

SUMMARY
Cancer elimination in humans can be achieved with immunotherapy that relies on T lymphocyte-mediated recognition of tumor antigens. Several types of these antigens have been recognized based on their cellular origins and expression patterns, while their detection has been greatly facilitated by recent achievements in next-generation sequencing and immunopeptidomics. Some of them have been targeted in clinical trials with various immunotherapy approaches, while many others remain untested. Here, we discuss molecular identification of different tumor antigen types, and the clinical safety and efficacy of targeting them with immunotherapy. Additionally, we suggest strategies to increase the efficacy and availability of antigen-directed immunotherapies for treatment of patients with metastatic cancer.
INTRODUCTION
Sporadic cancer arises from normal tissues through either spontaneous or environmentally induced accumulation of genetic and epigenetic aberrations. This process leads to production of proteins that differ, quantitatively or qualitatively, from those made by normal cells. When they are recognized by the adaptive immune system and thus provoke an immune attack against the cancer, these proteins can be classified as tumor antigens. Although this attack most often fails to control the growth of clinically apparent cancers, the molecular identity of tumor antigens can be exploited to improve the effectiveness of cancer immunotherapy.
The anti-cancer response of the adaptive immune system, with conventional T cells as its major mediator, is both induced and amplified by various cell types of the innate immune system. For instance, professional antigen-presenting cells (APCs), such as dendritic cells, phagocytize dying tumor cells and present processed tumor antigens to the cognate naive T cells, which subsequently causes their activation. Concomitantly, other types of innate immune cells, such as innate lymphoid cells or unconventional T cells, may directly eliminate cancer cells based on the presence or lack of certain membrane-bound ligands (Bruchard and Ghiringhelli, 2019; Godfrey et al., 2018).
Conventional T lymphocytes utilize their T cell receptors (TCRs), which are membrane-bound molecules composed of alpha and beta chains, to recognize antigens expressed by the target cell. Specifically, TCRs recognize peptides that are bound to major histocompatibility complex (MHC) molecules on the target cell surface. In this context, peptide sequences that are recognized are called “epitopes,” while their parent proteins are referred to as “antigens.” Upon epitope recognition, molecules associated with TCRs transmit the activation signal through their intracellular signaling domains. This, together with activation of various costimulatory receptors by ligands expressed in the tumor microenvironment (Chen and Flies, 2013), stimulates the lymphocyte to initiate an immune reaction.
The two main types of T cells, cluster of differentiation (CD) 8+ and CD4+, utilize their TCRs to recognize cognate epitopes in two different ways. TCRs on CD8+ cells can recognize 8–10 amino acid long peptides, which are derived from a variety of cytoplasmatic proteins via proteasomal digestion and are bound to MHC class I molecules. The latter are expressed on the membrane of almost all cells in the body, with the exception of germ cells and placental trophoblast. In contrast, TCRs on CD4+ T cells recognize longer peptides that are predominantly derived from both endosomal and ingested proteins via lysosomal digestion. These peptides are bound to MHC class II molecules, which are normally found on the surface of APCs but can also appear on a variety of other cells in the context of stress or inflammation. Upon activation, both CD8+ and CD4+ cells initiate a cascade of reactions that ultimately leads to destruction of target cells.
Recently, chimeric antigen receptors (CARs) have been engineered by covalently combining the antigen-binding domains of monoclonal antibodies with intracellular T cell activation domains. This provides CAR-transduced lymphocytes with the recognition attributes of antibodies, in contrast to TCR-mediated recognition by the conventional T cells (Figure 1). Both antibodies and CARs recognize the three-dimensional structure of intact membrane-bound molecules on the cancer cell surface, independently of MHC molecules. Furthermore, they may also recognize non-protein molecules such as gangliosides (Rossig et al., 2018). However, despite major research efforts, antibodies that can recognize cancer cells specifically have very rarely been identified, as virtually all intact membrane molecules can also be expressed on normal tissues. Therefore, as has been documented, targeting these molecules with CARs in solid tumors comes with an inherent risk of severe toxicities to normal tissues. Some of these toxicities can still be tolerated if the targeted normal tissues perform non-vital or medically replaceable functions, as has been demonstrated by studies in which CAR T cells have been used to effectively treat hematological malignancies that express molecules such as CD19 or BCMA, which are also found on the surface of normal B cells or plasma cells, respectively (Holstein and Lunning, 2020; Majzner and Mackall, 2019).
Figure 1. General Principles of Antigen Recognition by the Adaptive Immune System.
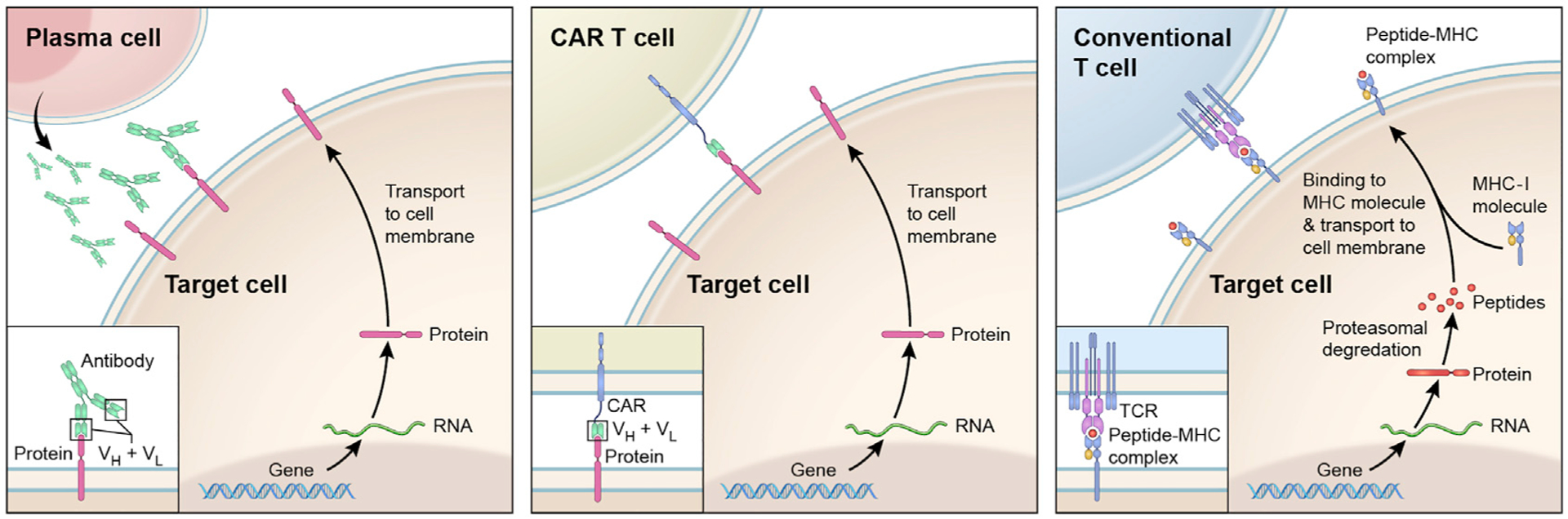
Upon stimulation, B lymphocytes differentiate into plasma cells (left panel) to produce antibodies (immunoglobulins). These can bind to a variety of intact proteins (but also other molecules) that are exposed on the cell surface. The antigen-binding portions of an antibody are composed of variable light (VL) and variable heavy chain (VH) domains (insert). CAR T cells (middle panel) recognize the same types of molecules as do the antibodies. These cells have been modified to express CARs, which are constructed by fusing antibody-derived antigen-binding domains (VH + VL) with intracellular T cell signaling domains (insert). In contrast, conventional T cells (right panel) recognize peptides that are derived from cellular proteins and are displayed on cell surface in complex with MHC molecules. For clarity, only the presentation on MHC class I molecules is depicted, in a simplified manner.
The role of conventional T cells in mediating antitumor immune responses has been well established, and the antigens they recognize have emerged as superior determinants that can distinguish cancer from normal cells. Therefore, this review will focus on tumor antigens recognized by conventional TCRs. Those that are derived from unmutated cellular proteins, which may also be expressed in normal tissues, will be referred to here as tumor-associated antigens. In contrast, those that are found exclusively in cancer cells as a result of cancer-specific mutations or other alterations will be referred to as tumor-specific antigens. In this context, peptides expressing aberrant sequences that are recognized by the T cells are called “neoepitopes,” and the molecules from which they are derived are called “neoantigens.”
T CELL-BASED CANCER IMMUNOTHERAPIES
Several types of T cell-based cancer immunotherapies have been used to treat patients with metastatic solid cancer (Table 1). Three types entail nonspecific (antigen-agnostic) stimulation of a broad variety of the endogenous T cells, including those that are capable of recognizing the cancer but are rendered ineffective due to a variety of immune escape mechanisms. Additionally, allogeneic hematopoietic stem cell transplantation, a form of cellular immunotherapy that allows reconstitution of the immune system with donor T cells that may exert graft-vs-tumor effect, has been successfully used in treatment of hematological malignancies and has been attempted in treatment of selected solid tumors (Bregni et al., 2016).
Table 1.
Overview of T Cell-based Immunotherapies for Treatment of Solid Cancers
| Therapy Type | Formulation | Administration | Mechanism of Action | ||
|---|---|---|---|---|---|
| Antigennonspecific | IL-2 | human recombinant IL-2 (main T lymphocyte growth factor) | intravenous | stimulating endogenous T cells that may recognize cancer (unspecified tumor antigens) | |
| ICIs | monoclonal antibodies targeting molecules that inhibit T cell function (i.e., CTLA-4, PD-1, PD-L1) | intravenous | disinhibiting endogenous T cells that may recognize cancer (unspecified tumor antigens) | ||
| oncolytic viruses | attenuated, native or genetically modified viral particles that selectively infect cancer cells | intratumoral, intravenous | inducing immunogenic cell death and potential release of new tumor antigens | ||
| Antigen specific | cancer vaccinesa | selected tumor antigens that can be loaded onto the APCs, embedded in a viral vector, or represented as peptides or nucleic acids; usually in combination with an adjuvant | cutaneous, subcutaneous, intramuscular, intravenous | generating/stimulating endogenous T cell responses to selected tumor antigens | |
| ACT | TILs | unmodified T cells expanded from a patient’s tumor (or PBL), selected for recognition of cancer cells or specific tumor antigens | intravenous (following preparative lymphodepleting chemotherapy) | transferred T cells seek and destroy cancer cells that present the targeted antigens on specific MHC molecules | |
| TCR-transduced T cells | autologous lymphocytes obtained by leukapheresis and transduced to express a TCR directed against a specific tumor antigen | intravenous (following preparative lymphodepleting chemotherapy) | transferred T cells seek and destroy cancer cells that present the targeted antigens on specific MHC molecules | ||
| CAR-transduced T cells | autologous lymphocytes obtained by leukapheresis and transduced to express a CAR directed against a specific membrane-bound antigen | intravenous (with or without preparative lymphodepleting chemotherapy) | transferred CAR T cells seek and destroy cancer cells that express the targeted antigen on the cell surface, independently of MHC molecules | ||
Some cancer vaccines entail administration of attenuated or lysed tumor cells. These can be classified as antigen nonspecific.
The first type of nonspecific immunotherapy is the systemic administration of interleukin-2 (IL-2), the main T lymphocyte growth factor. Clinical studies using IL-2 were the first to provide evidence that manipulation of the human immune system could reproducibly lead to durable tumor regressions (Rosenberg, 2014). This ultimately led to its US Food and Drug Administration (FDA) approvals for treatment of metastatic renal cell carcinoma in 1992 and for metastatic melanoma in 1998.
The second nonspecific type of immunotherapy is the blockade of T cell inhibitory signaling with immune checkpoint inhibitors (ICIs). These are monoclonal antibodies directed against inhibitory molecules, such as CTLA-4 or PD-1, which are expressed on the surface of T cells, or the PD-1 ligand (PD-L1), which is expressed on the surface of tumor cells or APCs (Wei et al., 2018). In recent years, ICIs have emerged as an efficacious therapy for various types of human cancers, with a continuously expanding list of FDA-approved indications. However, the majority of patients with metastatic epithelial cancers, which account for 90% of cancer-related deaths, still do not experience cancer regressions following this treatment.
Finally, the third type of an antigen-nonspecific approach to controlling cancer growth is the administration of oncolytic viruses. These are attenuated, native or genetically modified viral particles that selectively infect cancer cells and induce them to undergo immunogenic cell death. This in turn may generate or stimulate endogenous T cell responses to cancer (Kaufman et al., 2015).
Research that followed the first discovery of a human tumor antigen (van der Bruggen et al., 1991) has led to development of antigen-specific immunotherapies for cancer. One such type of therapy is anti-cancer vaccines, which entail the administration of specific proteins, peptides, or corresponding nucleic acids to patients in an attempt to boost or induce activation of endogenous antitumor T lymphocytes (Hu et al., 2018). Because of the ease of administration and the relative lack of toxicity, cancer vaccines are a popular immunotherapy approach. However, perhaps owing to their inability both to generate large numbers of T cells with high affinity for tumor antigens and to overcome an immunosuppressive tumor microenvironment, there has been limited evidence that cancer vaccines can reproducibly lead to clinically meaningful regressions of established cancers (Klebanoff et al., 2011; Rosenberg et al., 2004).
Another version of antigen-specific immunotherapy is the adoptive T cell transfer (ACT) of lymphocytes that exhibit antitumor activity. In one approach to ACT, which has been pioneered in patients with metastatic melanoma, tumor-infiltrating lymphocytes (TILs) are cultured from the resected cancer, then selected for antitumor activity and expanded to very large numbers ex vivo, and finally reinfused back into the patient (Rosenberg and Restifo, 2015). Alternatively, similar treatment can be performed using peripheral blood-derived T lymphocytes (PBLs) that have been expanded following exposure to a select antigen in vitro (Chapuis et al., 2016).
In another ACT approach, patients are infused with autologous T lymphocytes that were genetically engineered to express a TCR that confers recognition of a specific tumor antigen (Chandran and Klebanoff, 2019). Both approaches entail pretreating the patient with lymphodepleting chemotherapy, which enables “engraftment” of transferred lymphocytes, followed by intravenous administration of IL-2 to stimulate their survival and proliferation. Concomitantly, therapy with CAR T cells, which is not discussed here, can also be considered as a type of ACT.
The advantages of ACT include the ability to overcome the T cell-suppressive impact of the tumor microenvironment by transferring a very large number (up to 1011) of cells that exhibit antitumor activity. Furthermore, pretreatment with lymphodepleting chemotherapy may remove certain T cell suppressive mechanisms, such as regulatory T lymphocytes and/or myeloid-derived suppressor cells. Because ACT requires individual preparation of patients’ own T cells for infusion, it can be performed only in a few specialized treatment centers. However, as discussed in the following sections, examples exist that demonstrate a potential of this approach to mediate complete destruction of selected solid tumors.
ANTIGEN DISCOVERY METHODS
Identification of specific tumor antigens, as well as T cells that recognize them, is essential for the design and execution of both vaccine- and ACT-based immunotherapy approaches. Regardless of which of the methods is used, immunogenicity of each newly discovered antigen needs to be validated in functional assays. This is accomplished by demonstrating that T cell activation occurs only upon encountering a specific epitope, but not the corresponding control (e.g., wild-type peptide for mutant antigens) that is bound to the same MHC molecule. A mere finding that a peptide is bound or even predicted to bind to an MHC molecule expressed by cancer is not a proof of its immunogenicity and can be misleading.
cDNA Expression Library Screening
This method was the first one to be used for tumor antigen identification (Kawakami et al., 1994b; van der Bruggen et al., 1991). It typically starts with isolation of a tumor-reactive T cell population, either from a patient’s tumor or from the PBL. Next, total RNA is isolated from tumor cells and converted into pools of cDNA plasmids, which are transfected into recipient cells (such as COS-7 or 293-HEK cells), often together with plasmids encoding specific MHC molecules. T cells are then co-cultured with the transfected cells, and assayed for recognition of cDNA pools and, subsequently, individual cDNA plasmids. Those that elicit recognition by T cells are used to define the encoded epitopes (peptides), which are then synthesized, pulsed onto the MHC-transfected cells, and validated in a reaction with the same T cell population.
Although it allows identification of most tumor antigen types, this method is laborious and time consuming, and therefore inappropriate for high-throughput antigen screening. Furthermore, due to difficulties in cloning of GC-rich sequences and large or poorly expressed RNA transcripts, it may be insufficiently sensitive to detect some types of mutated tumor antigens (Garcia-Garijo et al., 2019).
Next-Generation Sequencing-Based Screening Methods
Autologous T cells can be screened using peptides that arise from various types of cellular proteins, including those that harbor tumor-specific mutations. The sequences of these peptides can be determined by interrogating tumor and normal DNA or RNA by next-generation sequencing (NGS) methods (Ding et al., 2014). To facilitate screening, especially in malignancies that harbor a vast number of mutations, such as melanoma, candidate peptides can be filtered by using algorithms that predict their immunogenicity or by directly assessing their presentation on the tumor cell surface using immunopeptidomics.
Prediction Algorithm-Based Screening Methods
Various algorithms have been developed to predict whether peptides derived from a specified protein, either wild-type or mutant, are available to interact with TCRs on T cells. This is most commonly done by predicting their ability to bind to specific MHC molecules that are expressed by the cancer, as exemplified by various iterations of the NetMHCpan algorithm (Jurtz et al., 2017; Reynisson et al., 2020). These and other algorithms have been trained on data resulting from in vitro binding assays involving peptides with defined amino acid sequences, or on data obtained by immunopeptidomics (see below). To obtain binding predictions, researchers input protein or peptide sequences, specify the desired peptide length, and select an MHC molecule of choice. The algorithms then generate a list of possible resulting peptides ranked by their MHC-binding affinities, which are usually expressed as either the half maximal inhibitory concentration (IC50) or as a percentile rank. Peptides passing a certain consensus threshold (i.e., <500 nM or ≤2, respectively) are generally considered MHC binders and are selected for antigen screening.
In one such screening approach, whole-exome sequencing (WES) data from matched tumor and normal DNA is coupled with translation in silico to identify peptides containing tumor-specific non-synonymous mutations. A portion of peptides that is predicted to strongly bind to patients’ own MHC class I molecules is then synthesized, pulsed onto the APCs, and tested for recognition by the autologous CD8+ T lymphocytes (Robbins et al., 2013).
In a variation of this approach, peptide-pulsed APCs are replaced with artificial multimeric peptide-MHC complexes (e.g., MHC tetramers), which are generated by joining a variable number of fluorescently labeled or genetically barcoded MHC molecules and loading them with candidate peptides. These complexes can bind to complementary TCRs and thus enable quantification of T cells that recognize candidate antigens (Cohen et al., 2015; van Rooij et al., 2013). This approach can be efficient for identifying epitopes that are predicted to bind to prevalent MHC class I molecules, but it is still of limited use for identification of those that bind to class II or rarer class I MHC molecules (Vollers and Stern, 2008). Furthermore, as it entails testing peptide libraries tied to selected MHC molecules, this method generally fails to assess all the potential antigens expressed by the cancer.
Prediction-based screening methods have several important limitations, and only a few of the predicted peptides are found to be immunogenic in validation experiments (Garcia-Garijo et al., 2019). These limitations include suboptimal algorithm performance with less common class I and most class II MHC molecules, lack of ability to identify post-translationally modified or spliced peptides, and propensity to miss some de facto immunogenic peptides. To overcome some of these limitations, various bioinformatical approaches also incorporate algorithms that predict other protein/peptide characteristics implicated in immunogenicity. For instance, a model with an increased ability to predict immunogenic mutated peptides has recently been developed by combining predictions of peptide-MHC binding affinity, wild-type-over-mutant affinity ratios and the stability of given peptide-MHC complexes, together with data on the expression of cognate genes (Gartner et al., unpublished data).
Unbiased Tumor Antigen Screening
To bypass the limitations of prediction algorithms, another WES-based approach has been developed to enable unbiased screening of all candidate antigens; i.e., without restricting the analysis to specific MHC molecules (Tran et al., 2017). In this approach, metastatic tumors are surgically removed and used both to generate TIL cultures and to perform WES to identify tumor-specific non-synonymous mutations, namely single-nucleotide variants (SNVs) and small (<50 bp) insertion and deletions (INDELs). These sequences are used as templates to synthesize two types of screening libraries. One type is prepared by synthesizing and pooling 25-mer peptides harboring a tumor-specific mutation in their center. The second type is prepared by designing minigenes that represent the mutant 25-mers and concatenating them into tandem minigenes (TMGs), which are ultimately transcribed into RNA in vitro. Next, autologous APCs are pulsed with peptide pools or electroporated with TMGs, allowing processing and presentation of candidate antigens on all possible autologous MHC molecules, and then co-cultured with a panel of TILs. Peptide pools or TMGs that elicit T cell activation are further deconvoluted to identify specific tumor antigens.
As described in the following sections, this approach has been used to identify a number of tumor antigens arising from missense SNVs (mSNVs) or INDELs. However, it does not allow detection of antigens that arise from unmutated genes, gene fusions (some bioinformatic approaches can still enable this, but with limitations), or from aberrant RNA processing or translation. These limitations could be overcome by utilization of RNA sequencing or whole-genome sequencing (WGS) in a similar paired tumor-vs-normal fashion.
Immunopeptidomics
Finally, tumor antigens can be identified by direct interrogation of the tumor immunopeptidome; i.e., all endogenous peptides that are presented by MHC molecules on the cell surface. In this approach, after extraction from tumor cells, peptides are eluted from their complexes with MHC molecules and then subjected to liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS). Next, in order to identify tumor-specific peptides, MS spectra are compared with customized databases, which are generated by combining NGS data from patients’ tumors with the reference protein sequences (Bassani-Sternberg et al., 2016). Although this approach has the potential to uncover all of the possible classes of tumor antigens, including products of post-translational modifications that could be missed by the aforementioned sequencing approaches, its use is still limited by overall low sensitivity, especially for MHC class II-bound peptides.
TARGETING TUMOR-ASSOCIATED ANTIGENS WITH IMMUNOTHERAPY
Several classes of tumor-associated antigens (TAAs) have been identified, mostly using cDNA screening-based approaches (Table 2). These antigens are encoded by unmutated genes that may also be expressed in select normal tissues. Although they are shared among patients, which has made targeting them with “off-the-shelf” immunotherapies attractive, doing so has significant limitations.
Table 2.
Characteristics of Different Tumor Antigen Types
| Tumor Antigen | Role in Oncogenesis | Expression in Normal Tissues | Structural Similarity to Normal Proteins | Shared Among Patients? | Clinical Experience Class Type with Targeting? | |
|---|---|---|---|---|---|---|
| Class | Type | |||||
| TAAs | CGAs | uncertain | limited (germ cells, placenta) | high | yes | yes |
| HERVs | uncertain | variable (type dependent) | low | yes | limited | |
| TDAs | uncertain | yes | high | yes | yes | |
| overexpressed antigens | uncertain | yes | high | yes | yes | |
| TSAs | mSNVs | rarely drivers | no | moderate | rarely | yes |
| INDELs | rarely drivers | no | low | rarely | limited | |
| gene fusions | rarely drivers | no | low | rarely | limited | |
| viral oncoproteins | drivers | no | low | yes | yes | |
| UCAs | splice variants alternative ORFs post-translational modifications | unexplored | unexplored; some are expressed in normal cells | variable | unexplored | no |
TSAs, tumor-specific antigens; UCAs, unconventional antigens.
Firstly, as has been documented in clinical studies, targeting these antigens by T cells can result in serious and sometimes fatal toxicities. In a phenomenon called “on-target toxicity,” administration of T lymphocytes bearing high-affinity TCRs can lead to destruction of normal tissues that express even a miniscule amount of the targeted tumor antigen. Additionally, targeting these antigens increases the chances of unintended cross-reactivity with normal tissues expressing structurally similar normal proteins, a phenomenon called “off-target toxicity.”
Secondly, T cells that bear TCRs with high affinity toward normal cellular proteins are usually eliminated during negative selection in the thymus. This theoretically limits the effectiveness of vaccine approaches that aim to boost the activity of endogenous T cells that recognize these tumor antigens, and also curtails successful isolation of high-affinity TCRs for purposes of ACT. However, the process of negative selection is not perfectly efficient. Some auto-reactive T cells, even those that bear high-affinity TCRs, can still escape the thymus and can be found elsewhere in the body, where they are restrained by various regulatory mechanisms (Davis, 2015; Maeda et al., 2014).
Cancer Germline Antigens
Cancer germline antigens (CGAs), also known as cancer-testis antigens, are derived from genes that are expressed during fetal development. These genes are epigenetically silenced in most normal adult tissues, except germ and placental trophoblast cells, which do not express MHC molecules and are thereby “invisible” to the immune system. However, they can be expressed in a variety of tumors as a result of DNA demethylation (De Smet et al., 1999) and can yield peptide fragments that elicit recognition by T cells. A large number of CGAs were identified using cDNA library screening approaches, including the NYESO-1 and members of MAGE-A family of proteins, both encoded by the X chromosome and expressed in several types of cancer (Almeida et al., 2009).
The implications of CGA expression on responses to ICIs have been largely unexplored. However, in one study involving melanoma patients, increased expression of MAGE-A family members was associated with poor anti-CTLA-4 but not anti-PD-1 responses, possibly due to interference with processing and presentation of other tumor antigens (Shukla et al., 2018).
Many attempts have been made to treat cancer by immunizing patients with CGA-based vaccines. However, despite generating high frequencies of reactive T cells in the blood, these vaccines failed to achieve tumor regressions in the vast majority of treated patients (Rosenberg et al., 2004). Furthermore, this approach has also failed to prevent cancer recurrences in patients with surgically excised tumors, as demonstrated by a recent phase III clinical trial of a MAGE-A3-based vaccine in treatment of localized melanoma (Dreno et al., 2018). The negative results of these studies could be explained by the inability of the vaccine-stimulated T cells to overcome the immunosuppressive tumor environment, or by the aforementioned consequences of the negative thymic selection against the cells that could efficiently target these antigens.
Several ACT clinical studies used autologous lymphocytes transduced with an affinity-enhanced and MHC class I-restricted TCR to target NY-ESO-1, a CGA expressed in a wide range of malignancies (Chen et al., 1997). This led to objective, largely transient responses in more than 50% of patients with NY-ESO-1-positive metastatic melanoma and synovial cell sarcoma, with no evidence of toxicities to normal tissues (D’Angelo et al., 2018; Nowicki et al., 2019; Robbins et al., 2011).
Targeting MAGE-A3 with CD4+ T cells engineered to express an unmodified MHC class II-restricted TCR yielded responses in patients with different types of MAGE-A3-positive cancers, while producing no off-target toxicities (Lu et al., 2017). However, targeting the same protein with MHC class I-restricted TCR led to off-target neurological toxicities due to previously unknown MAGE-A3 cross-reactivity with the MAGE-A12, which is expressed in the brain (Morgan et al., 2013). Furthermore, targeting MAGE-A3 with an affinity-enhanced TCR led to severe cardiac toxicities due to cross-reactivity to titin, a striated-muscle protein expressed in the myocardium (Cameron et al., 2013). These examples highlight the fact that targeting some CGAs, even when they are considered relatively tumor specific, can be unsafe due to off-target toxicities.
The overall benefit of ACT targeting either NY-ESO-1 or MAGE-A3 may also be limited by low prevalence of metastatic cancers that express these antigens, as well as by incomplete antigen expression in the tumors. In an immunohistochemistry-based study, only a small portion of metastatic carcinomas exhibited NY-ESO-1 or MAGE-A expression in >50% of tumor cells (Kerkar et al., 2016). Apart from these proteins, the efficacy and safety of targeting other CGAs with TCR-based therapies has been largely unexplored in patients with solid tumors.
Human Endogenous Retroviruses
Human endogenous retroviruses (HERVs) are remnants of ancient retroviral integration events, which are located throughout the human genome (Lander et al., 2001). Although they make up approximately 8.5% of the genomic DNA, only a small fraction of them harbor intact sequences that can be successfully translated. They are epigenetically silenced in normal tissues, predominantly through DNA methylation.
Similarly to CGAs, HERVs can become expressed in tumors due to DNA demethylation and can generate peptides that elicit recognition by patients’ own T cells. This was demonstrated by several reports, including a cDNA screening-based study of a patient with melanoma (Schiavetti et al., 2002), and further research in breast and ovarian cancers (Rycaj et al., 2015; Wang-Johanning et al., 2008). In addition to being immunogenic themselves, HERVs may lead to formation of double-stranded RNAs that trigger cellular immune responses similar to those seen in viral infections, a phenomenon called viral mimicry (Roulois et al., 2015).
Several studies have suggested that HERVs may help mediate tumor regression after immunotherapy. In one, a patient with metastatic renal cell cancer (RCC) who experienced a complete tumor regression following hematopoietic stem cell transplantation was found to have circulating donor T cells that recognized an HERV-E-derived peptide (Takahashi et al., 2008). Additionally, tumoral expression of select HERVs in ICI-treated patients with metastatic RCC was found to be significantly higher in responders than in non-responders (Panda et al., 2018; Pignon et al., 2019; Smith et al., 2018).
There is very limited clinical experience of direct HERV targeting. Although virtually all cancers may express them (Smith et al., 2018), directed immunotherapy may be limited to select cases due to their expression in normal tissues. For instance, one RNA sequencing-based study found that only three out of 66 analyzed HERVs were expressed exclusively in tumors (Rooney et al., 2015). Among these was the aforementioned HERV-E, the expression of which had already been known to occur specifically in RCC. The clinical safety and efficacy of TCR-transduced T cells directed against this particular HERV are currently being tested in patients with metastatic RCC (NCT03354390).
Tissue Differentiation Antigens
Expression of genes that encode tissue differentiation antigens (TDAs) is restricted to tumors and tissues of tumor origin. For instance, both MART-1 and gp100, the first TDAs identified, are expressed in melanoma but also in normal melanocytes in the skin, inner ear, and the eye (Kawakami et al., 1994a, 1994b).
Many clinical studies have attempted to treat cancer patients with TDA-based vaccines, but they failed to provide meaningful clinical benefits in the vast majority of cases (Rosenberg et al., 2004). The best results have been reported for sipuleucel-T, a vaccine based on prostate acid phosphatase (a prostate-specific TDA). In a phase III clinical trial that ultimately led to its FDA approval, this vaccine led to a modest prolongation of overall survival in patients with asymptomatic or minimally symptomatic metastatic prostate cancer (Kantoff et al., 2010). However, as there were no objective tumor regressions or even a significant decrease in specific tumor markers, the use of this therapy has not been widely adopted.
Due to their more promiscuous pattern of expression, targeting TDAs with ACT may carry a significant risk of toxicities affecting normal tissues. For example, in a study of 36 patients with metastatic melanoma, targeting MART-1 and gp100 with high-affinity TCRs resulted in cancer regression in 30% and 19% of patients, respectively. However, it also induced destruction of normal melanocytes, which led to vitiligo and serious ocular and auditory symptoms that required treatment with high doses of local steroids (Johnson et al., 2009).
Overexpressed Tumor Antigens
These antigens are products of normal genes that are expressed in several normal tissues and are overexpressed in certain types of cancer; e.g., HER2 in ovarian and breast cancer (Fisk et al., 1995) and carcinoembryonic antigen (CEA) in a variety of epithelial cancers (Parkhurst et al., 2009). As the least tumor-specific class of TAAs, they pose the most significant risk of on-target toxicities in ACT trials. For instance, in a study involving three patients with colorectal cancer, ACT with T cells bearing a high-affinity TCR against CEA resulted in an objective cancer regression in one patient, but also induced severe irreversible destruction of normal colonic mucosa in all three cases (Parkhurst et al., 2011).
In addition to conventional T cells, overexpressed tumor antigens have also been targeted with CAR T cells. In one report, targeting carbonic anhydrase IX (CAIX) in patients with metastatic kidney cancer resulted in unintended toxicities due to CAIX expression in the biliary duct epithelium (Lamers et al., 2013). These results further exemplify the safety concerns regarding targeting this antigen class.
TARGETING TUMOR-SPECIFIC ANTIGENS (NEOANTIGENS)
These antigens result mainly from genomic perturbations that occur exclusively in tumor cells and can be detected by either cDNA-, NGS-, or immunopeptidomics-based screening approaches. Unlike TAAs, neoantigens exhibit entirely novel amino acid sequences, which are rarely shared among patients. This may explain why their targeting with immunotherapy has produced no toxicities to normal tissues in clinical trials. Moreover, T cells bearing high-affinity TCRs against these antigens are presumably unaffected by negative thymic selection and so can readily be isolated from patients’ tumors or peripheral blood. Although the vast majority of previous studies have focused on products of SNVs, an increasing body of work indicates that other types of mutations may also be immunogenic (Table 2).
In contrast to sporadic mutations, which comprise the majority of cancer-associated mutations and are discussed here, germline mutations that may lead to formation of familial cancers are present in both cancerous and normal tissues. Thereby, they are expected to induce immunologic tolerance, resulting in a lack of endogenous T cell responses, as recently demonstrated in a study on individuals harboring a germline mutation in the tumor suppressor APC (Majumder et al., 2018).
Neoantigens Arising from mSNVs
mSNVs are the predominant form of mutations across different types of solid tumors (Vogelstein et al., 2013). Expression of mSNV-affected genes may lead to production of cellular proteins that harbor a single amino acid change in their sequence. These proteins may become immunogenic if the peptides that result from their fragmentation retain the mutated sequence and thus either acquire the property to bind to surface MHC molecules (Lurquin et al., 1989) or become recognizable by a TCR (Sibille et al., 1990). Products of mSNVs in MUM1 (Coulie et al., 1995), CDK4 (Wolfel et al., 1995), and CTNNB1 (Robbins et al., 1996) genes were among the first neoantigens ever discovered, all using cDNA-based screening approaches in patients with metastatic melanoma.
The development of the unbiased WES-based antigen screening method has enabled systematic evaluation of mSNV immunogenicity in different types of cancer (Table 3). In a study that focused on various types of metastatic gastrointestinal cancers, autologous TILs from 62 out of 75 (83%) patients were able to recognize at least one cancer-specific mutation (Parkhurst et al., 2019). Out of 7,654 mutations that were screened, a total of 124 were recognized by patients’ TILs. Most of these mutations were SNVs (122 out of ~7,200), which indicated that only a small proportion of SNVs were immunogenic (~1.6%). With the exception of two patients with colorectal cancer whose CD8+ T cells recognized the same product of a KRASG12D mutation, the vast majority (99%) of mSNV neoantigens were unique to each patient.
Table 3.
Neoantigens Recognized by TILs from Patients with Select Epithelial Cancers
| Cancer Type | # Of Patients Screened | # Of Patients with Neoantigen Reactivity (%) | # Of Mutations Screened | # Of Immunogenic Mutations (%) |
|---|---|---|---|---|
| All gastrointestinal | 75 | 62 (83) | 7,654 | 124 (1.6) |
| Colorectal | 51 | 45 (88) | 5,833 | 94 (1.6) |
| Biliary | 12 | 8 (67) | 866 | 12 (1.4) |
| Pancreatic | 7 | 5 (71) | 352 | 8 (2.3) |
| Gastric | 3 | 2 (67) | 378 | 6 (1.6) |
| Esophageal | 2 | 2 (100) | 225 | 4 (1.8) |
| Ovarian | 7 | 5 (71) | 1714 | 8 (0.5) |
In a study of ovarian cancer, autologous TILs from five out of seven patients recognized at least one tumor-specific mSNV (Deniger et al., 2018). Collectively, only eight out of 1714 (~0.5%) SNVs were recognized, and none of them were shared among the patients. Similar findings had also been reported in a study that focused on melanoma patients and utilized a different methodology: MHC multimer-based screening. Here, nine out of 459 (~2%) SNV-derived peptides that were predicted to bind to autologous MHC-I molecules were recognized by tumor- and PBL-derived T cells, and none of them were shared among patients (Cohen et al., 2015).
Together, these studies indicate that immunogenic SNVs represent only a small fraction of all cancer mutations. However, they can still be detected in a majority of patients with different types of cancer, including those with low overall mutational burden (e.g., the majority of gastrointestinal tumors).
Targeting mSNVs with Immunotherapy
Patients with metastatic melanoma and non-small cell lung cancer (NSCLC), two types of tumors with abundance of somatic mutations, were among the first to benefit from administration of ICIs. The clinical benefit was found to correlate with the total number of non-synonymous mutations (mostly mSNVs) in both types of cancers (Hugo et al., 2016; Rizvi et al., 2015; Van Allen et al., 2015). Following similar reports in other malignancies, a meta-analysis that included 27 tumor types treated with anti-PD-1 or anti-PD-L1 agents showed a significant correlation between the tumor mutation burden and the objective response rate in most types of cancer (Yarchoan et al., 2017). Collectively, these findings have led to a hypothesis that malignancies with a higher mutation burden are more likely to produce neoantigens, which in turn may be recognized by T cells that are activated by the administration of ICIs. In support of this, studies have found an association between the benefit from ICIs and the predicted neoantigen load (Rizvi et al., 2015; Van Allen et al., 2015), which was even stronger when clonality of the predicted neoantigens was considered (McGranahan et al., 2016).
mSNV-derived neoantigens have been targeted in personalized cancer vaccine trials, wherein patients were immunized with peptides (or peptide-encoding synthetic RNAs) that were identified by NGS-based approaches and were predicted to bind to patient’s MHC molecules. Despite the lack of their clinical effectiveness, recent trials in melanoma (Carreno et al., 2015; Ott et al., 2017; Sahin et al., 2017) and glioblastoma (Hilf et al., 2019; Keskin et al., 2019) have demonstrated that such vaccines can both augment and generate responses of circulating T cells to mSNV-derived peptides.
At the Surgery Branch of the National Cancer Institute, several ACT trials were performed in patients with metastatic melanoma using TILs that were enriched for recognition of autologous cancer cells. These led to an objective response rate in 55% and a complete response rate in 23% of treated patients (Goff et al., 2016). Most of the complete responses were durable, as only two recurrences were seen in a cohort of 46 complete responders with a median patient follow-up of over 7 years. The lack of toxicities to normal tissues in these trials suggested that the transferred T cells targeted molecules unique to the cancer. Indeed, a series of retrospective studies that focused on exceptional responders revealed that the infused TILs frequently targeted mSNV-derived neoantigens (Lu et al., 2013, 2014; Prickett et al., 2016; Robbins et al., 2013; Zhou et al., 2005).
In several other ACT studies, patients with ICI-refractory types of epithelial cancers were treated with TILs that were selected for their ability to recognize various mSNVs. In the first such case, a patient with chemo-refractory metastatic cholangiocarcinoma was treated twice with an autologous TIL product that contained CD4+ T cells targeting a neoantigen derived from a unique mSNV, the putative tumor suppressor ERBB2IP (Tran et al., 2014). The treatment with the first product, which contained approximately 25% ERBB2IP-reactive T cells, led to transient disease stabilization. However, the second treatment, which contained approximately 95% of reactive cells, resulted in substantial regression of lung and liver metastases, which has been ongoing for more than 60 months.
In the second case, a patient with mismatch repair-proficient metastatic colon cancer was treated with an autologous TIL product that contained approximately 75% of CD8+ cells recognizing a neoantigen derived from a hotspot (G12D) mutation in the KRAS gene (Tran et al., 2016). This resulted in the initial regression of all metastatic lung lesions. One of these progressed 9 months later and was subsequently resected, rendering the patient disease free (Figure 2A). Genomic analysis of the removed tumor revealed a loss of chromosome 6 haplotype encoding the MHC molecule that was required for recognition of the KRASG12D epitope, exemplifying one of the mechanisms that tumors may utilize to escape an immune attack.
Figure 2. Examples of Durable Tumor Regressions in Patients Who Were Treated with TILs That Recognized Tumor Neoantigens.
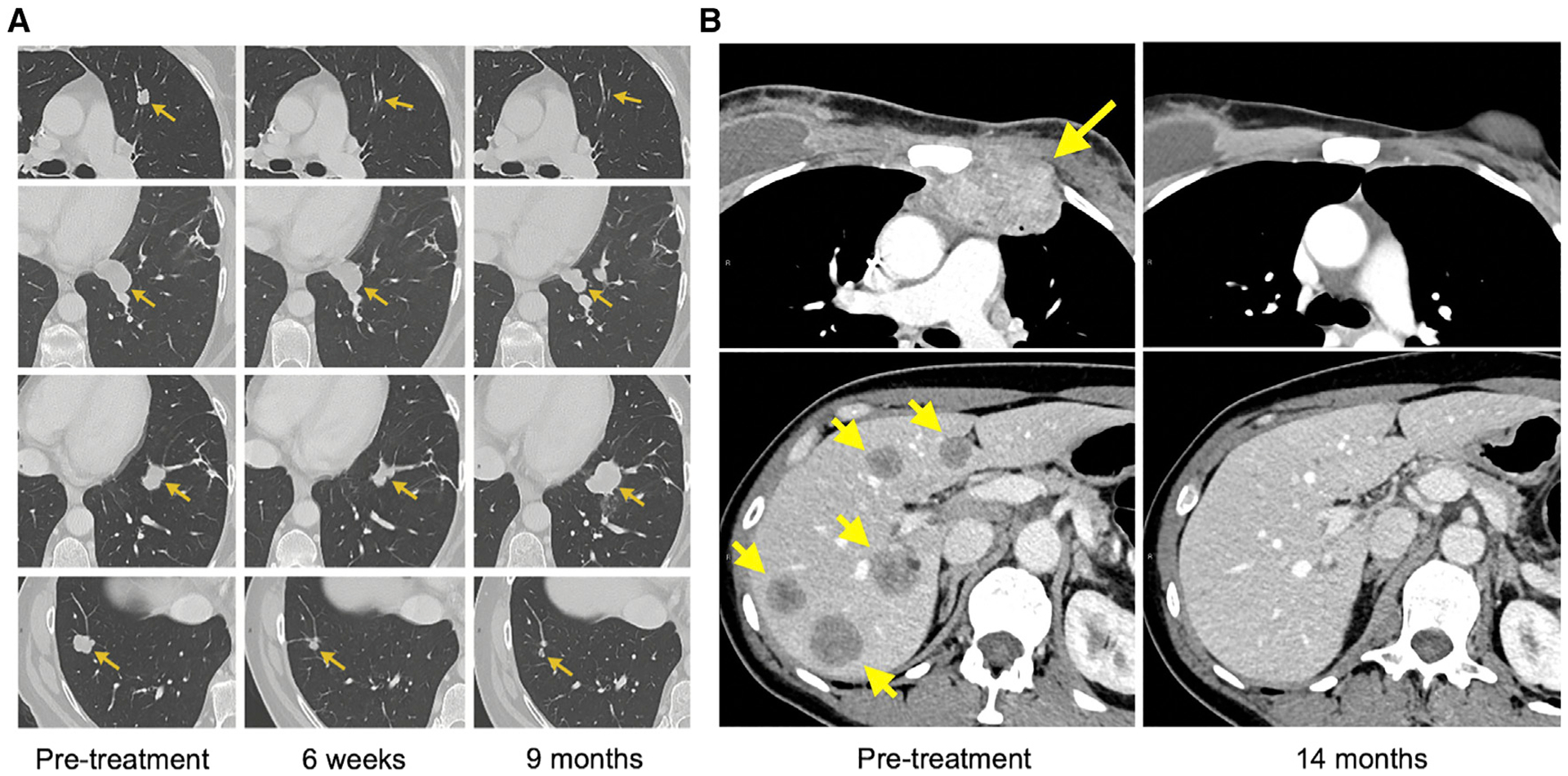
(A) Regression of lung tumors in a patient with metastatic colorectal carcinoma who received KRASG12D-reactive TILs. The tumor in the second row exhibited a partial response and was surgically resected; the pathology showed no viable tumor cells. The tumor in the third row progressed 9 months after the therapy and was then surgically resected. The pathology demonstrated loss of the MHC molecule necessary for the presentation of the KRASG12D.
(B) Complete regression of a chest wall mass (upper panels) and multiple liver tumors (lower panels) in a patient with metastatic breast cancer who received TILs directed against four different tumor mutations.
In another case, a patient with refractory hormone receptor-positive breast cancer was treated with an autologous TIL product that contained several T cell clones recognizing products of four different mSNVs (SLC3A2, KIAA0368, CADPS2, and CTSB) and comprising a total of 23% of the infused product (Zacharakis et al., 2018). This yielded complete regression of multiple lymph node, chest wall, and liver metastases, which has been ongoing for >30 months after the treatment (Figure 2B). Of note, the patient had also received pembrolizumab, a PD-1 inhibitor, but this therapy likely had no impact on the clinical outcome due to the lack of PD-1 ligand expression in the tumor, and due to the very modest benefit of pembrolizumab in patients with this type of cancer (Rugo et al., 2018).
These case reports have provided the most direct evidence that targeting mSNV-derived neoantigens with T cells can mediate durable tumor regressions in humans, without causing any toxicities to normal tissues. It should be noted, however, that most of the treated patients, at least when gastrointestinal cancers are concerned, still do not respond to this type of treatment (Parkhurst et al., 2019); potential reasons for this are discussed later.
Targeting Driver mSNVs
Only a small fraction of all mSNVs play a role in cancer evolution and are considered to be “driver mutations.” In contrast, “passenger mutations,” which represent the overwhelming majority of mSNVs, have no functional implications in cancer biology. Theoretically, targeting products of driver mutations with T cell-based therapies will be more advantageous for the following reasons.
Firstly, driver mutations are expected to be clonal, rendering all (or the vast majority) of tumor cells targetable. Secondly, as they are essential for the malignant phenotype, they are less likely to get lost or downregulated during an attempt by a tumor to evade the immune attack. This particularly applies to recurrent (“hotspot”) mSNVs that affect various oncogenes. Finally, the epitopes resulting from hotspot mutations can be shared among patients (and are thereby called “public epitopes”). This may enable development of immunotherapies in which off-the-shelf vaccines or TCRs could be used to treat a larger number of cancer patients.
Several hotspot mSNVs may affect the TP53 gene, which is mutated in almost 50% cases of all human cancers (Bykov et al., 2018). In a recent study, TILs from patients whose tumors harbored TP53 hotspots were screened against a panel of peptides and minigenes representing these mutations (Malekzadeh et al., 2019). The recognition of several different TP53 hotspot epitopes was demonstrated in 11 out of 28 (39%) patients harboring cancers of different histologies. These epitopes were restricted to a variety of MHC molecules and were all unique, with the exception of a TP53R175H peptide that was restricted to HLA*02:01 and was detected in two patients in this study. These results, which were later confirmed by a study that tested T lymphocytes from the PBL (Malekzadeh et al., 2020), indicate that TP53 hotspots are naturally immunogenic. Furthermore, they provide a rationale for creation of libraries of TCRs against TP53 hotspots, which may be used to treat a large number of cancer patients in ACT trials.
Discovery of immunogenic hotspot mSNVs that affect KRAS, an oncogene that is frequently mutated in pancreatic and colorectal cancer, presents another opportunity to create a library of TCRs that could be employed in ACT trials. Previous research had already identified TCRs against KRASG12D and KRASG12V mutations restricted to HLA-A 11:01 (Wang et al., 2016), both of which are now being tested in clinical trials (NCT03190941 and NCT03745326 ). To further expand these efforts with TCRs restricted to different MHC molecules, a screening model similar to the one described for TP53 is currently being employed in patients with KRAS-mutated colorectal and pancreatic cancer at the Surgery Branch. In addition to TP53 and KRAS hotspots, this approach could be used to create TCR libraries against other driver mutations, such as BRAF, PIK3CA, and EGFR.
Neoantigens Derived from INDELs
Depending on the number of affected base pairs, nucleotide insertions and deletions (collectively called INDELs) may result in formation of proteins with a gain or loss of one or more amino acids (non-frameshift INDELs). Alternatively, they may generate proteins with frameshifted sequences, which are often truncated due to a premature stop codon (frameshift INDELs [fsINDELs]).
Several studies in humans have provided evidence that INDEL-derived peptides can be immunogenic. For instance, a frameshift deletion in TGFBRII, a tumor suppressor gene that is frequently inactivated in colorectal cancer (Grady et al., 1999; Markowitz et al., 1995), was found to generate peptides recognized by both CD8+ and CD4+ cells (Linnebacher et al., 2001; Saeterdal et al., 2001a, 2001b). Furthermore, an analysis of TILs that mediated a near-complete response in a patient with metastatic melanoma revealed the presence of a CD8+ T cell clone that recognized a product of a frameshift deletion in the tumor suppressor CDKN2A (Huang et al., 2004).
Overall, INDELs are less prevalent in cancer than mSNVs. In pan-cancer analyses, the median proportion of INDELs among all non-synonymous mutations was around 5%, with RCC being a notable outlier (12%) (Turajlic et al., 2017; Vogelstein et al., 2013). However, fsINDELs were predicted to generate at least three times the number of high-affinity MHC binders as mSNVs, confirming findings of an earlier study in colorectal cancers (Giannakis et al., 2016).
In a study of patients with metastatic gastrointestinal cancers (Parkhurst et al., 2019), similar proportions of mSNVs and non-fsINDELs were recognized by the autologous TILs (1.6% and 1.7%, respectively), while none of the fsINDELs were recognized. However, the overall number of fsINDELs was low, and only 40% were found to be expressed by RNA sequencing, which precluded definite conclusions about their immunogenicity. In a study that screened RCC-derived TILs using predicted peptides coupled with MHC multimers, fractions of SNVs (3.2%) and fsINDELs (4.5%) that were recognized were not significantly different, while non-fsINDELs were not recognized (Hansen et al., 2020). Given these conflicting findings, further research is needed to establish the exact proportion of immunogenic INDELs in different cancer types.
Tumoral fsINDEL burden was found to be associated with ICI responses in three cohorts of melanoma patients, and was a better response predictor than SNV burden in two of these cohorts (Turajlic et al., 2017). Positive correlation between INDELs and clinical benefit from ICIs was also reported in patients with NSCLC (Chae et al., 2019) and MSI-high gastrointestinal tumors (Mandal et al., 2019). Although a preliminary report in patients with RCC treated with anti-PD-1 therapy found a significant association of the number of fsINDELs with overall survival (Voss et al., 2018), a larger, randomized trial of atezolizumab, an anti-PD-L1 agent, failed to confirm such an association (McDermott et al., 2018).
Predictive value of fsINDEL burden can be further refined after accounting for nonsense-mediated decay (NMD), a mechanism that protects cells from toxic accumulation of truncated proteins resulting from mRNAs with premature stop codons. Although the majority of these transcripts are expected to be degraded via NMD, some, especially when encoding tumor suppressors, are still translated. In one analysis, only fsINDELs that were predicted to escape NMD correlated with clinical benefit in ICI-treated patients affected by various types of cancer (Lindeboom et al., 2019).
Despite their low prevalence and very limited clinical experience with direct targeting, INDELs could still be valuable targets for ACT or vaccination therapies. Firstly, they recurrently affect tumor suppressors, such as TGFBRII in colorectal or EGFR in lung cancer (Lynch et al., 2004). Furthermore, in comparison with SNVs, their products are less similar to normal peptides and have been shown to exhibit a higher predicted MHC-binding capacity over their WT counterparts (Hansen et al., 2020). This could minimize the chances of their cross-reactivities to normal proteins.
Neoantigens Derived from Gene Fusions
Gene fusions, which can arise through large-scale genomic rearrangements such as chromosomal translocations, interstitial deletions, or chromosomal inversions, may produce fusion proteins that are unique to cancer. Peptides spanning the breakpoint regions of these proteins can be recognized by patients’ T cells, as first demonstrated in the case of BCR-ABL, a fusion protein resulting from a translocation between chromosomes 9 and 22 in patients with chronic myelogenous leukemia (Yotnda et al., 1998). This also proved to be true for solid malignancies, as in the case of SYT-SSX1 fusion arising from the pathognomonic X:18 translocation in patients with synovial cell sarcoma (Sato et al., 2002).
The development of various NGS-based computational algorithms has greatly facilitated detection of tumor-specific gene fusions. In a recent report, an RNA sequencing-based approach was used to identify immunogenic gene fusions in two cases of solid cancers with low SNV burden (Yang et al., 2019). In one case, circulating CD8+ T cells from a patient with head and neck cancer who had an exceptional response to an ICI were found to recognize a peptide derived from a novel DEK-AFF2 gene fusion. In the second case, CD8+ cells from a patient with adenoid cystic carcinoma (ACC) recognized a product of MYB-NFIB gene fusion, which occurs in 60% of ACC cases.
Although the overall gene fusion burden was found to be substantially lower than the total SNV burden across different types of solid tumors, gene fusions were predicted to produce a larger number of potential neoantigens (Gao et al., 2018; Wei et al., 2019). However, no studies have systematically tested the immunogenicity of fusion proteins and compared them with SNVs. Furthermore, the evidence from the correlative studies has been inconclusive. The aforementioned study of an exceptional ICI responder with head and neck cancer suggested that fusion neoantigens may mediate tumor regression in a setting of low SNV burden. Additionally, the same study reported a decrease in the number of predicted fusion neoantigens after anti-PD-1 therapy in melanoma biopsy samples, indicating that cells harboring select gene fusions may have been eliminated due to an immune attack. In contrast, a different study failed to show a correlation between predicted fusion neoantigen burden and benefit from administering ICIs to patients with metastatic melanoma (Wei et al., 2019).
Attempts to target fusion neoantigens with immunotherapy have been limited to a few vaccine-based approaches (Kawaguchi et al., 2012; Mackall et al., 2008). Given the lack of positive clinical outcomes in these studies, the utility of targeting fusion neoantigens can only be hypothesized. Although the vast majority of gene fusions represent byproducts of genetic instability, without known function (Mertens et al., 2015), some result in fusion proteins that are shared among patients and may function as oncogenes. These may prove to be valuable immunotherapy targets. Examples of such proteins include CCDC6-RET in thyroid cancers and TMPRSS2-ERG in prostate cancer (which results from only a subset of all TMPRSS2-ERG gene fusions), or FGFR3-TACC3, which is seen in 1%–2% of patients with various cancer types (Gao et al., 2018).
Furthermore, fusion neoantigens could be particularly valuable targets in malignancies that generally exhibit low SNV burden and suboptimal responsiveness to ICIs, such as prostate cancer. In a study by The Cancer Genome Atlas (TCGA), 87% of prostate cancer cases were found to harbor gene fusions, and 41% of fusion-positive tumors were predicted to have fusion breakpoints translated into amino acid sequences. The majority of these sequences were unique and were predicted to yield at least one MHC class I binding epitope (Kalina et al., 2017). However, their immunogenicity remains to be validated in assays with the autologous T cells.
Tumor Antigens Derived from Viral Oncoproteins
A subset of solid cancers arise as a result of viral infection, wherein the integration of viral genes into the cellular genome leads to expression of viral proteins with oncogenic properties. For instance, expression of E6 and E7 oncoproteins, which results from an infection with high-risk strains of the human papilloma virus (HPV), drives the evolution of a subset of head and neck, cervical, and anal cancers (Moody and Laimins, 2010). Concomitantly, peptides derived from these “foreign” proteins can be recognized by patients’ T cells (Draper et al., 2015; Jin et al., 2018).
This has also been shown for other virus-driven solid cancers, including Merkel cell carcinomas that result from a Merkel cell polyomavirus (MCPyV) infection (Iyer et al., 2011), and nasopharyngeal carcinomas that result from an Epstein-Barr virus (EBV) infection (Lee et al., 2000). Generally, viral antigens are capable of eliciting high-affinity TCR responses due to their marked dissimilarity to normal cellular proteins, as was shown in a study that compared the affinities of anti-viral TCR with those against TAAs (Aleksic et al., 2012).
Virus-derived tumor antigens have been targeted in several immunotherapy trials. In an ACT study with TILs selected for reactivity against HPV, durable tumor regressions were reported in two of nine patients with HPV-positive metastatic cancers (Draper et al., 2015). However, a later study found that HPV-reactive cells in the infusion product were outnumbered by cells recognizing other types of tumor antigens (Stevanovic et al., 2017). In two separate studies, administration of autologous T cells transduced with anti-E7 TCR led to responses in four out of 12 treated patients (Norberg et al., 2018), while the administration of anti-E6 TCR-transduced T cells led to responses in two out of 12 patients (Doran et al., 2019). Trials with these TCRs are still in progress (NCT02280811 and NCT02858310) and will hopefully provide more conclusive evidence regarding the utility of targeting HPV epitopes.
Similarly, targeting MCPyV- and EBV-driven cancers with ACT also yielded clinical responses, although the interpretation of these results has been confounded by co-administration of other effective therapies or by administration of cells with low reactivity against expressed viral antigens, respectively (Paulson et al., 2018; Kwong et al., 2019).
Importantly, no significant toxicities to normal tissues were observed in either of these clinical trials. Collectively, they provide evidence that targeting oncogenic viral proteins could safely mediate regression of selected types of tumors, and provide a rationale for further therapy optimization. Given the vital role that these antigens play in oncogenesis, and the fact that they are shared among the patients, they remain attractive targets for cancer immunotherapy.
UNCONVENTIONAL TUMOR ANTIGENS
The vast majority of previously described tumor antigens are classified as conventional tumor antigens, as they are generated from the coding regions of the genome via conventional transcription, translation, and proteasomal digestion. In contrast, unconventional antigens are those that arise from either non-coding regions of the genome or from coding regions (normal or mutated) by means of aberrant transcription, translation, or post-translational modifications (Table 2). Some of these processes, however, may not be entirely tumor specific and can also occur in normal tissues. Because of this, some unconventional antigens may assume the properties of TAAs, while others may behave as neoantigens.
Tumor Antigens Associated with Aberrant mRNA Splicing
Aberrant mRNA splicing in tumors may result in intron retention. Subsequently, sequences of the retained introns can be translated and can generate peptides that bind to the surface MHC molecules and elicit recognition by the T cells. Several such intronic antigens have been identified using cDNA library screening approaches, including a peptide encoded by an exon-intron junction of the MUM-1 gene in melanoma. Here, the intron retention itself was not tumor specific, but the intron harbored a tumor-specific mutation, making this a true neoantigen (Coulie et al., 1995). In another example from a melanoma patient, T cells recognized a peptide encoded by a retained intron of the gp100 gene. This intron, however, harbored no mutations and was found to be expressed at similar levels in normal melanocytes (Robbins et al., 1997). In addition to these examples, an intronic tumor antigen that was generated by transcription from a cryptic promoter within the intron has also been described (Guilloux et al., 1996).
A recent study suggested that the overall repertoire of candidate tumor antigens could be significantly expanded by including those produced by intron retention (Smart et al., 2018). Using an RNA sequencing-based approach, the investigators combined non-synonymous mutation data with intron retention analysis, which roughly doubled the total predicted neoantigen load in two cohorts of melanoma patients treated with ICIs. However, tumoral intronic retention was not directly compared with the normal cells, limiting the extent to which these peptides could potentially be classified as neoantigens. Furthermore, in contrast to SNVs, this study revealed no correlation between retained introns and the clinical benefit from ICIs.
Aberrant RNA splicing in tumors may also result in formation of novel exon-exon junctions (EEJs), which could potentially produce immunogenic proteins/peptides. In a study that compared tumor WES and RNA sequencing data, a subset of novel EEJs was detected only in cancer cells, but not in corresponding normal tissue, with some EEJs recurrently detected in different cancer types (Kahles et al., 2018). Moreover, in a cohort of patients with ovarian and breast cancer, peptides spanning these EEJs were found to be a more abundant source of predicted epitopes than SNVs. In a different study, mSNVs that induced aberrant splicing and thus formation of novel EEJs were predicted to generate more immunogenic epitopes than regular missense mutations (Jayasinghe et al., 2018). However, neither of these studies has tested the immunogenicity of EEJs in T cell-based assays.
Given the lack of the relevant clinical experience, the safety of targeting antigens resulting from aberrant RNA splicing by ACT is speculative. If this process occurs exclusively in cancer cells, then such antigens could be valuable targets for immunotherapy.
Tumor Antigens Associated with Aberrant RNA Translation
These antigens, also called cryptic antigens, are produced either by translation of protein-coding genes in alternative open reading frames (aORFs) or by translation of presumably non-coding sequences. Therefore, previously described intronic antigens can also be classified as cryptic antigens. As with the other unconventional antigens, cryptic antigens can be either tumor-associated or tumor-specific.
Several tumor antigens encoded in aORFs of previously described protein-coding genes have been discovered using cDNA screening approaches. The first one to be identified was a peptide encoded in an aORF of the gp75 gene (Wang et al., 1996). This gene was found to be translated in two overlapping ORFs, which resulted in formation of two completely different proteins. Only one of these proteins yielded an immunogenic peptide, which was recognized by TILs that mediated an objective tumor regression in a patient with metastatic melanoma. This finding was followed by other similar examples, both in melanoma and in other types of cancer (Aarnoudse et al., 1999; Probst-Kepper et al., 2001; Rosenberg et al., 2002; Shichijo et al., 1998; Wang et al., 1998).
In most of these cases, aberrant translation resulted from a phenomenon called ribosomal scan-through or “leaky scanning,” wherein the ribosomes continue to scan mRNA past the first ATG and initiate translation from the next available ATG (Bullock and Eisenlohr, 1996). However, other mechanisms of translation initiation have also been implicated in formation of cryptic tumor antigens. These include translation from a non-ATG aORF (Ronsin et al., 1999), translation from an IRES sequence (Carbonnelle et al., 2013), and translation from an aORF that is coupled with aberrant RNA splicing, as in the case of the AIM-2 antigen that was encoded in an exon-intron junction and translated from only the aORF (Harada et al., 2001).
Other mechanisms that may lead to translational changes appear to be poor generators of tumor antigens (Laumont and Perreault, 2018). These include stop codon read-through events and programmed ribosomal frameshifting (PRF). In the former, the ribosome proceeds to translate 3′- UTR down to the next in-frame stop codon (Bullock et al., 1997). In the latter, the ribosome encounters a “slippery site” and changes the reading frame during translation, which may result in production of chimeric peptides that can be recognized by T cells (Saulquin et al., 2002).
A recent immunopeptidomic study using human B lymphoblastoid cell lines revealed that cryptic peptides constituted between 6.5% and 13% of all the MHC class I-associated peptides (MAPs) (Laumont et al., 2016). Aside from translation in aORFs, a substantial portion of cryptic MAPs arose from non-annotated antisense transcripts and from regions that were previously assumed to be non-coding (i.e., 5′ or 3′ UTRs, exon-intron or UTR-exon junctions, retained introns, and intergenic regions).
Although not validated by T cell assays in solid cancers, these results suggest that cryptic MAPs could be a valuable source of tumor antigens, particularly because they are enriched with non-synonymous SNPs, and because 99% of cancer-specific mutations are located in the non-coding regions of the genome (Khurana et al., 2016). Moreover, non-canonical protein translation can be favored under conditions of cellular stress (Gerashchenko et al., 2012; Starck et al., 2016), suggesting that translation in aORFs itself may be a tumor-enriched process.
Tumor Antigens Resulting from Post-translational Changes
A subset of tumor antigens may arise from post-translational modifications of cellular proteins or peptides. One such antigen type consists of fusion peptides, which result from proteasomal splicing of two separate peptide fragments, derived from either the same protein (cis-splicing) or from different proteins (trans-splicing). The first such antigen to be identified was a peptide derived by joining two peptide fragments (a 5- and a 4-mer) that were separated by 40 amino acids in the parent protein encoded by FGF5, a gene that is frequently overexpressed in RCC (Hanada et al., 2004). This study was followed by discovery of immunogenic peptides spliced from other proteins, including the gp100 (Michaux et al., 2014), SP110 (Warren et al., 2006), and tyrosinase (Dalet et al., 2011).
Several immunopeptidomic studies found that spliced peptides accounted for at least 25% of all MAPs, with some proteins being represented only as spliced variants (Faridi et al., 2018; Liepe et al., 2016, 2019). Although the immunogenicity of these peptides was not studied in T cell-based assays, these reports suggest that fusion peptides may be abundantly presented on the tumor cell surface. However, normal cells may also produce these peptides (Dalet et al., 2011), which stresses the need for paired tumor-normal testing.
Another subset of posttranslationally modified tumor antigens consists of proteins that underwent enzymatic amino acid modifications, such as phosphorylation, acetylation, citrullination, glycosylation, or deamidation. These proteins can be fragmented into peptides that retain these modifications, which in turn may be recognized by patients’ T cells. This was first determined by a study of a patient with melanoma, in which a peptide that was derived from tyrosinase protein by deamidation of an asparagine into an aspartic acid was specifically recognized by patient’s T cells (Skipper et al., 1996).
The most abundant of the enzymatically modified tumor proteins are phosphoproteins, which result directly from dysregulation of kinase-mediated signaling pathways in cancer. Phosphopeptides derived from these proteins have been successfully eluted from the surface MHC molecules in both cancer tissue and cancer cell lines (Meyer et al., 2009; Zarling et al., 2006). Furthermore, a peptide derived from a phosphorylated but not from the unphosphorylated MART1 protein was found to elicit recognition by circulating CD4+ T cells from a patient with melanoma (Depontieu et al., 2009). These results indicate that antigens derived by phosphorylation, as well as from the other amino acid modifications, may be attractive targets for immunotherapy, providing that they occur exclusively in cancer cells and that the unmodified peptides cannot be recognized by T cells.
FUTURE STRATEGIES TO ADDRESS LIMITATIONS OF TUMOR ANTIGEN-DIRECTED THERAPIES
Safety
Targeting unmutated proteins (TAAs) with high-affinity TCRs can cause destruction of normal tissues, and both on- and off-target toxicities have been well documented. However, these toxicities have not been reported when targeting neoantigens, indicating that their targeting provides a safer venue for future immunotherapy trials. The safety of targeting unconventional antigens is largely unexplored and will ultimately depend on their tumor specificity.
For future approaches that are still based on TAA targeting, careful examination of antigen expression in normal tissues and assessment of TCR cross-reactivity against normal proteins is of pivotal importance, as even a miniscule amount of normal protein recognized by high-affinity TCRs can lead to fatal toxicities. Sole targeting of these antigens with lower-affinity TCRs or cancer vaccines does potentially allow for a wider therapeutic window, but at the expense of compromised efficacy.
Efficacy
Because neoantigens that arise from mSNVs have emerged as a major determinant of durable tumor regressions in various immunotherapy trials, clinical studies have largely been focused on targeting them. However, durable clinical responses to these therapies have occurred in only a fraction of patients with cancers of epithelial origin, the most common cancers in humans. Among the factors that can negatively affect the treatment efficacy, poor or heterogeneous antigen expression in the tumor poses a significant problem.
Several strategies that address the selection of targeted neoantigens could be utilized to overcome these limitations. Firstly, targeting should be reserved for neoantigens whose expression and presentation in the tumor has been verified by tumor RNA sequencing or immunopeptidomics. Secondly, targeting should optimally focus on neoantigens that were determined to be clonal (i.e., present in all the cancer cells) by bioinformatical approaches (Roth et al., 2014). Whenever possible, neoantigens arising from driver mutations, such as previously discussed TP53 and KRAS antigens, should be prioritized, as their importance in maintenance of malignant phenotypes renders them more likely to be clonal and less likely to be lost during an immune attack. Finally, simultaneous targeting of multiple neoantigens, including both mSNVs and other neoantigen types, could minimize not only the effects of tumor heterogeneity but also the chances of tumor resistance through either antigen loss or loss of specific MHC molecules that are required for antigen presentation.
To accomplish the latter, the existing antigen screening platforms could be modified to allow detection of a greater neoantigen variety. This could be performed either by substituting or by complementing WES with a combination of immunopeptidomics and RNA sequencing or WGS. This would allow simultaneous detection of tumor-specific SNVs, INDELs, gene fusions, viral epitopes, and unconventional candidate antigens. These could then be used to generate TMG and peptide libraries for screening of the autologous TILs or peripheral blood T cells (Figure 3).
Figure 3. A Suggested Approach for Comprehensive Identification of Tumor Neoantigen-Reactive T Cells and Their Use for Personalized Immunotherapy.
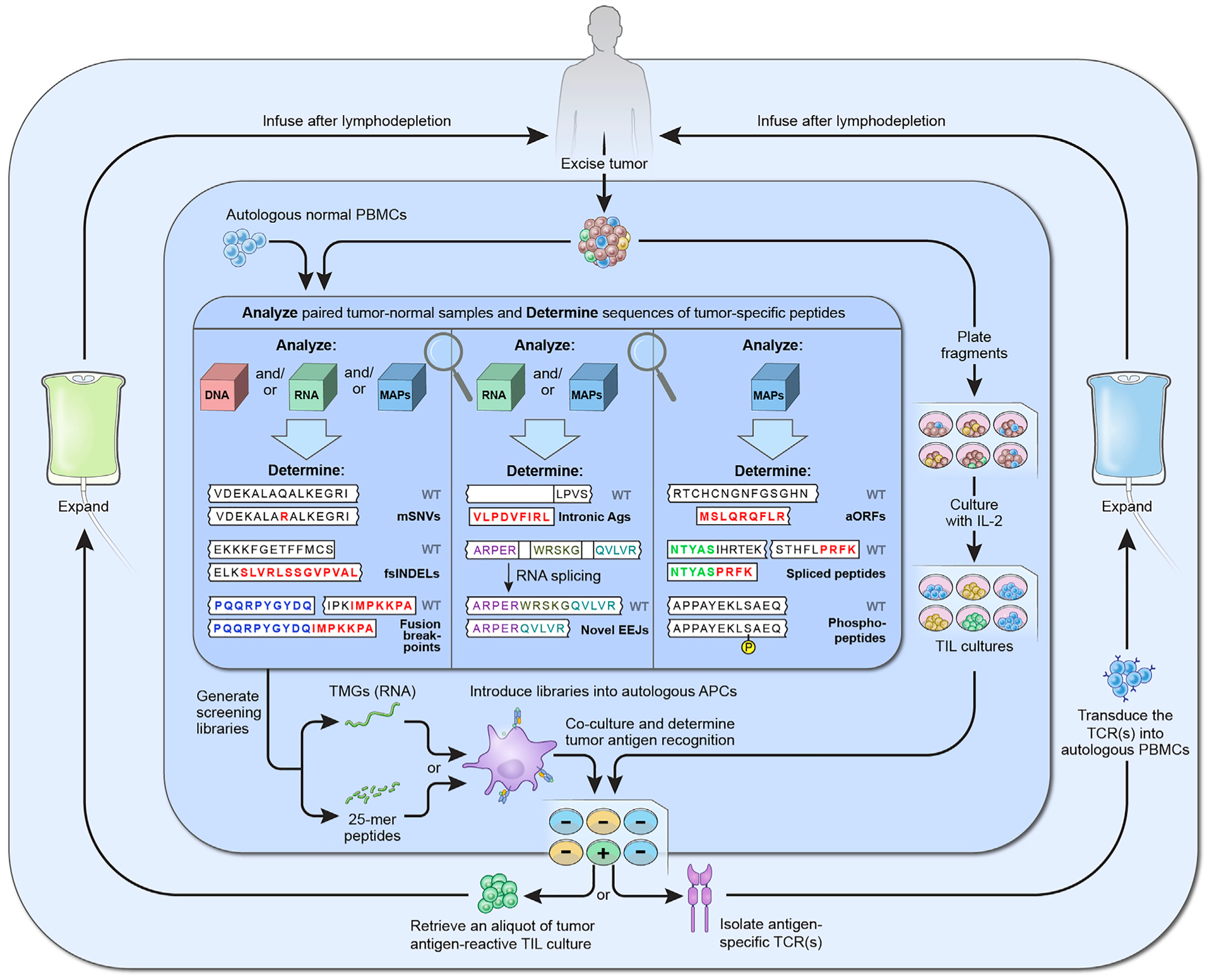
Three shaded areas in the figure represent suggested strategies for paired tumor-normal analysis to identify candidate tumor neoantigens (inner), TIL screening to identify neoantigen-reactive T cells (middle), and the use of reactive cells for immunotherapy (outer). Firstly, following surgical excision of one or more metastases, one tumor segment is further divided into 12–24 fragments, which are cultured with IL-2 in order to expand TILs (middle area, right). The other segment is analyzed together with a sample of normal cells, typically PBMCs, to identify sequences of candidate tumor antigens (inner area). This analysis can be performed on the cellular genome (DNA), transcriptome (RNA), and/or immunopeptidome (MAPs). Although most current analyses are limited to mSNVs and non-frameshift and fsINDELs, they could be further expanded to allow identification of other potential antigen types, such as fusion breakpoints, intronic antigens, novel EEJs, antigens translated from aORFs, and posttranslationally spliced or enzymatically modified peptides, such as phosphopeptides. Next, sequences of candidate antigens are used as templates to synthesize peptide and TMG screening libraries, which are then introduced to the autologous APCs (middle area, bottom). After the addition of TILs and the overnight co-cultures, such methods as measuring interferon-γ production or upregulation of T cell surface activation molecules (not depicted) are used to identify the TILs that recognize tumor neoantigens. Finally, reactive TILs are expanded to very large numbers ex vivo and reinfused back into the patient (outer area, left). Alternatively, TCRs that conferred tumor antigen recognition are retrovirally transduced into autologous PBMCs, which are then expanded and reinfused into the patient (outer area, right). Both approaches entail pretreating the patient with lymphodepleting chemotherapy, which enables engraftment of transferred lymphocytes, and are followed by intravenous IL-2 administration to stimulate their survival and proliferation.
These modifications could be further complemented with parallel analysis of multiple tumor samples for each patient, such as additional metastatic tumors, formalin-fixed and paraffin-embedded primary tumor samples, or even circulating tumor DNA. Additional improvements in mutation calling and antigen prediction algorithms, as well as improvements in sensitivity of the techniques that allow detection and isolation of antigen-reactive T cells, could also facilitate discovery of additional neoantigens. To this end, TIL-based screening could also be complemented (or even replaced) by screening with T cells from such alternative sources as the PBL or fresh tumor digests, or potentially by using nanoparticle-bound peptide-MHC tetramers (Peng et al., 2019)
Neoantigen screening could also be performed in conjunction with interventions that may stimulate immunological recognition of previously undetected tumor neoantigens. For instance, either stereotactic tumor radiation or intralesional or systemic delivery of immunogenic chemotherapies could be performed before the screening to induce immunogenic tumor cell death and potentially to increase the overall repertoire of discoverable neoantigens. Furthermore, pharmacological agents that inhibit NMD in tumor cells and thereby lead to the accumulation of aberrant truncated proteins could also render the tumors more immunogenic (Pastor et al., 2010). Finally, treatment with certain chemotherapies or other medications (e.g., aminoglycosides) could be used in an attempt to force the tumor cells to translate their mRNAs in an unconventional manner, which could further broaden the scope of detectable (and targetable) tumor antigens (Goodenough et al., 2014).
In addition to the properties of targeted antigens, several other factors could negatively affect the efficacy of neoantigen-directed therapies. These limitations are a product of complex immunological mechanisms that are outside the scope of this review and have been discussed elsewhere (Chandran and Klebanoff, 2019). Briefly, they include suboptimal properties of infused or in situ T cells, immunosuppressive tumor microenvironment, lack of appropriate dendritic cell stimulation in the tumor bed, intrinsic tumor resistance to T cell-mediated killing or the phenomenon of tumor “immunoediting” (O’Donnell et al., 2019), and defective antigen presentation in the tumor. Examples of ACT strategies that could overcome them include increasing the frequency of tumor antigen reactive cells in the infusion product (e.g., by selecting cells that upregulate surface activation markers upon antigen exposure), improving the functionality of the infused cells (e.g., by selecting cells with less differentiated phenotype or by blocking or reversing T cell exhaustion with pharmacologic or other manipulations), and combining different immunotherapies that could lead to superior stimulation of the transferred T cells in vivo (e.g., by combining ACT with ICIs or with cancer vaccines).
Availability
The overwhelming number of neoantigens are private; i.e., not shared between patients. Targeting these antigens with ACT or cancer vaccines mandates a highly individualized approach for each treated patient, which is accompanied by increased cost and is confined to a very small number of specialized treatment centers. These limitations could be overcome by creating libraries of neoantigen-reactive individual TCRs that target public neoantigens, such as the aforementioned products of TP53 and KRAS mutations. These TCRs could be transduced on demand into autologous peripheral blood mononuclear cells (PBMCs) for patient treatment.
CONCLUDING REMARKS
Targeting tumor antigens with immunotherapy can cure select cases of metastatic cancer but fails to provide clinical benefit to the majority of treated patients. Overcoming tumor mutational heterogeneity by increasing the repertoire of high-quality targeted antigens will be a crucial step in future attempts to improve the treatment’s efficacy. Although the current efforts within the realm of cancer vaccines and ACT have been focused predominantly on targeting neoantigens arising from mSNVs, novel research has revealed that other types of tumor antigens could be abundant in cancer cells and therefore also be utilized as immunotherapy targets. Future personalized immunotherapy trials will thus have to be redesigned to allow analysis of patients’ T cell responses to all possible classes of antigens and to test them as therapeutic targets. Furthermore, efforts to create libraries of TCRs that target public neoantigens arising from driver mutations will not only help to increase the efficacy of antigen-directed cancer immunotherapies but will also aid in making these therapies available to a larger number of patients with cancer.
ACKNOWLEDGMENTS
This work was supported by the Intramural Research Program of the National Cancer Institute.
REFERENCES
- Aarnoudse CA, van den Doel PB, Heemskerk B, and Schrier PI (1999). Interleukin-2-induced, melanoma-specific T cells recognize CAMEL, an unexpected translation product of LAGE-1. Int. J. Cancer 82, 442–448. [DOI] [PubMed] [Google Scholar]
- Aleksic M, Liddy N, Molloy PE, Pumphrey N, Vuidepot A, Chang KM, and Jakobsen BK (2012). Different affinity windows for virus and cancer-specific T-cell receptors: implications for therapeutic strategies. Eur. J. Immunol 42, 3174–3179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almeida LG, Sakabe NJ, deOliveira AR, Silva MC, Mundstein AS, Cohen T, Chen YT, Chua R, Gurung S, Gnjatic S, et al. (2009). CTdata-base: a knowledge-base of high-throughput and curated data on cancer-testis antigens. Nucleic Acids Res. 37, D816–D819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassani-Sternberg M, Braunlein E, Klar R, Engleitner T, Sinitcyn P, Audehm S, Straub M, Weber J, Slotta-Huspenina J, Specht K, et al. (2016). Direct identification of clinically relevant neoepitopes presented on native human melanoma tissue by mass spectrometry. Nat. Commun 7, 13404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bregni M, Badoglio M, Pedrazzoli P, and Lanza F (2016). Is allogeneic transplant for solid tumors still alive? Bone Marrow Transpl. 51, 751–752. [DOI] [PubMed] [Google Scholar]
- Bruchard M, and Ghiringhelli F (2019). Deciphering the roles of innate lymphoid cells in cancer. Front. Immunol 10, 656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullock TN, and Eisenlohr LC (1996). Ribosomal scanning past the primary initiation codon as a mechanism for expression of CTL epitopes encoded in alternative reading frames. J. Exp. Med 184, 1319–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullock TN, Patterson AE, Franlin LL, Notidis E, and Eisenlohr LC (1997). Initiation codon scanthrough versus termination codon readthrough demonstrates strong potential for major histocompatibility complex class I-restricted cryptic epitope expression. J. Exp. Med 186, 1051–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bykov VJN, Eriksson SE, Bianchi J, and Wiman KG (2018). Targeting mutant p53 for efficient cancer therapy. Nat. Rev. Cancer 18, 89–102. [DOI] [PubMed] [Google Scholar]
- Cameron BJ, Gerry AB, Dukes J, Harper JV, Kannan V, Bianchi FC, Grand F, Brewer JE, Gupta M, Plesa G, et al. (2013). Identification of a Titin-derived HLA-A1-presented peptide as a cross-reactive target for engineered MAGE A3-directed T cells. Sci. Transl Med 5, 197ra103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbonnelle D, Vignard V, Sehedic D, Moreau-Aubry A, Florenceau L, Charpentier M, Mikulits W, Labarriere N, and Lang F (2013). The melanoma antigens MELOE-1 and MELOE-2 are translated from a bona fide polycistronic mRNA containing functional IRES sequences. PLoS One 8, e75233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carreno BM, Magrini V, Becker-Hapak M, Kaabinejadian S, Hundal J, Petti AA, Ly A, Lie WR, Hildebrand WH, Mardis ER, and Linette GP (2015). Cancer immunotherapy. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science 348, 803–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chae YK, Viveiros P, Lopes G, Sukhadia B, Sheikh MM, Saravia D, Florou V, Sokol ES, Frampton GM, Chalmers ZR, et al. (2019). Clinical and immunological implications of frameshift mutations in lung cancer. J. Thorac. Oncol 14, 1807–1817. [DOI] [PubMed] [Google Scholar]
- Chandran SS, and Klebanoff CA (2019). T cell receptor-based cancer immunotherapy: emerging efficacy and pathways of resistance. Immunol. Rev 290, 127–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapuis AG, Roberts IM, Thompson JA, Margolin KA, Bhatia S, Lee SM, Sloan HL, Lai IP, Farrar EA, Wagener F, et al. (2016). T-cell therapy using interleukin-21-primed cytotoxic T-cell lymphocytes combined with cytotoxic T-cell lymphocyte antigen-4 blockade results in long-term cell persistence and durable tumor regression. J. Clin. Oncol 34, 3787–3795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, and Flies DB (2013). Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat. Rev. Immunol 13, 227–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YT, Boyer AD, Viars CS, Tsang S, Old LJ, and Arden KC (1997). Genomic cloning and localization of CTAG, a gene encoding an auto-immunogenic cancer-testis antigen NY-ESO-1, to human chromosome Xq28. Cytogenet. Cell Genet 79, 237–240. [DOI] [PubMed] [Google Scholar]
- Cohen CJ, Gartner JJ, Horovitz-Fried M, Shamalov K, Trebska-McGowan K, Bliskovsky VV, Parkhurst MR, Ankri C, Prickett TD, Crystal JS, et al. (2015). Isolation of neoantigen-specific T cells from tumor and peripheral lymphocytes. J. Clin. Invest 125, 3981–3991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulie PG, Lehmann F, Lethe B, Herman J, Lurquin C, Andrawiss M, and Boon T (1995). A mutated intron sequence codes for an antigenic peptide recognized by cytolytic T lymphocytes on a human melanoma. Proc. Natl. Acad. Sci. U S A 92, 7976–7980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Angelo SP, Melchiori L, Merchant MS, Bernstein D, Glod J, Kaplan R, Grupp S, Tap WD, Chagin K, Binder GK, et al. (2018). Antitumor activity associated with prolonged persistence of adoptively transferred NY-ESO-1 (c259)T cells in synovial sarcoma. Cancer Discov. 8, 944–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalet A, Robbins PF, Stroobant V, Vigneron N, Li YF, El-Gamil M, Hanada K, Yang JC, Rosenberg SA, and Van den Eynde BJ (2011). An antigenic peptide produced by reverse splicing and double asparagine deamidation. Proc. Natl. Acad. Sci. U S A 108, E323–E331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis MM (2015). Not-so-negative selection. Immunity 43, 833–835. [DOI] [PubMed] [Google Scholar]
- De Smet C, Lurquin C, Lethe B, Martelange V, and Boon T (1999). DNA methylation is the primary silencing mechanism for a set of germ line- and tumor-specific genes with a CpG-rich promoter. Mol. Cell Biol 19, 7327–7335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deniger DC, Pasetto A, Robbins PF, Gartner JJ, Prickett TD, Paria BC, Malekzadeh P, Jia L, Yossef R, Langhan MM, et al. (2018). T-cell responses to TP53 “hotspot” mutations and unique neoantigens expressed by human ovarian cancers. Clin. Cancer Res 24, 5562–5573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Depontieu FR, Qian J, Zarling AL, McMiller TL, Salay TM, Norris A, English AM, Shabanowitz J, Engelhard VH, Hunt DF, and Topalian SL (2009). Identification of tumor-associated, MHC class II-restricted phosphopeptides as targets for immunotherapy. Proc. Natl. Acad. Sci. U S A 106, 12073–12078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding L, Wendl MC, McMichael JF, and Raphael BJ (2014). Expanding the computational toolbox for mining cancer genomes. Nat. Rev. Genet 15, 556–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doran SL, Stevanović S, Adhikary S, Gartner JJ, Jia L, Kwong MLM, Faquin WC, Hewitt SM, Sherry RM, Yang JC, et al. (2019). T-cell receptor gene therapy for human papillomavirus-associated epithelial cancers: a first-in-human, phase I/II study. J. Clin. Oncol 37, 2759–2768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Draper LM, Kwong ML, Gros A, Stevanovic S, Tran E, Kerkar S, Raffeld M, Rosenberg SA, and Hinrichs CS (2015). Targeting of HPV-16+ epithelial cancer cells by TCR gene engineered T cells directed against E6. Clin. Cancer Res 21, 4431–4439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreno B, Thompson JF, Smithers BM, Santinami M, Jouary T, Gutzmer R, Levchenko E, Rutkowski P, Grob JJ, Korovin S, et al. (2018). MAGE-A3 immunotherapeutic as adjuvant therapy for patients with resected, MAGE-A3-positive, stage III melanoma (DERMA): a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 19, 916–929. [DOI] [PubMed] [Google Scholar]
- Faridi P, Li C, Ramarathinam SH, Vivian JP, Illing PT, Mifsud NA, Ayala R, Song J, Gearing LJ, Hertzog PJ, et al. (2018). A subset of HLA-I peptides are not genomically templated: evidence for cis- and trans-spliced peptide ligands. Sci. Immunol 3, eaar3947. [DOI] [PubMed] [Google Scholar]
- Fisk B, Blevins TL, Wharton JT, and Ioannides CG (1995). Identification of an immunodominant peptide of HER-2/neu protooncogene recognized by ovarian tumor-specific cytotoxic T lymphocyte lines. J. Exp. Med 181, 2109–2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Q, Liang WW, Foltz SM, Mutharasu G, Jayasinghe RG, Cao S, Liao WW, Reynolds SM, Wyczalkowski MA, Yao L, et al. (2018). Driver fusions and their implications in the development and treatment of human cancers. Cell Rep. 23, 227–238.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Garijo A, Fajardo CA, and Gros A (2019). Determinants for neoantigen identification. Front. Immunol 10, 1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerashchenko MV, Lobanov AV, and Gladyshev VN (2012). Genome-wide ribosome profiling reveals complex translational regulation in response to oxidative stress. Proc. Natl. Acad. Sci. U S A 109, 17394–17399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannakis M, Mu XJ, Shukla SA, Qian ZR, Cohen O, Nishihara R, Bahl S, Cao Y, Amin-Mansour A, Yamauchi M, et al. (2016). Genomic correlates of immune-cell infiltrates in colorectal carcinoma. Cell Rep. 17, 1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godfrey DI, Le Nours J, Andrews DM, Uldrich AP, and Rossjohn J (2018). Unconventional T cell targets for cancer immunotherapy. Immunity 48, 453–473. [DOI] [PubMed] [Google Scholar]
- Goff SL, Dudley ME, Citrin DE, Somerville RP, Wunderlich JR, Danforth DN, Zlott DA, Yang JC, Sherry RM, Kammula US, et al. (2016). Randomized, prospective evaluation comparing intensity of lymphodepletion before adoptive transfer of tumor-infiltrating lymphocytes for patients with metastatic melanoma. J. Clin. Oncol 34, 2389–2397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodenough E, Robinson TM, Zook MB, Flanigan KM, Atkins JF, Howard MT, and Eisenlohr LC (2014). Cryptic MHC class I-binding peptides are revealed by aminoglycoside-induced stop codon read-through into the 3′ UTR. Proc. Natl. Acad. Sci. U S A 111, 5670–5675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grady WM, Myeroff LL, Swinler SE, Rajput A, Thiagalingam S, Lutterbaugh JD, Neumann A, Brattain MG, Chang J, Kim SJ, et al. (1999). Mutational inactivation of transforming growth factor beta receptor type II in microsatellite stable colon cancers. Cancer Res. 59, 320–324. [PubMed] [Google Scholar]
- Guilloux Y, Lucas S, Brichard VG, Van Pel A, Viret C, De Plaen E, Brasseur F, Lethe B, Jotereau F, and Boon T (1996). A peptide recognized by human cytolytic T lymphocytes on HLA-A2 melanomas is encoded by an intron sequence of the N-acetylglucosaminyltransferase V gene. J. Exp. Med 183, 1173–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanada K, Yewdell JW, and Yang JC (2004). Immune recognition of a human renal cancer antigen through post-translational protein splicing. Nature 427, 252–256. [DOI] [PubMed] [Google Scholar]
- Hansen UK, Ramskov S, Bjerregaard AM, Borch A, Andersen R, Draghi A, Donia M, Bentzen AK, Marquard AM, Szallasi Z, et al. (2020). Tumor-infiltrating T cells from clear cell renal cell carcinoma patients recognize neoepitopes derived from point and frameshift mutations. Front. Immunol 11, 373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada M, Li YF, El-Gamil M, Ohnmacht GA, Rosenberg SA, and Robbins PF (2001). Melanoma-Reactive CD8+ T cells recognize a novel tumor antigen expressed in a wide variety of tumor types. J. Immunother 24, 323–333. [DOI] [PubMed] [Google Scholar]
- Hilf N, Kuttruff-Coqui S, Frenzel K, Bukur V, Stevanovic S, Gouttefangeas C, Platten M, Tabatabai G, Dutoit V, van der Burg SH, et al. (2019). Actively personalized vaccination trial for newly diagnosed glioblastoma. Nature 565, 240–245. [DOI] [PubMed] [Google Scholar]
- Holstein SA, and Lunning MA (2020). CAR T-cell therapy in hematologic malignancies: a voyage in progress. Clin. Pharmacol. Ther 107, 112–122. [DOI] [PubMed] [Google Scholar]
- Hu Z, Ott PA, and Wu CJ (2018). Towards personalized, tumour-specific, therapeutic vaccines for cancer. Nat. Rev. Immunol 18, 168–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, El-Gamil M, Dudley ME, Li YF, Rosenberg SA, and Robbins PF (2004). T cells associated with tumor regression recognize frameshifted products of the CDKN2A tumor suppressor gene locus and a mutated HLA class I gene product. J. Immunol 172, 6057–6064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu-Lieskovan S, Berent-Maoz B, Pang J, Chmielowski B, Cherry G, et al. (2016). Genomic and transcriptomic features of response to anti-PD-1 therapy in metastatic melanoma. Cell 165, 35–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer JG, Afanasiev OK, McClurkan C, Paulson K, Nagase K, Jing L, Marshak JO, Dong L, Carter J, Lai I, et al. (2011). Merkel cell polyomavirus-specific CD8⁺ and CD4⁺ T-cell responses identified in Merkel cell carcinomas and blood. Clin. Cancer Res 17, 6671–6680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayasinghe RG, Cao S, Gao Q, Wendl MC, Vo NS, Reynolds SM, Zhao Y, Climente-Gonzalez H, Chai S, Wang F, et al. (2018). Systematic analysis of splice-site-creating mutations in cancer. Cell Rep. 23, 270–281.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin BY, Campbell TE, Draper LM, Stevanovic S, Weissbrich B, Yu Z, Restifo NP, Rosenberg SA, Trimble CL, and Hinrichs CS (2018). Engineered T cells targeting E7 mediate regression of human papillomavirus cancers in a murine model. JCI Insight 3, e99488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson LA, Morgan RA, Dudley ME, Cassard L, Yang JC, Hughes MS, Kammula US, Royal RE, Sherry RM, Wunderlich JR, et al. (2009). Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood 114, 535–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurtz V, Paul S, Andreatta M, Marcatili P, Peters B, and Nielsen M (2017). NetMHCpan-4.0: improved peptide-MHC class I interaction predictions integrating eluted ligand and peptide binding affinity data. J. Immunol 199, 3360–3368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahles A, Lehmann KV, Toussaint NC, Huser M, Stark SG, Sachsenberg T, Stegle O, Kohlbacher O, Sander C, Cancer Genome Atlas Research, N., and Ratsch G (2018). Comprehensive analysis of alternative splicing across tumors from 8,705 patients. Cancer Cell 34, 211–224.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalina JL, Neilson DS, Lin YY, Hamilton PT, Comber AP, Loy EMH, Sahinalp SC, Collins CC, Hach F, and Lum JJ (2017). Mutational analysis of gene fusions predicts novel MHC class I-restricted T-cell epitopes and immune signatures in a subset of prostate cancer. Clin. Cancer Res 23, 7596–7607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, Penson DF, Redfern CH, Ferrari AC, Dreicer R, Sims RB, et al. (2010). Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N. Engl. J. Med 363, 411–422. [DOI] [PubMed] [Google Scholar]
- Kaufman HL, Kohlhapp FJ, and Zloza A (2015). Oncolytic viruses: a new class of immunotherapy drugs. Nat. Rev. Drug Discov 14, 642–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi S, Tsukahara T, Ida K, Kimura S, Murase M, Kano M, Emori M, Nagoya S, Kaya M, Torigoe T, et al. (2012). SYT-SSX breakpoint peptide vaccines in patients with synovial sarcoma: a study from the Japanese Musculoskeletal Oncology Group. Cancer Sci. 103, 1625–1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawakami Y, Eliyahu S, Delgado CH, Robbins PF, Sakaguchi K, Appella E, Yannelli JR, Adema GJ, Miki T, and Rosenberg SA (1994a). Identification of a human melanoma antigen recognized by tumor-infiltrating lymphocytes associated with in vivo tumor rejection. Proc. Natl. Acad. Sci. U S A 91, 6458–6462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawakami Y, Eliyahu S, Sakaguchi K, Robbins PF, Rivoltini L, Yannelli JR, Appella E, and Rosenberg SA (1994b). Identification of the immunodominant peptides of the MART-1 human melanoma antigen recognized by the majority of HLA-A2-restricted tumor infiltrating lymphocytes. J. Exp. Med 180, 347–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerkar SP, Wang ZF, Lasota J, Park T, Patel K, Groh E, Rosenberg SA, and Miettinen MM (2016). MAGE-A is more highly expressed than NY-ESO-1 in a systematic immunohistochemical analysis of 3668 cases. J. Immunother 39, 181–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keskin DB, Anandappa AJ, Sun J, Tirosh I, Mathewson ND, Li S, Oliveira G, Giobbie-Hurder A, Felt K, Gjini E, et al. (2019). Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature 565, 234–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khurana E, Fu Y, Chakravarty D, Demichelis F, Rubin MA, and Gerstein M (2016). Role of non-coding sequence variants in cancer. Nat. Rev. Genet 17, 93–108. [DOI] [PubMed] [Google Scholar]
- Klebanoff CA, Acquavella N, Yu Z, and Restifo NP (2011). Therapeutic cancer vaccines: are we there yet? Immunol. Rev 239, 27–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwong DLW, Lee VHF, and Nicholls JM (2019). Cancer immunotherapy for nasopharyngeal carcinoma In Nasopharyngeal Carcinoma, Lee W.M. Anne, Ng. Wai Tong, Lung., and Maria Li, eds. (Academic Press; ), pp. 337–351. [Google Scholar]
- Lamers CH, Sleijfer S, van Steenbergen S, van Elzakker P, van Krimpen B, Groot C, Vulto A, den Bakker M, Oosterwijk E, Debets R, and Gratama JW (2013). Treatment of metastatic renal cell carcinoma with CAIX CAR-engineered T cells: clinical evaluation and management of on-target toxicity. Mol. Ther 21, 904–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, et al. (2001). Initial sequencing and analysis of the human genome. Nature 409, 860–921. [DOI] [PubMed] [Google Scholar]
- Laumont CM, Daouda T, Laverdure JP, Bonneil E, Caron-Lizotte O, Hardy MP, Granados DP, Durette C, Lemieux S, Thibault P, and Perreault C (2016). Global proteogenomic analysis of human MHC class I-associated peptides derived from non-canonical reading frames. Nat. Commun 7, 10238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laumont CM, and Perreault C (2018). Exploiting non-canonical translation to identify new targets for T cell-based cancer immunotherapy. Cell Mol. Life Sci 75, 607–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SP, Chan AT, Cheung ST, Thomas WA, CroomCarter D, Dawson CW, Tsai CH, Leung SF, Johnson PJ, and Huang DP (2000). CTL control of EBV in nasopharyngeal carcinoma (NPC): EBV-specific CTL responses in the blood and tumors of NPC patients and the antigen-processing function of the tumor cells. J. Immunol 165, 573–582. [DOI] [PubMed] [Google Scholar]
- Liepe J, Marino F, Sidney J, Jeko A, Bunting DE, Sette A, Kloetzel PM, Stumpf MP, Heck AJ, and Mishto M (2016). A large fraction of HLA class I ligands are proteasome-generated spliced peptides. Science 354, 354–358. [DOI] [PubMed] [Google Scholar]
- Liepe J, Sidney J, Lorenz FKM, Sette A, and Mishto M (2019). Mapping the MHC class I-spliced immunopeptidome of cancer cells. Cancer Immunol. Res 7, 62–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindeboom RGH, Vermeulen M, Lehner B, and Supek F (2019). The impact of nonsense-mediated mRNA decay on genetic disease, gene editing and cancer immunotherapy. Nat. Genet 51, 1645–1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linnebacher M, Gebert J, Rudy W, Woerner S, Yuan YP, Bork P, and von Knebel Doeberitz M (2001). Frameshift peptide-derived T-cell epitopes: a source of novel tumor-specific antigens. Int. J. Cancer 93, 6–11. [DOI] [PubMed] [Google Scholar]
- Lu YC, Parker LL, Lu T, Zheng Z, Toomey MA, White DE, Yao X, Li YF, Robbins PF, Feldman SA, et al. (2017). Treatment of patients with metastatic cancer using a major histocompatibility complex class II-restricted T-cell receptor targeting the cancer germline antigen MAGE-A3. J. Clin. Oncol 35, 3322–3329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu YC, Yao X, Crystal JS, Li YF, El-Gamil M, Gross C, Davis L, Dudley ME, Yang JC, Samuels Y, et al. (2014). Efficient identification of mutated cancer antigens recognized by T cells associated with durable tumor regressions. Clin. Cancer Res 20, 3401–3410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu YC, Yao X, Li YF, El-Gamil M, Dudley ME, Yang JC, Almeida JR, Douek DC, Samuels Y, Rosenberg SA, and Robbins PF (2013). Mutated PPP1R3B is recognized by T cells used to treat a melanoma patient who experienced a durable complete tumor regression. J. Immunol 190, 6034–6042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lurquin C, Van Pel A, Mariame B, De Plaen E, Szikora JP, Janssens C, Reddehase MJ, Lejeune J, and Boon T (1989). Structure of the gene of tum- transplantation antigen P91A: the mutated exon encodes a peptide recognized with Ld by cytolytic T cells. Cell 58, 293–303. [DOI] [PubMed] [Google Scholar]
- Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat SM, Supko JG, Haluska FG, et al. (2004). Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N. Engl. J. Med 350, 2129–2139. [DOI] [PubMed] [Google Scholar]
- Mackall CL, Rhee EH, Read EJ, Khuu HM, Leitman SF, Bernstein D, Tesso M, Long LM, Grindler D, Merino M, et al. (2008). A pilot study of consolidative immunotherapy in patients with high-risk pediatric sarcomas. Clin. Cancer Res 14, 4850–4858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda Y, Nishikawa H, Sugiyama D, Ha D, Hamaguchi M, Saito T, Nishioka M, Wing JB, Adeegbe D, Katayama I, and Sakaguchi S (2014). Detection of self-reactive CD8(+) T cells with an anergic phenotype in healthy individuals. Science 346, 1536–1540. [DOI] [PubMed] [Google Scholar]
- Majumder S, Shah R, Elias J, Mistry Y, Coral K, Shah P, Maurya AK, Mittal B, D’Silva JK, Murugan S, et al. (2018). A neoepitope derived from a novel human germline APC gene mutation in familial adenomatous polyposis shows selective immunogenicity. PLoS One 13, e0203845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majzner RG, and Mackall CL (2019). Clinical lessons learned from the first leg of the CAR T cell journey. Nat. Med 25, 1341–1355. [DOI] [PubMed] [Google Scholar]
- Malekzadeh P, Pasetto A, Robbins PF, Parkhurst MR, Paria BC, Jia L, Gartner JJ, Hill V, Yu Z, Restifo NP, et al. (2019). Neoantigen screening identifies broad TP53 mutant immunogenicity in patients with epithelial cancers. J. Clin. Invest 129, 1109–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malekzadeh P, Yossef R, Cafri G, Paria BC, Lowery FJ, Jafferji M, Good ML, Sachs A, Copeland AR, Kim SP, et al. (2020). Antigen experienced T cells from peripheral blood recognize p53 neoantigens. Clin. Cancer Res 26, 1267–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal R, Samstein RM, Lee KW, Havel JJ, Wang H, Krishna C, Sabio EY, Makarov V, Kuo F, Blecua P, et al. (2019). Genetic diversity of tumors with mismatch repair deficiency influences anti-PD-1 immunotherapy response. Science 364, 485–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markowitz S, Wang J, Myeroff L, Parsons R, Sun L, Lutterbaugh J, Fan RS, Zborowska E, Kinzler KW, Vogelstein B, et al. (1995). Inactivation of the type II TGF-beta receptor in colon cancer cells with microsatellite instability. Science 268, 1336–1338. [DOI] [PubMed] [Google Scholar]
- McDermott DF, Huseni MA, Atkins MB, Motzer RJ, Rini BI, Escudier B, Fong L, Joseph RW, Pal SK, Reeves JA, et al. (2018). Clinical activity and molecular correlates of response to atezolizumab alone or in combination with bevacizumab versus sunitinib in renal cell carcinoma. Nat. Med 24, 749–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGranahan N, Furness AJ, Rosenthal R, Ramskov S, Lyngaa R, Saini SK, Jamal-Hanjani M, Wilson GA, Birkbak NJ, Hiley CT, et al. (2016). Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 351, 1463–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mertens F, Johansson B, Fioretos T, and Mitelman F (2015). The emerging complexity of gene fusions in cancer. Nat. Rev. Cancer 15, 371–381. [DOI] [PubMed] [Google Scholar]
- Meyer VS, Drews O, Günder M, Hennenlotter J, Rammensee HG, and Stevanovic S (2009). Identification of natural MHC class II presented phosphopeptides and tumor-derived MHC class I phospholigands. J. Proteome Res 8, 3666–3674. [DOI] [PubMed] [Google Scholar]
- Michaux A, Larrieu P, Stroobant V, Fonteneau JF, Jotereau F, Van den Eynde BJ, Moreau-Aubry A, and Vigneron N (2014). A spliced antigenic peptide comprising a single spliced amino acid is produced in the proteasome by reverse splicing of a longer peptide fragment followed by trimming. J. Immunol 192, 1962–1971. [DOI] [PubMed] [Google Scholar]
- Moody CA, and Laimins LA (2010). Human papillomavirus oncoproteins: pathways to transformation. Nat. Rev. Cancer 10, 550–560. [DOI] [PubMed] [Google Scholar]
- Morgan RA, Chinnasamy N, Abate-Daga D, Gros A, Robbins PF, Zheng Z, Dudley ME, Feldman SA, Yang JC, Sherry RM, et al. (2013). Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J. Immunother 36, 133–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norberg SM, Nagarsheth N, Doran S, Kanakry JA, Adhikary S, Schweitzer C, Astrow SH, Faquin W, Gkitsas N, Highfill S, and Hinrichs CS (2018). Regression of epithelial cancers following T cell receptor gene therapy targeting human papillomavirus-16 E7. Blood 132, 492.29866811 [Google Scholar]
- Nowicki TS, Berent-Maoz B, Cheung-Lau G, Huang RR, Wang X, Tsoi J, Kaplan-Lefko P, Cabrera P, Tran J, Pang J, et al. (2019). A pilot trial of the combination of transgenic NY-ESO-1-reactive adoptive cellular therapy with dendritic cell vaccination with or without ipilimumab. Clin. Cancer Res 25, 2096–2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Donnell JS, Teng MWL, and Smyth MJ (2019). Cancer immunoediting and resistance to T cell-based immunotherapy. Nat. Rev. Clin. Oncol 16, 151–167. [DOI] [PubMed] [Google Scholar]
- Ott PA, Hu Z, Keskin DB, Shukla SA, Sun J, Bozym DJ, Zhang W, Luoma A, Giobbie-Hurder A, Peter L, et al. (2017). An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 547, 217–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panda A, de Cubas AA, Stein M, Riedlinger G, Kra J, Mayer T, Smith CC, Vincent BG, Serody JS, Beckermann KE, et al. (2018). Endogenous retrovirus expression is associated with response to immune checkpoint blockade in clear cell renal cell carcinoma. JCI Insight 3, e121522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkhurst MR, Joo J, Riley JP, Yu Z, Li Y, Robbins PF, and Rosenberg SA (2009). Characterization of genetically modified T-cell receptors that recognize the CEA:691–699 peptide in the context of HLA-A2.1 on human colorectal cancer cells. Clin. Cancer Res 15, 169–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkhurst MR, Robbins PF, Tran E, Prickett TD, Gartner JJ, Jia L, Ivey G, Li YF, El-Gamil M, Lalani A, et al. (2019). Unique neoantigens arise from somatic mutations in patients with gastrointestinal cancers. Cancer Discov. 9, 1022–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkhurst MR, Yang JC, Langan RC, Dudley ME, Nathan DA, Feldman SA, Davis JL, Morgan RA, Merino MJ, Sherry RM, et al. (2011). T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis. Mol. Ther 19, 620–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastor F, Kolonias D, Giangrande PH, and Gilboa E (2010). Induction of tumour immunity by targeted inhibition of nonsense-mediated mRNA decay. Nature 465, 227–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulson KG, Voillet V, McAfee MS, Hunter DS, Wagener FD, Perdicchio M, Valente WJ, Koelle SJ, Church CD, Vandeven N, et al. (2018). Acquired cancer resistance to combination immunotherapy from transcriptional loss of class I HLA. Nat. Commun 9, 3868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng S, Zaretsky JM, Ng AHC, Chour W, Bethune MT, Choi J, Hsu A, Holman E, Ding X, Guo K, et al. (2019). Sensitive detection and analysis of neoantigen-specific T cell populations from tumors and blood. Cell Rep. 28, 2728–2738.e27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pignon J-C, Jegede O, Shukla SA, Braun DA, Horak C, Wind-Rotolo M, Ishii Y, Catalano PJ, Freeman GJ, Jennings RB, et al. (2019). Association of human endogenous retrovirus (hERV) expression with clinical efficacy of PD-1 blockade in metastatic clear cell renal cell carcinoma (mccRCC). J. Clin. Oncol 37, 4568. [Google Scholar]
- Prickett TD, Crystal JS, Cohen CJ, Pasetto A, Parkhurst MR, Gartner JJ, Yao X, Wang R, Gros A, Li YF, et al. (2016). Durable complete response from metastatic melanoma after transfer of autologous T cells recognizing 10 mutated tumor antigens. Cancer Immunol. Res 4, 669–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Probst-Kepper M, Stroobant V, Kridel R, Gaugler B, Landry C, Brasseur F, Cosyns JP, Weynand B, Boon T, and Van Den Eynde BJ (2001). An alternative open reading frame of the human macrophage colony-stimulating factor gene is independently translated and codes for an antigenic peptide of 14 amino acids recognized by tumor-infiltrating CD8 T lymphocytes. J. Exp. Med 193, 1189–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynisson B, Alvarez B, Paul S, Peters B, and Nielsen M (2020). NetMHCpan-4.1 and NetMHCIIpan-4.0: improved predictions of MHC antigen presentation by concurrent motif deconvolution and integration of MS MHC eluted ligand data. Nucleic Acids Res. 48, W449–W454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, Lee W, Yuan J, Wong P, Ho TS, et al. (2015). Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 348, 124–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins PF, El-Gamil M, Li YF, Fitzgerald EB, Kawakami Y, and Rosenberg SA (1997). The intronic region of an incompletely spliced gp100 gene transcript encodes an epitope recognized by melanoma-reactive tumor-infiltrating lymphocytes. J. Immunol 159, 303–308. [PubMed] [Google Scholar]
- Robbins PF, El-Gamil M, Li YF, Kawakami Y, Loftus D, Appella E, and Rosenberg SA (1996). A mutated beta-catenin gene encodes a melanoma specific antigen recognized by tumor infiltrating lymphocytes. J. Exp. Med 183, 1185–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins PF, Lu YC, El-Gamil M, Li YF, Gross C, Gartner J, Lin JC, Teer JK, Cliften P, Tycksen E, et al. (2013). Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells. Nat. Med 19, 747–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins PF, Morgan RA, Feldman SA, Yang JC, Sherry RM, Dudley ME, Wunderlich JR, Nahvi AV, Helman LJ, Mackall CL, et al. (2011). Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J. Clin. Oncol 29, 917–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronsin C, Chung-Scott V, Poullion I, Aknouche N, Gaudin C, and Triebel F (1999). A non-AUG-defined alternative open reading frame of the intestinal carboxyl esterase mRNA generates an epitope recognized by renal cell carcinoma-reactive tumor-infiltrating lymphocytes in situ. J. Immunol 163, 483–490. [PubMed] [Google Scholar]
- Rooney MS, Shukla SA, Wu CJ, Getz G, and Hacohen N (2015). Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 160, 48–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg SA (2014). IL-2: the first effective immunotherapy for human cancer. J. Immunol 192, 5451–5458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg SA, and Restifo NP (2015). Adoptive cell transfer as personalized immunotherapy for human cancer. Science 348, 62–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg SA, Tong-On P, Li Y, Riley JP, El-Gamil M, Parkhurst MR, and Robbins PF (2002). Identification of BING-4 cancer antigen translated from an alternative open reading frame of a gene in the extended MHC class II region using lymphocytes from a patient with a durable complete regression following immunotherapy. J. Immunol 168, 2402–2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg SA, Yang JC, and Restifo NP (2004). Cancer immunotherapy: moving beyond current vaccines. Nat. Med 10, 909–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossig C, Kailayangiri S, Jamitzky S, and Altvater B (2018). Carbohydrate targets for CAR T cells in solid childhood cancers. Front. Oncol 8, 513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth A, Khattra J, Yap D, Wan A, Laks E, Biele J, Ha G, Aparicio S, Bouchard-Cote A, and Shah SP (2014). PyClone: statistical inference of clonal population structure in cancer. Nat. Methods 11, 396–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roulois D, Loo Yau H, Singhania R, Wang Y, Danesh A, Shen SY, Han H, Liang G, Jones PA, Pugh TJ, et al. (2015). DNA-demethylating agents target colorectal cancer cells by inducing viral mimicry by endogenous transcripts. Cell 162, 961–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rugo HS, Delord JP, Im SA, Ott PA, Piha-Paul SA, Bedard PL, Sachdev J, Le Tourneau C, van Brummelen EMJ, Varga A, et al. (2018). Safety and antitumor activity of pembrolizumab in patients with estrogen receptor-positive/human epidermal growth factor receptor 2-negative advanced breast cancer. Clin. Cancer Res 24, 2804–2811. [DOI] [PubMed] [Google Scholar]
- Rycaj K, Plummer JB, Yin B, Li M, Garza J, Radvanyi L, Ramondetta LM, Lin K, Johanning GL, Tang DG, and Wang-Johanning F (2015). Cytotoxicity of human endogenous retrovirus K-specific T cells toward autologous ovarian cancer cells. Clin. Cancer Res 21, 471–483. [DOI] [PubMed] [Google Scholar]
- Saeterdal I, Bjorheim J, Lislerud K, Gjertsen MK, Bukholm IK, Olsen OC, Nesland JM, Eriksen JA, Moller M, Lindblom A, and Gaudernack G (2001a). Frameshift-mutation-derived peptides as tumor-specific antigens in inherited and spontaneous colorectal cancer. Proc. Natl. Acad. Sci. U S A 98, 13255–13260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saeterdal I, Gjertsen MK, Straten P, Eriksen JA, and Gaudernack G (2001b). A TGF betaRII frameshift-mutation-derived CTL epitope recognised by HLA-A2-restricted CD8+ T cells. Cancer Immunol. Immunother 50, 469–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahin U, Derhovanessian E, Miller M, Kloke BP, Simon P, Lower M, Bukur V, Tadmor AD, Luxemburger U, Schrors B, et al. (2017). Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature 547, 222–226. [DOI] [PubMed] [Google Scholar]
- Sato Y, Nabeta Y, Tsukahara T, Hirohashi Y, Syunsui R, Maeda A, Sahara H, Ikeda H, Torigoe T, Ichimiya S, et al. (2002). Detection and induction of CTLs specific for SYT-SSX-derived peptides in HLA-A24(+) patients with synovial sarcoma. J. Immunol 169, 1611–1618. [DOI] [PubMed] [Google Scholar]
- Saulquin X, Scotet E, Trautmann L, Peyrat MA, Halary F, Bonneville M, and Houssaint E (2002). +1 Frameshifting as a novel mechanism to generate a cryptic cytotoxic T lymphocyte epitope derived from human interleukin 10. J. Exp. Med 195, 353–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiavetti F, Thonnard J, Colau D, Boon T, and Coulie PG (2002). A human endogenous retroviral sequence encoding an antigen recognized on melanoma by cytolytic T lymphocytes. Cancer Res. 62, 5510–5516. [PubMed] [Google Scholar]
- Shichijo S, Nakao M, Imai Y, Takasu H, Kawamoto M, Niiya F, Yang D, Toh Y, Yamana H, and Itoh K (1998). A gene encoding antigenic peptides of human squamous cell carcinoma recognized by cytotoxic T lymphocytes. J. Exp. Med 187, 277–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla SA, Bachireddy P, Schilling B, Galonska C, Zhan Q, Bango C, Langer R, Lee PC, Gusenleitner D, Keskin DB, et al. (2018). Cancer-germline antigen expression discriminates clinical outcome to CTLA-4 blockade. Cell 173, 624–633.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sibille C, Chomez P, Wildmann C, Van Pel A, De Plaen E, Maryanski JL, de Bergeyck V, and Boon T (1990). Structure of the gene of tum- transplantation antigen P198: a point mutation generates a new antigenic peptide. J. Exp. Med 172, 35–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skipper JC, Hendrickson RC, Gulden PH, Brichard V, Van Pel A, Chen Y, Shabanowitz J, Wolfel T, Slingluff CL Jr., Boon T, et al. (1996). An HLA-A2-restricted tyrosinase antigen on melanoma cells results from post-translational modification and suggests a novel pathway for processing of membrane proteins. J. Exp. Med 183, 527–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smart AC, Margolis CA, Pimentel H, He MX, Miao D, Adeegbe D, Fugmann T, Wong KK, and Van Allen EM (2018). Intron retention is a source of neoepitopes in cancer. Nat. Biotechnol 36, 1056–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CC, Beckermann KE, Bortone DS, De Cubas AA, Bixby LM, Lee SJ, Panda A, Ganesan S, Bhanot G, Wallen EM, et al. (2018). Endogenous retroviral signatures predict immunotherapy response in clear cell renal cell carcinoma. J. Clin. Invest 128, 4804–4820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starck SR, Tsai JC, Chen K, Shodiya M, Wang L, Yahiro K, Martins-Green M, Shastri N, and Walter P (2016). Translation from the 5’ untranslated region shapes the integrated stress response. Science 351, aad3867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevanovic S, Pasetto A, Helman SR, Gartner JJ, Prickett TD, Howie B, Robins HS, Robbins PF, Klebanoff CA, Rosenberg SA, and Hinrichs CS (2017). Landscape of immunogenic tumor antigens in successful immunotherapy of virally induced epithelial cancer. Science 356, 200–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi Y, Harashima N, Kajigaya S, Yokoyama H, Cherkasova E, McCoy JP, Hanada K, Mena O, Kurlander R, Tawab A, et al. (2008). Regression of human kidney cancer following allogeneic stem cell transplantation is associated with recognition of an HERV-E antigen by T cells. J. Clin. Invest 118, 1099–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran E, Robbins PF, Lu YC, Prickett TD, Gartner JJ, Jia L, Pasetto A, Zheng Z, Ray S, Groh EM, et al. (2016). T-cell transfer therapy targeting mutant KRAS in cancer. N. Engl. J. Med 375, 2255–2262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran E, Robbins PF, and Rosenberg SA (2017). ‘Final common pathway’ of human cancer immunotherapy: targeting random somatic mutations. Nat. Immunol 18, 255–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran E, Turcotte S, Gros A, Robbins PF, Lu YC, Dudley ME, Wunderlich JR, Somerville RP, Hogan K, Hinrichs CS, et al. (2014). Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science 344, 641–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turajlic S, Litchfield K, Xu H, Rosenthal R, McGranahan N, Reading JL, Wong YNS, Rowan A, Kanu N, Al Bakir M, et al. (2017). Insertion-and-deletion-derived tumour-specific neoantigens and the immunogenic pheno-type: a pan-cancer analysis. Lancet Oncol. 18, 1009–1021. [DOI] [PubMed] [Google Scholar]
- Van Allen EM, Miao D, Schilling B, Shukla SA, Blank C, Zimmer L, Sucker A, Hillen U, Foppen MHG, Goldinger SM, et al. (2015). Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 350, 207–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Bruggen P, Traversari C, Chomez P, Lurquin C, De Plaen E, Van den Eynde B, Knuth A, and Boon T (1991). A gene encoding an antigen recognized by cytolytic T lymphocytes on a human melanoma. Science 254, 1643–1647. [DOI] [PubMed] [Google Scholar]
- van Rooij N, van Buuren MM, Philips D, Velds A, Toebes M, Heemskerk B, van Dijk LJ, Behjati S, Hilkmann H, El Atmioui D, et al. (2013). Tumor exome analysis reveals neoantigen-specific T-cell reactivity in an ipilimumab-responsive melanoma. J. Clin. Oncol 31, e439–e442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA Jr., and Kinzler KW (2013). Cancer genome landscapes. Science 339, 1546–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vollers SS, and Stern LJ (2008). Class II major histocompatibility complex tetramer staining: progress, problems, and prospects. Immunology 123, 305–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voss MH, Buros Novik J, Hellmann MD, Ball M, Hakimi AA, Miao D, Margolis C, Horak C, Wind-Rotolo M, De Velasco G, et al. (2018). Correlation of degree of tumor immune infiltration and insertion-and-deletion (indel) burden with outcome on programmed death 1 (PD1) therapy in advanced renal cell cancer (RCC). J. Clin. Oncol 36, 4518. [Google Scholar]
- Wang QJ, Yu Z, Griffith K, Hanada K, Restifo NP, and Yang JC (2016). Identification of T-cell receptors targeting KRAS-mutated human tumors. Cancer Immunol. Res 4, 204–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang RF, Johnston SL, Zeng G, Topalian SL, Schwartzentruber DJ, and Rosenberg SA (1998). A breast and melanoma-shared tumor antigen: T cell responses to antigenic peptides translated from different open reading frames. J. Immunol 161, 3598–3606. [PubMed] [Google Scholar]
- Wang RF, Parkhurst MR, Kawakami Y, Robbins PF, and Rosenberg SA (1996). Utilization of an alternative open reading frame of a normal gene in generating a novel human cancer antigen. J. Exp. Med 183, 1131–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang-Johanning F, Radvanyi L, Rycaj K, Plummer JB, Yan P, Sastry KJ, Piyathilake CJ, Hunt KK, and Johanning GL (2008). Human endogenous retrovirus K triggers an antigen-specific immune response in breast cancer patients. Cancer Res. 68, 5869–5877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren EH, Vigneron NJ, Gavin MA, Coulie PG, Stroobant V, Dalet A, Tykodi SS, Xuereb SM, Mito JK, Riddell SR, and Van den Eynde BJ (2006). An antigen produced by splicing of noncontiguous peptides in the reverse order. Science 313, 1444–1447. [DOI] [PubMed] [Google Scholar]
- Wei SC, Duffy CR, and Allison JP (2018). Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov. 8, 1069–1086. [DOI] [PubMed] [Google Scholar]
- Wei Z, Zhou C, Zhang Z, Guan M, Zhang C, Liu Z, and Liu Q (2019). The landscape of tumor fusion neoantigens: a pan-cancer analysis. iScience 21, 249–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfel T, Hauer M, Schneider J, Serrano M, Wolfel C, Klehmann-Hieb E, De Plaen E, Hankeln T, Meyer zum Buschenfelde KH, and Beach D (1995). A p16INK4a-insensitive CDK4 mutant targeted by cytolytic T lymphocytes in a human melanoma. Science 269, 1281–1284. [DOI] [PubMed] [Google Scholar]
- Yang W, Lee KW, Srivastava RM, Kuo F, Krishna C, Chowell D, Makarov V, Hoen D, Dalin MG, Wexler L, et al. (2019). Immunogenic neoantigens derived from gene fusions stimulate T cell responses. Nat. Med 25, 767–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarchoan M, Hopkins A, and Jaffee EM (2017). Tumor mutational burden and response rate to PD-1 inhibition. N. Engl. J. Med 377, 2500–2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yotnda P, Firat H, Garcia-Pons F, Garcia Z, Gourru G, Vernant JP, Lemonnier FA, Leblond V, and Langlade-Demoyen P (1998). Cytotoxic T cell response against the chimeric p210 BCR-ABL protein in patients with chronic myelogenous leukemia. J. Clin. Invest 101, 2290–2296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zacharakis N, Chinnasamy H, Black M, Xu H, Lu YC, Zheng Z, Pasetto A, Langhan M, Shelton T, Prickett T, et al. (2018). Immune recognition of somatic mutations leading to complete durable regression in metastatic breast cancer. Nat. Med 24, 724–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarling AL, Polefrone JM, Evans AM, Mikesh LM, Shabanowitz J, Lewis ST, Engelhard VH, and Hunt DF (2006). Identification of class I MHC-associated phosphopeptides as targets for cancer immunotherapy. Proc. Natl. Acad. Sci. U S A 103, 14889–14894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Dudley ME, Rosenberg SA, and Robbins PF (2005). Persistence of multiple tumor-specific T-cell clones is associated with complete tumor regression in a melanoma patient receiving adoptive cell transfer therapy. J. Immunother 28, 53–62. [DOI] [PMC free article] [PubMed] [Google Scholar]