

Abstract
Prader–Willi syndrome (PWS) is a genomic imprinting disorder characterized by infantile hypotonia with a poor suck and failure to thrive, hypogenitalism/hypogonadism, behavior and cognitive problems, hormone deficiencies, hyperphagia, and obesity. The Stanford Binet and Wechsler (WAIS-R; WISC-III) intelligence (IQ) tests were administered on 103 individuals with PWS from two separate cohorts [University of California, Irvine (UCI) (N = 56) and Vanderbilt University (N = 47)] and clinical information obtained including growth hormone (GH) treatment, PWS molecular classes, weight and height. Significantly higher IQ scores (p < .02) were found representing the vocabulary section of the Stanford Binet test in the growth hormone (GH) treated group when compared with non-GH treatment in the pediatric-based UCI PWS cohort with a trend for stabilization of vocabulary IQ scores with age in the GH treated maternal disomy (UPD) 15 subject group. Significant differences (p = .05) were also found in the adult-based Vanderbilt PWS cohort with 15q11-q13 deletion subjects having lower Verbal IQ scores compared with UPD 15. No difference in body mass index was identified based on the PWS molecular class or genetic subtype. Medical care and response to treatment with growth hormone may influence intelligence impacted by PWS genetic subtypes and possibly age, but more studies are needed.
Keywords: body mass index, growth hormone treatment, Prader–Willi syndrome, PWS molecular classes, Stanford Binet intelligence test, Wechsler intelligence test
1 |. INTRODUCTION
Prader–Willi syndrome (PWS) is a rare, complex neurobehavioral disorder due to errors in genomic imprinting that affects approximately 1/10,000–1/30,000 people. PWS is mostly detected during infancy or early childhood because of characteristic features including infantile hypotonia with a poor suck and failure to thrive, behavioral problems and developmental delay, specific facial findings (bitemporal narrowing, upslanted eyes, a high-palate, and small chin), gonadal hypoplasia, and small hands and feet with short stature related to growth and other hormone deficiencies. Hyperphagia occurs in early childhood with morbid obesity, if not externally controlled (Butler, 1990; Butler, 2016; Butler, Lee, & Whitman, 2006; Cassidy & Driscoll, 2009; Cassidy, Schwartz, Miller, & Driscoll, 2012).
All individuals with PWS show lack of expression of imprinted genes from the paternally inherited 15q11.2-q13 chromosomal region. There are three PWS molecular classes or genetic subtypes with the most common due to a typical paternal 15q11-q13 deletion seen in about 60% of individuals. The second most common cause is maternal uniparental disomy (UPD) 15 (seen in about 35% of cases) and the remaining subjects have a defect in the imprinting center controlling the activity of imprinted genes in the 15q11-q13 region, or by other chromosome 15 abnormalities (translocations, inversions) (Butler et al., 2019).
In most cases, PWS is characterized by a centrally-driven excessive appetite, hyperphagia, hypotonia, cognitive impairment, a distinct behavioral phenotype, hypogonadism, infantile failure to thrive and growth hormone deficiency (Butler, 2016; Butler et al., 2006; Cassidy et al., 2012; Cassidy & Driscoll, 2009). Clinical features evolve through several nutritional phases. Phase I is characterized by hypotonia with a poor suck from birth to the age of 2 years. Phase II then follows between 2 and 6 years of age when individuals begin food seeking with weight gain. Phase III is associated with hyperphagia and excessive weight gain between ages 6 and 12 years, leading to obesity, if uncontrolled (Miller et al., 2011). Growth hormone (GH) treatment is now the standard of care in PWS in developed countries and is initiated after establishing the diagnosis and evaluating for obstructive sleep apnea and endocrine disturbances (e.g., adrenal insufficiency, hypothyroidism). The nutritional phases have been altered with onset of growth hormone treatment.
At present, there is limited information regarding the benefit of GH treatment on intelligence. An earlier study by Carrel, Myers, Whitman, Eickhoff, and Allen (2010) compared 21 individuals (6–9 years of age) beginning at 4–32 months with initiation of human growth hormone treatment to another 27 individuals (5–9 years) without GH treatment. Early initiation and long-term GH therapy was noted to improve motor function, height and lipid profiles and contribute to better attention and increased memory performance.
Siemensma et al. (2012) later studied 50 randomized prepubertal children with PWS aged 3–14 years assessed with the Wechsler Intelligence Scale for Children Revised (WISC-R) and the Wechsler Pre-school and Primary Scale of Intelligence Revised (WPPSI-R) for children less than 7 years of age. Twenty-nine subjects with PWS were treated with GH and 21 were untreated. After 2 years, no significant differences were seen between the IQ subtest scores and total IQ for the GH treated individuals compared to their baseline visit. Individuals with PWS were then followed after 4 years of GH treatment and improvements were noted in abstract reasoning and visual spatial skills. The results were significant in the Similarities (p = .01) and Block Design (p = .01) sections of the test compared to the onset of GH treatment. Furthermore, GH treatment prevented deterioration of specific cognitive skills in children with PWS with improvements of abstract verbal reasoning.
Butler, Bittel, Kibiryeva, Talebizadeh, and Thompson (2004) also found that individuals with PWS having the 15q11-q13 deletion showed more behavioral and psychological problems than individuals with UPD 15; specifically, those with the deletion had poorer reading and math skills and visual-motor integration. They further analyzed individuals with the larger typical 15q11-q13 Type I deletion compared with the smaller 15q11-q13 Type II deletion and found that those with the larger typical deletion had more behavioral problems. An earlier study by Roof et al. (2000) also found that individuals with PWS having UPD 15 had significantly higher Verbal IQ scores than those with the deletion.
Jauregi et al. (2007) evaluated neuropsychological assessments in 16 individuals with PWS along with IQ, academic level and body mass index (BMI). The subtest scores for Full Scale IQ (Wechsler Adult Intelligence Scale) were significantly lower than in the normal population (p < .05). Their study also showed that individuals with PWS had improvement with repetition and the ability to retain and evoke the learned materials without difficulty in the short term. In many cases, PWS subjects had verbal deficits, which relate to the importance of using visual strategies with PWS individuals. Even so, there was a high possibility of an increased error rate during the test due to attention deficit. The authors suggested that a larger cohort was needed to examine for intelligence differences between the PWS deletion subtypes. More recently, Dykens, Roof, and Hunt-Hawkins (2017) showed that children who began GH treatment before 12 months of age had higher nonverbal and composite IQ scores than children with PWS who began treatment between 1 and 5 years of age.
The aim of the current study was to compare intelligence measures and BMI in individuals with PWS along with whether they had GH treatment and in relationship with PWS molecular classes or genetic subtypes. We hypothesize that individuals on GH treatment would have higher intelligence scores compared to those not on GH treatment and that significant differences in IQ (and subscales) may be present among the PWS genetic subtypes.
2 |. MATERIALS AND METHODS
2.1 |. Subjects
Fifty-six individuals with genetically confirmed PWS were analyzed from a March of Dimes sponsored pediatric-based study at the University of California, Irvine (UCI) as were 47 individuals with PWS from an adult-based cohort recruited for a National Institute of Health (NIH) funded study on genetics and behavior at Vanderbilt University. Research protocols were approved and informed consents obtained from all eligible participants or their legally responsible caregivers. Clinical, genetic and cognitive data were obtained at Case Western Reserve University, at UCI and/or at Vanderbilt University. Stanford Binet intelligence tests were used for the UCI PWS cohort which consisted of 29 males and 27 females with an age range of 3–38 years including 11 adults (≥18 years) and 45 children. Thirty-one individuals had the 15q11-q13 deletion and 25 with UPD 15. They were studied over a 5-year period from the early 2000s. Thirty individuals were GH treated, and by history, 26 never received GH treatment (see Table 1). One participant received thyroid hormone. Height (cm), weight (kg), and body mass index (BMI) were obtained on all individuals with PWS in the UCI cohort.
Table 1.
Demographics of individuals with Prader–Willi syndrome
| Variable | UCI cohort | Vanderbilt cohort | Total |
|---|---|---|---|
| Number of patients | 56 | 47 | 103 |
| No GH treatment | 26 (47%) | 45 (96%) | 71 (69%) |
| GH treatment | 30 (53%) | 2 (4%) | 32 (31%) |
| UPD 15 | 25 (45%) | 21 (45%) | 46 (45%) |
| Deletion | 31 (55%) | 26 (55%) | 57 (55%) |
| Age (mean) | 11.60 years (range 3.25–38 years) | 22.64 years (range 10–44 years) | 16.64 years |
| Female | 27 (48%) | 27 (57%) | 54 (52%) |
| Male | 29 (52%) | 20 (43%) | 49 (48%) |
| Children | 45 | 14 | 59 |
| Adults (>18 years) | 11 | 33 | 44 |
The demographics are from individuals in the UCI (56 individuals) and Vanderbilt (47 individuals) cohorts consisting of 103 individuals.
The Vanderbilt PWS cohort was older and studied over a 6-year period in the mid to late 1990s when growth hormone was used sparsely and only two participants in this cohort were treated. The Wechsler intelligence tests were utilized for cognitive measures (WISC-III for children up to 17 years and WAIS-R for those ≥17 years) (Reynolds, Wilson, & Clark, 1983; Wechsler, 1991). Forty-seven participants with PWS were recruited for the Vanderbilt PWS cohort with 20 males and 27 females, predominantly Caucasian (43 of 47 subjects). Their average age was 22.64 years ± 8.58 years and a minimum of 10.2 years with a maximum of44.4 years (median of 20.3 years). There were 11 individuals with the larger typical 15q11-q13 Type I deletion (average age 25.80 years ± 9.15 years); 15 with the smaller typical 15q11-q13 Type II deletion (average age = 18.77 years ± 5.91 years) and 21 individuals with UPD 15 (average age = 23.76 years ± 9.23 years) (see Table 1). Data were analyzed by one-way ANOVA and unpaired t tests with scatterplots generated, linear regression performed and trend lines developed. All data were continuous variables with normal distribution. Statistical significance was set at p-values of less than .05. The results were presented as mean ± standard deviation (SD).
2.2 |. Intelligence (IQ) testing for individuals with Prader–Willi syndrome
2.2.1 |. Stanford Binet intelligence testing for UCI cohort
The Stanford Binet test was used to measure the cognitive ability of individuals in the UCI cohort using standardized tools depending upon the age and extent of intellectual disability of the individual. The test requires approximately 5 min per subtest to administer with age range between 2 and 85 years. The Stanford Binet subtests include assessments of vocabulary, pattern analysis, quantitative, and bead memory. The test composite represents the sum of the subtests’ scores. The IQ score of the Stanford Binet test is divided into several categories of intelligence, which has been standardized by age (see Table 2). Composite scores allow the examiner to identify the intellect of the individual with mild or moderate intellectual disability by the age of 5 years. Verbal and nonverbal content are included in the test. This test is more animated than other intelligence tests with the use of colorful artwork, toys and manipulative skills. Depending on the condition of an individual such as communication disorders, deafness, or visual impairment, the nonverbal or verbal tests may be opted out (Delaney & Hopkins, 1987).
TABLE 2.
Stanford Binet test composite IQ, PWS genetic subtypes and growth hormone status in UCI cohort
| GH therapy | Non-GH therapy | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| IQ score | Categories | Del | UPD | GH therapy | Non-GH therapy | UPD | Del | UPD | Del |
| ≥132 | Very superior | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 121–131 | Superior | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 111–120 | High average | 2 | 0 | 2 | 0 | 0 | 2 | 0 | 0 |
| 89–110 | Average | 8 | 5 | 7 | 6 | 5 | 2 | 0 | 6 |
| 79–88 | Low average | 4 | 8 | 7 | 5 | 4 | 3 | 4 | 1 |
| 68–78 | Slow learner | 9 | 7 | 9 | 7 | 5 | 4 | 2 | 5 |
| ≤67 | Mentally retarded (intellectual disability) | 8 | 5 | 5 | 8 | 2 | 3 | 3 | 5 |
2.2.2 |. Wechsler intelligence testing for Vanderbilt cohort
The Wechsler intelligence test consists of a child version (Wechsler Intelligence Scale for Children—WISC) administered between ages 6 and 16 years while the adult version (Wechsler Adult Intelligence Scale—WAIS) was used at 17 years and older. The tests are individually administered over a 60–90 min session. The individual tests consist of subtests organized into Verbal IQ, Performance IQ, and Full Scale IQ. The WAIS-R is a comprehensive test of cognitive ability in adults with a mean score of 100. It consists of six verbal and five performance subtests and is recommended for use in the clinical and research settings. The verbal IQ subtests are: information, comprehension, arithmetic, digit span, similarities, and vocabulary. The performance subtests are: picture arrangement, picture completion, block design, object assembly, and digit symbol. The WISC-III is a collection of 13 distinct subtests divided into two scales (verbal and performance) for children. The six verbal scale tests use language-based items while the seven performance scale items are visual-motor based and less dependent on language. The WISC-III is an appropriate instrument for practitioners and clinical researchers in assessing childhood intelligence. Both the WAIS-R and WISC-III tests were administered in the Vanderbilt cohort.
3 |. RESULTS
For the Stanford Binet intelligence test administered to the UCI PWS cohort, the vocabulary IQ subtest showed a significantly higher average score for individuals who had GH treatment (p = .02). The subtests of pattern analysis, quantitative, and bead memory, and the test composite score did not show significant differences in the results (p = .41, .37, .79, .53, respectively) (see Table 3). The average subtests and test composite scores between UPD 15 and deletion genetic subtypes were varied but not sufficiently significant although the vocabulary scores were on an average 10% higher in the UPD 15 group compared with deletion (see Table 4). The test composite scores were divided into five intelligence levels such as high average, average, low average, slow learner, and mentally retarded (intellectually disabled) (see Table 5).
TABLE 3.
Growth hormone (GH) treatment versus non-GH treatment and Stanford Binet test scores
| UCI cohort | Non-growth hormone | Growth hormone | |||
|---|---|---|---|---|---|
| Average | St Dev | Average | St Dev | p value | |
| Vocabulary | 37.52 | 10.66 | 44.03 | 8.94 | .02* |
| Pattern analysis | 42.96 | 9.81 | 41.00 | 7.86 | 41 |
| Quantitative | 40.67 | 8.73 | 42.68 | 7.21 | .37 |
| Bead memory | 39.37 | 9.19 | 40.00 | 8.58 | 79 |
| Test composite | 78.31 | 15.65 | 80.90 | 14.73 | .53 |
p < .05 (significant).;
This table shows the comparison between GH and non-GH treated individuals for the UCI cohort as only two individuals with PWS in the Vanderbilt cohort were treated with GH. The Vanderbilt cohort was not included in the analysis.
TABLE 4.
Prader–Willi syndrome UPD 15 versus deletion subtypes and IQ test scores
| UPD 15 (N = 25) | Deletion (N = 31) | ||||
|---|---|---|---|---|---|
| UCI cohort | Average number | Standard deviation | Average number | Standard deviation | p value |
| Vocabulary | 43.1 | 12.8 | 39.3 | 7.6 | .17 |
| Pattern analysis | 41.3 | 6.9 | 42.7 | 10.1 | 54 |
| Quantitative | 41.4 | 8.2 | 42.2 | 7.9 | .70 |
| Bead memory | 38.3 | 7.3 | 40.5 | 9.9 | 35 |
| Test composite | 80.3 | 14.0 | 79.4 | 16.3 | .84 |
This table shows the comparison between UPD 15 and deletion individuals with PWS from the UCI cohort.
There was no significant difference in statistical analysis.
TABLE 5.
Prader–Willi syndrome UPD 15 versus deletion subtypes and IQ test composition score by percentage in UCI cohort
| UPD 15 (N = 25) | Deletion (N = 31) | |||
|---|---|---|---|---|
| Test composition | Number | Percentage | Number | Percentage |
| High average (111–120) | 0 | 0.00 | 2 | 6.45 |
| Average (89–110) | 5 | 20.00 | 8 | 25.81 |
| Low average (79–88) | 8 | 32.00 | 4 | 12.90 |
| Slow learner (68–78) | 7 | 28.00 | 9 | 29.03 |
| Mentally retarded/intellectual disability (≤67) | 5 | 20.00 | 8 | 25.81 |
This table shows the percentage of each category of IQ test composition results comparing individuals with UPD 15 and deletion in the UCI cohort.
Individuals with GH treatment versus non-GH treatment in the UCI PWS cohort showed similar high average and average intelligence scores and the p-values showed no significance when comparing the two treatments with 27% of both the combined GH treated and non-GH treated groups scoring as high average or average intelligence. The number of individuals with scores in the mentally retarded (intellectually disabled) range as the lowest intelligence level was higher (nearly doubled) for non-GH treated subjects in comparison with GH treatment (30.8% and 16.7%, respectively) (see Table 6).
TABLE 6.
Growth hormone (GH) treatment versus non-GH treatment and IQ test composition score by percentage in UCI cohort
| Test composition | Non-growth hormone (N = 26) | Growth hormone (N = 30) | ||
|---|---|---|---|---|
| Number | Percentage | Number | Percentage | |
| High average (111–120) | 0 | 0.00 | 2 | 6.67 |
| Average (89–110) | 6 | 23.08 | 7 | 23.33 |
| Low average (79–88) | 5 | 19.23 | 7 | 23.33 |
| Slow learner (68–78) | 7 | 26.92 | 9 | 30.00 |
| Mentally retarded/intellectual disability (≤67) | 8 | 30.77 | 5 | 16.67 |
Comparing the percentage of the test composition results of Stanford Binet test. There is no significant difference; however, there are higher percentages of individuals who did not take GH classified as intellectually disabled.
Figure 1 shows scatterplot data and linear regression R2 values with trend lines for the PWS genetic subtypes (UPD 15 and 15q11-q13 deletion) from Stanford Binet vocabulary and test composite IQ scores generated from PWS subjects in the UCI cohort with or without growth hormone therapy compared with age (in years). Separate trend lines were observed for UPD 15 and 15q11-q13 deletion subjects compared with age, possibly indicating lower test composite IQ scores with advancing age in the non-GH treated deletion subjects (R2 = 0.3256) and UPD 15 subjects (R2 = 0.4341) but less so in those UPD 15 subjects treated with growth hormone (R2 = 0.0068) in the UCI cohort. This was and similarly seen for vocabulary IQ scores. An R2 value or coefficient of determination represents the variation in percentage of dependent variables that is explained by a linear model and larger R2 values indicate a better regression model that fits the observations under study.
FIGURE 1.
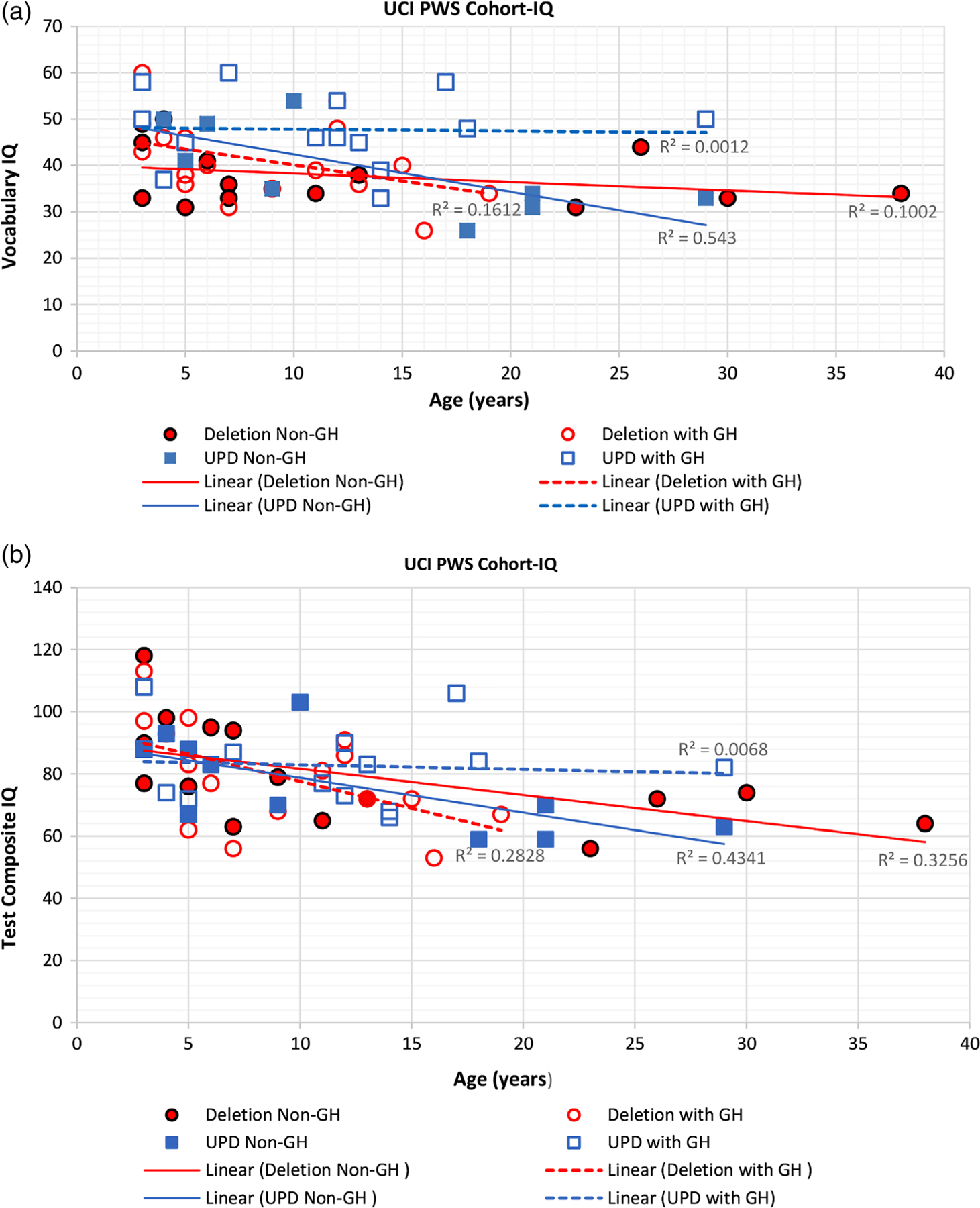
Shows scatterplots of data and linear regression (R2) values with trend lines for PWS genetic subtypes (UPD 15 and 15q11-q13 deletion) from Stanford Binet vocabulary IQ subscale and test composite (total) IQ scores generated from the UCI PWS cohort with and without growth hormone treatment compared with age. The trend lines for the non-GH treated PWS 15q11-q13 deletion and UPD 15 groups suggested a decrease with advanced age for both test composite and vocabulary IQ scores but those with UPD 15 and GH treatment did not show the same level of decline
In the Vanderbilt PWS cohort, intelligence was measured using the WISC-III instrument for 14 children from 10 up to 17 years. The remaining 33 individuals were 17 years or older and assessed using the WAIS-R instrument. The intelligence test data in comparison with the IQ test subscores and PWS genetic subtypes were statistically analyzed in this cohort. Significantly higher Verbal IQ scores (p = .002) were seen in the UPD 15 group compared with the combined typical deletion group (see Table 7). Figure 2 shows scatterplot data and R2 values with trend lines for the PWS genetic subtypes (UPD 15 and 15q11-q13 deletion) and Full Scale IQ, Performance IQ, and Verbal IQ scores and age (in years) for the Vanderbilt PWS cohort which indicated higher Verbal IQ scores in the UPD 15 group. Height, weight, and BMI data are shown in Table 8 from the Vanderbilt and UCI PWS cohorts, but no significant differences were found in relationship to PWS genetic subtypes. Additionally, no differences were seen in the number of males or females, adults or children and GH treatment or non-GH treatment in relationship to the PWS genetic subtype groups.
TABLE 7.
Wechsler IQ measures and Prader–Willi syndrome genetic subtypes in the Vanderbilt cohort
| Genetic subtype | Wechsler verbal IQ (average) | Wechsler performance IQ (average) | Wechsler full-scale IQ (average) |
|---|---|---|---|
| 15q11-q13 deletion type I (n = 11) | 62.91 ± 9.34 | 66.54 ± 10.79 | 63.45 ± 10.25 |
| 15q11-q13 deletion type II (n = 15) | 61.27 ± 9.76 | 65.47 ± 9.69 | 61.40 ± 10.15 |
| Maternal disomy 15 (n = 21) | 70.00 ± 6.20 | 62.95 ± 8.50 | 63.86 ± 7.26 |
| PWS deletion vs. maternal disomy 15; t test, p value | 0.002 | 0.21 | 0.42 |
Psychological Testing IQ (WISC-III; WAIS-R); (WISC-III, n = 14 children; WAIS-R, n = 33 adults).
FIGURE 2.
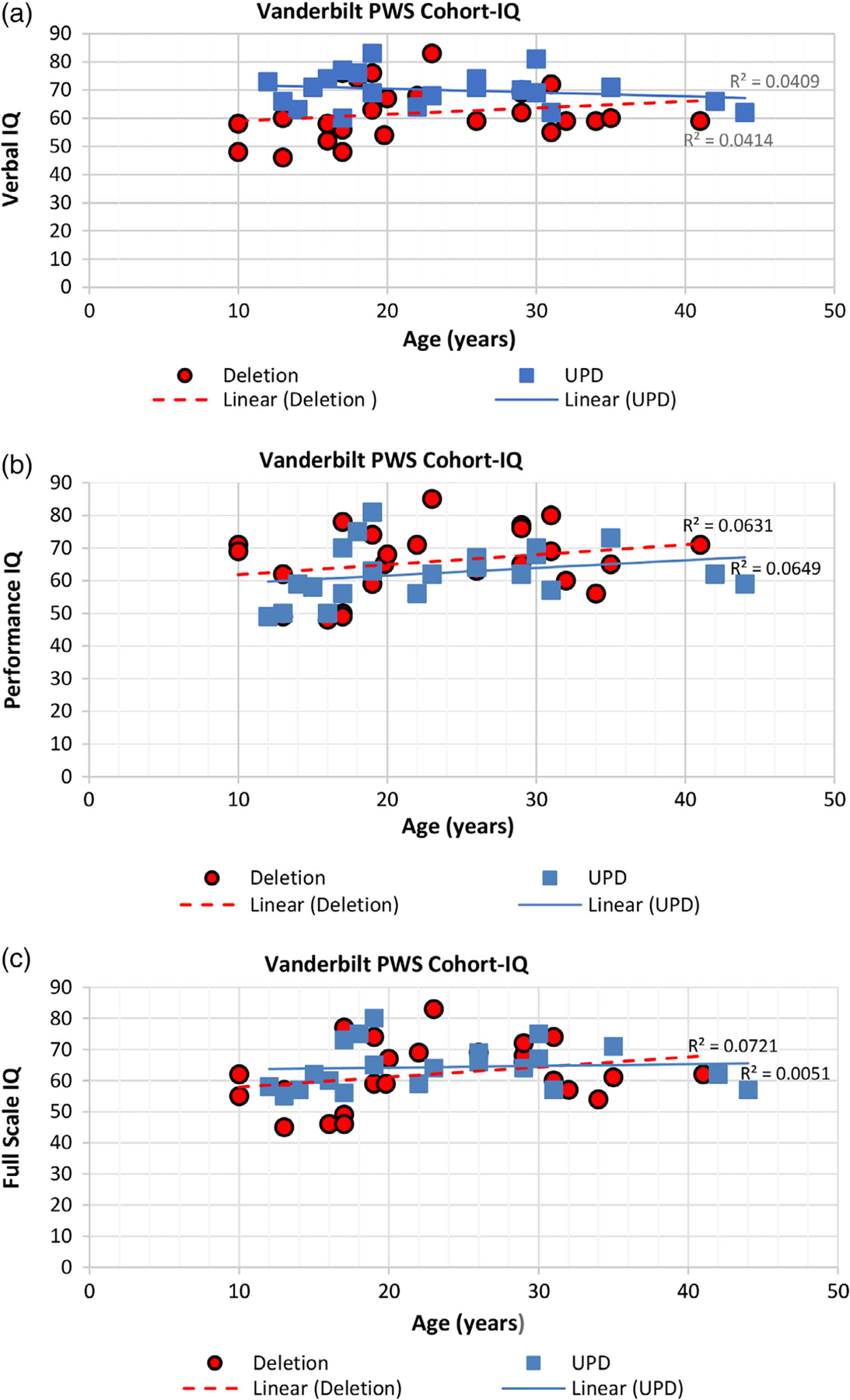
Shows scatterplots of data and linear regression (R2) values with trend lines for PWS genetic subtypes (UPD 15 and 15q11-q13 deletion) and full scale IQ, performance IQ and verbal IQ scores generated from the Wechsler intelligence test and compared with age from the Vanderbilt PWS cohort. Significant differences were seen in verbal IQ when comparing the genetic subtypes with the UPD 15 group having higher scores
TABLE 8.
Height, weight, and body mass index (BMI) in relationship with 15q11-q13 deletion or maternal disomy 15 in Vanderbilt and UCI cohorts of individuals with Prader–Willi syndrome
| Average height (cm) | Average weight (kg) | Average BMI (kg/M**2) | |
|---|---|---|---|
| Vanderbilt cohort (average age = 22.6 years) | |||
| 15q11-q13 deletion (n = 26) | 151.1 ± 8.0 | 76.84 ± 17.4 | 33.9 ± 8.2 |
| Maternal disomy 15 (n = 21) | 149.9 ± 8.3 | 75.9 ± 20.8 | 33.8 ± 9.1 |
| PWS deletion vs. maternal disomy 15; t test, p value | 0.67 | 0.88 | 0.94 |
| UCI cohort (average age = 11.6 years) | |||
| 15q11-q13 deletion (n = 31) | 128.7 ± 24.3 | 48.3 ± 28.6 | 26.6 ± 8.2 |
| Maternal disomy 15 (n = 25) | 131.8 ± 24.9 | 48.9 ± 26.7 | 23.7 ± 7.4 |
| PWS deletion vs. maternal disomy 15; t test, p value | 0.64 | 0.74 | 0.18 |
No significant differences were found in the number of males or females and GH treated or non-GH treated in relationship to the PWS genetic subtypes.
4 |. DISCUSSION
Prader–Willi syndrome (PWS) is a disorder in which symptoms are apparent in infancy and characterized by severe hypotonia, poor feeding, mild intellectual disability, behavior problems, hyperphagia and obesity with growth and other hormone deficiencies (Butler, 2016; Butler et al., 2006; Cassidy et al., 2012; Cassidy & Driscoll, 2009). By the age of 2 years, individuals can begin to gain weight and by 8 years of age, they can begin to exhibit hyperphagia leading to morbid obesity unless externally controlled by diet. Growth hormone treatment and exercise programs are often undertaken at this time but the importance of GH therapy on individual cognitive function and height with weight measures in relationship with PWS genetic subtypes has not been sufficiently studied.
We found that intelligence did not differ significantly between individuals with UPD 15 or deletion in the younger UCI PWS cohort using the Stanford Binet test. In contrast, statistically significant higher average Verbal scores were seen in the UPD 15 group in the older Vanderbilt PWS cohort using the Wechsler test. Also, those with 15q11-q13 type I deletions generally showed lower cognitive or adaptive behavior skills, whereas individuals with UPD 15 had better development of verbal than spatial skills.
In our study of 27 children with PWS treated with GH in the UCI cohort, the average (±SD) age for starting GH treatment was 7.5 ± 4.7 years with an average (±SD) duration of treatment of 2.5 ± 1.9 years. Their average test composite IQ score was 76.9 which fell in the slow learner category. There was a high possibility of bias due to some of the children having low cognitive skills and attention span deficits making type I deletions the most likely to gain from GH treatment.
We found a possible trend for raw IQ scores to be higher in individuals with GH treatment in the UCI cohort compared to non-GH treated individuals. There was a significant difference on the vocabulary subtest of the Stanford Binet test (p < .05) in the UCI cohort with higher levels in individuals on GH compared to the non-GH group, although differences did not reach statistical significance in the other subtests. There was a trend for lower vocabulary and test composite IQ scores with advancing age and potential stabilization with GH treatment particularly in the UPD 15 subjects. Hence, our data with GH treatment in the UCI PWS cohort, which is primarily pediatric-based, showed some evidence that GH treatment may lessen the decline in vocabulary and test composite IQ scores with age, particularly in the UPD 15 group, but more studies are needed.
Based on the literature and to further characterize GH effects, Oertel (2004) studied the impact of GH treatment on cognitive performance in 18 individuals with PWS in adulthood (21–63 years). Individuals were divided into two separate age groups when treated with GH or placebo for 6 months in a double-blind fashion with another 6 months of open label treatment. Cognitive performance such as attention, verbal memory and nonverbal intelligence were examined at baseline, 3, 6, 9, and 12 months. Speed of information processing was measured using a digit cancellation test (DCT) and a trail-making test (TMT). Verbal short-term memory (STM) and verbal long-term memory (LTM) were assessed using a text reproduction task. For the attention and memory test, a nonverbal intelligence test (Raven Standard Progressive Matrices) was used to assess the individuals’ intellect. After 3 and 6 months of closed label treatment, the GH treated group scored significantly higher on the DCT and TMT sections, while no differences were seen in the placebo group. After 3 and 6 months of GH treatment, an increase in an individuals’ short-term memory capacity was noted, although there was no significant improvement in long-term verbal memory and nonverbal intelligence compared to the baseline visit (Oertel, 2004). During the open label period, both PWS groups had significant results in the DCT, verbal LTM and STM scores with improved attention. Their study concluded that long-term GH intake would have benefit on the attention performance of individuals with GH deficiency and PWS. Similarly, in our UCI PWS cohort GH treatment appeared to increase verbal capacity. However, no significant results were seen for nonverbal intelligence measures for both the study reported by Oertel (2004) or in our UCI PWS cohort. Lo, Festen, Tummers-de Lind van Wijngaarden, Collin, and Hokken-Koelega (2015) also conducted a randomized control trial on 42 infants and 33 prepubertal children with PWS who were GH treatment naïve at the beginning of the study. A lower intelligence was correlated with more developmental delay in adaptive functioning with regards to communication, daily living skills, and socialization. After 7 years of GH treatment, a higher IQ was associated with less delay in adaptive functioning with the current level of IQ compared to baseline IQ. A study with a larger population focused on this issue would be essential for further understanding.
In a study by Hoybye, Thoren, and Bohm (2005), 19 individuals with PWS with a median age of 25 years were double blind treated and randomized as either with GH or placebo for the first 6 months and then followed by a 12-month open-label treatment period in all. Neuropsychological tests were performed at the baseline visit and after 18 months. Cognitive ability was assessed for verbal comprehension, perceptual organization, executive function, and mental speed. They found significant results on the subtests for Bender, Draw-A-Man, and Reaction Right. The Bender gestalt subtest assessed the individual’s visual perception and fine motor function in organizing materials. The Draw-A-Man subtest allowed individuals to draw a person with optional sex and other characteristics while the Reaction Right was used to measure mental speed with simple reaction-time measurement and auditory stimuli, while Finger Tapping measured an individual’s motor coordination (Hoybye et al., 2005). After 18 months, there were more significant improvements in the patients with PWS who had GH treatment; specifically, on the Block Design, Reaction Right, Left and Tapping right subtests. Block Design used WISC-III tested individuals on their perceptual organization. The study concluded that individuals with GH treatment had better mental speed, flexibility and motor performance.
Based on previous studies, starting GH treatment at an early age will maximize the benefits of treatment. Dykens et al. (2017) revealed a similar correlation with children who began GH treatment at <1 year of age having significantly higher nonverbal scores when compared to older age groups. The infant group had higher composite scores than children beginning GH treatment in the older age groups.
The lack of longitudinal or follow up data for analysis were limitations in our study. The UCI cohort was also younger (mean age 16.6 years) compared with the Vanderbilt cohort (mean age 27.6 years). Some individuals were not included for analysis and were young (<4 years old) or had severe learning disability and poor academic performance. English as a second language and level of education could also be limiting factors. The administered IQ tests did differ between the two cohorts but both test instruments are standardized and classically used for these types of cognitive measures and assessments in a wide range of studies, disorders and ages.
Our observations will require more investigation with larger number of subjects and length of duration of growth hormone treatment and age of onset to confirm whether GH treatment impacts IQ influenced by PWS genetic subtype or by advancing age. The authors encourage more research and further investigations with larger cohorts with Prader–Willi syndrome, a rare obesity-related genetic disorder.
5 |. CONCLUSION
Although there was no correlation between total composite and most subtest IQ scores between the 56 GH treated and untreated individuals with PWS in the UCI cohort, which had an average age of 11.6 years, there was a difference in the vocabulary subtest with a higher score in the GH treated group. The literature on PWS using follow up data on GH treatment (e.g., 4-year duration) indicates that GH treatment may prevent deterioration of cognitive skills particularly as noted in our UPD 15 subjects and historically improved abstract verbal reasoning in children with PWS. Possible improvement in IQ scores may occur with longer duration of GH treatment and by starting at an earlier age than 7 years as was seen in our UCI PWS cohort, which is supported by studies reported by Dykens et al. (2017). Their children with UPD 15 had lower visuospatial skills, but had larger improvements in skills within 4 years after GH treatment. Although no significant difference was found in total IQ scores in individuals with UPD 15 or deletion in our UCI cohort, individuals with PWS having the deletion began with better scores on the Block Design subtest compared to individuals with UPD 15. Individuals with a deletion also had better performance IQ scores. However, regression analysis and trend lines showed that GH treatment in the pediatric-based UCI PWS cohort showed less decline with age in test composite and vocabulary IQ scores in the UPD 15 group treated with GH compared with non-GH treatment. Differences were also found in the non-GH treated Vanderbilt PWS cohort with significantly higher Verbal IQ scores in the UPD 15 versus deletion subject groups but no difference in those with the typical 15q11-q13 deletion subtypes. The Vanderbilt cohort was also recruited during an earlier time period, had a higher average age (22.6 years) and subjects were not treated with GH, while the UCI cohort was generally younger (average age of 11.6 years) and the majority were treated with GH. Additional studies are needed to support the relationship between GH treatment and intelligence in PWS. Length and onset of GH treatment with its impact on IQ over time influenced by PWS genetic subtypes (UPD 15 vs. deletion) will require more investigation.
ACKNOWLEDGMENTS
We thank the individuals, families and their health care providers for participation in the study. We thank Charlotte Weber for preparation of the manuscript and Dr. Waheeda Hossain for statistical analysis. The study was supported by March of Dimes Foundation/Birth Defects Foundation grant (1999–2003), UC Irvine Institute for Clinical and Translational Science (ICTS) and Undergraduate Research Opportunity Program (UROP), National Institute of Child Health and Human Development (NICHD) grant PO1 HD30329 and Smith Intellectual and Developmental Disabilities Center grant NIH U54 HD090216 at University of Kansas Medical Center. We are thankful for the invaluable help from the staff at the University of California, Irvine, Case Western Reserve University and Vanderbilt University.
Footnotes
CONFLICT OF INTEREST
None.
REFERENCES
- Butler MG (1990). Prader–Willi syndrome: Current understanding of cause and diagnosis. American Journal of Medical Genetics, 35, 319–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler MG (2016). Single gene and syndromic causes of obesity: Illustrative examples. Progress in Molecular Biology and Translational Science, 140, 1–45. 10.1016/bs.pmbts.2015.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler MG, Bittel DC, Kibiryeva N, Talebizadeh Z, & Thompson T (2004). Behavioral differences among subjects with Prader–Willi syndrome and type I or type II deletion and maternal disomy. Pediatrics, 113, 565–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler MG, Hartin SN, Hossain WA, Manzardo AM, Kimonis V, Tamura R, … Driscoll DJ (2019). Molecular genetic classification in Prader–Willi syndrome: A multisite cohort study. Journal of Medical Genetics, 56(3), 149–153. 10.1136/jmedgenet-2018-105301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler MG, Lee PDK, & Whitman BY (2006). Management of Prader–Willi syndrome (3rd ed., pp. 1–550). New York, NY: Springer-Verlag. [Google Scholar]
- Carrel AL, Myers SB, Whitman BY, Eickhoff J, & Allen DB (2010). Long-term growth hormone therapy changes the natural history of body composition and motor function in children with Prader–Willi syndrome. Journal of Clinical Endocrinology & Metabolism, 95, 1131–1136. 10.1210/jc.2009-1389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassidy SB, & Driscoll DJ (2009). Prader–Willi syndrome. European Journal of Human Genetics, 17, 3–13. 10.1038/ejhg.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassidy SB, Schwartz S, Miller JL, & Driscoll DJ (2012). Prader–Willi syndrome. Genetics in Medicine, 14, 10–26. 10.1038/gim.0b013e31822bead0 [DOI] [PubMed] [Google Scholar]
- Delaney EA, & Hopkins TF (1987). The Stanford-Binet intelligence scale, fourth edition: Examiner’s handbook. Chicago, IL: Riverside Pub. [Google Scholar]
- Dykens EM, Roof E, & Hunt-Hawkins H (2017). Cognitive and adaptive advantages of growth hormone treatment in children with Prader–Willi syndrome. Journal of Child Psychology and Psychiatry, 58, 64–74. 10.1111/jcpp.12601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoybye C, Thoren M, & Bohm B (2005). Cognitive, emotional, physical and social effects of growth hormone treatment in adults with Prader–Willi syndrome. Journal of Intellectual Disability Research, 49, 245–252. [DOI] [PubMed] [Google Scholar]
- Jauregi J, Arias C, Vegas O, Alén F, Martinez S, Copet P, & Thuilleaux DA (2007). Neuropsychological assessment of frontal cognitive functions in Prader–Willi syndrome. Journal of Intellectual Disability Research, 51, 350–365. [DOI] [PubMed] [Google Scholar]
- Lo ST, Festen DA, Tummers-de Lind van Wijngaarden RF, Collin PJ, & Hokken-Koelega AC (2015). Beneficial effects of long-term growth hormone treatment on adaptive functioning in infants with Prader–Willi syndrome. American Journal on Intellectual and Developmental Disabilities, 120, 315–327. 10.1352/1944-7558-120.4.315 [DOI] [PubMed] [Google Scholar]
- Miller JL, Lynn CH, Driscoll DC, Goldstone AP, Gold JA, Kimonis V, … Driscoll DJ (2011). Nutritional phases in Prader–Willi syndrome. American Journal of Medical Genetics Part A, 155A, 1040–1049. 10.1002/ajmg.a.33951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oertel H (2004). The effect of growth hormone substitution on cognitive performance in adult patients with hypopituitarism. Psychoneuroendocrinology, 29, 839–850. [DOI] [PubMed] [Google Scholar]
- Reynolds CR, Wilson VI, & Clark PL (1983). A four-test form of WAIS-R for clinical screening. Clinical Neuropsychology, 3, 111–116. [Google Scholar]
- Roof E, Stone W, MacLean W, Feurer ID, Thompson T, & Butler MG (2000). Intellectual characteristics of Prader–Willi syndrome: Comparison of genetic subtypes. Journal of Intellectual Disability Research, 44, 25–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siemensma EP, Tummers-de Lind Van Wijngaarden RF, Festen DA, Festen DA, Troeman ZC, van Alfen-van der Velden AA, … Hokken-Koelega AC (2012). Beneficial effects of growth hormone treatment on cognition in children with Prader–Willi syndrome: A randomized controlled trial and longitudinal study. Journal of Clinical Endocrinology &Metabolism, 97, 2307–2314. 10.1210/jc.2012-1182 [DOI] [PubMed] [Google Scholar]
- Wechsler D (1991). The Wechsler scale for children-third edition. San Antonia, TX: The Psychology Corporation. [Google Scholar]