

Abstract
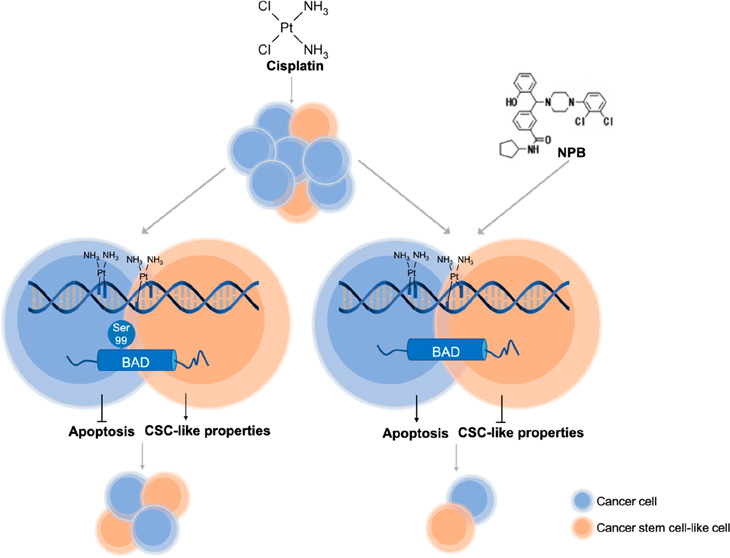
Platinum-based chemotherapy has been the standard treatment for ovarian cancer patients for approximately four decades. However, the prognosis of patients with advanced ovarian carcinoma remains dismal, mainly attributed to both dose-limiting toxicities of cisplatin and the high rate of chemo-resistant disease recurrence. Herein, both patient-derived and experimentally generated cisplatin-sensitive and -resistant ovarian cancer cell line models were used to delineate BADSer99 phosphorylation as an actionable target in ovarian cancer. BADSer99 phosphorylation was negatively associated with cisplatin sensitivity in ovarian cancer, and the inhibition of BADSer99 phosphorylation by point mutation induced apoptosis and reduced cisplatin IC50. In addition, BAD phosphorylation was also shown to be associated with cancer stem cell-like properties. Henceforth, a novel small molecule which inhibits BAD phosphorylation specifically at Ser99 (NPB) was utilized. NPB promoted apoptosis and reduced 3D growth of bulk cancer cells and inhibited cancer stem cell-like properties in both cisplatin-sensitive and -resistant ovarian cancer cells. The combination of cisplatin with NPB exhibited synergistic effects in vitro. NPB in combination with cisplatin also achieved an improved outcome compared to either monotreatment in vivo, including suppression of the cancer stem cell population, an effect not observed with cisplatin treatment. Furthermore, NPB exhibited strong synergistic effects with the AKT inhibitor AZD5363, and significantly reduced its IC50 in cells resistant to cisplatin treatment. These findings identify BADSer99 phosphorylation as an actionable and pharmacologically relevant target to improve outcomes of cisplatin treated ovarian cancer.
Keywords: BAD phosphorylation, NPB, ovarian cancer, cisplatin resistance, cancer stem cell
The 5-year overall survival rate of ovarian cancer has been stagnant at ∼40%, largely due to disease relapse associated with chemo-resistance.1 The standard front-line treatment for ovarian cancer, cytoreductive surgery combined with platinum- and taxane-based chemotherapy, renders a high initial response rate of approximately 80%.2 However, more than 70% of patients experience disease relapse with chemo-resistance within 18 months.1−3 Based on systemic therapy and the platinum-free interval (PFI) of the recurrent ovarian cancer, a number of chemotherapeutic drugs with diverse biologic mechanisms could be used, either as a single agent or in combination, at the discretion of clinicians.4,5 However, irrespective of treatment selected, recurrent ovarian cancer remains incurable.5 To address such issues, multiple chemotherapy agents in several combinations have been explored and clinical trials with the addition of a third cytotoxic agent have been conducted, but no extension of patient survival has been observed.6−9 Furthermore, four targeted therapeutic drugs (Olaparib, Niraparib, Rucaparib, and Bevacizumab) approved for clinical use in ovarian cancer, have only modestly prolonged progression-free survival in patients with naïve or recurrent disease.10,11 The development of new therapeutic approaches in ovarian cancer is thus an unmet clinical need, to address chemo-resistance and disease relapse and to therefore improve the clinical outcome of the disease.
The phosphorylation status of BCL-2 associated death promoter (BAD), a BCL-2 family pro-apoptotic protein, has been reported to be associated with cisplatin resistance and poor survival of ovarian cancer patients in several recent studies.12,13 A study that analyzed 147 ovarian cancer patient samples, revealed that patients with high BAD phosphorylation levels, indicative of inhibition of BAD apoptotic function, exhibited lower median survival rates.12 In the same study, a BAD-pathway gene expression signature was established, and a high BAD-pathway activity score was associated with a favorable survival trend.12 These findings were supported by another study that reported BAD phosphorylation to be crucial for regulating cisplatin sensitivity in vitro.13 In this study, we sought to address the mechanism of BAD phosphorylation associated reduction of cisplatin sensitivity in ovarian cancer, and explore the therapeutic application of inhibiting BAD phosphorylation by use of a novel small molecule.14
The apoptotic activity of BAD is essentially regulated by the phosphorylation of three serine residues: human BAD Ser75, Ser99, and Ser118 (equivalent to murine BAD Ser112, Ser136, and Ser155).15 Unphosphorylated BAD promotes apoptosis through binding and inhibition of the pro-survival BCL-2 proteins, while phosphorylated BAD, being sequestered in the cytosol, is antiapoptotic.16 Upstream kinases for BAD Ser75 include MAPK-activated protein kinase-1 (p90RSK)17 and protein kinase A (PKA),18 while Ser99 may be phosphorylated by protein kinase B (AKT),19 ribosomal protein S6 kinase beta-1 (p70S6K),20 and PKA.21 In addition, BAD phosphorylation has also been reported to be crucial for cancer stem cell (CSC) survival and self-renewal in mammary carcinoma and melanoma.22 With increasing evidence showing that CSC properties contribute to chemo-resistance and disease relapse,23,24 eradicating CSCs by inhibiting BAD phosphorylation holds the promise of preventing the development of chemo-resistance and achieving extended clinical remissions in ovarian cancer.
N-Cyclopentyl-3-((4-(2,3-dichlorophenyl) piperazin-1-yl) (2-hydroxyphenyl) methyl) benzamide (NPB), a novel small molecule which inhibits BAD phosphorylation specifically at Ser99 has been shown to be effective in inducing apoptosis and reducing cell viability in various human cancer cell lines, as well as in inhibiting tumor growth in mammary carcinoma xenograft models.14We sought to determine whether the inhibition of BADSer99 phosphorylation by NPB would enhance cisplatin sensitivity, as well as overcome acquired cisplatin resistance in ovarian cancer. Herein, we report that NPB suppressed cisplatin-induced BADSer99 phosphorylation, as well as the elevated level of BADSer99 phosphorylation in cisplatin-resistant ovarian cancer cells. In addition, NPB was shown to increase cisplatin sensitivity and partially overcome acquired cisplatin resistance through promotion of apoptosis and inhibition of CSC-like behavior in ovarian cancer cell lines and xenograft models. Furthermore, as AKT is an upstream mediator of BADSer99 phosphorylation, NPB combined synergistically with an AKT inhibitor to suppress BADSer99 phosphorylation in cisplatin-resistant ovarian cancer.
Results
BAD-Ser99 Phosphorylation Decreased Apoptosis and Cisplatin Sensitivity in EOC
The phosphorylation of human BAD at Ser99 (murine Ser136) through AKT activation and at Ser75 (murine S112) through ERK1/2 activation is known to suppress the pro-apoptotic effects of BAD.25 Hence, the inhibition of these pathways that lead to BAD phosphorylation was previously shown to increase cisplatin sensitivity of ovarian cancer cells.25 To determine the association of BAD phosphorylation with cisplatin sensitivity in EOC, a potential correlation of BAD phosphorylation levels (phospho-BAD/total BAD ratio) and cisplatin sensitivity utilizing a panel of EOC cell lines (Figure 1A, Figure S-1A) was examined. A significant negative correlation between BADSer99 phosphorylation and cisplatin sensitivity was observed (r = 0.87, p = 0.002; Figure 1B). In contrast, there was no significant correlation between BADSer75 phosphorylation and cisplatin sensitivity (r = 0.18, p = 0.61; Figure 1B).
Figure 1.

BADSer99 phosphorylation reduces apoptosis and is associated with cisplatin resistance. (A) Levels of phosphorylated and total BAD protein in a panel of epithelial ovarian cancer (EOC) cell lines were determined by Western blot analysis. β-ACTIN was used as input control. (B) Spearman correlation of BADSer99 phosphorylation levels in Figure 1A with cisplatin IC50 values in Figure S-1 in the panel of EOC cell lines. (C) Levels of phosphorylated and total BAD proteins in A2780 and A2780cis cells treated with the indicated concentrations of cisplatin for 24 h were determined by Western blot analysis. β-ACTIN was used as input control. (D) A2780 and A2780cis cells were transfected with either pcDNA3-BADS136A plasmid or empty vector. The phosphorylated and total levels of both endogenous and forced-expressed murine BAD protein in these cells were determined by Western blot analysis. β-ACTIN was used as input control. (E) Cell viability was determined by AlamarBlue assay 72 h after commencement of drug treatment. (F) Apoptosis of these cells were determined by caspase3/7 assay 24 h postdrug treatment. (G,H) Indicated cell lines were treated with cisplatin for 72 h, and cell viability was determined by AlamarBlue assay. Cisplatin IC50 values (G) and dose response curved (H) are shown. *P < 0.05, **P < 0.01 and ***P < 0.001.
Changes in BADSer75 and Ser99 phosphorylation levels upon acute cisplatin treatment and in acquired cisplatin resistance in EOC cells were next determined. Two pairs of cisplatin-sensitive and -resistant EOC cell lines were used: A2780 and A2780cis in which A2780cis was generated from A2780 by chronic exposure to cisplatin,26 and PEO1 and PEO4 which were obtained from the same patient before and after the development of chemo-resistance.27 The observed IC50 values of cisplatin in these four cell lines was 0.15 ± 0.02 μM in A2780, 2.7 ± 0.39 μM in A2780cis, 0.5 ± 0.05 μM in PEO1, and 3.9 ± 0.81 μM in PEO4 (Figure S-1C), confirming acquired cisplatin resistance in A2780cis and PEO4 cells. The levels of BADSer75 and Ser99 phosphorylation were observed to be higher in cisplatin-resistant EOC cells as compared to their respective parental cells (Figure 1C; Figure 1E). Furthermore, upon treatment with increasing concentrations of cisplatin, phosphorylation of BAD at Ser99 was observed to be increased in all four cell lines in a dose-dependent manner, while the increase of BAD Ser75 phosphorylation was less pronounced and observed only with a higher cisplatin dose in the cisplatin-sensitive cells (Figure 1C; Figure 1E). Therefore, compared to BAD Ser75 phosphorylation, BAD Ser99 phosphorylation is a more consistent hallmark of cisplatin resistance in ovarian cancer.
The role of BADSer99 phosphorylation in regulating EOC cell survival and cisplatin sensitivity was further assessed. The functional role of BADSer99 phosphorylation was determined through transfection of a mutated mouse (m)BADS136A construct or empty pcDNA3 vector as negative control into A2780 and A2780cis cells. The mutated BadS136A protein expressed has serine at position 136 substituted by alanine, rendering the specific site resistant to phosphorylation. The forced expression of mBadS136A was confirmed by Western blot analysis with bands (of higher molecular weight for mouse Bad) observed for phospho-BadSer112 and total Bad but not phospho-BadSer136 (Figure 1D). As shown in Figure 1E, the forced expression of mBadS136A significantly decreased viability of A2780 and A2780cis cells. The reduced cell viability observed was attributed to increased apoptosis with mBadS136A-expressing cells exhibiting an increase in caspase3/7 activity compared to vector transfected cells (Figure 1F). Moreover, the forced expression of mBadS136A reduced cell viability and enhanced apoptosis to a greater extent in A2780cis as compared to A2780, suggestive of a greater dependence of cisplatin-resistant EOC cells on BADSer99 phosphorylation. Notably, the forced expression of mBadS136A mutant increased cisplatin sensitivity in both cell lines: compared with vector control, the IC50 of cisplatin was 2.5-fold and 4.2-fold lower in A2780- and A2780cis-mBadS136A expressing cells respectively (Figure 1G; 0.1 ± 0.02 μM in A2780 Vector, 0.04 ± 0.01 μM in A2780 BADS136A, 1.82 ± 0.22 μM in A2780cis Vector, 0.43 ± 0.2 μM in A2780cis BADS136A, respectively). These data suggest that BADSer99 phosphorylation is associated with reduced cisplatin sensitivity in ovarian cancer.
BAD Phosphorylation Is Associated with Increased Stemness in EOC
As the enhancement of cancer stem cell (CSC) survival and self-renewal is one function of phosphorylated BAD,22 EOC cells were disaggregated and subjected to growth conditions selective for self-renewing, nonadherent spheroids (sphere formation assay). Spheroid-forming cells have been demonstrated to possess CSC-like properties in brain, breast, melanoma, and ovarian cancers, and function as tumor initiating cells.23,28−30 After 10 days of growth, cisplatin-resistant cells formed more, and larger spheroids compared to their respective parental cells (Figure 3A), and spheroids were harvested for subsequent assays. As a CSC marker, increased aldehyde–dehydrogenase 1A1 (ALDH1A1) expression has been reported to predict poor clinical outcome of ovarian cancer.31,32 Using Western blot analysis, it was observed that the protein level of ALDH1A1 was increased in spheroids compared to monolayer-cultured EOC cells, confirming the enrichment of CSC-like cells in the spheroids (Figure S-2A). Furthermore, cisplatin-resistant A2780cis cells exhibited a higher expression of ALDH1A1 as compared to A2780 cells (Figure S-2A). Importantly, a significantly decreased sensitivity to cisplatin was also observed in spheroid cells as compared to monolayer-cultured EOC cells, with marked increases in the IC50 of cisplatin for cells in sphere culture versus monolayer culture in both cisplatin-sensitive and -resistant cell lines (A2780 spheroids vs A2780: p = 0.0004; A2780cis spheroids vs A2780cis: p < 0.0001; Figure S-2B,C). This further supports the notion that CSC-like cells contribute to reduced cisplatin sensitivity in EOC cells as previously reported.24
Figure 3.

NPB reduces stemness properties in A2780 and A2780cis cells. (A) Spheroid cells were isolated by culturing parental A2780 and A2780cis bulk tumor cells under stem cell growth conditions for 10–14 days. Images of the spheroids were taken (left), and the number of spheroids in each well were counted and presented as percentages relative to the A2780 control group. Scale bar: 200 μm. (B) ALDH positive population in A2780 and A2780cis cells treated with the indicated concentrations of NPB for 72 h were determined using ALDEFLOUR assay. (C) Stemness markers were measured by qPCR after 24 h of NPB treatment in A2780 and A2780cis cells. GAPDH was used as input control. (D) EMT markers were measured by qPCR after 24 h of NPB treatment in A2780 and A2780cis cells. GAPDH was used as in input control. *P < 0.05; **P < 0.01, and ***P < 0.001.
It was next determined whether BADSer99 phosphorylation is associated with stemness in EOC CSC-like cells. Compared to monolayer-cultured (bulk) EOC cells, BADSer99 phosphorylation was elevated in CSC-like cell-enriched spheroids derived from cisplatin-sensitive cells and substantially so in cisplatin-resistant cells (Figure S-2D). Interestingly, BADSer75 phosphorylation was also increased in spheroids, despite not showing a significant correlation with cisplatin sensitivity (Figure 1D). Collectively, these data are suggestive of BADSer99 phosphorylation as a potential target to inhibit the CSC-like cell population in ovarian cancer and hence enhance cisplatin sensitivity.
NPB, a Novel BAD Ser99 Phosphorylation Inhibitor Suppressed EOC Cell Viability by Stimulating Apoptosis
To address the therapeutic potential of inhibiting BADSer99 phosphorylation, a novel small molecule (NPB) which specifically inhibits BADSer99 phosphorylation was used.24 As shown in Figure 2A, NPB decreased BAD BADSer99 phosphorylation in a dose dependent manner in both cisplatin-sensitive and -resistant EOC cells, without altering the phosphorylation levels at Ser75. The dose response and IC50 values (cell viability) of NPB in the four EOC cell lines were determined to be in the micromolar range (Figure 2B and Figure S-3A). The cisplatin-resistant EOC cells exhibited slightly higher IC50 values than their respective cisplatin-sensitive parental cells.
Figure 2.

NPB stimulates apoptosis and reduces cell cycle progression in both cisplatin-sensitive and -resistant ovarian cancer cells. (A) Levels of phosphorylated and total BAD after 24 h of treatment with NPB in A2780, A2780cis, PEO1, and PEO4 cells were determined by Western blot analysis. β-ACTIN was used as input control. (B) IC50 values of NPB after 72 h of treatment in both cisplatin-sensitive and-resistant EOC cell lines were determined by the AlamarBlue cell viability assay. (C) Apoptotic cell death was determined after 24 h of NPB treatment by flow cytometry in both cisplatin-sensitive and -resistant EOC cell lines. Q4: Early apoptotic population (PI–, FITC-Annexin V+). Q2: Late apoptotic population (PI+, FITC-Annexin V+). (D) Apoptosis of NPB treated cells was determined using Caspase 3/7 assay 24 h post-treatment. (E) Cell cycle progression in the cell lines after 24 h of treatment with NPB was determined using flow cytometry. (F) Colony formation was performed in respective cell lines with treatment of NPB. The cells were allowed to grow for 10 days for A2780/A2780cis and 14 days for PEO1/PEO4 before the number of colonies were counted under a microscope. (G) Cells were cultured in 2% FBS medium containing 4% Matrigel for 3 days prior to treatment with NPB for 6 days. Live cells were stained by calcein (green color) and dead cells were stained by PI (red color), scale bar: 200 μm. (H) Viability of cells grown in 3D Matrigel were determined using AlamarBlue cell viability assay. *P < 0.05, **P < 0.01 and ***P < 0.001.
The effect of NPB in promoting apoptotic cell death in both cisplatin-sensitive and -resistant EOC cells was assessed using ANNEXIN V-propidium iodide (PI) staining and caspase 3/7 assays. Consistently observed in all four cell lines, NPB treatment significantly increased early (PI–, FITC–Annexin V+) and late (PI+, FITC–Annexin V+) apoptosis compared to treatment with vehicle (Figure 2C; Figure S-3B). Likewise, NPB also induced caspase 3/7 activity in a dose-dependent manner in both cisplatin-sensitive and -resistant cells (Figure 2D). Cell cycle analysis showed that NPB treatment significantly increased the SubG1 population in all four cell lines (Figure 2E, Figure S-3C). Furthermore, NPB decreased cell cycle progression of EOC cells, as shown by the population of cells in S-phase (Figure 2E, Figure S-3C). Hence, in addition to promoting apoptosis, NPB also reduced proliferation of EOC cells through the inhibition of cell cycle progression. Furthermore, in colony formation assays, the number of colonies observed with 5 μM NPB was decreased by approximately 10-fold compared to vehicle treated cells, and almost no colonies were formed upon treatment with 10 μM NPB (Figure 2F; Figure S-4A). Moreover, to simulate in vivo therapy, the effect of NPB on pregrown colonies of the four EOC cell lines in 3D Matrigel was assessed. Consistent with its effects in 2D culture, NPB significantly suppressed the 3D growth of these four cell lines, with smaller and less 3D colonies observed along with reduction in cell viability upon NPB treatment (Figure 2G,H). “Live–dead” staining of the 3D colonies by Calcein green and PI also revealed a decrease of “live” colonies and an increase of “dead” colonies with NPB treatment (Figure S-4B). These results demonstrated that NPB suppressed cell viability, colony formation, and 3D growth of EOC cells.
NPB Decreased CSC-like Properties of EOC Cells
The chemo-resistant and tumor-initiating properties of CSCs indicate that treatment approaches targeting this cell population could potentially increase the efficacy of the current available treatment regimens and reduce the risk of tumor recurrence and metastasis.33 As phosphorylated BADSer99 was observed to be increased in the CSC-like population of EOC cells, the effect of NPB on the CSC-like behavior of EOC cells was evaluated. NPB treatment resulted in a significant dose dependent decrease in the number of spheroids formed from cisplatin-sensitive EOC cells as compared to the vehicle control group (Figure 3A). Although the spheroid-forming efficiency of cisplatin-resistant EOC cells was higher as compared to their respective parental cells, NPB treatment decreased it to levels similar to the cisplatin-sensitive EOC cells (Figure 3A). Changes in the ALDH+ population with NPB treatment were also assessed using the ALDEFLUOR assay in the four EOC cell lines. Consistently, as shown in Figure 3B, the ALDH+ cell population was markedly higher in cisplatin-resistance, as compared to their respective cisplatin-sensitive EOC cells. Moreover, NPB significantly decreased the ALDH+ cell population in both cisplatin-sensitive and -resistant EOC cells (Figure 3B and Figure S-4C).
Gene expression analysis of stemness and EMT markers in cisplatin-sensitive and -resistant EOC cells ± NPB was also performed using RT-qPCR. The expression of stem cell markers (ALDH1A1, NANOG, OCT4, and BMI1) and signaling pathway mediators regulating stemness (WNT5B, NOTCH1, and EGFR) in A2780 and A2780cis cells was examined after 24 h of NPB treatment. Consistent with the observation of an increased CSC-like population in cisplatin-resistant EOC cells, these cells also exhibited markedly higher expression of NANOG, OCT4, WNT5B, NOTCH1, and ALDH1A1, as compared to their respective parental cells (Figure 3C). The expression of BMI1, NOTCH1, EGFR, and ALDH1A1 were significantly reduced by NPB treatment in A2780 cells (Figure 3C). Notably, the markedly high expressions of NANOG, OCT4, WNT5B, NOTCH1, and ALDH1A1 in A2780cis cells were decreased by NPB to levels similar to those of A2780 cells (Figure 3C). Increasing evidence indicates that the activation of the epithelial mesenchymal transition (EMT) program in non-CSCs enables their entrance into the CSC state.34,35 The expression of mesenchymal markers (CDH2, SNAIL2, ZEB2, VIM and CTNNB1) and a signaling pathway mediator that promotes EMT (IGF1) were therefore examined. The expression of CDH2 and IGF1 mRNAs were significantly higher in A2780cis cells compared toA2780 cells (Figure 3D), and such higher expression was decreased by NPB (Figure 3D). Hence, NPB inhibited the CSC-like properties of EOC cells.
NPB Enhanced Cisplatin Sensitivity in EOC in a Synergistic Manner
Given the observation that BAD Ser99 phosphorylation is associated with reduced cisplatin sensitivity in EOC, it was hypothesized that by inhibiting BADSer99 phosphorylation, NPB would enhance cisplatin sensitivity in EOC cells. The level of BADSer99 phosphorylation upon cisplatin and NPB treatment in EOC cells was therefore examined. BADSer99 phosphorylation was increased by cisplatin treatment in cisplatin-sensitive but not cisplatin-resistant cell lines, and it was decreased with NPB in both sensitive and resistant lines (Figure 4A). Importantly, NPB markedly inhibited the cisplatin induced increase in BADSer99 phosphorylation in A2780 and PEO1 cells and maintained suppression of BADSer99 phosphorylation in A2780cis and PEO4 when treated with cisplatin (Figure 4A). The effect of NPB on cell viability when used in combination with cisplatin was therefore evaluated. EOC cells were treated with NPB, cisplatin, or the combination using the indicated folds of the individual drugs’ IC50 values (Figure S-5A). Cell viability was measured after 72 h of drug exposure, and the combination index (CI) along with dose reduction index (DRI) values were calculated using the Chou-Talalay method.36 The viability of the EOC cell lines decreased to a greater extent with combination treatment as compared to either monotreatment across the range of concentrations (Figure S-5A). This was observed at concentrations starting from as low as 0.125-fold IC50 of either drug in A2780 and A2780cis cells, and at concentrations lower than IC50 of either drug in PEO1 and PEO4 cells (Figure S-5A). Importantly, the Chou-Talalay analysis revealed strong synergy of NPB and cisplatin in both pairs of EOC cell lines as indicated by CI < 1, with decreasing CI values at increasing fraction affected (Fa) (Figure 4B,C). Consistently, NPB promoted the reduction of the effective dose of cisplatin in both cisplatin-sensitive and -resistant EOC cells with DRI > 1 (Figure 4D). Similarly, cisplatin also promoted reduction of the NPB dose required for the same inhibitory effect in the four cell lines as indicated by the DRI values (Figure S-5B). Furthermore, NPB treatment decreased cisplatin IC50 in A2780cis cells in a dose-dependent manner with an approximate 1.5-fold decrease observed at NPB concentration as low as IC30 (Figure S-5C).
Figure 4.

NPB exhibited synergistic effects with cisplatin in both cisplatin-sensitive and -resistant cell lines. (A) Levels of BAD phosphorylation and total BAD were determined using Western blot analysis after the respective cells were treated with vehicle, NPB, cisplatin, or NPB and cisplatin combination for 24 h. β-ACTIN was used as input control. (B) Respective cell lines were treated with NPB, cisplatin, or NPB and cisplatin combination for 72 h, with the concentration of NPB and cisplatin constant at the ratio of their IC50 values. Subsequently, cell viability was determined using AlamarBlue cell viability assay, and combination indices were calculated using the Chou–Talalay method. Representative CI curves are shown. (C) The CI values at Fa50, Fa75, and Fa90 were determined and shown as mean ± SEM. (D) Dose reduction curves for cisplatin were generated using Compusyn and representative curves are shown. (E) Apoptotic cell death upon treatment with NPB, cisplatin, and NPB and cisplatin combination for 24 h was determined by caspase3/7 assay. (FA) Cells were cultured in 3D Matrigel for 72 h before being exposed to the indicated treatments. Ten days later, cell viability was quantified using AlamarBlue cell viability assay. (G) Respective cell lines were seeded for colony formation for 48 h before 10–14 days of drug treatment. The number of colonies were counted under a microscope after crystal violet staining. *P < 0.05; **P < 0.01, and ***P < 0.001.
In addition, the effect of NPB on cisplatin-induced apoptosis, and cisplatin-mediated inhibition of colony formation and 3D Matrigel growth were examined. As expected, cisplatin treatment significantly increased caspase 3/7 activity, and significantly inhibited colony formation and 3D growth of cisplatin-sensitive but not cisplatin-resistant EOC cells (Figure 4E–G; Figure 5D,E). Notably, NPB further enhanced cisplatin-induced apoptosis in cisplatin-sensitive EOC cells as measured by caspase 3/7 activity (Figure 4E). The combination treatment also resulted in increased apoptosis as compared to NPB alone in A2780cis cells, wherein NPB increased apoptosis in combination with cisplatin to a level similar to that of cisplatin-induced apoptosis in A2780 cells (Figure 4E). Similarly, NPB further enhanced the cisplatin-mediated inhibition of colony formation and 3D Matrigel growth of cisplatin-sensitive EOC cells (Figures 4F,G; Figure 5D,E). Importantly, in cisplatin-resistant EOC cells, the combination treatment also resulted in a greater reduction of colony formation and 3D Matrigel growth than NPB treatment alone, where NPB potentiated the inhibitory effects of cisplatin to levels similar to those of cisplatin-treated cisplatin-sensitive EOC cells (Figure 4F,G; Figure 5D,E). Hence, inhibition of BAD Ser99 phosphorylation by NPB synergistically enhanced the inhibitory effect of cisplatin in cisplatin-sensitive and -resistant EOC cells, and at least partially resensitized cisplatin-resistant cells.
Figure 5.

NPB and cisplatin combination reduced tumor volume and tumor weight in ovarian cancer xenografts. (A) Tumor volumes in mice were recorded daily with NPB alone, cisplatin alone, and NPB+cisplatin combination treatment during the first and the second cycle of therapy. Red arrows in panel A indicate the time points at which cisplatin was administered. Blue arrows indicate the time points described in the main text. (B) Body weights of the animals were recorded daily with NPB alone, cisplatin alone, and NPB+cisplatin combination treatment during the first and second cycle of therapy. (C) Individual value plots of final tumor weight after NPB alone, cisplatin alone, and NPB+cisplatin combination treatment were recorded at the end of cycle 1 and cycle 2. Results were statistically analyzed with one-way ANOVA followed by Dunnett’s test post hoc. ($/I̵) NPB treated group; (¶) cis treated group; (§) NPB+cis-treated group. Statistical analysis for tumor volume data, $/ ¶ /§P < 0.05, $ $ /¶ ¶/§ §P < 0.01 and $ $ $/ ¶ ¶ ¶ /§ § §P < 0.001, compared with the vehicle-treated group in cycle 1, and compared with the combination-treated group in cycle 2. Statistical analysis for tumor weight data: (cycle 1) */I̵P < 0.05, **P < 0.01 and ***P < 0.001, compared with the vehicle-treated group; I̵P < 0.05, ##P < 0.01 compared with combination group; (cycle 2) ##P < 0.01 compared with the combination-treated group.
NPB Increased the Efficacy of Cisplatin in EOC Xenografts
A2780 and A2780cis cell-derived xenograft models were generated to examine the efficacy of NPB in potentiating the effect of cisplatin in cisplatin-sensitive and -resistant EOC cells in vivo. The schematic of the workflow is shown in Figure S-6A. Vehicle treated xenografts derived from both A2780 and A2780cis cells reached 1000 mm3 or greater on day 9 of drug treatment (7 days after the cisplatin dose). Hence, all mice in the vehicle groups (n = 8), and half of the mice (n = 4) in each of the treated groups were sacrificed at this time point (cycle 1), whereas the remainder (n = 4 for each treatment group) were sacrificed on day 15 after commencement of drug treatment (cycle 2). No significant changes in body weight (Figure 5B) nor relative important organ weights (Figure S-6B) were observed in treatment groups compared with the vehicle group during the experimental period, indicating that the treatments were well tolerated. The tumor volumes measured in both A2780 and A2780cis cell-derived xenograft models after the start of treatment are presented in Figure 5A: Vehicle treated xenografts derived from both A2780 and A2780cis had reached 1000 mm3 or greater after 9 days with the growth rate in A2780cis cell-derived tumors being slightly faster. Tumors derived from both cell lines in all treated groups (NPB, cisplatin, and NPB+cisplatin) exhibited significant reduction in volumes after 3 days of treatment. Among all groups, tumors in the NPB+cisplatin groups exhibited the slowest increase in volume. After the second cisplatin treatment on day 9, tumor volumes in the cisplatin or in the cisplatin in combination with NPB groups decreased in volume for 2 to 4 days in cisplatin-sensitive A2780 cell-derived tumors. Tumor volumes then started to recover in the cisplatin group and at a faster rate than the minimal recovery in the combination group. However, no significant effect of the second cisplatin injection on tumor volume was observed in cisplatin-resistant A2780cis cell-derived tumors in either the cisplatin or combination groups although the combination group was maintained at a lower volume than NPB treatment alone.
Additionally, the increase in tumor volume in the combination group (shown by §) slowed more markedly than that in the NPB or cisplatin alone groups from A2780 or A2780cis cells-derived tumors after day 6 or day 5, respectively (P < 0.01). After completing 2 cycles, there were significant differences in the tumor volume between the NPB+cisplatin and NPB alone groups from day 10, and between the NPB+cisplatin and cisplatin alone groups from day 14 to day 15 in A2780 cells-derived tumors (Figure 5A, Figure S-7A, representative images in Figure S-7B). There were no significant differences between the NPB+cisplatin, NPB alone, or cisplatin alone groups after day 13 in A2780cis cells-derived tumors. All treated groups from both A2780 and A2780cis cell-derived tumors (NPB, cisplatin, and NPB+cisplatin) decreased tumor weight as compared to the vehicle-treated group, with the combination group exhibiting lower tumor weights compared to the NPB or cisplatin alone groups in cycle 1 (Figure 5B, Figure S-7C). Furthermore, the combination group also exhibited lower tumor weights as compared to the NPB or cisplatin alone groups in cycle 2, albeit it did not reach statistical significance for the cisplatin-resistant A2780cis cell-derived xenograft (Figure 5B).
Immunohistochemical analyses of tumor specimens resected from the animals were performed to examine the levels of pBADSer99, Ki67, TUNEL positivity, and the CSC marker ALDH1A1. A significant reduction in pBADSer99-positive cells and immunoreactive score was observed with NPB treatment in both A2780 and A2780cis derived tumors; however, increased phosphorylation of BADSer99 was not observed with cisplatin treatment. Notably, in cycle 2, a further significant decrease of BADSer99 phosphorylation was observed with the combination treatment as compared to either monotreatment. (Figure 6A,B; Figure S-8A). Moreover, while cisplatin treatment only led to a modest decrease in Ki67-positive cells and increase in TUNEL immunoreactivity score in cisplatin-sensitive A2780cis cell-derived tumors, NPB and the combination treatment resulted in significantly decreased percentages of Ki67- and TUNEL-positive cells in both A2780 and A2780cis cell-derived tumors compared with the vehicle-treated group in cycle 1 (Figure 6A,C,D; Figure S-8B,C). The combination-treated group exhibited decreased Ki67 marker expression and increased TUNEL positivity as compared to the cisplatin-treated group in cycle 2 (Figure 6A,C,D; Figure S-8B,C). NPB alone, and in combination with cisplatin, decreased the expression of the CSC marker ALDH1A1 in both A2780 and A2780cis cell-derived xenografts, an effect not observed with cisplatin treatment alone (Figure 7A,B; Figure S-8D). Hence, NPB, administered with cisplatin, further inhibited cell proliferation and CSC-like properties, in both cisplatin-sensitive and -resistant EOC xenografts associated with reduction in BADSer99 phosphorylation, with a more significant effect in cycle 2. In addition, NPB in combination with cisplatin further enhanced apoptosis in cisplatin-sensitive EOC xenografts compared to either monotreatment, albeit not showing a significant effect in cisplatin-resistant EOC xenografts.
Figure 6.

Efficacy of the NPB and cisplatin combination treatment in ovarian cancer xenografts. Representative images (A) and quantification (B–D) of immunohistochemistry analyses of positive cell (%) in the tumor tissues for selected markers, pBAD (Ser99), Ki67, and TUNEL. Results were statistically analyzed with one-way ANOVA followed by Dunnett’s test post hoc. (Cycle 1) I̵/&P < 0.05, I̵ I̵ /&&P < 0.01 and I̵ I̵ I̵/&&&P < 0.001, compared with the combination-treated group; *P < 0.05, **P < 0.01 and ***P < 0.001, compared with the vehicle-treated group. Cycle 2: #P < 0.05, ##P < 0.01 and ###P < 0.001, compared with the combination-treated group.
Figure 7.

NPB and the cisplatin and NPB combination decreases ALDH1A1 positive cells in xenografts. Representative images (A) and quantification (B) of immunohistochemistry analyses of ALDH1A1 positive cells (%) in xenograft tissues from either A2780 or A2780cis cell-derived tumors. Results were statistically analyzed with one-way ANOVA followed by Dunnett’s test post hoc. Cycle 1: I̵/&P < 0.05, I̵ I̵ /&&P < 0.01 and I̵ I̵ I̵/&&&P < 0.001, compared with the combination-treated group; *P < 0.05, **P < 0.01 and ***P < 0.001, compared with the vehicle-treated group. Cycle 2: #P < 0.05, and ###P < 0.001, compared with the combination-treated group.
NPB Enhanced AKT Inhibitor (AZD5363) Sensitivity in EOC by Inducing Apoptosis and Reducing CSC-like Behavior
AKT is a crucial upstream mediator of BADSer99 phosphorylation.15 The activation of the AKT pathway has also been implicated in cisplatin resistance, and AKT inhibition has been reported to reverse cisplatin resistance.37 AKT inhibitors have not been clinically approved for the treatment of cancer yet, being limited by low clinical activity as a single agent and dose-dependent adverse effects.38 Hence, it was further investigated whether combination with NPB could lower the effective dose of AKT inhibitors. The combination of an AKT inhibitor (AZD5363) or a PI3K inhibitor (GDC0032) with NPB was first examined (Figure S-9A,B). AZD5363, but not GDC0032, exhibited a favorable combination with NPB, resulting in a further decrease in cell viability of A2780cis cells as compared to either monotreatment (Figure S-9A,B). The inhibition of BADSer99 phosphorylation by AZD5363-mediated inhibition of AKT, as well as by NPB, was confirmed by Western blot analysis (Figure 8A). The combination of NPB and AZD5363 resulted in a further decrease in the level of phosphorylated BADSer99 (Figure 8A). Notably, the elevated level of phosphorylated BADSer99 in cisplatin-resistant A2780cis cells was reduced lower than that of the cisplatin-sensitive A2780 cells by the combination treatment (Figure 8A).
Figure 8.

NPB exhibited synergistic effects with AZD5363 in A2780 and A2780cis cells. (A) Levels of p-BAD Ser99 and total BAD was determined by Western blot analysis 24 h after the indicated treatment. (B) A2780 and A2780cis cells were treated with AZD5363 and with increasing dose of NPB for 72 h. Cell viability was determined using AlamarBlue cell viability assay and IC50 was calculated using GraphPad Prism 5. Data are shown as mean ± SEM. Representative dose response curves are shown in panel C. (D,E) A2780 and A2780cis cells were treated with AZD5363, NPB, or AZD5363 and NPB in combination for 72 h, with the concentration of AZD5363 and NPB constant at the same ratio as their respective IC50 value. Cell viability was determined using AlamarBlue cell viability assay, and the combination index was calculated using the Chou–Talalay method. Representative CI curves were shown in panel D, and CI values at Fa50, Fa75, and Fa90 were determined and shown as means ± SEM in panel E. (F) Apoptotic cell death in A2780 and A2780cis cells upon treatment with AZD5363, NPB, or the combination of AZD5363 and NPB was determined by caspase3/7 assay. (G,H) A2780 and A2780cis cells were cultured in 3D Matrigel for 72 h before being exposed to the indicated treatment for 10 days. Representative images of the colonies are shown in panel G, and cell viability was quantified using AlamarBlue cell viability assay in panel H. (I) A2780 and A2780cis cells were seeded at low density for 48 h before being exposed to the indicated treatment for 10 days. The number of colonies were counted under a microscope after crystal violet staining. (J) A2780 and A2780cis cells were subjected to spheroid selective growth conditions and with the indicated treatments. Representative images of the spheroids were taken after 10 days of incubation. (K) Levels of AKT phosphorylation and total AKT in 2D cultured ovarian cancer cells and their respective spheroids derived were determined using Western blot analysis. β-ACTIN was used as input control. (L) The ALDH positive cell population in A2780cis cells were quantified using ALDEFLOUR assay after 72 h of treatment with AZD5363, NPB, or AZD5363 in combination with NPB. *P < 0.05; **P < 0.01, and ***P < 0.001.
NPB treatment increased the sensitivity of both A2780 and A2780cis cells to AZD5363 in a dose-dependent manner, as shown by progressive decreases in the IC50 values (cell viability) of AZD5363 with increasing concentrations of NPB (Figure 8B,C). Chou–Talalay analysis revealed a favorable interaction between NPB and AZD5363 in both A2780 and A2780cis cells with CI values below 1 and decreasing at higher Fa values (Figure 8D,E; Figure S-9C). Furthermore, DRI values of each drug were greater than 1 and increasing with higher Fa values, indicative that NPB resulted in substantial reduction of the dose of AZD5363 required for the same inhibitory effect, and vice versa (Figure S-9D). Functionally, the combination of AZD5363 and NPB led to a significant increase in apoptosis when compared to either treatment alone in both A2780 and A2780cis cells, as measured by caspase 3/7 activity (Figure 8F). Furthermore, NPB, in combination with AZD5363, also significantly reduced colony formation and 3D growth (Figure 8G–I; Figure S-9E) of both A2780 and A2780cis cells, showing a greater inhibitory effect than either treatment alone.
Similar to BADSer99 phosphorylation, a significant increase of AKT phosphorylation was also observed in spheroid cells as compared to the monolayer-cultured bulk tumor cell population in the four EOC cell lines (Figure 8J). To test the effect of the combination of NPB and AZD5363 in modulating the CSC-like population of cisplatin-sensitive and -resistant EOC cells, sphere formation and ALDEFLUOR assays were performed. AZD5363 decreased sphere formation of both A2780 and A2780cis cells as a single agent, while a significant further decrease was observed when utilized in combination with NPB (Figure 8K), supporting the observation that AZD5363 inhibition of AKT decreased the CSC-like behavior of cisplatin-sensitive and -resistant EOC cells. Consistently, ALDEFLOUR assay also showed that the combination of AZD5363 and NPB substantially decreased the ALDH+ cell population in A2780cis as compared to either monotreatment (Figure 8L, Figure S-9F). These results suggest that the combination of AZD5363 and NPB synergistically decreased cell viability and CSC-like behavior, and enhanced apoptosis in both cisplatin-sensitive and -resistant EOC cells.
Discussion
Despite in vitro and clinical evidence of the association between BAD phosphorylation and cisplatin resistance, the preclinical utility of specific (nonupstream) and direct inhibition of BAD phosphorylation has not yet been addressed. In this study, we first verified the correlation between BAD phosphorylation and cisplatin resistance, and demonstrated that a novel small molecule, NPB, which specifically inhibits BADSer99 phosphorylation, exhibits efficacy in both cisplatin-sensitive and cisplatin-resistant ovarian cancer. Such findings are meaningful to address the current incurability of recurrent ovarian cancer. The choice of treatment regimen for recurrent ovarian cancer depends on the previous systemic therapy, platinum-free interval (PFI), previous treatment with bevacizumab, and BRCA status of the patients. For patients with platinum-sensitive (PFI > 12 months) or partially sensitive (6 months ≤ PFI ≤ 12 months) relapse, a platinum-based combination chemotherapy is currently considered the treatment of choice.39,40 However, its prolongation of PFS is modest, and patients may suffer from several adverse effects. With the introduction of Bevacizumab and Olaparib in the treatment of this disease, it is hoped that more platinum-based combination regimens with targeted therapy will prolong PFS and improve the quality-of-life of patients. Herein, this study has demonstrated a significant synergistic effect of NPB and cisplatin in cisplatin-sensitive ovarian cancer in vitro, and the in vivo data also indicated that the combination achieved a better outcome compared with either drug used as a single agent. Hence, NPB or similar inhibitors may be promising for further development as combination therapy with cisplatin in cisplatin naïve EOC, or cisplatin-sensitive recurrent EOC, not only to reduce dose-dependent toxicity of cisplatin, but also to inhibit the CSC-like population to delay the development of resistance and prevent disease relapse.
For patients with resistant (PFI < 6 months) or refractory (disease develops during platinum treatment) ovarian cancer, the prognosis is unfavorable, and most patients succumb to the disease within one year. Recently, bevacizumab has been approved by the FDA for platinum-resistant recurrent EOC patients used in combination with chemotherapies;41 and Olaparib and Niraparib have been approved for EOC patients after three lines of chemotherapies, subject to BRCA/HRD status.42,43 Results herein indicated that although NPB partially overcame cisplatin resistance, it did not fully resensitize cisplatin-resistant cells to cisplatin. However, the combination of an AKT inhibitor, AZD5363, and NPB exhibited synergistic effects in cisplatin-resistant cell lines. AZD5363 is currently in a phase Ib clinical trial in combination with Olaparib for the treatment of recurrent ovarian cancer.44 Although AZD5363 showed promising effects as a single agent in clinical trial, patients receiving AZD5363 as a single agent reported side effects such as diarrhea, hyperglycemia, and nausea.45,46 The observation that the combination of AZD5363 and NPB reduced the ALDH+ population in ovarian cancer cells is consistent with previous reports that AKT activation contributes to the maintenance of CSC-like population in various cancers, including liver,47 prostate,48 breast,49 and brain cancers.50 Hence, the combination of AZD5363 and NPB may be effective to reduce dose-dependent toxicities and improve the quality of life of EOC patients who have developed cisplatin-resistance and yet still reduce the chemo-resistant CSC-like cell population in the tumor. Another combination option for NPB in ovarian cancer is with a PARP inhibitor and studies in progress indicate this combination may be particularly effective for cisplatin-resistant ovarian cancer (Zhang et al., in progress).
It is well recognized that there are five different histological types of ovarian cancer with different risk factors, cell of origin, genomic landscape, clinical features, and efficacy of response to therapy.3 In addition to developing treatment regimens for histological subtypes that are sensitive to chemotherapy, such as high grade serous ovarian (HGSOC) and endometrioid cancer, clinical trials of targeted therapy in rarer subtypes are also being undertaken. Compared to a response rate of 5% to chemotherapy, a 15%–20% response rate to drugs such as Selumetinib has been reported in low grade serous ovarian carcinoma.51 The two pairs of cisplatin-sensitive and -resistant ovarian cancer cell lines used in this study belong to different histological subtypes. PEO1 and PEO4 are HGSOC cell lines derived from the same patient before and after the development of chemo-resistance; A2780 and A2780cis have long been categorized as HGSOC, but recent studies have provided evidence of derivation from ovarian endometrioid adenocarcinoma.52 Despite belonging to different histological subtypes, the combination of cisplatin and NPB exhibited similar effects in both pairs. However, the IC50 values of AZD5363 in PEO1 and PEO4 cells were not obtainable, despite the highest concentration of 100 μM used in the serial dilution. It is reported that PIK3CA and AKT mutations are not frequently found in HGSOC; in contrast, as high as 12–20% of endometrioid ovarian carcinoma harbors gain of function mutations in PIK3CA.53 This is consistent with the clinical data showing a relatively modest single-agent activity of AKT inhibitors and which implied that improved efficacy might be achieved by patient selection. However, it is noteworthy that a recent study pointed out that although necessary, AKT activation alone was not sufficient to confer sensitivity to AKT inhibitors in ovarian cancer, and combination of AKT inhibitors with other targeted agents is required to elicit optimized efficiency.54
The cancer stem cell (CSC) concept has proposed that there exists a subpopulation of tumor cells with self-renewal and differentiation properties that can sustain tumor growth and entirely regenerate a heterogeneous tumor.55 Evidence have shown that CSCs are more resistant to conventional chemotherapeutic regimens, and with their tumor-initiating capacity, CSCs are responsible for disease relapse. For this reason, treatment regimens that eradicate both bulk tumor cells and CSCs hold the promise of achieving extended, if not permanent, clinical remissions. In EOC patients, the early steps of disease progression involve the shedding of malignant cells from the primary tumors into the peritoneal cavity, where they survive singly or as multicellular aggregates (also known as spheroids) in ascites.3 In addition, it has been suggested that ovarian tumor cells isolated from ascites of chemo-resistant patients exhibited mRNA profiles that were characteristic of CSCs,56 which indicates the importance of targeting “spheroid” cells in preventing ovarian cancer relapse and further spread.57,58 Furthermore, considering that ascites results in deterioration of the quality of life of patients, therapeutic strategies eliminating peritoneal spread are needed for efficacious disease treatment.58 Although p-AKT and p-GSK3β levels in ovarian cancer cells from ascites did not exhibit a significant correlation with patient response to subsequent chemotherapy, phospho-p70S6K was significantly higher in cancer cells isolated from ascites in patients who did not respond to subsequent chemotherapy compared to those who did.59,60 As p70S6K also phosphorylates BADSer99,13 the importance of targeting BADSer99 phosphorylation, rather than AKT, in inhibiting ascitic growth of CSC-like cells is exemplified. Consistently, the results herein demonstrated that BADSer99 phosphorylation is critical for spheroids formation and NPB inhibits ovarian cancer spheroids formation. Moreover, it was observed herein that EMT associated genes were decreased by NPB treatment of ovarian cancer cells. This suggests a possible role of phosphorylated BAD in regulating EMT in ovarian cancer. Ovarian cancer, like many other epithelial-derived tumors, undergoes EMT during cancer progression and metastasis.61,62 Furthermore, it has also been reported that the increased expression of SNAIL and ZEB2 (with ZEB2 markedly decreased by NPB) is associated with poor prognosis of ovarian cancer.63 As EMT is tightly correlated with CSC properties, a more detailed study on the function and mechanisms of BAD phosphorylation in EMT in ovarian cancer would provide further insights into mechanisms of ovarian cancer progression.
In summary, this study highlights the potential of inhibiting BADSer99 phosphorylation to be used concurrent with platinum-based chemotherapy, or an AKT inhibitor. Inhibitors of specific BAD phosphorylation in combination with cisplatin may provide a further benefit to patients in terms of prolonging the progression-free interval and increasing the interval between lines of chemotherapy, hence delaying further hospitalization and the cumulative toxicities associated with chemotherapy. The inhibition of BADSer99 phosphorylation by NPB has also demonstrated promising results in combination with AZD5363, the use of which in ovarian cancer has been limited by dose-limiting toxicities. Hence, the clinical development of inhibitors of BAD phosphorylation may be beneficial to both primary and recurrent ovarian cancer patients.
Materials and Methods
Cell Culture and Drugs
Twelve epithelial ovarian cancer (EOC) cell lines from the SGOCL cell line library64,65 were used in this study, including two pairs of cisplatin-sensitive and -resistant EOC cell lines: A2780 and A2780cis, and PEO1 and PEO4. The maintenance conditions for each cell line are listed in Supplementary Table 1. Cisplatin (Sigma-Aldrich, St. Louis, MO, USA) was initially dissolved in H2O or saline with similar results, AZD5363 (Selleck Chem, Houston, TX, USA) and NPB were initially dissolved in DMSO; all three drugs were stored at −20 °C with no light exposure.
Transient Transfection
For forced expression of murine BADS136A mutant, pcDNA3-BADS136A plasmid (Addgene, Watertown, MA, USA) or pcDNA3 vector was transfected to A2780 or A2780cis using X-tremeGENE HP DNA Transfection Reagent (Roche, Basel, Switzerland).
Cell Viability Assay
A total of 2 × 103 cells/well were plated in a 96-well cell culture plate before drug treatment. At 24 h later, cells were treated with drugs that were diluted to indicated concentrations in cell culture medium. At 72 h after drug administration, culture medium was removed and replaced with AlamarBlue (Invitrogen, Carlsbad, CA, USA) diluted in cell culture medium at the ratio of 1:10. After 4–6 h of incubation, fluorescence was detected with Tecan fluorescence/luminescence reader, using an excitation between 530 and 560 nm, and emission at 590 nm.
RT-qPCR
Total RNA of EOC cell lines was extracted using RNeasy mini kit (Qiagen, Hilden, Germany). A 1 μg aliquot of purified RNA was reverse transcribed using SuperScript VILOTM cDNA Synthesis Kit (Invitrogen, Carlsbad, CA, USA), and the resulting cDNAs were mixed with Fast SYBR Green Master Mix (Invitrogen, Carlsbad, CA, USA). qPCR and its analysis were performed using the ABI 7900T Real-time PCR system (Applied Biosystems, Foster City, CA, USA). Two housekeeping genes ACTB and GAPDH were used for normalization. Each reaction was performed in triplicate, and the mRNA expression level of each gene was normalized to the expression of housekeeping genes and presented as average fold change (2–ΔΔCt) with respect to control.
Western Blot Analysis
Cell lysates were extracted using lysis buffer (2% SDS, 0.1% Bromophenol blue, 10% glycerol, 10 mM dithiothreitol, 50 mM Tris pH7.6), assessed by standard Western blotting, and visualized with SuperSignal West Pico Chemiluminescent Substrate (Thermo Fisher Scientific, Watertown, MA, USA). Primary antibodies used include Phospho-BAD (Ser136) (D25H8) (Cell Signaling Technology, Danvers, MA, USA), Phospho-BAD (Ser112) (40A9) (Cell Signaling Technology, Danvers, MA, USA), BAD (Santa Cruz Biotechnology, Dallas, TX, USA), Phospho-AKT (S473) (D7F10) (Cell Signaling Technology, Danvers, MA, USA), Phospho-AKT (T308) (244F9) (Cell Signaling Technology, Danvers, MA, USA), pan-AKT (Abcam, Cambridge, UK), ALDH1A1(L-15) (Santa Cruz Biotechnology, Dallas, TX, USA), β-ACTIN (Santa Cruz Biotechnology, Dallas, TX, USA).
Cell Function Assays
Cell cycle analysis, three-dimensional (3D) Matrigel growth, and ALDEFLUOR assays were performed as previously described.66,67 Apoptotic cell death was determined using Annexin-V AlexaFluor &488 Propidium Iodide (PI) Dead Cell Apoptosis Kit (Life Technologies, Carlsbad, CA, USA) and Caspase-Glo Caspase 3/7 kit (Promega, Madison, WI, United States) according to the recommended protocols. The staining of live and dead cells in Matrigel was performed using the LIVE/DEAD Cell Imaging Kit (Invitrogen, Carlsbad, CA, USA). Colony formation assay was performed with 1 × 103 to 2 × 103 cells (in single cell suspension) seeded in 6-well plates 24 h before drug treatment, followed by incubation of the cells with drug for 10–14 days, and staining with crystal violet solution (1% crystal violet, 80 mL of distilled H2O, and 20 mL of methanol); quantification was achieved using ImageJ software.
Isolation and Culture of EOC Spheroids
Spheroids were allowed to form in 96-well or 6-well Corning Costar Ultra-Low Attachment plates, and DMEM/F12 (Hyclone, Logan, UT, United States) media was used with the supplement of 2% B27 (Gibco, Waltham, MA, USA), 20 ng/mL recombinant human EGF (Sigma-Aldrich, St. Louis, MO, USA), and 10 ng/mL recombinant human basic FGF (BD Biosciences, Franklin Lakes, NJ, USA). For mRNA and protein collection, EOC cells were seeded at the density of 5 × 103 to 10 × 103 cells/well into 6-well ultralow attachment plates. After 10–14 days of incubation, spheroids formed were collected and pelleted for mRNA and protein extraction. For analyzing sphere formation efficiency, 500–1000 cells/well were seeded into 96-well ultralow attachment plates with or without drug treatment. At 10–14 days later, the number of spheroids (diameter greater than 60 μm) were recorded under the microscope.
To assess chemo-sensitivity of spheroid cells to cisplatin, spheroids were dissociated and seeded at 2 × 103 cells/well. In parallel, parental EOC cells were seeded at the same density under standard 2D culture conditions. After 24 h, both groups were treated for 72 h with serially diluted cisplatin. Relative cell viability was determined using AlamarBlue (Invitrogen, Carlsbad, CA, USA) cell viability assay. IC50 values were determined using Prism 5 (GraphPad Software).
In Vivo Xenograft Studies
All protocols used in this study for animal experiments was approved by Institutional Animal Care and Use Committee of the Laboratory Animal Center of Peking University Shenzhen Graduate School (the permit number is YW). The xenograft experiments were performed with two cell lines: A2780 and A2780cis. A2780 or A2780cis cells were counted, and 200 μL of cell suspension (5 × 106 cells) were injected subcutaneously (s.c.) into the right flanks of six-week-old female nude mice. Tumor growth was monitored by measuring the tumor size using calipers; tumor volume (V) was determined using the following formula: V = 1/2 × larger diameter × (smaller diameter)2. Tumors were allowed to grow to reach ∼100 mm3, and then mice bearing tumors from the same cell line were randomized into four groups and subsequently injected with indicated drugs: group 1, vehicle administrated with the respective solvents of cisplatin and NPB at the same time as group 4; group 2, NPB injected daily i.p. at the dose of 20 mg; group 3, cisplatin injected by once a week i.p. at the dose of 5 mg/kg; group 4, combination of NPB and cisplatin (summarized in Figure 1). Vehicle consisted of 4.6% DMSO, 14.3% PEG400, 9.7% water pH 5.0, and 71.4% N-saline. All mice received vehicle in volumes equivalent to those used for injection of the NPB and/or cisplatin (Figure S-5). Tumor volume and body weight were measured daily. Mice were sacrificed according to the schematic in Figure S-5. Liver, kidney, and spleen were excised and weighed. The dissected tumors were placed in 10% buffered formalin, embedded in paraffin, and cut in 4 μm sections for hematoxylin and eosin (H&E) and immunohistochemistry (IHC).
Statistical Analysis
Prism 5 (GraphPad Software) and Microsoft Excel were used to generate graphical presentations and for statistical analysis. All in vitro experiments were reported as mean ± SEM. Statistical significance was assessed using unpaired student’s t tests or one-way ANOVA followed by Dunnett’s test post hoc.
Acknowledgments
This work was supported by grants from the National Medical Research Council of Singapore [R-713-000-163-511]; the Singapore Ministry of Education under its Research Centres of Excellence initiative to the Cancer Science Institute of Singapore, National University of Singapore; Tsinghua Berkeley Shenzhen Institute Faculty Start-Up Fund, the Shenzhen Development and Reform Commission Subject Construction Project [2017]1434, the Shenzhen Bay Laboratory; and VGST, GOK, India.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsptsci.0c00064.
Histological subtype and maintenance of EOC cell lines; supplemental figures as described in the text (PDF)
Author Contributions
Y.W., V.P., B., and P.E.L. designed research; Y.W., Y.C., Q.C., and M.Z. performed research; K.S.R. and B. synthesized NPB; Y.W., Y.C., Q.C., L.M., T.Z., A.P.K., R.Y.H, B., and P.E.L. analyzed data; and Y.W. and P.E.L. wrote the paper.
The authors declare the following competing financial interest(s): K.S.R., V.P., B., and P.E.L. are listed as inventors on a patent application for NPB which is used in this work (WO/2019/194520). P.E.L. is an equity holder in Sinotar Pharmaceuticals Ltd which currently holds the license for this patent.
Supplementary Material
References
- Cornelison R.; Llaneza D. C.; Landen C. N. (2017) Emerging Therapeutics to Overcome Chemoresistance in Epithelial Ovarian Cancer: A Mini-Review. Int. J. Mol. Sci. 18 (10), 2171. 10.3390/ijms18102171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J. Y.; Cho C. H.; Song H. S. (2017) Targeted therapy of ovarian cancer including immune check point inhibitor. Korean J. Intern. Med. 32 (5), 798–804. 10.3904/kjim.2017.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero I.; Bast R. C. Jr. (2012) Minireview: human ovarian cancer: biology, current management, and paths to personalizing therapy. Endocrinology 153 (4), 1593–602. 10.1210/en.2011-2123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markman M.; Bookman M. A. (2000) Second-line treatment of ovarian cancer. Oncologist 5 (1), 26–35. 10.1634/theoncologist.5-1-26. [DOI] [PubMed] [Google Scholar]
- Foley O. W.; Rauh-Hain J. A.; del Carmen M. G. (2013) Recurrent epithelial ovarian cancer: an update on treatment. Oncology (Williston Park) 27 (4), 288. [PubMed] [Google Scholar]
- Bookman M. A.; Brady M. F.; McGuire W. P.; Harper P. G.; Alberts D. S.; Friedlander M.; Colombo N.; Fowler J. M.; Argenta P. A.; De Geest K.; Mutch D. G.; Burger R. A.; Swart A. M.; Trimble E. L.; Accario-Winslow C.; Roth L. M. (2009) Evaluation of new platinum-based treatment regimens in advanced-stage ovarian cancer: a Phase III Trial of the Gynecologic Cancer Intergroup. J. Clin. Oncol. 27 (9), 1419–25. 10.1200/JCO.2008.19.1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- du Bois A.; Weber B.; Rochon J.; Meier W.; Goupil A.; Olbricht S.; Barats J. C.; Kuhn W.; Orfeuvre H.; Wagner U.; Richter B.; Lueck H. J.; Pfisterer J.; Costa S.; Schroeder W.; Kimmig R.; Pujade-Lauraine E. (2006) Addition of epirubicin as a third drug to carboplatin-paclitaxel in first-line treatment of advanced ovarian cancer: a prospectively randomized gynecologic cancer intergroup trial by the Arbeitsgemeinschaft Gynaekologische Onkologie Ovarian Cancer Study Group and the Groupe d’Investigateurs Nationaux pour l’Etude des Cancers Ovariens. J. Clin. Oncol. 24 (7), 1127–35. 10.1200/JCO.2005.03.2938. [DOI] [PubMed] [Google Scholar]
- Pfisterer J.; Weber B.; Reuss A.; Kimmig R.; du Bois A.; Wagner U.; Bourgeois H.; Meier W.; Costa S.; Blohmer J. U.; Lortholary A.; Olbricht S.; Stahle A.; Jackisch C.; Hardy-Bessard A. C.; Mobus V.; Quaas J.; Richter B.; Schroder W.; Geay J. F.; Luck H. J.; Kuhn W.; Meden H.; Nitz U.; Pujade-Lauraine E. (2006) Randomized phase III trial of topotecan following carboplatin and paclitaxel in first-line treatment of advanced ovarian cancer: a gynecologic cancer intergroup trial of the AGO-OVAR and GINECO. J. Natl. Cancer Inst 98 (15), 1036–45. 10.1093/jnci/djj296. [DOI] [PubMed] [Google Scholar]
- Hoskins P.; Vergote I.; Cervantes A.; Tu D.; Stuart G.; Zola P.; Poveda A.; Provencher D.; Katsaros D.; Ojeda B.; Ghatage P.; Grimshaw R.; Casado A.; Elit L.; Mendiola C.; Sugimoto A.; D’Hondt V.; Oza A.; Germa J. R.; Roy M.; Brotto L.; Chen D.; Eisenhauer E. A. (2010) Advanced ovarian cancer: phase III randomized study of sequential cisplatin-topotecan and carboplatin-paclitaxel vs carboplatin-paclitaxel. J. Natl. Cancer Inst 102 (20), 1547–56. 10.1093/jnci/djq362. [DOI] [PubMed] [Google Scholar]
- Ledermann J.; Harter P.; Gourley C.; Friedlander M.; Vergote I.; Rustin G.; Scott C.; Meier W.; Shapira-Frommer R.; Safra T.; et al. (2012) Olaparib maintenance therapy in platinum-sensitive relapsed ovarian cancer. N. Engl. J. Med. 366 (15), 1382–1392. 10.1056/NEJMoa1105535. [DOI] [PubMed] [Google Scholar]
- Perren T. J.; Swart A. M.; Pfisterer J.; Ledermann J. A.; Pujade-Lauraine E.; Kristensen G.; Carey M. S.; Beale P.; Cervantes A.; Kurzeder C.; et al. (2011) A phase 3 trial of bevacizumab in ovarian cancer. N. Engl. J. Med. 365 (26), 2484–2496. 10.1056/NEJMoa1103799. [DOI] [PubMed] [Google Scholar]
- Marchion D. C.; Cottrill H. M.; Xiong Y.; Chen N.; Bicaku E.; Fulp W. J.; Bansal N.; Chon H. S.; Stickles X. B.; Kamath S. G.; Hakam A.; Li L.; Su D.; Moreno C.; Judson P. L.; Berchuck A.; Wenham R. M.; Apte S. M.; Gonzalez-Bosquet J.; Bloom G. C.; Eschrich S. A.; Sebti S.; Chen D. T.; Lancaster J. M. (2011) BAD phosphorylation determines ovarian cancer chemosensitivity and patient survival. Clin. Cancer Res. 17 (19), 6356–66. 10.1158/1078-0432.CCR-11-0735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayakawa J.; Ohmichi M.; Kurachi H.; Kanda Y.; Hisamoto K.; Nishio Y.; Adachi K.; Tasaka K.; Kanzaki T.; Murata Y. (2000) Inhibition of BAD phosphorylation either at serine 112 via extracellular signal-regulated protein kinase cascade or at serine 136 via Akt cascade sensitizes human ovarian cancer cells to cisplatin. Cancer research 60 (21), 5988–94. [PubMed] [Google Scholar]
- Pandey V.; Wang B.; Mohan C. D.; Raquib A. R.; Rangappa S.; Srinivasa V.; Fuchs J. E.; Girish K. S.; Zhu T.; Bender A.; Ma L.; Yin Z.; Basappa; Rangappa K. S.; Lobie P. E. (2018) Discovery of a small-molecule inhibitor of specific serine residue BAD phosphorylation. Proc. Natl. Acad. Sci. U. S. A. 115 (44), E10505–e10514. 10.1073/pnas.1804897115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danial N. N. (2008) BAD: undertaker by night, candyman by day. Oncogene 27 (Suppl 1), S53–70. 10.1038/onc.2009.44. [DOI] [PubMed] [Google Scholar]
- Masters S. C.; Yang H.; Datta S. R.; Greenberg M. E.; Fu H. (2001) 14–3-3 inhibits Bad-induced cell death through interaction with serine-136. Mol. Pharmacol. 60 (6), 1325–31. 10.1124/mol.60.6.1325. [DOI] [PubMed] [Google Scholar]
- Tan Y.; Ruan H.; Demeter M. R.; Comb M. J. (1999) p90(RSK) blocks bad-mediated cell death via a protein kinase C-dependent pathway. J. Biol. Chem. 274 (49), 34859–67. 10.1074/jbc.274.49.34859. [DOI] [PubMed] [Google Scholar]
- Harada H.; Becknell B.; Wilm M.; Mann M.; Huang L. J.; Taylor S. S.; Scott J. D.; Korsmeyer S. J. (1999) Phosphorylation and inactivation of BAD by mitochondria-anchored protein kinase A. Mol. Cell 3 (4), 413–22. 10.1016/S1097-2765(00)80469-4. [DOI] [PubMed] [Google Scholar]
- Datta S. R.; Dudek H.; Tao X.; Masters S.; Fu H.; Gotoh Y.; Greenberg M. E. (1997) Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell 91 (2), 231–41. 10.1016/S0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- Harada H.; Andersen J. S.; Mann M.; Terada N.; Korsmeyer S. J. (2001) p70S6 kinase signals cell survival as well as growth, inactivating the pro-apoptotic molecule BAD. Proc. Natl. Acad. Sci. U. S. A. 98 (17), 9666–70. 10.1073/pnas.171301998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta S. R.; Katsov A.; Hu L.; Petros A.; Fesik S. W.; Yaffe M. B.; Greenberg M. E. (2000) 14–3-3 proteins and survival kinases cooperate to inactivate BAD by BH3 domain phosphorylation. Mol. Cell 6 (1), 41–51. 10.1016/S1097-2765(05)00012-2. [DOI] [PubMed] [Google Scholar]
- Sastry K. S.; Al-Muftah M. A.; Li P.; Al-Kowari M. K.; Wang E.; Ismail Chouchane A.; Kizhakayil D.; Kulik G.; Marincola F. M.; Haoudi A.; Chouchane L. (2014) Targeting proapoptotic protein BAD inhibits survival and self-renewal of cancer stem cells. Cell Death Differ. 21 (12), 1936–49. 10.1038/cdd.2014.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bapat S. A.; Mali A. M.; Koppikar C. B.; Kurrey N. K. (2005) Stem and progenitor-like cells contribute to the aggressive behavior of human epithelial ovarian cancer. Cancer Res. 65 (8), 3025–9. 10.1158/0008-5472.CAN-04-3931. [DOI] [PubMed] [Google Scholar]
- Dean M.; Fojo T.; Bates S. (2005) Tumour stem cells and drug resistance. Nat. Rev. Cancer 5 (4), 275–84. 10.1038/nrc1590. [DOI] [PubMed] [Google Scholar]
- Dasari S.; Tchounwou P. B. (2014) Cisplatin in cancer therapy: molecular mechanisms of action. Eur. J. Pharmacol. 740, 364–78. 10.1016/j.ejphar.2014.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahne J. C.; Honig A.; Meyer S. R.; Gambaryan S.; Walter U.; Wischhusen J.; Haussler S. F.; Segerer S. E.; Fujita N.; Dietl J.; Engel J. B. (2012) Downregulation of AKT reverses platinum resistance of human ovarian cancers in vitro. Oncol. Rep. 28 (6), 2023–8. 10.3892/or.2012.2041. [DOI] [PubMed] [Google Scholar]
- Sakai W.; Swisher E. M.; Jacquemont C.; Chandramohan K. V.; Couch F. J.; Langdon S. P.; Wurz K.; Higgins J.; Villegas E.; Taniguchi T. (2009) Functional restoration of BRCA2 protein by secondary BRCA2 mutations in BRCA2-mutated ovarian carcinoma. Cancer Res. 69 (16), 6381–6. 10.1158/0008-5472.CAN-09-1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bez A.; Corsini E.; Curti D.; Biggiogera M.; Colombo A.; Nicosia R. F.; Pagano S. F.; Parati E. A. (2003) Neurosphere and neurosphere-forming cells: morphological and ultrastructural characterization. Brain Res. 993 (1–2), 18–29. 10.1016/j.brainres.2003.08.061. [DOI] [PubMed] [Google Scholar]
- Dontu G.; Abdallah W. M.; Foley J. M.; Jackson K. W.; Clarke M. F.; Kawamura M. J.; Wicha M. S. (2003) In vitro propagation and transcriptional profiling of human mammary stem/progenitor cells. Genes Dev. 17 (10), 1253–70. 10.1101/gad.1061803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang D.; Nguyen T. K.; Leishear K.; Finko R.; Kulp A. N.; Hotz S.; Van Belle P. A.; Xu X.; Elder D. E.; Herlyn M. (2005) A tumorigenic subpopulation with stem cell properties in melanomas. Cancer Res. 65 (20), 9328–37. 10.1158/0008-5472.CAN-05-1343. [DOI] [PubMed] [Google Scholar]
- Silva I. A.; Bai S.; McLean K.; Yang K.; Griffith K.; Thomas D.; Ginestier C.; Johnston C.; Kueck A.; Reynolds R. K.; Wicha M. S.; Buckanovich R. J. (2011) Aldehyde dehydrogenase in combination with CD133 defines angiogenic ovarian cancer stem cells that portend poor patient survival. Cancer Res. 71 (11), 3991–4001. 10.1158/0008-5472.CAN-10-3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nwani N. G.; Condello S.; Wang Y.; Swetzig W. M.; Barber E.; Hurley T.; Matei D. (2019) A Novel ALDH1A1 Inhibitor Targets Cells with Stem Cell Characteristics in Ovarian Cancer. Cancers 11 (4), 502. 10.3390/cancers11040502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal S.; Rodriguez-Bravo V.; Galsky M.; Cordon-Cardo C.; Domingo-Domenech J. (2014) Targeting cancer stem cells to suppress acquired chemotherapy resistance. Oncogene 33 (36), 4451. 10.1038/onc.2013.411. [DOI] [PubMed] [Google Scholar]
- Morel A.-P.; Lièvre M.; Thomas C.; Hinkal G.; Ansieau S.; Puisieux A. (2008) Generation of breast cancer stem cells through epithelial-mesenchymal transition. PLoS One 3 (8), e2888. 10.1371/journal.pone.0002888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mani S. A.; Guo W.; Liao M.-J.; Eaton E. N.; Ayyanan A.; Zhou A. Y.; Brooks M.; Reinhard F.; Zhang C. C.; Shipitsin M.; et al. (2008) The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 133 (4), 704–715. 10.1016/j.cell.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou T.-C.; Talalay P. (1984) Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv. Enzyme Regul. 22, 27–55. 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- Fraser M.; Bai T.; Tsang B. K. (2008) Akt promotes cisplatin resistance in human ovarian cancer cells through inhibition of p53 phosphorylation and nuclear function. Int. J. Cancer 122 (3), 534–546. 10.1002/ijc.23086. [DOI] [PubMed] [Google Scholar]
- Dienstmann R.; Rodon J.; Serra V.; Tabernero J. (2014) Picking the point of inhibition: a comparative review of PI3K/AKT/mTOR pathway inhibitors. Mol. Cancer Ther. 13 (5), 1021–1031. 10.1158/1535-7163.MCT-13-0639. [DOI] [PubMed] [Google Scholar]
- Pfisterer J.; Plante M.; Vergote I.; du Bois A.; Hirte H.; Lacave A. J.; Wagner U.; Stahle A.; Stuart G.; Kimmig R.; Olbricht S.; Le T.; Emerich J.; Kuhn W.; Bentley J.; Jackisch C.; Luck H. J.; Rochon J.; Zimmermann A. H.; Eisenhauer E. (2006) Gemcitabine plus carboplatin compared with carboplatin in patients with platinum-sensitive recurrent ovarian cancer: an intergroup trial of the AGO-OVAR, the NCIC CTG, and the EORTC GCG. J. Clin. Oncol. 24 (29), 4699–707. 10.1200/JCO.2006.06.0913. [DOI] [PubMed] [Google Scholar]
- Parmar M. K.; Ledermann J. A.; Colombo N.; du Bois A.; Delaloye J. F.; Kristensen G. B.; Wheeler S.; Swart A. M.; Qian W.; Torri V.; Floriani I.; Jayson G.; Lamont A.; Trope C. (2003) Paclitaxel plus platinum-based chemotherapy versus conventional platinum-based chemotherapy in women with relapsed ovarian cancer: the ICON4/AGO-OVAR-2.2 trial. Lancet 361 (9375), 2099–106. 10.1016/S0140-6736(03)13718-X. [DOI] [PubMed] [Google Scholar]
- McClung E. C.; Wenham R. M. (2016) Profile of bevacizumab in the treatment of platinum-resistant ovarian cancer: current perspectives. Int. J. Women's Health 8, 59. 10.2147/IJWH.S78101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim G.; Ison G.; McKee A. E.; Zhang H.; Tang S.; Gwise T.; Sridhara R.; Lee E.; Tzou A.; Philip R.; et al. (2015) FDA approval summary: olaparib monotherapy in patients with deleterious germline BRCA-mutated advanced ovarian cancer treated with three or more lines of chemotherapy. Clin. Cancer Res. 21 (19), 4257–4261. 10.1158/1078-0432.CCR-15-0887. [DOI] [PubMed] [Google Scholar]
- Moore K. N.; Secord A. A.; Geller M. A.; Miller D. S.; Cloven N.; Fleming G. F.; Hendrickson A. E. W.; Azodi M.; DiSilvestro P.; Oza A. M.; et al. (2019) Niraparib monotherapy for late-line treatment of ovarian cancer (QUADRA): a multicentre, open-label, single-arm, phase 2 trial. Lancet Oncol. 20 (5), 636–648. 10.1016/S1470-2045(19)30029-4. [DOI] [PubMed] [Google Scholar]
- Boussios S.; Karihtala P.; Moschetta M.; Karathanasi A.; Sadauskaite A.; Rassy E.; Pavlidis N. (2019) Combined Strategies with Poly (ADP-Ribose) Polymerase (PARP) Inhibitors for the Treatment of Ovarian Cancer: A Literature Review. Diagnostics 9 (3), 87. 10.3390/diagnostics9030087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerji U.; Dean E. J.; Pérez-Fidalgo J. A.; Batist G.; Bedard P. L.; You B.; Westin S. N.; Kabos P.; Garrett M. D.; Tall M.; et al. (2018) A phase I open-label study to identify a dosing regimen of the Pan-AKT inhibitor AZD5363 for evaluation in solid tumors and in PIK3CA-mutated breast and gynecologic cancers. Clin. Cancer Res. 24 (9), 2050–2059. 10.1158/1078-0432.CCR-17-2260. [DOI] [PubMed] [Google Scholar]
- Tamura K.; Hashimoto J.; Tanabe Y.; Kodaira M.; Yonemori K.; Seto T.; Hirai F.; Arita S.; Toyokawa G.; Chen L.; Yamamoto H.; Kawata T.; Lindemann J.; Esaki T. (2016) Safety and tolerability of AZD5363 in Japanese patients with advanced solid tumors. Cancer Chemother. Pharmacol. 77 (4), 787–95. 10.1007/s00280-016-2987-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma S.; Lee T.; Zheng B.; Chan K.; Guan X. (2008) CD133+ HCC cancer stem cells confer chemoresistance by preferential expression of the Akt/PKB survival pathway. Oncogene 27 (12), 1749. 10.1038/sj.onc.1210811. [DOI] [PubMed] [Google Scholar]
- Dubrovska A.; Kim S.; Salamone R. J.; Walker J. R.; Maira S.-M.; García-Echeverría C.; Schultz P. G.; Reddy V. A. (2009) The role of PTEN/Akt/PI3K signaling in the maintenance and viability of prostate cancer stem-like cell populations. Proc. Natl. Acad. Sci. U. S. A. 106 (1), 268–273. 10.1073/pnas.0810956106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.; Zhou B. P. (2011) Activation of β-catenin and Akt pathways by Twist are critical for the maintenance of EMT associated cancer stem cell-like characters. BMC Cancer 11 (1), 49. 10.1186/1471-2407-11-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyler C. E.; Foo W. C.; LaFiura K. M.; McLendon R. E.; Hjelmeland A. B.; Rich J. N. (2008) Brain cancer stem cells display preferential sensitivity to Akt inhibition. Stem Cells 26 (12), 3027–3036. 10.1634/stemcells.2007-1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farley J.; Brady W. E.; Vathipadiekal V.; Lankes H. A.; Coleman R.; Morgan M. A.; Mannel R.; Yamada S. D.; Mutch D.; Rodgers W. H.; Birrer M.; Gershenson D. M. (2013) Selumetinib in women with recurrent low-grade serous carcinoma of the ovary or peritoneum: an open-label, single-arm, phase 2 study. Lancet Oncol. 14 (2), 134–40. 10.1016/S1470-2045(12)70572-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaufort C. M.; Helmijr J. C.; Piskorz A. M.; Hoogstraat M.; Ruigrok-Ritstier K.; Besselink N.; Murtaza M.; van I. W. F.; Heine A. A.; Smid M.; Koudijs M. J.; Brenton J. D.; Berns E. M.; Helleman J. (2014) Ovarian cancer cell line panel (OCCP): clinical importance of in vitro morphological subtypes. PLoS One 9 (9), e103988. 10.1371/journal.pone.0103988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philp A. J.; Campbell I. G.; Leet C.; Vincan E.; Rockman S. P.; Whitehead R. H.; Thomas R. J.; Phillips W. A. (2001) The phosphatidylinositol 3′-kinase p85alpha gene is an oncogene in human ovarian and colon tumors. Cancer Res. 61 (20), 7426–9. [PubMed] [Google Scholar]
- Hanrahan A. J.; Schultz N.; Westfal M. L.; Sakr R. A.; Giri D. D.; Scarperi S.; Janikariman M.; Olvera N.; Stevens E. V.; She Q.-B.; et al. (2012) Genomic complexity and AKT dependence in serous ovarian cancer. Cancer Discovery 2 (1), 56–67. 10.1158/2159-8290.CD-11-0170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan C. T.; Guzman M. L.; Noble M. (2006) Cancer stem cells. N. Engl. J. Med. 355 (12), 1253–61. 10.1056/NEJMra061808. [DOI] [PubMed] [Google Scholar]
- Latifi A.; Luwor R. B.; Bilandzic M.; Nazaretian S.; Stenvers K.; Pyman J.; Zhu H.; Thompson E. W.; Quinn M. A.; Findlay J. K.; Ahmed N. (2012) Isolation and characterization of tumor cells from the ascites of ovarian cancer patients: molecular phenotype of chemoresistant ovarian tumors. PLoS One 7 (10), e46858. 10.1371/journal.pone.0046858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolomainen D. F.; A’Hern R.; Coxon F. Y.; Fisher C.; King D. M.; Blake P. R.; Barton D. P.; Shepherd J. H.; Kaye S. B.; Gore M. E. (2003) Can patients with relapsed, previously untreated, stage I epithelial ovarian cancer be successfully treated with salvage therapy?. J. Clin. Oncol. 21 (16), 3113–8. 10.1200/JCO.2003.06.119. [DOI] [PubMed] [Google Scholar]
- Tan D. S.; Agarwal R.; Kaye S. B. (2006) Mechanisms of transcoelomic metastasis in ovarian cancer. Lancet Oncol. 7 (11), 925–34. 10.1016/S1470-2045(06)70939-1. [DOI] [PubMed] [Google Scholar]
- Carden C. P.; Stewart A.; Thavasu P.; Kipps E.; Pope L.; Crespo M.; Miranda S.; Attard G.; Garrett M. D.; Clarke P. A.; Workman P.; de Bono J. S.; Gore M.; Kaye S. B.; Banerji U. (2012) The association of PI3 kinase signaling and chemoresistance in advanced ovarian cancer. Mol. Cancer Ther. 11 (7), 1609–17. 10.1158/1535-7163.MCT-11-0996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pullen N.; Dennis P. B.; Andjelkovic M.; Dufner A.; Kozma S. C.; Hemmings B. A.; Thomas G. (1998) Phosphorylation and activation of p70s6k by PDK1. Science 279 (5351), 707–710. 10.1126/science.279.5351.707. [DOI] [PubMed] [Google Scholar]
- Huang R. Y.; Chung V. Y.; Thiery J. P. (2012) Targeting pathways contributing to epithelial-mesenchymal transition (EMT) in epithelial ovarian cancer. Curr. Drug Targets 13 (13), 1649–53. 10.2174/138945012803530044. [DOI] [PubMed] [Google Scholar]
- (2011) Integrated genomic analyses of ovarian carcinoma. Nature 474 (7353), 609–15. 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan T. Z.; Miow Q. H.; Miki Y.; Noda T.; Mori S.; Huang R. Y.; Thiery J. P. (2014) Epithelial-mesenchymal transition spectrum quantification and its efficacy in deciphering survival and drug responses of cancer patients. EMBO Mol. Med. 6 (10), 1279–93. 10.15252/emmm.201404208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang R. Y.; Kuay K. T.; Tan T. Z.; Asad M.; Tang H. M.; Ng A. H.; Ye J.; Chung V. Y.; Thiery J. P. (2015) Functional relevance of a six mesenchymal gene signature in epithelial-mesenchymal transition (EMT) reversal by the triple angiokinase inhibitor, nintedanib (BIBF1120). Oncotarget 6 (26), 22098–113. 10.18632/oncotarget.4300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang R. Y.; Wong M. K.; Tan T. Z.; Kuay K. T.; Ng A. H.; Chung V. Y.; Chu Y. S.; Matsumura N.; Lai H. C.; Lee Y. F.; Sim W. J.; Chai C.; Pietschmann E.; Mori S.; Low J. J.; Choolani M.; Thiery J. P. (2013) An EMT spectrum defines an anoikis-resistant and spheroidogenic intermediate mesenchymal state that is sensitive to e-cadherin restoration by a src-kinase inhibitor, saracatinib (AZD0530). Cell Death Dis. 4, e915. 10.1038/cddis.2013.442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu T.; Starling-Emerald B.; Zhang X.; Lee K. O.; Gluckman P. D.; Mertani H. C.; Lobie P. E. (2005) Oncogenic transformation of human mammary epithelial cells by autocrine human growth hormone. Cancer Res. 65 (1), 317–24. [PubMed] [Google Scholar]
- Chong Q. Y.; You M. L.; Pandey V.; Banerjee A.; Chen Y. J.; Poh H. M.; Zhang M.; Ma L.; Zhu T.; Basappa S.; Liu L.; Lobie P. E. (2017) Release of HER2 repression of trefoil factor 3 (TFF3) expression mediates trastuzumab resistance in HER2+/ER+ mammary carcinoma. Oncotarget 8 (43), 74188–74208. 10.18632/oncotarget.18431. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.