

Abstract
Protein homeostasis is critical for maintaining eukaryotic cell function as well as responses to intrinsic and extrinsic stress. The proteasome is a major portion of the proteolytic machinery in mammalian cells and plays an important role in protein homeostasis. Multiple myeloma (MM) is a plasma cell malignancy with high production of immunoglobulins and is especially sensitive to treatments that impact protein catabolism. Therapeutic agents such as proteasome inhibitors have demonstrated significant benefit for myeloma patients in all treatment phases. Here, we demonstrate that the 11S proteasome activator PA28α is upregulated in MM cells and is key for myeloma cell growth and proliferation. PA28α also regulates MM cell sensitivity to proteasome inhibitors. Downregulation of PA28α inhibits both proteasomal load and activity, resulting in a change in protein homeostasis less dependent on the proteasome and leads to cell resistance to proteasome inhibitors. Thus, our findings suggest an important role of PA28α in MM biology, and also provides a new approach for targeting the ubiquitin-proteasome system and ultimately sensitivity to proteasome inhibitors.
Subject terms: Myeloma, Translational research
Introduction
Multiple myeloma (MM) is the second most common hematologic malignancy in the United States. Active treatments include CD38 antibodies, immunomodulatory imide drugs, dexamethasone, and proteasome inhibitors (PI), which have improved patient outcomes. Bortezomib, carfilzomib, and the oral proteasome inhibitor ixazomib are approved for the treatment of MM patients each with different formulations and adverse events1–3. Despite the clinical benefits of PIs, most patients inevitably develop resistance and relapse, and the underlying mechanisms of drug resistance remain unclear4–8.
Initial sensitivity to PIs is a consequence of aberrant protein homeostasis in MM cells. As a consequence of their high rate of immunoglobulin synthesis, myeloma cells are dependent upon protein folding, trafficking, and degradation systems to maintain survival. Perturbation of protein homeostasis can ultimately induce myeloma cell death9,10. The ubiquitin–proteasome system (UPS) is a central mechanism for protein homeostasis11 and its pivotal role in protein catabolism renders it an attractive target in myeloma therapy.
A functional proteasome is composed of a hollow cylindrical 20S proteasome (core particle, CP) with a regulatory particle (RP) on one or both ends12. Three different proteasome regulators can activate a 20S proteasome, namely, PA700 (19S), REG/PA28 family (α, β, γ), and PA20013–15. PA28γ (REGγ, 11Sγ, or PSME3) is localized in the nucleus and forms homo-heptameric rings. PA28γ is not IFN-γ inducible but participates in chromosomal stability maintenance16, as well as ATP and ubiquitin-independent degradation of some molecules, such as SRC-3 and P2117,18. PA28α (REGα, 11Sα, or PSME1) and PA28β (REGβ, 11Sβ, or PSME2) form hetero-heptameric rings binding to one or two ends of the 20S proteasome to form 11S/20S/11S or hybrid proteasome. PA28α and β are IFN-γ inducible and function as part of MHC class I antigen presentation19–21. PA28α and β are basally expressed in all tissues, but at higher levels in antigen-presenting cells. Our analysis of plasma cells from MM patients with healthy donors has demonstrated increased expression of the 11S proteasome in malignant plasma cells. Although PA28α and β form a heptameric complex to activate proteasome activity within cells, only recombinant PA28α can directly stimulate proteasome peptidase activity. PA28β does not exert a stimulatory effect but enhances the affinity of 11S with 20S proteasome22–25. Our recent finding has demonstrated that perturbation of 11S/20S interaction can regulate proteasome function and sensitivity to PIs in MM8. Given the importance of the 11S proteasome in malignant plasma cells, we sought to identify the function of the 11S subunits, especially the α subunit, in myeloma biology. We generated PA28α stable knockdown myeloma cells and have demonstrated that PA28α is important for myeloma cell proliferation and sensitivity to PIs. Silencing of PA28α suppresses MM cell proliferation and induces PI resistance. The mechanism of PI resistance is distinct from which has been reported with knocking down 20S or 19S proteasome subunits. Knockdown of PA28α inhibited proteasome activity, but also decreased proteasome load, resulting in a change in protein homeostasis which is less dependent on the proteasome. Together, these data demonstrate that the 11S proteasome is an important target in myeloma cell biology and provides a new opportunity to enhance the efficacy of currently available treatment methods.
Results
PA28α upregulation is a hallmark of human MM
We first evaluated proteasome subunit expression levels in a group of human myeloma cell lines, primary cells from two MM patients, and a few non-MM cell lines. Among all tested cells, the 20S proteasome subunit, PSMA2, PSMB5, and the 19S proteasome subunit Rpt5 were constitutively expressed, whereas the 11S subunit PA28α was specifically expressed at high levels only in MM cells (Fig.1a). We next evaluated the expression of PSME1 (which encodes PA28α) in plasma cells from healthy donors, monoclonal gammopathy of undetermined significance (MGUS), smoldering multiple myeloma (SMM), newly diagnosed multiple myeloma (NDMM), and relapsed/refractory multiple myeloma (RRMM) using data from Chng et al.26. This demonstrated that PSME1 was progressively upregulated with disease progression (Fig.1b), suggesting that PA28α is associated with MM cell transformation and tumor progression.
Fig. 1. PA28α is upregulated in human multiple myeloma cells.

a Western blot of proteasome subunits expression on cell lysate from a variety of cell lines and primary MM cells, β-actin as a loading control. b Boxplot of PSME1 expression data in normal plasma cells (PC, n = 15), monoclonal gammopathy of undetermined significance (MGUS, n = 22), smoldering multiple myeloma (SMM, n = 24), newly diagnosed multiple myeloma (NDMM, n = 73), and relapsed/refractory multiple myeloma (RRMM, n = 28) from Chng et al.25. *P < 0.05, **P < 0.01, ***P < 0.001, determined using linear regression with disease stage as a factor.
PA28α regulates multiple myeloma sensitivity to PI
Since PA28α can stimulate 20S proteasome activity both alone and in combination with the PA28β subunit as an 11S complex25, we initially anticipated that silencing of PA28α would enhance MM cells' sensitivity to PIs via a reduction in proteasome capacity. We transiently transfected LP1 cell line and primary cells from two MM patients with pooled PA28α siRNA and compared cell growth following exposure to bortezomib. Unexpectedly, silencing of PA28α protected cells from bortezomib-induced cell growth inhibition (Fig. 2a and Supplementary Fig. 1). We further confirmed the regulation of sensitivity to bortezomib through transient overexpression of PA28α in LP1 cells (Fig. 2b). To better characterize the function of PA28α, we generated LP1 and RPMI8226 PA28α knockdown stable cell lines. Consistent with transient transfection, stable knockdown of PA28α blocked bortezomib and carfilzomib-induced cell growth inhibition and apoptosis (Fig. 2c and Supplementary Fig. 2).
Fig. 2. PA28α regulates multiple myeloma sensitivity to PIs.

a LP1 cells were transfected with PA28α siRNA for 72 h, cells were then treated with bortezomib for 24 h and MTT assay was performed to analyze cell growth. b LP1 cells were transfected with Flag-PA28α for 48 h, cells were then treated with bortezomib for 24 h, and MTT assay was performed to analyze cell growth. c MTT assay and Annexin V/Propidium Iodide (PI) flow cytometry assay of LP1 PA28α knockdown stable cells under exposure to different concentrations of bortezomib or carfilzomib for 48 h. Western blot was performed to show PA28α expression. *P < 0.05, **P < 0.01, Student’s t test.
Silencing of PA28α inhibits proteasome activity
Proteasomes possess three different enzymatic activities in the 20S proteasome chamber, caspase-like (C-L), chymotrypsin-like (CT-L), and trypsin-like (T-L). Both bortezomib and carfilzomib directly target the proteasome CT-L enzymatic activity via binding the PSMB5 subunit. To explore mechanisms responsible for reduced PI sensitivity induced by PA28α knockdown, we measured proteasome enzymatic activities from the whole-cell lysates of the stable cell lines. In PA28α knockdown cells, C-L and CT-L peptidase activity of the proteasome was significantly reduced, while there was no change of T-L peptidase activity (Fig. 3a). We also compared the proteasome sensitivity to bortezomib by measuring the CT-L protease activity between LP1 PA28α knockdown and control cells. Two-hour treatment with 10 nM bortezomib caused 74% suppression in CT-L protease activity in control cells, but only 55% in PA28α knockdown cells (Fig. 3b). We further observed that under the exposure with escalating concentrations of bortezomib, LP1 PA28α knockdown cells demonstrated less susceptible to bortezomib (Fig. 3c). Transient knockdown of PA28α using pooled siRNA in LP1 cells also demonstrated suppressed C-L and CT-L proteasome activities (Fig. 3d). These data suggest that silencing of PA28α regulated proteasome activity which could manipulate myeloma cells' response to PIs.
Fig. 3. Downregulation of PA28α alters proteasome activity.

a Proteasome fluorogenic peptidase assays of cell lysates from LP1 and RPMI8226 PA28α knockdown and scrambled control stable cells. b LP1 PA28α knockdown stable cells were treated with 10 nM bortezomib for 2 h, cell lysates were obtained, and proteasome fluorogenic peptidase assay was performed to check the chymotrypsin-like proteasome activity, SUC-LLVY-AMC as substrate. c LP1 PA28α knockdown stable cells were treated with serial concentrations of bortezomib, chymotrypsin-like proteasome activity was checked as in b. d LP1 cells were transiently transfected with PA28α siRNA for 72 h, proteasome fluorogenic peptidase assays were performed as in a. **P < 0.01, Student’s t test.
Silencing of PA28α modulates proteasome subunit expression
To further dissect the underlying mechanism of alteration in proteasome activity by PA28α, we first examined proteasome abundancy in PA28α knockdown cells. Although proteasome enzymatic activity decreased in PA28α knockdown cells, expression of constitutive proteasome subunits bearing enzymatic activities such as PSMB5, PSMB6, PSMB7 were increased in PA28α silenced cells, while at the same time, the immunoproteasome subunit PSMB8 was decreased (Fig. 4a). Quantitative real-time PCR confirmed that constitutive proteasome subunits increased in LP1 PA28α knockdown cells, while immunoproteasome subunits decreased (Fig. 4b). We also found increased transcript levels of other proteasome subunits, such as PSMA3, PSMD4 (S5a), PSME3 but decreased expression of PSMB9 and PSMB10 (Fig. 4b). Although the transcript level of PA28β was not changed in PA28α knockdown cells, the protein level decreased significantly. This is consistent with reports from other groups showing that PA28α interacts with and stabilizes PA28β27. Transient knockdown of PA28α in LP1 cells also upregulated PSMB5 and PA28γ, with the downregulation of PSMB8 (Fig. 4c). To confirm the regulation of PSMB5 by PA28α, we transiently transfected HeLa, LP1, and U266 cells with a plasmid expressing Flag-PA28α. Consistently, overexpression of PA28α indeed downregulated PSMB5 (Fig. 4d).
Fig. 4. PA28α regulates proteasome subunits expression.

a Western blot of proteasome subunit expression from LP1 and RPMI8226 PA28α knockdown and control stable cells, β-actin as a loading control. b Real-time qPCR assay of proteasome subunits transcript levels from LP1 PA28α knockdown and control stable cells, β-actin as an endogenous control. c LP1 cells were transiently transfected with shRNA targeting PA28α or control vector, 48 h after transfection proteasome components expression were checked using western blot. d HeLa, LP1, and U266 cells were transiently transfected with Flag-PA28α vector or Flag empty vector, 48 h after transfection, cell lysates were collected, and western blot was performed to check PA28α and PSMB5 expression, β-actin as a loading control.
NRF1 is important for PA28α knockdown-induced proteasome regulation
In mammalian cells, short-term treatment with PIs induces transcriptional upregulation of proteasome synthesis mediated by nuclear factor erythroid 2-related factor 1 (NRF1 encoded by NFE2L1 also known as TCF11)-dependent proteasome recovery pathway (or proteasome bounce-back)28. In our model, we also observed increased expression of NRF1, suggesting the feedback could result from NRF1 activation (Figs. 4c and 5a). To verify the function of NRF1 in PA28α knockdown-induced proteasome regulation, we transiently transfected LP1 PA28α knockdown and control cells with NRF1 siRNA. Knockdown of NRF1 downregulated PSMB5 and PA28γ (Fig. 5b), and also enhanced cell sensitivity to bortezomib (Fig. 5c). The above data suggest that NRF1 is involved in PA28α knockdown-induced proteasome subunits regulation.
Fig. 5. Silencing NRF1 reverses PA28α knockdown-induced proteasome recovery and resensitizes to bortezomib treatment.

a Western blot analysis of NRF1 in LP1 and RPMI8226 PA28α knockdown and control stable cells. b LP1 PA28α knockdown and control stable cells were transiently transfected with NRF1 siRNA or control siRNA. 48 h after transfection, cell lysates were collected and analyzed for expression of proteasome subunits by western blot (b) or cells were treated with different concentrations of bortezomib for 24 h, MTT assay was performed to assess relative cell growth (c).
Silencing of PA28α modifies proteasome complex formation
The discrepancy between proteasome abundance and proteasome activity prompted us to explore how silencing of PA28α altered proteasome function. We examined proteasome formation by checking the proteasome pool using native PAGE from cell lysates of LP1 PA28α knockdown stable cell lines. In PA28α knockdown cells, the formation of 19S/20S/19S double-capped proteasome decreased, with the increased formation of PA28γ capped proteasomes (Fig. 6). Among the three 11S proteasome subunits, α and β can activate all three types of protease activities, while γ can only activate T-L protease activity29, the increased formation of PA28γ-bearing proteasomes explained our previous observation that there was a significant decrease in C-L and CT-L protease activities in knockdown cells but no difference on T-L protease activity.
Fig. 6. Downregulation of PA28α changes proteasome complex formation.
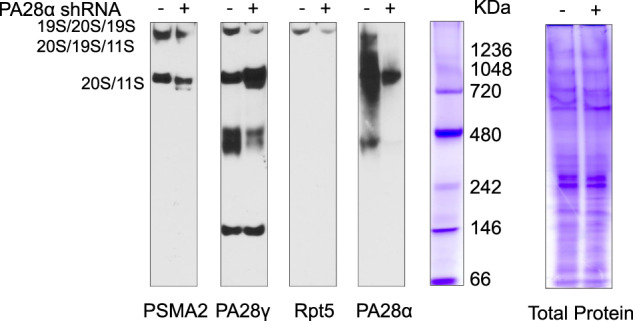
LP1 PA28α knockdown and control stable cells were lysed in proteasome lysis buffer, same amounts of cell lysates were loaded on native PAGE, and proteasome complex was checked followed by western blot. Total protein was also visualized by Coomassie Blue R-250 staining.
Silencing of PA28α decreases proteasome load
Since PA28α knockdown inhibited proteasome activity, we expected that impaired proteasome activity could induce proteasome stress with the accumulation of protein–ubiquitin conjugates in the cell. Although proteasome activity is suppressed in PA28α knockdown cells, we did not detect more accumulated ubiquitinated proteins (Fig. 7a). In MM cells, sensitivity to PIs is closely correlated with proteasome load versus capacity. Cells with higher proteasome load and lower proteasome capacity show more sensitivity to PIs30,31. We asked whether knockdown of PA28α could also decrease proteasome load within the cells. We performed a pulse-chase assay to compare the proteasomal degradation. Cells were labeled with O-propargyl-puromycin (OPP) for 30 min and chased for 2 h with or without 40 nM bortezomib after thoroughly washing. The ratio of labeled protein degradation through proteasome was significantly lower in PA28α knockdown cells (Fig. 7b). In multiple myeloma cells, the proteasomal load is contributed mainly by the extensive synthesis of immunoglobulin31,32. We also found that in PA28α knockdown cells, the expression of the immunoglobulin light chain was greatly inhibited (Fig. 7c). Reduction in proteasome load is quite interesting and requires further investigation. Based on our observation that PA28α knockdown activated eIF2α (Fig. 7d), the regulation could result from protein synthesis inhibition in PA28α knockdown cells. Importantly, OPP-labeling also demonstrated that nascent protein synthesis was suppressed in PA28α knockdown cells (Fig. 7b). We also observed that although silencing of PA28α did not induce autophagy, which is active in these cells, however, it did upregulate p62/SQSTM1 protein levels (Fig. 7d) which could protect cells under proteasome deficiency by enhancing the delivery of ubiquitinated proteins to the autophagy pathway. The above data demonstrated that knockdown of PA28α reduced both cellular proteasome activity and proteasome load, resulting in new protein homeostasis with less dependent on proteasome for protein degradation and subsequent PI resistance (Fig. 7e).
Fig. 7. Downregulation of PA28α decreases proteasome workload.

a Western blot analysis of ubiquitinated proteins in total protein lysates extracted from LP1 and RPMI8226 PA28α knockdown stable cells. β-actin as a loading control. b OPP pulse-chase assay of proteasome degradation and protein synthesis in LP1 PA28α knockdown stable cells. Western blot analysis of immunoglobulin lambda light chain (ƛ IgL) (c), eIF2α, p62/SQSTM1, and LC3B (d) in LP1 and RPMI8226 PA28α knockdown stable cells, β-actin as a loading control. **P < 0.01, Student’s t test. e Working model of PA28α knockdown in MM.
Discussion
Proteasome inhibitors are a standard part of front-line therapy in MM, and have continued to improve the outcomes for MM patients. Unfortunately, the intrinsic and acquired resistance to PIs limits their long-term benefit in the clinic. Many factors have been identified that contribute to PI resistance, including proteasome gene mutations and upregulation, alterations of protein expression involved in cell stress response, survival, and apoptotic pathways4–7, but many of the key underlying mechanisms remain unclear.
The proteasome load versus capacity ratio influences cell sensitivity to PIs, which has been verified both in intrinsic and acquired PI resistance. Increasing proteasome load or decreasing proteasome capacity could disturb the balance and result in PI sensitivity. Researchers have been focused on modulating the proteasome capacity portion of the equation by targeting specific proteasome subunits. Unfortunately, targeting the expression of different proteasome subunits results in distinct cell responses to PIs. Evidence from human cancer cell lines has indicated that silencing of the 20S core particle subunits reduced cell viability upon PI exposure. In contrast, downregulation of the 19S proteasome subunits protects cells from treatment with PIs. The mechanism of 19S reduction-induced PI resistance involves reduced proteotoxic stress and a broad change of proteasome substrates mediating cell survival and proliferation33,34. NRF1-dependent induction of proteasome subunit expression and subsequent rescue of proteasome function in response to PIs has been well studied. 19S subunit reduction does not activate NRF1 and the expression of 20S subunits is unchanged34. In our present study, we demonstrated that knockdown of the 11S proteasome activator, PA28α, activated NRF1-mediated proteasome recovery response. Moreover, although some of the proteasome subunits were upregulated in PA28α knockdown cells, the composition of the 26S proteasome complex was reduced, with an accordingly increased assembly of PA28γ capped proteasome. The reason why cells adapt to PA28α knockdown via assembly PA28γ incorporated proteasome is still unknown and warrants further investigation. Rapid formation of PA28 or PA200 bearing alternative proteasome has also been observed in rabbit reticulocytes lysate (RRL) and human lung fibroblasts under exposure to bortezomib35,36. Furthermore, alterations in 20S composition such as the α4–α4 proteasome, mediates cell resistance to toxic metal ions in human cells under the regulation of multiple oncogenic signals37. The underlying mechanism and function of such alternative proteasomes remains to be further determined but suggests an essential role in helping cells respond to environmental stresses.
In addition to the decreased proteasome activity, knockdown of PA28α also downregulated proteasome load which may result from the inhibition of protein synthesis and upregulation of p62SQSTM1. How cells responded to proteasome inhibition by lowered protein synthesis is quite important and under further investigation. In myeloma, excessive immunoglobulin production and accumulation cause severe complications in patients, eradicating the overproduction of immunoglobulin could provide a promising therapeutic strategy. Altogether, although knockdown of PA28α inhibits proteasome activity, proteasome load within the cells decreased concurrently, resulting in the new protein homeostasis.
Until now, the function of PA28α/β remains partly described. Some groups have reported PA28α/β participates in MHC class I antigen presentation, but the underlying mechanism is controversial and not clear20,38–41. Others have suggested that PA28α/β may play roles in protein degradation and antioxidant response27. Here, based on our observations of PA28α knockdown cell lines, the function of PA28α comprises at least two key aspects in MM cells. On the one hand, PA28α can regulate cell growth and proliferation. Expression of PA28α is upregulated in MM cells comparing to normal plasma cells as well as during disease progression in vivo. Silencing of PA28α significantly suppresses MM cell proliferation (Supplementary Fig. 3), suggesting that PA28α is important for myeloma progression. On the other hand, PA28α can regulate proteasome biogenesis and activity directly or indirectly. Silencing of PA28α reduced proteasome load and proteasome stress within the cell, sustained protein homeostasis with a lower level of proteasome activity leading to PI resistance. In our observation, the expression level of PA28α may not be correlated with intrinsic cell sensitivity to bortezomib. As we compared MM cell lines under the exposure to bortezomib, sensitivity to bortezomib was not associated with the basal level of PA28α (Supplementary Fig. 4). While manipulating the PA28α protein level could impact cell response to PIs as evidenced by data from transient knockdown or overexpression of PA28α within MM cells. Altogether, in this report, we describe the function of PA28α in MM cells, also provide novel insights into regulating PIs sensitivity through modulation of the 11S proteasome subunit PA28α.
Materials and methods
Reagents, constructs, and antibodies
Bortezomib and carfilzomib were purchased from selleck chemicals. Other reagents were purchased from puromycin (Sigma), cycloheximide (Sigma), SUC-LLVY-AMC (Enzo Life Science), Z-Leu-Leu-Glu-AMC (Enzo Life Science), Boc-Leu-Arg-Arg-AMC (Enzo Life Science). Antibodies used were as follows: PA28α (Cell Signaling), PSMA2 (Cell Signaling), S5a (Cell signaling), PA28β (Cell Signaling), Phospho-eIF2α (Ser51) (Cell signaling), eIF2α (Cell signaling), α-tubulin (Genetex), PA28γ (Genetex), PSMB5 (Genetex), PSMB6 (Enzo life science), PSMB7 (Genetex), PSMB8 (Genetex), PSMB9 (R&D systems), PSMB10 (R&D systems), Rpt5 (Enzo life science), Ubiquitin (Cell Signaling), β-actin (Santa Cruz Technology), TCF11/NRF1 (Cell Signaling), LC3B (Cell Signaling), p62/SQSTM1 (MBL International) human Ig lambda light chain (R&D systems), actin (Sigma). pLKO.1 empty vector, shRNA vector targeting human PA28α, NRF1 siRNA (ON-TARGETplus SMARTpool), PA28α siRNA (Accell SMARTpool), and control siRNA were purchased from Dharmacon. Native Mark unstained protein standard was purchased from life technologies.
Generation of PA28α knockdown stable cell lines
To generate PA28α knockdown stable cell lines, LP1 and RPMI8226 cells were infected with recombinant lentiviruses carrying PA28α knockdown shRNA generated in 293T cells. Sequences for PA28α knockdown were: TAACACAGCATAAGCATTGCG (shRNA #1), TTTCATTGCAGTTCACTGGGC (shRNA #2). Infected cells were then selected in a medium with 1.25 ng/ml puromycin. The puromycin-resistant cells were expanded and tested with PA28α antibody before experiments.
Cell growth assay
Cell growth was assessed using Cell Proliferation Assay kits from atcc.org following the manufacturer’s procedure.
Cell apoptosis assay
Cell apoptosis was analyzed using FITC-annexin V/PI staining (BD Biosciences) following the manufacturer’s protocol. Data were collected and analyzed using FlowJo (Treestar, Ashland, OR) software.
Western blot
Western blot was performed as described previously42.
Proteasome fluorogenic peptidase assays
Proteasome fluorogenic peptidase assays were performed on whole cell lysate. In brief, cells were collected and lysed in proteasome lysis buffer (50 mM Tris-HCl (pH 7.5), 5 mM MgCl2, 0.5 mM EDTA, 10% glycerol, 0.25% NP-40,1 mM DTT, 2 mM ATP). The extracts were cleared by centrifugation at 16,000×g for 30 min at 4 °C. The same amount of proteins were transferred to a 96-well microtiter plate (BD Falcon). To measure the proteasome peptidase activity, three different kinds of substrates were used, caspase-like: Z-Leu-Leu-Glu-AMC, chymotrypsin-like: Suc-Leu-Leu-Val-Tyr-AMC, trypsin-like: Boc-Leu-Arg-Arg-AMC. The substrate was added to the plate to a final concentration of 50 µM and mixed, incubated at 37 °C for 30 min. Fluorescence was monitored on a microplate fluorometer SpectraMAX (Molecular Devices) using wavelength 380-nm excitation, 460-nm emission.
Real-time qPCR
Real-time qPCR was performed as described previously42. Primers used were KiCqStart SYBR Green Primers obtained from Sigma.
Native gel electrophoretic mobility assays
Native gel electrophoresis was performed, as described previously8. Cell extracts and Native Mark unstained protein standard were also resolved in the gel and followed by Coomassie Blue R250 staining.
Proteasomal-mediated degradation assay
Proteasomal-mediated degradation was evaluated using a protein synthesis assay kit from Cayman chemical according to the manufacturer’s manual with a small modifications. Briefly, cells were labeled using O-propargyl-puromycin (OPP) for 30 min then washed and incubated with complete media with or without 40 nM bortezomib for 2 h. After the incubation, cells were fixed and stained with 5-FAM azide for 30 min. After washing, cells were collected and analyzed using flow cytometry. Proteasomal-mediated degradation was calculated using the percentage of bortezomib inhibited peptides degradation relative to the peptides at the end of OPP labeling.
Patient samples
Bone marrow aspirates from MM patients were prepared following Emory University Institutional Review Board approved consent. Buffy coat cells were collected as described previously42 and applied in experiments.
Analysis of plasma cell and myeloma gene expression
Affymetrix microarray data were downloaded from GEO data sets GSE6477 and log2 transformed. Data were plotted in R/Bioconductor43 using the function “boxplot”. Statistical significance between groups was determined relative to plasma cells from healthy donors using linear regression where disease stage (e.g., MGUS, SMM, NDMM, RRMM) was treated as a factor.
Supplementary information
Acknowledgements
The work is supported by Takeda and Celgene to S.L. Y.G. is supported by Research Fellow Award from the Multiple Myeloma Research Foundation (MMRF). B.G.B. is supported by Developmental Funds from the Winship Cancer Institute of Emory University, post-doctoral fellowship PF-17-109-1-TBG from the American Cancer Society, and a Research Scholar Fellow Award from the Multiple Myeloma Research Foundation (MMRF).
Conflict of interest
S.L. is a consultant for Takeda, Celgene, Amgen, Novartis, and BMS. L.H.B. is a consultant and has received research funding from AstraZeneca and served on an advisory board for Genentech. C.C.H. received research grants from Takeda, Oncolytics Biotech, research and personal grants from Janssen, BMS, Sanofi, Nektar, and Karyopharm, and personal grants from Imbrium and Oncopeptides, all outside the submitted work. The remaining authors declare no competing financial interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information accompanies this paper at (10.1038/s41408-020-00393-0).
References
- 1.Richardson PG, et al. Single-agent bortezomib in previously untreated multiple myeloma: efficacy, characterization of peripheral neuropathy, and molecular correlations with response and neuropathy. J. Clin. Oncol. 2009;27:3518–3525. doi: 10.1200/JCO.2008.18.3087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lonial S, Boise LH. Current advances in novel proteasome inhibitor-based approaches to the treatment of relapsed/refractory multiple myeloma. Oncology. 2011;25(Suppl 2):25–31. [PubMed] [Google Scholar]
- 3.Richardson PG, et al. Ixazomib for the treatment of multiple myeloma. Expert Opin. Pharmacother. 2018;19:949–1968. doi: 10.1080/14656566.2018.1528229. [DOI] [PubMed] [Google Scholar]
- 4.McConkey DJ, Zhu K. Mechanisms of proteasome inhibitor action and resistance in cancer. Drug Resist. Updat. 2008;11:164–179. doi: 10.1016/j.drup.2008.08.002. [DOI] [PubMed] [Google Scholar]
- 5.Gutman D, Morales AA, Boise LH. Acquisition of a multidrug-resistant phenotype with a ⦸ proteasome inhibitor in multiple myeloma. Leukemia. 2009;23:2181–2183. doi: 10.1038/leu.2009.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rückrich T, et al. Characterization of the ubiquitin-proteasome system in bortezomib- adapted cells. Leukemia. 2009;23:1098–1105. doi: 10.1038/leu.2009.8. [DOI] [PubMed] [Google Scholar]
- 7.Oerlemans R, et al. Molecular basis of bortezomib resistance: proteasome subunit beta5 (PSMB5) gene mutation and overexpression of PSMB5 protein. Blood. 2008;112:2489–2499. doi: 10.1182/blood-2007-08-104950. [DOI] [PubMed] [Google Scholar]
- 8.Gu Y, et al. 14-3-3ζ binds the proteasome, limits proteolytic function and enhances sensitivity to proteasome inhibitors. Leukemia. 2018;32:744–751. doi: 10.1038/leu.2017.288. [DOI] [PubMed] [Google Scholar]
- 9.Hideshima T, Anderson KC. Biologic impact of proteasome inhibition in multiple myeloma cells-from the aspects of preclinical studies. Semin. Hematol. 2012;49:223–227. doi: 10.1053/j.seminhematol.2012.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boise LH, Kaufman JL, Bahlis NJ, Lonial S, Lee KP. The Tao of myeloma. Blood. 2014;124:1873–1879. doi: 10.1182/blood-2014-05-578732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Glickman MH, Ciechanover A. The ubiquitin-proteasome proteolytic pathway: destruction for the sake of construction. Physiol. Rev. 2002;82:373–428. doi: 10.1152/physrev.00027.2001. [DOI] [PubMed] [Google Scholar]
- 12.Voges D, Zwickl P, Baumeister W. The 26S proteasome: a molecular machine designed for controlled proteolysis. Annu. Rev. Biochem. 1999;68:1015–1068. doi: 10.1146/annurev.biochem.68.1.1015. [DOI] [PubMed] [Google Scholar]
- 13.Chu-Ping M, Vu JH, Proske RJ, Slaughter CA, DeMartino GN. Identification, purification, and characterization of a high molecular weight, ATP-dependent activator (PA700) of the 20S proteasome. J. BiolChem. 1994;269:3539–3547. [PubMed] [Google Scholar]
- 14.Ma CP, Slaughter CA, DeMartino GN. Identification, purification, and characterization of a protein activator (PA28) of the 20S proteasome (macropain) J. Biol. Chem. 1992;267:10515–10523. [PubMed] [Google Scholar]
- 15.Ustrell V, Hoffman L, Pratt G, Rechsteiner M. PA200, a nuclear proteasome activator involved in DNA repair. EMBO J. 2002;21:3516–3525. doi: 10.1093/emboj/cdf333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zannini L, et al. REGgamma proteasome activator is involved in the maintenance of chromosomal stability. Cell Cycle. 2008;7:504–512. doi: 10.4161/cc.7.4.5355. [DOI] [PubMed] [Google Scholar]
- 17.Li X, et al. The SRC-3/AIB1 coactivator is degraded in a ubiquitin- and ATP-independent manner by the REG gamma proteasome. Cell. 2006;124:381–392. doi: 10.1016/j.cell.2005.11.037. [DOI] [PubMed] [Google Scholar]
- 18.Li X, et al. Ubiquitin- and ATP-independent proteolytic turnover of p21 by the REG gamma-proteasome pathway. Mol. Cell. 2007;26:831–842. doi: 10.1016/j.molcel.2007.05.028. [DOI] [PubMed] [Google Scholar]
- 19.Realini C, Dubiel W, Pratt G, Ferrell K, Rechsteiner M. Molecular cloning and expression of a gamma-interferon-inducible activator of the multicatalytic protease. J. Biol. Chem. 1994;269:20727–20732. [PubMed] [Google Scholar]
- 20.Rechsteiner M, Realini C, Ustrell V. The proteasome activator 11S REG (PA28) and class I antigen presentation. Biochem. J. 2000;345:1–15. doi: 10.1042/bj3450001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ahn JY, et al. Primary structures of two homologous subunits of PA28, a gamma-interferon- inducible protein activator of the 20S proteasome. FEBS Lett. 1995;366:37–42. doi: 10.1016/0014-5793(95)00492-R. [DOI] [PubMed] [Google Scholar]
- 22.Groettrup M, et al. A role for the proteasome regulator PA28alpha in antigen presentation. Nature. 1996;381:166–168. doi: 10.1038/381166a0. [DOI] [PubMed] [Google Scholar]
- 23.Song X, von Kampen J, Slaughter CA, DeMartino GN. Relative functions of the alpha and beta subunits of the proteasome activator, PA28. J. Biol. Chem. 1997;272:27994–28000. doi: 10.1074/jbc.272.44.27994. [DOI] [PubMed] [Google Scholar]
- 24.Cascio P. PA28αβ: the enigmatic magic ring of the proteasome? Biomolecules. 2014;4:566–584. doi: 10.3390/biom4020566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kuehn L, Dahlmann B. Reconstitution of proteasome activator PA28 from isolated subunits: optimal activity is associated with an alpha, beta-heteromultimer. FEBS Lett. 1996;394:183–186. doi: 10.1016/0014-5793(96)00946-5. [DOI] [PubMed] [Google Scholar]
- 26.Chng WJ, et al. Molecular dissection of hyperdiploid multiple myeloma by gene expression profiling. Cancer Res. 2007;67:2982–2989. doi: 10.1158/0008-5472.CAN-06-4046. [DOI] [PubMed] [Google Scholar]
- 27.Li J, Powell SR, Wang X. Enhancement of proteasome function by PA28a overexpression protects against oxidative stress. FASEB J. 2011;25:883–893. doi: 10.1096/fj.10-160895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Radhakrishnan SK, et al. Transcription factor Nrf1 mediates the proteasome recovery pathway after proteasome inhibition in mammalian cells. Mol. Cell. 2010;38:17–28. doi: 10.1016/j.molcel.2010.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Realini C, et al. Characterization of recombinant REG alpha, REG beta, and REG gamma proteasome activators. J. Biol. Chem. 1997;272:25483–25492. doi: 10.1074/jbc.272.41.25483. [DOI] [PubMed] [Google Scholar]
- 30.Bianchi G, et al. The proteasome load versus capacity balance determines apoptotic sensitivity of multiple myeloma cells to proteasome inhibition. Blood. 2009;113:3040–3049. doi: 10.1182/blood-2008-08-172734. [DOI] [PubMed] [Google Scholar]
- 31.Obeng EA, et al. Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood. 2006;107:4907–4916. doi: 10.1182/blood-2005-08-3531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Meister S, et al. Extensive immunoglobulin production sensitizes myeloma cells for proteasome inhibition. Cancer Res. 2007;67:1783–1792. doi: 10.1158/0008-5472.CAN-06-2258. [DOI] [PubMed] [Google Scholar]
- 33.Acosta-Alvear D, et al. Paradoxical resistance of multiple myeloma to proteasome inhibitors by decreased levels of 19S proteasomal subunits. eLife. 2015;4:e08153. doi: 10.7554/eLife.08153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tsvetkov P, et al. Compromising the 19S proteasome complex protects cells from reduced flux through the proteasome. eLife. 2015;4:e08467. doi: 10.7554/eLife.08467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Welk V, et al. Inhibition of proteasome activity induces formation of alternative proteasome complexes. J. Biol. Chem. 2016;291:13147–13159. doi: 10.1074/jbc.M116.717652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shibatani T, et al. Global organization and function of mammalian cytosolic proteasome pools: implications for PA28 and 19S regulatory complexes. Mol. Biol. Cell. 2006;17:4962–4971. doi: 10.1091/mbc.e06-04-0311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Padmanabhan A, Vuong SA, Hochstrasser M. Assembly of an evolutionarily conserved alternative proteasome isoform in human cells. Cell Rep. 2016;14:2962–2974. doi: 10.1016/j.celrep.2016.02.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Preckel T, et al. Impaired immunoproteasome assembly and immune responses in PA28-/- mice. Science. 1999;286:2162–2165. doi: 10.1126/science.286.5447.2162. [DOI] [PubMed] [Google Scholar]
- 39.Van Hall T, et al. Differential influence on cytotoxic T lymphocyte epitope presentation by controlled expression of either proteasome immunosubunits or PA28. J. Exp. Med. 2000;192:483–494. doi: 10.1084/jem.192.4.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Murata S, et al. Immunoproteasome assembly and antigen presentation in mice lacking both PA28alpha and PA28beta. EMBO J. 2001;20:5898–5907. doi: 10.1093/emboj/20.21.5898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Raule M, et al. PA28alphabeta reduces size and increases hydrophilicity of 20S immunoproteasome peptide products. Chem. Biol. 2014;21:470–480. doi: 10.1016/j.chembiol.2014.02.006. [DOI] [PubMed] [Google Scholar]
- 42.Gu Y, et al. MLN4924, an NAE inhibitor, suppresses AKT and mTOR signaling via upregulation of REDD1 in human myeloma cells. Blood. 2014;123:3269–3276. doi: 10.1182/blood-2013-08-521914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gentleman RC, et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.