

Abstract
Age-related hearing loss (AHL) is a progressive sensorineural hearing loss in elderly people. Although no prevention or treatments have been established for AHL, recent studies have demonstrated that oxidative stress is closely related to pathogenesis of AHL, suggesting that suppression of oxidative stress leads to inhibition of AHL progression. NRF2 is a master transcription factor that regulates various antioxidant proteins and cytoprotection factors. To examine whether NRF2 pathway activation prevents AHL, we used Keap1-knockdown (Keap1FA/FA) mice, in which KEAP1, a negative regulator of NRF2, is decreased, resulting in the elevation of NRF2 activity. We compared 12-month-old Keap1FA/FA mice with age-matched wild-type (WT) mice in the same breeding colony. In the Keap1FA/FA mice, the expression levels of multiple NRF2 target genes were verified to be significantly higher than the expression levels of these genes in the WT mice. Histological analysis showed that cochlear degeneration at the apical and middle turns was ameliorated in the Keap1FA/FA mice. Auditory brainstem response (ABR) thresholds in the Keap1FA/FA mice were significantly lower than those in the WT mice, in particular at low–mid frequencies. Immunohistochemical detection of oxidative stress markers suggested that oxidative stress accumulation was attenuated in the Keap1FA/FA cochlea. Thus, we concluded that NRF2 pathway activation protects the cochlea from oxidative damage during aging, in particular at the apical and middle turns. KEAP1-inhibiting drugs and phytochemicals are expected to be effective in the prevention of AHL.
Subject terms: Transcription, Ageing, Auditory system
Introduction
Age-related hearing loss (AHL) is the most common sensorineural hearing loss in the elderly and is caused by degenerative and irreversible changes in the inner ear1–4. Age-related histological impairment has been reported in hair cells, spiral ganglion neurons (SGNs), the spiral ligament (SL), the stria vascularis (SV), and synaptic connections between hair cells and SGNs5–9. Although AHL is thought to be caused by various factors, such as exposure to noise10,11, ototoxic chemicals12, systemic disorders13,14, and genetic predispositions15–18, the precise molecular mechanisms of AHL are not well understood. Accordingly, neither effective prevention nor treatments for AHL have been established. As longevity increases worldwide, AHL is becoming an increasingly serious problem that negatively affects the quality of life in the aged population.
Recent studies have suggested that reactive oxygen species (ROS) are closely related to the pathogenesis of AHL19,20. ROS are mainly produced by NADPH oxidases and mitochondrial respiration in the cochlea21,22. Excessive ROS production or decreased antioxidant capacity induces oxidative damage in the cochlea23–25. Although a few antioxidant reagents have been shown to protect the cochlea by decreasing ROS levels26–29, no drugs have ever been shown to prevent the progression of AHL.
The transcription factor NRF2 is a master regulator of various detoxifying and antioxidant genes, and is systemically activated in response to oxidative and electrophilic stress30,31. Under normal conditions, NRF2 is ubiquitinated by the KEAP1-CUL3 ubiquitin E3 ligase complex in the cytoplasm, resulting in the NRF2 protein degradation through the proteasome. Upon exposure to ROS or electrophiles, which inactivate KEAP1, NRF2 is stabilized and subsequently translocated into the nucleus where it activates many cytoprotective genes, including NAD(P):quinone oxidoreductase 1 (Nqo1), Thioredoxin reductase 1 (Txnrd1), Glutamate-cysteine ligase, catalytic subunit (Gclc), and Glutamate-cysteine ligase, modifier subunit (Gclm), by binding to antioxidant response element (GCnnnG/CTCAT/C). In addition, recent studies clarified that NRF2 exerts potent anti-inflammatory functions32,33. Appropriate activation of NRF2 has been shown beneficial for our health preventing and alleviating various pathological conditions, such as ischemia-reperfusion injury, neurodegenerative diseases, and chronic inflammation30,31,34. Regarding the protective role of NRF2 in the cochlea, we previously reported that noise-induced hearing loss (NIHL) was exacerbated in Nrf2-knockout mice and prevented by pretreatment of NRF2 inducers35. Gsta4, one of the typical NRF2 target genes, was shown to play a critical role in the protection from drug ototoxicity in female mice36. In addition, AHL progression was reported to be accelerated in Nrf2-knockout mice. The numbers of hair cells and spiral ganglion cells were reduced earlier in Nrf2-knockout mice than they were in wild-type (WT) mice37. Thus, endogenous NRF2 is regarded as necessary for resisting the progression of age-related pathology of the inner ear.
In the current study, we investigated whether NRF2 pathway activation suppresses AHL progression. To this end, we examined Keap1-knockdown (Keap1FA/FA) mice, in which NRF2 pathway is systemically activated due to the decreased expression of Keap138,39. We compared congenic Keap1FA/FA mice with WT control mice on a C57BL/6 genetic background at the ages of 2, 5, and 12 months. As the C57BL/6 strain exhibits early onset of AHL due to a single-nucleotide polymorphism (SNP) of Cdh23 gene, we evaluated the mice at 12 months of age, which is generally considered as middle age40–42. As expected, the 12-month-old Keap1FA/FA mice were well protected from oxidative damage and degenerative alterations of the inner ear, and retained better hearing ability than the WT mice at the same age. These results strongly suggest that chemicals and stimuli that activate NRF2 pathway, or KEAP1 inhibitors, are effective for the prevention of AHL.
Results
AHL in the WT C57BL/6 mice
The C57BL/6 mouse is a well-known model of early onset of AHL, and functional and structural impairment of cochlea within 1 year have been documented42–44. We first examined AHL progression in the WT C57BL/6 mice by comparing those at 2, 5 and 12 months of age in our breeding colony. Auditory brainstem response (ABR) thresholds were significantly elevated in the 5-month-old mice compared with those in the 2-month-old mice at frequencies of 16 and 32 kHz, respectively (Fig. 1a). Significant elevation of the ABR thresholds at all frequencies from 4 to 32 kHz were observed by 12 months of age (Fig. 1a). These results suggest that the hearing ability became gradually impaired, and that AHL was fully apparent by 12 months of age. To assess the SGN function, we examined latency and amplitude of the ABR wave I induced by a stimulus intensity of 100 dB sound pressure level (SPL). The latencies were comparable irrespective of mouse age (Fig. 1b), whereas the amplitudes were mostly reduced in 5- and 12-month-old mice (Fig. 1c). These results imply that functional impairment of the SGN activity was already started by 5 months of age.
Fig. 1. Progression of hearing impairment in the WT C57BL/6 mice during aging.

ABR thresholds (a), ABR wave I latencies (b), and ABR wave I amplitudes (c) of the wild-type (WT) C57BL/6 mice at the age of 2, 5, and 12 months (n = 6 for 2-month-old mice, n = 3 for 5-month-old mice, and n = 6 for 12-month-old mice). The data represent the mean ± SEM. *P < 0.05, **P < 0.01. Two-way analysis of variance (ANOVA) followed by Tukey’s multiple comparison test was applied. As the ABR wave I latencies and amplitudes that were induced by a stimulus intensity of 100 dB SPL were measured, one and two 12-month-old mice that exhibited ABR thresholds above 100 dB SPL at 4 and 32 kHz, respectively, were omitted from the waveform analysis shown in b and c.
Histological alterations of the cochleae were apparent in the SGNs and SL at 12 months of age but not in SV (Fig. 2a–c). The SGN density was significantly decreased in the 12-month-old mice compared with that in the 2- and 5-month-old mice at all cochlear turns (Fig. 2a). The density of the SL fibrocytes, in particular those defined as type IV fibrocytes45, was lower in the 12-month-old mice than it was in the 2- and 5-month-old mice at all cochlear turns (Fig. 2b). In contrast, the thickness of the SV showed no significant differences among the three groups (Fig. 2c). Surface preparation analysis of the hair cells revealed a dramatic difference between the 2- and 12-month-old mice. Although occasional loss of outer hair cells (OHCs) was observed at only the basal turns in the 2-month-old mice, a high degree of OHC loss was apparent at all the turns in the 12-month-old mice (Fig. 2d). These results indicated that the WT mice had fully developed AHL by 12 months of age, both in functional and structural aspects.
Fig. 2. Progression of cochlear degeneration in the WT C57BL/6 mice during aging.

a–c Histological analysis of spiral ganglion neurons (SGNs) (a), spiral ligaments (SL) (b), and stria vascularis (SV) (c) at the apical, middle, and basal turns in the 2-, 5-, and 12-month-old mice (n = 6 in each group). SGN density (a), type IV fibrocyte density in the SL (b), and SV thickness (c) were quantified. Green arrows indicate neuronal soma (a), fibrocytes (b), and SV width (c). Areas of type IV fibrocytes are indicated by dashed green triangles. The data represent the mean ± SEM. *P < 0.05, **P < 0.01. Two-way ANOVA followed by Tukey’s multiple comparison test was applied. d Images of the surface preparation of the hair cells at 2 and 12 months. Missing outer hair cells (OHCs) are indicated with white arrowheads. Most OHCs in the basal turns at 12 months were lost, as indicated in the area circumscribed by the dashed line. IHCs, inner hair cells. Missing OHCs were quantitatively analyzed by evaluating 90 OHCs at each turn (n = 5 in each group). The data represent the mean ± SEM. **P < 0.01. Unpaired two-tailed Student’s t-test was applied. Scale bars correspond to 50 μm (a–c) and 40 μm (d).
Cochlear gene expression in WT C57BL/6 mice
We next compared NRF2 pathway activity in WT mice at 2, 5, and 12 months of age by examining the expression levels of representative NRF2 target genes Nqo1, Gclc, Gclm, Txnrd1, and Hmox1, as well as Nrf2. The expression levels were nearly the same among the three groups and no apparent age-related changes were observed, except for Gclc and Gclm, which were higher in the 5-month-old cochlea than the rest for unknown reasons (Fig. 3a). As the pathogenesis of NIHL correlates with inflammation46–48 and, as previous reports suggested, inflammation also contributes to AHL49,50, we additionally investigated the expression of the proinflammatory cytokine genes Il1b, Il6, and Tnfa. However, the expression of these proinflammatory cytokine genes was not significantly different among the three groups (Fig. 3b). These results suggested that either deterioration of NRF2 pathway activity or inner ear inflammation was not directly related to AHL in the WT mice in our experimental setting.
Fig. 3. Gene expression in the WT C57BL/6 cochleae during aging.
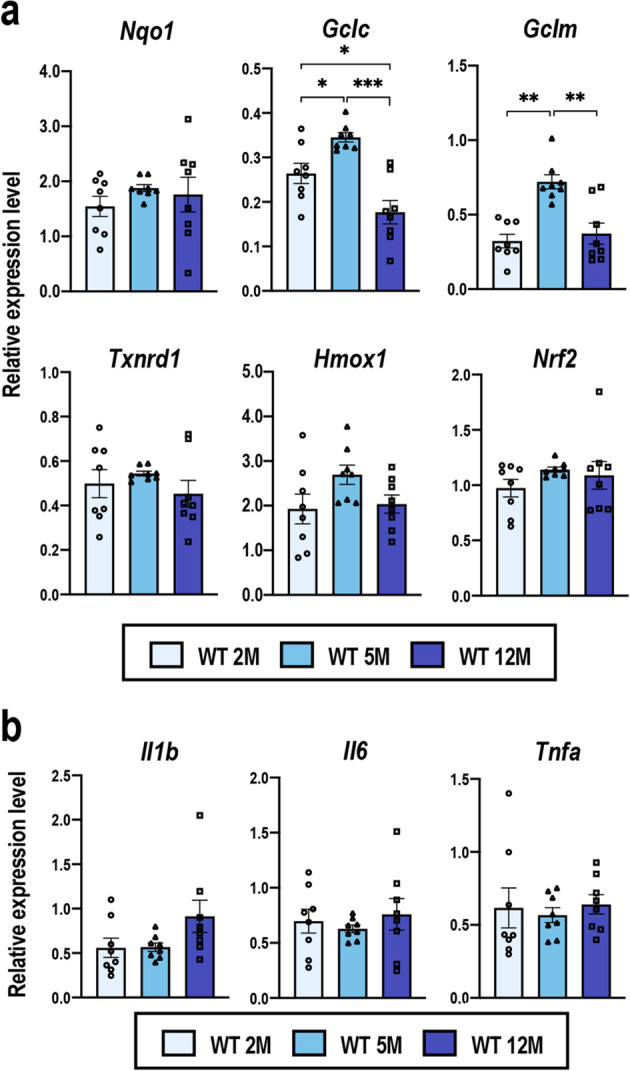
Expression levels of Nrf2, NRF2 target genes (a), and proinflammatory cytokine genes (b) in whole cochleae of the WT C57BL/6 mice at 2, 5, and 12 months of age were examined by quantitative real-time PCR. All the samples were quantified by using the same standard curve and each expression level was normalized to that of the Hprt or Beta-Actin expression level. Data represent the mean ± SEM (n = 8 in each group). *P < 0.05, **P < 0.01, ***P < 0.001. Two-way ANOVA followed by Tukey’s multiple comparison test was applied.
Comparison of the Keap1FA/FA mice and WT mice at 2 months of age
The C57BL/6 strain of mice possesses an SNP in the Cdh23 gene, Cdh23ahl allele, which is thought to cause an early decline in hearing ability in this strain of mice41,51. To verify the genotype of Cdh23 in the Keap1FA/FA and WT mice, which had been backcrossed into the C57BL/6 background, we sequenced their tail DNA samples obtained from our breeding colony through random sampling. All the Keap1FA/FA and WT mice had the Cdh23ahl allele (Cdh23753A/753A) (Fig. 4a).
Fig. 4. Gene expression in cochleae of the WT and Keap1FA/FA mice at 2 months of age.

a Detection of an SNP in the Cdh23 gene (n = 6 in each group). Red arrowheads indicate the SNP of the Cdh23753A allele. Expression levels of Keap1 (b) and NRF2 target genes and Nrf2 (c) in the whole cochleae were measured by quantitative real-time PCR (n = 8 in each group). All the samples were quantified by using the same standard curve and each expression level was normalized to Hprt expression. Average expression levels of WT cochlea are set as 1. Data represent the mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001. Unpaired two-tailed Student’s t-test was applied. A part of data on the WT mice at 2 months of age are the same as those shown in Fig. 3.
We next characterized the inner ears of the Keap1FA/FA mice at 2 months of age. Although the Keap1FA/FA mice were shown to exhibit activation of the NRF2 pathway due to reduced expression of the Keap1 gene in various organs38,39, no information was available on the Keap1 expression status of the cochlea in the Keap1FA/FA mice. We found decreased expression of Keap1 (Fig. 4b) and significantly increased expression of Nqo1, Txnrd1, Gclc, Gstp1, Pgd, and Idh1, 6 out of 18 representative NRF2 target genes (Fig. 4c), in Keap1FA/FA cochlea compared with WT cochlea. Expression of Gclm, Prdx1, and Gpx2 tended to be increased in Keap1FA/FA cochlea, but without reaching statistical significance (Fig. 4c). Nrf2 mRNA expression were comparable between the two genotypes (Fig. 4c). These results suggested that the NRF2 pathway was activated in the cochlea by decreased expression of Keap1, although we could not detect a clear increase in NRF2 protein level in the Keap1FA/FA cochlear extracts (data not shown), probably because NRF2 protein accumulation induced by ~50% reduction of Keap1 mRNA was rather mild. Histological and morphological examination showed that the cochleae were not apparently different between the mice of the two genotypes (Fig. 5a–c).
Fig. 5. Comparison of cochlear structure in the WT and Keap1FA/FA mice at 2 months of age.

a, b Cochlear histology at each turn. The SGN density (a) and density of the SL fibrocytes (b) are shown (n = 6 in each group). c Surface preparation images of the hair cells. Missing OHCs are indicated with white arrowheads. Missing OHCs were quantitatively analyzed by evaluating 90 OHCs at each turn (n = 5 in each group). Data represent the mean ± SEM. Unpaired two-tailed Student’s t-test was applied. Differences between WT and Keap1FA/FA mice were not statistically significant. The samples of the WT mice at 2 months of age are the same as those shown in Fig. 2. Scale bars correspond to 50 μm (a, b) and 40 μm (c).
The ABR thresholds were comparable between Keap1FA/FA and WT mice at all frequencies (Fig. 6a). Similarly, the ABR wave I latencies were comparable between the two genotypes (Fig. 6b). Intriguingly, the ABR wave I amplitudes were larger in Keap1FA/FA mice than WT mice at 2 months of age (Fig. 6c), implying functional robustness of SGNs in Keap1FA/FA cochlea.
Fig. 6. Comparison of ABR of the WT and Keap1FA/FA mice at 2 months of age.

ABR thresholds (a), ABR wave I latencies (b), and ABR wave I amplitudes (c) of the WT and Keap1FA/FA mice at the age of 2 months (n = 6). The data represent the mean ± SEM. *P < 0.05. Unpaired two-tailed Student’s t-test was applied. The samples of the WT mice at 2 months of age are the same as those shown in Fig. 1.
Comparison of the Keap1FA/FA mice and WT mice at 5 months of age
At 5 months of age, when elevation of ABR thresholds was already detectable in WT mice (see Fig. 1a), the ABR thresholds of Keap1FA/FA mice tended to be lower than those of WT mice, although the difference did not reach statistical significance (Fig. 7a). The ABR wave I latencies were comparable between Keap1FA/FA and WT mice as observed at 2 months of age (Fig. 7b). The ABR wave I amplitudes were larger in Keap1FA/FA mice than WT mice as observed at 2 months of age (Fig. 7c), although the amplitudes in 5-month-old mice were lower than those in 2-month-old mice in both genotypes (see Fig. 6c). Histological differences were not obvious between Keap1FA/FA and WT mice (Fig. 7d, e).
Fig. 7. Comparison of ABR and cochleae in the WT and Keap1FA/FA mice at 5 months of age.

ABR thresholds (a), ABR wave I latencies (b), and ABR wave I amplitudes (c) of the WT and Keap1FA/FA mice at the age of 5 months (n = 3 for WT mice and n = 5 for Keap1FA/FA mice). d, e Cochlear histology at each turn. The SGN density (d) and density of the SL fibrocytes (e) are shown (n = 6 for WT mice and n = 5 for Keap1FA/FA mice). The data represent the mean ± SEM. *P < 0.05. Unpaired two-tailed Student’s t-test was applied. The samples of the WT mice at 5 months of age are the same as those shown in Figs. 1 and 2. Scale bars correspond to 50 μm (d, e).
Protection of the cochleae from age-related degeneration by Keap1 knockdown
We then examined histological and morphological alterations in the Keap1FA/FA and WT cochleae at 12 months of age, when AHL fully developed in WT mice, in terms of SGN density, SL fibrocyte density, and OHC loss. The SGN density at the apical and middle turns in the Keap1FA/FA mice was retained significantly better than it was in the WT mice (Fig. 8a). No differences were apparent in the SGNs at the basal turns in the two mouse genotypes, as the basal SGNs were highly degenerated in the mice of both genotypes. The SL fibrocyte density in the Keap1FA/FA mice tended to be higher than it was in the WT mice and the difference in the middle turns reached statistical significance (Fig. 8b). According to the surface preparation images, the OHCs were remarkably well retained in the Keap1FA/FA cochleae at the apical and middle turns (Fig. 8c). The OHCs at the basal turns were highly degenerated in both groups. Thus, the morphological and histological integrity of the cochleae at the apical and middle turns was better retained in the Keap1FA/FA mice than it was in the WT mice at 12 months of age.
Fig. 8. Comparison of cochleae in the WT and Keap1FA/FA mice at 12 months of age.

a, b Cochlear histology at each turn. The SGN density (a) and density of the SL fibrocytes (b) are shown (n = 6 in each group). c Images of the surface preparation of the hair cells. Missing OHCs are indicated with white arrowheads. Almost all OHCs at the basal turns are missing, as indicated by the area circumscribed by the dashed line. Missing OHCs were quantitatively analyzed by evaluating 90 OHCs at each turn (n = 5 in each group). Data represent the mean ± SEM. *P < 0.05, **P < 0.01. Unpaired two-tailed Student’s t-test was applied. The samples of the WT mice at 12 months of age are the same as those shown in Fig. 2. Scale bars correspond to 50 μm (a, b) and 40 μm (c).
Attenuation of AHL in the Keap1FA/FA mice
To examine whether histological and morphological preservation of Keap1FA/FA cochlea resulted in the attenuation of AHL, we measured the ABR thresholds of the Keap1FA/FA and WT mice at 12 months of age (Fig. 9a). The ABR thresholds were lower in the 12-month-old Keap1FA/FA mice, especially at 4, 8, and 12 kHz, than they were in the 12-month-old WT mice, indicating that hearing ability was better preserved in the Keap1FA/FA mice than it was in the WT mice for low- and mid-frequency sounds. Thresholds at 32 kHz were elevated in both the 12-month-old Keap1FA/FA and WT mice, indicating that the hearing ability at high frequencies was impaired similarly in the mice of both genotypes. These results are in good agreement with the morphological and histological observations: basal turns that are critical for sensing high-frequency sounds were profoundly degenerated irrespective of Keap1 status, whereas the apical and middle turns that are critical for sensing low- and mid-frequencies were better preserved in the Keap1FA/FA mice than they were in the WT mice.
Fig. 9. Comparison of ABR of the WT and Keap1FA/FA mice at 12 months of age.

ABR thresholds (a, n = 6 for each group), ABR wave I latencies (b, n = 6 for WT mice and n = 5 for Keap1FA/FA mice), and ABR wave I amplitudes (c, n = 6 for WT mice and n = 5 for Keap1FA/FA mice) of the WT and Keap1FA/FA mice at the age of 12 months. One and two WT mice that exhibited ABR thresholds above 100 dB SPL at 4 and 32 kHz, respectively, were omitted from the waveform analysis shown in b and c. Likewise, one Keap1FA/FA mouse that exhibited an ABR threshold above 100 dB SPL at 32 kHz was omitted from the waveform analysis shown in b and c. The data represent the mean ± SE. *P < 0.05. Unpaired two-tailed Student’s t-test was applied. The samples of the WT mice at 12 months of age are the same as those shown in Fig. 1.
The ABR wave I latencies and amplitudes were comparable between the two genotypes at 12 months of age (Fig. 9b, c). Of note, the amplitudes, which were larger in Keap1FA/FA mice than in WT mice at 2 and 5 months of age, were reduced to the level similar to those of WT mice by 12 months of age, at which age the cochlear histology and ABR thresholds were still better preserved in Keap1FA/FA mice than WT mice. As wave I amplitudes are reported to be more sensitive indicators for damages of hair cells and/or SGNs than their histological loss and ABR thresholds52, beneficial impacts of Keap1 inhibition on wave I amplitudes might have disappeared prior to those on histological signs and ABR thresholds. Keap1FA/FA cochlea, although delayed, might eventually exhibit functional and structural declines similar to those of WT cochlea.
Decrease in oxidative stress in the Keap1FA/FA cochleae
We examined whether NRF2 target genes were elevated in the Keap1FA/FA cochleae in mice at 12 months of age (Fig. 10a). Eleven genes out of 18 representative NRF2 target genes were significantly upregulated in the Keap1FA/FA cochleae. Expression levels of the remaining seven genes tended to be also higher in the Keap1FA/FA cochleae than WT cochleae, but without reaching statistical significance. Thus, NRF2 pathway was regarded to be activated. The expression of proinflammatory cytokine genes was not significantly different between the Keap1FA/FA and WT mice or rather increased in the former at 12 months of age (Fig. 10b). Combining this finding with the observation that aging did not induce the proinflammatory cytokine gene expression in the WT cochleae (see Fig. 3b), we surmise that AHL attenuation in Keap1FA/FA mice is less attributable to the control of inflammation.
Fig. 10. Gene expression in the WT and Keap1FA/FA cochleae at 12 months of age.

Expression levels of NRF2 target genes and Nrf2 (a), and proinflammatory cytokine genes (b) in whole cochleae were evaluated by quantitative real-time PCR (n = 8 in each group). All the samples were quantified by using the same standard curve and each expression level was normalized to the Hprt or Beta-Actin expression level. Average expression levels of WT cochlea are set as 1. The data represent the mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001. Unpaired two-tailed Student’s t-test was applied. A part of data on the WT mice at 12 months of age are the same as those shown in Fig. 3.
As smoldering inflammation was unlikely to be the direct cause of the AHL in the mice used in this study, we hypothesized that NRF2 pathway activation due to Keap1 knockdown suppressed oxidative stress accumulation and protected the cochlea from the oxidative damage during aging. We examined the accumulation of 4-hydroxy-2-nonenal (4-HNE) as an indicator of oxidative stress in the cochlea by immunohistochemistry. The 4-HNE staining was almost undetectable in both the 2-month-old Keap1FA/FA and WT cochleae (Fig. 11a, b). A slight increase in the 4-HNE staining was observed in 5-month-old WT cochlea compared with Keap1FA/FA cochlea (Fig. 11c–e). Clear accumulation of 4-HNE was observed in both the 12-month-old Keap1FA/FA and WT cochleae but was more remarkable in the latter (Fig. 11f–h). Intense 4-HNE staining was observed in various portions of the cochlea and their intensity levels were nearly similar in all the cochlear turns in the WT mice. 4-HNE accumulation was consistently reduced in various cochlear portions of the Keap1FA/FA mice.
Fig. 11. Accumulation of 4-HNE in the WT and Keap1FA/FA cochleae during aging.

Immunohistochemical staining with 4-HNE in the cochleae at each turn of the Keap1FA/FA and WT mice at 2 (a), 5 (c), and 12 months (f). High-power field images are shown for each region (SGNs, SL, and organ of Corti (OC)) in the cochleae at the middle turns in the Keap1FA/FA and WT mice at 2 (b), 5 (d), and 12 months (g) of age. The experiments were performed for five samples each of 2- and 5-month-old mice and six samples of 12-month-old mice. Relative intensities of the staining at each turn of 5-month-old cochlea (e) and 12-month-old cochlea (h) were semi-quantified. Average staining intensities of WT mice are set as 1. The data represent the mean ± SEM. *P < 0.05, **P < 0.01. Unpaired two-tailed Student’s t-test was applied. Scale bars correspond to 100 μm (a, c, f) and 50 μm (b, d, g).
As an alternative oxidative stress marker, the accumulation of 8-hydroxydeoxyguanosine (8-OHdG) was measured by immunofluorescence in the cochleae of the 5- and 12-month-old mice. At 5 months of age, intensity of 8-OHdG was similarly low in the cochleae of both genotypes (Fig. 12a, b). At 12 months of age, intense 8-OHdG staining was detected in the SGNs and hair cells at the apical and middle turns of the WT cochlea, whereas the Keap1FA/FA mice exhibited less accumulation of 8-OHdG at the corresponding turns (Fig. 12c, d). The accumulation of 8-OHdG at the basal turns was not obvious, probably because the SGNs and hair cells had mostly degenerated (data not shown). These results suggest that NRF2 pathway activation maintains the functional and structural integrity of the cochlea by suppressing oxidative stress accumulation during aging.
Fig. 12. Accumulation of 8-OHdG in the WT and Keap1FA/FA cochleae at 5 and 12 months of age.

Immunofluorescence detection of 8-OHdG in the SGNs (a, c) and hair cells (b, d) at each turn in the Keap1FA/FA and WT mice at 5 (a, b) and 12 (c, d) months of age. 8-OHdG signals are shown as red fluorescence. Nuclei were counterstained with DAPI (blue). OHCs, outer hair cells; IHC, inner hair cell. The experiments were performed for five samples in each group. Relative intensities of the 8-OHdG signals at each turn were semi-quantified. Average staining intensities of WT mice are set as 1. The data represent the mean ± SEM. *P < 0.05. Unpaired two-tailed Student’s t-test was applied. Scale bars correspond to 50 μm (a, c) and 20 μm (b, d).
Discussion
In this study, we found that KEAP1 inhibition was effective for attenuating AHL progression in C57BL/6 mice. Suppression of oxidative stress by KEAP1 inhibition was considered to underlie the alleviation of the age-related functional decline of the cochlea. These results are in a good agreement with previous reports describing oxidative damage significantly related to AHL19,20. In particular, OHCs were surprisingly well protected in the Keap1FA/FA mice, suggesting that OHCs are susceptible to oxidative stress and that enhancement of antioxidant capacity has a large impact on the maintenance of OHC integrity during aging. Consistently, previous studies that analyzed mouse models subjected to oxidative stress described hearing impairment accompanied by loss of OHCs, as well as SGNs24,53,54. Although ROS level and antioxidant response of each cochlear component was not precisely evaluated, our study clearly demonstrated that the enhancement of antioxidant capacity is favorable for antagonizing age-related degenerative changes in the cochleae. Considering the strictly specific relationship between KEAP1 and NRF2, namely all apparent phenotypes caused by KEAP1 inhibition, are reversed by simultaneous NRF2 inhibition55–60, it is most likely that NRF2 activation due to Keap1 knockdown is crucial for the prevention of AHL in the Keap1FA/FA mice.
The redox steady state depends on the balance between the rates of ROS production and ROS elimination61. Similar to the findings of previous studies62–65, our results revealed oxidative stress accumulation in the cochleae of the old mice, in which the ROS production level is thought to override the ROS elimination levels. It has been the consensus that, during aging, mitochondrion-derived ROS production increases due to the functional impairment of the mitochondria64,66,67. In contrast, changes in ROS elimination caused by aging are likely to vary depending on the tissues and contexts. Antioxidant defense activities were reported to decrease in cochlea and brain during aging68,69; however, other reports showed that antioxidant defense activities in the muscle and brain remained unchanged or increased65,70–72. In this study, we found that NRF2 pathway activity was not changed in the cochleae for as long as 12 months. We surmise that ROS accumulation during physiological aging is not sufficient to induce NRF2 pathway activation. This supposition explains the reason that active induction of NRF2 pathway by KEAP1 inhibition is effective for the attenuation of AHL.
Notably, NRF2 pathway activation has a dominant protective effect on the apical and middle turns of the cochlea during aging. Currently, mechanisms are unknown how enhanced NRF2 pathway activity exerts differential impacts on each cochlear turn. One of the possible explanations would be that NRF2 pathway may be more active at the apical and middle turns than the basal turns, considering analogy with SOD2 whose antioxidant activity was shown different among cochlear turns73. SOD2, which is involved in the NRF2-independent antioxidant pathway, is expressed in the SGNs and their expression levels were found to be higher at the apical and middle turns than it was at basal turns. Antioxidant defense capacities might be generally limited in the basal turns of cochleae. Another possibility is that the basal turns may be affected by molecular mechanisms different from those of oxidative stress, such as mechanical stress. A previous study clearly showed that Cdh23 SNP inherent to C57BL/6 strain of mice is a cause of the early onset of AHL particularly at high frequencies24. As CDH23, a product of Cdh23 gene, is a component of tip link in stereocilia controlling mechanoelectrical transduction by hair cells74, the Cdh23 SNP is likely to augment mechanical stress, especially in basal turns, which is responsible for sensing of high-frequency stimuli. The impact of antioxidant capacity enhancement by NRF2 pathway activation might be thus diminished in basal turns.
In humans, auditory thresholds typically increase from high frequency at the beginning of AHL, followed by hearing impairment at low–mid frequencies3,20. As auditory capacity at mid-frequency is critical for conversation comprehension, preventing the progression of the initial stage AHL, before it advances toward the stage with mid-frequency loss, has a large impact on the quality of life of elderly people. NRF2 activation was shown to be effective for the prevention of NIHL35. Administration of an NRF2 inducer, or a KEAP1 inhibitor, before noise exposure, but not after exposure, successfully prevented NIHL. This result implies that early intervention with NRF2 inducers is favorable for the effective prevention of AHL. We propose to start medication for AHL with NRF2 inducers from its initial stage, which is expected to protect the apical and middle turns of the cochlea effectively to maintain auditory capacity at low–mid frequencies. Although currently available NRF2 inducers are all KEAP1 inhibitors, NRF2 could be also activated independently of KEAP1 function by a compound that directly bound to the N terminus of NRF2 and hindered the interaction with KEAP1. To explore and make use of such a compound, further verification is required for the substantial contribution of NRF2 to the prevention of AHL, because we could not obtain a direct evidence of NRF2 nuclear translocation in the Keap1FA/FA cochlear cells in this study due to the technical difficulty. Simultaneous disruption of Nrf2 gene in Keap1FA/FA mice will overcome this limitation and give us the answer in the future study.
Methods
Animals
Male WT and Keap1-knockdown (Keap1FA/FA) mice38,39 on a C57BL/6 genetic background were used in this study. Keap1FA is a hypomorphic allele due to the insertion of loxP sequences. Keap1FA/FA mice are homozygous for this hypomorphic allele. Mice were genotyped by PCR using the following primers: Keap1FA forward, 5′-CAG CAG TTA AGG GCA CCA ATG C-3′ and Keap1FA reverse, 5′-CCT GCC TCA GCT TCC CAT CA-3′. The mice were maintained on a normal diet with water ad libitum and housed under a standard 12 h light/12 h dark schedule. All the mice were treated in accordance with guidelines presented in The Standards for Human Care and Use of Laboratory Animals of Tohoku University and Guidelines for Proper Conduct of Animal Experiments by the Ministry of Education, Culture, Sports, Science, and Technology of Japan. The mouse experiments were approved by Institutional Laboratory Animal Care and Use Committee of Tohoku University and Safety Committee for Recombinant DNA Experiments of Tohoku University.
Hearing function test
The mice were anesthetized using ketamine (100 mg/kg body weight) and xylazine (20 mg/kg body weight) by intraperitoneal administration. ABR recording was conducted using a TDT System 3 auditory-evoked potential workstation and BioSigRP software (Tucker-Davis Technologies). ABR responses were evoked using bursts of pure tones at frequencies of 4, 8, 12, 16, and 32 kHz. Evoked responses were averaged across 1000 sweeps. The responses were recorded for each stimulus level in 5 dB steps from 100 dB SPL to 10 dB SPL. The ABR threshold was defined as the lowest sound intensity sufficient to elicit at least one peak against the averaged ABR value.
The ABR wave I amplitude was determined by measuring the voltage difference between the highest positive value and lowest negative value for the first wave at 100 dB SPL. The ABR wave I latency was measured as the length of time between the onset of the stimulus and the peak of the first wave at 100 dB SPL.
Histological analysis
Cochleae were quickly dissected from the skull and immediately soaked in 4% paraformaldehyde (PFA). Small holes were made at the round window, oval window, and apex of the cochleae. The cochleae were fixed with 4% PFA at 4 °C overnight and then decalcified in 10% EDTA for 2 days at 4 °C. The decalcified cochleae were embedded in paraffin and 3 μm coronal sections were made. These sections were stained with hematoxylin and eosin, and were visualized using a light microscope (BZ-9000, Keyence, Osaka, Japan). Three cochlear turns (apical, middle, and basal) were used for histological evaluation of each cochlea. Three sections per animal were used for calculation of the mean numbers. Area measurements and cell counts were performed using BZ-H1C software (Keyence).
Hair cell count
For counting of hair cells, surface preparations of the organ of Corti and the basilar membrane were prepared. The hair cells were stained for F-actin with rhodamine-conjugated phalloidin (1 : 100, Invitrogen) at room temperature for 30 min under light-protected conditions. High-power fluorescence images were obtained using a microscope (BZ-9000, Keyence) and BZ-H1C software (Keyence). The quantitative results were obtained by evaluating 90 OHCs for each turn in a given microscopic field.
Quantitative real-time PCR analysis
The cochleae were quickly dissected from the skull, placed on ice, and stored at −80 °C. The cochleae were homogenized in ISOGEN (Nippon gene) and, for each sample, the total RNA was purified from whole cochleae. cDNA was synthesized using reverse transcriptase (ReverTra Ace, Toyobo). Quantitative PCR was performed on a QuantStudio3 sequence detector system using Thunderbird SYBR qPCR mix (Toyobo) for the SYBR green system and Thunderbird probe qPCR mix (Toyobo) for the TaqMan probe system. The primers utilized in this study are shown in Supplementary Table 1.
Immunohistochemistry
Cochlear samples were prepared using a procedure similar to that used for the histological analysis. The decalcified cochleae were embedded in paraffin and 3 μm coronal sections were made. Immunohistochemistry with anti-4-HNE antibody was performed using a Histofine®⃞ mouse stain kit (#424021, Nichirei). Tissue sections were deparaffinized, and endogenous peroxidase was blocked by 3% hydrogen peroxide/phosphate-buffered saline (PBS). Sections were subsequently blocked with 10% rabbit serum for 10 min at room temperature and incubated overnight with a primary antibody (anti-4-HNE; 1 : 200, MHN-020P, JaICA) at 4 °C. All sections were washed three times in PBS and incubated with a secondary biotin-conjugated antibody (anti-mouse IgG in Histofine®⃞ mouse stain kit; 1 : 500) for 10 min at room temperature. Then, the sections were incubated with peroxidase-conjugated streptavidin and the 3,3′-diaminobenzidine (DAB) substrate was added. Hematoxylin was used for counterstaining.
Immunofluorescence was performed with an anti-8-OHdG antibody. After deparaffinization, we conducted antigen retrieval by autoclaving for 10 min at 121 °C. The tissue sections were blocked with 1% fetal bovine serum (FBS)/PBS for 30 min at room temperature and incubated overnight with a primary antibody (anti-8-OHdG; 1 : 200, bs-1278R, Bioss) diluted in 1% FBS/PBS at 4 °C. All the sections were subsequently incubated with a secondary antibody (Alexa Fluor 594 anti-rabbit; 1 : 300, A11012, Thermo Fisher Scientific) for 1.5 h at room temperature in light-protected conditions. The nuclei were counterstained with 4′,6-diamidino-2-phenylindole (1 : 1000).
Semi-quantification of 4-HNE staining intensity was conducted using ImageJ Fiji software as described previously75. Color deconvolution was applied to images, which were converted to black and white images. Subsequently, the maximum threshold was adjusted so that background signal was removed, and DAB signals were measured and expressed as the intensity per area of the cochlear section from each cochlear turn. Immunofluorescence intensity of 8-OHdG was measured using a BZ-9000 microscope equipped with Dynamic cell count software BZ-H1C (Keyence).
Statistical analysis
All data are presented as the mean ± SEM. Student’s t-test and two-way analysis of variance followed by Tukey’s multiple comparison test were utilized. For all tests, P-values of < 0.05 were considered significant.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Supplementary information
Acknowledgements
We thank Professor Masi Yamamoto for providing Keap1FA/FA mice and constructive advice, Ms. N. Shibata for technical support regarding histological analysis, and the Biomedical Research Core of the Tohoku University Graduate School of Medicine for its technical support. This work was supported by JSPS under grant numbers 19K07361 (D.M.), 18K16826 (Y.H.), 18H02949 (Y.K.), 18H02621 (H.M.), and 18H04794 (H.M.), the Naito Foundation (H.M.), a research grant from the Princess Takamatsu Cancer Research Fund 15-24728 (H.M.), the Uehara Memorial Foundation (H.M.), and AMED under grant number JP20gm5010002 (H.M.). The funders had no role in the study design, data collection and analysis, decision to publish, or manuscript preparation.
Author contributions
T.O. designed the study, conducted the experiments, analyzed the data, and wrote the paper. N.O. conducted the experiments. D.M., H.K., T.Z., and Y.H. provided critical biomaterials and analyzed the data. Y.K. provided critical instruments and an analysis platform for the study and analyzed the data. H.M. designed the study, supervised the research, analyzed the data, and wrote the paper.
Data availability
The data generated or analyzed during this study are available from the corresponding author on request. Keap1FA/FA mice used in this study are available from RIKEN BRC (https://knowledge.brc.riken.jp/resource/animal/card?brc_no=RBRC09595&__lang__=en).
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary information is available for this paper at 10.1038/s41514-020-00053-4.
References
- 1.Gates GA, Mills JH. Presbycusis. Lancet. 2005;366:1111–1120. doi: 10.1016/S0140-6736(05)67423-5. [DOI] [PubMed] [Google Scholar]
- 2.Yamasoba T, et al. Current concepts in age-related hearing loss: epidemiology and mechanistic pathways. Hear Res. 2013;303:30–38. doi: 10.1016/j.heares.2013.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yang CH, Schrepfer T, Schacht J. Age-related hearing impairment and the triad of acquired hearing loss. Front. Cell Neurosci. 2015;9:276. doi: 10.3389/fncel.2015.00276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lin FR, Thorpe R, Gordon-Salant S, Ferrucci L. Hearing loss prevalence and risk factors among older adults in the United States. J. Gerontol. A Biol. Sci. Med. Sci. 2011;66:582–590. doi: 10.1093/gerona/glr002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schuknecht HF, Gacek MR. Cochlear pathology in presbycusis. Ann. Otol. Rhinol. Laryngol. 1993;102:1–16. doi: 10.1177/00034894931020S101. [DOI] [PubMed] [Google Scholar]
- 6.Nelson EG, Hinojosa R. Presbycusis: a human temporal bone study of individuals with downward sloping audiometric patterns of hearing loss and review of the literature. Laryngoscope. 2006;116:1–12. doi: 10.1097/01.mlg.0000236089.44566.62. [DOI] [PubMed] [Google Scholar]
- 7.Keithley, E. M. Pathology and mechanisms of cochlear aging. J. Neurosci. Res. 98, 1674–1684 (2020). [DOI] [PMC free article] [PubMed]
- 8.Sergeyenko Y, Lall K, Liberman MC, Kujawa SG. Age-related cochlear synaptopathy: an early-onset contributor to auditory functional decline. J. Neurosci. 2013;33:13686–13694. doi: 10.1523/JNEUROSCI.1783-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boero LE, et al. Preventing presbycusis in mice with enhanced medial olivocochlear feedback. Proc. Natl Acad. Sci. USA. 2020;117:11811–11819. doi: 10.1073/pnas.2000760117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kujawa SG, Liberman MC. Acceleration of age-related hearing loss by early noise exposure: evidence of a misspent youth. J. Neurosci. 2006;26:2115–2123. doi: 10.1523/JNEUROSCI.4985-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Alvarado JC, Fuentes-Santamaria V, Gabaldón-Ull MC, Juiz JM. Age-related hearing loss is accelerated by repeated short-duration loud sound stimulation. Front. Neurosci. 2019;13:77. doi: 10.3389/fnins.2019.00077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Joo Y, et al. The contribution of ototoxic medications to hearing loss among older adults. J. Gerontol. A Biol. Sci. Med. Sci. 2020;75:561–566. doi: 10.1093/gerona/glz166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mitchell P, et al. Relationship of type 2 diabetes to the prevalence, incidence and progression of age-related hearing loss. Diabet. Med. 2009;26:483–488. doi: 10.1111/j.1464-5491.2009.02710.x. [DOI] [PubMed] [Google Scholar]
- 14.Gates GA, Cobb JL, D’Agostino RB, Wolf PA. The relation of hearing in the elderly to the presence of cardiovascular disease and cardiovascular risk factors. Arch. Otolaryngol. Head Neck Surg. 1993;119:156–161. doi: 10.1001/archotol.1993.01880140038006. [DOI] [PubMed] [Google Scholar]
- 15.Van Eyken E, et al. Contribution of the N-acetyltransferase 2 polymorphism NAT2*6A to age-related hearing impairment. J. Med. Genet. 2007;44:570–578. doi: 10.1136/jmg.2007.049205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nolan LS, Cadge BA, Gomez-Dorado M, Dawson SJ. A functional and genetic analysis of SOD2 promoter variants and their contribution to age-related hearing loss. Mech. Ageing Dev. 2013;134:298–306. doi: 10.1016/j.mad.2013.02.009. [DOI] [PubMed] [Google Scholar]
- 17.Morgan A, et al. Next-generation sequencing identified SPATC1L as a possible candidate gene for both early-onset and age-related hearing loss. Eur. J. Hum. Genet. 2019;27:70–79. doi: 10.1038/s41431-018-0229-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wells, H. R. R., Newman, T. A. & Williams, F. M. K. Genetics of age-related hearing loss. J. Neurosci. Res.10.1002/jnr.24549 (2020). [DOI] [PubMed]
- 19.Fujimoto C, Yamasoba T. Oxidative stresses and mitochondrial dysfunction in age-related hearing loss. Oxid. Med. Cell Longev. 2014;2014:582849. doi: 10.1155/2014/582849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang J, Puel JL. Presbycusis: an update on cochlear mechanisms and therapies. J. Clin. Med. 2020;9:218. doi: 10.3390/jcm9010218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bánfi B, et al. NOX3, a superoxide-generating NADPH oxidase of the inner ear. J. Biol. Chem. 2004;279:46065–46072. doi: 10.1074/jbc.M403046200. [DOI] [PubMed] [Google Scholar]
- 22.Böttger EC, Schacht J. The mitochondrion: a perpetrator of acquired hearing loss. Hear. Res. 2013;303:12–19. doi: 10.1016/j.heares.2013.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.White K, et al. Loss of IDH2 Accelerates age-related hearing loss in male mice. Sci. Rep. 2018;8:5039. doi: 10.1038/s41598-018-23436-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Johnson KR, et al. Separate and combined effects of Sod1 and Cdh23 mutations on age-related hearing loss and cochlear pathology in C57BL/6J mice. Hear. Res. 2010;268:85–92. doi: 10.1016/j.heares.2010.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Someya S, et al. Sirt3 mediates reduction of oxidative damage and prevention of age-related hearing loss under caloric restriction. Cell. 2010;143:802–812. doi: 10.1016/j.cell.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Seidman MD. Effects of dietary restriction and antioxidants on presbycusis. Laryngoscope. 2000;110:727–738. doi: 10.1097/00005537-200005000-00003. [DOI] [PubMed] [Google Scholar]
- 27.Ding D. N-acetyl-cysteine prevents age-related hearing loss and the progressive loss of inner hair cells in γ-glutamyl transferase 1 deficient mice. Aging. 2016;8:730–750. doi: 10.18632/aging.100927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim YR, et al. Therapeutic potential of the mitochondria-targeted antioxidant MitoQ in mitochondrial-ROS induced sensorineural hearing loss caused by Idh2 deficiency. Redox Biol. 2019;20:544–555. doi: 10.1016/j.redox.2018.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Someya S, et al. Age-related hearing loss in C57BL/6J mice is mediated by Bak-dependent mitochondrial apoptosis. Proc. Natl Acad. Sci. USA. 2009;106:19432–19437. doi: 10.1073/pnas.0908786106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Motohashi H, Yamamoto M. Nrf2-Keap1 defines a physiologically important stress response mechanism. Trends Mol. Med. 2004;10:549–557. doi: 10.1016/j.molmed.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 31.Yamamoto M, Kensler TW, Motohashi H. The KEAP1-NRF2 system: a thiol-based sensor-effector apparatus for maintaining redox homeostasis. Physiol. Rev. 2018;98:1169–1203. doi: 10.1152/physrev.00023.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kobayashi EH, et al. Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nat. Commun. 2016;7:11624. doi: 10.1038/ncomms11624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Suzuki T, et al. Systemic activation of NRF2 alleviates lethal autoimmune inflammation in scurfy mice. Mol. Cell Biol. 2017;37:e00063–17. doi: 10.1128/MCB.00063-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Uruno A, et al. Nrf2 suppresses oxidative stress and inflammation in App knock-in Alzheimer’s disease model mice. Mol. Cell Biol. 2020;40:e00467–19. doi: 10.1128/MCB.00467-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Honkura Y, et al. NRF2 is a key target for prevention of noise-Induced hearing loss by reducing oxidative damage of cochlea. Sci. Rep. 2016;6:19329. doi: 10.1038/srep19329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Park HJ, et al. GSTA4 mediates reduction of cisplatin ototoxicity in female mice. Nat. Commun. 2019;10:4150. doi: 10.1038/s41467-019-12073-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hoshino T, et al. Protective role of Nrf2 in age-related hearing loss and gentamicin ototoxicity. Biochem. Biophys. Res. Commun. 2011;415:94–98. doi: 10.1016/j.bbrc.2011.10.019. [DOI] [PubMed] [Google Scholar]
- 38.Okawa H, et al. Hepatocyte-specific deletion of the keap1 gene activates Nrf2 and confers potent resistance against acute drug toxicity. Biochem. Biophys. Res. Commun. 2006;339:79–88. doi: 10.1016/j.bbrc.2005.10.185. [DOI] [PubMed] [Google Scholar]
- 39.Taguchi K, et al. Genetic analysis of cytoprotective functions supported by graded expression of Keap1. Mol. Cell Biol. 2010;30:3016–3026. doi: 10.1128/MCB.01591-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bowl MR, Dawson SJ. The mouse as a model for age-related hearing loss - a mini-review. Gerontology. 2015;61:149–157. doi: 10.1159/000368399. [DOI] [PubMed] [Google Scholar]
- 41.Noben-Trauth K, Zheng Q, Johnson K. Association of cadherin 23 with polygenic inheritance and genetic modification of sensorineural hearing loss. Nat. Genet. 2003;35:21–23. doi: 10.1038/ng1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Henry KR. Males lose hearing earlier in mouse models of late-onset age-related hearing loss; females lose hearing earlier in mouse models of early-onset hearing loss. Hear. Res. 2004;190:141–148. doi: 10.1016/S0378-5955(03)00401-5. [DOI] [PubMed] [Google Scholar]
- 43.Park SN, et al. Comparison of cochlear morphology and apoptosis in mouse models of presbycusis. Clin. Exp. Otorhinolaryngol. 2010;3:126–135. doi: 10.3342/ceo.2010.3.3.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Han C, et al. Sirt1 deficiency protects cochlear cells and delays the early onset of age-related hearing loss in C57BL/6 mice. Neurobiol. Aging. 2016;43:58–71. doi: 10.1016/j.neurobiolaging.2016.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hequembourg S, Liberman MC. Spiral ligament pathology: a major aspect of age-related cochlear degeneration in C57BL/6 mice. J. Assoc. Res. Otolaryngol. 2001;2:118–129. doi: 10.1007/s101620010075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fredelius L, Rask-Andersen H. The role of macrophages in the disposal of degeneration products within the organ of corti after acoustic overstimulation. Acta Otolaryngol. 1990;109:76–82. doi: 10.3109/00016489009107417. [DOI] [PubMed] [Google Scholar]
- 47.Fujioka M, et al. Proinflammatory cytokines expression in noise-induced damaged cochlea. J. Neurosci. Res. 2006;83:575–583. doi: 10.1002/jnr.20764. [DOI] [PubMed] [Google Scholar]
- 48.Kalinec GM, Lomberk G, Urrutia RA, Kalinec F. Resolution of cochlear inflammation: novel target for preventing or ameliorating drug-, noise- and age-related hearing loss. Front. Cell Neurosci. 2017;11:192. doi: 10.3389/fncel.2017.00192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Frye MD, Yang W, Zhang C, Xiong B, Hu BH. Dynamic activation of basilar membrane macrophages in response to chronic sensory cell degeneration in aging mouse cochleae. Hear. Res. 2017;344:125–134. doi: 10.1016/j.heares.2016.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Verschuur C, Agyemang-Prempeh A, Newman TA. Inflammation is associated with a worsening of presbycusis: evidence from the MRC national study of hearing. Int. J. Audiol. 2014;53:469–475. doi: 10.3109/14992027.2014.891057. [DOI] [PubMed] [Google Scholar]
- 51.Johnson K, et al. Effects of Cdh23 single nucleotide substitutions on age-related hearing loss in C57BL/6 and 129S1/Sv mice and comparisons with congenic strains. Sci. Rep. 2017;7:44450. doi: 10.1038/srep44450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kujawa SG, Liberman MC. Adding insult to injury: cochlear nerve degeneration after “temporary” noise-induced hearing loss. Version 2. J. Neurosci. 2009;29:14077–14085. doi: 10.1523/JNEUROSCI.2845-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Someya S, et al. The role of mtDNA mutations in the pathogenesis of age-related hearing loss in mice carrying a mutator DNA polymerase gamma. Neurobiol. Aging. 2008;29:1080–1092. doi: 10.1016/j.neurobiolaging.2007.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.McFadden SL, et al. Cu/Zn SOD deficiency potentiates hearing loss and cochlear pathology in aged 129,CD-1 mice. J. Comp. Neurol. 1999;413:101–112. doi: 10.1002/(SICI)1096-9861(19991011)413:1<101::AID-CNE7>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 55.Wakabayashi N, et al. Keap1-null mutation leads to postnatal lethality due to constitutive Nrf2 activation. Nat. Genet. 2003;35:238–245. doi: 10.1038/ng1248. [DOI] [PubMed] [Google Scholar]
- 56.Mitsuishi Y, et al. Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell. 2012;22:66–79. doi: 10.1016/j.ccr.2012.05.016. [DOI] [PubMed] [Google Scholar]
- 57.Taguchi K, et al. Nrf2 enhances cholangiocyte expansion in Pten-deficient livers. Mol. Cell Biol. 2014;34:900–913. doi: 10.1128/MCB.01384-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Murakami S, et al. NRF2 activation impairs quiescence and bone marrow reconstitution capacity of hematopoietic stem cells. Mol. Cell Biol. 2017;37:e00086–17. doi: 10.1128/MCB.00086-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Suzuki T, et al. Hyperactivation of Nrf2 in early tubular development induces nephrogenic diabetes insipidus. Nat. Commun. 2017;8:14577. doi: 10.1038/ncomms14577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kitamura H, Onodera Y, Murakami S, Suzuki T, Motohashi H. IL-11 contribution to tumorigenesis in an NRF2 addiction cancer model. Oncogene. 2017;36:6315–6324. doi: 10.1038/onc.2017.236. [DOI] [PubMed] [Google Scholar]
- 61.Davalli P, Mitic T, Caporali A, Lauriola A, D’Arca D. ROS, cell senescence, and novel molecular mechanisms in aging and age-related diseases. Oxid. Med. Cell Longev. 2016;2016:3565127. doi: 10.1155/2016/3565127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sen T, et al. Depolarization and cardiolipin depletion in aged rat brain mitochondria: relationship with oxidative stress and electron transport chain activity. Neurochem. Int. 2007;50:719–725. doi: 10.1016/j.neuint.2007.01.007. [DOI] [PubMed] [Google Scholar]
- 63.Chabi B, et al. Mitochondrial function and apoptotic susceptibility in aging skeletal muscle. Aging Cell. 2008;7:2–12. doi: 10.1111/j.1474-9726.2007.00347.x. [DOI] [PubMed] [Google Scholar]
- 64.Chakrabarti S, et al. Mitochondrial dysfunction during brain aging: role of oxidative stress and modulation by antioxidant supplementation. Aging Dis. 2011;2:242–256. [PMC free article] [PubMed] [Google Scholar]
- 65.Miwa S, et al. Decreased mTOR signalling reduces mitochondrial ROS in brain via accumulation of the telomerase protein TERT within mitochondria. Aging. 2016;8:2551–2567. doi: 10.18632/aging.101089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kudryavtseva AV, et al. Mitochondrial dysfunction and oxidative stress in aging and cancer. Oncotarget. 2016;7:44879–44905. doi: 10.18632/oncotarget.9821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yakes FM, Van HB. Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc. Natl Acad. Sci. USA. 1997;94:514–519. doi: 10.1073/pnas.94.2.514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jiang H, Talaska AE, Schacht J, Sha SH. Oxidative imbalance in the aging inner ear. Neurobiol. Aging. 2007;28:1605–1612. doi: 10.1016/j.neurobiolaging.2006.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Li Y, et al. Age-associated decline in Nrf2 signaling and associated mtDNA damage may be involved in the degeneration of the auditory cortex: implications for central presbycusis. Int. J. Mol. Med. 2018;42:3371–3385. doi: 10.3892/ijmm.2018.3907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Payne BA, Chinnery PF. Mitochondrial dysfunction in aging: Much progress but many unresolved questions. Biochim. Biophys. Acta. 2015;1847:1347–1353. doi: 10.1016/j.bbabio.2015.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Leutner S, Eckert A, Müller WE. ROS generation, lipid peroxidation and antioxidant enzyme activities in the aging brain. J. Neural Transm. 2001;108:955–967. doi: 10.1007/s007020170015. [DOI] [PubMed] [Google Scholar]
- 72.Barreiro E, et al. Aging, sex differences, and oxidative stress in human respiratory and limb muscles. Free Radic. Biol. Med. 2006;41:797–809. doi: 10.1016/j.freeradbiomed.2006.05.027. [DOI] [PubMed] [Google Scholar]
- 73.Ying YL, Balaban CD. Regional distribution of manganese superoxide dismutase 2 (Mn SOD2) expression in rodent and primate spiral ganglion cells. Hear. Res. 2009;253:116–124. doi: 10.1016/j.heares.2009.04.006. [DOI] [PubMed] [Google Scholar]
- 74.Kazmierczak P, et al. Cadherin 23 and protocadherin 15 interact to form tip-link filaments in sensory hair cells. Nature. 2007;449:87–91. doi: 10.1038/nature06091. [DOI] [PubMed] [Google Scholar]
- 75.Crowe AR, Yue W. Semi-quantitative determination of protein expression using immunohistochemistry staining and analysis: an integrated protocol. Bio Protoc. 2019;9:e3465. doi: 10.21769/BioProtoc.3465. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data generated or analyzed during this study are available from the corresponding author on request. Keap1FA/FA mice used in this study are available from RIKEN BRC (https://knowledge.brc.riken.jp/resource/animal/card?brc_no=RBRC09595&__lang__=en).