

Abstract
Dysfunction in regulation of mRNA translation is an increasingly recognized characteristic of many diseases and disorders, including cancer, diabetes, autoimmunity, neurodegeneration, and chronic pain. Approximately 50 million adults in the United States experience chronic pain. This economic burden is greater than annual costs associated with heart disease, cancer, and diabetes combined. Treatment options for chronic pain are inadequately efficacious and riddled with adverse side effects. There is thus an urgent unmet need for novel approaches to treating chronic pain. Sensitization of neurons along the nociceptive pathway causes chronic pain states driving symptoms that include spontaneous pain and mechanical and thermal hypersensitivity. More than a decade of preclinical research demonstrates that translational mechanisms regulate the changes in gene expression that are required for ongoing sensitization of nociceptive sensory neurons. This review will describe how key translation regulation signaling pathways, including the integrated stress response, mammalian target of rapamycin, AMP-activated protein kinase (AMPK), and mitogen-activated protein kinase–interacting kinases, impact the translation of different subsets of mRNAs. We then place these mechanisms of translation regulation in the context of chronic pain states, evaluate currently available therapies, and examine the potential for developing novel drugs. Considering the large body of evidence now published in this area, we propose that pharmacologically manipulating specific aspects of the translational machinery may reverse key neuronal phenotypic changes causing different chronic pain conditions. Therapeutics targeting these pathways could eventually be first-line drugs used to treat chronic pain disorders.
Significance Statement
Translational mechanisms regulating protein synthesis underlie phenotypic changes in the sensory nervous system that drive chronic pain states. This review highlights regulatory mechanisms that control translation initiation and how to exploit them in treating persistent pain conditions. We explore the role of mammalian/mechanistic target of rapamycin and mitogen-activated protein kinase–interacting kinase inhibitors and AMPK activators in alleviating pain hypersensitivity. Modulation of eukaryotic initiation factor 2α phosphorylation is also discussed as a potential therapy. Targeting specific translation regulation mechanisms may reverse changes in neuronal hyperexcitability associated with painful conditions.
I. The Unmet Need of Analgesics
The International Association for the Study of Pain defines pain as an unpleasant experience associated with actual or potential tissue damage (Nicholas et al., 2019). At its core, acute pain is an adaptive response intended to protect and improve the survivability of the organism. However, when pain transitions into a chronic state, it loses its adaptive value and becomes a disease in and of itself. Prevalence of chronic pain in the United States varies from 11% to 40% depending on the cohort studied (Dahlhamer et al., 2018). A recent study performed by the Centers for Disease Control and Prevention estimates that over 50 million adults (20.6%) in the United States are affected by chronic pain (Dahlhamer et al., 2018). Roughly 20 million people from among patients suffering from chronic pain in the United States identify chronic pain as a significant limiting factor in their normal day-to-day activities (Dahlhamer et al., 2018).
Because of its wide prevalence, the economic burden of chronic pain in the United States is estimated to be as high as $635 billion, which is greater than annual costs associated with heart disease, cancer, and diabetes (Gaskin and Richard, 2012). Current pharmacological treatment options to manage chronic pain typically include a concoction of nonsteroidal anti-inflammatory drugs, antidepressants, gabapentinoids, and opioids (Lynch and Watson, 2006). These medications for chronic pain demonstrate modest efficacy at best, notwithstanding the numerous adverse side effects associated with chronic drug use (Lynch and Watson, 2006). Hence, understanding the processes governing pain pathophysiology is crucial to the development of novel and effective therapeutics against persistent pain while leaving the adaptive acute pain response intact.
II. Physiology of Pain
Nociceptive information arising from exposure to intense pressure, temperature extremes, or chemical irritants is transduced, transmitted, and modulated into the sensation of pain by a multineuronal circuit that begins in the periphery (Basbaum et al., 2009). Specialized sensory neurons known as nociceptors innervate most tissues in the body, and direct damage to their axons or their environment can trigger action potential generation and the transmittance of nociceptive information to the central nervous system. Nociceptive information is processed in the spinal cord and then sent onto the brain where complex circuits eventually lead to the perception of pain (Koch et al., 2018).
After injury, nociceptors become sensitized, leading to enhanced activity and pain responsiveness after exposure to normally innocuous (allodynia) and noxious (hyperalgesia) stimuli (Woller et al., 2017). Studies in squid with fin-crush injuries have demonstrated the importance of nociceptor sensitization in species survival, as it serves as a warning mechanism to escape danger and protect ourselves after tissue insult (Crook et al., 2014). However, sensitization that persists past the healing phase of an injury is maladaptive and is widely viewed as a leading cause of chronic pain conditions (Meacham et al., 2017; Price and Gold, 2018; Price et al., 2018). Regulation of the collective translational capacity of the cell, or the translatome, underlies at least part of these persistent changes in neuronal phenotype driving sensitization in these cells (Khoutorsky and Price, 2018).
Sensory neurons are particularly susceptible to sensitization because of their physiologic role as sensors of the external environment. In this regard, these neurons must be able to detect and communicate whether a stimulus is injurious or benign, and, as a result, sensory neurons must demonstrate versatile and dynamic response properties (Bennett et al., 2019). As such, nociceptors are capable of changing phenotypes by altering their gene expression profiles via regulation of translational control pathways (Khoutorsky and Price, 2018). In addition to sensitization, these nociceptors can also generate spontaneous activity after injury, and this is likely a major cause of the ongoing pain that is an important feature of neuropathic pain states (North et al., 2019). Changes in gene expression allow these neurons to become increasingly excitable, resulting in a hypersensitive state and the generation of spontaneous pain, which ultimately drives the pathology of chronic pain (Haroutounian et al., 2014, 2018; Vaso et al., 2014).
III. Why Target Translation for Pain Treatment
A key driver of many chronic pain states is increased excitability and/or spontaneous activity in nociceptors. This physiologic change can be very long-lasting and usually persists after an injury has healed (Price and Gold, 2018). This strongly suggests that changes in gene expression drive this change. A key question is how these changes in gene expression occur—do they happen at the transcriptional or translational level or do both occur simultaneously?
A great number of studies have attempted to addresses this question, and the development of RNA sequencing technologies have had substantial impact on advances in this area of research. One thing is absolutely clear: Nerve injury causes transcriptional reprogramming in sensory neurons. However, this reprogramming seems to be better associated with the regeneration response to axonal injury than it does to altered excitability. The best evidence for this comes from two types of studies: single cell sequencing and translating ribosome affinity purification [targeted purification of polysomal mRNA (TRAP)]. In an elegant study done on trigeminal (TG) sensory neurons, Nguyen, Le Pichon, and Ryba (2019) demonstrated that injury to TG neuron axons causes a convergent change in transcription across neuronal subtypes that was best described as a response to injury. Key evidence that this was associated with axonal regeneration and not pain comes from the finding that scratching the skin, which does not cause a pain state, causes this stereotyped transcriptional change in a subset of TG neurons that innervate the skin. Many chemotherapeutics cause neuropathic pain through a direct action on sensory neurons. A hallmark of the neuropathy caused by these drugs is axonal injury localized to the distal tips of axons resulting in “die back” from the epidermis (Ma et al., 2018). TRAP sequencing on DRG nociceptors taken from animals with neuropathic pain from the chemotherapeutic drug paclitaxel demonstrates that there are few changes at the transcriptional level, but the translation of hundreds of mRNAs is changed at the peak of the neuropathic pain state (Megat et al., 2019a). Examining these data reveal how translational changes in nociceptors drive the neuropathic pain state at the cellular signaling level. Collectively, these and other studies discussed and cited below indicate that translational changes in gene expression are crucial for the generation and persistence of neuropathic pain states.
Another reason to prioritize translational changes in gene expression for pain therapy is based simply on the cellular anatomy of nociceptors (Woolf and Ma, 2007). These neurons are among the largest cells in the entire body, with axons that are up to a meter in length in large mammals. The nucleus of these cells is at a great length from where many changes in excitability occur, and these changes in excitability are often selective for certain axonal sub-branches of these highly complex neurons (Devor, 1999). This has been shown using microneurography in human sensory neurons exposed to nerve growth factor (NGF) (Obreja et al., 2018). Such a selective change in axonal excitability is hard to explain through transcriptional changes that would presumably invade the entirety of axonal branches for any given nociceptor. In this way, localized changes in gene expression controlled by translation signaling are likely crucial for nociceptor plasticity (Price and Géranton, 2009) in much the same way that localized translational control is critical for synaptic plasticity at specific dendritic spines in the CNS (Steward and Schuman, 2003; Sutton and Schuman, 2005; Biever et al., 2019).
A final reason to focus on translation regulation is the growing opportunity to manipulate gene expression at this level with very specific pharmacological interventions. This will be the focus for the rest of this review. First, we will describe signaling pathways that regulate translation, highlighting how these pathways are distinct and therefore affording control over subsets of mRNAs by targeting individual pathways. Next, we will highlight how these signaling pathways are implicated in chronic pain, providing evidence for how different types of chronic pain may be driven by distinct types of translation regulation. Finally, we will discuss how these pathways can be perturbed with drugs and how these drugs might help patients who suffer from chronic pain in the not-too-distant future.
An important point to emphasize before delving into the basic mechanisms of translation regulation is that it is now clear that specific subsets of mRNAs are regulated by distinct signaling pathways. For instance, the mammalian/mechanistic target of rapamycin (mTOR) pathway is likely the best-known translation regulation signaling pathway and is often referred to as a master control for cap-dependent translation, which encompasses most translation in cells. However, we now know that mTOR exerts very specific control over the translation of mRNAs that contain sequences in their 5′ untranslated regions (UTRs) called terminal oligopyrimidine tracts (TOPs) (Thoreen et al., 2012). Only about 500 mRNAs contain these TOP sequences in their 5′ UTRs (K. B. Jensen et al., preprint, DOI: https://doi.org/10.1101/2020.04.18.047571). Similar specificity exists for many other signaling pathways, including the major pathways we will discuss here. A key to translating this basic science knowledge into effective treatments for chronic pain will be to align our understanding of changes in gene expression in chronic pain states with gene expression control pathways that cause these pain states to persist. We are only now beginning to scratch the surface of this detailed level of insight, but tools are readily available to achieve rapid advances in this area of therapeutic opportunity.
IV. Basics of Gene Expression
Two cellular processes govern the expression of genes: transcription and translation. During transcription, a portion of the genome is transcribed into RNA by RNA polymerases driven by transcription factors and enhancers. Translation refers to the synthesis of amino acid polypeptide chains based on the mRNA code. Translation is a tightly regulated process that is increasing viewed as a potential target for therapeutic development in a variety of disease states (Sonenberg and Hinnebusch, 2009; Bhat et al., 2015; Khoutorsky and Price, 2018).
Mature mRNA transcripts contain a 7-methylguanosine “cap” at their 5′ end and a poly-adenosine [poly(A)] tail usually composed of approximately 250 adenosine bases at the 3′ end of the transcript. Both of these modifications to the transcripts prevent mRNA degradation by exonucleases, allow for mRNA export from the nucleus, and are crucial regulators of mRNA translation (Sonenberg and Hinnebusch, 2009). In between the 5′ cap and the poly(A) tail are the 5′ UTR, the subsequent coding sequence for the polypeptide, and a 3′ UTR. Before the mRNA can exit the nucleus, spliceosomes excise introns or noncoding intermittent regions of the mRNA. The remaining RNA segments, known as exons, are merged and exported and potentially bound for translation. Variations in mRNA splicing provides further diversity in the proteome of the cell. Mechanisms of RNA splicing and its implication in disease have been reviewed elsewhere (Lee and Rio, 2015; Montes et al., 2019).
Translation is divided into three phases: initiation, elongation, and termination. Eukaryotic translation varies significantly from prokaryotic translation, especially at the initiation stage (Kozak, 1999; Rodnina and Wintermeyer, 2009; Hershey et al., 2019). The eukaryotic initiation factor (eIF) 2 is a trimeric complex that requires GTP for initiation (Kozak, 1999; Rodnina and Wintermeyer, 2009). The GTP-bound eIF2 (eIF2-GTP) along with the initiator transfer RNA, Met-tRNAiMet and the small 40S ribosome create the 43S preinitiation complex, which is recruited to the 5′ cap of the mRNA by the eIF4F complex (Sonenberg and Hinnebusch, 2009; Khoutorsky and Price, 2018). The preinitiation complex then scans the 5′ UTR for the start codon, which is typically AUG (noncanonical start codons are sometime found in upstream open reading frames), which codes for methionine. Upon encountering the start codon, the large 60S ribosomal subunit is recruited to the preinitiation complex, creating an active 80S ribosome that is ready for elongation. Along this process, the GTP bound to eIF2 is hydrolyzed to GDP, dissociating the initiation factors from the ribosome for recycling. eIF2B, a guanine nucleotide exchange factor, replenishes eIF2-GTP for another round of initiation.
Unlike initiation, elongation is a well conserved process between eukaryotes and prokaryotes (Rodnina and Wintermeyer, 2009). Elongation requires the assembly of the 80S ribosome as well as numerous elongation factors. After initiation, the second codon lies in the acceptor (A) site of the 80S ribosome awaiting the docking of the next aminoacyl-tRNA. The eukaryotic elongation factor (eEF) 1A binds to the aminoacyl-tRNA coded by the second codon and recruits it to the ribosome. eEF1A hydrolyzes GTP to release the tRNA on the A site of the ribosome. Well-positioned by the larger 60S subunit, peptidyl transferase catalyzes peptide bonds between amino acids. The GTPase activity of eEF2 is required to translocate mRNA and tRNA in the ribosome to E and P sites to continue elongation. Like many cellular processes, elongation is a complex and well regulated mechanism that has been extensively reviewed elsewhere (Dever and Green, 2012).
Spatiotemporal regulation of translation allows for a rapid response to a variety of internal and external stimuli, and hence, protein synthesis has evolved to be highly regulated in localized compartments throughout the cell, in particular in neurons (Martin et al., 2000; Khoutorsky and Price, 2018; Biever et al., 2019; Megat and Price, 2018). Translation initiation is the rate-limiting step in the translation process. Signaling pathways, such as the mTOR pathway, mitogen-activated protein kinase [MAPK; extracellular signal-regulated kinase (ERK); p38 and MAPK-interacting kinase are kinases belonging to this family] pathway, AMP-activated protein kinase (AMPK) pathway, and the integrated stress response (ISR), tightly regulate the function of eukaryotic initiation factors and the formation of initiation complexes and have now been studied broadly in pain neuroscience. This review will focus on these pathways, how they are regulated, how they are involved in pain signaling, and how they might be targeted with therapeutics. Importantly, each of these signaling pathways preferentially controls the translation of a subset of mRNAs. This demonstrates the complexities and specificity of translation initiation but also affords therapeutic opportunities since these signaling pathways appear to be preferentially involved in different types of chronic pain.
V. Regulation of mRNA Translation—Linking Signaling to Translation of Subsets of mRNAs
A. The Mammalian/Mechanistic Target of Rapamycin Pathway
Rapamycin, an antifungal macrolide compound produced by the bacterium Streptomyces hygroscopius, was initially discovered in 1972 on the island of Rapa Nui (Seto, 2012). Rapamycin’s antifungal properties were soon overshadowed by its potent immunosuppressive effects. Since then, rapamycin has been adapted to prevent organ transplant rejection and restenosis after coronary stent insertion as well as the treatment of a rare, progressing condition affecting the lungs: lymphangioleiomyomatosis (Seto, 2012). In the 1990s, reports of yeast mutants resistant to rapamycin led to the identification of target of rapamycin in these organisms with subsequent discovery of mTOR as the direct target of rapamycin (Heitman et al., 1991; Brown et al., 1994).
The mTOR signaling pathway regulates essential metabolic processes, including autophagy, lipogenesis, mitochondrial function, and protein synthesis. mTOR is a serine/threonine protein kinase belonging to the phosphatidylinositol 3-kinase related kinase superfamily (Zoncu et al., 2011). Structural analysis of mTOR revealed various regulatory domains on mTOR that modulate its catalytic function; however, the FRAP, ATM, and TRRAP C-terminal (FATC) domain is essential to the kinase activity of the enzyme (Yang et al., 2013; Showkat et al., 2014). Rapamycin binds to a cytosolic protein, FK-binding protein 12, and interacts with the FK-binding protein 12–rapamycin binding domain of mTOR, thereby suppressing its enzymatic activity in an allosteric fashion. mTOR forms two multimeric complexes, mTOR complex (mTORC) 1 and mTORC2, each with a distinct set of substrates and effectors and rapamycin sensitivity (Showkat et al., 2014).
The mTORC1 complex consists of mTOR, regulatory associated protein of mTOR (RAPTOR), G-protein β-subunit–like protein (GβL), proline-rich AKT/protein kinase B substrate 40 kDa (PRAS40), and death domain containing mTOR interacting protein (Showkat et al., 2014). Raptor binds to the N-terminal domain of mTOR and supports the linking of 4E-binding protein (4E-BP) 1 and S6 ribosomal kinase (S6K) 1 to mTOR (Hara et al., 2002). Phosphorylation of raptor at S863 enhances mTORC1 activity, whereas phosphorylation of the S722/792 residue by AMPK suppresses mTORC1 (Gwinn et al., 2008; Foster et al., 2010). PRAS40 is largely considered to be an inhibitory subunit of mTORC1 that may be phosphorylated at various sites by mTORC1, further promoting the dissociation of the inhibitory subunit and enhancing mTORC1 activity (Nascimento et al., 2010). In contrast, other studies report that PRAS40 is necessary for mTORC1 activity such that silencing PRAS40 reduces the phosphorylation of downstream targets of mTORC1 (Fonseca et al., 2007; Hong-Brown et al., 2010). Post-translational modification of PRAS40 has been suggested to mediate this seemingly contradictory function of the subunit (Pallares-Cartes et al., 2012; Wiza et al., 2012). Although the exact effects of GβL on mTORC1 are not well studied, it is known that loss of GβL in vivo has negligible impact on mTORC1 activity under physiologic conditions (Guertin et al., 2006). However, in cancer cells GβL was found to supresses mTORC1 mediated 4E-BP1 phosphorylation (Kakumoto et al., 2015).
mTORC2 also consists of the catalytic mTOR but contains the rapamycin-insensitive companion of mTOR (RICTOR), mammalian stress-activated protein kinase interacting protein 1 (mSin1), protein observed with RICTOR, GβL, death domain containing mTOR-interacting protein, and PRAS40 (Showkat et al., 2014). As the name suggests, RICTOR-associated mTORC2 is not affected by acute rapamycin treatment (Jacinto et al., 2004). However, chronic application of rapamycin and rapamycin derivatives has been shown to reduce mTORC2 assembly and activity (Sarbassov et al., 2006; Zeng et al., 2007). Unlike mTORC1, mTORC2 requires the presence of GβL for proper functioning (Guertin et al., 2006; Kakumoto et al., 2015). mSin1 is also integral to the formation of the mTORC2 complex because it guides the proper binding of RICTOR to mTOR (Frias et al., 2006). Splice variants of mSin1 give rise to distinct mTORC2 signatures (Frias et al., 2006). mTORC2 regulates cellular metabolism, growth, and apoptosis by phosphorylating AKT and in turn enhancing mTORC1 activity (Sarbassov et al., 2005; Jhanwar-Uniyal et al., 2019). mTORC1-activated S6K1 directly phosphorylates RICTOR at T1135, promoting mTORC2 signaling (Julien et al., 2010). Hence, both mTOR complexes are intricately linked by a variety of regulators and substrates, which are further reviewed by Jhanwar-Uniyal et al. (2019).
1. Mammalian/Mechanistic Target of Rapamycin Complex 1 Regulates Translation Initiation for Terminal Oligopyrimidine Tract–Containing mRNAs
eIF4F is a trimeric complex consisting of eIF4E, eIF4A, and eIF4G. As a scaffolding protein, eIF4G binds to the cap-binding eIF4E protein as well as the DEAD-box RNA helicase enzyme eIF4A (Roux and Topisirovic, 2018). Once bound to the 5′ cap of the mRNA transcript, the eIF4F complex unwinds the 5′ UTR, exposing the RNA to the preinitiation complex. The 40S ribosomal subunit, as a constituent of the preinitiation complex, scans for the start codon, AUG, and, when found, recruits the 60S ribosomal subunit for the next phase of translation (i.e., elongation) (Sonenberg and Hinnebusch, 2009; Khoutorsky and Price, 2018). Because of its relatively low expression, eIF4E determines the rate of 5′ cap-mediated protein synthesis. Various homeostatic mechanisms regulate the function of eIF4E (Tahmasebi et al., 2018).
mTORC1 enhances translation by inhibiting 4E-binding proteins (4E-BPs) and promoting the activity of S6Ks. There are three isoforms of 4E-BPs (4E-BP1, 4E-BP2, and 4E-BP3) encoded by separate genes but sharing considerable homology (Pause et al., 1994; Poulin et al., 1998). 4E-BPs act as translational inhibitors that prevent the formation of the eIF4F complex by interacting with eIF4E. Since each 4E-BP protein contains a binding motif for eIF4E, their role in translation initiation overlaps substantially (Mader et al., 1995; Siddiqui et al., 2012). 4E-BP1 is the most well studied of the three isoforms, whereas literature on 4E-BP3 is sparse (Musa et al., 2016). mTORC1 phosphorylates 4E-BP1 and 4E-BP2 similarly. However, post-translational modifications, like deamidation of asparagine, of 4E-BP2 can further increase its phosphorylation by mTORC1 (Bidinosti et al., 2010). Hypo-phosphorylated 4E-BPs strongly bind to eIF4E, rendering it incapable of binding with eIF4G (Khoutorsky and Price, 2018). Phosphorylation of 4E-BPs by mTORC1 weakens its affinity toward eIF4E, allowing for the eIF4F complex to be formed and thereby increasing 5′ cap-dependent translation.
Another downstream effector of mTORC1 is a group of serine/threonine kinases, S6Ks, that regulate cellular metabolism, cytoskeletal organization, transcription, and protein synthesis (Tavares et al., 2015). In mammals, S6Ks are encoded by two genes, RPS6KB1 and RPS6KB2, giving rise to two protein isoforms, S6K1 and S6K2, respectively. S6K kinases belong to the AGC kinase family, which includes the cyclic-nucleotide–dependent protein kinase A, G, and C. Different translational start sites of S6K1 and S6K2 predominantly give rise to two isoforms of each protein: p70-S6K1, p85-S6K1, p54-S6K2, and p56-S6K2. Among these, p70S6K1 is the most widely studied and heavily associated with mTORC1 activation (Tavares et al., 2015). Upon phosphorylation by mTORC1, p70-S6K1 mobilizes from the cytosol into the nucleus especially in response to growth factor signaling (Rosner and Hengstschläger, 2011). The primary target of phosphorylation for the S6Ks is the ribosomal protein S6 (rpS6) (Pende et al., 2004). Since rpS6 is part of the 40S ribosomal subunit, phosphorylation of rpS6 regulates the translation of proteins (Kawasome et al., 1998; Chauvin et al., 2014). However, the exact mechanism by which phosphorylation of rpS6 by S6Ks modulates the activity of ribosomes is not well understood. Additionally, phospho-S6K1 targets programmed cell death protein 4 (PDCD4) and eIF4B. The phosphorylation of PDCD4 dissociates it from eIF4A, allowing the initiation factor to join the eIF4F complex via eIF4G (Yang et al., 2003). S6K1 phosphorylation of eIF4B facilitates its binding to the initiation complex, enhancing eIF4A activity and promoting translation (Holz et al., 2005; Shahbazian et al., 2006). Furthermore, phosphorylated eIF4B acts as a scaffold for S6K1-mediated phosphorylation of eIF3, thereby enhancing the interaction between eIF3 and poly(A)-binding protein (PABP)-interacting protein 1 and increasing translation (Holz et al., 2005; Martineau et al., 2008, 2014). By phosphorylating rpS6, PDCD4, eIF3, and eIF4B, mTORC1-S6K1 promotes translation initiation and protein synthesis.
This discussion of the biochemical targets of mTOR leaves the impression that this kinase controls all cap-dependent translation, but this does not seem to be the case. Elegant translation profiling studies demonstrate that mTORC1 specifically regulates the translation of mRNAs that contain TOP sequences in their 5′ UTR near the cap of the mRNA (Thoreen et al., 2012). A relatively small number of mRNAs contain these TOP sequences. A very recent study suggests that as few as 500 mRNAs are targeted by mTORC1 (K. B. Jensen et al., preprint, DOI: https://doi.org/10.1101/2020.04.18.047571). Many of these mRNAs encode proteins that are critical for translation regulation processes, such as ribosomal proteins and other proteins involved in ribosomal biogenesis. This is likely a key reason that mTORC1 activity appears to play an outsized role in translation regulation. By targeting the translation of a small number of mRNAs that are critical for overall cap-dependent translation, mTORC1 activation can lead to profound changes in cellular translation capacity.
2. Extracellular Signals Modulate Mammalian/Mechanistic Target of Rapamycin Complex 1 Activity
mTORC complexes act as a gateway to integrate extracellular signals with intracellular signaling mechanisms, allowing the cell to respond appropriately to its environment. Both variants of mTORC respond to extracellular stimuli; however, the bulk of literature has focused on mTORC1 signaling. It is only beginning to become apparent that mTORC2 activation is mediated by growth factor signaling (Jhanwar-Uniyal et al., 2019). Various receptors on the plasma membrane activate secondary messengers that converge upon mTORC1, some of which enhance, whereas others inhibit, mTORC1 activity. This allows for a fine-tuned response to a changing extracellular environment.
Phosphoinositide 3-kinase (PI3K)-Akt pathway is an intracellular pathway that regulates cell cycle and growth. Through this pathway, cells are able to discern the current metabolic state of the organism and react accordingly. As such, growth factors, such as insulin, activate the PI3K-Akt axis, which stimulates mTORC1 activity (Fig. 1). The binding of insulin or insulin-like growth factor to their respective receptors activates PI3K, which in turn leads to the phosphorylation and plasma membrane localization of Akt (Showkat et al., 2014; Zhao et al., 2015). Among other effects, activated Akt phosphorylates tuberous sclerosis protein (TSC) 2 and suppresses the GTPase-activating capability of the TSC complex consisting of TSC1 and TSC2 (Inoki et al., 2002; Tahmasebi et al., 2018). The TSC complex activates the GTPase activity of Ras homolog enriched in brain (RHEB) protein, converting the mTORC1 activating RHEB-GTP to RHEB-GDP (Inoki et al., 2003a,b). As a result, Akt-mediated repression of TSC sustains RHEB-GTP, thereby promoting mTORC1 activity. Akt also phosphorylates and inhibits PRAS40, the inhibitory subunit of mTORC1 (Oshiro et al., 2007; Wiza et al., 2012). By alleviating the braking mechanisms (i.e., inhibition of the TSC complex and PRAS40), PI3K-Akt activation activates mTORC1-mediated translation.
Fig. 1.

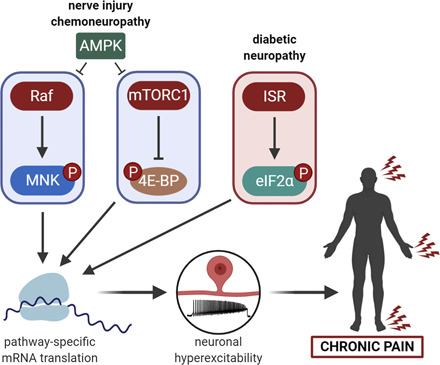
Regulation of eIF4E-mediated translation initiation. Extracellular factors, like cytokines and growth factors, induce intracellular signaling pathways that modulate eIF4E-dependent protein synthesis in a pathway-specific manner. Ras/Raf-MNK signaling phosphorylates eIF4E enhancing the expression of certain genes. Activation of mTORC1 relieves 4E-BP inhibition and thereby increases eIF4E-mediated translation. AMPK stimulation further fine-tunes translation initiation by inhibiting mTORC1 and Raf signaling. MEK, mitogen-activated protein (MAP) kinase/extracellular signal-regulated kinase (ERK) kinase.
Efficient cap-dependent mRNA translation is enhanced by the circulation of the mRNA, during which the scaffolding protein, eIF4G, interacts with both the 5′ cap as well as the poly(A) tail of the mRNA (Khoutorsky and Price, 2018). This circularization is mediated by PABP. The poly(A) tail is further lengthened by the binding of the cytoplasmic polyadenylation element–binding protein to a cytoplasmic polyadenylation element region in the 3′ UTR (Richter, 2007). The elongated poly(A) tail protects the mRNA from degradation, enhances circularization, and promotes translation (Kim and Richter, 2007; Martineau et al., 2008). Recent work demonstrates that PABP is expressed extensively throughout the peripheral nervous system, and competitively inhibiting PABP reduces nascent protein synthesis and pain hypersensitivity after injury and inflammation (Barragán-Iglesias et al., 2018).
B. Mitogen-Activated Protein Kinase Signaling and Translation
The MAPKs are serine/threonine kinases that are grouped into three families: ERK1/2, c-Jun N-terminal protein kinase, and p38 kinase (Cargnello and Roux, 2011; Roux and Topisirovic, 2018). Each family further consists of highly conserved, sequentially acting kinases: an MAPK, an MAPK kinase (MAPKK), and an MAPKK kinase. The three-tier MAPK cascade is usually initiated by the phosphorylation of MAPKK kinase and/or the binding of a Ras/Rho GTPase followed by phosphorylation and activation of MAPKK and subsequent phosphorylation of MAPK on threonine and tyrosine residues (Cargnello and Roux, 2011; Roux and Topisirovic, 2018). Activated MAPK phosphorylates MAPK-activated protein kinases, among which p90 ribosomal S6 protein kinases and MAPK-interacting kinases (MNKs) directly regulate mRNA translation.
1. Mitogen-Activated Protein Kinase–Interacting Kinases Phosphorylate Eukaryotic Elongation Factor 4E to Regulate the Translation of a Distinct Subset of mRNAs
MNK proteins are encoded by two genes, MKNK1 and MKNK2, with two splice variants each, giving rise to four isoforms (MNK1a, MNK1b, MNK2a, and MNK2b) that vary at their C termini (Scheper et al., 2003; O’Loghlen et al., 2004). The long isoforms, MNK1a and MNK2a, are predominantly expressed in the cytosol, whereas the shorter isoforms, MNK1b and MNK2b, show equal nuclear and cytoplasmic expression (Buxade et al., 2008). In mice, only MNK1a and MNK2a isoforms have been identified (Scheper et al., 2003; Proud, 2015). MNK1 binds to both ERK and p38 MAP kinase, whereas the more constitutively active isoform, MNK2, interacts with ERK and mTORC1 (Waskiewicz et al., 1997; Ueda et al., 2004; Xie et al., 2020). Upon binding, ERK phosphorylates at least two threonine residues (T197, T202) on MNK1/2 to increase its catalytic activity (Waskiewicz et al., 1999), but mTORC1 phosphorylation of MNK2 at Ser274 (Xie et al., 2020) and Ser437 (Stead and Proud, 2013) suppresses MNK2 activity. The most well known target of MNK1/2 is the S209 site on the eIF4E subunit of the eIF4F complex (Waskiewicz et al., 1999) (Fig. 1). The scaffolding subunit of eIF4F, eIF4G, facilitates the binding of MNK1/2 to eIF4E, suggesting that MNK-mediated phosphorylation of eIF4E may occur after or during the formation of the eIF4F complex (Pyronnet et al., 1999). Although both MNK1 and MNK2 phosphorylate eIF4E, MNK1-mediated phosphorylation of eIF4E is considered “inducible” since MNK1 basal activity is typically low in a cell (Scheper et al., 2003), and activation of either ERK or p38 MAPK induces a strong MNK1 response (Wang et al., 1998).
The phosphorylation of eIF4E is typically not correlated with any change in global translation (McKendrick et al., 2001). As such, nonphosphorylatable eIF4E mutant (S209A) mice develop normally (Furic et al., 2010), suggesting that eIF4E phosphorylation serves a more nuanced regulatory function in translation rather than influencing bulk translation. It was further found that eIF4E phosphorylation selectively increases the translation efficiency of certain genes, particularly those involved in synaptic plasticity (Gkogkas et al., 2014; Amorim et al., 2018a), oncogenesis (Musa et al., 2016), inflammation (Su et al., 2015), and, more recently, pain (Moy et al., 2017, 2018b). Genes sensitive to changes in eIF4E levels contain long, structured secondary components in their 5′ UTR, which require the helicase activity of eIF4A prior to initiation (Koromilas et al., 1992; Svitkin et al., 2001). The recruitment of eIF4A to the initiation complex by eIF4E is thought to link the translation sensitivity of these genes to changes in eIF4E expression. However, the effect of eIF4E phosphorylation in recruiting eIF4A and/or other initiation factors has not been demonstrated. Structural and biophysical data suggest that phosphorylation of eIF4E reduces its affinity to the 5′ cap (Scheper and Proud, 2002; Khan and Goss, 2018). Excessive phosphorylation of eIF4E reduces cap-dependent translation while increasing cap-independent translation possible via internal ribosome entry site structures (Knauf et al., 2001; Scheper and Proud, 2002; Thompson and Gilbert, 2017). Phosphorylated eIF4E also stabilizes the binding of the γ interferon–activated inhibitor of translation complex with eIF4F, which in turn represses the translation of a subset of proinflammatory circular mRNAs (Amorim et al., 2018a). Dephosphorylation of eIF4E removes the γ interferon–activated inhibitor of translation complex by reducing its affinity for eIF4F and subsequently enhances translation of these mRNAs (Amorim et al., 2018b). Thus, it is likely that phosphorylation of eIF4E influences multiple mechanisms of translation that impact only a small subset of mRNAs. In any case, inhibiting MNK-mediated phosphorylation of eIF4E has proven beneficial in various pain models, suggesting that MNK is an important pain target (see below).
C. The AMP-Activated Protein Kinase Pathway
The hydrolysis of phosphate bonds of ATP provides the energy required for many cellular processes (Camici et al., 2018). As a nucleoside triphosphate, ATP is an important precursor to the nucleic acid backbone of DNA and RNA molecules. Monitoring levels of ATP and its reduced forms, ADP and AMP, is crucial to the survival of the cell. With only one phosphate group, AMP does not possess any high-energy phosphoanhydride bonds, unlike ADP and ATP, and hence represents a state of depleted energy (Jeon, 2016). AMP-activated protein kinase (AMPK) was initially identified as a sensor of AMP. Under energetic stress, homeostatic processes, such as glycolysis and mitochondrial respiration recover ATP by metabolizing nutrients like glucose (Burke, 2017).
AMPK is a trimeric complex composed of a catalytic α subunit and regulatory β and γ subunits (Davies et al., 1994; Xiao et al., 2011). There are two isoforms of α and β subunits and three isoforms of the γ subunit, each denoted by 1, 2, and 3 after the Greek letter, respectively. Each subunit is slightly different in function, and the combination of various subunits provides diversity in AMPK activity (Ross et al., 2016). The most prevalent isoforms of AMPK subunits are α1, β1, and γ1, although other isoforms are known to be highly expressed in specific cell populations like skeletal muscle (Thornton et al., 1998; Kim et al., 2016). Allosteric modulation and phosphorylation of AMPK work in tandem to regulate the kinase activity of the α subunit. Phosphorylation of the α subunit at T172 enhances AMPK kinase activity (Woods et al., 2003). ATP competes with AMP and ADP to bind to the cystathionine β-synthetase (CBS) 3 site on the γ subunit to allow phosphatase access to the T172 residue on the α subunit, preventing the activation of the catalytic subunit (Oakhill et al., 2010; Jeon, 2016). In this manner, the ratios of AMP to ATP and ADP to ATP determine the activation state of AMPK. Under energetic stress, high levels of AMP and ADP outcompete ATP for the CBS3 site on the γ subunit preventing phosphatases access to T172 on the α subunit, whereby a phosphorylated, active state is maintained (Jeon, 2016). In addition, AMP and ADP binding to CBS3 enhances phosphorylation of AMPK by liver kinase B1, which depends on the myristoylation of the β subunit (Oakhill et al., 2010). AMP also acts as an allosteric activator of AMPK when bound to the CBS1 motif of the γ subunit (Suter et al., 2006). An increase in intracellular Ca2+, an indication of cellular activity, also promotes AMPK activity via phosphorylation by calcium/calmodulin-dependent kinase kinase 2. A recent report found that calcium/calmodulin-dependent kinase kinase 2–AMPK interaction was supported by stromal interaction molecule 2, a Ca2+ sensor for store-operated Ca2+-entry, in a Ca2+-dependent manner (Chauhan et al., 2019).
1. AMP-Activated Protein Kinase Suppresses Mammalian/Mechanistic Target of Rapamycin and Mitogen-Activated Protein Kinase Signaling
Protein synthesis is an energy-intensive process requiring high-energy phosphate bonds at each step, starting from transcription to translation to protein folding and other post-translational modifications. Neuronal cells, especially sensory neurons, are plastic and undergo phenotypic changes in response to metabolic stress, hypoxia, ischemia, and glucose deprivation. As such, AMPK is a critical component in maintaining energy homeostasis in every cell, in particular when faced with metabolic challenge (Jeon, 2016). AMPK activation results in impaired protein synthesis by affecting transcription (Shin et al., 2016; Ke et al., 2018), ribosomal RNA synthesis (Hoppe et al., 2009), and translation elongation (Deng et al., 2017). This is because AMPK is crucial for stimulating ATP-producing catabolic pathways, suppressing ATP-consuming pathways, and regulating protein synthesis via its control over mTOR and MAPK signaling.
Upon activation, AMPK phosphorylates raptor, a regulatory subunit of mTORC1, at S722/S792 residues as well as the mTOR kinase at the T2446 site (Cheng et al., 2004). Phosphorylation of raptor promotes the binding of the inhibitory 14-3-3 protein to raptor, whereas phosphorylation of mTOR can suppress its catalytic activity (Cheng et al., 2004; Gwinn et al., 2008). Additionally, AMPK also activates TSC2 by phosphorylating S1387 and T1271 residues, indirectly inhibiting mTORC1 by stimulating the GTPase activity of RHEB (Inoki et al., 2003a,b; Huang and Manning, 2008). In this manner, AMPK powerfully inhibits mTOR activity (Fig. 1).
Moreover, AMPK also regulates MAPK pathway via its phosphorylation of B-rapidly accelerated fibrosarcoma kinase, a member of the MAPK pathway (Shen et al., 2013; Hardie, 2014) (Fig. 1). Phosphorylation of B-rapidly accelerated fibrosarcoma kinase allows it to associate with 14-3-3 proteins, which blocks its interaction with kinase suppressor of Ras 1 scaffolding protein and disrupts downstream signaling (Shen et al., 2013). Therefore, AMPK activation can lead to suppression of mTOR and MAPK activity, limiting protein synthesis. This is an important factor for treating the pathology of chronic pain, which will be discussed in later sections.
D. The Integrated Stress Response Shapes Unconventional Translation
The ISR refers to a collection of homeostatic cellular processes converging upon the phosphorylation of eIF2α at Ser51 [extensively reviewed by Pakos-Zebrucka et al. (2016)]. The ISR acts as a sensor for internal and external stressors, such as accumulation of unfolded proteins, viral infection, lack of amino acids, and heme deficiency. Four serine/threonine kinases phosphorylate eIF2α, leading to a reduction in global translation while increasing the translation of stress-resistant genes, like activating transcription factor (ATF) 4 (Baird and Wek, 2012; Bretin et al., 2016; Rozpedek et al., 2016). The ISR also promotes cell death under severe conditions (Sano and Reed, 2013; Chan et al., 2015; Rozpedek et al., 2016). eIF2α kinases share considerable homology and are encoded by EIF2AK1, EIF2AK2, EIF2AK3, and EIF2AK4, each giving rise to heme-regulated inhibitor (HRI), RNA-dependent protein kinase R (PKR), PKR-like ER kinase (PERK), and general control nonderepressible 2 (GCN2) kinases, respectively (Pakos-Zebrucka et al., 2016) (Fig. 2). Upon stimulation, these kinases oligomerize and autophosphorylate, activating secondary signaling cascades (Bertolotti et al., 2000; Vattem et al., 2001; Narasimhan et al., 2004; Igarashi et al., 2011; Wang et al., 2018). Although each kinase is preferentially activated by a specific stressor, there is appreciable overlap in their response to phosphorylate eIF2α, suggesting that ISR is a cooperative endeavor. For example, all four kinases become activated by oxidative stress (Pyo et al., 2008; Baker et al., 2012; Suragani et al., 2012; Verfaillie et al., 2012), and PERK and GCN2 compensate for each other when either of them is knocked down in models of cellular stress (Devi and Ohno, 2013; Lehman et al., 2015).
Fig. 2.

Modulation of the integrated stress response. (A) The ternary complex, consisting of GTP-bound eIF2 and the initiator Met-tRNAi, aids in the recognition of the start codon. The GTP bound to eIF2 is hydrolyzed upon encountering the start codon. The resulting GDP-eIF2 dissociates from the initiation complex and is recycled by eIF2B for the next round of initiation. Under cellular stress, four kinases (PERK, PKR, GCN2, and HRI) phosphorylate eIF2α at Ser51 and initiate the ISR, which inhibits global translation by sequestering available eIF2B. (B) Various small molecules are known to modulate the ISR, particularly by maintaining phosphorylation of eIF2α (e.g., salubrinal, guanabenz, and sephin1) or by activating eIF2B (e.g., ISRIB). (Ci) Phosphorylation of eIF2α stabilizes the interaction of two molecules of eIF2 with eIF2Bα and eIF2Bδ subunits of eIF2B, inducing a conformational rearrangement that prevents the GDP to GTP exchange and competes with the binding of Met-tRNAi. In this manner, phospho-eIF2α acts as a noncompetitive inhibitor of eIF2B [Gordiyenko et al. (2019), PDB: 6QG0]. (Cii) ISRIB restores eIF2B levels by facilitating the binding of two tetramer subunits, particularly by interacting with eIF2Bδ and eIF2Bβ [Zyryanova et al. (2018), PDB: 6EZO]. (D) Although phosphorylation of eIF2α suppresses global translation, it induces the expression of certain genes, such as ATF4, CHOP, and GADD34. GADD34 is a stress-induced regulatory subunit of PP1, which dephosphorylates phospho-eIF2α to normalize translation. Recent structural and functional analysis shows that GADD34 promotes binding of PP1 and phospho-eIF2α via its lysine (K), valine (V), arginine (R), and phenylalanine (F).(KVRF) and proline (P), glutamic acid (E), serine (S), and threonine (T) (PEST) motifs, respectively [Choy et al. (2015), PDB: 4XPN]. The PEST sequences in GADD34 are not found in CreP, a constitutively active PP1 regulatory subunit, and may represent a novel target for the suppression of ISR-induced GADD34.
eIF2α is a regulatory subunit of the eIF2 complex consisting of additional β and γ subunits (Pakos-Zebrucka et al., 2016; Khoutorsky and Price, 2018) (Fig. 2A). The eIF2 complex binds to GTP and then catalyzes hydrolysis of GTP to GDP to regulate the initiation of mRNA translation. Conversion of the now inactive eIF2-GDP complex to the active eIF2-GTP is mediated by eIF2B, a guanine nucleotide exchange factor (Khoutorsky and Price, 2018; Anand and Walter, 2020). Under conditions of cellular stress, phosphorylation of eIF2α stabilizes the inactive eIF2-GDP-eIF2B complex, preventing the recycling of GDP and reducing the ability of eIF2 to further initiate translation (Fig. 2A). The concentration of eIF2α is many-fold higher than eIF2B, such that phosphorylation of even a few molecules of eIF2α has a drastic impact on translation. Ultimately, induction of phospho-eIF2α leads to a reduction in general translation while increasing the translation of mRNAs with an upstream open reading frame (uORF) in their 5′ UTR (Barbosa et al., 2013) (Fig. 3). Many of these genes allow the cell to appropriately respond to stress, such as ATF4, whereas other genes may code for other translational repressors (Martínez et al., 2018). ATF4, the most widely studied transcription factor induced by phosphorylation of eIF2α, increases the expression of ER chaperones to mitigate ER stress (McQuiston and Diehl, 2017). It also induces the expression of CHOP, a proapoptotic transcription factor, which in turn enhances the expression of more apoptotic genes (Harding et al., 2000). ATF4 and CHOP expression negatively regulate the phosphorylation of eIF2α through the expression of protein phosphatase 1 (PP1), allowing the cell to recover general translation (Han et al., 2013). In this manner, stressors, like accumulation of unfolded proteins, may shape the translatome and, hence, the fate of a cell.
Fig. 3.

Other mechanisms of translation regulation. Despite a loss in the ternary complex after ISR, the synthesis of certain proteins, like CHOP and ATF4, is enhanced by leaky scanning (A) and delayed reinitiation (B) mechanism, respectively. The reduced availability of eIF2 in stressed conditions allows for the scanning ribosomes to bypass (i.e., leaky scanning) the uORF in the 5′ UTR and translate the mORF. This is the case for CHOP and GADD34. Some genes, like ATF4, contain multiple uORFs in which one of the uORFs overlaps with the mORF in an out-of-frame manner, thereby suppressing the expression of the main gene. During ISR, the delay in eIF2 recycling allows for the scanning ribosome to skip the start codon of uORF and instead translate the mORF.
From among the four kinases of eIF2α, PERK signaling is the most well characterized and associated with a variety of intracellular and extracellular stressors, including accumulation of unfolded protein (Hetz et al., 2013), Ca2+ dyshomeostasis (van Vliet et al., 2017), mitochondrial dysfunction (Verfaillie et al., 2012; Lebeau et al., 2018), oxidative stress (Verfaillie et al., 2012), and inflammation (Meares et al., 2014). PERK is a transmembrane ER resident protein bound to a luminal chaperone, glucose-regulated protein 78-kDa [GRP78; also known as binding immunoglobulin protein (BiP)]. With the accumulation of unfolded proteins in the ER, BiP dissociates from PERK’s luminal domain and binds to hydrophobic domains of nascent/unfolded proteins, freeing PERK to oligomerize and autophosphorylate (Bertolotti et al., 2000). The dissociation of BiP from PERK and other ER transmembrane proteins, inositol-requiring enzyme 1 and ATF6, initiates homeostatic mechanisms in a process known as unfolded protein response (Hetz et al., 2013). Recent evidence also suggests that misfolded proteins directly bind to the luminal domain of PERK and induce oligomerization and activation of the kinase (Wang et al., 2018). Activation of PERK and subsequent eIF2α phosphorylation reduces the synthesis of new proteins and thereby lessens the ER load.
As noted above, despite the suppression of general translation during bouts of stress, the cell continues to synthesize proteins to address the stressor, such as chaperones (Starck et al., 2016), and ensure the survival of the cell. This observation shows that alternative mechanisms of protein synthesis in addition to 5′ cap-mediated translation initiation exist. A recent study profiled ribosome-associated mRNA in cultured human cells stressed with sodium arsenite, a chemical stressor and a potent inducer of eIF2 phosphorylation (Andreev et al., 2015). The authors found that nearly all stress-resistant genes had at least one uORF in their 5′ UTR that repressed the coding gene product under normal conditions. Such is the case for ATF4 and DDIT3 (i.e., transcript encoding CHOP) translation, which relies on delayed reinitiation and leaky translation, respectively (Fig. 3). uORF-mediated translation is extensively reviewed by Barbosa et al. (2013), Young and Wek (2016), and Jaud et al. (2020). Under normal conditions, when eIF2 is constantly replenished, a 5′ uORF sequence (uORF1) on the ATF4 mRNA codes for a short peptide, leading to the dissociation of the eIF2-containing ternary complex, whereas the 40S ribosome continues to scan the transcript until it encounters another uORF (uORF2) (Vattem and Wek, 2004). The coding sequence of uORF2 overlaps out of frame with the main ORF (mORF) coding sequence. Because of ample GTP-bound eIF2 levels, the uORF2 sequence is efficiently translated, and the ribosomal complex dissociates midway through the mORF, suppressing ATF4 translation. When phospho-eIF2α levels are high, the scanning ribosome skips the initiation site of the second uORF because of the delay in the restoration of the ternary complex with eIF2-GTP. The ribosome continues scanning and reaches the ATF4 mORF coding sequence, by which time the eIF2 complex becomes available, ultimately allowing for the expression of ATF4 (Vattem and Wek, 2004). The DDIT3 transcript also contains a uORF in its 5′ UTR, which codes for a 34–amino acid peptide that stalls scanning ribosomes and prevents translation of the mORF (Jousse et al., 2001; Palam et al., 2011; Young et al., 2016). This indicates that the presence of a uORF can lead to translational repression of mRNAs in the absence of cellular stress. Under conditions of cellular stress, the scanning ribosome bypasses the short uORF sequence (e.g., a “leaky” ribosome) and instead translates the DDIT3 coding sequence. Yet, other accounts suggest that stress-induced translation requires the presence of a uORF in the 5′ UTR to translate the coding sequence. Stress-resistant genes like PPP1R15B and IFRD1 required the uORF to confer resistance to eIF2 suppression. Mutations in the 5′ uORF were able to abrogate the stress-resistance phenotype, particularly that of PPP1R15B, which codes for growth arrest and DNA damage-inducible protein 34 (GADD34), a phosphatase required to dephosphorylate eIF2α and alleviate the ISR (Andreev et al., 2015). Why and how some uORFs inhibit translation of the mORF coding sequence when others stimulate translation still remain unknown. However, it has been proposed that the translation of uORFs depends upon the distance of the start site from the 5′ cap, the context of the start codon, the length of the uORF, secondary structures in the mRNA, conservation across species, the amount of uORFs in the 5′ UTR, the position of the stop codon, and the distance between the uORF and the mORF (Barbosa et al., 2013). Altogether, these observations suggest that uORFs in the 5′ UTR finely regulate translation of genes in a complex yet unconventional manner.
eIF2A, not to be confused with eIF2α, is a 65-kDa protein that recruits methionine-bound initiator tRNA to the 40S ribosomal subunit [see review by Komar and Merrick (2020)] (Fig. 3). The function of mammalian eIF2A is similar to yeast eIF2A and prokaryotic initiation factor 2, suggesting that eIF2A is the primeval mechanism of translation initiation (Zoll et al., 2002). Although canonical translation with the eIF2 complex requires GTP-dependent binding of eIF2 to Met-tRNAi, eIF2A-mediated binding of Met-tRNAi is codon-dependent and GTP-independent (Komar and Merrick, 2020). In addition to using AUG as a start codon, eIF2A-mediated translation initiation uses CUG and UUG codons (Kearse and Wilusz, 2017). The viability and overall healthiness of the recently characterized eIF2A−/− mice suggest that eIF2A-mediated translation does not have a major impact on protein synthesis under physiologic conditions (Golovko et al., 2016). How eIF2A−/− animals respond to stressors remains to be investigated. Unlike the prokaryotic yeast initiation factor 2, eIF2A does not physically bind to the 40S ribosome and does not hydrolyze GTP, which is required for the dissociation of the protein from the ribosome and to resume the next phase of translation (Zoll et al., 2002). Recently, eIF5B, a GTPase that stabilizes ribosomal docking to the start codon, was shown to function synergistically with eIF2A under ER stress but not under normal conditions, suggesting that stress-induced eIF2A-eIF5B interaction may induce the translation of stress-resistant genes (Kim et al., 2018). Furthermore, ER stress-induced BiP translation was shown to be mediated by a uORF in the HSPA5 gene that required initiation by eIF2A at a UUG start site (Starck et al., 2016). eIF2A has also been demonstrated to interact with internal ribosome entry site structures within the mRNA to recruit ribosomes and translate genes, particularly c-Src, a transcription factor associated with cellular survival and proliferation (Kwon et al., 2017b) (Fig. 3). Taken together, these observations suggest that multiple mechanisms of unconventional translation work synergistically to synthesize proteins under conditions of stress despite the suppression of eIF2α-mediated global translation.
VI. Targeting Translation in Chronic Pain
A. Evidence for Injury-Induced Translational Dysregulation in Sensory Neurons
After injury, cells release a variety of signaling ligands that act on nociceptors, and injury to nociceptor axons themselves also induces changes in cellular signaling in these neurons (Reichling et al., 2013; Kandasamy and Price, 2015; Pinho-Ribeiro et al., 2017; Price and Gold, 2018). Axonal endings can become sensitized to thermal, chemical, and mechanical stimulation, likely as a means to protect the injured area from further damage. However, in many cases, sensitization persists even after the injury resolves, leading to persistent hyperalgesia. These neurons can also become spontaneously active, which is likely an underlying cause of many neuropathic pain states (Campbell and Meyer, 2006; Devor, 2006; Serra et al., 2012; North et al., 2019). Many of these changes in nociceptor sensitivity and excitability are associated with translation regulation signaling, resulting in altered gene expression.
Elevated levels of translational control pathway components, such as eIF4E, P13K, AKT, ERK, mTOR, S6, and p38, are found in their active, phosphorylated forms in sensory neurons of rodents with peripheral nerve injury and persistent inflammation (Jiménez-Díaz et al., 2008; Géranton et al., 2009; Melemedjian et al., 2011; Persson et al., 2011; Moy et al., 2017, 2018b; Megat et al., 2019a). Some of these findings have been recapitulated in humans, in which elevated levels of activated p38 and ERK have been detected in neuromas of human amputees (Black et al., 2008). A likely driver of these changes in signaling is release of inflammatory mediators from cells at the injury site or from immune cells that may infiltrate or proliferate within an injured nerve. Inflammatory cytokines, chemokines, and neuropeptides bind to neuronal receptors that signal to activation of the mTOR and MAPK pathways, leading to changes in translation that are connected to downstream events engaged by these signaling pathways. Examples of this are NGF and brain-derived neurotropic factor (BDNF), which bind to neuronal TRK receptors to trigger downstream translation cascades via the mTORC1 and MAPK pathways. Positive feedback loops in this type of signaling may also play a role in the exacerbation of nociceptive sensitization by driving peripheral and central sensitization. For instance, DRG neurons exposed to NGF show increased expression of BDNF mRNA and protein (Kerr et al., 1999; Mannion et al., 1999), and it was recently discovered that BDNF translation is dependent on MAPK-mediated eIF4E phosphorylation (Moy et al., 2018a). Because eIF4E phosphorylation is increased in sensory neurons after injury (Megat et al., 2019a; Melemedjian et al., 2011), it is likely that BDNF overexpression is mediated by activation of eIF4E-dependent translation, which may in turn cause central sensitization by acting on the first synapses of the nociceptive pathway in the spinal dorsal horn (Kerr et al., 1999; Thompson et al., 1999; Zhao et al., 2006; Sikandar et al., 2018).
Increased signaling through translation control pathways also causes expression differences in proteins that directly contribute to the functional properties of sensory neurons. For example, neuropathic pain is associated with increased expression of voltage-gated sodium channel 1.8 (Nav1.8) at the lesion site, which potentially gives rise to ectopic activity (Gold et al., 2003). Pharmacological inhibition or knockdown of Nav1.8 also attenuates neuropathic pain behaviors in rodent models (Lai et al., 2004; Hameed, 2019). Nav1.8 protein is also significantly upregulated in neuromas from human patients with limb amputations (Black et al., 2008). Interestingly, there is a decrease in Nav1.8 mRNA transcripts in the soma (Okuse et al., 1997; Thakor et al., 2009) but a marked increase in mRNA encoding the channel in axotomized fibers (Thakor et al., 2009; Ruangsri et al., 2011), suggesting that trafficking of mRNA from the soma to the fibers is occurring after nerve injury. Nav1.8 shRNA treatment reduces Nav1.8 mRNA in the injured nerve but not the soma and reverses pain behaviors after peripheral nerve injury (Ruangsri et al., 2011). Interestingly, increased axonal trafficking of Nav1.8 transcripts after nerve injury occurs via an injury-induced splicing that lengthens the 3′ UTR, presumably altering RNA-binding protein association (Hirai et al., 2017). These findings around one of the most widely studied ion channels in nociceptors reveal how mRNA trafficking and localized mRNA translation in injured axons are instrumental in key aspects of underlying neuronal plasticity that drives neuropathic pain.
B. Mammalian/Mechanistic Target of Rapamycin Complex 1 and Mammalian/Mechanistic Target of Rapamycin Kinase Inhibitors
One of the first translation regulators to be implicated in pain neurobiology was mTORC1 (Price et al., 2007; Jiménez-Díaz et al., 2008; Asante et al., 2009; Géranton et al., 2009). As reviewed above, mTORC1 controls phosphorylation of 4E-binding proteins (4E-BPs) and p70-S6 ribosomal kinase (S6K1/2). When phosphorylated, 4E-BPs initiate translation by allowing assembly of the eIF4F complex and its binding to the mRNA cap, whereas S6K1/2 activates other proteins that control translation initiation and elongation (Zoncu et al., 2011) (Fig. 1). Importantly, mTOR activity is enhanced in the peripheral nervous system and in CNS pain-neuromodulatory regions, such as the outer lamina of the spinal dorsal horn, in a large number of rodent pain models (Jiménez-Díaz et al., 2008; Géranton et al., 2009; Melemedjian et al., 2011; Terenzio et al., 2018; Megat et al., 2019a). These range from acute pain models, such as the capsaicin (Megat et al., 2019b) and formalin test (Price et al., 2007; Asante et al., 2009; Xu et al., 2011), to inflammation (Norsted Gregory et al., 2010; Liang et al., 2013; Martin et al., 2017) and neuropathic pain (Géranton et al., 2009; Asante et al., 2010; Melemedjian et al., 2011; Uttam et al., 2018; Inyang et al., 2019c; Megat et al., 2019a) models and even into opioid-induced hyperalgesia models (Xu et al., 2014). Therefore, mTORC1 is a core signaling molecule causing neuronal plasticity that mediates pain sensitization in animal models. This makes it an enticing candidate for therapeutic intervention.
Indeed, short-term inhibition of mTOR using the specific inhibitor, rapamycin, attenuates nociceptive behaviors and nociceptor plasticity in many models of inflammatory and neuropathic pain (Price et al., 2007; Géranton et al., 2009; Duan et al., 2018; Melemedjian et al., 2010, 2013; Norsted Gregory et al., 2010; Obara et al., 2011; Xing et al., 2020). Microinjections of rapamycin and other mTOR inhibitors (rapalogues) into CNS pain-neuromodulatory regions, such as the anterior cingulate cortex and insular cortex, also have profound effects on pain behaviors, neuronal excitability, and synaptic plasticity in rodents (Kwon et al., 2017a; Um et al., 2019; Kim et al., 2020). However, chronic treatment with mTORC1 inhibitors in patients can cause higher incidences of pain (Carracedo et al., 2008; Budde et al., 2011; McCormack et al., 2011), even leading in some cases to severe pain conditions that are similar to complex regional pain syndrome (Witzig et al., 2005; Molina et al., 2008; Massard et al., 2010). These findings have been mechanistically replicated in rodent models in which sham-treated animals display nociceptive behaviors after chronic dosing with rapamycin (Melemedjian et al., 2013; Lisi et al., 2015). At the molecular level, rapamycin inhibition of mTORC1 causes disinhibition of the S6K1/2–insulin receptor substrate 1, resulting in activation of a feedback circuit that drives MAPK activation (Carracedo et al., 2008; Melemedjian et al., 2013). A longstanding finding that rapamycin treatment enhances eIF4E phosphorylation was thought to be solely driven by this compensatory hyperactivation of MAPK signaling. However, cells treated with rapamycin display enhanced eIF4E phosphorylation even when MNK1 is knocked down, suggesting that mTOR activity directly contributes to MNK2 phosphorylation of eIF4E (Wang et al., 2007). Indeed, recent work has revealed that mTORC1 directly phosphorylates MNK2 to shut down its constitutive activity (Xie et al., 2020).
ATP-competitive inhibitors of the mTOR kinase have also been developed. These drugs inhibit both mTORC1 and mTORC2. These inhibitors, such as AZD8055, have been extensively studied because of their antitumor activity and are currently in clinical trials for cancer (Chresta et al., 2010; Pike et al., 2013). Although these compounds have not yet been investigated widely in preclinical pain models, their efficacy will likely be complicated by similar feedback signaling issues. For example, AZD8055 causes inhibition of AKT, leading to increased expression and activation of epidermal growth factor receptor, which can trigger downstream signaling in the MAPK and Janus kinase/signal transducer and activator of transcription proteins signaling pathways (Wei et al., 2015). Our view is that therapeutics that allosterically target mTORC1 or directly target the mTOR kinase are poor candidates for pain therapeutics for two reasons. The first is because of their potential for exacerbating pain or other symptoms due to the feedback mechanisms discussed above. Investigators in the cancer field have proposed that mTOR inhibitors could be coupled with MAPK blockers to prevent activation of the insulin receptor substrate 1–MAPK negative feedback circuit (Carracedo et al., 2008), or that cotreatment with epidermal growth factor receptor inhibitors could be used to block engagement of other feedback mechanisms (Wei et al., 2015). However, these approaches may be fraught with other issues related to adverse side effects. The second is related to the immune suppression caused by mTOR inhibitors. This approach is an advantage when preventing organ transplant rejection but is likely not tolerable for most patients with chronic pain. Because these pathways influence the translation of distinct subsets of mRNAs, the physiologic responses to drugs targeting different translation regulation pathways diverge. AMPK activators and MNK inhibitors do not appear to cause immune suppression.
Investigating the role of mTOR signaling in chronic pain states has unquestionably advanced the field in terms of gaining a better understanding of the underlying neurobiology that causes chronic pain. However, this is unlikely to be an approach that can be directly adapted into a viable treatment option.
C. AMP-Activated Protein Kinase Activators
AMPK has emerged as a prime therapeutic target for pain treatment because of its ability to simultaneously suppress mTOR and MAPK/ERK signaling (Fig. 1) (Melemedjian et al., 2011; Tillu et al., 2012; Khoutorsky and Price, 2018) without engaging feedback loops (Melemedjian et al., 2013) or causing immunosuppression. Indirect and direct AMPK activators have shown efficacy in preclinical models of chronic pain, although these drugs do, in some cases, differ in their efficacy between sexes (Melemedjian et al., 2011; Mao-Ying et al., 2014; Burton et al., 2017; Afshari et al., 2018; Shiers et al., 2018; Das et al., 2019; Inyang et al., 2019a,b,c). Indirect AMPK activators stimulate AMPK without binding to the kinase, whereas direct AMPK activators bind directly to α, β, or γ subunits of the kinase to augment AMPK activation (Hardie et al., 2012; Hardie, 2015). Indirect AMPK activators can stimulate AMPK through a variety of mechanisms, but one of the best understood involves the class of molecules like metformin that affect mitochondrial complex I activity, resulting in changes in AMP/ATP levels in the cell (Shaw et al., 2005; Xiao et al., 2013). Interestingly, a variety of preclinical studies have now shown that indirect AMPK activators are more efficacious at alleviating both acute and chronic pain measures in rodent models (Burton et al., 2017; Inyang et al., 2019a,b,c). This likely occurs because injury decreases AMPK phosphorylation in sensory neurons (Roy Chowdhury et al., 2012; Inyang et al., 2019a), resulting in a decreased ability for direct AMPK activators to enhance kinase activity. A core feature of indirect AMPK activators is that they drive upstream activation of kinases, like liver kinase B1 that phosphorylate the α subunit of AMPK. This phosphorylation is needed to fully activate the kinase. Direct activators of AMPK are at least partially dependent on the phosphorylation state of AMPK to enhance kinase activity (Timmermans et al., 2014; Cameron and Kurumbail, 2016). If kinase phosphorylation has been decreased through other mechanisms, this would be expected to result in increased efficacy of indirect (versus direct) AMPK activators, as is seen in many preclinical pain models (Melemedjian et al., 2011; Roy Chowdhury et al., 2012; Tillu et al., 2012; Han et al., 2014; Mao-Ying et al., 2014; Alcocer-Gómez et al., 2015; Maixner et al., 2015; Mejia et al., 2016; Inyang et al., 2019a,b).
One of the most interesting features of indirect AMPK activator effects in preclinical models is that they appear to be disease-modifying. For example, metformin, an already FDA-approved type II diabetes drug that is safe to use in humans and an indirect AMPK activator, resolves pain behaviors for several months after only 1 week of dosing in neuropathic mice (Melemedjian et al., 2011, 2013; Inyang et al., 2019c). Additionally, certain comorbidities of chronic pain that develop in preclinical models—particularly cognitive impairment—are reversed after a week-long treatment regimen in mice is completed (Shiers et al., 2018). There is also evidence that metformin can be effective for the treatment of pain in humans. In a retrospective study, metformin usage was linked to reduced lumbar radiculopathy pain (Taylor et al., 2013), but another larger retrospective study did not find a positive effect of metformin (Smith and Ang, 2015). Metformin treatment has also been linked to mobility improvements and reductions in cartilage degradation in rodents and humans with osteoarthritis, which may be a consequence of improved pain outcomes (Wang et al., 2019; Li et al., 2020a,b). Finally, positive results have been reported for pain in a polycystic ovary syndrome trial (Kiałka et al., 2016), and a preventative trial for oxaliplatin neuropathy showed positive results for metformin cotreatment (El-Fatatry et al., 2018). The latter clinical finding is consistent with preclinical literature showing efficacy for metformin in prevention of chemotherapy-induced neuropathy (Mao-Ying et al., 2014; Inyang et al., 2019b; Pereira et al., 2019). The clinical results in pain trials with metformin to date are promising and warrant further clinical trials.
An important unresolved question is whether the antipain effects of metformin are mediated by AMPK. Although the effects of metformin are consistent with the effects of structurally different AMPK activators in preclinical pain models, there are important differences that we have cataloged in several recent studies. These include sex differences and improved efficacy for metformin versus other AMPK activators (Burton et al., 2017; Inyang et al., 2019a,b,c). We cannot rule out metformin effects on other targets as an important part of the mechanism of action of this compound. An interesting possibility is that metformin can act as a reactive glycation product scavenger (Mehta et al., 2009). These reactive molecules play an important role in pain sensitization (Bierhaus et al., 2012; Brings et al., 2017; Liu et al., 2017; Barragán-Iglesias et al., 2019) and may underlie some of the beneficial effects of this compound in pain models and in patients.
Altogether, there is now a very large body of preclinical literature and emerging clinical literature indicating that indirect AMPK activators can be efficacious in treating a wide range of chronic pain conditions. This plethora of findings establishes AMPK as a promising therapeutic target that should be vigorously pursued. Compounds targeting AMPK already exist, are FDA-approved, and are readily being used in humans (i.e., metformin), creating an opportunity for rapid clinical translation of the basic science advances described above. Newer generation and more specific AMPK activators may eventually make their way to the clinic however, their induction of left ventricular hypertrophy may be problematic for systemic exposure to these drugs (Feng et al., 2017; Myers et al., 2017).
D. Mitogen-Activated Protein Kinase–Interacting Kinase Inhibitors
MNK1/2 inhibitors are emerging as promising therapeutic targets for pain management because of their ability to inhibit eIF4E phosphorylation. Longstanding evidence supports that eIF4E phosphorylation is abnormally elevated in sensory neurons after injury (Melemedjian et al., 2011; Moy et al., 2017; Megat et al., 2019a) and gives rise to the enhanced translation of a subset of mRNAs. Some of these mRNAs, such as Bdnf and Mmp9, are known to encode key molecules in neuronal plasticity and nociceptor sensitization in chronic pain (Furic et al., 2010; Gkogkas et al., 2014; Amorim et al., 2018a; Moy et al., 2018a). Over a decade of preclinical work supports that disruption of MNK-eIF4E signaling dampens translation dysregulation and nociceptor sensitization and treats chronic pain. Transgenic mice that lack MNK isoforms and/or eIF4E phosphorylation show diminished pain sensitization in response to inflammatory mediators (Moy et al., 2017, 2018a), injury to nerves (Shiers et al., 2020), chemotherapeutic agents (Megat et al., 2019a), and even interferons induced by viral infection (Barragán-Iglesias et al., 2020). In this section, we will discuss the currently available or in-development MNK inhibitors, evidence for MNK inhibition as a strategy for pain treatment, and the future for drug development of MNK inhibitors.
One of the first identified compounds that targets MNK1/2 is the antifungal agent known as cercosporamide (Sussman et al., 2004; Konicek et al., 2011). Although this drug does inhibit MNK1/2 and subsequent eIF4E phosphorylation, it shows a higher selectivity for MNK2 (IC50 of 11 nM) over MNK1 (IC50 of 116 nM) (Konicek et al., 2011). Additionally, its effects may be compromised by its ability to inhibit other kinases, including Janus kinase 3, activin receptor-like kinase 4, GSK3B, and Pim1, although these are in the low micromolar range (Konicek et al., 2011). Nevertheless, the first preclinical experiments using cercosporamide demonstrate that its use has profound effects on nociceptor activity and analgesia. The first line of evidence is based on in vitro work demonstrating that sensory neuron hyperactivity caused by exposure to inflammatory mediators like NGF or interleukin-6 is blocked by cercosporamide (Moy et al., 2017). In a second set of experiments, mice treated with cercosporamide display reduced mechanical and thermal hypersensitivity because of inflammation induced by NGF and interleukin-6 and are protected from hyperalgesic priming (Moy et al., 2017). Similar in vitro and in vivo findings have been replicated genetically in mice with a knock-in null mutation in the phosphorylation site of eIF4E [eukaryotic initiation factor 4E (4EKI)] and in MNK1 and 2 double-KO animals (Moy et al., 2017).
Although promising, the selectivity concerns of cercosporamide limit its usefulness as a tool compound for probing MNK signaling, as it could be accompanied by multiple off-target effects with acute or chronic dosing. This and other factors have prompted the development of a new generation of MNK inhibitors. Tomivosertib, also known as eFT508, is a highly potent and specific MNK inhibitor developed by eFFECTOR Therapeutics for the treatment of cancer that is now in phase III clinical trials (Thompson et al., 2017). Tomivosertib has an IC50 of 2.4 nM for MNK1 and an IC50 of 1 nM for MNK2 with excellent oral bioavailability (Reich et al., 2018). Oral dosing of tomivosertib significantly reduces eIF4E phosphorylation in the DRG and sciatic nerve as well as in the CNS (Megat et al., 2019a; Shiers et al., 2020). Similar to effects seen with cercosporamide, DRG neurons cultured from animals with chemotherapy-induced peripheral neuropathy or exposed to inflammatory cytokines show reduced excitability after in vitro exposure to tomivosertib (Megat et al., 2019a; Shiers et al., 2020). Because nociceptor hyperactivity is among the most salient features of spontaneous pain in peripheral neuropathy, these data support the notion that tomivosertib could be used to treat neuropathic pain conditions. Indeed, 4EKI mice and/or tomivosertib-treated mice with spared nerve injury (SNI), a model of neuropathic pain, do not display any behavioral signs of having spontaneous pain (Shiers et al., 2020). Similar results have been obtained in 4EKI mice with peripheral neuropathy due to chemotherapeutics (Megat et al., 2019a).
Importantly, inhibition of MNK-eIF4E signaling also prevents the development of pain-related cognitive comorbidities related to attention in mouse models of neuropathic pain. 4EKI mice and mice treated with tomivosertib display no signs of deficits in attentional set shifting measured in a T-maze–based behavioral task after having SNI for 3 weeks (Shiers et al., 2020). This is relevant to neuropathic pain treatment because cognitive comorbidities are a prominent feature of the clinical picture for this disease. The question remains whether these positive effects are peripherally or centrally mediated. Published results to date, as cited above, suggest that inhibition of ectopic peripheral neuron activity normalizes ascending pain inputs to the parabrachial nucleus, amygdala, and prefrontal regions, resulting in recovery of pain-related cognitive dysfunction. However, changes in translation control pathways in pain models have been identified in the amygdala (Carrasquillo and Gereau, 2007; Missig et al., 2017) and anterior cingulate cortex (Wei and Zhuo, 2008; Dai et al., 2011; Um et al., 2019), which do have projections into prefrontal regions that control cognitive function. Therefore, it is possible that restoration of cognitive dysfunction in pain by MNK inhibitors is centrally mediated. This question can be addressed by development and testing of peripherally restricted MNK inhibitors.
The evidence described above makes a compelling case for MNK inhibitor use in several types of neuropathic pain; however, results in inflammatory pain models are mixed. Although some inflammatory mediators fail to evoke pain behaviors in mice lacking eIF4E phosphorylation, complete Freund’s adjuvant (CFA) provokes inflammatory mechanical hyperalgesia that is similar in WT and 4EKI mice (Moy et al., 2017). Interestingly, this observation is quite different in mice more than 18 months old. In aged 4EKI mice, the inflammatory pain response is greatly reduced, and CFA-evoked pain resolves more rapidly in these aged mice (Mody et al., 2020). This suggests that MNK inhibitors may be advantageously used for certain types of persistent inflammatory pain in older populations, in which these problems are actually more severe (Price et al., 2018).
Altogether, MNK inhibitors are promising therapeutic candidates because of their ability to diminish expression of a variety of proinflammatory and neuroplasticity-related genes that drive persistent nociceptor activity and chronic pain. A growing body of preclinical evidence has substantiated MNK as an important analgesic target, particularly for neuropathic pain; however, there are several important considerations for further investigation of MNK as a pain target moving forward. In our view, the most important of these is that MNK inhibitors designed for the treatment of pain should likely be designed to be peripherally restricted. The rationale for this is that disruption of MNK-eIF4E signaling through genetic or pharmacological manipulation causes some CNS-mediated phenotypes that would not be desirable for a pain therapeutic. 4EKI and MNK1 KO mice display abnormal obsessive compulsive disorder–like behaviors, such as stereotypic jumping and increased marble burying (Shiers et al., 2020). Moreover, 4EKI, MNK1/2 double KO, and mice treated with cercosporamide display anxiety- and depression-like behaviors coupled with impaired serotonergic tone in the cortex (Aguilar-Valles et al., 2018; Amorim et al., 2018b). It is noteworthy that most of these studies have been conducted in genetically modified animals in which eIF4E phosphorylation is disrupted from development. However, treatment with cercosporamide did cause CNS symptoms after only 5 days of treatment (Aguilar-Valles et al., 2018), and it has yet to be determined whether these effects worsen after more chronic dosing. Interestingly, tomivosertib has only been noted to cause gastrointestinal side effects in humans (Hubbard et al., 2019), and no depression-like symptoms have been reported. However, it is not known whether this drug crosses the blood-brain barrier in humans as it does in mice (Shiers et al., 2020). Restriction of future MNK inhibitors to the periphery would be predicted to beneficially reduce sensory neuron sensitization by targeting peripheral axons and nociceptors in the dorsal root ganglion without eliciting CNS-mediated adverse reactions.
1. Mitogen-Activated Protein Kinase–Interacting Kinase Inhibitors in the Clinic
A broad array of small-molecule MNK1/2 inhibitors have been identified to date that encompass diverse chemotypes as represented by Dreas et al. (2017), Matsui et al. (2018), Jin et al. (2019), and Mishra et al. (2019). Well studied MNK inhibitors include staurosporine, CGP57380, and the natural product, cercosporamide (Fig. 4). These compounds lack selectivity for MNK1/2, however, and are mainly used as chemical tools for MNK target validation (Hou et al., 2012). This section will focus on eFT508, ETC-206, and BAY 1143269, three MNK1/2 inhibitors that have entered clinical trials (Fig. 4), and an overview of the medicinal chemistry optimization process for eFT508 and ETC-206 will be provided. The full chemical structure of BAY 1143269 as well as details surrounding its development have not been disclosed; as such, a brief discussion of the structure activity relationships of published analogs of BAY 1143269 will be presented. We have chosen to detail medicinal chemistry progress for MNK inhibitors here because of the great potential for this target for neuropathic pain treatment and because there are few existing reviews on the topic. Comprehensive, recent reviews have been published on mTOR (Roychowdhury et al., 2010; Schenone et al., 2011) and AMPK (Cameron and Kurumbail, 2016) medicinal chemistry and pharmacology.
Fig. 4.

Chemical structures of MNK inhibitors. LE, ligand efficiency; MW, molecular weight; TPSA, topological polar surface area.
eFT508 (tomivosertib) is being developed by eFFECTOR Therapeutics as an orally dosed drug for the treatment of multiple cancer types, including nonsmall cell lung cancer (NSCLC), metastatic breast cancer, and castrate-resistant prostate cancer (NCT03616834, NCT03690141, NCT03258398). The drug discovery effort leading to eFT508 commenced with a broad search to identify optimal fragments that would provide a scaffold for constructing collections of compounds for biologic screening. In particular, molecular fragments were selected that would be expected to facilitate favorable stereoelectronic interactions with Phe159 and Cys225, atypical residues in the MNK1/2-ATP binding site (Reich et al., 2018). This approach was expected to generate compounds having both enhanced MNK1/2 potency and kinome selectivity.
The benzamide 1 was identified early as a key fragment because of its modest MNK1 inhibitory activity and favorable ligand efficiency and ClogP (Fig. 5). Constraining the primary amide of 1 as the lactam generated the isoindolinone scaffold 2 (Fig. 5), which improved MNK1 potency while maintaining ligand efficiency and ClogP. The reduced number of H-bond donors in 2 was expected to have a favorable effect on permeability (Reich et al., 2018). Lead fragment optimization was ultimately achieved by making modifications to fragment 2 that maintained or improved upon drug-like properties while only marginally reducing MNK1 potency. These changes involved deconstruction of the purine to the pyrimidine ring, replacement of the benzene ring with a pyridone ring, and introduction of alkyl groups at the C3 benzylic carbon, affording pyridone aminal 3. Cocrystal structural analysis of 2 and its C3 methylated analog (unpublished data) indicate that the presence of C3 alkyl groups induce a ring flip to relieve steric interactions (Reich et al., 2018). This change of binding mode enables out-of-plane C3 alkyl groups to be oriented toward the MNK p-loop, an interaction that facilitates a boost in potency. The amide functional group within the pyridone ring system of three imparts a favorable effect on potency that has been attributed to a stereoelectronic interaction between the lone pairs of the sulfur atom of Cys225 (Dunitz interaction) and the amide carbonyl. The pyridone ring incorporated into scaffold 3 also provides the benefit of reduced lipophilicity relative to the analogous benzene analog 2. The pyrimidine-tethered pyridone aminal template, exemplified by compound 3 (Fig. 5), was thus selected as the optimal starting point for designing novel MNK inhibitors (Reich et al., 2018). Pyridone aminal analogs of the general structure 4 were designed by making modifications at three diversification points (Fig. 5), which included the R1 of the pyrimidine ring, the R2 of the pyridone ring, and R3/R4 of the imidazolidinone ring. After preparing over 200 analogs with varying combinations of functional groups, it was discovered that compounds having a free amino group at R1, a methyl group at R2, and a spirocyclic cycloalkyl group at R3/R4 were found to deliver the most favorable blend of MNK1/2 potency, minimal off-target activity, and adequate permeability and metabolic stability [WO2015200481, Reich et al. (2018)]. From the set of optimized clinical candidates, eFT508 (R1 = NH2; R2 = CH3; R3/R4 = spirocyclohexane) emerged as eFFECTOR Therapeutics’ drug candidate and is currently being evaluated in multiple phase II clinical trials (NCT03690141, NCT03616834, NCT03258398). As mentioned above, this compound has pronounced effects in many preclinical neuropathic pain models, making it an excellent starting point for development of a novel neuropathic pain drug.
Fig. 5.

(A) Key fragments and pyridone aminal 3. (B) Diversification points on pyridone aminal 4 and selected properties of eFT508. MW, molecular weight; TPSA, topological polar surface area.
Under development by AUM Biosciences, ETC-206 is an orally bioavailable inhibitor of MNK1/2 that was recently evaluated in phase I clinical trials for the treatment of blood cancers (NCT03414450). The medicinal chemistry approach leading to the discovery of ETC-206 began by selecting three previously identified bicyclic fragments (5–7) having MNK1 inhibitory activity with desirable ligand efficiency values (Oyarzabal et al., 2010; Yang et al., 2018) (Fig. 6). Guided by ligand-protein–docking studies, collections of compounds were prepared comprising C3- and C6-functionalized imidazolopyrazines 5, imidazolopyridazines 6, and imidazolopyridines 7 (Fig. 6). It is noteworthy that aryl groups were identified as optimal C3 substituents, as they provided superior MNK potency relative to other groups that were surveyed. Molecular modeling studies indicate that the enhanced activity is attributed to the favorable interaction of the aryl group with a hydrophobic pocket in the MNK1 and MNK2 binding sites (Yang et al., 2018). The optimal C6 substituents included piperazinobenzamides and morpholinobenzamides, as is exemplified by analogs 8–10 and ETC-206 (Figs. 4 and 6). The morpholine analogs 10 and ETC-206 were prioritized because they had better drug-like properties and performed well in in vitro drug metabolism and pharmacokinetics assays. The activity of ETC-206 was superior to that of 10 in cellular eIF4E phosphorylation inhibition assays. Moreover, ETC-206 demonstrated more favorable physicochemical properties and improved kinase selectivity and was therefore selected as AUM Biosciences’ drug candidate for leukemia. ETC-206 was recently found to be safe in healthy human volunteers (Teneggi et al., 2020). This compound has not been evaluated in any preclinical pain models.
Fig. 6.

Key scaffolds 5–7, optimized analogs 8–10, and selected properties of ETC-206.
BAY 1143269 is a small-molecule inhibitor of MNK1/2 that was discovered by Bayer using high-throughput screening and subsequent chemistry optimization. The compound is unique among known MNK inhibitors owing to its unusually high selectivity for MNK1 over MNK2 (MNK1 IC50 = 40 nM; MNK2 IC50 = 904 nM). BAY 1143269 induces apoptosis in multiple NSCLC cell lines, blunts the release of proinflammatory cytokines, and exhibits antitumor activity in patient-derived xenograft models. Because of these favorable attributes, a phase I clinical trial was launched for the treatment of NSCLC. However, because of project deprioritization the clinical study was not completed (NCT02439346).
The full chemical structure of BAY 1143269 has not been disclosed, but it has been depicted as a disubstituted imidazolopyridazine having the general structure 11. Analogs in the Bayer collection consist of C3-substituted 2-benzofuranyl groups or derivatives thereof, whereas C6 substituents are ethers (Fig. 7; 11: X = O) or anilines (Fig. 7; 11: X = NH) (WO2013034570; WO2013087581; and WO2013149909). A number of analogs of BAY 1143269 inhibit MNK1 with single-digit nanomolar potency, and this activity appears to be easily modulated through tuning of the steric and electronic properties of the pendant R group on 11 (Fig. 7). For example, when the potent aminocyclobutyl ether group in compound 12 (MNK1 IC50 = 5 nM) is replaced with an (aminomethyl)pyridyl ether group as in 13 (MNK1 IC50 = 94 nM), an almost 20-fold reduction in potency is observed. Indeed, amino ether R groups (Fig. 7; X = O) devoid of aromatic rings generally exhibit the best MNK1 inhibitory activity. In contrast to the aryl ether series, many aniline derivatives (Fig. 7; 11: X = NH) that contain aromatic rings within the R1 group of 11 exhibit excellent MNK1 activity, as exemplified by 14 (MNK1 IC50 = 1 nM). The tertiary alcohol 15 represents a notable exception, as the additional methyl group elicits a substantial decrease (60-fold) in MNK1 activity.
Fig. 7.

The general structure of BAY 1143269 (11) and related analogs 12–15.
A small molecule that inhibits MNK as its primary mechanism of action has yet to be FDA-approved for any indication. This is, at least in part, due to the relatively recent understanding of the role of MNK in cancer and other diseases like neuropathic pain (Moy et al., 2017, 2018b; Megat and Price, 2018; Megat et al., 2019a; Barragán-Iglesias et al., 2020; Shiers et al., 2020). Moreover, a majority of known MNK inhibitors developed prior to eFT508 suffer from modest potency (high nanomolar/low micromolar) while exhibiting extensive off-target kinase activity. However, the discovery of eFT508 demonstrates that highly potent and selective small-molecule MNK inhibitors can indeed be developed, and ongoing clinical trials with both eFT508 and ETC-206 indicate that MNK inhibitors have good safety profiles in humans. This bodes well for the future vetting of this target in neuropathic pain clinical trials.
An unresolved question is whether a specific MNK1 inhibitor would be preferable over a dual MNK1/2 inhibitor for neuropathic pain. Some results with MNK1-specific knockout in mice suggest that MNK1 is the key isoform for neuropathic pain (Megat et al., 2019a; Shiers et al., 2020); however, MNK2 knockout mice have never been tested in any pain assays, and MNK2 selective compounds have not been described. It is also important to note that most nociceptors appear to express both MNK isoforms (Zeisel et al., 2018). At this stage it is not possible to definitively discern which of these approaches (MNK1 selective or dual inhibitor) would be best for the development of a neuropathic pain drug targeting MNK(s).
E. Integrated Stress Response Modulators
The ISR regulates translation initiation by phosphorylating eIF2α, blocking eIF2B, and preventing the recycling of GTP bound to eIF2 (Fig. 2A). As a result, the phosphorylation of eIF2α leads to a repression of general translation while enhancing translation of certain mRNAs. Some of these induced gene products, like ATF4 and CHOP, feed back onto the ISR to recover from stress or, in dire cases, induce cell death. The dephosphorylation of eIF2α is mediated by PP1 and its regulatory subunits, constitutive repressor of eIF2α phosphorylation (CreP) and GADD34 (Pakos-Zebrucka et al., 2016) (Fig. 2A). Although CreP is constitutively active in physiologic conditions, the expression of GADD34 is dramatically induced by ISR and functions to restore general translation.
Various pharmacotherapies have been developed to target the phosphorylation of eIF2α to promote or inhibit the ISR (Hetz et al., 2019) (Fig. 2, B and C). Salubrinal, one of the first ISR modulators to be developed, is a nonselective inhibitor of the PP1 phosphatase and thus prevents the dephosphorylation of eIF2α, thereby maintaining or delaying the ISR (Boyce et al., 2005). Salubrinal administration in naïve animals produced heat hypersensitivity without any change in mechanosensation (Khoutorsky et al., 2016). On the contrary, salubrinal treatment after nerve injury and chronic inflammation resolved pain hypersensitivity (Yang et al., 2014; Yamaguchi et al., 2018). In other words, inducing ISR in naïve animals resulted in heat hypersensitivity, whereas inducing ISR after injury or inflammation resolved pain hypersensitivity. It is possible that inhibiting protein synthesis under normal circumstances leads to a reduction in proteins important for proper cellular functioning, whereas repression of global protein synthesis after injury or disease may suppress expression of detrimental proteins and/or reduce ER load. These observations further suggest that the context of eIF2α phosphorylation may represent a fine balance between adaptive nociception and maladaptive chronic pain.
Salubrinal’s nonspecific effect on the PP1 holoenzyme makes it a poor choice as a therapeutic and makes its effects difficult to interpret as a tool compound. Since GADD34 is potently induced after cellular stress, selectively inhibiting GADD34 then would maintain ISR specifically in stressed cells and tissues without interfering with the constitutively active CreP in healthy cells. This strategy is thereby predicted to limit adverse side effects. In this regard, two small molecules, guanabenz and Sephin1, have been used to target the GADD34:PP1 complex while sparing the CreP:PP1 holoenzyme (Tsaytler et al., 2011). Guanabenz was initially developed as an α-adrenergic agonist and is clinically used as such to treat hypertension (Holmes et al., 1983). It was repurposed for use as an ISR promoter. However, clinical trials employing guanabenz oral therapy in treating multiple sclerosis had to be terminated prematurely because of severe side effects associated with the adrenergic system, despite promising effects in rodent models (Way et al., 2015; Clayton and Popko, 2016). Sephin1 is preferred over guanabenz owing to its better potency at promoting ISR, a lack of adrenergic effect, and fewer adverse effects (Das et al., 2015). Although no clinical trials have yet been performed with Sephin1, preclinical studies investigating cancer (Cao et al., 2019), amyotrophic lateral sclerosis (Das et al., 2015), multiple sclerosis (Chen et al., 2019), viral infections (Fusade-Boyer et al., 2019), and prion diseases (Thapa et al., 2020) demonstrate encouraging results. The efficacy of Sephin1 at resolving chronic pain remains to be investigated.
The molecular targets of guanabenz and Sephin1 and how they selectively inhibit GADD34:PP1 over CreP:PP1 are still debated in the literature. One hypothesis is that Sephin1 and guanabenz directly bind to the N terminus of GADD34, inducing a conformational change that lowers the ability of GADD34:PP1 to bind phospho-eIF2α (Carrara et al., 2017). Structural and biochemical analysis of the GADD34:PP1 holoenzyme suggests that GADD34 functions as a scaffold for PP1 and phospho-eIF2α interaction (Choy et al., 2015) (Fig. 2D). Neither salubrinal nor guanabenz treatment can dissociate the GADD34:PP1 holoenzyme or impair its interaction with phospho-eIF2α (Choy et al., 2015). Other reports argue against direct binding of Sephin1 and guanabenz to GADD34 altogether (Crespillo-Casado et al., 2017, 2018). Furthermore, through an unknown mechanism, Sephin1 treatment induces ATF4 expression but, ironically, does not enhance the expression of CHOP, a well established proapoptotic target of ATF4, and subsequent cell death (Das et al., 2015; Hetz et al., 2019). More recently, guanabenz and Sephin1 were shown to interact with and allosterically modulate acid-sensing ion channel 3, which has been implicated in inflammatory pain, proposing a novel mechanism of action for these drugs (Callejo et al., 2020). From these studies, it is evident that our current understanding of the available pharmacological tools that target GADD34:PP1 is poor. The identification of internal PEST repeats [a sequence rich in proline (P), glutamic acid (E), serine (S), and threonine (T)] on GADD34, a lack of these repeats in CreP, and its importance in the binding of the GADD34:PP1 holoenzyme to phospho-eIF2α may represent a novel target for future drug design (Brush and Shenolikar, 2008; Choy et al., 2015) (Fig. 2D).
Another approach to target the ISR has been to dampen its effect on general translation. ISR inhibitor (ISRIB) is commonly used to reverse the effects of phospho-eIF2α (Sidrauski et al., 2013) (Fig. 2B). Although ISRIB can reduce the translation of ATF4, a well known target of the ISR, it does not immediately reverse the phosphorylation of eIF2α, implying that ISRIB’s effects on translation may be eIF2α-independent. Recent biochemical and structural analysis has revealed that ISRIB targets the β- and δ-subunits of eIF2B and enhances its GDP exchange activity, replenishing the ternary complex and reducing ATF4 translation (Sekine et al., 2015; Sidrauski et al., 2015; Wong et al., 2018). Moreover, ISRIB aids in the generation of new eIF2B complexes from its subunits by binding together two eIF2B tetrameric subcomplexes (Costa-Mattioli and Walter, 2020) (Fig. 2C). However, ISRIB’s activity is limited by the amount of free eIF2B that is not already sequestered by phospho-eIF2α (Hetz et al., 2019). In conditions in which phosphorylation of eIF2α far exceeds eIF2B levels, the potency of ISRIB significantly decreases, suggesting that ISRIB works ideally in mild ISR conditions (Rabouw et al., 2019). Furthermore, ISRIB is hard to formulate and not orally bioavailable, making it a suboptimal choice for clinical applications in its current form (Sidrauski et al., 2013; Halliday et al., 2017; Hetz et al., 2019). However, owing to its safety profile, high potency, and the ability to penetrate the CNS, ISRIB is still an invaluable tool to study ISR-regulated translation (Hetz et al., 2019; Costa-Mattioli and Walter, 2020). Other compounds, including eIF2B activator (2BAct) (Wong et al., 2019), trazodone (Halliday et al., 2017), and dibenzoylmethane (Halliday et al., 2017), have recently been identified as eIF2B activators but more research is needed to identify their molecular targets and characterize their pharmacological properties in multiple experimental models, including chronic pain.
Cancer, diabetes, inflammation, and neurodegeneration have all been associated with stimulation of the ISR (Pakos-Zebrucka et al., 2016). However, the effect of phosphorylation of eIF2α in mediating pain hypersensitivity has only recently begun to be uncovered. The first study we are aware of that associated eIF2α phosphorylation with nerve injury–induced neuropathic pain found a clear sign of increased ISR signaling in the injured axons of rats and mice. However, this study did not investigate a causative role of the pathway in the SNI or spinal nerve ligation models in which these observations were made (Melemedjian et al., 2011). The first evidence for a causative role of the ISR in neuropathic pain came from diabetic neuropathy models. In the streptozotocin-induced type I diabetic rat model, phosphorylation of eIF2α, along with its ER kinase, PERK, and markers of unfolded protein response, were significantly elevated in sciatic nerves, contributing to mechanical allodynia in these animals (Inceoglu et al., 2015). Alleviation of ER stress using a chemical chaperone, 4-phenylbutyric acid, reduced the phosphorylation of eIF2α and rapidly resolved pain hypersensitivity (Inceoglu et al., 2015). Similarly, intrathecal administration of the ISR inhibitor, ISRIB, alleviated mechanical hyperalgesia in streptozotocin-induced diabetic mice and rats (Barragán-Iglesias et al., 2019). The exact by-product of diabetes that mediates ISR and hyperexcitability in nociceptors remains to be pin-pointed. One such candidate is methylglyoxal (MGO), a metabolite of glycolysis that is elevated in diabetes and is implicated in pain associated with diabetic neuropathy. In addition to activating transient receptor potential ankyrin 1 cation channel channels at high concentrations, MGO, even at low concentrations, increases the phosphorylation of PERK and eIF2α particularly in isolectin B4+ nociceptors and induces mechanical hypersensitivity (Barragán-Iglesias et al., 2019). In vitro, low-concentration MGO (1 µM) sensitizes DRG neurons in addition to activating the ISR. Resolution of ISR activation using systemic delivery of ISRIB and/or 4-phenylbutyric acid ameliorates mechanical hypersensitivity induced by low-dose MGO treatment. Altogether, these results suggest that targeting the ISR is a viable strategy for combating pain in diabetic neuropathy.
In addition, chronic inflammation after intraplantar administration of CFA induces the ISR as well as mechanical and thermal hyperalgesia in rodents (Khoutorsky et al., 2016). Mutation of the serine 51 residue to alanine prevents the phosphorylation of eIF2α, and homozygous mutation (eIF2αS51A/S51A) is lethal (Scheuner et al., 2001). Naïve heterozygous mutants (eIF2α+/S51A) have increased threshold to noxious heat stimulation without any change in mechanical sensitivity (Khoutorsky et al., 2016). This loss of heat sensitivity is a result of reduced TRPV1 currents despite no change in TRPV1 protein levels, suggesting that phospho-eIF2α controls TRPV1 activity by possibly modulating regulatory or accessory proteins (Khoutorsky et al., 2016). Upstream kinase mutants of PERK, PKR, and GCN2 show a similar phenotype (Khoutorsky et al., 2016). Although these data demonstrate a novel role of eIF2α and its kinases in mediating heat nociception via TRPV1, they do not entirely address the role of ISR in modulating CFA-induced thermal and mechanical hyperalgesia. Roughly 45% of A-fibers (neurofilament 200+) and 40% of nonpeptidergic sensory (isolectin B4+) express phospho-eIF2α after CFA administration compared with about 20% of TRPV1+ nociceptors, suggesting that eIF2α phosphorylation may also be contributing to inflammation-induced tactile hypersensitivity via distinct mechanisms (Khoutorsky et al., 2016). Hence, more work is needed to better understand whether the ISR is a tractable pain target for chronic pain disorders other than diabetic neuropathy.
1. Inhibiting Eukaryotic Initiation Factor 2α Kinases
Four kinases (PKR, PERK, GCN2, and HRI) phosphorylate eIF2α, among which the bulk of the literature has focused on inhibitors of PERK and PKR and their role in pain hypersensitivity. Consistent with effects observed in eIF2α phosphorylation mutant mice, systemic administration of PKR inhibitor (PKRi) in healthy mice produces hypoalgesia to radiant heat stimulation with no effect on mechanical sensitivity (Khoutorsky et al., 2016). Although PKRi inhibits PKR-mediated phosphorylation of eIF2α, off-target inhibition of cyclin-dependent kinases (CDKs) has also been reported in the literature (Chen et al., 2008). CDKs have previously been implicated in pain pathophysiology (Pareek et al., 2006; Liu et al., 2015; Rozas et al., 2016; Hsieh et al., 2019). Thus, with the use of PKRi to study the effect of PKR and eIF2α on pain pathophysiology, its effect on CDKs must also be considered.
GSK2606414 and GSK2656157 are two small molecules that have been widely used to inhibit PERK activation (Hetz et al., 2019). A recent study found that GSK2606414 attenuated mechanical and thermal hyperalgesia associated with neuroinflammation in a bone cancer pain model (Mao et al., 2020). Both of these compounds have excellent potency and show promising pharmacokinetics in vivo. However, recent reports demonstrate that GSK2606414 and GSK2656157 also inhibit receptor-interacting protein kinase 1 [receptor-interacting serine/threonine protein kinase 1 (RIPK1)] and the tyrosine kinase receptor KIT, again questioning the specificity of these compounds (Smith et al., 2015; Rojas-Rivera et al., 2017; Mahameed et al., 2019). A screen for a novel compound with better selectivity for PERK identified two compounds, AMG44 and AMG52, albeit with lower potency than the previously identified GSK inhibitors (Smith et al., 2015; Hetz et al., 2019). AMG44 and AMG52 are comparable to GSK inhibitors in preventing PERK activation, and they are also orally bioavailable (Smith et al., 2015). These compounds bind to the ATP-binding region of PERK and induce a conformational change that traps PERK in an inactive state. In light of favorable pharmacodynamics and kinetics of AMG44 and AMG52 along with better specificity for PERK (Smith et al., 2015; Rojas-Rivera et al., 2017), AMG44 and AMG52 should be preferred when studying PERK-eIF2α signaling. Tissue distribution of systemic AMG44 and AMG52 treatment remains to be investigated, so an open question is whether these compounds efficiently cross the blood-brain barrier.
F. Localized Translation in Peripheral Axons as a Therapeutic Target
Spatial control of mRNA localization and trafficking is dictated by specific sequences in the 3′ UTR known as zipcodes, which interact with RNA-binding proteins. These proteins can then shuttle mRNAs to synaptic sites along axons and/or dendrites, allowing for localized translation of proteins at distal sites (Kiebler and Bassell, 2006). Localized translation accompanied by post-translational modifications (i.e., phosphorylation, methylation, or sumoylation) allows individual synapses to remodel their synaptic proteome in an activity-dependent manner, a key feature of synaptic plasticity (Biever et al., 2019).
The same holds true in the peripheral nervous system. Translation machinery, including mRNA, translation initiation factors (eIF2α, eIF4E, and eIF5), and free ribosomes, which cluster into polyribosomal plaques, have been identified in peripheral sensory axons in both invertebrates (Giuditta et al., 1968, 1991; Piper and Holt, 2004; Eyman et al., 2007; Mathur et al., 2018) and vertebrates (Droz and Leblond, 1963; Koenig et al., 2000; Zheng et al., 2001; Verma et al., 2005; Court et al., 2011) (Fig. 8). It is thought that this axonally localized protein synthesis machinery prepares sensory neurons to rapidly respond to injurious stimuli via the rapid synthesis of proteins that then regulate the excitability of the peripheral terminal or injured site along the axon. One point supporting this conclusion is the sheer distance between the sensory neuron soma and axon terminal, which can be up to 1 m long. Given what is known about axonal transport rates for proteins synthesized in the soma, it is simply not possible for gene expression at this location to explain the rapid, local effects that are seen in most preclinical models and in human volunteer studies. It is also clear that local blockade of protein synthesis at the site of peripheral terminals has a profound effect on inhibiting nociceptive sensitization, whereas transcriptional inhibitors have no effect ( Price et al., 2007; Melemedjian et al., 2010, 2014; Ferrari et al., 2013, 2015a,b; Moy et al., 2017; Barragán-Iglesias et al., 2018). In further support of this idea, DRG neurons in vitro form axonal growth cones within 20 minutes of axotomy, an effect that is impaired after application of protein synthesis but not transcriptional inhibitors (Verma et al., 2005). These collective findings indicate that rapid changes in protein synthesis can happen within the peripheral axons of sensory neurons, and these localized changes in gene expression likely control plasticity events that alter nociceptive thresholds. There is, however, a major issue with this idea—although ribosomes have been observed in peripheral axons, they are sparse and do not have the arrangement that is found, for instance, in dendritic spines that links their subcellular anatomy to plasticity events (Steward and Schuman, 2003; Biever et al., 2019).
Fig. 8.

Evidence for localized translation in sensory axons in neuropathic pain. 1) Translation machinery is present in sensory neuron axons, such as mRNA, ribosomes, translation initiation factors (eIF2α, eIF4E, and eIF5), and translation regulators (ERK, p38, mTOR). 2) Sensory neuron mRNAs (ex: Nav1.8) are trafficked and translated locally in peripheral axons leading to ectopic activity. 3) Ribosomes and exosomes from Schwann cells (SCs) are trafficked into sensory axons (blue arrows). In myelinated axons, these ribosomes form ribosomal plaques that are located near the myelin-axon border. We hypothesize the same occurs between nonmyelinating SCs and nociceptive axons [i.e., in Remak bundles (top right panel)]. It is possible that the SC-axon cytoplasms become continuous after axotomy, forming a syncytium in which SCs aid in axonal translation during recovery.
Interestingly, recent work supports that translational activity in sensory neuron axons may be regulated by Schwann cells. In addition to providing myelin and other supporting functions in normal conditions and controlling axonal regeneration via the recruitment of the immune system and phagocytosis of axonal debris after injury, Schwann cells can transfer their ribosomes to sensory neuron axons after axotomy (Court et al., 2008, 2011) (Fig. 8). Polyribosomal plaques in sensory axons are organized along the boundary between the neuronal membrane and myelin sheath, which may be reflective of Schwann-cell ribosomal transfer (Koenig et al., 2000). Although unknown, it is possible that other Schwann-cell translation machinery is transferred to axons, such as mRNAs and protein. Exosomes are secreted from Schwann cells and internalized by peripheral axons after injury and are hypothesized to contain mRNA transcripts (Lopez-Verrilli et al., 2013). Accordingly, it is difficult to delineate neuron-specific mRNAs in sensory nerves because the majority of the transcripts are also present in Schwann cells.
Recently, a new pain organ composed of a cutaneous glial network was found in the subepidermal layer of the skin (Abdo et al., 2019). This glial matrix is comprised of SOX10-positive Schwann cells that appear to ensheath free nerve endings and convey noxious thermal and mechanical information (Abdo et al., 2019). Importantly, SOX10-positive Schwann cells are also found along the length of peripheral nerves and may function similarly to those in the skin. It is hypothesized that Schwann cells share a cytoplasm with axons (López-Leal et al., 2016), forming a syncytium that merges the transcriptomes of both Schwann cell and sensory neuron. This syncytium may be important in localized protein synthesis after nerve injury. Together, these findings may offer a new avenue for therapeutic development. This is obviously speculative at this point; however, if some of the mysteries of local translation in long sensory axons are ultimately explained by Schwann cell–axon interactions in which mRNA, ribosomes, and other material are exchanged, these mechanisms could potentially be exploited for therapeutic benefit.
An area ripe for future discovery is determining how localized translation is linked to nociceptor excitability and spontaneous activity. The differential contribution of translation in the axonal compartment versus in the soma in persistent pain conditions has not been determined. It is possible that both are important, but more detailed insight would be useful. For instance, if localized translation at the site of nociceptor endings in neuromas is a major contributor to neuropathic pain in this clinical setting, localized applied drugs may be highly efficacious. This improves some aspects of the safety window compared with a systemically administered drug. However, if translation regulation signaling along the neuron axon and into the soma is required to drive neuropathic pain in patients with neuromas then the locally administered drug approach will obviously not work. Advancing our understanding of how localized changes in gene expression control nociceptor excitability in chronic pain conditions will move the field forward.
VII. Future Opportunities
Aside from the development of nonaddictive, nonopioid analgesics, one of the most significant unmet needs in pain clinical care is for improved individualized approaches to pain management. There are no existing clinical diagnostic tools to predict whether a person will develop chronic pain and/or which medications will work for any given patient. Moreover, we do not have reliable tools to gain insight into underlying molecular disease mechanisms in individual patients with chronic pain. Developing such tools would have obvious utilities. They would increase our ability to identify people at risk of developing chronic pain, they would enhance our ability to effectively treat certain types of chronic pain disorders, and they would aid future drug development efforts because molecular mechanisms of chronic pain in people would be better understood. Above, we have argued that translation regulation signaling is a key feature of many types of chronic pain. We have also argued that targeting translation signaling pathways is a viable route to treat different types of chronic pain. Although dysregulated translation in peripheral neurons is a common theme of many chronic pain types, the specific alterations in signaling observed in different conditions offer unique opportunities to link diagnostics with therapeutics.
One way that this can be exploited is through the use of skin biopsies as a window into molecular pathology driving chronic pain in individual patients (Fig. 9). Skin biopsies are invasive but can be performed, and the samples are filled with the free nerve endings of sensory neurons. As we have described in detail above, alterations in translation regulation signaling appear to occur along the course of the nociceptor, so these molecular mechanisms can be examined in these biopsies. Samples could be assessed for panels of translation signaling molecules to gain insight into changes in signaling between painful and nonpainful skin or between patients with standardized pain and nonpain populations. Another possibility is that changes in mRNA trafficking may be linked to specific types of chronic pain, and these could be assessed in individual patients using microarrays or RNA sequencing. To that end, we have recently shown that many sensory neuronal mRNAs are readily detected in distal human nerves using an RNA sequencing approach (Ray et al., 2019). Although preliminary, because of small sample sizes and inexact medical records, these mRNA signatures seem to change in diseases like diabetes that cause neuropathic pain (Ray et al., 2019).
Fig. 9.

Personalized treatment approaches for chronic pain. After injury and disease, translational regulation mechanisms alter protein synthesis, which forms the basis of enhanced excitability in sensory neurons and potentially also in central networks. As a result, patients with chronic pain experience evoked and spontaneous pain. Since the plasticity after injury and disease is unique to its pathology, we propose that individualized treatment avenues that specifically target the underlying pathology would best alleviate pain. To determine the course of treatment, skin biopsies can be obtained and analyzed for inflammatory mediators and translational regulators as well as sequenced for RNA profiling. Based on these personalized analyses of underlying molecular mechanisms, therapeutic development and decision making can be improved.
Based on the current state of the science, we propose that there are three near-term opportunities for development of novel, nonopioid pain therapeutics that manipulate translation regulation signaling. The first of these is metformin. The preclinical and clinical rationales for pivotal clinical trials for this drug are strong. Given the current state of the science, there are tremendous opportunities for metformin to be very useful in the prevention of neuropathic pain caused by chemotherapeutic drugs and in the treatment of pain diseases like fibromyalgia. The second are MNK inhibitors. As stated above, we think that peripherally restricted MNK inhibitors likely need to be developed, but this class of drugs has tremendous potential as a treatment of nondiabetic neuropathic pain. Finally, there is a clear rationale for developing inhibitors of the ISR as diabetic neuropathic pain drugs. A variety of approaches can be taken to achieve ISR inhibition in this context. As these drugs move toward the clinic for a variety of diseases, we will learn more about their safety and tolerability. It is possible that a triumvirate of translation regulation targeting therapeutics will transform chronic pain treatment in the coming decade.
Abbreviations
- AKT
protein kinase B
- AMPK
AMP-activated protein kinase
- ATF
activating transcription factor
- BDNF
brain-derived neurotrophic factor
- BiP
binding immunoglobulin protein
- CBS
cystathionine β-synthetase
- CDK
cyclin-dependent kinase
- CFA
complete Freund's adjuvant
- CHOP
C/EBP-homologous protein
- CNS
central nervous system
- CreP
constitutive repressor of eIF2α phosphorylation
- DDIT3
DNA damage inducible transcript 3
- DRG
dorsal root ganglia
- 4E-BP
4E-binding protein
- eEF
eukaryotic elongation factor
- 4EKI
4E knock-in with null mutation
- eIF
eukaryotic initiation factor
- ER
endoplasmic reticulum
- ERK
extracellular signal-regulated kinase
- FDA
Food and Drug Administration
- GADD34
growth arrest and DNA damage-inducible protein 34
- GβL
G-protein β-subunit–like protein
- GCN2
general control nonderepressible 2
- GSK
glycogen synthase kinase
- HRI
heme-regulated inhibitor kinase
- IF2
yeast initiation factor 2
- ISR
integrated stress response
- ISRIB
ISR inhibitor
- KO
knockout
- MAPK
mitogen-activated protein kinase
- MAPKK
MAPK kinase
- Met-tRNAiMet
methionine-bound initiator tRNA
- MGO
methylglyoxal
- MNK
MAPK-interacting kinase
- mORF
main ORF
- mSin1
mammalian stress-activated protein kinase interacting protein 1
- mTOR
mammalian/mechanistic target of rapamycin
- mTORC
mTOR complex
- Nav1.8
voltage-gated sodium channel 1.8
- NGF
nerve growth factor
- NSCLC
nonsmall cell lung cancer
- ORF
open reading frame
- PABP
poly(A)-binding protein
- PDB
Protein Data Bank
- PDCD4
programmed cell death protein 4
- PERK
PKR-like endoplasmic reticulum kinase
- PEST repeats
sequence rich in proline, glutamic acid, serine, and threonin
- PI3K
phosphoinositide 3-kinase
- PKR
protein kinase R
- PKRi
PKR inhibitor
- poly(A)
poly-adenosine
- PP1
protein phosphatase 1
- PRAS40
proline-rich AKT/protein kinase B substrate 40 kDa
- RAPTOR
regulatory associated protein of mTOR
- RHEB
Ras homolog enriched in brain
- RICTOR
rapamycin-insensitive companion of mTOR
- rpS6
ribosomal protein S6
- S6K
S6 ribosomal kinase
- SNI
spared nerve injury
- TG
trigeminal ganglia
- TOP
terminal oligopyrimidine tract
- tRNA
transfer RNA
- TRPV1
transient receptor potential vanilloid 1 cation channel
- TSC
tuberous sclerosis protein
- uORF
upstream ORF
- UTR
untranslated region
Authorship Contributions
Wrote or contributed to the writing of the manuscript: Yousuf, Shiers, Sahn, Price.
Footnotes
This work was supported by Postdoctoral Fellowship Program of Natural Sciences and Engineering Research Council of Canada and by National Institutes of Health National Institute of Neurological Disorders and Stroke [Grant R01-NS102161], [Grant R01-NS065926], and [Grant U44-NS115692].
The authors declare financial relationships. J.J.S. is an employee of 4E Therapeutics, and T.J.P. is a cofounder of 4E Therapeutics.
References
- Abdo H, Calvo-Enrique L, Lopez JM, Song J, Zhang MD, Usoskin D, El Manira A, Adameyko I, Hjerling-Leffler J, Ernfors P. (2019) Specialized cutaneous Schwann cells initiate pain sensation. Science 365:695–699. [DOI] [PubMed] [Google Scholar]
- Afshari K, Dehdashtian A, Haddadi NS, Haj-Mirzaian A, Iranmehr A, Ebrahimi MA, Tavangar SM, Faghir-Ghanesefat H, Mohammadi F, Rahimi N, et al. (2018) Anti-inflammatory effects of Metformin improve the neuropathic pain and locomotor activity in spinal cord injured rats: introduction of an alternative therapy. Spinal Cord 56:1032–1041. [DOI] [PubMed] [Google Scholar]
- Aguilar-Valles A, Haji N, De Gregorio D, Matta-Camacho E, Eslamizade MJ, Popic J, Sharma V, Cao R, Rummel C, Tanti A, et al. (2018) Translational control of depression-like behavior via phosphorylation of eukaryotic translation initiation factor 4E. Nat Commun 9:2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alcocer-Gómez E, Garrido-Maraver J, Bullón P, Marín-Aguilar F, Cotán D, Carrión AM, Alvarez-Suarez JM, Giampieri F, Sánchez-Alcazar JA, Battino M, et al. (2015) Metformin and caloric restriction induce an AMPK-dependent restoration of mitochondrial dysfunction in fibroblasts from Fibromyalgia patients. Biochim Biophys Acta 1852:1257–1267. [DOI] [PubMed] [Google Scholar]
- Amorim IS, Kedia S, Kouloulia S, Simbriger K, Gantois I, Jafarnejad SM, Li Y, Kampaite A, Pooters T, Romanò N, et al. (2018a) Loss of eIF4E phosphorylation engenders depression-like behaviors via selective mRNA translation. J Neurosci 38:2118–2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amorim IS, Lach G, Gkogkas CG. (2018b) The role of the eukaryotic translation initiation factor 4E (eIF4E) in neuropsychiatric disorders. Front Genet 9:561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anand AA, Walter P. (2020) Structural insights into ISRIB, a memory-enhancing inhibitor of the integrated stress response. FEBS J 287:239–245. [DOI] [PubMed] [Google Scholar]
- Andreev DE, O’Connor PB, Fahey C, Kenny EM, Terenin IM, Dmitriev SE, Cormican P, Morris DW, Shatsky IN, Baranov PV. (2015) Translation of 5′ leaders is pervasive in genes resistant to eIF2 repression. eLife 4:e03971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asante CO, Wallace VC, Dickenson AH. (2009) Formalin-induced behavioural hypersensitivity and neuronal hyperexcitability are mediated by rapid protein synthesis at the spinal level. Mol Pain 5:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asante CO, Wallace VC, Dickenson AH. (2010) Mammalian target of rapamycin signaling in the spinal cord is required for neuronal plasticity and behavioral hypersensitivity associated with neuropathy in the rat. J Pain 11:1356–1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baird TD, Wek RC. (2012) Eukaryotic initiation factor 2 phosphorylation and translational control in metabolism. Adv Nutr 3:307–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker BM, Nargund AM, Sun T, Haynes CM. (2012) Protective coupling of mitochondrial function and protein synthesis via the eIF2α kinase GCN-2. PLoS Genet 8:e1002760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbosa C, Peixeiro I, Romão L. (2013) Gene expression regulation by upstream open reading frames and human disease. PLoS Genet 9:e1003529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barragán-Iglesias P, Franco-Enzástiga U, Jeevakumar V, Shiers S, Wangzhou A, Granados-Soto V, Campbell ZT, Dussor G, Price TJ. (2020) Type I interferons act directly on nociceptors to produce pain sensitization: implications for viral infection-induced pain. J Neurosci 40:3517–3532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barragán-Iglesias P, Kuhn J, Vidal-Cantú GC, Salinas-Abarca AB, Granados-Soto V, Dussor GO, Campbell ZT, Price TJ. (2019) Activation of the integrated stress response in nociceptors drives methylglyoxal-induced pain. Pain 160:160–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barragán-Iglesias P, Lou TF, Bhat VD, Megat S, Burton MD, Price TJ, Campbell ZT. (2018) Inhibition of Poly(A)-binding protein with a synthetic RNA mimic reduces pain sensitization in mice. Nat Commun 9:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basbaum AI, Bautista DM, Scherrer G, Julius D. (2009) Cellular and molecular mechanisms of pain. Cell 139:267–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett DL, Clark AJ, Huang J, Waxman SG, Dib-Hajj SD. (2019) The role of voltage-gated sodium channels in pain signaling. Physiol Rev 99:1079–1151. [DOI] [PubMed] [Google Scholar]
- Bertolotti A, Zhang Y, Hendershot LM, Harding HP, Ron D. (2000) Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat Cell Biol 2:326–332. [DOI] [PubMed] [Google Scholar]
- Bhat M, Robichaud N, Hulea L, Sonenberg N, Pelletier J, Topisirovic I. (2015) Targeting the translation machinery in cancer. Nat Rev Drug Discov 14:261–278. [DOI] [PubMed] [Google Scholar]
- Bidinosti M, Ran I, Sanchez-Carbente MR, Martineau Y, Gingras AC, Gkogkas C, Raught B, Bramham CR, Sossin WS, Costa-Mattioli M, et al. (2010) Postnatal deamidation of 4E-BP2 in brain enhances its association with raptor and alters kinetics of excitatory synaptic transmission. Mol Cell 37:797–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bierhaus A, Fleming T, Stoyanov S, Leffler A, Babes A, Neacsu C, Sauer SK, Eberhardt M, Schnölzer M, Lasitschka F, et al. (2012) Methylglyoxal modification of Nav1.8 facilitates nociceptive neuron firing and causes hyperalgesia in diabetic neuropathy [published correction appears in Nat Med (2012) 18:1445]. Nat Med 18:926–933. [DOI] [PubMed] [Google Scholar]
- Biever A, Donlin-Asp PG, Schuman EM. (2019) Local translation in neuronal processes. Curr Opin Neurobiol 57:141–148. [DOI] [PubMed] [Google Scholar]
- Black JA, Nikolajsen L, Kroner K, Jensen TS, Waxman SG. (2008) Multiple sodium channel isoforms and mitogen-activated protein kinases are present in painful human neuromas. Ann Neurol 64:644–653. [DOI] [PubMed] [Google Scholar]
- Boyce M, Bryant KF, Jousse C, Long K, Harding HP, Scheuner D, Kaufman RJ, Ma D, Coen DM, Ron D, et al. (2005) A selective inhibitor of eIF2alpha dephosphorylation protects cells from ER stress. Science 307:935–939. [DOI] [PubMed] [Google Scholar]
- Bretin A, Carrière J, Dalmasso G, Bergougnoux A, B’chir W, Maurin A-C, Müller S, Seibold F, Barnich N, Bruhat A, et al. (2016) Activation of the EIF2AK4-EIF2A/eIF2α-ATF4 pathway triggers autophagy response to Crohn disease-associated adherent-invasive Escherichia coli infection. Autophagy 12:770–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brings S, Fleming T, De Buhr S, Beijer B, Lindner T, Wischnjow A, Kender Z, Peters V, Kopf S, Haberkorn U, et al. (2017) A scavenger peptide prevents methylglyoxal induced pain in mice. Biochim Biophys Acta Mol Basis Dis 1863:654–662. [DOI] [PubMed] [Google Scholar]
- Brown EJ, Albers MW, Shin TB, Ichikawa K, Keith CT, Lane WS, Schreiber SL. (1994) A mammalian protein targeted by G1-arresting rapamycin-receptor complex. Nature 369:756–758. [DOI] [PubMed] [Google Scholar]
- Brush MH, Shenolikar S. (2008) Control of cellular GADD34 levels by the 26S proteasome. Mol Cell Biol 28:6989–7000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budde K, Becker T, Arns W, Sommerer C, Reinke P, Eisenberger U, Kramer S, Fischer W, Gschaidmeier H, Pietruck F, ZEUS Study Investigators (2011) Everolimus-based, calcineurin-inhibitor-free regimen in recipients of de-novo kidney transplants: an open-label, randomised, controlled trial [published correction appears in Lancet (2011) 377:2006; Lancet (2012) 380:1994. Lancet 377:837–847. [DOI] [PubMed] [Google Scholar]
- Burke PJ. (2017) Mitochondria, bioenergetics and apoptosis in cancer. Trends Cancer 3:857–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton MD, Tillu DV, Mazhar K, Mejia GL, Asiedu MN, Inyang K, Hughes T, Lian B, Dussor G, Price TJ. (2017) Pharmacological activation of AMPK inhibits incision-evoked mechanical hypersensitivity and the development of hyperalgesic priming in mice. Neuroscience 359:119–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buxade M, Parra-Palau JL, Proud CG. (2008) The Mnks: MAP kinase-interacting kinases (MAP kinase signal-integrating kinases). Front Biosci 13:5359–5373. [DOI] [PubMed] [Google Scholar]
- Callejo G, Pattison LA, Greenhalgh JC, Chakrabarti S, Andreopoulou E, Hockley JRF, Smith ESJ, Rahman T. (2020) In silico screening of GMQ-like compounds reveals guanabenz and sephin1 as new allosteric modulators of acid-sensing ion channel 3. Biochem Pharmacol 174:113834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron KO, Kurumbail RG. (2016) Recent progress in the identification of adenosine monophosphate-activated protein kinase (AMPK) activators. Bioorg Med Chem Lett 26:5139–5148. [DOI] [PubMed] [Google Scholar]
- Camici M, Garcia-Gil M, Tozzi MG. (2018) The inside story of adenosine. Int J Mol Sci 19:784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell JN, Meyer RA. (2006) Mechanisms of neuropathic pain. Neuron 52:77–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao X, Dang L, Zheng X, Lu Y, Lu Y, Ji R, Zhang T, Ruan X, Zhi J, Hou X, et al. (2019) Targeting super-enhancer-driven oncogenic transcription by CDK7 inhibition in anaplastic thyroid carcinoma. Thyroid 29:809–823. [DOI] [PubMed] [Google Scholar]
- Cargnello M, Roux PP. (2011) Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol Mol Biol Rev 75:50–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carracedo A, Ma L, Teruya-Feldstein J, Rojo F, Salmena L, Alimonti A, Egia A, Sasaki AT, Thomas G, Kozma SC, et al. (2008) Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J Clin Invest 118:3065–3074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrara M, Sigurdardottir A, Bertolotti A. (2017) Decoding the selectivity of eIF2α holophosphatases and PPP1R15A inhibitors. Nat Struct Mol Biol 24:708–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrasquillo Y, Gereau RW., IV (2007) Activation of the extracellular signal-regulated kinase in the amygdala modulates pain perception. J Neurosci 27:1543–1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan JY, Luzuriaga J, Maxwell EL, West PK, Bensellam M, Laybutt DR. (2015) The balance between adaptive and apoptotic unfolded protein responses regulates β-cell death under ER stress conditions through XBP1, CHOP and JNK. Mol Cell Endocrinol 413:189–201. [DOI] [PubMed] [Google Scholar]
- Chauhan AS, Liu X, Jing J, Lee H, Yadav RK, Liu J, Zhou Y, Gan B. (2019) STIM2 interacts with AMPK and regulates calcium-induced AMPK activation. FASEB J 33:2957–2970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chauvin C, Koka V, Nouschi A, Mieulet V, Hoareau-Aveilla C, Dreazen A, Cagnard N, Carpentier W, Kiss T, Meyuhas O, et al. (2014) Ribosomal protein S6 kinase activity controls the ribosome biogenesis transcriptional program. Oncogene 33:474–483. [DOI] [PubMed] [Google Scholar]
- Chen HM, Wang L, D’Mello SR. (2008) A chemical compound commonly used to inhibit PKR, 8-(imidazol-4-ylmethylene)-6H-azolidino[5,4-g] benzothiazol-7-one, protects neurons by inhibiting cyclin-dependent kinase. Eur J Neurosci 28:2003–2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Podojil JR, Kunjamma RB, Jones J, Weiner M, Lin W, Miller SD, Popko B. (2019) Sephin1, which prolongs the integrated stress response, is a promising therapeutic for multiple sclerosis. Brain 142:344–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng SW, Fryer LG, Carling D, Shepherd PR. (2004) Thr2446 is a novel mammalian target of rapamycin (mTOR) phosphorylation site regulated by nutrient status. J Biol Chem 279:15719–15722. [DOI] [PubMed] [Google Scholar]
- Choy MS, Yusoff P, Lee IC, Newton JC, Goh CW, Page R, Shenolikar S, Peti W. (2015) Structural and functional analysis of the GADD34:PP1 eIF2α phosphatase. Cell Rep 11:1885–1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chresta CM, Davies BR, Hickson I, Harding T, Cosulich S, Critchlow SE, Vincent JP, Ellston R, Jones D, Sini P, et al. (2010) AZD8055 is a potent, selective, and orally bioavailable ATP-competitive mammalian target of rapamycin kinase inhibitor with in vitro and in vivo antitumor activity. Cancer Res 70:288–298. [DOI] [PubMed] [Google Scholar]
- Clayton BLL, Popko B. (2016) Endoplasmic reticulum stress and the unfolded protein response in disorders of myelinating glia. Brain Res 1648 (Pt B):594–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa-Mattioli M, Walter P. (2020) The integrated stress response: from mechanism to disease. Science 368:eaat5314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Court FA, Hendriks WT, MacGillavry HD, Alvarez J, van Minnen J. (2008) Schwann cell to axon transfer of ribosomes: toward a novel understanding of the role of glia in the nervous system. J Neurosci 28:11024–11029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Court FA, Midha R, Cisterna BA, Grochmal J, Shakhbazau A, Hendriks WT, Van Minnen J. (2011) Morphological evidence for a transport of ribosomes from Schwann cells to regenerating axons. Glia 59:1529–1539. [DOI] [PubMed] [Google Scholar]
- Crespillo-Casado A, Chambers JE, Fischer PM, Marciniak SJ, Ron D. (2017) PPP1R15A-mediated dephosphorylation of eIF2α is unaffected by Sephin1 or Guanabenz. eLife 6:e26109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crespillo-Casado A, Claes Z, Choy MS, Peti W, Bollen M, Ron D. (2018) A Sephin1-insensitive tripartite holophosphatase dephosphorylates translation initiation factor 2α. J Biol Chem 293:7766–7776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crook RJ, Dickson K, Hanlon RT, Walters ET. (2014) Nociceptive sensitization reduces predation risk. Curr Biol 24:1121–1125. [DOI] [PubMed] [Google Scholar]
- Dahlhamer J, Lucas J, Zelaya C, Nahin R, Mackey S, DeBar L, Kerns R, Von Korff M, Porter L, Helmick C. (2018) Prevalence of chronic pain and high-impact chronic pain among adults - United States, 2016. MMWR Morb Mortal Wkly Rep 67:1001–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai RP, Li CQ, Zhang JW, Li F, Shi XD, Zhang JY, Zhou XF. (2011) Biphasic activation of extracellular signal-regulated kinase in anterior cingulate cortex distinctly regulates the development of pain-related anxiety and mechanical hypersensitivity in rats after incision. Anesthesiology 115:604–613. [DOI] [PubMed] [Google Scholar]
- Das I, Krzyzosiak A, Schneider K, Wrabetz L, D’Antonio M, Barry N, Sigurdardottir A, Bertolotti A. (2015) Preventing proteostasis diseases by selective inhibition of a phosphatase regulatory subunit. Science 348:239–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das V, Kroin JS, Moric M, McCarthy RJ, Buvanendran A. (2019) AMP-activated protein kinase (AMPK) activator drugs reduce mechanical allodynia in a mouse model of low back pain. Reg Anesth Pain Med DOI: 10.1136/rapm-2019-100839 [published ahead of print]. [DOI] [PubMed] [Google Scholar]
- Davies SP, Hawley SA, Woods A, Carling D, Haystead TA, Hardie DG. (1994) Purification of the AMP-activated protein kinase on ATP-gamma-sepharose and analysis of its subunit structure. Eur J Biochem 223:351–357. [DOI] [PubMed] [Google Scholar]
- Deng Z, Luo P, Lai W, Song T, Peng J, Wei HK. (2017) Myostatin inhibits eEF2K-eEF2 by regulating AMPK to suppress protein synthesis. Biochem Biophys Res Commun 494:278–284. [DOI] [PubMed] [Google Scholar]
- Dever TE, Green R. (2012) The elongation, termination, and recycling phases of translation in eukaryotes. Cold Spring Harb Perspect Biol 4:a013706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devi L, Ohno M. (2013) Deletion of the eIF2α Kinase GCN2 fails to rescue the memory decline associated with Alzheimer’s disease. PLoS One 8:e77335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devor M. (1999) Unexplained peculiarities of the dorsal root ganglion. Pain 82:S27–S35. [DOI] [PubMed] [Google Scholar]
- Devor M. (2006) Sodium channels and mechanisms of neuropathic pain. J Pain 7 (1 Suppl 1):S3–S12. [DOI] [PubMed] [Google Scholar]
- Dreas A, Mikulski M, Milik M, Fabritius CH, Brzózka K, Rzymski T. (2017) Mitogen-activated protein kinase (MAPK) interacting kinases 1 and 2 (MNK1 and MNK2) as targets for cancer therapy: recent progress in the development of MNK inhibitors. Curr Med Chem 24:3025–3053. [DOI] [PubMed] [Google Scholar]
- Droz B, Leblond CP. (1963) Axonal migration of proteins in the central nervous system and peripheral nerves as shown by radioautography. J Comp Neurol 121:325–346. [DOI] [PubMed] [Google Scholar]
- Duan Z, Li J, Pang X, Wang H, Su Z. (2018) Blocking mammalian target of rapamycin (mTOR) alleviates neuropathic pain induced by chemotherapeutic bortezomib. Cell Physiol Biochem 48:54–62. [DOI] [PubMed] [Google Scholar]
- El-Fatatry BM, Ibrahim OM, Hussien FZ, Mostafa TM. (2018) Role of metformin in oxaliplatin-induced peripheral neuropathy in patients with stage III colorectal cancer: randomized, controlled study. Int J Colorectal Dis 33:1675–1683. [DOI] [PubMed] [Google Scholar]
- Eyman M, Cefaliello C, Ferrara E, De Stefano R, Lavina ZS, Crispino M, Squillace A, van Minnen J, Kaplan BB, Giuditta A. (2007) Local synthesis of axonal and presynaptic RNA in squid model systems. Eur J Neurosci 25:341–350. [DOI] [PubMed] [Google Scholar]
- Feng D, Biftu T, Romero FA, Kekec A, Dropinski J, Kassick A, Xu S, Kurtz MM, Gollapudi A, Shao Q, et al. (2017) Discovery of MK-8722: a systemic, direct pan-activator of AMP-activated protein kinase. ACS Med Chem Lett 9:39–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari LF, Araldi D, Levine JD. (2015a) Distinct terminal and cell body mechanisms in the nociceptor mediate hyperalgesic priming. J Neurosci 35:6107–6116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari LF, Bogen O, Chu C, Levine JD. (2013) Peripheral administration of translation inhibitors reverses increased hyperalgesia in a model of chronic pain in the rat. J Pain 14:731–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari LF, Bogen O, Reichling DB, Levine JD. (2015b) Accounting for the delay in the transition from acute to chronic pain: axonal and nuclear mechanisms. J Neurosci 35:495–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonseca BD, Smith EM, Lee VH, MacKintosh C, Proud CG. (2007) PRAS40 is a target for mammalian target of rapamycin complex 1 and is required for signaling downstream of this complex. J Biol Chem 282:24514–24524. [DOI] [PubMed] [Google Scholar]
- Foster KG, Acosta-Jaquez HA, Romeo Y, Ekim B, Soliman GA, Carriere A, Roux PP, Ballif BA, Fingar DC. (2010) Regulation of mTOR complex 1 (mTORC1) by raptor Ser863 and multisite phosphorylation. J Biol Chem 285:80–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frias MA, Thoreen CC, Jaffe JD, Schroder W, Sculley T, Carr SA, Sabatini DM. (2006) mSin1 is necessary for Akt/PKB phosphorylation, and its isoforms define three distinct mTORC2s. Curr Biol 16:1865–1870. [DOI] [PubMed] [Google Scholar]
- Furic L, Rong L, Larsson O, Koumakpayi IH, Yoshida K, Brueschke A, Petroulakis E, Robichaud N, Pollak M, Gaboury LA, et al. (2010) eIF4E phosphorylation promotes tumorigenesis and is associated with prostate cancer progression. Proc Natl Acad Sci USA 107:14134–14139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fusade-Boyer M, Dupré G, Bessière P, Khiar S, Quentin-Froignant C, Beck C, Lecollinet S, Rameix-Welti MA, Eléouët JF, Tangy F, et al. (2019) Evaluation of the antiviral activity of sephin1 treatment and its consequences on eIF2α phosphorylation in response to viral infections. Front Immunol 10:134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaskin DJ, Richard P. (2012) The economic costs of pain in the United States. J Pain 13:715–724. [DOI] [PubMed] [Google Scholar]
- Géranton SM, Jiménez-Díaz L, Torsney C, Tochiki KK, Stuart SA, Leith JL, Lumb BM, Hunt SP. (2009) A rapamycin-sensitive signaling pathway is essential for the full expression of persistent pain states. J Neurosci 29:15017–15027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giuditta A, Dettbarn WD, Brzin M. (1968) Protein synthesis in the isolated giant axon of the squid. Proc Natl Acad Sci USA 59:1284–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giuditta A, Menichini E, Perrone Capano C, Langella M, Martin R, Castigli E, Kaplan BB. (1991) Active polysomes in the axoplasm of the squid giant axon. J Neurosci Res 28:18–28. [DOI] [PubMed] [Google Scholar]
- Gkogkas CG, Khoutorsky A, Cao R, Jafarnejad SM, Prager-Khoutorsky M, Giannakas N, Kaminari A, Fragkouli A, Nader K, Price TJ, et al. (2014) Pharmacogenetic inhibition of eIF4E-dependent Mmp9 mRNA translation reverses fragile X syndrome-like phenotypes. Cell Rep 9:1742–1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold MS, Weinreich D, Kim CS, Wang R, Treanor J, Porreca F, Lai J. (2003) Redistribution of Na(V)1.8 in uninjured axons enables neuropathic pain. J Neurosci 23:158–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golovko A, Kojukhov A, Guan BJ, Morpurgo B, Merrick WC, Mazumder B, Hatzoglou M, Komar AA. (2016) The eIF2A knockout mouse. Cell Cycle 15:3115–3120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordiyenko Y, Llácer JL, Ramakrishnan V. (2019) Structural basis for the inhibition of translation through eIF2α phosphorylation. Nat Commun 10:2640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guertin DA, Stevens DM, Thoreen CC, Burds AA, Kalaany NY, Moffat J, Brown M, Fitzgerald KJ, Sabatini DM. (2006) Ablation in mice of the mTORC components raptor, rictor, or mLST8 reveals that mTORC2 is required for signaling to Akt-FOXO and PKCalpha, but not S6K1. Dev Cell 11:859–871. [DOI] [PubMed] [Google Scholar]
- Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, Turk BE, Shaw RJ. (2008) AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell 30:214–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halliday M, Radford H, Zents KAM, Molloy C, Moreno JA, Verity NC, Smith E, Ortori CA, Barrett DA, Bushell M, et al. (2017) Repurposed drugs targeting eIF2α-P-mediated translational repression prevent neurodegeneration in mice. Brain 140:1768–1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hameed S. (2019) Nav1.7 and Nav1.8: role in the pathophysiology of pain. Mol Pain 15:1744806919858801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J, Back SH, Hur J, Lin YH, Gildersleeve R, Shan J, Yuan CL, Krokowski D, Wang S, Hatzoglou M, et al. (2013) ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat Cell Biol 15:481–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Y, Jiang C, Tang J, Wang C, Wu P, Zhang G, Liu W, Jamangulova N, Wu X, Song X. (2014) Resveratrol reduces morphine tolerance by inhibiting microglial activation via AMPK signalling. Eur J Pain 18:1458–1470. [DOI] [PubMed] [Google Scholar]
- Hara K, Maruki Y, Long X, Yoshino K, Oshiro N, Hidayat S, Tokunaga C, Avruch J, Yonezawa K. (2002) Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell 110:177–189. [DOI] [PubMed] [Google Scholar]
- Hardie DG. (2014) AMPK--sensing energy while talking to other signaling pathways. Cell Metab 20:939–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardie DG. (2015) AMPK: positive and negative regulation, and its role in whole-body energy homeostasis. Curr Opin Cell Biol 33:1–7. [DOI] [PubMed] [Google Scholar]
- Hardie DG, Ross FA, Hawley SA. (2012) AMP-activated protein kinase: a target for drugs both ancient and modern. Chem Biol 19:1222–1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding HP, Novoa I, Zhang Y, Zeng H, Wek R, Schapira M, Ron D. (2000) Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol Cell 6:1099–1108. [DOI] [PubMed] [Google Scholar]
- Haroutounian S, Ford AL, Frey K, Nikolajsen L, Finnerup NB, Neiner A, Kharasch ED, Karlsson P, Bottros MM. (2018) How central is central poststroke pain? The role of afferent input in poststroke neuropathic pain: a prospective, open-label pilot study. Pain 159:1317–1324. [DOI] [PubMed] [Google Scholar]
- Haroutounian S, Nikolajsen L, Bendtsen TF, Finnerup NB, Kristensen AD, Hasselstrøm JB, Jensen TS. (2014) Primary afferent input critical for maintaining spontaneous pain in peripheral neuropathy. Pain 155:1272–1279. [DOI] [PubMed] [Google Scholar]
- Heitman J, Movva NR, Hall MN. (1991) Targets for cell cycle arrest by the immunosuppressant rapamycin in yeast. Science 253:905–909. [DOI] [PubMed] [Google Scholar]
- Hershey JWB, Sonenberg N, Mathews MB. (2019) Principles of translational control. Cold Spring Harb Perspect Biol 11:a032607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetz C, Axten JM, Patterson JB. (2019) Pharmacological targeting of the unfolded protein response for disease intervention. Nat Chem Biol 15:764–775. [DOI] [PubMed] [Google Scholar]
- Hetz C, Chevet E, Harding HP. (2013) Targeting the unfolded protein response in disease. Nat Rev Drug Discov 12:703–719. [DOI] [PubMed] [Google Scholar]
- Hirai T, Mulpuri Y, Cheng Y, Xia Z, Li W, Ruangsri S, Spigelman I, Nishimura I. (2017) Aberrant plasticity of peripheral sensory axons in a painful neuropathy. Sci Rep 7:3407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes B, Brogden RN, Heel RC, Speight TM, Avery GS. (1983) Guanabenz. A review of its pharmacodynamic properties and therapeutic efficacy in hypertension. Drugs 26:212–229. [DOI] [PubMed] [Google Scholar]
- Holz MK, Ballif BA, Gygi SP, Blenis J. (2005) mTOR and S6K1 mediate assembly of the translation preinitiation complex through dynamic protein interchange and ordered phosphorylation events. Cell 123:569–580. [DOI] [PubMed] [Google Scholar]
- Hong-Brown LQ, Brown CR, Kazi AA, Huber DS, Pruznak AM, Lang CH. (2010) Alcohol and PRAS40 knockdown decrease mTOR activity and protein synthesis via AMPK signaling and changes in mTORC1 interaction. J Cell Biochem 109:1172–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoppe S, Bierhoff H, Cado I, Weber A, Tiebe M, Grummt I, Voit R. (2009) AMP-activated protein kinase adapts rRNA synthesis to cellular energy supply. Proc Natl Acad Sci USA 106:17781–17786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou J, Lam F, Proud C, Wang S. (2012) Targeting Mnks for cancer therapy. Oncotarget 3:118–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh MC, Peng HY, Ho YC, Lai CY, Cheng JK, Chen GD, Lin TB. (2019) Transcription repressor Hes1 contributes to neuropathic pain development by modifying CDK9/RNAPII-dependent spinal mGluR5 transcription. Int J Mol Sci 20:4177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Manning BD. (2008) The TSC1-TSC2 complex: a molecular switchboard controlling cell growth. Biochem J 412:179–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbard JM, Patel MR, Bekaii-Saab TS, Falchook GS, Freilich BL, Dasari A, Knisely BT, Anderson M, Chiang GG, Webster KR, et al. (2019) A phase II, open label, randomized, noncomparative study of eFT508 (tomivosertib) alone or in combination with avelumab in subjects with relapsed/refractory microsatellite stable colorectal cancer (MSS CRC). J Clin Oncol 37:e14145. [Google Scholar]
- Igarashi J, Sasaki T, Kobayashi N, Yoshioka S, Matsushita M, Shimizu T. (2011) Autophosphorylation of heme-regulated eukaryotic initiation factor 2α kinase and the role of the modification in catalysis. FEBS J 278:918–928. [DOI] [PubMed] [Google Scholar]
- Inceoglu B, Bettaieb A, Trindade da Silva CA, Lee KS, Haj FG, Hammock BD. (2015) Endoplasmic reticulum stress in the peripheral nervous system is a significant driver of neuropathic pain. Proc Natl Acad Sci USA 112:9082–9087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoki K, Li Y, Xu T, Guan KL. (2003a) Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev 17:1829–1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoki K, Li Y, Zhu T, Wu J, Guan KL. (2002) TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol 4:648–657. [DOI] [PubMed] [Google Scholar]
- Inoki K, Zhu T, Guan KL. (2003b) TSC2 mediates cellular energy response to control cell growth and survival. Cell 115:577–590. [DOI] [PubMed] [Google Scholar]
- Inyang KE, Burton MD, Szabo-Pardi T, Wentworth E, McDougal TA, Ramirez ED, Pradhan G, Dussor G, Price TJ. (2019a) Indirect AMP-activated protein kinase activators prevent incision-induced hyperalgesia and block hyperalgesic priming, whereas positive allosteric modulators block only priming in mice. J Pharmacol Exp Ther 371:138–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inyang KE, McDougal TA, Ramirez ED, Williams M, Laumet G, Kavelaars A, Heijnen CJ, Burton M, Dussor G, Price TJ. (2019b) Alleviation of paclitaxel-induced mechanical hypersensitivity and hyperalgesic priming with AMPK activators in male and female mice. Neurobiol Pain 6:100037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inyang KE, Szabo-Pardi T, Wentworth E, McDougal TA, Dussor G, Burton MD, Price TJ. (2019c) The antidiabetic drug metformin prevents and reverses neuropathic pain and spinal cord microglial activation in male but not female mice. Pharmacol Res 139:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacinto E, Loewith R, Schmidt A, Lin S, Rüegg MA, Hall A, Hall MN. (2004) Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat Cell Biol 6:1122–1128. [DOI] [PubMed] [Google Scholar]
- Jaud M, Philippe C, Di Bella D, Tang W, Pyronnet S, Laurell H, Mazzolini L, Rouault-Pierre K, Touriol C. (2020) Translational regulations in response to endoplasmic reticulum stress in cancers. Cells 9:540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon SM. (2016) Regulation and function of AMPK in physiology and diseases. Exp Mol Med 48:e245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jhanwar-Uniyal M, Wainwright JV, Mohan AL, Tobias ME, Murali R, Gandhi CD, Schmidt MH. (2019) Diverse signaling mechanisms of mTOR complexes: mTORC1 and mTORC2 in forming a formidable relationship. Adv Biol Regul 72:51–62. [DOI] [PubMed] [Google Scholar]
- Jiménez-Díaz L, Géranton SM, Passmore GM, Leith JL, Fisher AS, Berliocchi L, Sivasubramaniam AK, Sheasby A, Lumb BM, Hunt SP. (2008) Local translation in primary afferent fibers regulates nociception. PLoS One 3:e1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin X, Merrett J, Tong S, Flower B, Xie J, Yu R, Tian S, Gao L, Zhao J, Wang X, et al. (2019) Design, synthesis and activity of Mnk1 and Mnk2 selective inhibitors containing thieno[2,3-d]pyrimidine scaffold. Eur J Med Chem 162:735–751. [DOI] [PubMed] [Google Scholar]
- Jousse C, Bruhat A, Carraro V, Urano F, Ferrara M, Ron D, Fafournoux P. (2001) Inhibition of CHOP translation by a peptide encoded by an open reading frame localized in the chop 5’UTR. Nucleic Acids Res 29:4341–4351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Julien LA, Carriere A, Moreau J, Roux PP. (2010) mTORC1-activated S6K1 phosphorylates Rictor on threonine 1135 and regulates mTORC2 signaling. Mol Cell Biol 30:908–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakumoto K, Ikeda J, Okada M, Morii E, Oneyama C. (2015) mLST8 promotes mTOR-mediated tumor progression. PLoS One 10:e0119015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandasamy R, Price TJ. (2015) The pharmacology of nociceptor priming. Handb Exp Pharmacol 227:15–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawasome H, Papst P, Webb S, Keller GM, Johnson GL, Gelfand EW, Terada N. (1998) Targeted disruption of p70(s6k) defines its role in protein synthesis and rapamycin sensitivity. Proc Natl Acad Sci USA 95:5033–5038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke R, Xu Q, Li C, Luo L, Huang D. (2018) Mechanisms of AMPK in the maintenance of ATP balance during energy metabolism. Cell Biol Int 42:384–392. [DOI] [PubMed] [Google Scholar]
- Kearse MG, Wilusz JE. (2017) Non-AUG translation: a new start for protein synthesis in eukaryotes. Genes Dev 31:1717–1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr BJ, Bradbury EJ, Bennett DL, Trivedi PM, Dassan P, French J, Shelton DB, McMahon SB, Thompson SW. (1999) Brain-derived neurotrophic factor modulates nociceptive sensory inputs and NMDA-evoked responses in the rat spinal cord. J Neurosci 19:5138–5148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan MA, Goss DJ. (2018) Kinetic analyses of phosphorylated and non-phosphorylated eIFiso4E binding to mRNA cap analogues. Int J Biol Macromol 106:387–395. [DOI] [PubMed] [Google Scholar]
- Khoutorsky A, Price TJ. (2018) Translational control mechanisms in persistent pain. Trends Neurosci 41:100–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoutorsky A, Sorge RE, Prager-Khoutorsky M, Pawlowski SA, Longo G, Jafarnejad SM, Tahmasebi S, Martin LJ, Pitcher MH, Gkogkas CG, et al. (2016) eIF2α phosphorylation controls thermal nociception. Proc Natl Acad Sci USA 113:11949–11954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiałka M, Milewicz T, Sztefko K, Rogatko I, Majewska R. (2016) Metformin increases pressure pain threshold in lean women with polycystic ovary syndrome. Drug Des Devel Ther 10:2483–2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiebler MA, Bassell GJ. (2006) Neuronal RNA granules: movers and makers. Neuron 51:685–690. [DOI] [PubMed] [Google Scholar]
- Kim E, Kim JH, Seo K, Hong KY, An SWA, Kwon J, Lee SV, Jang SK. (2018) eIF2A, an initiator tRNA carrier refractory to eIF2α kinases, functions synergistically with eIF5B. Cell Mol Life Sci 75:4287–4300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Yang G, Kim Y, Kim J, Ha J. (2016) AMPK activators: mechanisms of action and physiological activities. Exp Mol Med 48:e224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JH, Richter JD. (2007) RINGO/cdk1 and CPEB mediate poly(A) tail stabilization and translational regulation by ePAB. Genes Dev 21:2571–2579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K, Choi S, Cha M, Lee BH. (2020) Effects of mTOR inhibitors on neuropathic pain revealed by optical imaging of the insular cortex in rats. Brain Res 1733:146720. [DOI] [PubMed] [Google Scholar]
- Knauf U, Tschopp C, Gram H. (2001) Negative regulation of protein translation by mitogen-activated protein kinase-interacting kinases 1 and 2. Mol Cell Biol 21:5500–5511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch SC, Acton D, Goulding M. (2018) Spinal circuits for touch, pain, and itch. Annu Rev Physiol 80:189–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koenig E, Martin R, Titmus M, Sotelo-Silveira JR. (2000) Cryptic peripheral ribosomal domains distributed intermittently along mammalian myelinated axons. J Neurosci 20:8390–8400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komar AA, Merrick WC. (2020) A retrospective on eIF2A-and not the alpha subunit of eIF2. Int J Mol Sci 21:2054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konicek BW, Stephens JR, McNulty AM, Robichaud N, Peery RB, Dumstorf CA, Dowless MS, Iversen PW, Parsons S, Ellis KE, et al. (2011) Therapeutic inhibition of MAP kinase interacting kinase blocks eukaryotic initiation factor 4E phosphorylation and suppresses outgrowth of experimental lung metastases. Cancer Res 71:1849–1857. [DOI] [PubMed] [Google Scholar]
- Koromilas AE, Lazaris-Karatzas A, Sonenberg N. (1992) mRNAs containing extensive secondary structure in their 5′ non-coding region translate efficiently in cells overexpressing initiation factor eIF-4E. EMBO J 11:4153–4158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozak M. (1999) Initiation of translation in prokaryotes and eukaryotes. Gene 234:187–208. [DOI] [PubMed] [Google Scholar]
- Kwon M, Han J, Kim UJ, Cha M, Um SW, Bai SJ, Hong SK, Lee BH. (2017a) Inhibition of mammalian target of rapamycin (mTOR) signaling in the insular cortex alleviates neuropathic pain after peripheral nerve injury. Front Mol Neurosci 10:79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon OS, An S, Kim E, Yu J, Hong KY, Lee JS, Jang SK. (2017b) An mRNA-specific tRNAi carrier eIF2A plays a pivotal role in cell proliferation under stress conditions: stress-resistant translation of c-Src mRNA is mediated by eIF2A. Nucleic Acids Res 45:296–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai J, Porreca F, Hunter JC, Gold MS. (2004) Voltage-gated sodium channels and hyperalgesia. Annu Rev Pharmacol Toxicol 44:371–397. [DOI] [PubMed] [Google Scholar]
- Lebeau J, Saunders JM, Moraes VWR, Madhavan A, Madrazo N, Anthony MC, Wiseman RL. (2018) The PERK arm of the unfolded protein response regulates mitochondrial morphology during acute endoplasmic reticulum stress. Cell Rep 22:2827–2836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Rio DC. (2015) Mechanisms and regulation of alternative pre-mRNA splicing. Annu Rev Biochem 84:291–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehman SL, Ryeom S, Koumenis C. (2015) Signaling through alternative Integrated Stress Response pathways compensates for GCN2 loss in a mouse model of soft tissue sarcoma. Sci Rep 5:11781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Ding X, Terkeltaub R, Lin H, Zhang Y, Zhou B, He K, Li K, Liu Z, Wei J, et al. (2020a) Exploration of metformin as novel therapy for osteoarthritis: preventing cartilage degeneration and reducing pain behavior. Arthritis Res Ther 22:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Zhang B, Liu WX, Lu K, Pan H, Wang T, Oh CD, Yi D, Huang J, Zhao L, et al. (2020b) Metformin limits osteoarthritis development and progression through activation of AMPK signalling. Ann Rheum Dis 79:635–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang L, Tao B, Fan L, Yaster M, Zhang Y, Tao YX. (2013) mTOR and its downstream pathway are activated in the dorsal root ganglion and spinal cord after peripheral inflammation, but not after nerve injury. Brain Res 1513:17–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisi L, Aceto P, Navarra P, Dello Russo C. (2015) mTOR kinase: a possible pharmacological target in the management of chronic pain. BioMed Res Int 2015:394257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu CC, Zhang XS, Ruan YT, Huang ZX, Zhang SB, Liu M, Luo HJ, Wu SL, Ma C. (2017) Accumulation of methylglyoxal increases the advanced glycation end-product levels in DRG and contributes to lumbar disk herniation-induced persistent pain. J Neurophysiol 118:1321–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Du J, Yang Y, Wang Y. (2015) Phosphorylation of TRPV1 by cyclin-dependent kinase 5 promotes TRPV1 surface localization, leading to inflammatory thermal hyperalgesia. Exp Neurol 273:253–262. [DOI] [PubMed] [Google Scholar]
- López-Leal R, Alvarez J, Court FA. (2016) Origin of axonal proteins: is the axon-schwann cell unit a functional syncytium? Cytoskeleton (Hoboken) 73:629–639. [DOI] [PubMed] [Google Scholar]
- Lopez-Verrilli MA, Picou F, Court FA. (2013) Schwann cell-derived exosomes enhance axonal regeneration in the peripheral nervous system. Glia 61:1795–1806. [DOI] [PubMed] [Google Scholar]
- Lynch ME, Watson CP. (2006) The pharmacotherapy of chronic pain: a review. Pain Res Manag 11:11–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J, Kavelaars A, Dougherty PM, Heijnen CJ. (2018) Beyond symptomatic relief for chemotherapy-induced peripheral neuropathy: targeting the source. Cancer 124:2289–2298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mader S, Lee H, Pause A, Sonenberg N. (1995) The translation initiation factor eIF-4E binds to a common motif shared by the translation factor eIF-4 gamma and the translational repressors 4E-binding proteins. Mol Cell Biol 15:4990–4997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahameed M, Wilhelm T, Darawshi O, Obiedat A, Tommy WS, Chintha C, Schubert T, Samali A, Chevet E, Eriksson LA, et al. (2019) The unfolded protein response modulators GSK2606414 and KIRA6 are potent KIT inhibitors. Cell Death Dis 10:300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maixner DW, Yan X, Gao M, Yadav R, Weng HR. (2015) Adenosine monophosphate-activated protein kinase regulates interleukin-1β expression and glial glutamate transporter function in rodents with neuropathic pain. Anesthesiology 122:1401–1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannion RJ, Costigan M, Decosterd I, Amaya F, Ma QP, Holstege JC, Ji RR, Acheson A, Lindsay RM, Wilkinson GA, et al. (1999) Neurotrophins: peripherally and centrally acting modulators of tactile stimulus-induced inflammatory pain hypersensitivity. Proc Natl Acad Sci USA 96:9385–9390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao Y, Wang C, Tian X, Huang Y, Zhang Y, Wu H, Yang S, Xu K, Liu Y, Zhang W, et al. (2020) Endoplasmic reticulum stress contributes to nociception via neuroinflammation in a murine bone cancer pain model. Anesthesiology 132:357–372. [DOI] [PubMed] [Google Scholar]
- Mao-Ying QL, Kavelaars A, Krukowski K, Huo XJ, Zhou W, Price TJ, Cleeland C, Heijnen CJ. (2014) The anti-diabetic drug metformin protects against chemotherapy-induced peripheral neuropathy in a mouse model. PLoS One 9:e100701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin KC, Barad M, Kandel ER. (2000) Local protein synthesis and its role in synapse-specific plasticity. Curr Opin Neurobiol 10:587–592. [DOI] [PubMed] [Google Scholar]
- Martin LJ, Smith SB, Khoutorsky A, Magnussen CA, Samoshkin A, Sorge RE, Cho C, Yosefpour N, Sivaselvachandran S, Tohyama S, et al. (2017) Epiregulin and EGFR interactions are involved in pain processing. J Clin Invest 127:3353–3366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martineau Y, Derry MC, Wang X, Yanagiya A, Berlanga JJ, Shyu AB, Imataka H, Gehring K, Sonenberg N. (2008) Poly(A)-binding protein-interacting protein 1 binds to eukaryotic translation initiation factor 3 to stimulate translation. Mol Cell Biol 28:6658–6667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martineau Y, Wang X, Alain T, Petroulakis E, Shahbazian D, Fabre B, Bousquet-Dubouch MP, Monsarrat B, Pyronnet S, Sonenberg N. (2014) Control of Paip1-eukayrotic translation initiation factor 3 interaction by amino acids through S6 kinase. Mol Cell Biol 34:1046–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez G, Khatiwada S, Costa-Mattioli M, Hetz C. (2018) ER proteostasis control of neuronal physiology and synaptic function. Trends Neurosci 41:610–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massard C, Fizazi K, Gross-Goupil M, Escudier B. (2010) Reflex sympathetic dystrophy in patients with metastatic renal cell carcinoma treated with everolimus. Invest New Drugs 28:879–881. [DOI] [PubMed] [Google Scholar]
- Mathur C, Johnson KR, Tong BA, Miranda P, Srikumar D, Basilio D, Latorre R, Bezanilla F, Holmgren M. (2018) Demonstration of ion channel synthesis by isolated squid giant axon provides functional evidence for localized axonal membrane protein translation. Sci Rep 8:2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui Y, Yasumatsu I, Yoshida KI, Iimura S, Ikeno Y, Nawano T, Fukano H, Ubukata O, Hanzawa H, Tanzawa F, et al. (2018) A novel inhibitor stabilizes the inactive conformation of MAPK-interacting kinase 1. Acta Crystallogr F Struct Biol Commun 74:156–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormack FX, Inoue Y, Moss J, Singer LG, Strange C, Nakata K, Barker AF, Chapman JT, Brantly ML, Stocks JM, et al. National Institutes of Health Rare Lung Diseases Consortium; MILES Trial Group (2011) Efficacy and safety of sirolimus in lymphangioleiomyomatosis. N Engl J Med 364:1595–1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKendrick L, Morley SJ, Pain VM, Jagus R, Joshi B. (2001) Phosphorylation of eukaryotic initiation factor 4E (eIF4E) at Ser209 is not required for protein synthesis in vitro and in vivo. Eur J Biochem 268:5375–5385. [DOI] [PubMed] [Google Scholar]
- McQuiston A, Diehl JA. (2017) Recent insights into PERK-dependent signaling from the stressed endoplasmic reticulum. F1000 Res 6:1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meacham K, Shepherd A, Mohapatra DP, Haroutounian S. (2017) Neuropathic pain: central vs. peripheral mechanisms. Curr Pain Headache Rep 21:28. [DOI] [PubMed] [Google Scholar]
- Meares GP, Liu Y, Rajbhandari R, Qin H, Nozell SE, Mobley JA, Corbett JA, Benveniste EN. (2014) PERK-dependent activation of JAK1 and STAT3 contributes to endoplasmic reticulum stress-induced inflammation. Mol Cell Biol 34:3911–3925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Megat S, Price TJ. (2018) Therapeutic opportunities for pain medicines via targeting of specific translation signaling mechanisms. Neurobiol Pain 4:8–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Megat S, Ray PR, Moy JK, Lou TF, Barragán-Iglesias P, Li Y, Pradhan G, Wanghzou A, Ahmad A, Burton MD, et al. (2019a) Nociceptor translational profiling reveals the ragulator-Rag GTPase complex as a critical generator of neuropathic pain. J Neurosci 39:393–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Megat S, Ray PR, Tavares-Ferreira D, Moy JK, Sankaranarayanan I, Wanghzou A, Fang Lou T, Barragan-Iglesias P, Campbell ZT, Dussor G, et al. (2019b) Differences between dorsal root and trigeminal ganglion nociceptors in mice revealed by translational profiling. J Neurosci 39:6829–6847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta R, Wong L, O’Brien PJ. (2009) Cytoprotective mechanisms of carbonyl scavenging drugs in isolated rat hepatocytes. Chem Biol Interact 178:317–323. [DOI] [PubMed] [Google Scholar]
- Mejia GL, Asiedu MN, Hitoshi Y, Dussor G, Price TJ. (2016) The potent, indirect adenosine monophosphate- activated protein kinase activator R419 attenuates mitogen-activated protein kinase signaling, inhibits nociceptor excitability, and reduces pain hypersensitivity in mice. Pain Rep 1:e562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melemedjian OK, Asiedu MN, Tillu DV, Peebles KA, Yan J, Ertz N, Dussor GO, Price TJ. (2010) IL-6- and NGF-induced rapid control of protein synthesis and nociceptive plasticity via convergent signaling to the eIF4F complex. J Neurosci 30:15113–15123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melemedjian OK, Asiedu MN, Tillu DV, Sanoja R, Yan J, Lark A, Khoutorsky A, Johnson J, Peebles KA, Lepow T, et al. (2011) Targeting adenosine monophosphate-activated protein kinase (AMPK) in preclinical models reveals a potential mechanism for the treatment of neuropathic pain. Mol Pain 7:70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melemedjian OK, Khoutorsky A, Sorge RE, Yan J, Asiedu MN, Valdez A, Ghosh S, Dussor G, Mogil JS, Sonenberg N, et al. (2013) mTORC1 inhibition induces pain via IRS-1-dependent feedback activation of ERK. Pain 154:1080–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melemedjian OK, Tillu DV, Moy JK, Asiedu MN, Mandell EK, Ghosh S, Dussor G, Price TJ. (2014) Local translation and retrograde axonal transport of CREB regulates IL-6-induced nociceptive plasticity. Mol Pain 10:45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra RK, Clutter MR, Blyth GT, Kosciuczuk EM, Blackburn AZ, Beauchamp EM, Schiltz GE, Platanias LC. (2019) Discovery of novel Mnk inhibitors using mutation-based induced-fit virtual high-throughput screening. Chem Biol Drug Des 94:1813–1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Missig G, Mei L, Vizzard MA, Braas KM, Waschek JA, Ressler KJ, Hammack SE, May V. (2017) Parabrachial pituitary adenylate cyclase-activating polypeptide activation of amygdala endosomal extracellular signal-regulated kinase signaling regulates the emotional component of pain. Biol Psychiatry 81:671–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mody PH, Dos Santos NL, Barron LR, Price TJ, Burton MD. (2020) eIF4E phosphorylation modulates pain and neuroinflammation in the aged. Geroscience DOI: 10.1007/s11357-020-00220-1 [published ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molina MG, Diekmann F, Burgos D, Cabello M, Lopez V, Oppenheimer F, Navarro A, Campistol J. (2008) Sympathetic dystrophy associated with sirolimus therapy. Transplantation 85:290–292. [DOI] [PubMed] [Google Scholar]
- Montes M, Sanford BL, Comiskey DF, Chandler DS. (2019) RNA splicing and disease: animal models to therapies. Trends Genet 35:68–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moy JK, Khoutorsky A, Asiedu MN, Black BJ, Kuhn JL, Barragán-Iglesias P, Megat S, Burton MD, Burgos-Vega CC, Melemedjian OK, et al. (2017) The MNK-eIF4E signaling axis contributes to injury-induced nociceptive plasticity and the development of chronic pain. J Neurosci 37:7481–7499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moy JK, Khoutorsky A, Asiedu MN, Dussor G, Price TJ. (2018a) eIF4E phosphorylation influences Bdnf mRNA translation in mouse dorsal root ganglion neurons. Front Cell Neurosci 12:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moy JK, Kuhn JL, Szabo-Pardi TA, Pradhan G, Price TJ. (2018b) eIF4E phosphorylation regulates ongoing pain, independently of inflammation, and hyperalgesic priming in the mouse CFA model. Neurobiol Pain 4:45–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musa J, Orth MF, Dallmayer M, Baldauf M, Pardo C, Rotblat B, Kirchner T, Leprivier G, Grünewald TG. (2016) Eukaryotic initiation factor 4E-binding protein 1 (4E-BP1): a master regulator of mRNA translation involved in tumorigenesis. Oncogene 35:4675–4688. [DOI] [PubMed] [Google Scholar]
- Myers RW, Guan HP, Ehrhart J, Petrov A, Prahalada S, Tozzo E, Yang X, Kurtz MM, Trujillo M, Gonzalez Trotter D, et al. (2017) Systemic pan-AMPK activator MK-8722 improves glucose homeostasis but induces cardiac hypertrophy. Science 357:507–511. [DOI] [PubMed] [Google Scholar]
- Narasimhan J, Staschke KA, Wek RC. (2004) Dimerization is required for activation of eIF2 kinase Gcn2 in response to diverse environmental stress conditions. J Biol Chem 279:22820–22832. [DOI] [PubMed] [Google Scholar]
- Nascimento EB, Snel M, Guigas B, van der Zon GC, Kriek J, Maassen JA, Jazet IM, Diamant M, Ouwens DM. (2010) Phosphorylation of PRAS40 on Thr246 by PKB/AKT facilitates efficient phosphorylation of Ser183 by mTORC1. Cell Signal 22:961–967. [DOI] [PubMed] [Google Scholar]
- Nguyen MQ, Le Pichon CE, Ryba N. (2019) Stereotyped transcriptomic transformation of somatosensory neurons in response to injury. eLife 8:e49679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholas M, Vlaeyen JWS, Rief W, Barke A, Aziz Q, Benoliel R, Cohen M, Evers S, Giamberardino MA, Goebel A, et al. IASP Taskforce for the Classification of Chronic Pain (2019) The IASP classification of chronic pain for ICD-11: chronic primary pain. Pain 160:28–37. [DOI] [PubMed] [Google Scholar]
- Norsted Gregory E, Codeluppi S, Gregory JA, Steinauer J, Svensson CI. (2010) Mammalian target of rapamycin in spinal cord neurons mediates hypersensitivity induced by peripheral inflammation. Neuroscience 169:1392–1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- North RY, Li Y, Ray P, Rhines LD, Tatsui CE, Rao G, Johansson CA, Zhang H, Kim YH, Zhang B, et al. (2019) Electrophysiological and transcriptomic correlates of neuropathic pain in human dorsal root ganglion neurons. Brain 142:1215–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakhill JS, Chen ZP, Scott JW, Steel R, Castelli LA, Ling N, Macaulay SL, Kemp BE. (2010) β-Subunit myristoylation is the gatekeeper for initiating metabolic stress sensing by AMP-activated protein kinase (AMPK). Proc Natl Acad Sci USA 107:19237–19241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obara I, Tochiki KK, Géranton SM, Carr FB, Lumb BM, Liu Q, Hunt SP. (2011) Systemic inhibition of the mammalian target of rapamycin (mTOR) pathway reduces neuropathic pain in mice. Pain 152:2582–2595. [DOI] [PubMed] [Google Scholar]
- Obreja O, Rukwied R, Nagler L, Schmidt M, Schmelz M, Namer B. (2018) Nerve growth factor locally sensitizes nociceptors in human skin. Pain 159:416–426. [DOI] [PubMed] [Google Scholar]
- Okuse K, Chaplan SR, McMahon SB, Luo ZD, Calcutt NA, Scott BP, Akopian AN, Wood JN. (1997) Regulation of expression of the sensory neuron-specific sodium channel SNS in inflammatory and neuropathic pain. Mol Cell Neurosci 10:196–207. [DOI] [PubMed] [Google Scholar]
- O’Loghlen A, González VM, Piñeiro D, Pérez-Morgado MI, Salinas M, Martín ME. (2004) Identification and molecular characterization of Mnk1b, a splice variant of human MAP kinase-interacting kinase Mnk1. Exp Cell Res 299:343–355. [DOI] [PubMed] [Google Scholar]
- Oshiro N, Takahashi R, Yoshino K, Tanimura K, Nakashima A, Eguchi S, Miyamoto T, Hara K, Takehana K, Avruch J, et al. (2007) The proline-rich Akt substrate of 40 kDa (PRAS40) is a physiological substrate of mammalian target of rapamycin complex 1. J Biol Chem 282:20329–20339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oyarzabal J, Zarich N, Albarran MI, Palacios I, Urbano-Cuadrado M, Mateos G, Reymundo I, Rabal O, Salgado A, Corrionero A, et al. (2010) Discovery of mitogen-activated protein kinase-interacting kinase 1 inhibitors by a comprehensive fragment-oriented virtual screening approach. J Med Chem 53:6618–6628. [DOI] [PubMed] [Google Scholar]
- Pakos-Zebrucka K, Koryga I, Mnich K, Ljujic M, Samali A, Gorman AM. (2016) The integrated stress response. EMBO Rep 17:1374–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palam LR, Baird TD, Wek RC. (2011) Phosphorylation of eIF2 facilitates ribosomal bypass of an inhibitory upstream ORF to enhance CHOP translation. J Biol Chem 286:10939–10949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pallares-Cartes C, Cakan-Akdogan G, Teleman AA. (2012) Tissue-specific coupling between insulin/IGF and TORC1 signaling via PRAS40 in Drosophila. Dev Cell 22:172–182. [DOI] [PubMed] [Google Scholar]
- Pareek TK, Keller J, Kesavapany S, Pant HC, Iadarola MJ, Brady RO, Kulkarni AB. (2006) Cyclin-dependent kinase 5 activity regulates pain signaling. Proc Natl Acad Sci USA 103:791–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pause A, Belsham GJ, Gingras AC, Donzé O, Lin TA, Lawrence JC, Jr., Sonenberg N. (1994) Insulin-dependent stimulation of protein synthesis by phosphorylation of a regulator of 5′-cap function. Nature 371:762–767. [DOI] [PubMed] [Google Scholar]
- Pende M, Um SH, Mieulet V, Sticker M, Goss VL, Mestan J, Mueller M, Fumagalli S, Kozma SC, Thomas G. (2004) S6K1(-/-)/S6K2(-/-) mice exhibit perinatal lethality and rapamycin-sensitive 5′-terminal oligopyrimidine mRNA translation and reveal a mitogen-activated protein kinase-dependent S6 kinase pathway. Mol Cell Biol 24:3112–3124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira AF, Pereira LMS, Silva CMP, Freitas Alves BW, Barbosa JS, Pinto FMM, Pereira AC, Silva KO, Pontes RB, Alencar NMN, et al. (2019) Metformin reduces c-Fos and ATF3 expression in the dorsal root ganglia and protects against oxaliplatin-induced peripheral sensory neuropathy in mice. Neurosci Lett 709:134378. [DOI] [PubMed] [Google Scholar]
- Persson AK, Gasser A, Black JA, Waxman SG. (2011) Nav1.7 accumulates and co-localizes with phosphorylated ERK1/2 within transected axons in early experimental neuromas. Exp Neurol 230:273–279. [DOI] [PubMed] [Google Scholar]
- Pike KG, Malagu K, Hummersone MG, Menear KA, Duggan HM, Gomez S, Martin NM, Ruston L, Pass SL, Pass M. (2013) Optimization of potent and selective dual mTORC1 and mTORC2 inhibitors: the discovery of AZD8055 and AZD2014. Bioorg Med Chem Lett 23:1212–1216. [DOI] [PubMed] [Google Scholar]
- Pinho-Ribeiro FA, Verri WA, Jr., Chiu IM. (2017) Nociceptor sensory neuron-immune interactions in pain and inflammation. Trends Immunol 38:5–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piper M, Holt C. (2004) RNA translation in axons. Annu Rev Cell Dev Biol 20:505–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulin F, Gingras AC, Olsen H, Chevalier S, Sonenberg N. (1998) 4E-BP3, a new member of the eukaryotic initiation factor 4E-binding protein family. J Biol Chem 273:14002–14007. [DOI] [PubMed] [Google Scholar]
- Price TJ, Basbaum AI, Bresnahan J, Chambers JF, De Koninck Y, Edwards RR, Ji RR, Katz J, Kavelaars A, Levine JD, et al. (2018) Transition to chronic pain: opportunities for novel therapeutics. Nat Rev Neurosci 19:383–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price TJ, Géranton SM. (2009) Translating nociceptor sensitivity: the role of axonal protein synthesis in nociceptor physiology. Eur J Neurosci 29:2253–2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price TJ, Gold MS. (2018) From mechanism to cure: renewing the goal to eliminate the disease of pain. Pain Med 19:1525–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price TJ, Rashid MH, Millecamps M, Sanoja R, Entrena JM, Cervero F. (2007) Decreased nociceptive sensitization in mice lacking the fragile X mental retardation protein: role of mGluR1/5 and mTOR. J Neurosci 27:13958–13967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proud CG. (2015) Mnks, eIF4E phosphorylation and cancer. Biochim Biophys Acta 1849:766–773. [DOI] [PubMed] [Google Scholar]
- Pyo CW, Lee SH, Choi SY. (2008) Oxidative stress induces PKR-dependent apoptosis via IFN-gamma activation signaling in Jurkat T cells. Biochem Biophys Res Commun 377:1001–1006. [DOI] [PubMed] [Google Scholar]
- Pyronnet S, Imataka H, Gingras AC, Fukunaga R, Hunter T, Sonenberg N. (1999) Human eukaryotic translation initiation factor 4G (eIF4G) recruits mnk1 to phosphorylate eIF4E. EMBO J 18:270–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabouw HH, Langereis MA, Anand AA, Visser LJ, de Groot RJ, Walter P, van Kuppeveld FJM. (2019) Small molecule ISRIB suppresses the integrated stress response within a defined window of activation. Proc Natl Acad Sci USA 116:2097–2102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray PR, Khan J, Wangzhou A, Tavares-Ferreira D, Akopian AN, Dussor G, Price TJ. (2019) Transcriptome analysis of the human tibial nerve identifies sexually dimorphic expression of genes involved in pain, inflammation, and neuro-immunity. Front Mol Neurosci 12:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reich SH, Sprengeler PA, Chiang GG, Appleman JR, Chen J, Clarine J, Eam B, Ernst JT, Han Q, Goel VK, et al. (2018) Structure-based design of pyridone-aminal eFT508 targeting dysregulated translation by selective mitogen-activated protein kinase interacting kinases 1 and 2 (MNK1/2) inhibition. J Med Chem 61:3516–3540. [DOI] [PubMed] [Google Scholar]
- Reichling DB, Green PG, Levine JD. (2013) The fundamental unit of pain is the cell. Pain 154 (Suppl 1):S2–S9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter JD. (2007) CPEB: a life in translation. Trends Biochem Sci 32:279–285. [DOI] [PubMed] [Google Scholar]
- Rodnina MV, Wintermeyer W. (2009) Recent mechanistic insights into eukaryotic ribosomes. Curr Opin Cell Biol 21:435–443. [DOI] [PubMed] [Google Scholar]
- Rojas-Rivera D, Delvaeye T, Roelandt R, Nerinckx W, Augustyns K, Vandenabeele P, Bertrand MJM. (2017) When PERK inhibitors turn out to be new potent RIPK1 inhibitors: critical issues on the specificity and use of GSK2606414 and GSK2656157. Cell Death Differ 24:1100–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosner M, Hengstschläger M. (2011) Nucleocytoplasmic localization of p70 S6K1, but not of its isoforms p85 and p31, is regulated by TSC2/mTOR. Oncogene 30:4509–4522. [DOI] [PubMed] [Google Scholar]
- Ross FA, Jensen TE, Hardie DG. (2016) Differential regulation by AMP and ADP of AMPK complexes containing different γ subunit isoforms. Biochem J 473:189–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux PP, Topisirovic I. (2018) Signaling pathways involved in the regulation of mRNA translation. Mol Cell Biol 38:e00070-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roychowdhury A, Sharma R, Kumar S. (2010) Recent advances in the discovery of small molecule mTOR inhibitors. Future Med Chem 2:1577–1589. [DOI] [PubMed] [Google Scholar]
- Roy Chowdhury SK, Smith DR, Saleh A, Schapansky J, Marquez A, Gomes S, Akude E, Morrow D, Calcutt NA, Fernyhough P. (2012) Impaired adenosine monophosphate-activated protein kinase signalling in dorsal root ganglia neurons is linked to mitochondrial dysfunction and peripheral neuropathy in diabetes. Brain 135:1751–1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozas P, Lazcano P, Piña R, Cho A, Terse A, Pertusa M, Madrid R, Gonzalez-Billault C, Kulkarni AB, Utreras E. (2016) Targeted overexpression of tumor necrosis factor-α increases cyclin-dependent kinase 5 activity and TRPV1-dependent Ca2+ influx in trigeminal neurons. Pain 157:1346–1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozpedek W, Pytel D, Mucha B, Leszczynska H, Diehl JA, Majsterek I. (2016) The role of the PERK/eIF2α/ATF4/CHOP signaling pathway in tumor progression during endoplasmic reticulum stress. Curr Mol Med 16:533–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruangsri S, Lin A, Mulpuri Y, Lee K, Spigelman I, Nishimura I. (2011) Relationship of axonal voltage-gated sodium channel 1.8 (NaV1.8) mRNA accumulation to sciatic nerve injury-induced painful neuropathy in rats. J Biol Chem 286:39836–39847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sano R, Reed JC. (2013) ER stress-induced cell death mechanisms. Biochim Biophys Acta 1833:3460–3470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarbassov DD, Ali SM, Sengupta S, Sheen JH, Hsu PP, Bagley AF, Markhard AL, Sabatini DM. (2006) Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell 22:159–168. [DOI] [PubMed] [Google Scholar]
- Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. (2005) Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 307:1098–1101. [DOI] [PubMed] [Google Scholar]
- Schenone S, Brullo C, Musumeci F, Radi M, Botta M. (2011) ATP-competitive inhibitors of mTOR: an update. Curr Med Chem 18:2995–3014. [DOI] [PubMed] [Google Scholar]
- Scheper GC, Parra JL, Wilson M, Van Kollenburg B, Vertegaal AC, Han ZG, Proud CG. (2003) The N and C termini of the splice variants of the human mitogen-activated protein kinase-interacting kinase Mnk2 determine activity and localization. Mol Cell Biol 23:5692–5705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheper GC, Proud CG. (2002) Does phosphorylation of the cap-binding protein eIF4E play a role in translation initiation? Eur J Biochem 269:5350–5359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheuner D, Song B, McEwen E, Liu C, Laybutt R, Gillespie P, Saunders T, Bonner-Weir S, Kaufman RJ. (2001) Translational control is required for the unfolded protein response and in vivo glucose homeostasis. Mol Cell 7:1165–1176. [DOI] [PubMed] [Google Scholar]
- Sekine Y, Zyryanova A, Crespillo-Casado A, Fischer PM, Harding HP, Ron D. (2015) Stress responses. Mutations in a translation initiation factor identify the target of a memory-enhancing compound. Science 348:1027–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serra J, Bostock H, Solà R, Aleu J, García E, Cokic B, Navarro X, Quiles C. (2012) Microneurographic identification of spontaneous activity in C-nociceptors in neuropathic pain states in humans and rats. Pain 153:42–55. [DOI] [PubMed] [Google Scholar]
- Seto B. (2012) Rapamycin and mTOR: a serendipitous discovery and implications for breast cancer. Clin Transl Med 1:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahbazian D, Roux PP, Mieulet V, Cohen MS, Raught B, Taunton J, Hershey JW, Blenis J, Pende M, Sonenberg N. (2006) The mTOR/PI3K and MAPK pathways converge on eIF4B to control its phosphorylation and activity. EMBO J 25:2781–2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw RJ, Lamia KA, Vasquez D, Koo SH, Bardeesy N, Depinho RA, Montminy M, Cantley LC. (2005) The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science 310:1642–1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen CH, Yuan P, Perez-Lorenzo R, Zhang Y, Lee SX, Ou Y, Asara JM, Cantley LC, Zheng B. (2013) Phosphorylation of BRAF by AMPK impairs BRAF-KSR1 association and cell proliferation. Mol Cell 52:161–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiers S, Mwirigi J, Pradhan G, Kume M, Black B, Barragan-Iglesias P, Moy JK, Dussor G, Pancrazio JJ, Kroener S, et al. (2020) Reversal of peripheral nerve injury-induced neuropathic pain and cognitive dysfunction via genetic and tomivosertib targeting of MNK. Neuropsychopharmacology 45:524–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiers S, Pradhan G, Mwirigi J, Mejia G, Ahmad A, Kroener S, Price T. (2018) Neuropathic pain creates an enduring prefrontal cortex dysfunction corrected by the type II diabetic drug metformin but not by gabapentin. J Neurosci 38:7337–7350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin HJ, Kim H, Oh S, Lee JG, Kee M, Ko HJ, Kweon MN, Won KJ, Baek SH. (2016) AMPK-SKP2-CARM1 signalling cascade in transcriptional regulation of autophagy. Nature 534:553–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Showkat M, Beigh MA, Andrabi KI. (2014) mTOR signaling in protein translation regulation: implications in cancer genesis and therapeutic interventions. Mol Biol Int 2014:686984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddiqui N, Tempel W, Nedyalkova L, Volpon L, Wernimont AK, Osborne MJ, Park HW, Borden KL. (2012) Structural insights into the allosteric effects of 4EBP1 on the eukaryotic translation initiation factor eIF4E. J Mol Biol 415:781–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidrauski C, Acosta-Alvear D, Khoutorsky A, Vedantham P, Hearn BR, Li H, Gamache K, Gallagher CM, Ang KK, Wilson C, et al. (2013) Pharmacological brake-release of mRNA translation enhances cognitive memory. eLife 2:e00498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidrauski C, Tsai JC, Kampmann M, Hearn BR, Vedantham P, Jaishankar P, Sokabe M, Mendez AS, Newton BW, Tang EL, et al. (2015) Pharmacological dimerization and activation of the exchange factor eIF2B antagonizes the integrated stress response. eLife 4:e07314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikandar S, Minett MS, Millet Q, Santana-Varela S, Lau J, Wood JN, Zhao J. (2018) Brain-derived neurotrophic factor derived from sensory neurons plays a critical role in chronic pain. Brain 141:1028–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AL, Andrews KL, Beckmann H, Bellon SF, Beltran PJ, Booker S, Chen H, Chung YA, D’Angelo ND, Dao J, et al. (2015) Discovery of 1H-pyrazol-3(2H)-ones as potent and selective inhibitors of protein kinase R-like endoplasmic reticulum kinase (PERK). J Med Chem 58:1426–1441. [DOI] [PubMed] [Google Scholar]
- Smith B, Ang D. (2015) Metformin: potential analgesic? Pain Med 16:2256–2260. [DOI] [PubMed] [Google Scholar]
- Sonenberg N, Hinnebusch AG. (2009) Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell 136:731–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starck SR, Tsai JC, Chen K, Shodiya M, Wang L, Yahiro K, Martins-Green M, Shastri N, Walter P. (2016) Translation from the 5′ untranslated region shapes the integrated stress response. Science 351:aad3867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stead RL, Proud CG. (2013) Rapamycin enhances eIF4E phosphorylation by activating MAP kinase-interacting kinase 2a (Mnk2a). FEBS Lett 587:2623–2628. [DOI] [PubMed] [Google Scholar]
- Steward O, Schuman EM. (2003) Compartmentalized synthesis and degradation of proteins in neurons. Neuron 40:347–359. [DOI] [PubMed] [Google Scholar]
- Su X, Yu Y, Zhong Y, Giannopoulou EG, Hu X, Liu H, Cross JR, Rätsch G, Rice CM, Ivashkiv LB. (2015) Interferon-γ regulates cellular metabolism and mRNA translation to potentiate macrophage activation. Nat Immunol 16:838–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suragani RN, Zachariah RS, Velazquez JG, Liu S, Sun CW, Townes TM, Chen JJ. (2012) Heme-regulated eIF2α kinase activated Atf4 signaling pathway in oxidative stress and erythropoiesis. Blood 119:5276–5284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sussman A, Huss K, Chio LC, Heidler S, Shaw M, Ma D, Zhu G, Campbell RM, Park TS, Kulanthaivel P, et al. (2004) Discovery of cercosporamide, a known antifungal natural product, as a selective Pkc1 kinase inhibitor through high-throughput screening. Eukaryot Cell 3:932–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suter M, Riek U, Tuerk R, Schlattner U, Wallimann T, Neumann D. (2006) Dissecting the role of 5′-AMP for allosteric stimulation, activation, and deactivation of AMP-activated protein kinase. J Biol Chem 281:32207–32216. [DOI] [PubMed] [Google Scholar]
- Sutton MA, Schuman EM. (2005) Local translational control in dendrites and its role in long-term synaptic plasticity. J Neurobiol 64:116–131. [DOI] [PubMed] [Google Scholar]
- Svitkin YV, Pause A, Haghighat A, Pyronnet S, Witherell G, Belsham GJ, Sonenberg N. (2001) The requirement for eukaryotic initiation factor 4A (elF4A) in translation is in direct proportion to the degree of mRNA 5′ secondary structure. RNA 7:382–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahmasebi S, Khoutorsky A, Mathews MB, Sonenberg N. (2018) Translation deregulation in human disease. Nat Rev Mol Cell Biol 19:791–807. [DOI] [PubMed] [Google Scholar]
- Tavares MR, Pavan IC, Amaral CL, Meneguello L, Luchessi AD, Simabuco FM. (2015) The S6K protein family in health and disease. Life Sci 131:1–10. [DOI] [PubMed] [Google Scholar]
- Taylor A, Westveld AH, Szkudlinska M, Guruguri P, Annabi E, Patwardhan A, Price TJ, Yassine HN. (2013) The use of metformin is associated with decreased lumbar radiculopathy pain. J Pain Res 6:755–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teneggi V, Novotny-Diermayr V, Lee LH, Yasin M, Yeo P, Ethirajulu K, Gan SBH, Blanchard SE, Nellore R, Umrani DN, et al. (2020) First-in-human, healthy volunteers integrated protocol of ETC-206, an oral Mnk 1/2 kinase inhibitor oncology drug. Clin Transl Sci 13:57–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terenzio M, Koley S, Samra N, Rishal I, Zhao Q, Sahoo PK, Urisman A, Marvaldi L, Oses-Prieto JA, Forester C, et al. (2018) Locally translated mTOR controls axonal local translation in nerve injury. Science 359:1416–1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakor DK, Lin A, Matsuka Y, Meyer EM, Ruangsri S, Nishimura I, Spigelman I. (2009) Increased peripheral nerve excitability and local NaV1.8 mRNA up-regulation in painful neuropathy. Mol Pain 5:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thapa S, Abdelaziz DH, Abdulrahman BA, Schatzl HM. (2020) Sephin1 reduces prion infection in prion-infected cells and animal model. Mol Neurobiol 57:2206–2219. [DOI] [PubMed] [Google Scholar]
- Thompson MK, Gilbert WV. (2017) mRNA length-sensing in eukaryotic translation: reconsidering the “closed loop” and its implications for translational control. Curr Genet 63:613–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson PA, Chen WJ, Appleman J. (2017) inventors, Effector Therapeutics Inc, assignee. Mnk Biomarkers and Uses Therof. U.S. patent US20170191136A1.
- Thompson SW, Bennett DL, Kerr BJ, Bradbury EJ, McMahon SB. (1999) Brain-derived neurotrophic factor is an endogenous modulator of nociceptive responses in the spinal cord. Proc Natl Acad Sci USA 96:7714–7718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoreen CC, Chantranupong L, Keys HR, Wang T, Gray NS, Sabatini DM. (2012) A unifying model for mTORC1-mediated regulation of mRNA translation. Nature 485:109–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornton C, Snowden MA, Carling D. (1998) Identification of a novel AMP-activated protein kinase beta subunit isoform that is highly expressed in skeletal muscle. J Biol Chem 273:12443–12450. [DOI] [PubMed] [Google Scholar]
- Tillu DV, Melemedjian OK, Asiedu MN, Qu N, De Felice M, Dussor G, Price TJ. (2012) Resveratrol engages AMPK to attenuate ERK and mTOR signaling in sensory neurons and inhibits incision-induced acute and chronic pain. Mol Pain 8:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmermans AD, Balteau M, Gélinas R, Renguet E, Ginion A, de Meester C, Sakamoto K, Balligand JL, Bontemps F, Vanoverschelde JL, et al. (2014) A-769662 potentiates the effect of other AMP-activated protein kinase activators on cardiac glucose uptake. Am J Physiol Heart Circ Physiol 306:H1619–H1630. [DOI] [PubMed] [Google Scholar]
- Tsaytler P, Harding HP, Ron D, Bertolotti A. (2011) Selective inhibition of a regulatory subunit of protein phosphatase 1 restores proteostasis. Science 332:91–94. [DOI] [PubMed] [Google Scholar]
- Ueda T, Watanabe-Fukunaga R, Fukuyama H, Nagata S, Fukunaga R. (2004) Mnk2 and Mnk1 are essential for constitutive and inducible phosphorylation of eukaryotic initiation factor 4E but not for cell growth or development. Mol Cell Biol 24:6539–6549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Um SW, Kim MJ, Leem JW, Bai SJ, Lee BH. (2019) Pain-relieving effects of mTOR inhibitor in the anterior cingulate cortex of neuropathic rats. Mol Neurobiol 56:2482–2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uttam S, Wong C, Amorim IS, Jafarnejad SM, Tansley SN, Yang J, Prager-Khoutorsky M, Mogil JS, Gkogkas CG, Khoutorsky A. (2018) Translational profiling of dorsal root ganglia and spinal cord in a mouse model of neuropathic pain. Neurobiol Pain 4:35–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Vliet AR, Giordano F, Gerlo S, Segura I, Van Eygen S, Molenberghs G, Rocha S, Houcine A, Derua R, Verfaillie T, et al. (2017) The ER stress sensor PERK coordinates ER-plasma membrane contact site formation through interaction with filamin-A and F-actin remodeling. Mol Cell 65:885–899.e6. [DOI] [PubMed] [Google Scholar]
- Vaso A, Adahan HM, Gjika A, Zahaj S, Zhurda T, Vyshka G, Devor M. (2014) Peripheral nervous system origin of phantom limb pain. Pain 155:1384–1391. [DOI] [PubMed] [Google Scholar]
- Vattem KM, Staschke KA, Wek RC. (2001) Mechanism of activation of the double-stranded-RNA-dependent protein kinase, PKR: role of dimerization and cellular localization in the stimulation of PKR phosphorylation of eukaryotic initiation factor-2 (eIF2). Eur J Biochem 268:3674–3684. [DOI] [PubMed] [Google Scholar]
- Vattem KM, Wek RC. (2004) Reinitiation involving upstream ORFs regulates ATF4 mRNA translation in mammalian cells. Proc Natl Acad Sci USA 101:11269–11274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verfaillie T, Rubio N, Garg AD, Bultynck G, Rizzuto R, Decuypere JP, Piette J, Linehan C, Gupta S, Samali A, et al. (2012) PERK is required at the ER-mitochondrial contact sites to convey apoptosis after ROS-based ER stress. Cell Death Differ 19:1880–1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma P, Chierzi S, Codd AM, Campbell DS, Meyer RL, Holt CE, Fawcett JW. (2005) Axonal protein synthesis and degradation are necessary for efficient growth cone regeneration. J Neurosci 25:331–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P, Li J, Tao J, Sha B. (2018) The luminal domain of the ER stress sensor protein PERK binds misfolded proteins and thereby triggers PERK oligomerization. J Biol Chem 293:4110–4121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Flynn A, Waskiewicz AJ, Webb BL, Vries RG, Baines IA, Cooper JA, Proud CG. (1998) The phosphorylation of eukaryotic initiation factor eIF4E in response to phorbol esters, cell stresses, and cytokines is mediated by distinct MAP kinase pathways. J Biol Chem 273:9373–9377. [DOI] [PubMed] [Google Scholar]
- Wang X, Yue P, Chan CB, Ye K, Ueda T, Watanabe-Fukunaga R, Fukunaga R, Fu H, Khuri FR, Sun SY. (2007) Inhibition of mammalian target of rapamycin induces phosphatidylinositol 3-kinase-dependent and Mnk-mediated eukaryotic translation initiation factor 4E phosphorylation. Mol Cell Biol 27:7405–7413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Hussain SM, Wluka AE, Lim YZ, Abram F, Pelletier JP, Martel-Pelletier J, Cicuttini FM. (2019) Association between metformin use and disease progression in obese people with knee osteoarthritis: data from the Osteoarthritis Initiative-a prospective cohort study. Arthritis Res Ther 21:127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waskiewicz AJ, Flynn A, Proud CG, Cooper JA. (1997) Mitogen-activated protein kinases activate the serine/threonine kinases Mnk1 and Mnk2. EMBO J 16:1909–1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waskiewicz AJ, Johnson JC, Penn B, Mahalingam M, Kimball SR, Cooper JA. (1999) Phosphorylation of the cap-binding protein eukaryotic translation initiation factor 4E by protein kinase Mnk1 in vivo. Mol Cell Biol 19:1871–1880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Way SW, Podojil JR, Clayton BL, Zaremba A, Collins TL, Kunjamma RB, Robinson AP, Brugarolas P, Miller RH, Miller SD, et al. (2015) Pharmaceutical integrated stress response enhancement protects oligodendrocytes and provides a potential multiple sclerosis therapeutic. Nat Commun 6:6532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei F, Zhang Y, Geng L, Zhang P, Wang G, Liu Y. (2015) mTOR inhibition induces EGFR feedback activation in association with its resistance to human pancreatic cancer. Int J Mol Sci 16:3267–3282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei F, Zhuo M. (2008) Activation of Erk in the anterior cingulate cortex during the induction and expression of chronic pain. Mol Pain 4:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witzig TE, Geyer SM, Ghobrial I, Inwards DJ, Fonseca R, Kurtin P, Ansell SM, Luyun R, Flynn PJ, Morton RF, et al. (2005) Phase II trial of single-agent temsirolimus (CCI-779) for relapsed mantle cell lymphoma. J Clin Oncol 23:5347–5356. [DOI] [PubMed] [Google Scholar]
- Wiza C, Nascimento EB, Ouwens DM. (2012) Role of PRAS40 in Akt and mTOR signaling in health and disease. Am J Physiol Endocrinol Metab 302:E1453–E1460. [DOI] [PubMed] [Google Scholar]
- Woller SA, Eddinger KA, Corr M, Yaksh TL. (2017) An overview of pathways encoding nociception. Clin Exp Rheumatol 35 (5 Suppl 107):40–46. [PMC free article] [PubMed] [Google Scholar]
- Wong YL, LeBon L, Basso AM, Kohlhaas KL, Nikkel AL, Robb HM, Donnelly-Roberts DL, Prakash J, Swensen AM, Rubinstein ND, et al. (2019) eIF2B activator prevents neurological defects caused by a chronic integrated stress response. eLife 8:e42940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong YL, LeBon L, Edalji R, Lim HB, Sun C, Sidrauski C. (2018) The small molecule ISRIB rescues the stability and activity of Vanishing White Matter Disease eIF2B mutant complexes. eLife 7:e32733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods A, Vertommen D, Neumann D, Turk R, Bayliss J, Schlattner U, Wallimann T, Carling D, Rider MH. (2003) Identification of phosphorylation sites in AMP-activated protein kinase (AMPK) for upstream AMPK kinases and study of their roles by site-directed mutagenesis. J Biol Chem 278:28434–28442. [DOI] [PubMed] [Google Scholar]
- Woolf CJ, Ma Q. (2007) Nociceptors--noxious stimulus detectors. Neuron 55:353–364. [DOI] [PubMed] [Google Scholar]
- Xiao B, Sanders MJ, Carmena D, Bright NJ, Haire LF, Underwood E, Patel BR, Heath RB, Walker PA, Hallen S, et al. (2013) Structural basis of AMPK regulation by small molecule activators. Nat Commun 4:3017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao B, Sanders MJ, Underwood E, Heath R, Mayer FV, Carmena D, Jing C, Walker PA, Eccleston JF, Haire LF, et al. (2011) Structure of mammalian AMPK and its regulation by ADP. Nature 472:230–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie J, Shen K, Jones AT, Yang J, Tee AR, Shen MH, Yu M, Irani S, Wong D, Merrett JE, et al. (2020) Reciprocal signaling between mTORC1 and MNK2 controls cell growth and oncogenesis. Cell Mol Life Sci DOI: 10.1007/s00018-020-03491-1 [published ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing X, Wu K, Dong Y, Zhou Y, Zhang J, Jiang F, Hu WP, Li JD. (2020) Hyperactive Akt-mTOR pathway as a therapeutic target for pain hypersensitivity in Cntnap2-deficient mice. Neuropharmacology 165:107816. [DOI] [PubMed] [Google Scholar]
- Xu JT, Zhao JY, Zhao X, Ligons D, Tiwari V, Atianjoh FE, Lee CY, Liang L, Zang W, Njoku D, et al. (2014) Opioid receptor-triggered spinal mTORC1 activation contributes to morphine tolerance and hyperalgesia. J Clin Invest 124:592–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Q, Fitzsimmons B, Steinauer J, O’Neill A, Newton AC, Hua XY, Yaksh TL. (2011) Spinal phosphinositide 3-kinase-Akt-mammalian target of rapamycin signaling cascades in inflammation-induced hyperalgesia. J Neurosci 31:2113–2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi Y, Oh-Hashi K, Matsuoka Y, Takemura H, Yamakita S, Matsuda M, Sawa T, Amaya F. (2018) Endoplasmic reticulum stress in the dorsal root ganglion contributes to the development of pain hypersensitivity after nerve injury. Neuroscience 394:288–299. [DOI] [PubMed] [Google Scholar]
- Yang ES, Bae JY, Kim TH, Kim YS, Suk K, Bae YC. (2014) Involvement of endoplasmic reticulum stress response in orofacial inflammatory pain. Exp Neurobiol 23:372–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Chennamaneni LR, Ho MWT, Ang SH, Tan ESW, Jeyaraj DA, Yeap YS, Liu B, Ong EH, Joy JK, et al. (2018) Optimization of selective mitogen-activated protein kinase interacting kinases 1 and 2 inhibitors for the treatment of blast crisis leukemia. J Med Chem 61:4348–4369. [DOI] [PubMed] [Google Scholar]
- Yang H, Rudge DG, Koos JD, Vaidialingam B, Yang HJ, Pavletich NP. (2013) mTOR kinase structure, mechanism and regulation. Nature 497:217–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang HS, Jansen AP, Komar AA, Zheng X, Merrick WC, Costes S, Lockett SJ, Sonenberg N, Colburn NH. (2003) The transformation suppressor Pdcd4 is a novel eukaryotic translation initiation factor 4A binding protein that inhibits translation. Mol Cell Biol 23:26–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young SK, Palam LR, Wu C, Sachs MS, Wek RC. (2016) Ribosome elongation stall directs gene-specific translation in the integrated stress response. J Biol Chem 291:6546–6558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young SK, Wek RC. (2016) Upstream open reading frames differentially regulate gene-specific translation in the integrated stress response. J Biol Chem 291:16927–16935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeisel A, Hochgerner H, Lönnerberg P, Johnsson A, Memic F, van der Zwan J, Häring M, Braun E, Borm LE, La Manno G, et al. (2018) Molecular architecture of the mouse nervous system. Cell 174:999–1014.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng Z, Sarbassov D, Samudio IJ, Yee KW, Munsell MF, Ellen Jackson C, Giles FJ, Sabatini DM, Andreeff M, Konopleva M. (2007) Rapamycin derivatives reduce mTORC2 signaling and inhibit AKT activation in AML. Blood 109:3509–3512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Seereeram A, Nassar MA, Levato A, Pezet S, Hathaway G, Morenilla-Palao C, Stirling C, Fitzgerald M, McMahon SB, et al. London Pain Consortium (2006) Nociceptor-derived brain-derived neurotrophic factor regulates acute and inflammatory but not neuropathic pain. Mol Cell Neurosci 31:539–548. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Lin Y, Zhang H, Mañas A, Tang W, Zhang Y, Wu D, Lin A, Xiang J. (2015) Ubl4A is required for insulin-induced Akt plasma membrane translocation through promotion of Arp2/3-dependent actin branching. Proc Natl Acad Sci USA 112:9644–9649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng JQ, Kelly TK, Chang B, Ryazantsev S, Rajasekaran AK, Martin KC, Twiss JL. (2001) A functional role for intra-axonal protein synthesis during axonal regeneration from adult sensory neurons. J Neurosci 21:9291–9303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoll WL, Horton LE, Komar AA, Hensold JO, Merrick WC. (2002) Characterization of mammalian eIF2A and identification of the yeast homolog. J Biol Chem 277:37079–37087. [DOI] [PubMed] [Google Scholar]
- Zoncu R, Efeyan A, Sabatini DM. (2011) mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol 12:21–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zyryanova AF, Weis F, Faille A, Alard AA, Crespillo-Casado A, Sekine Y, Harding HP, Allen F, Parts L, Fromont C, et al. (2018) Binding of ISRIB reveals a regulatory site in the nucleotide exchange factor eIF2B. Science 359:1533–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]