD) and is therefore probably linked to processes involving the destabilization of RNA secondary structures. Moreover, a group of RTs within clade 3 appeared to be associated with other small (91–110 aa) proteins (cluster 2_1) (type VI) predicted to be DNA-binding proteins with a Cro/C1-type HTH domain (Figure 5 and Table 1) potentially targeting transcriptional regulation. This retron system is also associated with loci encoding small (75–79 aa) auxiliary proteins from cluster 2075_1, 2760_1 or 3272_1 containing no identifiable domain, but with a similar 3D structure with a three-helix bundle fold, as predicted by trRosetta models (Supplementary Figure S14). Strikingly, these proteins had a predicted three-dimensional structure similar to that of epsilon antitoxin proteins from postsegregational killing (PSK) systems (71), as shown by a DALI search against PDB, suggesting a plausible common role for the clusters in a retron-associated toxin-antitoxin system.
The type X system (Figure 5 and Table 1) was found to be associated with a gene encoding a cluster 2273 protein upstream from the RT, predicted to contain a central integration host factor (IHF)-like DNA-binding domain (Pfam00216) and a C-terminal von Hippel-Landau (VHL) beta domain (Pfam01847) separated by a single TM helix, with the VHL beta domain on the cytoplasmic side. The function of VHL beta domains in prokaryotic proteins is unknown, but, by analogy to the pVHL tumor suppressor protein, we speculate that the VHL beta domain of cluster 2273 proteins bound to DNA by the IHF-like domain may recruit other proteins for the formation of nucleoprotein complexes associated with the cell membrane.
Finally, there are two retron systems, the type VIII system carrying a clade 4 RT and the type XI system carrying a clade 8 RT, both lacking recognizable ncRNA modules, associated with putative proteases, a serine esterase (DUF626), and with the C-terminus of the RT fused to a trypsin-like serine protease. We hypothesize that these two systems may be involved in cleavage of targeting peptides.
DISCUSSION
The biological role of retrons has remained a mystery for almost three decades, despite the identification of these elements as the first type of RT to be discovered in prokaryotes. Some observations, such as the presence of additional open reading frames (ORFs) downstream from the RT gene or between the msr and msd regions in one third of annotated retrons, have raised questions about their implications for retron biology prompting further speculation about their possible involvement in the production of unusual msDNAs (28). Here, we describe a computational pipeline for the systematic prediction of genes specifically associated with retron RTs based on a previously published large dataset of annotated RTs representative of current diversity in prokaryotes. We therefore avoid any bias relating to the particular phylogenetic group of the RT, the genomic context of the RT gene, the distance to the RT gene or hypothetical possible functions, making it possible to predict the functional association of genes with retrons with confidence. Using this computational strategy, we found that the (msr-msd)-RT cassette of retrons could be predicted to be functionally linked to a single locus encoding putative nucleases, genes involved in nucleotide metabolism, genes encoding proteins binding DNA or RNA, or proteins associated with the cell membrane. In some cases, the retron unit is linked to additional accessory protein-encoding genes, or the RT has additional fused domains. Based on the associated protein or RT-fused domain, we have classified retrons into 13 distinct systems, some with subtypes or variants. We would expect the vast majority of retron systems to be of the types identified here, but it remains plausible that other rare types and variants may be discovered in the future, particularly in metagenomic analyses. Our results demonstrate that retrons can be broadly considered to be tripartite systems consisting of the ncRNA (msr-msd), the RT and additional proteins with diverse enzymatic functions.
We also found a few retron RT clades/subclades that did not appear to be associated with any specific protein cluster, such as the branches found in clades 2 and 11. Clade 2 includes the RT of the Retron-Eco5 (Ec107) intron, which is located adjacent to an annotated orotate phosphoribosyltransferase. However, only two of the 59 RTs belonging to this branch are associated with such an additional protein. This branch has a node in common with a cluster of RTs linked to PRTase-like proteins. It is therefore plausible that most of the members of the Retron-Eco5 (Ec107) branch have lost the associated protein. Interestingly, Retron-Eco5 is the only experimentally studied E. coli retron not related to prophages (72). It is, therefore, also conceivable that, in this particular case, the adjacent phosphoribosyltransferase is not functionally linked to the retron unit. Moreover, most of the host genomes harboring the orphan retrons lack homologs of the identified retron associated proteins. It therefore remains to be determined whether such retron systems have evolved to function with currently unknown protein effectors in trans, or whether they have acquired a different mechanistic function.
Interestingly, these tripartite units display extraordinary modularity, and distantly related RTs and different ncRNAs appear to be associated with genes encoding proteins of similar function. Conversely, particular RTs and ncRNAs appear to be linked to functionally different proteins. Associated protein-coding genes of similar types are present in different clades of the RT phylogeny, indicating a probable exchange of modules between systems. These phylogenetic studies reveal a complex evolutionary scenario for retrons, in which the different modules of the systems have been exchanged on multiple occasions. For example, types I-A and II-A1 have an ncRNA-RT module in common, but have different putative ‘effector’ modules. It seems that, at a certain point in evolution, type II-A1 retrons acquired a different effector module from the ‘Septu’ defense system (66). The presence of an ncRNA-RT module may add new features and complexity to the regulatory mechanism of previous existing systems, as for the RTs embedded in RT/CRISPR-Cas systems, which made it possible to acquire spacers from RNA molecules (14,16,19). The diversity of associated proteins and the modularity of retron systems are expanding the known functional diversity of the retron unit and the range of cellular processes underlying retron activity. A similar cassette organization with diverse accessory modules has recently been described for DGRs, linking RTs to functions other than diversifying attachment proteins (73)
The dynamic gain-loss is frequent for genes present in mobile genetic elements (MGEs) and defense systems, such as toxin-antitoxin modules (74). Autonomous mobility has not been demonstrated for retrons, but there is evidence for the horizontal transfer of these units (75) even with insertion into the same genomic site in closely related strains (76), and retrons seem to be components of rapidly evolving genomic islands under strong selective pressure, probably due to the constant threat posed by new bacteria.
Consistent with gain-loss and HGT, we found that the most retron RTs, albeit more abundant in the phylum Proteobacteria, show a patchy distribution into distinct bacterial phyla. Nevertheless, we also found that some retron systems like the type XIII have a relevant correlation with the 16S rRNA gene of the host genome consistent with the vertical inheritance reported in Myxobacteria (77). Moreover, two RT clades show a significant restriction in their taxonomic distribution. Clade 2 is mostly found in Gammaproteobacteria and clade 11 is essentially restricted to the phylum Actinobacteria. Thus, some retrons appear to display a vertical inheritance and may have been domesticated to perform useful cellular functions other than defense.
Sequence divergent retrons such as Vc95 (Retron-Vch1), Vc81 (Retron-Vch2) and Vc137 (Retron-Vch3) are inserted into the same site on chromosome I of V. cholerae (76). The first retron is almost exclusively found in pathogenic serogroups of V. cholerae whilst the latter two are found in non-pathogenic serotypes. These observations have led to speculation that either the retron (msr-msd-RT cassette) or the additional ORFs of unknown function are determinants of pathogenicity, and that the latter may associate with retrons to move and exchange between genomes (28). According to the RT phylogeny and specific associated proteins, Vc95 and Vc85 belong to the type I-A and type II-A1 systems, respectively, and their RTs are closely related (clade 1) sharing a ncRNA (IA/IIA1) structure. In our retron dataset Vc137 correspond in Supplementary Table S1 to entry 1575 (fig|1343738.3.peg.2232) and belongs to the type II-A3 with a more divergent RT (clade 9). Vc81 and Vc137 are associated to NDT-like proteins whereas Vc95 has associated ATPase and HNH domains. It seems plausible that these retrons share similar mechanistic functions and insertion mechanism that likely explain their presence in the same locus, but the distinctive putative effector proteins of Vc95 may be the determinant of its predicted pathogenicity also depending on the host genome.
The idea that retrons (msr-msd-RT cassette) and associated proteins function as a unit is also supported by transcriptional analyses showing that the retron and the putative associated effector protein function as an operon, expressed under the control of a single promoter (78). The exact mechanism of retron functioning and the possible interaction of the (msr-msd)-RT cassette with the linked protein remain to be elucidated, but we speculate that the conserved ncRNA and RT components form the sensing/recognition module, and that the additional associated protein acts as the response module in the retron system, to deal with different cellular stresses and regulatory processes, potentially including anti-phage defense. Retrons have been reported to be involved in bacterial responses to stress conditions, such as starvation and anaerobic conditions (79,80).
Recently, it was reported that some retrons members of the type systems I-A (Retron+ATPase+HNH), II-A3 (Nuc_deoxy+Retron), I-C1 (Retron-Toprim) and XII (Retron-TIR) defined here mediate antiphage defense and that the additional proteins or RT-fused domains are also required for activity (81). Moreover, a recent report (82) and two pre-prints (83,84) have also suggested that retrons may be novel anti-phage defense systems and that they may function as a toxin/antitoxin system. The first of these roles is thought to be mediated by the RT and msDNA, with the linked effector protein responsible for toxin/antitoxin functions. Furthermore, according to these reports (81,82), a large number of retrons, but not all are associated with defense genes in defense islands. It would be interesting to know whether there are specific retron systems consistently unrelated the known and novel candidate defense systems recently described (81). In summary, our findings further our understanding of the biological role of retrons and provide a basis for future studies on their function and potential biotechnological applications.
Supplementary Material
ACKNOWLEDGEMENTS
We thank all members of the NTG laboratory for helpful discussions during the development of this project. We acknowledge support of the publication fee by the CSIC Open Access Publication Support Initiative through its Unit of Information Resources for Research (URICI).
Contributor Information
Mario Rodríguez Mestre, Structure, Dynamics and Function of Rhizobacterial Genomes, Grupo de Ecología Genética de la Rizosfera, Department of Soil Microbiology and Symbiotic Systems, Estación Experimental del Zaidín, Consejo Superior de Investigaciones Científicas, C/ Profesor Albareda 1, 18008 Granada, Spain.
Alejandro González-Delgado, Structure, Dynamics and Function of Rhizobacterial Genomes, Grupo de Ecología Genética de la Rizosfera, Department of Soil Microbiology and Symbiotic Systems, Estación Experimental del Zaidín, Consejo Superior de Investigaciones Científicas, C/ Profesor Albareda 1, 18008 Granada, Spain.
Luis I Gutiérrez-Rus, Departamento de Química Física. Facultad de Ciencias, Universidad de Granada, 18071 Granada, Spain.
Francisco Martínez-Abarca, Structure, Dynamics and Function of Rhizobacterial Genomes, Grupo de Ecología Genética de la Rizosfera, Department of Soil Microbiology and Symbiotic Systems, Estación Experimental del Zaidín, Consejo Superior de Investigaciones Científicas, C/ Profesor Albareda 1, 18008 Granada, Spain.
Nicolás Toro, Structure, Dynamics and Function of Rhizobacterial Genomes, Grupo de Ecología Genética de la Rizosfera, Department of Soil Microbiology and Symbiotic Systems, Estación Experimental del Zaidín, Consejo Superior de Investigaciones Científicas, C/ Profesor Albareda 1, 18008 Granada, Spain.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Spanish Ministerio de Ciencia, Innovación y Universidades; ERDF (European Regional Development Funds) [BIO2017-82244-P]; A.G.-D. was supported by a FPU predoctoral fellowship grant from the Ministerio de Economía y Competitividad [FPU15/02714]; L.I.G.-R. was supported by a FPU predoctoral fellowship grant from the Ministerio de Ciencia, Innovación y Universidades [FPU17/05087].
Conflict of interest statement. None declared.
REFERENCES
- 1. Baltimore D. RNA-dependent DNA polymerase in virions of RNA tumour viruses. Nature. 1970; 226:1209–1211. [DOI] [PubMed] [Google Scholar]
- 2. Temin H.M., Mizutani S.. RNA-dependent DNA polymerase in virions of Rous sarcoma virus. Nature. 1970; 226:1211–1213. [DOI] [PubMed] [Google Scholar]
- 3. Finnegan D.J. Retrotransposons. Curr. Biol. 2012; 22:R432–R437. [DOI] [PubMed] [Google Scholar]
- 4. Menéndez-Arias L., Sebastián-Martín A., Álvarez M.. Viral reverse transcriptases. Virus Res. 2017; 234:153–176. [DOI] [PubMed] [Google Scholar]
- 5. Lampson B.C., Inouye M., Inouye S.. Reverse transcriptase with concomitant ribonuclease H activity in the cell-free synthesis of branched RNA-linked msDNA of Myxococcus xanthus. Cell. 1989; 56:701–707. [DOI] [PubMed] [Google Scholar]
- 6. Lim D., Maas W.K.. Reverse transcriptase-dependent synthesis of a covalently linked, branched DNA-RNA compound in E. coli B. Cell. 1989; 56:891–904. [DOI] [PubMed] [Google Scholar]
- 7. Liu M., Deora R., Doulatov S.R., Gingery M., Eiserling F.A., Preston A., Maskell D.J., Simons R.W., Cotter P.A., Parkhill J. et al.. Reverse transcriptase-mediated tropism switching in Bordetella bacteriophage. Science. 2002; 295:2091–2094. [DOI] [PubMed] [Google Scholar]
- 8. Doulatov S., Hodes A., Dai L., Mandhana N., Liu M., Deora R., Simons R.W., Zimmerly S., Miller J.F.. Tropism switching in Bordetella bacteriophage defines a family of diversity-generating retroelements. Nature. 2004; 431:476–481. [DOI] [PubMed] [Google Scholar]
- 9. Fortier L.C., Bouchard J.D., Moineau S.. Expression and site-directed mutagenesis of the lactococcal abortive phage infection protein AbiK. J. Bacteriol. 2005; 187:3721–3730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Odegrip R., Nilsson A.S., Haggård-Ljungquist E.. Identification of a gene encoding a functional reverse transcriptase within a highly variable locus in the P2-like coliphages. J. Bacteriol. 2006; 188:1643–1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Durmaz E., Klaenhammer T.R.. Abortive phage resistance mechanism AbiZ speeds the lysis clock to cause premature lysis of phage-infected Lactococcus lactis. J. Bacteriol. 2007; 189:1417–1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kojima K.K., Kanehisa M.. Systematic survey for novel types of prokaryotic retroelements based on gene neighborhood and protein architecture. Mol. Biol. Evol. 2008; 25:1395–1404. [DOI] [PubMed] [Google Scholar]
- 13. Toro N., Nisa-Martínez R.. Comprehensive phylogenetic analysis of bacterial reverse transcriptases. PLoS One. 2014; 9:e114083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Silas S., Mohr G., Sidote D.J., Markham L.M., Sanchez-Amat A., Bhaya D., Lambowitz A.M., Fire A.Z.. Direct CRISPR spacer acquisition from RNA by a natural reverse transcriptase-Cas1 fusion protein. Science. 2016; 351:aad4234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Toro N., Martínez-Abarca F., González-Delgado A., Mestre M.R.. On the origin and evolutionary relationships of the reverse transcriptases associated with type III CRISPR-Cas systems. Front. Microbiol. 2018; 9:1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Schmidt F., Cherepkova M.Y., Platt R.J.. Transcriptional recording by CRISPR spacer acquisition from RNA. Nature. 2018; 562:380–385. [DOI] [PubMed] [Google Scholar]
- 17. Toro N., Martínez-Abarca F., Mestre M.R., González-Delgado A.. Multiple origins of reverse transcriptases linked to CRISPR-Cas systems. RNA Biol. 2019; 16:1486–1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Toro N., Mestre M.R., Martínez-Abarca F., González-Delgado A.. Recruitment of reverse transcriptase-Cas1 fusion proteins by type VI-A CRISPR-Cas systems. Front. Microbiol. 2019; 10:2160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. González-Delgado A., Mestre M.R., Martínez-Abarca F., Toro N.. Spacer acquisition from RNA mediated by a natural reverse transcriptase-Cas1 fusion protein associated with a type III-D CRISPR-Cas system in Vibrio vulnificus. Nucleic Acids Res. 2019; 47:10202–10211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Michel F., Ferat J.L.. Structure and activities of group II introns. Annu. Rev. Biochem. 1995; 64:435–461. [DOI] [PubMed] [Google Scholar]
- 21. Dai L., Zimmerly S.. ORF-less and reverse-transcriptase-encoding group II introns in archaebacteria, with a pattern of homing into related group II intron ORFs. RNA. 2003; 9:14–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Toro N., Martínez-Abarca F., Fernández-López M., Muñoz-Adelantado E.. Diversity of group II introns in the genome of Sinorhizobium meliloti strain 1021: splicing and mobility of RmInt1. Mol. Genet. Genomics. 2003; 268:628–636. [DOI] [PubMed] [Google Scholar]
- 23. Lambowitz A.M., Zimmerly S.. Mobile group II introns. Annu. Rev. Genet. 2004; 38:1–35. [DOI] [PubMed] [Google Scholar]
- 24. Yee T., Furuichi T., Inouye S., Inouye M.. Multicopy single-stranded DNA isolated from a gram-negative bacterium. Myxococcus xanthus. Cell. 1984; 38:203–209. [DOI] [PubMed] [Google Scholar]
- 25. Hsu M.Y., Eagle S.G., Inouye M., Inouye S.. Cell-free synthesis of the branched RNA-linked msDNA from retron-Ec67 of Escherichia coli. J. Biol. Chem. 1992; 267:13823–13829. [PubMed] [Google Scholar]
- 26. Shimamoto T., Inouye M., Inouye S.. The formation of the 2′, 5′-phosphodiester linkage in the cDNA priming reaction by bacterial reverse transcriptase in a cell-free system. J. Biol. Chem. 1995; 270:581–588. [DOI] [PubMed] [Google Scholar]
- 27. Lampson B.C., Inouye M., Inouye S.. Retrons, msDNA, and the bacterial genome. Cytogenet. Genome Res. 2005; 110:491–499. [DOI] [PubMed] [Google Scholar]
- 28. Simon A.J., Ellington A.D., Finkelstein I.J.. Retrons and their applications in genome engineering. Nucleic Acids Res. 2019; 47:11007–11019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Xie X., Yang R.. Multi-copy single-stranded DNA in Escherichia coli. Microbiology. 2017; 163:1735–1739. [DOI] [PubMed] [Google Scholar]
- 30. Rychlik I., Sebkova A., Gregorova D., Karpiskova R.. Low-molecular-weight plasmid of Salmonella enterica serovar Enteritidis codes for retron reverse transcriptase and influences phage resistance. J. Bacteriol. 2001; 183:2852–2858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Inouye M. The first demonstration of the existence of reverse transcriptases in bacteria. Gene. 2017; 597:76–77. [DOI] [PubMed] [Google Scholar]
- 32. Steinegger M., Söding J.. MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat. Biotechnol. 2017; 35:1026–1028. [DOI] [PubMed] [Google Scholar]
- 33. Katoh K., Standley D.M.. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 2013; 30:772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zimmermann L., Stephens A., Nam S.Z., Rau D., Kübler J., Lozajic M., Gabler F., Söding J., Lupas A.N., Alva V.. A completely reimplemented MPI Bioinformatics toolkit with a new HHpred server at its core. J. Mol. Biol. 2018; 430:2237–2243. [DOI] [PubMed] [Google Scholar]
- 35. Remmert M., Biegert A., Hauser A., Söding J.. HHblits: lightning-fast iterative protein sequence searching by HMM-HMM alignment. Nat. Methods. 2011; 9:173–175. [DOI] [PubMed] [Google Scholar]
- 36. Mitchell A.L., Attwood T.K., Babbitt P.C., Blum M., Bork P., Bridge A., Brown S.D., Chang H.Y., El-Gebali S., Fraser M.I. et al.. InterPro in 2019: improving coverage, classification and access to protein sequence annotations. Nucleic Acids Res. 2019; 47:D351–D360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. El-Gebali S., Mistry J., Bateman A., Eddy S.R., Luciani A., Potter S.C., Qureshi M., Richardson L.J., Salazar G.A., Smart A. et al.. The Pfam protein families database in 2019. Nucleic Acids Res. 2019; 47:D427–D432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Letunic I., Bork P.. 20 years of the SMART protein domain annotation resource. Nucleic Acids Res. 2018; 46:D493–D496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wang S., Li W., Liu S., Xu J.. RaptorX-Property: a web server for protein structure property prediction. Nucleic Acids Res. 2016; 44:W430–W435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kelley L.A., Mezulis S., Yates C.M., Wass M.N., Sternberg M.J.. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015; 10:845–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yang J., Zhang Y.. I-TASSER server: new development for protein structure and function predictions. Nucleic Acids Res. 2015a; 43:W174–W181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yang J., Yan R., Roy A., Xu D., Poisson J., Zhang Y.. The I-TASSER Suite: protein structure and function prediction. Nat. Methods. 2015b; 12:7–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Yang J., Anishchenko I., Park H., Peng Z., Ovchinnikov S., Baker D.. Improved protein structure prediction using predicted interresidue orientations. Proc. Natl. Acad. Sci.i USA. 2020; 117:1496–1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Holm L. DALI and the persistence of protein shape. Protein Sci. 2020; 29:128–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Krissinel E., Henrick K.. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr. D. Biol. Crystallogr. 2004; 60:2256–2268. [DOI] [PubMed] [Google Scholar]
- 46. Robert X., Gouet P.. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014; 42:W320–W324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Price M.N., Dehal P.S., Arkin A.P.. FastTree 2–approximately maximum-likelihood trees for large alignments. PLoS One. 2010; 5:e9490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Nguyen L.T., Schmidt H.A., von H.A., Minh B.Q.. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015; 32:268–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kalyaanamoorthy S., Minh B.Q., Wong T.K.F., von H.A., Jermiin L.S.. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat. Methods. 2017; 14:587–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Weinberg Z., Lünse C.E., Corbino K.A., Ames T.D., Nelson J.W., Roth A., Perkins K.R., Sherlock M.E., Breaker R.R.. Detection of 224 candidate structured RNAs by comparative analysis of specific subsets of intergenic regions. Nucleic Acids Res. 2017; 45:10811–10823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Rivas E. RNA structure prediction using positive and negative evolutionary information. PLoS Comput. Biol. 2020; 16:e1008387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Nawrocki E.P., Eddy S.R.. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics. 2013; 29:2933–2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lorenz R., Bernhart S.H., Höner Z.S.C., Tafer H., Flamm C., Stadler P.F., Hofacker I.L.. ViennaRNA Package 2.0. Algorithms Mol. Biol. 2011; 6:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Quast C., Pruesse E., Yilmaz P., Gerken J., Schweer T., Yarza P., Peplies J., Glöckner F.O.. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 2013; 41:D590–D596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ochoa D., Pazos F.. Practical aspects of protein co-evolution. Front. Cell. Dev. Biol. 2014; 2:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Galili T. dendextend: an R package for visualizing, adjusting and comparing trees of hierarchical clustering. Bioinformatics. 2015; 31:3718–3720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Shmakov S.A., Makarova K.S., Wolf Y.I., Severinov K.V., Koonin E.V.. Systematic prediction of genes functionally linked to CRISPR-Cas systems by gene neighborhood analysis. Proc. Natl. Acad. Sci. U.S.A. 2018; 115:E5307–E5316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Wu L., Gingery M., Abebe M., Arambula D., Czornyj E., Handa S., Khan H., Liu M., Pohlschroder M., Shaw K.L. et al.. Diversity-generating retroelements: natural variation, classification and evolution inferred from a large-scale genomic survey. Nucleic Acids Res. 2018; 46:11–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Aravind L., Leipe D.D., Koonin E.V.. Toprim–a conserved catalytic domain in type IA and II topoisomerases, DnaG-type primases, OLD family nucleases and RecR proteins. Nucleic Acids Res. 1998; 26:4205–4213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Yang W. Nucleases: diversity of structure, function and mechanism. Q. Rev. Biophys. 2011; 44:1–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Hyjek M., Figiel M., Nowotny M.. RNases H: Structure and mechanism. DNA Repair (Amst.). 2019; 84:102672. [DOI] [PubMed] [Google Scholar]
- 62. Anantharaman V., Makarova K.S., Burroughs A.M., Koonin E.V., Aravind L.. Comprehensive analysis of the HEPN superfamily: identification of novel roles in intra-genomic conflicts, defense, pathogenesis and RNA processing. Biol. Direct. 2013; 8:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Makarova K.S., Aravind L., Koonin E.V.. SWIM, a novel Zn-chelating domain present in bacteria, archaea and eukaryotes. Trends Biochem. Sci. 2002; 27:384–386. [DOI] [PubMed] [Google Scholar]
- 64. Schiltz C.J., Lee A., Partlow E.A., Hosford C.J., Chappie J.S.. Structural characterization of Class 2 OLD family nucleases supports a two-metal catalysis mechanism for cleavage. Nucleic Acids Res. 2019; 47:9448–9463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Schiltz C.J., Adams M.C., Chappie J.S.. The full-length structure of Thermus scotoductus OLD defines the ATP hydrolysis properties and catalytic mechanism of Class 1 OLD family nucleases. Nucleic Acids Res. 2020; 48:2762–2776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Doron S., Melamed S., Ofir G., Leavitt A., Lopatina A., Keren M., Amitai G., Sorek R.. Systematic discovery of antiphage defense systems in the microbial pangenome. Science. 2018; 359:eaar4120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Jiang W., Samai P., Marraffini L.A.. Degradation of phage transcripts by CRISPR-associated RNases enables type III CRISPR-Cas immunity. Cell. 2016; 164:710–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Liu L., Li X., Wang J., Wang M., Chen P., Yin M., Li J., Sheng G., Wang Y.. Two distant catalytic sites are responsible for C2c2 RNase activities. Cell. 2017; 168:121–134. [DOI] [PubMed] [Google Scholar]
- 69. Pillon M.C., Goslen K.H., Gordon J., Wells M.L., Williams J.G., Stanley R.E.. It takes two (Las1 HEPN endoribonuclease domains) to cut RNA correctly. J. Biol. Chem. 2020; 295:5857–5870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Hua-Van A., Capy P.. Analysis of the DDE motif in the mutator superfamily. J. Mol. Evol. 2008; 67:670–681. [DOI] [PubMed] [Google Scholar]
- 71. Meinhart A., Alonso J.C., Sträter N., Saenger W.. Crystal structure of the plasmid maintenance system epsilon/zeta: functional mechanism of toxin zeta and inactivation by epsilon 2 zeta 2 complex formation. Proc. Natl. Acad. Sci. U.S.A. 2003; 100:1661–1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Mao J.R., Inouye S., Inouye M.. msDNA-Ec48, the smallest multicopy single-stranded DNA from Escherichia coli. J. Bacteriol. 1997; 179:7865–7868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Vallota-Eastman A., Arrington E.C., Meeken S., Roux S., Dasari K., Rosen S., Miller J.F., Valentine D.L., Paul B.G.. Role of diversity-generating retroelements for regulatory pathway tuning in cyanobacteria. BMC Genomics. 2020; 21:664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Koonin E.V., Makarova K.S., Wolf Y.I. et al.. Evolutionary entanglement of mobile genetic elements and host defence systems: guns for hire. Nat. Rev. Genet. 2020; 21:119–131. [DOI] [PubMed] [Google Scholar]
- 75. Ahmed A.M., Shimamoto T.. msDNA-St85 a multicopy single-stranded DNA isolated from Salmonella enterica serovar Typhimurium LT2 with the genomic analysis of its retron. FEMS Microbiol. Lett. 2003; 224:291–297. [DOI] [PubMed] [Google Scholar]
- 76. Inouye K., Tanimoto S., Kamimoto M., Shimamoto T., Shimamoto T.. Two novel retron elements are replaced with retron-Vc95 in Vibrio cholerae. Microbiol Immunol. 2011; 55:510–513. [DOI] [PubMed] [Google Scholar]
- 77. Rice S.A., Lampson B.C.. Phylogenetic comparison of retron elements among the myxobacteria: evidence for vertical inheritance. J. Bacteriol. 1995; 177:37–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Kim S., Jeong H., Kim E.Y., Kim J.F., Lee S.Y., Yoon S.H.. Genomic and transcriptomic landscape of Escherichia coli BL21(DE3). Nucleic Acids Res. 2017; 45:5285–5293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Herzer P.J. Starvation-induced expression of retron-Ec107 and the role of ppGpp in multicopy single-stranded DNA production. J. Bacteriol. 1996; 178:4438–4444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Elfenbein J.R., Knodler L.A., Nakayasu E.S., Ansong C., Brewer H.M., Bogomolnaya L., Adams L.G., McClelland M., Adkins J.N., Andrews-Polymenis H.L.. Multicopy single-stranded DNA directs intestinal colonization of enteric pathogens. PLos Genet. 2015; 11:e1005472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Gao L., Altae-Tran H., Böhning F., Makarova K.S., Segel M., Schmid-Burgk J.L., Koob J., Wolf Y.I., Koonin E.V., Zhang F.. Diverse enzymatic activities mediate antiviral immunity in prokaryotes. Science. 2020; 369:1077–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Millman A., Bernheim A., Stokar-Avihail A., Fedorenko T., Voichek M., Leavitt A., Sorek R.. Bacterial retrons function in anti-phage defense. Cell. 2020; doi:10.1016/j.cell.2020.09.065. [DOI] [PubMed] [Google Scholar]
- 83. Bobonis J., Mateus A., Pfalz B., Garcia-Santamarina S., Galardini M., Kobayashi C., Stein F., Savitski M.M., Elfenbein J.R., Andrews-Polymenis H. et al.. Bacterial retrons encode tripartite toxin/antitoxin systems. 2020; bioRxiv doi:22 June 2020, preprint: not peer reviewed 10.1101/2020.06.22.160168. [DOI]
- 84. Bobonis J., Mitosch K., Mateus A., Kritikos G., Elfenbein J.R., Savitski M.M., Andrews-Polymenis H., Typas A.. Phage proteins block and trigger retron toxin/antitoxin systems. 2020; biorXiv doi:22 June 2020, preprint: not peer reviewed 10.1101/2020.06.22.160242. [DOI]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.