

Abstract
The DNA binding protein AT-rich interacting domain 3a (ARID3a)2 is expressed in healthy human hematopoietic cord blood progenitors where its modulation influences myeloid versus B lineage development. ARID3a is also variably expressed in subsets of adult peripheral blood hematopoietic progenitors where the consequences of ARID3a expression are unknown. In B lymphocytes, Toll-like receptor (TLR)3 signaling induces ARID3a expression in association with Type I interferon inflammatory cytokines. We hypothesized that TLR ligand stimulation of peripheral blood hematopoietic progenitors would induce ARID3a expression resulting in interferon production, and potentially influencing lineage decisions. Our data revealed that the TLR9 agonist CpG induces ARID3a expression with interferon alpha synthesis in human hematopoietic progenitors. However, ARID3a expression was not associated with increased B lineage development. These results demonstrate the need for further experiments to better define how pathogen-associated responses influence hematopoiesis.
Keywords: Hematopoiesis, ARID3a, Innate immunity
1. Introduction
Both mouse and human hematopoietic stem progenitor cells (HSPCs) are responsive to external stimuli including TLR ligand ligation and these signals alter gene expression pathways and can alter hematopoiesis resulting in increased myeloid lineage development [1–3]. Understanding how these external pathogenic signals influence these progenitors is therefore an important area of investigation. We previously found that over-expression of the DNA-binding protein ARID3a (A + T rich interaction domain protein 3a) in human cord blood hematopoietic stem progenitor cells (HSPCs) led to diminished myeloid differentiation and enhanced B lineage development, while knockdown of ARID3a expression significantly reduced B lineage developmental capacity in culture [4]. Knockdown of ARID3a in cord blood hematopoietic stem cells increased RNA expression of several myeloid lineage-associated genes while decreasing genes associated with both lymphoid and erythroid lineage cells [4], implicating an association between ARID3a expression and genes associated with hematopoietic lineage decisions. These data also suggested that modulation of ARID3a levels in circulating adult human hematopoietic precursors might also influence their developmental potential.
While healthy human cord blood HSPCs revealed ARID3a frequencies as high as 74% [4], we found that ARID3a expression is variable and low in adult peripheral blood HSPCs [5]. We also discovered that TLR9 stimulation induces ARID3a expression in healthy B cells that do not normally express it where its expression is associated with production of the inflammatory Type I interferon, interferon alpha (IFNα) and inflammatory responses [6,7]. IFNα promoted developmental maturation of embryonic mouse hematopoietic stem cells (HSCs), and those responses were also associated with ARID3a expression [8]. Human hematopoietic progenitors can express multiple types of TLR receptors and responses vary depending on lineage development (reviewed in [9]). In addition, human HSPCs can respond to type I interferons although the results of several studies appear contradictory regarding their role in inducing cell cycle entry versus quiescence. Therefore, we asked if TLR ligation would increase ARID3a expression in adult peripheral blood-derived HSPCs, and if increased ARID3a expression in those cells was associated with developmental pathways and innate inflammatory responses in these cells.
2. Materials and methods
2.1. Progenitor cells
Peripheral blood mononuclear cells from twelve healthy female donors (ages 18–53) were isolated from heparinized peripheral blood with Ficoll-Paque Plus (GE Healthcare). Donors were recruited after informed consent as part of the Oklahoma Medical Research Foundation (OMRF) Rheumatology Center of Excellence under OMRF (IRB compliance #06–19) and OUHSC (IRB compliance 5946) Institutional Review Board approvals and in accordance with the Declaration of Helsinki. CD34+ hematopoietic progenitor cells were enriched from mononuclear cells using the EasySep Human CD34 Positive Selection Kit (StemCell Technologies). CD34-enriched cells were used immediately or cryopreserved in RPMI-based freezing medium containing 20% FCS and 5% DMSO at −80 °C until further use.
2.2. TLR stimulation
CD34+ cells were seeded into QBSF®60 (Quality Biological, Inc.) supplemented with 10% FBS (Atlanta Biologicals), 100U Penicillin/Streptomycin (Gibco), 10% human adipose stem cell (hASC) conditioned media (LaCell LLC), 10 ng/ml stem cell factor (Biolegend), 10 ng/ml granulocyte stimulating factor (R&D), 5 ng/ml FLT-3 ligand (R&D), and 5 ng/ml IL-7 (Biolegend), and wells were inoculated with one of the following TLR ligands: CpG (2 μg/ml), imiquimod (IMQ, 20 μg/ml), lipopolysaccharide (LPS, 2 μg/ml), poly I:C (pIC, 4 μg/ml), Pam3GSK4 (200 ng/ml), or B. anthracis peptidoglycan (BA PGN, 10 μg/ml), and cultured for six days. Concentrations were used based on published reports [1,10–13]. For analyses of ARID3a expression and IFNα production, CD34+ cells were first treated with lentivirus containing short hairpin RNA (shRNA) for ARID3a or an unrelated scrambled control shRNA, as previously described [4,14], using a multiplicity of infection of 1 with 6 μg/ml of polybrene for 48 hrs. Media controls also contained polybrene. The cells were then washed and seeded into CpG stimulation cultures as described above. Brefeldin A (BFA) was added to wells 4 h prior to harvest to allow visualization of intracellular IFNα. For vivo-morpholino treated cultures, an ARID3a-specific vivo-morpholino targeted against the ATG region of human ARID3a and purchased from GeneTools, LLC (Philomath, Oregon) [15,16], was added at 5 μM at the beginning of culture along with CpG, and cultures were allowed to develop for four weeks as described above. Harvested cells were fixed with 2% paraformaldehyde or Cytofix, permeabilized with 0.1% Tween-20 or Cytofix/Cytoperm and stained with anti-human IFNα antibody (Miltenyi) and goat anti-human ARID3a antibody [17], followed by rabbit anti-goat FITC secondary antibody (Invitrogen). Data were collected using an LSRII (BD Biogenics) with FACSDiva (BD Biosciences) software version 4.1, or Stratedigm S1200Ex and CellCapTure acquisition software, and were analyzed using FlowJo (Tree Star) software version 10.
2.3. In vitro cultures
Unstimulated and TLR-Stimulated CD34+ hematopoietic progenitors were plated at 10,000 cells/well for liquid, stromal cell-free B lineage cultures, or 1000 cells per sample in Methocult H4434 Classic (Stemcell Technologies), according to the manufacturer’s directions, as previously described [4,5,18,19]. For B lineage liquid culture, cells were grown in QBSF®60 (Quality Biological, Inc.) supplemented with 10% FBS (Atlanta Biologicals), 100U Penicillin/Streptomycin (Gibco), 10% hASC conditioned media (LaCell LLC), 10 ng/ml stem cell factor, 10 ng/ml granulocyte stimulating factor, 5 ng/ml FLT-3 ligand, and 5 ng/ml IL-7 (as above). Half of the media was replaced once a week. After 4 weeks, cells were harvested, counted and assessed by flow cytometry using CD34-PE, CD10-Pacific Blue, CD19-PE-Cy5 (BioLegend), CD33-APC (BD Biosciences), and goat anti-ARID3a with a FITC-conjugated anti-goat secondary. Methocult cultures were incubated for 14 days and colonies were analyzed visually for monocytic, granulocytic and erythrocytic lineage cell colonies and counted using a Nikon Eclipse TS100 inverted microscope. Methocult cultures were harvested with warm 1X PBS, washed twice with warm PBS and once with ice cold PBS and then assessed by flow cytometry for the following surface markers: CD34, CD33, CD14, CD16, and CD41 with intracellular ARID3a.
2.4. Data analyses
Data were statistically evaluated using the paired student T test or repeated-measures ANOVA to compare distribution of variables between pairs of groups. Statistical analyses were performed using Prism (Graphpad) software version 8.2. P values of < 0.05 were considered significant.
3. Results
3.1. ARID3a is induced by TLR stimulation
To determine if TLRs could induce ARID3a expression in HSPCs, adult peripheral blood HSPCs were stimulated with various TLR ligands for 6 days and assessed for numbers of ARID3a-expressing CD34+ progenitors by flow cytometry (Fig. 1A). TLR9 engagement (CpG) stimulated the highest levels of ARID3a expression (average 36%); although TLR4 (LPS), TLR3 (Poly I:C, pIC), and TLR7 (IMQ) ligands also induced ARID3a expression to a lesser extent (Fig. 1B). TLR 1/2 (Pam3GSK4) and TLR2 (BA PGN) ligands did not induce ARID3a expression above background levels (Fig. 1B). Numbers of ARID3a-expressing cells were gradually increased over the course of 6 days, as shown for CpG and LPS (Fig. 1C). Together, these data indicate that some, but not all, TLR agonists can induce ARID3a expression in adult HSPCs.
Fig. 1.
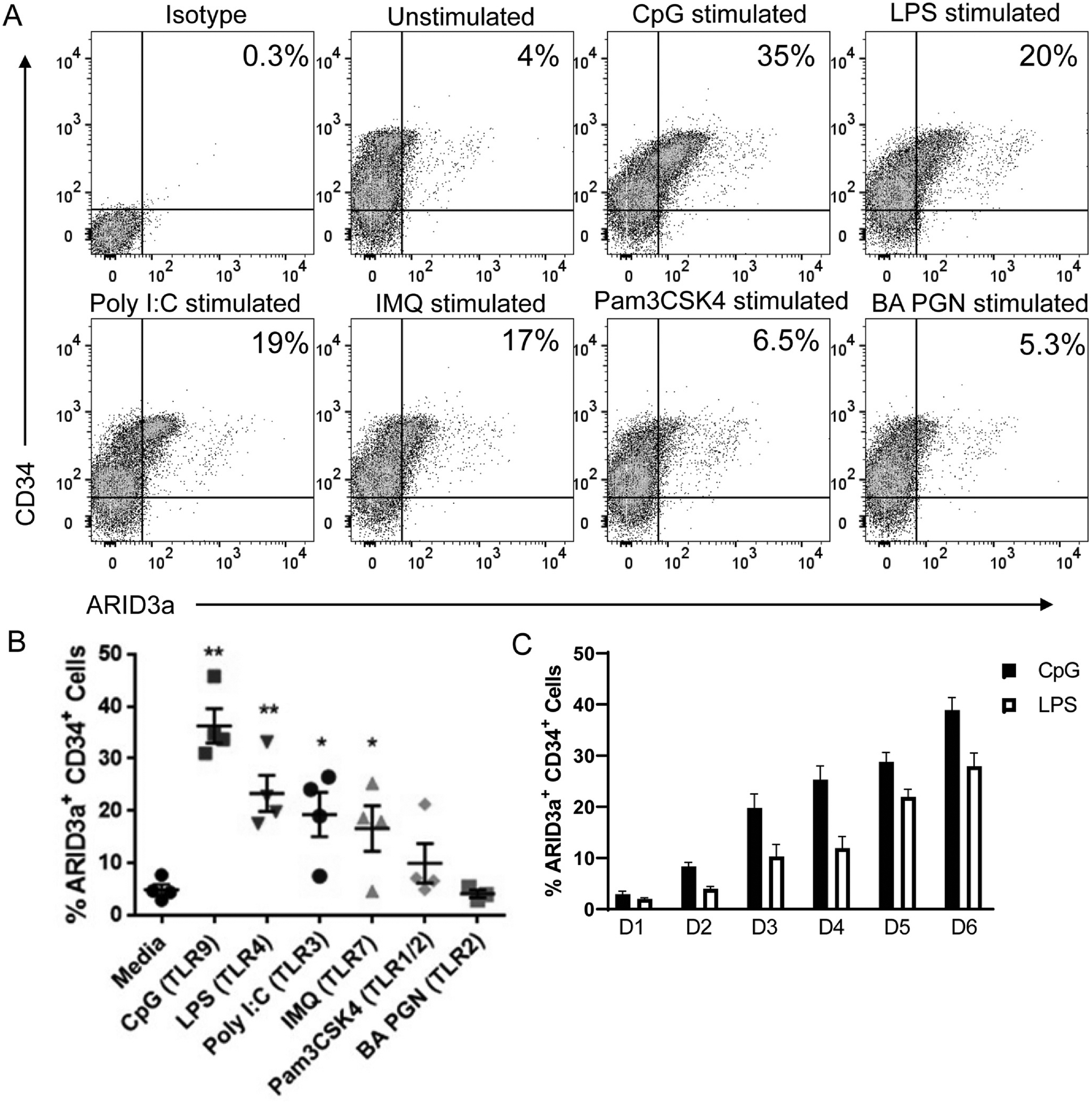
TLR ligands induce ARID3a expression in HSPCs. (A) HSPCs were stimulated with various TLR ligands for 6 days and analyzed by flow cytometry for ARID3a and CD34 expression. Representative plots are shown with ARID3a frequencies. (B) Cumulative data from 4 experiments are indicated. Each point represents averages of technical triplicates from a different individual. (C) Daily increases in ARID3a expression frequencies are shown after CpG or LPS stimulation over the six days, D1-D6, from 4 independent experiments. Averages, standard error bars and significance, assessed by paired t test, * p < 0.05; ** p < 0.01 are shown.
3.2. Pre-stimulation of HSPCs with TLRs increases myeloid lineage development in vitro.
We previously reported that ARID3a expression drives B lineage development in cord blood progenitors [4], but nothing is known regarding the potential developmental effects of ARID3a expression on adult hematopoietic progenitors. TLR stimuli typically induce differentiation along the myeloid pathway [1,3,20]. We therefore asked if TLR9 stimulation gave rise to increased numbers of B lymphocytes compared to stimulation with other TLR ligands in developmental assays. To assess consequences of TLR ligation on B lineage development, HSPCs were stimulated with each of the four ligands (CpG, LPS, pIC or IMQ) for six days and seeded into feeder free liquid cultures that allow B lineage development [4,5]. B lineage liquid cultures can also support development of a small proportion of myeloid cells, as we have previously shown [5]. Therefore, we assessed both the CD33 myeloid marker and the early stem cell progenitor marker, CD34, in these cultures. Reduced total cell numbers compared to media controls were observed in all TLR stimulated cultures, with imiquimod causing significant reductions (Fig. 2A). Gating for myeloid, and B cell subsets, as well as for CD34 are shown in Fig. 2B. Viabilities were similar in all cultures (unstimulated- 91%, CpG-77%, IMQ-70% and pIC 84%). While total B lineage cells were decreased after TLR stimulation with each of the TLR ligands (Fig. 2C), numbers of CD33+ myeloid cells were dramatically increased in TLR stimulated cultures compared to media controls (Fig. 2D). ARID3a was present in all B lineage cells (not shown), but expression of ARID3a in CD33+ cells was highly variable (Fig. 2E), but was present in TLR-ligand stimulated cultures at levels above that observed in media controls. Maintenance of the stem cell progenitor marker CD34 was significantly reduced in all TLR pre-stimulated cultures compared to unstimulated controls (Fig. 2E), suggesting TLR stimulation initiated differentiation of progenitors. Together, these data indicate that pre-stimulation with CpG prior to initiation of B lineage cultures failed to increase B lineage developmental potential.
Fig. 2.
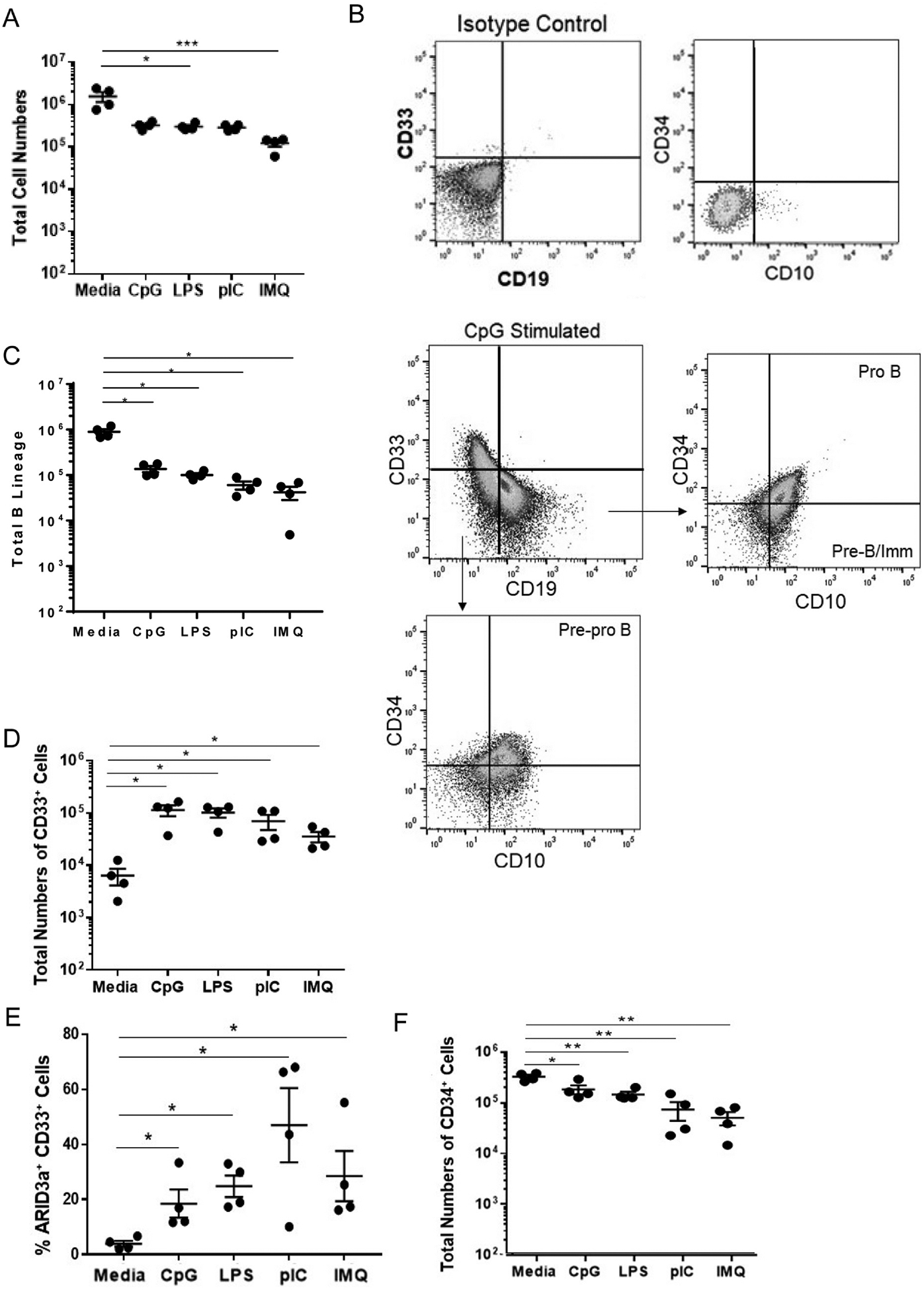
TLR treatment of HSPCs leads to increased myeloid versus B lineage cell production despite induction of ARID3a. Liquid B cell-supporting cultures were initiated in triplicate with 10,000 TLR-stimulated HSPCs from 4 donors and were assessed by flow cytometry for surface lineage markers after 4 weeks. (A) Total cell numbers from four experiments are presented. (B) A representative gating scheme for myeloid and B lineage cells is shown with gating for B lineage subsets. (C) Total numbers of CD33−CD19+ B lineage cells are shown. (D) Total numbers of CD33+ CD19− cells are shown with percentages of those cells expressing ARID3a (E). (F) Numbers of cells retaining CD34 in each culture are shown. Each point represents one sample. Means and standard error bars are shown. Significance was assessed by paired T test, *, p < 0.05; **, p < 0.01; ***, p = 0.0002.
To determine if TLR stimulation affected myeloid colony formation and differentiation into specific myeloid lineage precursors, TLR-stimulated HSPCs were seeded into myeloid lineage development cultures. Cultures were evaluated microscopically for development of erythroid (CFU-E), monocyte (CFU-M), granulocyte-monocyte (CFU-GM), mixed granulocyte-erythroid -megakaryocyte-monocyte (CFU-GEMM) and erythroid burst-forming unit (BFU-E) colonies by morphology and color two weeks later, as previously described [4,21]. Pre-stimulation of cells with CpG resulted in only marginally lower colony numbers, while stimulation with the other three TLR ligands resulted in significantly fewer average total colony numbers than the unstimulated media control (Fig. 3A). LPS-stimulation significantly inhibited monocyte colony development, while pIC treatment increased CFU-GEMM granulocytic colonies (Fig. 3B). Pretreatment with all four TLR ligands significantly inhibited development of erythroid colonies (Fig. 3B). Because we noted differences in colony sizes in some cultures, particularly the CFU-M colonies pre-treated with LPS, cultures were harvested and evaluated at the single cell level for myeloid, monocyte, granulocyte and megakaryocyte markers, in conjunction with ARID3a, by flow cytometry. Total numbers of myeloid cells did not significantly differ from the numbers present in the media controls for any pre-treatment stimulation with TLR ligands (Fig. 3C), despite the differences observed in total colony numbers observed for LPS stimulated cultures. Likewise, percentages of ARID3a-expressing myeloid cells, as shown for the total myeloid lineage cells, did not differ appreciably from media controls, but represented < 25% of the total myeloid cells (Fig. 3D). Similar observations were made in total cell numbers of monocytes, granulocytes and megakaryocytes (Fig. 3E, F G). These data indicate that TLR pre-treatment of HSPCs did not grossly affect total myeloid lineage cell numbers or grossly redirect maturation down a specific granulocytic pathway in these semisolid colony assays.
Fig. 3.
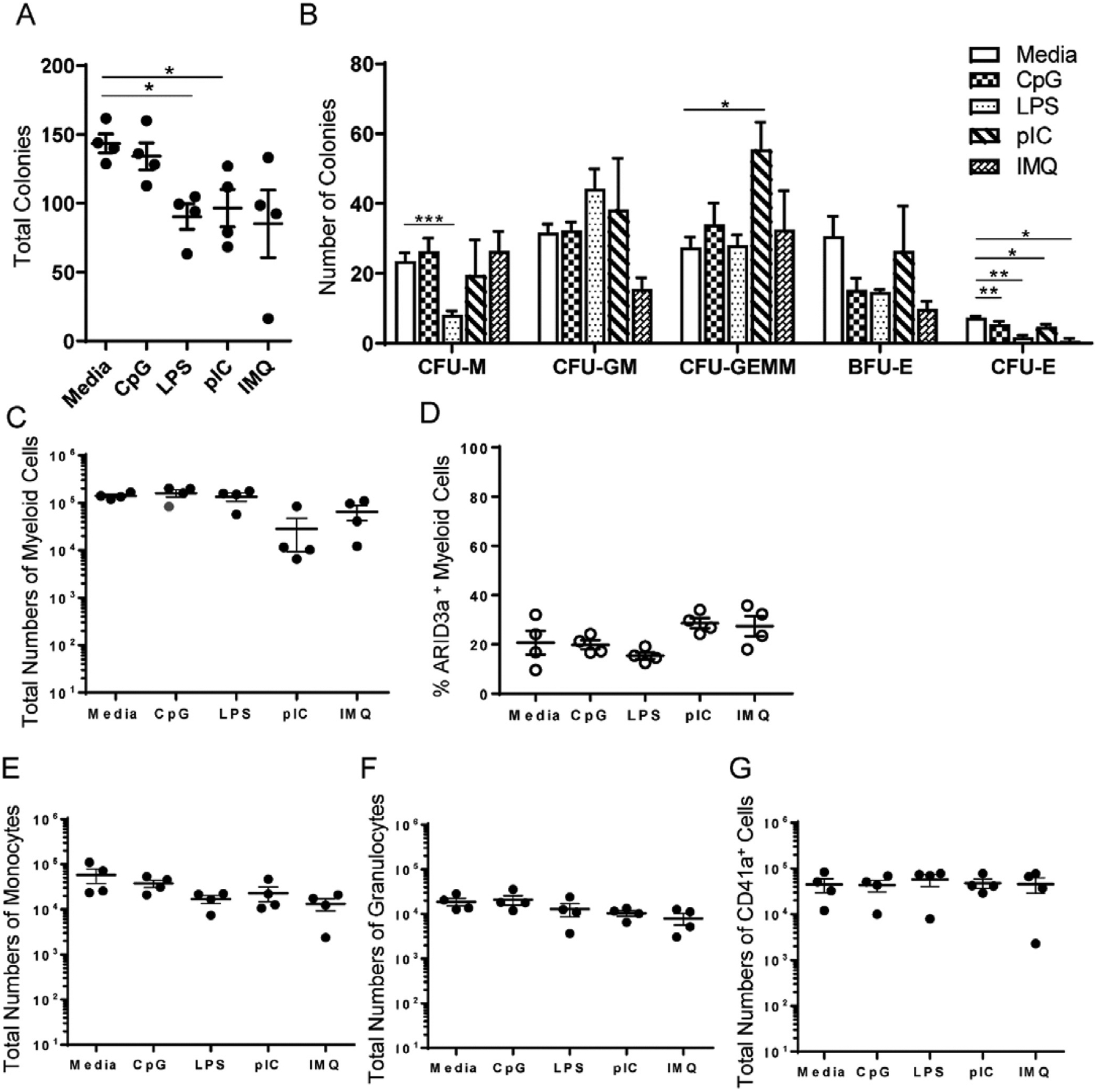
TLR-ligation of HSPCs did not grossly affect granulocyte maturation in vitro. HSPCs were stimulated with various TLR ligands for 6 days prior to seeding into semisolid methylcellulose media (1000 cells/well). (A)Total colony numbers are presented, (B) with numbers of erythroid burst forming units (BFU-E), erythroid colony forming units (CFU-E), monocyte colony forming units (CFU-M), granulocyte-monocyte colony forming units (CFU-GM), and granulocyte-erythrocyte-megakaryocyte-monocyte colony forming unit (CFU-GEMM). (C) Flow cytometric analyses of harvested colonies indicate total myeloid numbers, (D) frequencies of ARID3a+ myeloid cells, and numbers of (E) CD33+ CD14+ CD16+ monocytes, (F) CD33+ CD14− CD16+ granulocytes, and (G) CD41+ megakaryocytes. Means and standard errors are indicated. Significance was assessed by paired T test, *, p < 0.03, **, p < 0.005.
3.3. Inhibition of ARID3a expression in CpG-stimulated cells does not alter lymphoid versus myeloid developmental ratios.
To more directly evaluate the effect of ARID3a pre-stimulation on B cell development, we used an ARID3a-specific vivo morpholino to inhibit ARID3a expression at the beginning of cultures simultaneously with TLR stimulation. Vivo-morpholinos do not stimulate TLRs, but act to suppress protein translation by passively diffusing into cells [15]. Furthermore, use of morpholinos allows reversible inhibition of ARID3a so that B cell development can occur when it is washed out. CpG induced the highest ARID3a expression, so we focused on that agonist for the remainder of the studies. The ARID3a-targeting vivo-morpholino effectively inhibited ARID3a protein production in CpG stimulated cells by as much as 90% as shown by flow cytometry (Fig. 4A). After five days of CpG induction with and without ARID3a specific morpholinos, total numbers of ARID3a-expressing precursors from 4 individuals were significantly reduced (Fig. 4B). Evaluation of relative numbers of lymphoid lineage versus myeloid lineage cultures revealed that both the morpholino and CpG treated cells showed reduced numbers of lymphoid cells and slightly increased numbers of myeloid cells after only five days of stimulation (Fig. 4C and D). In addition, cultures that received both anti-ARID3a morpholinos and CpG revealed similar numbers of lymphoid and myeloid progenitors that did not differ significantly from either the CpG-treated or morpholino control treated cultures. Together, these data suggest that development trends toward myeloid development occur by five days of culture. Furthermore, inhibition of ARID3a at culture initiation also resulted in reduced lymphoid cells and increased myeloid lineage development.
Fig. 4.
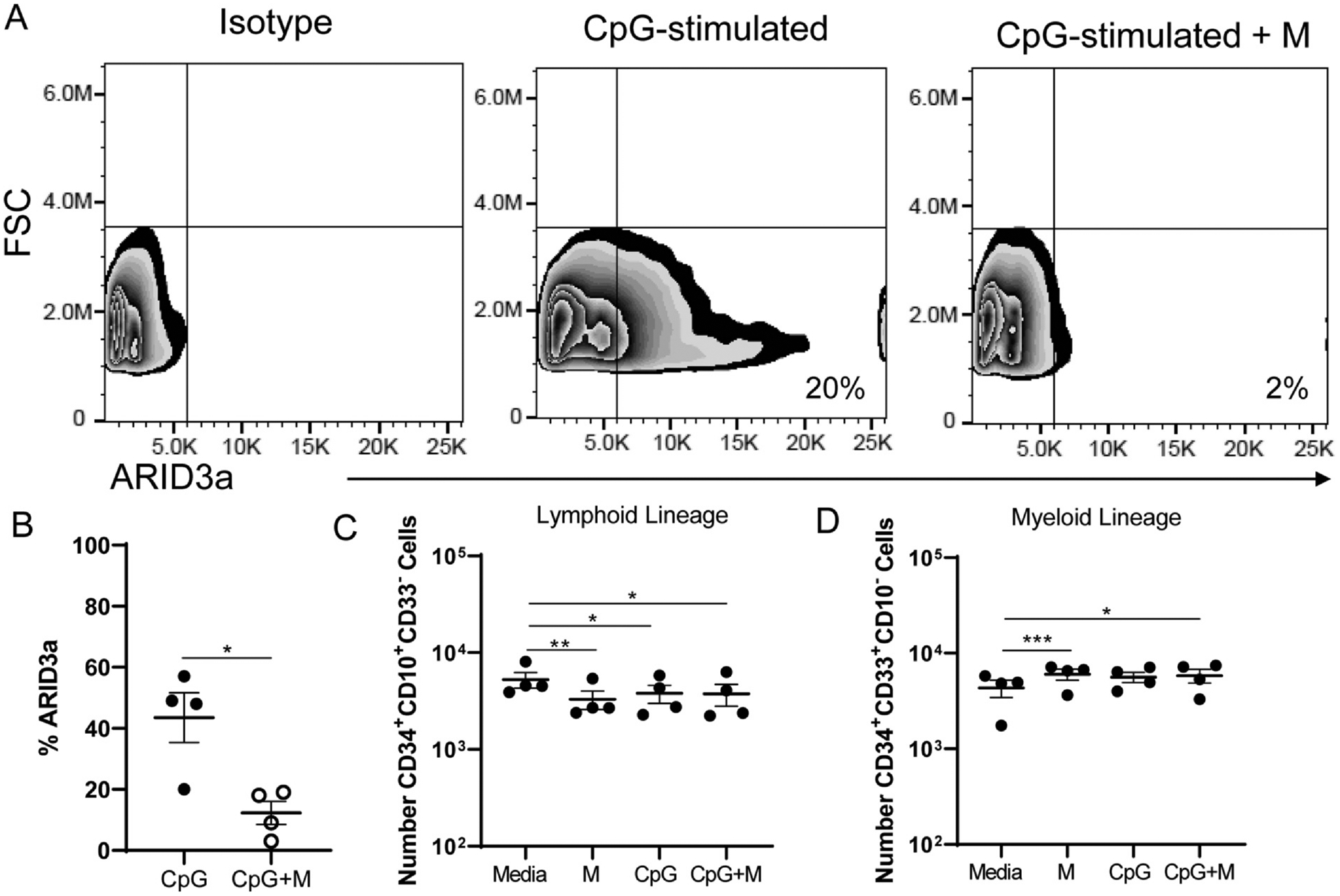
Short-term loss of ARID3a expression impairs B lineage development but does not alter CpG-related lineage commitment in liquid cultures. HSPCs were treated as in Fig. 2 in the presence or absence of an ARID3a-specific morpholino during CpG stimulation and assessed for B lineage and myeloid lineage cell generation by flow cytometric analyses after 5 days of culture. (A) Representative flow cytometry data from one sample treated with CpG with and without ARID3a inhibiting morpholinos are shown. (B) Cumulative data show reductions in ARID3a expression frequency in CpG and morpholino-treated cultures. Total numbers of lymphoid lineage cells (C) and myeloid lineage cells (D) are shown. Each point represents one sample. Means and standard error bars are shown. Significance was assessed by paired T test, *, p < 0.014, **, p = 0.0054, ***, p = 0.0003.
After four weeks of B lineage culture allowing expansion of those progenitors, similar effects were observed. Both morpholino-treated and CpG treated cultures, as well as cultures treated with both CpG and inhibited for ARID3a expression gave rise to reduced numbers of B lineage cells (Fig. 5A) and increased numbers of myeloid lineage cells (Fig. 5B). Numbers of CD34-expressing progenitor cells were similar among all four culture conditions (Fig. 5C). Indeed, examination of B lymphoid progenitor subsets suggested that reduction in B lineage cell production occurred as early as the pre-pro-B cell and pro-B cell stages (Fig. 5D and E), but became less apparent at the pre-B to immature B cell stages (Fig. 5F). However, these effects were small, and are best explained by failure of CpG and ARID3a-inhibition, either singly or together, to allow expansion of B lineage cells versus myeloid cells when compared to media controls (Fig. 5G). These data suggest that stimulation of ARID3a by CpG was insufficient to increase B lineage development in these in vitro cultures consistent with data obtained in Fig. 1.
Fig. 5.
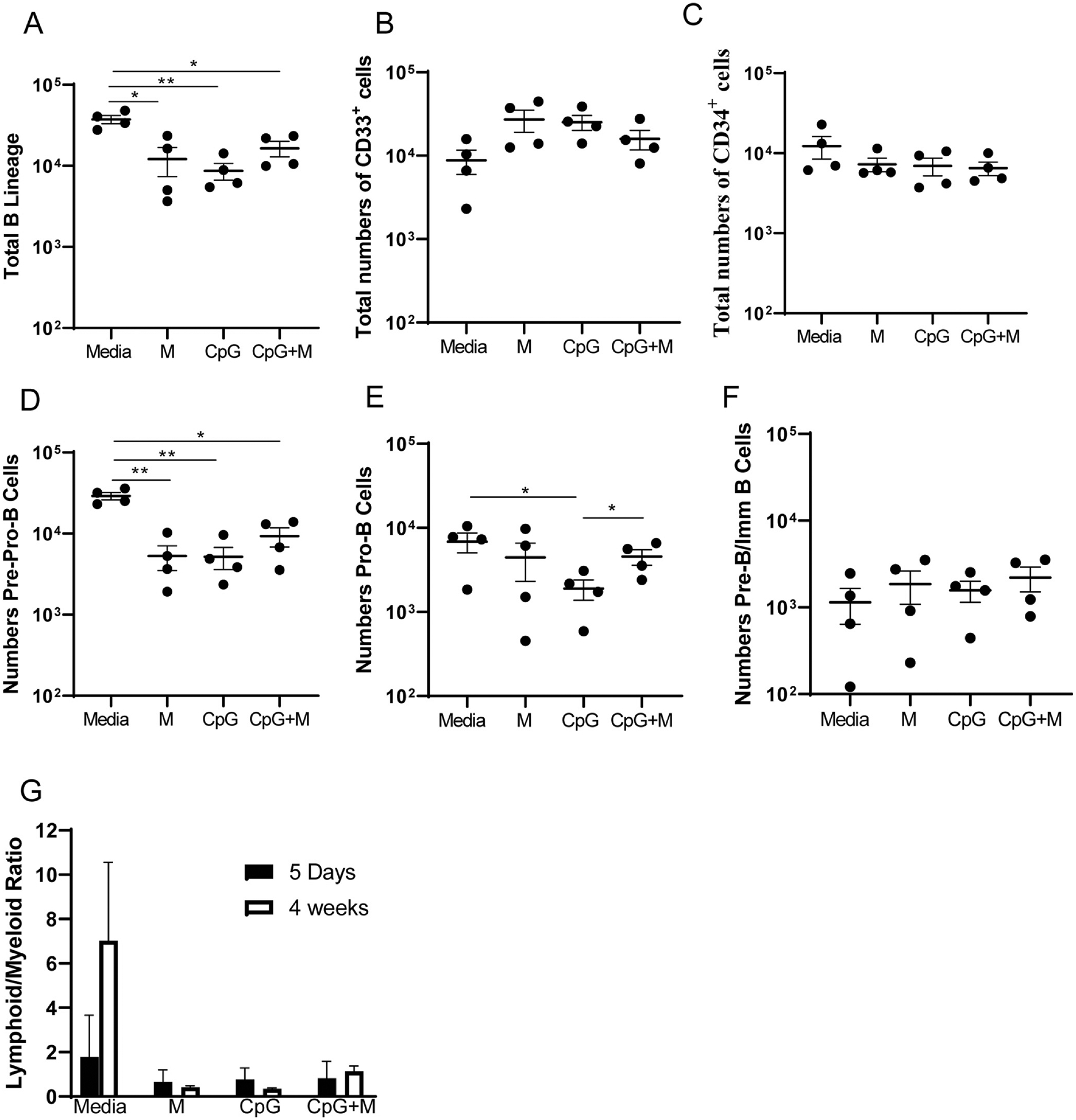
CpG stimulation and ARID3a-inhbition resulted in reduced expansion and development of B lineage cells. Cultures from Fig. 4 were plated in triplicate cultures and allowed to develop in B lineage expanding conditions for four weeks prior to harvesting for flow cytometric analyses. (A) Total numbers of B lineage (CD10 + CD19−CD33−, CD10+CD19+CD33−, CD10−CD19+CD33−), (B) myeloid lineage myeloid cells (CD33+CD19−CD10−), and (C) CD34-expressing cells were enumerated from media only, morpholino treated (M), CpG-treated, and ARID3a-inhibited CpG-treated cultures (CpG + M). (D) Numbers of pre-pro-B (CD34+ CD10+ CD19− CD33−), (E) pro-B (CD34+ CD33− CD10+ CD19), and (F) pre- and immature B cells (CD34− CD33− CD10+ CD19+) are shown for each treatment condition. (G) Ratios of total numbers of lymphoid versus myeloid cells were calculated for each individual culture and are plotted as averages with standard error bars for samples after five days of treatment, and after four weeks of B lineage expansion. Each point represents one sample. Means and standard error bars are shown. Significance was assessed by paired T test, *, p < 0.05, **, p < 0.0046.
3.4. IFNα protein expression in HSPCs is associated with ARID3a expression.
We next determined if CpG stimulation induced IFNα protein expression. ARID3a and IFNα protein levels in CpG-stimulated HSPCs were evaluated by flow cytometry. ARID3a and IFNα expression were co-expressed in the majority of cells (Fig. 6A). To determine if ARID3a is required for IFNα induction in response to CpG, fresh HSPCs were infected with lentivirus producing shRNA against ARID3a or an unrelated control shRNA and were then stimulated with CpG. ARID3a was efficiently knocked down in HSPCs, while there were no significant differences in ARID3a expression between the scramble control-treated samples and the media only samples (Fig. 6B). Cultures that were inhibited for ARID3a expression also showed striking decreases in expression of IFNα compared to both media and scramble shRNA controls (Fig. 6C). These data suggest that ARID3a is required for IFNα production in HSPCs, and support our previous results indicating strong associations of ARID3a expression with IFNα in multiple mature cell types in the peripheral blood [6,7,22].
Fig. 6.
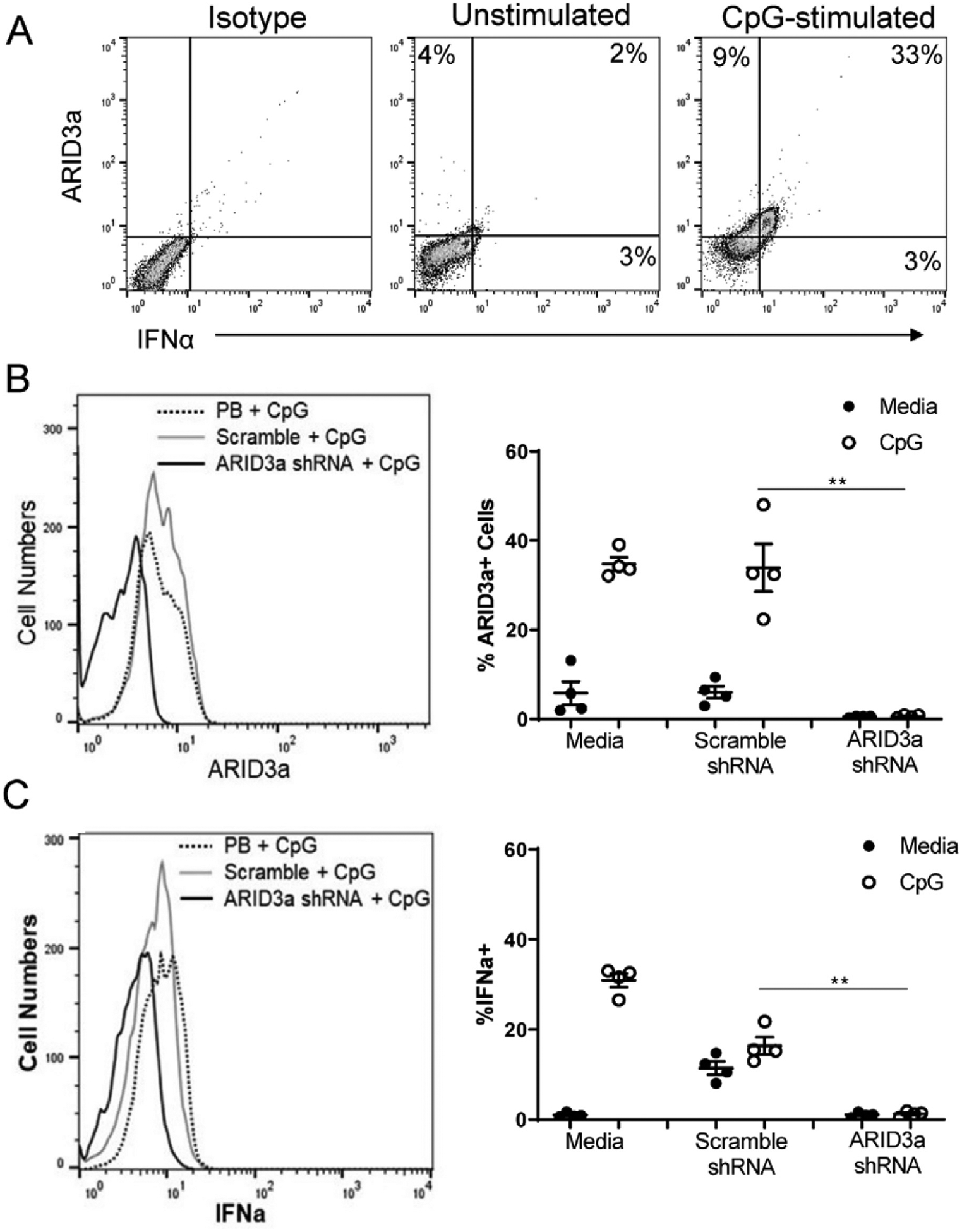
ARID3a is Required for CpG-Induced Expression of IFNα. HSPCs were stimulated with CpG for 6 days and assessed by flow cytometry for ARID3a and IFNα.(A) Representative flow cytometric plots of ARID3a and IFNα expression with and without CpG stimulation show percentages in each quadrant. (B) Cells were infected with ARID3a shRNA or unrelated scramble shRNA-expressing lentivirus prior to stimulation with CpG and analyzed by flow cytometry for ARID3a and IFNα expression. A representative histogram showing ARID3a inhibition (left panel). MFIs: CpG-9.74, Scramble-11.3, ARID3a shRNA- 3.78. Data from 4 samples were evaluated for numbers of ARID3a expressing cells (right panel). (C) Similar analyses as shown in B are presented for IFNa. MFIs: CpG-11.2, Scramble-9.37, ARID3a shRNA- 5.48. Open circles are cultures stimulated with CpG. Averages and error bars are shown. Significance was assessed by paired T test, *, p < 0.05; **, p < 0.01, ***, p < 0.001.
4. Discussion
Here we demonstrate that several TLR agonists, and particularly the TLR9 agonist CpG, induce expression of ARID3a in healthy adult HSPCs derived from peripheral blood. Our data indicate that despite associations of ARID3a expression with B lineage development in cord blood cells in vitro, that B lineage cells were not appreciably increased as the result of prior exposure to CpG in in vitro cultures. Nor did inhibition of ARID3a in CpG-stimulated cultures result in further increases in myeloid versus lymphoid cells. However, CpG stimulation of HSPCs induced the inflammatory cytokine, Type I interferon. Further, induction of IFNα in CpG-stimulated HSPCs is associated with ARID3a expression, suggesting ARID3a is necessary for IFNα expression in these cells, although it may not be sufficient.
Understanding how external stimuli in circulating hematopoietic progenitors may affect their developmental potential is important both for understanding how HSPCs are induced to differentiate in individuals exposed to pathogens, but also because CD34+ HSPCs are a source of blood cells used for bone marrow transplants. ARID3a induction by CpG in HSPCs was associated with increased inflammatory responses and IFNα production as we observed in peripheral blood B lymphocytes stimulated with CpG. ARID3a is also induced in human B cells in response to Epstein Barr Virus exposure where it was required for maintenance of the EBNA C promoter [23], further supporting a role for ARID3a expression in innate immune responses. LPS also increased ARID3a expression somewhat in HSPCs, suggesting ARID3a induction is not limited to viral triggers. Others reported that in vivo stimulation of mouse hematopoietic progenitors with LPS and pIC induced IFNα production [24], but ARID3a expression was not examined in those studies. While there are notable differences in cytokine induction and function between mice and humans [25], reports indicated that human natural killer cells and macrophages produce IFNα in response to LPS [26,27], suggesting that there could be IFNα induction in circulating human HSPCs following TLR4 engagement. Although a small percentage of circulating HSPCs in healthy adults already express ARID3a, it is unclear if this expression is the result of prior exposure to TLR or pathogenic signaling, or if ARID3a expression occurs as a reflection of developmental lineage decisions.
There are 13 distinct genes for IFNα (reviewed in [28,29]) with the subtypes binding IFN receptors at differing affinities [29]. Our anti-IFNα antibodies recognized the majority, but not all IFNα subtypes, perhaps explaining the lack of perfect associations of ARID3a with IFNα in Fig. 6. Other IFNα subtypes may also be induced in HSPCs without dependence on ARID3a expression. Type 1 interferons have been shown to activate quiescent HSCs in vivo [30]. Intriguingly, duration of type I IFN exposure on hematopoietic stem cells may differentially regulate quiescence. Acute exposure to type I IFNs in wild type mice was found to be pro-proliferative for HSC, but chronic exposure led to HSCs reentering quiescence and largely protecting those cells from the apoptotic effects of chronic IFN [31]. Others found that inflammatory cytokines reduced HSC self-renewal and increased myeloid differentiation [32]. Additional experiments will be required to assess the effects of IFN production on the cells in these cultures.
Healthy HSPCs represent a heterogeneous population of progenitors with the capacity to differentiate down multiple hematopoietic pathways and each of these progenitors, are capable of expressing ARID3a at varying levels [4]. The small numbers of HSPCs available in peripheral blood precluded the ability to better define lineage committed precursors via assessment of surface markers prior to their use in experiments. Lower expression of the progenitor cell marker CD34 was observed in TLR-treated HSPCs, with enhanced differentiation of those cells in comparison to untreated cultures. We previously demonstrated that both common lymphoid and common myeloid progenitors from SLE patients had increased numbers of ARID3a-expressing cells compared to healthy controls [5], revealing that early myeloid progenitors can express ARID3a, and raising the possibility that CD34+ CD33+ cells we observed may reflect such progenitors.
Although forced expression of ARID3a in total cord blood HSPCs lead to B lymphoid versus myeloid development [4] and ARID3a is necessary for B1 lineage development in mice [4,33,34], increasing ARID3a levels in circulating adult HSPCs via CpG stimulation prior to culture initiation did not increase B lineage development. Rather, all TLR stimuli resulted in increases in myeloid cell development, consistent with previous observations by others [1,3,20]. Reducing ARID3a expression at the initiation of cultures also reduced B lineage cells, as we expected, but did not further enhance myeloid versus B lineage development significantly compared to CpG-treated cultures. These data suggest that ARID3a expression alone is insufficient to skew peripheral blood progenitor development toward the B lineage. Rather, our data indicate that TLR9 engagement induces the upregulation of several genes associated with myeloid lineage development and the down regulation of genes associated with lymphoid lineage development and maintenance of stem cell quiescence. The majority of TLR stimulation studies performed to date have used mice instead of human HSPCs [20]; however, De Luca et al. showed TLR 1/2 stimulation of human cord blood led to myeloid development at the expense of lymphoid development [1]. TLR1/2 stimulation did not induce ARID3a and was not further investigated in our experiments. Others used bone marrow derived human HSPCs in liquid cultures lacking B lineage cytokines to study effects of TLR 7/8 ligand stimulation, and also observed enhanced myeloid development in those cultures [3]. Humanized mice engrafted with cord blood HSPCs and exposed to persistent low doses of LPS also resulted in diminished HSC and lymphoid progenitor cell numbers in favor of myeloid progenitors [35]. Our data further support previously observed effects of TLR stimulation on hematopoietic progenitors and underscore the need to further explore responses of individual precursor subsets to TLR signals.
TLRs, and particularly TLR9, the receptor for CpG, have been proposed to contribute to inflammatory processes in multiple autoimmune disorders [36–38]. We previously observed increased ARID3a expression in multiple peripheral blood cell types from SLE patients compared with healthy controls [22,39], including HSPCs [5]. Increased expression of ARID3a in B lymphocytes, plasmacytoid dendritic cells and low density neutrophils was associated with increased Type I IFN responses in SLE patient blood samples [6,7,22]. Although the external signals responsible for ARID3a induction in SLE HSPCs and other cells are unknown, TLR signaling may contribute to those responses. Chronic inflammation, including that associated with persistent TLR ligation, has been implicated in acquisition of mutations that lead to and maintain leukemic diseases [40,41], but associations with ARID3a expression in most of those diseases are unknown. Experiments to directly explore these possibilities would be of interest.
5. Conclusion
TLR signaling and inflammatory responses can influence steady-state hematopoiesis. Here we demonstrate that the TLR9 agonist CpG induces expression of the transcription factor ARID3a in healthy adult HSPCs, and that expression of ARID3a in those cells is associated with induction of IFNα and inflammatory responses. These data have important implications for understanding how inflammatory responses may influence hematopoietic progenitors used clinically.
Acknowledgements
The authors wish to thank Mr. M. D. Barron for critically reading the manuscript. We also thank the OMRF Flow Cytometry Core, the Flow Cytometry Core Facility at OUHSC, and the OMRF Arthritis & Clinical Immunology Oklahoma Rheumatic Disease Research Cores Center Clinical Characterization and Biorepository Core and Human Phenotyping Core and the Oklahoma Center for Translational Research clinical core for healthy donor recruitment and analysis support.
Funding
These studies were supported by the National Institutes of Health (grant numbers AI118836 and AI123951 (CFW), in part by K99AG055717 (MLR), and P30AR053483 and U54GM104938 (JAJ)).
Abbreviations:
- ARID3a
AT rich interacting domain 3a
- TLR
toll-like receptor
- HSPCs
hematopoietic stem and progenitor cells
- IFNα
interferon alpha
- HSCs
hematopoietic stem cells
- hASCs
human adipose stem cells
- IMQ
imiquimod
- LPS
lipopolysaccharide
- pIC
poly (I:C)
- BA PGN
Bacillus anthracis peptidoglycan
- BFA
brefeldin A
- BFU-E
erythroid burst forming unit
- CFU-E
erythroid colony forming unit
- CFU-GEMM
granulocyte-erythrocyte-monocyte-megakaryocyte colony forming unit
- CFU-GM
granulocyte-monocyte colony forming unit
- CFU-M
monocyte colony forming unit
Footnotes
ARID3a, AT-rich interacting domain 3a
TLR, Toll-like receptor
Reference
- [1].De Luca K, Frances-Duvert V, Asensio MJ, Ihsani R, Debien E, Taillardet M, Verhoeyen E, Bella C, Lantheaume S, Genestier L, Defrance T, The TLR1/2 agonist PAM(3)CSK(4) instructs commitment of human hematopoietic stem cells to a myeloid cell fate, Leukemia 23 (2009) 2063–2074. [DOI] [PubMed] [Google Scholar]
- [2].Megias J, Yanez A, Moriano S, O’Connor JE, Gozalbo D, Gil ML, Direct Toll-like receptor-mediated stimulation of hematopoietic stem and progenitor cells occurs in vivo and promotes differentiation toward macrophages, Stem Cells 30 (2012) 1486–1495. [DOI] [PubMed] [Google Scholar]
- [3].Sioud M, Floisand Y, Forfang L, Lund-Johansen F, Signaling through toll-like receptor 7/8 induces the differentiation of human bone marrow CD34+ progenitor cells along the myeloid lineage, J. Mol. Biol 364 (2006) 945–954. [DOI] [PubMed] [Google Scholar]
- [4].Ratliff ML, Mishra M, Frank MB, Guthridge JM, Webb CF, The transcription factor ARID3a is important for in vitro differentiation of human hematopoietic progenitors, J. Immunol 196 (2016) 614–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Ratliff ML, Ward JM, Merrill JT, James JA, Webb CF, Differential expression of the transcription factor ARID3a in lupus patient hematopoietic progenitor cells,J. Immunol 194 (2015) 940–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Ward JM, Ratliff ML, Dozmorov MG, Wiley G, Guthridge JM, Gaffney PM, James JA, Webb CF, Human effector B lymphocytes express ARID3a and secrete interferon alpha, J. Autoimmun 75 (2016) 130–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Ward JM, Ratliff ML, Dozmorov MG, Wiley G, Guthridge JM, Gaffney PM, James JA, Webb CF, Expression and methylation data from SLE patient and healthy control blood samples subdivided with respect to ARID3a levels, Data Brief 9 (2016) 213–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Kim PG, Canver MC, Rhee C, Ross SJ, Harriss JV, Tu HC, Orkin SH, Tucker HO, Daley GQ, Interferon-alpha signaling promotes embryonic HSC maturation, Blood (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Boettcher S, Manz MG, Regulation of Inflammation- and Infection-Driven Hematopoiesis, Trends Immunol. 38 (2017) 345–357. [DOI] [PubMed] [Google Scholar]
- [10].Liu J, Guo YM, Hirokawa M, Iwamoto K, Ubukawa K, Michishita Y, Fujishima N, Tagawa H, Takahashi N, Xiao W, Yamashita J, Ohteki T,Sawada K, A synthetic double-stranded RNA, poly I:C, induces a rapid apoptosis of human CD34(+) cells, Exp. Hematol 40 (2012) 330–341. [DOI] [PubMed] [Google Scholar]
- [11].Weichold FF, Zella D, Barabitskaja O, Maciejewski JP, Dunn DE, Sloand EM, Young NS, Neither human immunodeficiency virus-1 (HIV-1) nor HIV-2 infects most-primitive human hematopoietic stem cells as assessed in long-term bone marrow cultures, Blood 91 (1998) 907–915. [PubMed] [Google Scholar]
- [12].Kirshenbaum AS, Swindle E, Kulka M, Wu Y, Metcalfe DD, Effect of lipopolysaccharide (LPS) and peptidoglycan (PGN) on human mast cell numbers, cytokine production, and protease composition, BMC Immunol 9 (2008) 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Nagai Y, Garrett KP, Ohta S, Bahrun U, Kouro T, Akira S, Takatsu K, Kincade PW, Toll-like receptors on hematopoietic progenitor cells stimulate innate immune system replenishment, Immunity 24 (2006) 801–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].An G, Miner CA, Nixon JC, Kincade PW, Bryant J, Tucker PW, Webb CF, Loss of bright/ARID3a function promotes developmental plasticity, Stem Cells 28 (2010) 1560–1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Moulton JD, Using morpholinos to control gene expression, Curr. Protoc. Nucleic Acid Chem 68 (2017) 4 30 31–34 30 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Subbotina E, Koganti SR, Hodgson-Zingman DM, Zingman LV, Morpholino-driven gene editing: A new horizon for disease treatment and prevention, Clin. Pharmacol. Ther 99 (2016) 21–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Nixon JC, Rajaiya JB, Ayers N, Evetts S, Webb CF, The transcription factor, Bright, is not expressed in all human B lymphocyte subpopulations, Cell. Immunol 228 (2004) 42–53. [DOI] [PubMed] [Google Scholar]
- [18].Ichii M, Oritani K, Yokota T, Zhang Q, Garrett KP, Kanakura Y, Kincade PW, The density of CD10 corresponds to commitment and progression in the human B lymphoid lineage, PLoS ONE 5 (2010) e12954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Ichii M, Oritani K, Yokota T, Schultz DC, Holter JL, Kanakura Y, Kincade PW, Stromal cell-free conditions favorable for human B lymphopoiesis in culture, J. Immunol. Methods 359 (2010) 47–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Yanez A, Goodridge HS, Gozalbo D, Gil ML, TLRs control hematopoiesis during infection, Eur. J. Immunol 43 (2013) 2526–2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Pereira C, Clarke E, Damen J, Hematopoietic colony-forming cell assays, Methods Mol. Biol 407 (2007) 177–208. [DOI] [PubMed] [Google Scholar]
- [22].Ratliff ML, Garton J, Garman L, Barron MD, Georgescu C, White KA, Chakravarty E, Wren JD, Montgomery CG, James JA, Webb CF, ARID3a gene profiles are strongly associated with human interferon alpha production, J. Autoimmun 96 (2019) 158–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Borestrom C, Forsman A, Ruetschi U, Rymo L, E2F1, ARID3A/Bright and Oct-2 factors bind to the Epstein-Barr virus C promoter, EBNA1 and oriP, participating in long-distance promoter-enhancer interactions, J. Gen. Virol 93 (2012) 1065–1075. [DOI] [PubMed] [Google Scholar]
- [24].Zhang H, Rodriguez S, Wang L, Wang S, Serezani H, Kapur R, Cardoso AA, Carlesso N, Sepsis induces hematopoietic stem cell exhaustion and myelosuppression through distinct contributions of TRIF and MYD88, Stem Cell Rep. 6 (2016) 940–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Mestas J, Hughes CC, Of mice and not men: differences between mouse and human immunology, J. Immunol 172 (2004) 2731–2738. [DOI] [PubMed] [Google Scholar]
- [26].Lindemann RA, Roles of interferon and cellular adhesion molecules in bacterial activation of human natural killer cells, Infect. Immun 57 (1989) 1702–1706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Malcolm KC, Worthen GS, Lipopolysaccharide stimulates p38-dependent induction of antiviral genes in neutrophils independently of paracrine factors, J. Biol. Chem 278 (2003) 15693–15701. [DOI] [PubMed] [Google Scholar]
- [28].McNab F, Mayer-Barber K, Sher A, Wack A, O’Garra A, Type I interferons in infectious disease, Nat. Rev. Immunol 15 (2015) 87–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Gibbert K, Schlaak JF, Yang D, Dittmer U, IFN-alpha subtypes: distinct biological activities in anti-viral therapy, Br. J. Pharmacol 168 (2013) 1048–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Essers MA, Offner S, Blanco-Bose WE, Waibler Z, Kalinke U, Duchosal MA, Trumpp A, IFNalpha activates dormant haematopoietic stem cells in vivo, Nature 458 (2009) 904–908. [DOI] [PubMed] [Google Scholar]
- [31].Pietras EM, Lakshminarasimhan R, Techner JM, Fong S, Flach J, Binnewies M, Passegue E, Re-entry into quiescence protects hematopoietic stem cells from the killing effect of chronic exposure to type I interferons, J. Exp. Med 211 (2014) 245–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Schuettpelz LG, Link DC, Regulation of hematopoietic stem cell activity by inflammation, Front. Immunol 4 (2013) 204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Nixon JC, Ferrell S, Miner C, Oldham AL, Hochgeschwender U, Webb CF, Transgenic mice expressing dominant-negative Bright exhibit defects in B1 B cells, J. Immunol 181 (2008) 6913–6922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Hayakawa K, Li YS, Shinton SA, Bandi SR, Formica AM, Brill-Dashoff J, Hardy RR, Crucial role of increased Arid3a at the Pre-B and immature B cell stages for B1a cell generation, Front. Immunol 10 (2019) 457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Liu A, Wang Y, Ding Y, Baez I, Payne KJ, Borghesi L, Cutting edge: hematopoietic stem cell expansion and common lymphoid progenitor depletion require hematopoietic-derived, cell-autonomous TLR4 in a model of chronic endotoxin, J. Immunol 195 (2015) 2524–2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Farrugia M, Baron B, The role of toll-like receptors in autoimmune diseases through failure of the self-recognition mechanism, Int J Inflam 2017 (2017) 8391230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Devarapu SK, Anders HJ, Toll-like receptors in lupus nephritis, J. Biomed. Sci 25 (2018) 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Duffy L, O’Reilly SC, Toll-like receptors in the pathogenesis of autoimmune diseases: recent and emerging translational developments, Immunotargets Ther 5 (2016) 69–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Ward JM, Rose K, Montgomery C, Adrianto I, James JA, Merrill JT, Webb CF, Disease activity in systemic lupus erythematosus correlates with expression of the transcription factor AT-rich-interactive domain 3A, Arthritis Rheumatol. 66 (2014) 3404–3412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Camacho V, McClearn V, Patel S, Welner RS, Regulation of normal and leukemic stem cells through cytokine signaling and the microenvironment, Int. J. Hematol 105 (2017) 566–577. [DOI] [PubMed] [Google Scholar]
- [41].Manso BA, Zhang H, Mikkelson MG, Gwin KA, Secreto CR, Ding W, Parikh SA, Kay NE, Medina KL, Bone marrow hematopoietic dysfunction in untreated chronic lymphocytic leukemia patients, Leukemia 33 (2019) 638–652. [DOI] [PMC free article] [PubMed] [Google Scholar]